
GBA1 and The Immune System: A Potential Role in Parkinson’s Disease?
Abstract
It is clear that the immune system and inflammation have a role in Parkinson’s disease (PD), including sporadic PD and some genetic forms such as LRRK2-associated PD. One of the most important genes associated with PD is GBA1, as variants in this gene are found in 5–20% of PD patients in different populations worldwide. Biallelic variants in GBA1 may cause Gaucher disease, a lysosomal storage disorder with involvement of the immune system, and other lines of evidence link GBA1 to the immune system and inflammation. In this review, we discuss these different pieces of evidence and whether the interplay between GBA1 and the immune system may have a role in PD.
INTRODUCTION
The immune system and inflammation seem to have an important role in Parkinson’s disease (PD), with multiple lines of evidence from pathological, genetic, serological, and experimental studies [1]. From the genetic perspective, the association with specific human leukocyte antigen (HLA) types and amino acid changes [2, 3] and with immune-related genes including LRRK2 and BST1 [4, 5] supports a role for the immune system in the pathogenesis of PD.
GBA1 variants are among the most common and most important genetic risk factors of PD. Depending on the population, these variants are found in 5–20% of all PD patients [6] and are associated with a more severe, rapidly progressive phenotype [7]. Specifically, PD patients who carry GBA1 variants tend to have faster motor deterioration and cognitive decline compared to non-carriers, as well as higher frequencies of other non-motor symptoms such as hyposmia, rapid eye movement (REM)-sleep behavior disorder, hallucinations and others [7]. GBA1 encodes glucocerebrosidase (GCase), a lysosomal enzyme responsible for the degradation of glucocerebrosides and Glucosylsphingosines. The activity of GCase is reduced among individuals who carry GBA1 variants, and also in a subset of PD patients who do not have detectable GBA1 variants [8, 9], implying that GCase may also be important in some non-carriers of GBA1 variants with PD.
Gaucher disease (GD) is an autosomal recessive lysosomal storage disorder caused by biallelic GBA1 variants, with a very large spectrum of clinical presentations [10]. The association between GBA1 and PD was discovered after several clinical observations of GD patients who also developed PD [10–13]. Some patients may have very severe forms of GD with death in early childhood, whereas others may have very mild disease with almost no symptoms [14]. The variants in GBA1 can therefore be divided accordingly: “severe” variants are those associated with the severe forms of GD (type 2 and type 3), and “mild” GBA1 variants are those associated with the mild form of GD (type 1) [15]. This classification for severe and mild seems to be relevant for PD as well, since carriers of severe GBA1 variants may have higher risk for PD compared to mild variants carriers, have an earlier age at onset of PD [6], and faster cognitive decline [16, 17]. Some GBA1 variants, such as p.E326K and p.T369M, do not cause clinical GD, but are associated with PD [18, 19]. It is important to note that while GBA1 variants are common risk factors for PD, their penetrance (the probability to develop PD for someone who carries these variants) is low, between 10–30% in different studies [20–22]. In other words, most carriers of GBA1 variants will not develop PD, and the penetrance may be dependent on both genetic [23, 24] and other factors, some of which may be related to the immune system and inflammation.
One of the characteristics of GD is the presence of Gaucher cells: dysfunctional macrophages with accumulated glucocerebrosides [25]. These macrophages can accumulate in different tissues such as the spleen, liver, lung, and bone marrow. The infiltration of Gaucher cells affect the reticuloendothelial system of the respective organs and may lead to inflammation and organ dysfunction [25, 26]. Inflammation may have a key role in GD, possibly through complement activation [27] or other mechanisms, such as abnormality of cellular immunity [28], microglial activation and neuroinflammation in the neuronopathic forms of GD [29], among others.
The mechanism by which GBA1 variants increase the risk of PD is still unknown, although different hypotheses have been made, including lysosomal membrane dysfunction, the interaction between GCase substrates and α-synuclein, impaired mitochondrial function, endoplasmic reticulum stress, and impaired immune response [30]. However, the interplay between GBA1 variants, immune response, and their potential role in PD is still unclear. In this review, we summarize findings and discuss several hypotheses potentially linking GBA1, the immune system and PD.
GBA1 VARIANTS AND THE IMMUNE SYSTEM: LESSONS FROM GAUCHER’S DISEASE
GD is a rare, autosomal recessive lysosomal storage disease caused by biallelic variants in the GBA1 gene, leading to markedly reduced GCase activity and accumulation of glycosphingolipids such as glucosylceramide and glucosylsphingosine in macrophages and other cell types [29, 31] [32]. GD has a prevalence of about 1/100,000 individuals and is overrepresented in particular ethnicities, such as Ashkenazi Jews (prevalence of about 1/800 individuals) [33]. There are over 300 variants in GBA1 including point variants, deletions and insertions, and recombination variants with the nearby pseudogene [31]. GD can be classified into three types, depending on its clinical presentation: The most common form, GD type 1, has mainly peripheral signs and symptoms including splenomegaly, hepatomegaly, anemia, and acute bone pain in the pelvis and lower limbs and no apparent neuronal involvement. GD types 2 and 3, on the other hand, are characterized by severe neurological impairment, which can be acute (in type 2) or more chronic (in type 3), in addition to the typical GD symptoms seen in GD type 1 [34, 35]. However, the clinical spectrum within each type is highly variable, and it is possible to view GD more as a continuum of phenotypes rather than distinct subtypes [35].
The immune system and inflammation have an important role in the pathophysiology of all types of GD. The cells known as Gaucher cells are macrophages with extensive accumulation of glycosphingolipids, mainly glucosylceramide and glucosylsphingosine. GD patients, on top of the classical symptoms of GD, can develop a plethora of immune related symptoms including gammopathies, inflammation, and other types of immunological-related dysfunction [36]. While some of these immune-related phenomena, such as Gaucher cells, may only be relevant in GD but not in PD (where they are not typically present), it is possible that other immune-related impairments involved in GD may also have a role in PD. In this section, we will discuss several important aspects of the immune system in GD that may also be relevant in PD.
Cytokines have an important role in GD, with multiple lines of evidence from humans and different GD models showing increased pro-inflammatory cytokines [37]. For example, a study of 24 GD patients demonstrated elevated levels of different cytokines (e.g., IL-1β, IL-1 receptor antagonist, IL-6, and others) compared to control, also showing differences in cytokine levels between those who had GD type 3 compared to type 1 [38]. Whether differences in cytokine levels between severe and mild GD may explain some of the clinical differences seen in PD patients with severe versus mild GBA1 variants require additional studies. A similar study comparing 22 GD patients and 22 age- and sex-matched controls reported elevated levels of IL-6 and IL-10 in GD patients [39]. In induced macrophages stimulated with the bacterial endotoxin lipopolysaccharide (LPS), the levels of induction of the cytokine tumor necrosis factor (TNF) were significantly higher in cells induced from GD patients compared to controls [40]. Not only cytokines may be different between GD patients and healthy individuals, but also the counts of specific cell types. In a study of 18 children with GD type 1, the authors reported an increase in the number of total lymphocytes, and a specific increase in CD19+, CD3+, CD4+, and CD8+ cells [41]. In addition, another study reported a reduced number of monocytes, with decreased capacity for migration, comparing monocytes from 24 untreated GD patients and 18 controls [42]. These results and the higher levels of immunoglobulins also seen in GD patients suggest that chronic stimulation of the humoral immune system is an important characteristic of GD [43].
On top of peripheral immune response, there is accumulating evidence for neuroinflammation in GD, with a few reports on microglial activation in humans [44, 45] and in different animal models [29, 46]. Overall, the neuropathological data for GD in humans is limited, and a study reported microglial activation only in GD type 2, and its absence in GD type 3 [45]. In a neuropathological study on 14 subjects with GD, including seven with GD type 1, three with type 2 and four with type 3, no microgliosis was reported; however, analysis of microglia was not included in the study [47]. Since most of the evidence of microglial activation in GD comes from animal models, it remains to be determined if the same phenomenon is common in GD in humans, especially in GD type 1. However, in the latter study, marked astrogliosis was reported in almost all patients [47]. Astrogliosis on its own can be an important modulator of neuroinflammation [48], although this can be a reaction to neurodegeneration [49] rather than a primary part of the pathogenic process. Nevertheless, even if neuroinflammation is reactive to a neurodegenerative process, i.e., it is an outcome of neurodegeneration rather than a primary cause, it can still contribute to the neurodegenerative process as a secondary factor.
A recent, interesting imaging study used 11C-(R)-PK11195 BPND positron emission tomography (PET) scan to determine whether there is increased neuroinflammation and microglial activation in GBA1 variant carriers. Since PK11195 is a transporter protein (TSPO) ligand, and since TSPO is increased in both microglia and astrocytes during neuroinflammation, increased binding of this ligand is a marker of neuroinflammation in general, rather than only of microgliosis. In this study, the authors compared nine GBA variant carriers (five biallelic carriers and four heterozygous carriers) who do not have PD and 29 age-matched controls and reported increased neuroinflammation in brain areas that are susceptible for α-synuclein accumulation [50]. One of the interesting aspects of this observation, is that the biallelic carriers of GBA1 variants all had at least one mild variant (i.e., they would have GD type 1), and that the increased neuroinflammation was observed also in the heterozygous GBA1 variant carriers. Since at the time of the study none of the GBA1 variant carriers had PD, the results might imply that neuroinflammation or increased microglial activation is a general feature of carrying GBA1 variants. Whether or not these features have a role in developing PD in GBA1 variant carriers is yet to be determined, and since this study was small and without replication, additional studies are required to confirm this observation.
INFLAMMATION MARKERS AND CYTOKINES IN GBA1-ASSOCIATED PARKINSON’S DISEASE
In contrast to the evidence from GD patients with homozygous or compound heterozygous GBA1 variants, it is still unclear whether heterozygous GBA1 variants play a role in modulating global levels of inflammatory markers. Several studies aimed to address this question (Table 1), yet they often analyzed different cytokines with different methods and reached inconsistent results. A recent study analyzed different cytokines using two methods: ELISA was used for analyzing eight GBA1-PD patients, 28 sporadic PD patients and 28 controls, and a multiplex assay was used for analyzing eight GBA1-PD patients, 23 sporadic PD patients and 29 controls [51]. In the ELISA analysis, levels of IL-1β and TNFα were both significantly higher among the GBA1-PD group, and IFNγ was lower in GBA1-PD compared to sporadic PD. However, in the multiplex assay, only the results for IL-1β were replicated. TNFα was not different between the two groups of patients, and it was lower compared to controls. IFNγ, which in the ELISA analysis was lower in GBA1-PD compared to sporadic PD, was much higher in the multiplex assay in GBA1-PD compared to both sporadic PD patients and controls. The multiplex assay also analyzed cytokines that were not measured using ELISA, and suggested that the levels of IL-2 and MCP-1 (monocyte chemoattractant protein-1, also known as chemokine (c-c motif) ligand 2, CCL2), a cytokine that regulates migration and infiltration of monocytes [52] are increased in GBA1-PD.
Table 1
Summary of studies analyzing different inflammation markers and cytokines in GBA1-PD
| Study | Population | Main results | Ref |
| Miliukhina et al. | ELISA analysis: | ELISA analysis: | [51] |
| GBA1-PD – 8 | IL-1β and TNFα elevated in GBA1- | ||
| Sporadic PD – 28 | PD, IFNγ reduced. | ||
| Controls – 28 | Multiplex assay: | ||
| Multiplex assay: | IL-1β elevated in GBA1-PD. TNFα similar in PD groups and lower compared to controls. IFNγ, IL-2 and MCP-1 elevated in GBA1-PD. | ||
| GBA1-PD – 8 | |||
| Sporadic PD – 23 | |||
| Controls – 29 | |||
| Chahine et al. | Discovery cohort: | Discovery cohort: | [53] |
| GBA1-PD – 20 (17 het, 3 hom) | MCP-1, IL-8, MIP1α, PARC, and stem cell factor elevated in GBA1-PD. | ||
| Sporadic PD – 242 | Replication cohort: | ||
| Replication cohort: | IL-8 elevated in GBA1-PD. | ||
| GBA1-PD – 19 (17 het, 2 hom) | |||
| Sporadic PD – 41 | |||
| Thaler et al. | GBA1-PD – 77 | No associations (tested TNF-α, IL-1, IL-2, IL-4, IL-6, IL-8, IL-10, and INF-γ). | [54] |
| Sporadic PD – 31 | |||
| Galper et al. | GBA1-PD with biallelic variants – 6 | Ferritin, CCL18, and MIP1α elevated in GBA1-PD with biallelic variants. | [55] |
| GBA1-PD heterozygous – 129 | |||
| GBA1 heterozygous without PD – 83 | |||
| Controls without GBA1 variants – 77 |
ELISA, enzyme-linked immunosorbent assay; PD, Parkinson’s disease; IL, interleukin; TNF, tumor necrosis factor; IFN, interferon; MCP-1, monocyte chemoattractant protein-1, also called CCL2; MIP1α, macrophage inflammatory protein 1α, also called CCL3; PARC, Pulmonary and activation-regulated chemokine, also called CCL18.
A previous report comparing 20 GBA1-PD patients (17 heterozygous and three homozygous) and 242 sporadic PD patients also reported increased plasma levels of MCP-1 in GBA1-PD [53]. The same study also reported increased levels of IL-8, MIP1α (macrophage inflammatory protein 1α, also known as chemokine (c-c motif) ligand 3, CCL3) and several other cytokines in GBA1-PD. However, in a replication cohort of 19 GBA1-PD patients (17 heterozygous and two homozygous) and 41 sporadic PD patients, only IL-8 showed increased levels in GBA1-PD [53]. A recent study comparing 77 GBA1-PD patients with 31 idiopathic PD patients, however, did not identify association between GBA1-PD and any of the tested cytokines, including TNF-α, IL-1, IL-2, IL-4, IL-6, IL-8, IL-10, and INF-γ [54]. A different study evaluated the levels of established GD-associated cytokines in biallelic carriers of GBA1 variants with PD (n = 6, four of which were also diagnosed with GD), heterozygous carriers of GBA1 variants with PD (n = 129), individuals with GBA1 variants without PD (n = 83) and controls without GBA1 variants (n = 77) [55]. The analysis demonstrated levels of ferritin, CCL18, and MIP1α were only elevated in the group of biallelic GBA1 variants with PD. Of note, some of the chemokines reported in the studies above to potentially be involved in GBA1-PD, including IL-8, MIP1α, TNF-α, and MIP1α, have been suggested to predict the progression of LRRK2-associated PD, another common genetic form of PD [56]. Their putative role in GBA1-PD progression has not been studied yet, and direct comparisons between GBA1-PD and LRRK2-PD will be of much interest and importance.
There are many differences between the studies mentioned above that can explain the contradicting results, including differences in sample size, methods used for analysis, ethnicity, environmental factors and individual genetic factors that can affect the levels of cytokines. A recent study aimed to standardize different assays in immune cells, including measurement of cytokines, suggested that performing stimulation-dependent analysis will yield more consistent results across different centers [57]. A coordinated, large effort of multiple centers that will use standardized methods and analysis, accounting for as many confounders as possible, is required to better understand whether specific cytokines have a role or can serve as biomarkers in GBA1-PD. Furthermore, not all the aforementioned studies considered the different types of GBA1 variants, and therefore included all variant carriers as a single group. It will be important in future studies to separately analyze the effects of different types of GBA1 variants (severe, mild, risk factors, specific variants) and to detail exactly which variants were analyzed, in order to fully understand how they might specifically contribute to PD through immune response.
GLYCOSPHINGOLIPIDS AND NEUROINFLAMMATION
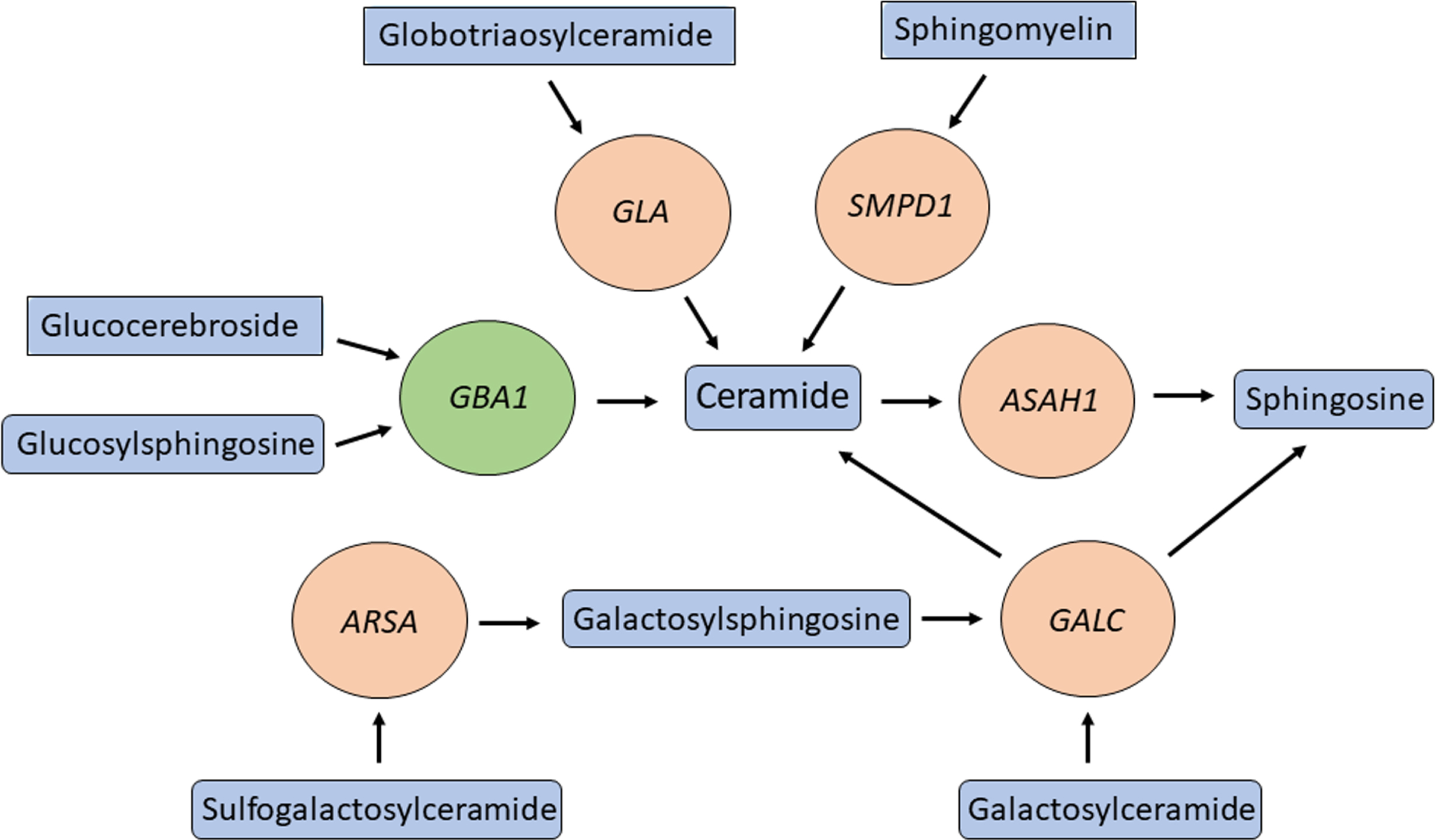
Several genes in the lysosomal sphingolipid metabolism pathway have been suggested to be involved in PD. These include, GBA1, SMPD1, ASAH1 [59], GALC [5], ARSA [60], and GLA [61], and their main substrates, products, related lysosomal storge disorders and suggested involvement in PD are detailed in Table 2. All these genes work closely together in the sphingolipid metabolism pathway in the lysosome (Fig. 1), suggesting that dysregulation of the flux in this pathway may be important in PD pathogenesis. Other genes that regulate GBA1 have also been implicated in α-synucleinopathies, including PSAP [62, 63], SCARB2 [64, 65], and CTSB [23].
Table 2
Genes for known lysosomal storage diseases and their respective genetic associations to Parkinson’s disease
| Gene Name | Substrate | Products | Lysosomal Storage Disorder | Association with PD | References |
| GBA1 | Glucosylceramide, glucosylsphingosine | Ceramide, sphingosine and glucose | Gaucher’s Disease | Multiple gene variants shown to confer greater risk for developing PD | [99–101] |
| SMPD1 | Sphingomyelin | ceramide and phosphorylcholine | Niemann-Pick type A/B | Gene variants p.L302P and p.fsP330 associated with PD | [58] |
| ASAH1 | Ceramide | Sphingosine | Farber Lipogranulomatosis | [59] | |
| GALC | Galactosylceramide, galactosylsphingosine psychosine | Galactose, sphingosine, ceramide | Krabbe disease | Gene identified as risk factor for PD | [102] |
| ARSA | sulfogalactosylceramide | Galactosylceramide | Metachromatic leukodystrophy | Multiple variants shown to modify PD | [60] |
| GLA | Globotriaosylceramide | Galactose, ceramide | Fabry Disease | No genetic evidence, but enzymatically linked with PD risk | [103] |
Fig. 1
Multiple genes involved in ceramide metabolism and in PD. GBA1 (green circle) is one of many genes (orange circles) encoding for enzymes in the ceramide metabolism pathway that are both responsible for a lysosomal storage disease and modifying the risk of PD. Important substrates and products of these enzymes are in blue rectangles.
At the same time, the sphingolipids that are substrates and/or products of this pathway have important immune-related and inflammatory roles. This topic has been thoroughly reviewed in relation to PD [66], therefore here we provide some important details and takes. Glucocerebrosides (or glucosylceramides) are the main substrates of all glycosphingolipids in the brain. In general, glycosphingolipids regulate multiple processes in the brain, including immune response and neuroinflammation. One mechanism linking glycosphingolipids and neuroinflammation is activation of inflammasomes— a complex of proteins activating caspase-1, which is responsible for the maturation of the proinflammatory cytokines IL1-beta and IL18. Autophagy and the inflammasome regulate each other [67], and efficient autophagy is critically regulated by the composition of membranes, which is determined by the levels of the different sphingolipids [68–70]. Whether this link between GCase and other enzymes in the sphingolipids metabolism pathway, autophagy and inflammasome activation has a role in PD remains hypothetical and requires additional studies.
Another potential link between the lysosomal membrane composition and the immune system in PD is through antigen presentation. There is a clear association between the HLA genomic region and specific HLA types and PD [2, 3], and the process of antigen processing and presentation is regulated by autophagy and the lysosome [71]. It is therefore possible that GBA1 variants, as well as variants in other genes in this pathway, affect the lysosomal membrane composition, which affects autophagy and antigen presentation. This hypothesis, as does the former one, require additional studies in humans and different models. Other mechanisms by which glycosphingolipids may affect neuroinflammation include their effects on calcium homeostasis, blood-brain-barrier permeability and autoantibodies production [66].
Lastly, there is a large body of evidence on the interaction between α-synuclein and membranes, which has been extensively reviewed before [72]. Since GBA1 variants affect membrane composition, it is possible that they influence the interaction between these membranes and α-synuclein. These hypothesized effects should be further studied in the context of GBA1-PD in different PD-relevant models and cell types.
GBA1, α-SYNUCLEIN, AND THE IMMUNE SYSTEM
Unlike another common genetic form of PD, LRRK2-associated neurodegeneration, GBA1-associated PD is almost exclusively characterized by α-synuclein neuropathology [73]. Numerous studies in multiple human and animal show that reduced GCase activity, whether due to variants, chemical inhibition, knockdown or knockout, leads to α-synuclein accumulation [74–76]. Furthermore, it was suggested that GCase, its substrate glucocerebrosidase and α-synuclein directly interact as part of the pathogenic process in GBA1-associated PD [77, 78].
Despite decades of research, it is still unclear what the normal, physiological role of α-synuclein is. It is possible that one of its potential roles is as a part of the immune system [79], and whether this hypothesis is true or not, years of evidence clearly show that α-synuclein can trigger the immune system, and that it may have a role in the pathogenic process of PD [80]. Specifically, aggregated α-synuclein can activate microglia in vitro and in vivo [81], which in turn may contribute to the pathogenic process of PD. This activation may occur through binding to the Toll-like receptor 2 (TLR2), as mice lacking TLR2 or treated with TLR2 blocking antibody did not show microglial activation [82]. Other lines of evidence show that α-synuclein can also trigger the activation of T cells. Certain epitopes of α-synuclein may act as antigens and stimulate helper and cytotoxic T cell responses in PD patients [83], perhaps due to their homology to some viruses such as the herpes simplex virus 1 (HSV1) [84]. It is still unclear whether this activation is part of the normal function of α-synuclein in attempt to activate the immune system as a defence mechanism, or a pathogenic process that in some individuals may contribute to the development of PD, or a combination of both scenarios. While this is not a unique feature of GBA1-associated PD and may occur in other forms of PD, it represents another path by which the immune system participates in this specific PD subtype.
ADDITIONAL EVIDENCE POTENTIALLY LINKING GBA1 AND THE IMMUNE SYSTEM WITH PD
The complement system plays an important role in assisting immune cells with clearing infections and modulating immunity. Dysregulation of the complement system leads to various effects from autoimmune disorders to failures in host immunity. Furthermore, the complement system has been implicated in normal central nervous system development and in different neurological disorders, including PD [85, 86]. Activated complement components have been reported in neuropathological studies of brains of PD patients and observed within Lewy bodies. Changes in complement levels in blood of PD patients has also been reported, and in vitro studies suggest that α-synuclein may have a role in their activation [87].
A few studies pointed towards a potential involvement of the complement system in GBA1-mediated dysregulation of the immune system. GBA1 mutations may result in activation of the complement pathway via increased production of C5a and activation of the C5aR1 receptor which in turn results in the production of pro-inflammatory cytokines. Compared to wildtype, mice with heteroallelic mutations (p.Asp409Val/knockout) in GBA1 had a marked increase in C5a, and increased levels of inflammatory cells and cytokines including IFNγ, TNF, IL-1β, IL-6, and IL-17A/F [27]. Of note, this specific mutation has been reported only once in GD, and never in PD, and its severity is unknown. Similarly, GD-derived macrophages (using induced pluripotent stem cells) showed marked production of TNF in response to C5aR1 activation in the presence of recombinant C5a [88]. Both of these studies point towards a possible role for the complement system in potentiating an inflammatory environment due to GBA1 variants.
Another potential link between GBA1 variants and immune response was demonstrated in mice, where GCase inhibition with conduritol-β-epoxide (CBE) led to impaired response of the nuclear factor erythroid 2-related factor (Nfe2l2), a key transcription factor involved in multiple processes, including neuroinflammation. Using neuronal-microglial co-cultures derived from the same mouse models revealed that the inhibition of the response was mediated by inhibition of GCase activity in microglia and not in neurons themselves. Furthermore, GCase inhibition in microglia was sufficient and necessary for ablation of the Nfe2l2 in neurons [89]. Another study showed that mice treated with continuous administration of GCase inhibitor resulted in prolonged activation of microglia, the complement pathway, and increased expression of autophagy-related proteins in the substantia nigra after 28 days of treatment compared to mice treated with vehicle [90]. In long-lived transgenic mouse models with homozygous p.L444P (associated with GD type 3) or p.R463C (associated with GD type 1) GBA1 variants had increased activation of astrocytes and accumulation of alpha-synuclein in the nigrostriatal pathway. Furthermore, in the same models, subchronic administration of GCase inhibitor also resulted in activation of microglia and astrocytes in the cortex, hippocampus, substantia nigra and striatum, as well as marked increases in alpha-synuclein accumulation in the substantia nigra [91].
One interesting aspect of the potential involvement of GBA1 in immune response and inflammation, is that its substrates and products may also have a role. For example, in monocytes, inhibition of ceramide kinase (CERK) leads to reduction in expression of inflammatory markers CD11c and HLA-DR by monocytes in response to TNF-α stimulation [92]. Glucosylceramide, one of the main substrates of GCase, can activate myeloid cells and increase the levels of inflammatory cytokines [93]. Conversely, reduction of glucosylceramide can reduce LPS-induced inflammatory response in human macrophages and in mice [94]. Other studies have shown that the activity of the GCase itself might be specifically reduced in monocytes, even in non-carriers of GBA1 variants, and that this reduced activity may correlate with the motor severity of PD [95, 96]. These are just a few examples, but multiple other studies link glucosylceramide and other glycosphingolipids to immune homeostasis [97], providing an additional potential link between GBA1 and inflammation. Due to contradicting results it is still unclear whether in PD with heterozygous GBA1 variant there is an elevation of glucosylceramide, glucosylsphingosine, or other GCase substrates [98], suggesting that other mechanisms may be involved.
CONCLUDING REMARKS
The goal of this review was to provide an overview of the current evidence linking GBA1, the immune system and PD. While there is some suggestive evidence that the immune system and/or inflammation may be involved in GBA1-associated PD (Fig. 2), it is far from being conclusive. The differences between the different studies which examined cytokine levels in GBA1-PD patients require additional studies that at the very least should be: a) larger, b) use the same methodology, and c) take into account other confounding factors like genetics, co-morbidities and different exposures. Even if there are differences in cytokine levels, showing that they have a role in the pathogenic process of GBA1-associated PD will require additional studies. Similar questions remain regarding all other evidence that we discussed in this review, and their potential role in GBA1-associated PD. Since there are many drugs that can modulate the immune system and inflammation, it will be important to address these questions.
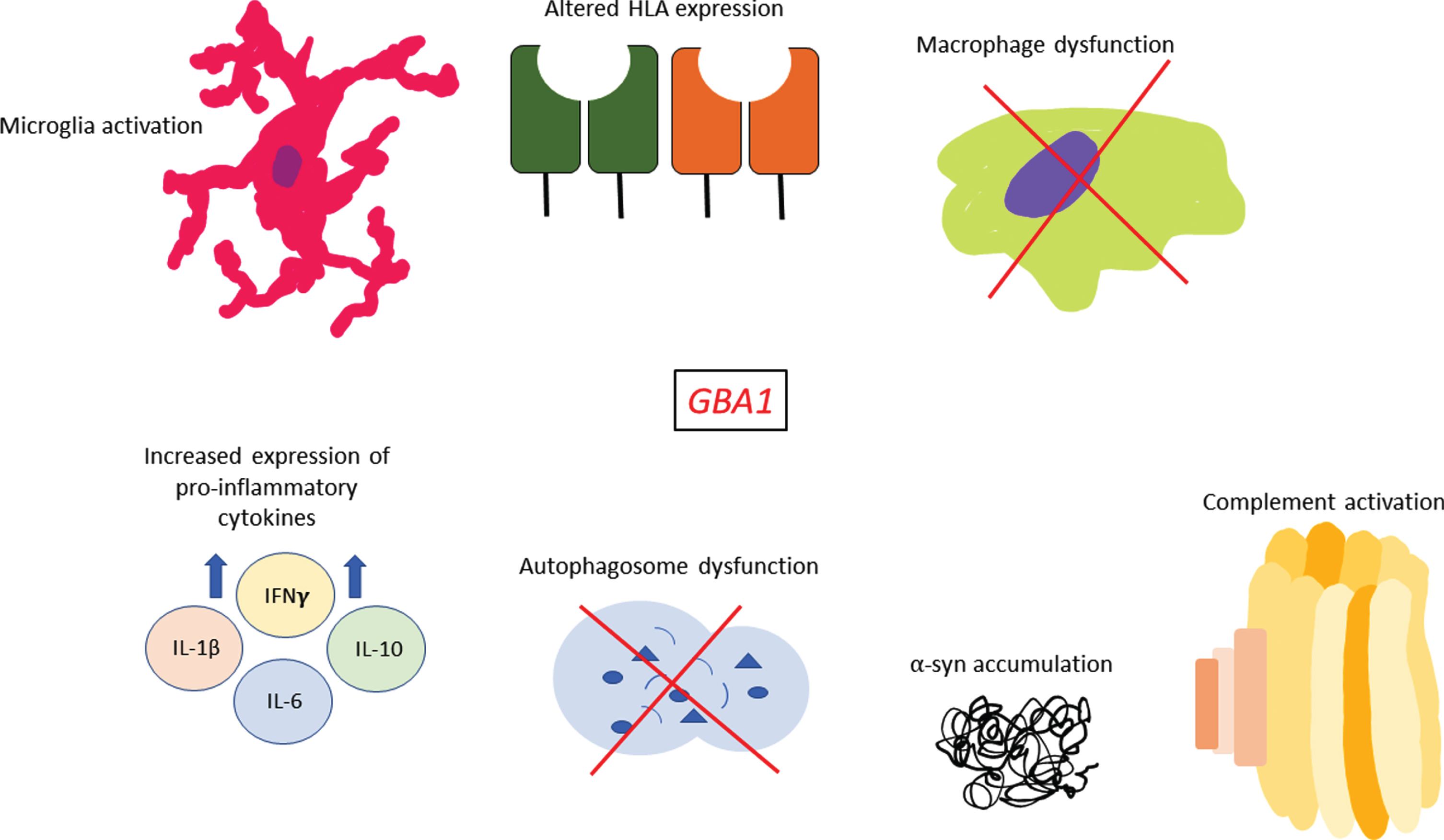
Fig. 2
Sequelae of immune events associated with GBA1 variants. GBA1 variants have been shown to affect multiple facets of the immune system such as increased pro-inflammatory cytokine expression, complement activation, potentially altered HLA expression, microglia activation and macrophage dysfunction. In addition, many studies have shown that GBA1 variants are associated with greater α-synuclein accumulation within cells and marked autophagosome dysfunction which may also have immune-related effects in PD.
ACKNOWLEDGMENTS
This work was financially supported by the Michael J. Fox Foundation, the Canadian Consortium on Neurodegeneration in Aging (CCNA), and the Canada First Research Excellence Fund (CFREF) awarded to McGill University for the Healthy Brains for Healthy Lives (HBHL) program.
CONFLICT OF INTEREST
Z.G.O. is supported by the Fonds de recherche du Quebec Sante (FRQS) Chercheurs-boursiers award. He received consultancy fees from Ono Therapeutics, Handl Therapeutics, UCB, Neuron23, Lysosomal Therapeutics Inc., Bial Biotech Inc., Deerfield, Lighthouse, Gudiepoint, and Idorsia.
REFERENCES
[1] | Tansey MG , Wallings RL , Houser MC , Herrick MK , Keating CE , Joers V ((2022) ) Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol. doi: 10.1038/s41577-022-00684-6. |
[2] | Yu E , Ambati A , Andersen MS , Krohn L , Estiar MA , Saini P , Senkevich K , Sosero YL , Sreelatha AAK , Ruskey JA , Asayesh F , Spiegelman D , Toft M , Viken MK , Sharma M , Blauwendraat C , Pihlstrøm L , Mignot E , Gan-Or Z ((2021) ) Fine mapping of the HLA locus in Parkinson’s disease in Europeans. NPJ Parkinsons Dis 7: , 84. |
[3] | Naito T , Satake W , Ogawa K , Suzuki K , Hirata J , Foo JN , Tan EK , Toda T , Okada Y ((2021) ) Trans-ethnic fine-mapping of the major histocompatibility complex region linked to Parkinson’s disease. Mov Disord 36: , 1805–1814. |
[4] | Kline EM , Houser MC , Herrick MK , Seibler P , Klein C , West A , Tansey MG ((2021) ) Genetic and environmental factors in Parkinson’s disease converge on immune function and inflammation. Mov Disord 36: , 25–36. |
[5] | Nalls MA , Blauwendraat C , Vallerga CL , Heilbron K , Bandres-Ciga S , Chang D , Tan M , Kia DA , Noyce AJ , Xue A , Bras J , Young E , von Coelln R , Simón-Sánchez J , Schulte C , Sharma M , Krohn L , Pihlstrøm L , Siitonen A , Iwaki H , Leonard H , Faghri F , Gibbs JR , Hernandez DG , Scholz SW , Botia JA , Martinez M , Corvol JC , Lesage S , Jankovic J , Shulman LM , Sutherland M , Tienari P , Majamaa K , Toft M , Andreassen OA , Bangale T , Brice A , Yang J , Gan-Or Z , Gasser T , Heutink P , Shulman JM , Wood NW , Hinds DA , Hardy JA , Morris HR , Gratten J , Visscher PM , Graham RR , Singleton AB ((2019) ) Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol 18: , 1091–1102. |
[6] | Gan-Or Z , Amshalom I , Kilarski LL , Bar-Shira A , Gana-Weisz M , Mirelman A , Marder K , Bressman S , Giladi N , Orr-Urtreger A ((2015) ) Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 84: , 880–887. |
[7] | Gan-Or Z , Liong C , Alcalay RN ((2018) ) GBA-associated Parkinson’s disease and other synucleinopathies. Curr Neurol Neurosci Rep 18: , 44. |
[8] | Alcalay RN , Levy OA , Waters CC , Fahn S , Ford B , Kuo SH , Mazzoni P , Pauciulo MW , Nichols WC , Gan-Or Z , Rouleau GA , Chung WK , Wolf P , Oliva P , Keutzer J , Marder K , Zhang X ((2015) ) Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 138: , 2648–2658. |
[9] | Gegg ME , Burke D , Heales SJ , Cooper JM , Hardy J , Wood NW , Schapira AH ((2012) ) Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol 72: , 455–463. |
[10] | Roshan Lal T , Sidransky E ((2017) ) The spectrum of neurological manifestations associated with Gaucher disease. Diseases 5: , 10. |
[11] | Neudorfer O , Giladi N , Elstein D , Abrahamov A , Turezkite T , Aghai E , Reches A , Bembi B , Zimran A ((1996) ) Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 89: , 691–694. |
[12] | Várkonyi J , Rosenbaum H , Baumann N , MacKenzie JJ , Simon Z , Aharon-Peretz J , Walker JM , Tayebi N , Sidransky E ((2003) ) Gaucher disease associated with parkinsonism: Four further case reports. Am J Med Genet A 116a: , 348–351. |
[13] | Machaczka M , Rucinska M , Skotnicki AB , Jurczak W ((1999) ) Parkinson’s syndrome preceding clinical manifestation of Gaucher’s disease. Am J Hematol 61: , 216–217. |
[14] | Sidransky E ((2012) ) Gaucher disease: Insights from a rare Mendelian disorder. Discov Med 14: , 273–281. |
[15] | Beutler E , Gelbart T , Scott CR ((2005) ) Hematologically important mutations: Gaucher disease. Blood Cells Mol Dis 35: , 355–364. |
[16] | Liu G , Boot B , Locascio JJ , Jansen IE , Winder-Rhodes S , Eberly S , Elbaz A , Brice A , Ravina B , van Hilten JJ , Cormier-Dequaire F , Corvol JC , Barker RA , Heutink P , Marinus J , Williams-Gray CH , Scherzer CR ((2016) ) Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann Neurol 80: , 674–685. |
[17] | Cilia R , Tunesi S , Marotta G , Cereda E , Siri C , Tesei S , Zecchinelli AL , Canesi M , Mariani CB , Meucci N , Sacilotto G , Zini M , Barichella M , Magnani C , Duga S , Asselta R , Soldà G , Seresini A , Seia M , Pezzoli G , Goldwurm S ((2016) ) Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann Neurol 80: , 662–673. |
[18] | Huang Y , Deng L , Zhong Y , Yi M ((2018) ) The association between E326K of GBA and the risk of Parkinson’s disease. Parkinsons Dis 2018: , 1048084. |
[19] | Mallett V , Ross JP , Alcalay RN , Ambalavanan A , Sidransky E , Dion PA , Rouleau GA , Gan-Or Z ((2016) ) GBA p.T369M substitution in Parkinson disease: Polymorphism or association? A meta-analysis. Neurol Genet 2: , e104. |
[20] | Anheim M , Elbaz A , Lesage S , Durr A , Condroyer C , Viallet F , Pollak P , Bonaïti B , Bonaïti-Pellié C , Brice A ((2012) ) Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 78: , 417–420. |
[21] | Rana HQ , Balwani M , Bier L , Alcalay RN ((2013) ) Age-specific Parkinson disease risk in GBA mutation carriers: Information for genetic counseling. Genet Med 15: , 146–149. |
[22] | Balestrino R , Tunesi S , Tesei S , Lopiano L , Zecchinelli AL , Goldwurm S ((2020) ) Penetrance of glucocerebrosidase (GBA) mutations in Parkinson’s disease: A kin cohort study. Mov Disord 35: , 2111–2114. |
[23] | Blauwendraat C , Reed X , Krohn L , Heilbron K , Bandres-Ciga S , Tan M , Gibbs JR , Hernandez DG , Kumaran R , Langston R , Bonet-Ponce L , Alcalay RN , Hassin-Baer S , Greenbaum L , Iwaki H , Leonard HL , Grenn FP , Ruskey JA , Sabir M , Ahmed S , Makarious MB , Pihlstrøm L , Toft M , van Hilten JJ , Marinus J , Schulte C , Brockmann K , Sharma M , Siitonen A , Majamaa K , Eerola-Rautio J , Tienari PJ , Pantelyat A , Hillis AE , Dawson TM , Rosenthal LS , Albert MS , Resnick SM , Ferrucci L , Morris CM , Pletnikova O , Troncoso J , Grosset D , Lesage S , Corvol JC , Brice A , Noyce AJ , Masliah E , Wood N , Hardy J , Shulman LM , Jankovic J , Shulman JM , Heutink P , Gasser T , Cannon P , Scholz SW , Morris H , Cookson MR , Nalls MA , Gan-Or Z , Singleton AB ((2020) ) Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 143: , 234–248. |
[24] | Straniero L , Rimoldi V , Monfrini E , Bonvegna S , Melistaccio G , Lake J , Soldà G , Aureli M , Shankaracharya , Keagle P , Foroud T , Landers JE , Blauwendraat C , Zecchinelli A , Cilia R , Di Fonzo A , Pezzoli G , Duga S , Asselta R ((2022) ) Role of lysosomal gene variants in modulating GBA-associated Parkinson’s disease risk. Mov Disord 37: , 1202–1210. |
[25] | Cox TM ((2001) ) Gaucher disease: Understanding the molecular pathogenesis of sphingolipidoses. J Inherit Metab Dis 24: (Suppl 2), 106–121; discussion 187-108. |
[26] | Panicker LM , Miller D , Awad O , Bose V , Lun Y , Park TS , Zambidis ET , Sgambato JA , Feldman RA ((2014) ) Gaucher iPSC-derived macrophages produce elevated levels of inflammatory mediators and serve as a new platform for therapeutic development. Stem Cells 32: , 2338–2349. |
[27] | Pandey MK , Burrow TA , Rani R , Martin LJ , Witte D , Setchell KD , McKay MA , Magnusen AF , Zhang W , Liou B , Köhl J , Grabowski GA ((2017) ) Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature 543: , 108–112. |
[28] | Burstein Y , Zakuth V , Rechavi G , Spirer Z ((1987) ) Abnormalities of cellular immunity and natural killer cells in Gaucher’s disease. J Clin Lab Immunol 23: , 149–151. |
[29] | Vitner EB , Farfel-Becker T , Eilam R , Biton I , Futerman AH ((2012) ) Contribution of brain inflammation to neuronal cell death in neuronopathic forms of Gaucher’s disease. Brain 135: , 1724–1735. |
[30] | Senkevich K , Gan-Or Z ((2020) ) Autophagy lysosomal pathway dysfunction in Parkinson’s disease; evidence from human genetics. Parkinsonism Relat Disord 73: , 60–71. |
[31] | Smith L , Mullin S , Schapira AHV ((2017) ) Insights into the structural biology of Gaucher disease. Exp Neurol 298: , 180–190. |
[32] | Baris HN , Cohen IJ , Mistry PK ((2014) ) Gaucher disease: The metabolic defect, pathophysiology, phenotypes and natural history. Pediatr Endocrinol Rev 12 Suppl 1: , 72–81. |
[33] | Burrow TA , Barnes S , Grabowski GA ((2011) ) Prevalence and management of Gaucher disease. Pediatric Health Med Ther 2: , 59–73. |
[34] | Stirnemann J , Belmatoug N , Camou F , Serratrice C , Froissart R , Caillaud C , Levade T , Astudillo L , Serratrice J , Brassier A , Rose C , Billette de Villemeur T , Berger MG ((2017) ) A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci 18: , 441. |
[35] | Sidransky E ((2004) ) Gaucher disease: Complexity in a “simple” disorder. Mol Genet Metab 83: , 6–15. |
[36] | Aflaki E , Westbroek W , Sidransky E ((2017) ) The complicated relationship between Gaucher disease and parkinsonism: Insights from a rare disease. Neuron 93: , 737–746. |
[37] | Rigante D , Cipolla C , Basile U , Gulli F , Savastano MC ((2017) ) Overview of immune abnormalities in lysosomal storage disorders. Immunol Lett 188: , 79–85. |
[38] | Barak V , Acker M , Nisman B , Kalickman I , Abrahamov A , Zimran A , Yatziv S ((1999) ) Cytokines in Gaucher’s disease. Eur Cytokine Netw 10: , 205–210. |
[39] | Allen MJ , Myer BJ , Khokher AM , Rushton N , Cox TM ((1997) ) Pro-inflammatory cytokines and the pathogenesis of Gaucher’s disease: Increased release of interleukin-6 and interleukin-10. QJM 90: , 19–25. |
[40] | Panicker LM , Miller D , Park TS , Patel B , Azevedo JL , Awad O , Masood MA , Veenstra TD , Goldin E , Stubblefield BK , Tayebi N , Polumuri SK , Vogel SN , Sidransky E , Zambidis ET , Feldman RA ((2012) ) Induced pluripotent stem cell model recapitulates pathologic hallmarks of Gaucher disease. Proc Natl Acad Sci U S A 109: , 18054–18059. |
[41] | Zahran AM , Eltayeb AA , Elsayh KI , Saad K , Ahmad FA , Ibrahim AIM ((2017) ) Activated and memory T lymphocytes in children with Gaucher disease. Arch Immunol Ther Exp (Warsz) 65: , 263–269. |
[42] | Bettman N , Avivi I , Rosenbaum H , Bisharat L , Katz T ((2015) ) Impaired migration capacity in monocytes derived from patients with Gaucher disease. Blood Cells Mol Dis 55: , 180–186. |
[43] | Shoenfeld Y , Gallant LA , Shaklai M , Livni E , Djaldetti M , Pinkhas J ((1982) ) Gaucher’s disease: A disease with chronic stimulation of the immune system. Arch Pathol Lab Med 106: , 388–391. |
[44] | Lloyd OC , Norman RM , Urich H ((1956) ) The neuropathology of infantile Gaucher’s disease. J Pathol Bacteriol 72: , 121–131. |
[45] | Kaye EM , Ullman MD , Wilson ER , Barranger JA ((1986) ) Type 2 and type 3 Gaucher disease: A morphological and biochemical study. Ann Neurol 20: , 223–230. |
[46] | Farfel-Becker T , Vitner EB , Pressey SN , Eilam R , Cooper JD , Futerman AH ((2011) ) Spatial and temporal correlation between neuron loss and neuroinflammation in a mouse model of neuronopathic Gaucher disease. Hum Mol Genet 20: , 1375–1386. |
[47] | Wong K , Sidransky E , Verma A , Mixon T , Sandberg GD , Wakefield LK , Morrison A , Lwin A , Colegial C , Allman JM , Schiffmann R ((2004) ) Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab 82: , 192–207. |
[48] | Colombo E , Farina C ((2016) ) Astrocytes: Key regulators of neuroinflammation. Trends Immunol 37: , 608–620. |
[49] | Barkhuizen M , Renggli K , Gubian S , Peitsch MC , Mathis C , Talikka M ((2022) ) Causal biological network models for reactive astrogliosis: A systems approach to neuroinflammation. Sci Rep 12: , 4205. |
[50] | Mullin S , Stokholm MG , Hughes D , Mehta A , Parbo P , Hinz R , Pavese N , Brooks DJ , Schapira AHV ((2021) ) Brain microglial activation increased in glucocerebrosidase (GBA) mutation carriers without Parkinson’s disease. Mov Disord 36: , 774–779. |
[51] | Miliukhina IV , Usenko TS , Senkevich KA , Nikolaev MA , Timofeeva AA , Agapova EA , Semenov AV , Lubimova NE , Totolyan AA , Pchelina SN ((2020) ) Plasma cytokines profile in patients with Parkinson’s disease associated with mutations in GBA gene. Bull Exp Biol Med 168: , 423–426. |
[52] | Deshmane SL , Kremlev S , Amini S , Sawaya BE ((2009) ) Monocyte chemoattractant protein-1 (MCP-1): An overview. J Interferon Cytokine Res 29: , 313–326. |
[53] | Chahine LM , Qiang J , Ashbridge E , Minger J , Yearout D , Horn S , Colcher A , Hurtig HI , Lee VM , Van Deerlin VM , Leverenz JB , Siderowf AD , Trojanowski JQ , Zabetian CP , Chen-Plotkin A ((2013) ) Clinical and biochemical differences in patients having Parkinson disease with vs without GBA mutations. JAMA Neurol 70: , 852–858. |
[54] | Thaler A , Omer N , Giladi N , Gurevich T , Bar-Shira A , Gana-Weisz M , Goldstein O , Kestenbaum M , Shirvan JC , Cedarbaum JM , Orr-Urtreger A , Regev K , Shenhar-Tsarfaty S , Mirelman A ((2021) ) Mutations in GBA and LRRK2 are not associated with increased inflammatory markers. J Parkinsons Dis 11: , 1285–1296. |
[55] | Galper J , Balwani M , Fahn S , Waters C , Krohn L , Gan-Or Z , Dzamko N , Alcalay RN ((2021) ) Cytokines and Gaucher biomarkers in glucocerebrosidase carriers with and without Parkinson disease. Mov Disord 36: , 1451–1455. |
[56] | Ahmadi Rastegar D , Ho N , Halliday GM , Dzamko N ((2019) ) Parkinson’s progression prediction using machine learning and serum cytokines. NPJ Parkinsons Dis 5: , 14. |
[57] | Wallings RL , Hughes LP , Staley HA , Simon ZD , McFarland NR , Alcalay RN , Garrido A , Martí MJ , Sarró ET , Dzamko N , Tansey MG ((2022) ) WHOPPA enables parallel assessment of leucine-rich repeat kinase 2 and glucocerebrosidase enzymatic activity in Parkinson’s disease monocytes. Front Cell Neurosci 16: , 892899. |
[58] | Alcalay RN , Mallett V , Vanderperre B , Tavassoly O , Dauvilliers Y , Wu RYJ , Ruskey JA , Leblond CS , Ambalavanan A , Laurent SB , Spiegelman D , Dionne-Laporte A , Liong C , Levy OA , Fahn S , Waters C , Kuo SH , Chung WK , Ford B , Marder KS , Kang UJ , Hassin-Baer S , Greenbaum L , Trempe JF , Wolf P , Oliva P , Zhang XK , Clark LN , Langlois M , Dion PA , Fon EA , Dupre N , Rouleau GA , Gan-Or Z ((2019) ) SMPD1 mutations, activity, and α-synuclein accumulation in Parkinson’s disease. Mov Disord 34: , 526–535. |
[59] | Robak LA , Jansen IE , van Rooij J , Uitterlinden AG , Kraaij R , Jankovic J , Heutink P , Shulman JM ((2017) ) Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140: , 3191–3203. |
[60] | Lee JS , Kanai K , Suzuki M , Kim WS , Yoo HS , Fu Y , Kim DK , Jung BC , Choi M , Oh KW , Li Y , Nakatani M , Nakazato T , Sekimoto S , Funayama M , Yoshino H , Kubo SI , Nishioka K , Sakai R , Ueyama M , Mochizuki H , Lee HJ , Sardi SP , Halliday GM , Nagai Y , Lee PH , Hattori N , Lee SJ ((2019) ) Arylsulfatase A, a genetic modifier of Parkinson’s disease, is an α-synuclein chaperone. Brain 142: , 2845–2859. |
[61] | Wise AH , Yang A , Naik H , Stauffer C , Zeid N , Liong C , Balwani M , Desnick RJ , Alcalay RN ((2018) ) Parkinson’s disease prevalence in Fabry disease: A survey study. Mol Genet Metab Rep 14: , 27–30. |
[62] | Oji Y , Hatano T , Ueno SI , Funayama M , Ishikawa KI , Okuzumi A , Noda S , Sato S , Satake W , Toda T , Li Y , Hino-Takai T , Kakuta S , Tsunemi T , Yoshino H , Nishioka K , Hattori T , Mizutani Y , Mutoh T , Yokochi F , Ichinose Y , Koh K , Shindo K , Takiyama Y , Hamaguchi T , Yamada M , Farrer MJ , Uchiyama Y , Akamatsu W , Wu YR , Matsuda J , Hattori N ((2020) ) Variants in saposin D domain of prosaposin gene linked to Parkinson’s disease. Brain 143: , 1190–1205. |
[63] | Sosero YL , Yu E , Estiar MA , Krohn L , Mufti K , Rudakou U , Ruskey JA , Asayesh F , Laurent SB , Spiegelman D , Trempe JF , Quinnell TG , Oscroft N , Arnulf I , Montplaisir JY , Gagnon JF , Desautels A , Dauvilliers Y , Gigli GL , Valente M , Janes F , Bernardini A , Sonka K , Kemlink D , Oertel W , Janzen A , Plazzi G , Antelmi E , Biscarini F , Figorilli M , Puligheddu M , Mollenhauer B , Trenkwalder C , Sixel-Döring F , Cochen De Cock V , Monaca CC , Heidbreder A , Ferini-Strambi L , Dijkstra F , Viaene M , Abril B , Boeve BF , Postuma RB , Rouleau GA , Ibrahim A , Stefani A , Högl B , Hu MTM , Gan-Or Z ((2022) ) Rare PSAP variants and possible interaction with GBA in REM sleep behavior disorder. J Parkinsons Dis 12: , 333–340. |
[64] | Do CB , Tung JY , Dorfman E , Kiefer AK , Drabant EM , Francke U , Mountain JL , Goldman SM , Tanner CM , Langston JW , Wojcicki A , Eriksson N ((2011) ) Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet 7: , e1002141. |
[65] | Rothaug M , Zunke F , Mazzulli JR , Schweizer M , Altmeppen H , Lüllmann-Rauch R , Kallemeijn WW , Gaspar P , Aerts JM , Glatzel M , Saftig P , Krainc D , Schwake M , Blanz J ((2014) ) LIMP-2 expression is critical for β-glucocerebrosidase activity and α-synuclein clearance. Proc Natl Acad Sci U S A 111: , 15573–15578. |
[66] | Belarbi K , Cuvelier E , Bonte MA , Desplanque M , Gressier B , Devos D , Chartier-Harlin MC ((2020) ) Glycosphingolipids and neuroinflammation in Parkinson’s disease. Mol Neurodegener 15: , 59. |
[67] | Sun Q , Fan J , Billiar TR , Scott MJ ((2017) ) Inflammasome and autophagy regulation - a two-way street. Mol Med 23: , 188–195. |
[68] | Roney JC , Li S , Farfel-Becker T , Huang N , Sun T , Xie Y , Cheng XT , Lin MY , Platt FM , Sheng ZH ((2021) ) Lipid-mediated impairment of axonal lysosome transport contributing to autophagic stress. Autophagy 17: , 1796–1798. |
[69] | Kaushik S , Bandyopadhyay U , Sridhar S , Kiffin R , Martinez-Vicente M , Kon M , Orenstein SJ , Wong E , Cuervo AM ((2011) ) Chaperone-mediated autophagy at a glance. J Cell Sci 124: , 495–499. |
[70] | Kaushik S , Massey AC , Cuervo AM ((2006) ) Lysosome membrane lipid microdomains: Novel regulators of chaperone-mediated autophagy. EMBO J 25: , 3921–3933. |
[71] | Crotzer VL , Blum JS ((2009) ) Autophagy and its role in MHC-mediated antigen presentation. J Immunol 182: , 3335–3341. |
[72] | Runwal G , Edwards RH ((2021) ) The membrane interactions of synuclein: Physiology and pathology. Annu Rev Pathol 16: , 465–485. |
[73] | Poulopoulos M , Levy OA , Alcalay RN ((2012) ) The neuropathology of genetic Parkinson’s disease. Mov Disord 27: , 831–842. |
[74] | Manning-Boğ AB , Schüle B , Langston JW ((2009) ) Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: A biological link between Gaucher disease and parkinsonism. Neurotoxicology 30: , 1127–1132. |
[75] | Jo J , Yang L , Tran HD , Yu W , Sun AX , Chang YY , Jung BC , Lee SJ , Saw TY , Xiao B , Khoo ATT , Yaw LP , Xie JJ , Lokman H , Ong WY , Lim GGY , Lim KL , Tan EK , Ng HH , Je HS ((2021) ) Lewy body-like inclusions in human midbrain organoids carrying glucocerebrosidase and α-synuclein mutations. Ann Neurol 90: , 490–505. |
[76] | Kuo SH , Tasset I , Cheng MM , Diaz A , Pan MK , Lieberman OJ , Hutten SJ , Alcalay RN , Kim S , Ximénez-Embún P , Fan L , Kim D , Ko HS , Yacoubian T , Kanter E , Liu L , Tang G , Muñoz J , Sardi SP , Li A , Gan L , Cuervo AM , Sulzer D ((2022) ) Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci Adv 8: , eabm6393. |
[77] | Mazzulli JR , Xu YH , Sun Y , Knight AL , McLean PJ , Caldwell GA , Sidransky E , Grabowski GA , Krainc D ((2011) ) Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146: , 37–52. |
[78] | Yap TL , Gruschus JM , Velayati A , Westbroek W , Goldin E , Moaven N , Sidransky E , Lee JC ((2011) ) Alpha-synuclein interacts with Glucocerebrosidase providing a molecular link between Parkinson and Gaucher diseases. J Biol Chem 286: , 28080–28088. |
[79] | Barbut D , Stolzenberg E , Zasloff M ((2019) ) Gastrointestinal immunity and alpha-synuclein. J Parkinsons Dis 9: , S313–s322. |
[80] | Allen Reish HE , Standaert DG ((2015) ) Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease. J Parkinsons Dis 5: , 1–19. |
[81] | Zhang W , Wang T , Pei Z , Miller DS , Wu X , Block ML , Wilson B , Zhang W , Zhou Y , Hong JS , Zhang J ((2005) ) Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J 19: , 533–542. |
[82] | Kim C , Ho DH , Suk JE , You S , Michael S , Kang J , Joong Lee S , Masliah E , Hwang D , Lee HJ , Lee SJ ((2013) ) Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 4: , 1562. |
[83] | Sulzer D , Alcalay RN , Garretti F , Cote L , Kanter E , Agin-Liebes J , Liong C , McMurtrey C , Hildebrand WH , Mao X , Dawson VL , Dawson TM , Oseroff C , Pham J , Sidney J , Dillon MB , Carpenter C , Weiskopf D , Phillips E , Mallal S , Peters B , Frazier A , Lindestam Arlehamn CS , Sette A ((2017) ) T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 546: , 656–661. |
[84] | Caggiu E , Paulus K , Galleri G , Arru G , Manetti R , Sechi GP , Sechi LA ((2017) ) Homologous HSV1 and alpha-synuclein peptides stimulate a T cell response in Parkinson’s disease. J Neuroimmunol 310: , 26–31. |
[85] | Presumey J , Bialas AR , Carroll MC ((2017) ) Complement system in neural synapse elimination in development and disease. Adv Immunol 135: , 53–79. |
[86] | Carpanini SM , Torvell M , Morgan BP ((2019) ) Therapeutic inhibition of the complement system in diseases of the central nervous system. Front Immunol 10: , 362. |
[87] | Ziabska K , Ziemka-Nalecz M , Pawelec P , Sypecka J , Zalewska T ((2021) ) Aberrant complement system activation in neurological disorders. Int J Mol Sci 22: , 4675. |
[88] | Serfecz JC , Saadin A , Santiago CP , Zhang Y , Bentzen SM , Vogel SN , Feldman RA ((2021) ) C5a activates a pro-inflammatory gene expression profile in human Gaucher iPSC-derived macrophages. Int J Mol Sci 22: , 9912. |
[89] | Brunialti E , Villa A , Mekhaeil M , Mornata F , Vegeto E , Maggi A , Di Monte DA , Ciana P ((2021) ) Inhibition of microglial β-glucocerebrosidase hampers the microglia-mediated antioxidant and protective response in neurons. J Neuroinflammation 18: , 220. |
[90] | Rocha EM , Smith GA , Park E , Cao H , Graham AR , Brown E , McLean JR , Hayes MA , Beagan J , Izen SC , Perez-Torres E , Hallett PJ , Isacson O ((2015) ) Sustained systemic glucocerebrosidase inhibition induces brain α-synuclein aggregation, microglia and complement C1q activation in mice. Antioxid Redox Signal 23: , 550–564. |
[91] | Ginns EI , Mak SK , Ko N , Karlgren J , Akbarian S , Chou VP , Guo Y , Lim A , Samuelsson S , LaMarca ML , Vazquez-DeRose J , Manning-Boğ AB ((2014) ) Neuroinflammation and α-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol Genet Metab 111: , 152–162. |
[92] | Al-Rashed F , Ahmad Z , Snider AJ , Thomas R , Kochumon S , Melhem M , Sindhu S , Obeid LM , Al-Mulla F , Hannun YA , Ahmad R ((2021) ) Ceramide kinase regulates TNF-α-induced immune responses in human monocytic cells. Sci Rep 11: , 8259. |
[93] | NagataM, IzumiY, IshikawaE, KiyotakeR, DoiR, IwaiS, OmahdiZ, YamajiT, MiyamotoT, BambaT, YamasakiS ((2017) ) Intracellular metabolite β-glucosylceramide is an endogenous Mincle ligand possessing immunostimulatory activity. Proc Natl Acad Sci U S A 114: , E3285–e3294. |
[94] | Mobarak E , Håversen L , Manna M , Rutberg M , Levin M , Perkins R , Rog T , Vattulainen I , Borén J ((2018) ) Glucosylceramide modifies the LPS-induced inflammatory response in macrophages and the orientation of the LPS/TLR4 complex in silico. Sci Rep 8: , 13600. |
[95] | Hughes LP , Pereira MMM , Hammond DA , Kwok JB , Halliday GM , Lewis SJG , Dzamko N ((2021) ) Glucocerebrosidase activity is reduced in cryopreserved Parkinson’s disease patient monocytes and inversely correlates with motor severity. J Parkinsons Dis 11: , 1157–1165. |
[96] | Atashrazm F , Hammond D , Perera G , Dobson-Stone C , Mueller N , Pickford R , Kim WS , Kwok JB , Lewis SJG , Halliday GM , Dzamko N ((2018) ) Reduced glucocerebrosidase activity in monocytes from patients with Parkinson’s disease. Sci Rep 8: , 15446. |
[97] | Ilan Y ((2019) ) β-glycosphingolipids as mediators of both inflammation and immune tolerance: A manifestation of randomness in biological systems. Front Immunol 10: , 1143. |
[98] | Milenkovic I , Blumenreich S , Futerman AH ((2022) ) GBA mutations, glucosylceramide and Parkinson’s disease. Curr Opin Neurobiol 72: , 148–154. |
[99] | Clark LN , Ross BM , Wang Y , Mejia-Santana H , Harris J , Louis ED , Cote LJ , Andrews H , Fahn S , Waters C , Ford B , Frucht S , Ottman R , Marder K ((2007) ) Mutations in the glucocerebrosidase gene are associated with early-onset Parkinson disease. Neurology 69: , 1270–1277. |
[100] | Sidransky E , Nalls MA , Aasly JO , Aharon-Peretz J , Annesi G , Barbosa ER , Bar-Shira A , Berg D , Bras J , Brice A , Chen CM , Clark LN , Condroyer C , De Marco EV , Dürr A , Eblan MJ , Fahn S , Farrer MJ , Fung HC , Gan-Or Z , Gasser T , Gershoni-Baruch R , Giladi N , Griffith A , Gurevich T , Januario C , Kropp P , Lang AE , Lee-Chen GJ , Lesage S , Marder K , Mata IF , Mirelman A , Mitsui J , Mizuta I , Nicoletti G , Oliveira C , Ottman R , Orr-Urtreger A , Pereira LV , Quattrone A , Rogaeva E , Rolfs A , Rosenbaum H , Rozenberg R , Samii A , Samaddar T , Schulte C , Sharma M , Singleton A , Spitz M , Tan EK , Tayebi N , Toda T , Troiano AR , Tsuji S , Wittstock M , Wolfsberg TG , Wu YR , Zabetian CP , Zhao Y , Ziegler SG ((2009) ) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361: , 1651–1661. |
[101] | Aharon-Peretz J , Rosenbaum H , Gershoni-Baruch R ((2004) ) Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med 351: , 1972–1977. |
[102] | Chang D , Nalls MA , Hallgrímsdóttir IB , Hunkapiller J , van der Brug M , Cai F , Kerchner GA , Ayalon G , Bingol B , Sheng M , Hinds D , Behrens TW , Singleton AB , Bhangale TR , Graham RR ((2017) ) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49: , 1511–1516. |
[103] | Alcalay RN , Wolf P , Levy OA , Kang UJ , Waters C , Fahn S , Ford B , Kuo SH , Vanegas N , Shah H , Liong C , Narayan S , Pauciulo MW , Nichols WC , Gan-Or Z , Rouleau GA , Chung WK , Oliva P , Keutzer J , Marder K , Zhang XK ((2018) ) Alpha galactosidase A activity in Parkinson’s disease. Neurobiol Dis 112: , 85–90. |


