
Role of NLRP3 Inflammasome in Parkinson’s Disease and Therapeutic Considerations
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disease, with two main pathological features: misfolded α-synuclein protein accumulation and neurodegeneration. Inflammation has recently been identified as a contributor to a cascade of events that may aggravate PD pathology. Inflammasomes, a group of intracellular protein complexes, play an important role in innate immune responses to various diseases, including infection. In PD research, accumulating evidence suggests that α-synuclein aggregations may activate inflammasomes, particularly the nucleotide-binding oligomerization domain-leucine-rich repeat-pyrin domain-containing 3 (NLRP3) type, which exacerbates inflammation in the central nervous system by secreting proinflammatory cytokines like interleukin (IL)-18 and IL-1β. Afterward, activated NLRP3 triggers local microglia and astrocytes to release additional IL-1β. In turn, the activated inflammatory process may contribute to additional α-synuclein aggregation and cell loss. This review summarizes current research evidence on how the NLRP3 inflammasome contributes to PD pathogenesis, as well as potential therapeutic strategies targeting the NLRP3 inflammasome in PD.
INTRODUCTION
Parkinson’s disease (PD) is a neurodegenerative disease characterized by movement disorders caused by aggregation of misfolded α-synuclein (αSyn) and neuronal loss, particularly of dopaminergic neurons [1]. The inflammation has long been understood as one of the pathogenesis of PD through postmortem study, but recently it has been recognized that the progressive nature of PD is attributed to immune dysfunction of central and peripheral systems [2].
Inflammasomes are intracellular protein complexes that act as sensors of innate immune responses, activating the inflammatory cascade during various disease conditions such as infection [3]. The following evidence suggests the neuroinflammatory role of inflammasomes in PD pathogenesis [4]. In a postmortem study, increased constituent proteins of the inflammasome were found in the substantia nigra of PD brains [5, 6]. Furthermore, protein components of inflammasomes, which are abundant in peripheral blood mononuclear cells (PBMCs) in patients with PD, are considered potential biomarkers for PD [7]. In terms of αSyn pathology, the inflammasome effector protein caspase-1 enhances αSyn aggregation by inducing truncation of the αSyn protein [8]. In an animal model, Gordon et al. found a direct link between nucleotide-binding oligomerization domain (NOD)-leucine-rich repeat (LRR)-pyrin domain (PYD)-containing (NLRP3) and PD pathogenesis induced by αSyn preformed fibrils [6]. They also confirmed this theory by treating the mice with the small molecule MCC950 and observing its inhibitory effect on the NLRP3 inflammasome, which further reduced motor dysfunction, neurodegeneration, and αSyn accumulation. The hypothesis of the PD–inflammasome axis suggests that the NLRP3 inflammasome may collaborate with other contributors to PD progression, inducing αSyn spread and exacerbating neuroinflammation, which leads to more cell death.
In this review, we will explain how the NLRP3 inflammasome is activated in PD by discussing its priming and activation pathways and summarize some proposed PD treatments targeting this NLRP3 inflammasome based on its molecular pathways.
WHAT IS AN INFLAMMASOME?
Inflammasomes were first identified as a molecular platform of the innate immune system in 2002 [9]. The inflammasome is a cytosolic multiprotein complex mostly present in immune cells that can be assembled in response to pathogen or damage-associated molecular pattern (PAMP or DAMP) signals by pattern recognition receptors [3]. The maturation of the inflammasome and release of proinflammatory cytokines guide immune cell reactions during inflammation [3].
Scientists discovered several types of inflammasomes, four of which were well characterized: NLRP1 in 2002, NLRP3 and NLR family caspase recruitment domain (CARD) domain-containing protein 4 (NLRC4) (also known as ice protease-activating factor) in 2006, and absent in melanoma 2 (AIM2) in 2009 [3]. A wide range of research has been conducted to better understand this protein complex, and it has become clear that these inflammasomes have been linked to various diseases, including neurodegenerative, autoimmune, metabolic, and infectious diseases [10]. Accordingly, clinical trials targeting the function of these proteins have been developed for various diseases [11]. In 2008, the Food and Drug Administration approved rilonacept, an interleukin (IL)-1 Trap, and canakinumab, an anti-IL-1β with an NLRP3 inhibitory effect, to treat cryopyrin-associated periodic syndrome (CAPS). Later in 2015, Coll et al. demonstrated that the MCC950, which was originally designed to block IL-1β production, can also inhibit the NLRP3 inflammasome and thus improve the symptoms of multiple sclerosis and CAPS [12]. These breakthroughs ushered in a new decade of inflammasome research, with scientists believing that inhibitors of inflammasome have beneficial effects on cardiovascular diseases, metabolic diseases, cancer, and neurodegenerative diseases [13].
INVOLVEMENT OF INFLAMMASOME IN PD PATHOGENESIS
The activation of inflammasome was studied in various PD models. Table 1 summarizes the postmortem, in vitro, and in vivo characteristics of various inflammasomes found in the PD model. As shown in Table 1, the NLRP3 inflammasome was consistently found to be activated in animals, cells, human biofluids, and postmortem human studies of PD. However, other types of inflammasomes, such as NLRP1, NLRP2, NLRC4, and AIM2, showed little or no difference in activity in PD models.
Table 1
Characteristics of Inflammasomes in various PD models
| Type of inflammasome | Species | Sources | Models | Change in component of inflammasomes | Evaluation method | References |
| NLRP3 | Rodent | Midbrain | MPTP | ↑NLRP3/↑pro-CASP1/↑CASP1/↑pro-IL-1β/↑IL-1β | WB | [14] |
| Striatum/Substantia nigra | 6-OHDA/αSyn fibril | ↑pro-CASP1/↑CASP1/↑ASC | WB | [6] | ||
| Substantia nigra | Transgenic αSyn mutated A30P*A53T | ↑pro-IL-1β/↑IL-1β | WB/RT-qPCR | [15] | ||
| Whole brain | AVV-αSyn | ↑NLRP3/↑CASP1/↑ASC | WB | [16] | ||
| Primary microglia | αSyn fibril | ↑NLRP3/↑CASP1/↑pro-IL-1β/ ↑IL-1β | WB/ELISA | [16] | ||
| ↑IL-1β/↑NLRP3 | ELISA/WB | [17] | ||||
| Monomeric/oligomeric/ribbons αSyn | ↑IL-1β/↑NLRP3/↑ASC | ELISA/WB | [18] | |||
| Primary astrocyte | MPP+ | ↑NLRP3/↑CASP1/↑pro-IL-1β/↑IL-1β | WB | [19] | ||
| Primary cortical neurons | Rotenone | ↑IL-1β/↑IL-18/↑CASP1 | WB/RT-qPCR/ELISA/cytology | [15] | ||
| Human | Monocytes | αSyn fibril | ↑NLRP3/↑CASP1/↑pro-IL-1β/↑IL-1β | RT-qPCR/WB/ELISA | [20] | |
| PBMCs | Patients with PD | ↑NLRP3/↑ASC/↑CASP1/↑IL-1β ↑NLRP3/ ↑CASP1 | WB/RT-qPCR RT-qPCR/cytology | [7] [58] | ||
| Monomeric/fibril αSyn + LPS | ↑CASP1/↑IL-1β | ELISA/cytology | [21] | |||
| Primary microglia | αSyn fibril | ↑NLRP3/↑ASC/↑CASP1/↑pro-IL-1β/↑IL-1β | WB/ELISA/cytology | [17] | ||
| Extracellular vesicle from plasma | Patients with PD | ↑NLRP3/↑ASC/↑pro-CASP1/↑pro-GSDMD/↑GSDMD | WB | [23] | ||
| CSF | Patients with PD | ↑IL-1β/↑IL-18 | ELISA | [15] | ||
| Postmortem substantia nigra | Patients with PD | ↑pro-CASP1/↑CASP1/↑ASC | WB/histology | [6] | ||
| Postmortem mesencephalic tissues | Patients with PD | ↑NLRP3/↑ASC | Histology | [23] | ||
| ↑NLRP3 | Histology/RT-qPCR | [5] | ||||
| NLRP1 | Rodent | Midbrain | MPTP | ↑NLRP1/↑pro-CASP1/↑CASP1/↑pro-IL-1β/↑IL-1β | WB | [14] |
| Human | PBMCs | Patients with PD | No differences | WB/RT-qPCR | [7] | |
| NLRP2 | Rodent | Brain | MPTP | No differences | WB | [14] |
| NLRC4 | Human | PBMCs | Patients with PD | No differences | WB/RT-qPCR | [7] |
WB, western blot; RT-qPCR, reverse transcription-quantitative polymerase chain reaction; ELISA, enzyme-linked immunosorbent assay; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; MPP+, 1-methyl-4-phenylpyridinium (ion); PD, Parkinson’s disease; PBMCs, peripheral blood mononuclear cells; CSF, cerebrospinal fluid; 6-OHDA, 6-hydroxydopamine hydrobromide; AAV-αSyn, adeno-associated virus expresses αSyn; NLRP, nucleotide-binding oligomerization domain-leucine-rich repeat-pyrin domain-containing; NLRC4, NLR family caspase recruitment domain (CARD) domain-containing protein 4; AIM2, absent in melanoma 2; IL-1β, interleukin 1 beta; IL-18, interleukin 18; ASC, apoptosis-associated speck-like protein containing a CARD; CASP1, caspase-1; GSDMD, gasdermin D.
It is unclear why only the specific phenotype of the inflammasome is activated, but it is assumed that different types of stimuli from specific diseases may cause distinct activations in the specific phenotype of the inflammasome [22].
Besides increased NLRP3 activation in PD, several studies support the link between the inflammasome and clinical features in the disease. Increased NLRP3 protein was found in the postmortem PD brain, particularly in the late disease stages, suggesting an association with disease severity [5]. To better understand the link between inflammasome activation and disease severity, Anderson et al. investigated the inflammasome in postmortem substantia nigra pars compacta at different stages of neurodegeneration [23]. Unexpectedly, immunoreactive NLRP3 constituent proteins were found in patients with the intermediate stage of cell loss, indicating that inflammasome activation is not merely a late event or a result of the disease. Fan et al. also reported that the plasma NLRP3 inflammasome effector protein and IL-1β levels in patients with PD were positively correlated with the severity of motor dysfunction assessed by the Unified Parkinson’s Disease Rating Scale part III scores and Hoehn and Yahr staging [7].
NLRP3: STRUCTURE AND ACTIVATION PATHWAY
The NLRP3 inflammasome is derived from the sensor protein NLRP3 (Fig. 1). NIMA-related kinase 7 (NEK7) has recently been shown to activate NLRP3 by binding to the LRR domain and NOD [24, 25]. The NLRP3 PYD interacts with the apoptosis-associated speck-like protein containing a CARD (ASC) adaptor PYD. This PYD then assembles ASC multimers to create a protein complex called ASC speck [26]. When activated, the ASC CARD recruits an effector pro-caspase-1, resulting in NLRP3 oligomerization and the formation of a full inflammasome structure. Mature caspase-1 then induces the release of proinflammatory cytokines IL-1β and IL-18, as well as pyroptosis, a type of programmed cell death, by cleaving gasdermin D (GSDMD) and forming a membrane pore [27, 28].
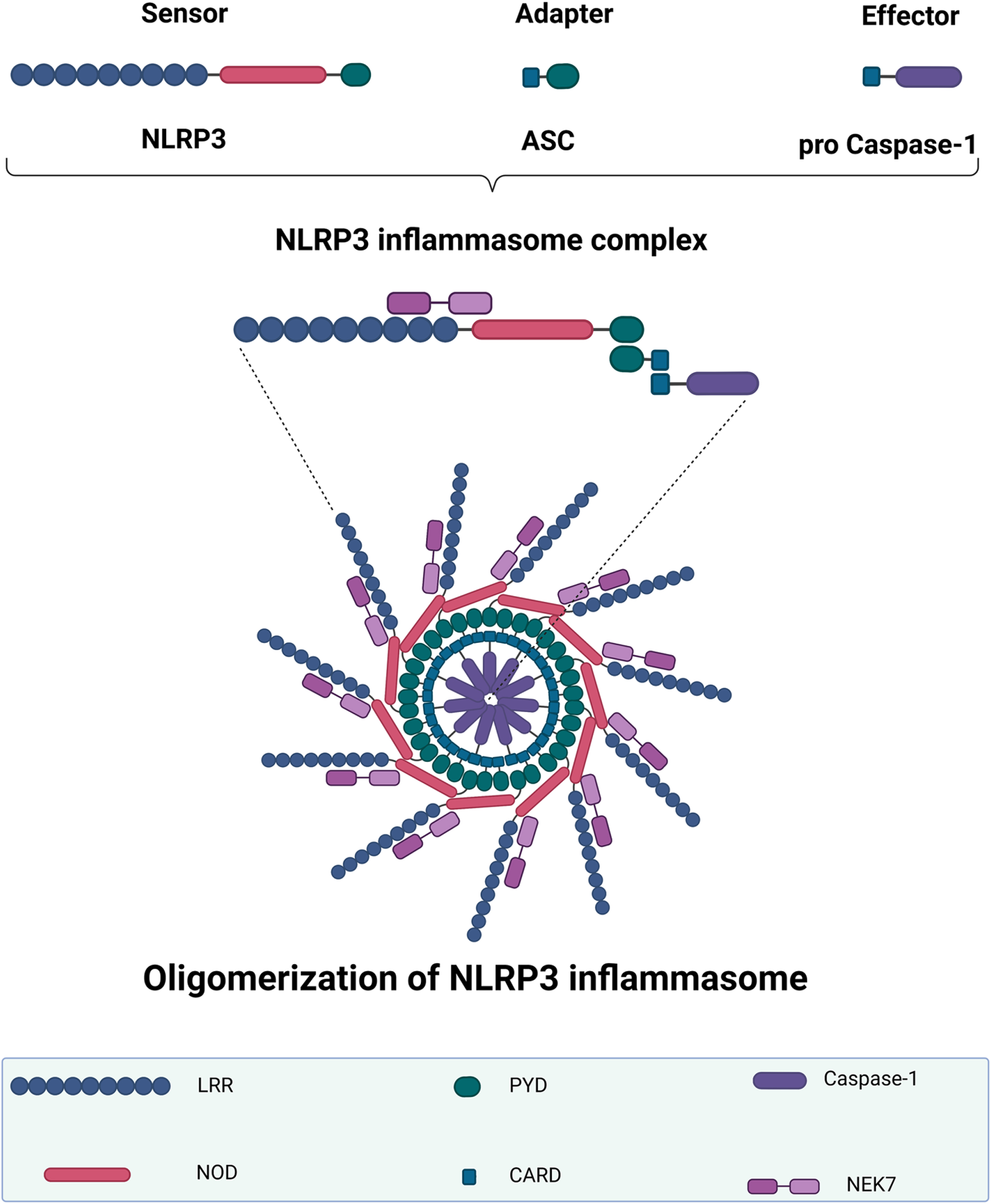
Fig. 1
NLRP3 inflammasome structure and oligomerization. The NLRP3 inflammasome complex includes a sensor NLRP3 protein, an adapter ASC protein, and a pro-caspase-1 protein. When the NLRP3 inflammasome is activated, the adaptor protein ASC binds to the sensor NLRP3 protein via PYD–PYD polymerization. NLRP3 oligomerization occurs after this ASC recruits the effector protein pro-caspase-1 via its CARD. The first half of NEK7 binds to the NLRP3 LRR domain, whereas the second half interacts with the NOD. NLRP3, NOD-like receptor family-pyrin domain-containing 3; LRR, leucine-rich repeat; NOD, nucleotide-binding oligomerization domain; PYD, pyrin domain; ASC, apoptosis-associated speck-like protein containing a CARD; CARD, caspase recruitment domain; NEK7, NIMA-related kinase 7. The graph was created using Biorender.com.
There are three types of NLRP3 inflammasome activation pathways: canonical, noncanonical, and alternative [29]. In the NLRP3-canonical pathway, two signals are required, corresponding to two processes: priming and activation. The priming pathway is produced when PAMPs or DAMPs activate the nuclear factor kappa B (NF-κB) pathway via the toll-like receptors (TLRs) or tumor necrosis factor receptor (TNFR) cytokine ligands, promoting pro-IL-1β and pro-IL-18 transcription [30, 31]. The activation pathway starts with PAMP or DAMP signals to assemble the full structure of inflammasomes with caspase-1 maturation. Pathogens that induce PAMPs include viruses, fungi, and bacterial pore-forming toxins [32–34]. Some factors have been identified as DAMPs, including amyloid-β, monosodium urate crystals, imidazoquinolinone compounds, and others [35–37].
Noncanonical NLRP3 activation, unlike canonical activation, is unrelated to caspase-1 activity. Caspase-11 in mice and its human orthologues, caspase-4/5, are involved in the intracellular lipopolysaccharide (LPS)-activating noncanonical pathway [38, 39]. When LPS activates a noncanonical NLRP3 pathway, caspases 4/5/11 cause pyroptotic cell death in a pattern similar to the canonical pathway, but proinflammatory cytokines are not directly secreted [40]. Ion flux following membrane pore formation could be the signal that recruits caspase-1 and cytokines [41].
Furthermore, for extracellular LPS, another activation pathway known as the “alternative pathway” exists in which caspase-8 (in humans) is activated via TLR4–adaptor protein TIR domain-containing adapter molecule 1 axis signals and directly triggers the assembly of NLRP3 without the involvement of a second signal [42].
NLRP3 CANONICAL PATHWAY IN PD
NLRP3 priming pathway in PD
In postmortem studies, TLR2 was found to be increased in the brains of patients with PD [43, 44]. Fitz et al. found that αSyn upregulated TLR gene expression in primary and BV-2 microglia [45]. They also found an increase in downstream MYD88 and NF-κB genes in primary microglia, indicating TLR activation. The hypothesis of αSyn as a priming signal via TLRs was supported once more when TLR4 was thought to be unreplaceable for αSyn-dependent activation of microglia and astrocytes [46]. TLR3 and TLR4 mRNA levels were higher in the brains of PD animal models, including 6-hydroxydopamine hydrobromide (6-OHDA), rotenone, and the LPS model [47]. The TLR4 deficiency in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model reduces NLRP3, ASC, pro-caspase-1, and IL-1β levels, supporting the role of TLRs and the NLRP3 axis in the PD context [48, 49]. Panicker et al. found a novel pathway in which αSyn binds to TLR2 and cluster of differentiation (CD36) receptors, priming NLRP3 via Fyn kinase, leading to NF-κB transcription, and finally assembling the full structure by inducing reactive oxygen species (ROS) release [16]. Notably, Scheiblich et al. demonstrated that αSyn monomer and oligomer could directly activate microglial NLRP3 to release IL-1β in primary microglia via TLR2/TLR5 and that the absence of NLRP3 also contributed to αSyn clearance [18]. Interestingly, they found that TLR2 contributed to priming, whereas the novel TLR5 acted on activation via an unknown mechanism.
Taken together, PD pathology such as αSyn may prime NLRP3 mainly via TLR2/4, leading to activation of NF-κB transcription and downstream pathways. Other priming pathways in PD that are mediated by different receptors, such as TNFR or IL-1R, have yet to be identified (Fig. 2).
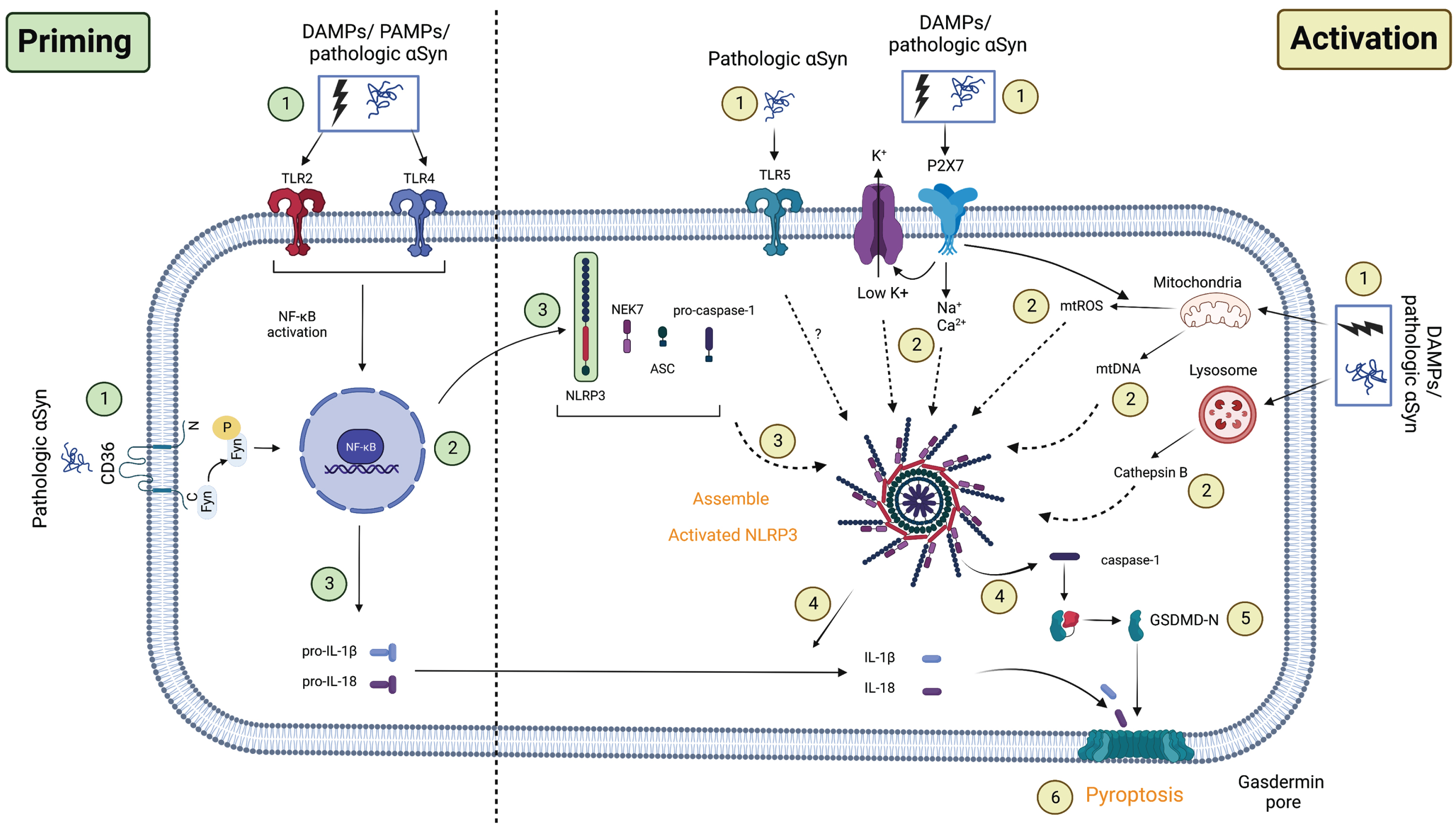
Fig. 2
Proposed NLRP3 priming and activation pathway in PD. Priming: (1) The first signals associated with the priming process may be caused by PAMPs/DAMPs or pathologic αSyn binding to TLRs, followed by (2) activation of downstream NF-κB transcription. This induces the upregulation of (3) NLRP3 and (3) pro-IL-1β/pro-IL-18 transcription in the cell. Activation: The second signal corresponding to the activation process may come from (1) the novel TLR5/ion flux in association with the P2X7 receptor/lysosome/mitochondrial dysfunction. These events induced (2) K+ efflux in addition to Na+/Ca+ influx, cathepsin B release, or mtROS/mtDNA production in the cell. When this process is completed, (3) NLRP3’s full structure will be recruited, releasing (4) caspase-1 and maturing (4) IL-1β/IL-18. Active caspase-1 now cleaves GSDMD (5) and forms gasdermin pores on the cell membrane, inducing programmed cell death known as (6) pyroptosis. Another novel pathway involves the facilitation of Fyn kinase, which allows (1) pathologic αSyn to directly prime and activate NLRP3 via the CD36 receptor and TLR2, as well as (2) mtROS secretion. PAMPs/DAMPs, pathogen/damage-associated molecular pattern; TLRs, toll-like receptors; NF-κB, nuclear factor kappa B; CD36, cluster of differentiation 36; P2X7, P2X purinoceptor 7; NEK7, NIMA-related kinase 7; NLRP3, NOD-like receptor family-pyrin domain-containing 3; mtROS, mitochondrial reactive oxygen species; mtDNA, mitochondrial DNA; ASC, apoptosis-associated speck-like protein containing a CARD; GSDMD, gasdermin D. The graph was created using Biorender.com.
NLRP3 activation pathway in PD
Via ion flux
The NLRP3 inflammasome is thought to be activated by ion fluxes, including K+ efflux, Na+ influx, and Ca2+ mobilization [50, 51]. The ion flux is linked to P2X purinoceptor 7 (P2X7), a member of the plasma membrane receptor family known as the P2 receptor family, which may be mediated by extracellular adenosine triphosphate (ATP) [52]. After stimulation, P2X7 collaborates with the two-pore-domain potassium channel K+ efflux channel to further mediate NLRP3-induced inflammation [53]. In a genetic study, the P2X7 receptor gene polymorphism (1513A>C) is associated with an increased risk of sporadic PD in the Han Chinese population [54]. This receptor is linked to the abundance of microglia and the inflammasomes put this link to PD into context [55]. Furthermore, P2X7 has recently been identified as a neuroinflammatory marker in PD [56]. In rats injected with 6-OHDA, neuroinflammation was marked by increased P2X7 colocalization with microglia, but no differences were observed in the αSyn over-expression models [57]. Recent research on PBMCs of patients with PD revealed NLRP3 activation via P2X7, which could trigger downstream pathways of NLRP3 inflammasome activation [58]. Blocking this receptor with an antagonist reduced IL-1β/IL-18 release in primary microglia from animal models or PBMCs from patients with PD [55, 58]. These findings indicate that PD pathology may activate the NLRP3 inflammasome via ion flux in relation to the P2X7 receptor.
Via lysosome
Several genes involved in autophagy–lysosomal function have been linked to PD pathogenesis, including GBA1, LRR kinase 2 (LRRK2), GALC, GLA, VPS35, SMPD1, and others [59, 60]. Furthermore, dysfunction in these genes may contribute to neuroinflammation. In Zebrafish, a loss-of-function mutation in GBA1 gene, which encodes for the lysosomal enzyme β-glucocerebrosidase, resulted in early microglial activation [61]. In the GBA1 knock-out mouse model, overexpressed human αSyn continued to induce microglial activation in the substantia nigra region [62]. In mouse primary microglia, LRRK2 could promote microglia activation and release neurotoxic mediators such as TNF-α and IL-6 [63]. These findings suggest that lysosomal damage may contribute to neuroinflammation in PD pathogenesis, although the precise pathway remains unknown.
A protease family of lysosomal cathepsins is thought to be involved in αSyn clearance and protein truncation [64, 65]. Furthermore, various types of cathepsin have been shown to function in microglia [66]. Interestingly, the relationship between cathepsin and NLRP3 activation was found in response to different types of DAMP or PAMP stimuli [67]. The absence of cathepsins in an in vitro or in vivo model reduced IL-1β release, indicating that they have an effect on the NLRP3 activation process [68–70]. In human monocytes, monomeric αSyn caused lysosomal leakage, resulting in the release of cathepsin B and the activation of NLRP3 [20]. These findings were confirmed in THP-1 cells, a human leukemia monocytic cell line, with the rupture of lysosome-induced cathepsin B and NLRP3 activation [71]. Cathepsin B, NLRP3, caspase-1, and IL-1β were found to be higher in A53T mutant microglial cells and animal models [72]. Overall, these studies proposed an activation mode of the NLRP3 inflammasome via cathepsin B release when PD pathology causes lysosomal dysfunction.
Via mitochondria
Like the lysosome, mitochondrial gene mutations, such as PINK1, PARK2, and VPS35, have been linked to PD pathology in rare familial cases [73]. In general, it is known that mitochondria contribute to the innate immune system during the disease process, especially by regulating NLRP3 inflammasome activation [74]. Interestingly, PINK1 and Parkin have been shown to modulate NLRP3 activation by affecting the TLR downstream pathway in microglia [75].
ROS is one of the by-products of the mitochondrial cellular process that may be the missing puzzle piece of the mitochondrial damage–inflammasome axis. Mitochondrial ROS (mtROS) regulates microglia activation during inflammation via mitogen-activated protein kinase and NF-κB [76]. In PD, αSyn caused mitochondrial damage-induced ROS release in human dopaminergic neurons [77]. Furthermore, αSyn could induce ROS release alongside NLRP3 activation by releasing caspase-1 and IL-1β in a THP-1 cell line [71]. Suppression of ROS by the fungus Antrodia amphorate polysaccharide in the 6-OHDA animal model significantly decreased NLRP3, ASC, and caspase-1 [78]. Interestingly, the P2X7 receptor was also involved in αSyn-induced mtROS release [79]. In addition to mtROS, mitochondrial DNA (mtDNA) is involved in the mitochondrial response to stress. The newly synthesized mtDNA, in particular, is responsible for oxidized mtDNA, which is thought to activate the NLRP3 inflammasome [80, 81]. The study of mtDNA release in serum revealed a significant difference between patients with idiopathic PD and those with PD due to the Parkin/PINK1 mutation [82]. Furthermore, a postmortem study of mtDNA levels in the cerebrospinal fluid revealed distinct levels between PD and other diseases [83]. However, direct evidence that mtDNA activates the NLRP3 inflammasome in PD using animal or cell models is still required for further investigation.
NONCANONICAL PATHWAY OF NLRP3 IN PD
In contrast to the canonical pathway, the noncanonical pathway of the NLRP3 in PD is poorly understood. For a decade, scientists assumed that αSyn only activated the NLRP3 inflammasome and released proinflammatory cytokines via caspase-1. In 2021, Pike et al. found that inhibiting caspase-1 activation did not reduce IL-1β in primary human microglia induced by fibrillar αSyn [17]. This was in contrast to their findings in primary mouse microglia, which were also induced by fibrillar αSyn but were entirely dependent on caspase-1. The authors suggested that the noncanonical NLRP3 activation via different caspases (caspase-8/11) was the pathway underlying the αSyn-induced PD model using human primary microglia. Caspase-11 was found to be involved in PD pathogenesis in vivo, but whether it is related to the inflammasome requires further investigation [84].
Taken together, we proposed that DAMPs (6-OHDA/MPTP/1-methyl-4-phenylpyridinium (ion)), PAMPs (LPS), or PD pathology (αSyn) may prime or activate the canonical NLRP3 inflammasome pathway via the aforementioned patterns, as shown in Fig. 2. Because the noncanonical NLRP3 activation and alternative activation pathways in PD remain unknown, we excluded them in this figure.
THERAPEUTIC TRIALS TARGETING THE NLRP3 INFLAMMASOME
Drug development for the NLRP3-related disease mechanism typically aims to prevent canonical activation of this inflammasome by (1) detaching the structure, (2) ceasing transcription during the priming pathway, (3) hindering activation signals (DAMPs) during the activation pathway, and (4) deactivating this inflammasome via post-translational modification. In terms of NLRP3 activation in PD, finding treatment targets should be based on the mechanism of NLRP3 activation. Therefore, in this section, we will discuss the potential treatment targets for inhibiting NLRP3 inflammasome activation that have been studied in other diseases as well as PD. Additionally, we will include the novel therapeutic candidates targeting the PD–NLRP3 axis with an unknown mechanism for future research consideration. Table 2 summarizes potential PD therapeutic candidates under consideration with known or unknown NLRP3 mechanisms.
Table 2
Current therapeutic trials targeting NLRP3 inflammasome in PD
| Mechanism | Target | Candidate | Model | Phase | References |
| Inhibiting NLRP3 constituents | NEK7 | MCC950 | Fibril αSyn | Preclinical | [6] |
| NLRP3 | MCC950 | Fibril αSyn | Preclinical | [6] | |
| Caspase-1 | Ac-YVAD-CMK | LPS/6-OHDA | Preclinical | [85] | |
| Inhibiting NLRP3 priming pathway | TLRs | Calycosin | MPTP | Preclinical | [86] |
| Cordycepin | MPTP | Preclinical | [87] | ||
| Glaucocalyxin B | LPS | Preclinical | [88] | ||
| NPT520-34 | Fibril αSyn | Phase I | (NCT03954600) [89] | ||
| NF-κB | Papaverine | MPTP+LPS | Launched | [90] | |
| Inhibiting NLRP3 activation pathway | P2X7 | Brilliant Blue G | MPTP | Preclinical | [91] |
| Memantine and dopamine receptor agonist | PBMCs of patients with PD | Launched | (NCT03918616) [58] | ||
| Cathepsin B | Ca074Me | N27/SH-SY5Y cells treated with fibril αSyn | Preclinical | [71] | |
| ROS | Antrodia camphorata polysaccharide | 6-OHDA | Preclinical | [78] | |
| Bushen–Yizhi | MPTP | Preclinical | [92] | ||
| Tenuiginen | MPTP | Preclinical | [93] | ||
| Fingolimod (FTY720) | MPTP/BV2 cell+mouse primary microglia treated with MPP+ | Preclinical | [94] | ||
| GSK-650394 | MPTP/overexpressing αSyn+fibril αSyn | Preclinical | [95] | ||
| Dl-3-n-butylphthalide | MPTP/6-OHDA | Preclinical | [96] | ||
| Inhibiting NLRP3 via post-translational modification | NLRP3 | Dopamine | MPTP/mouse BMDM treated with LPS | Launched | [97] |
| SP600125 | MPTP | Preclinical | [98] | ||
| ASC | Piceatannol | Fibril αSyn in vitro | Preclinical | [99] | |
| SIRT1 pathway | Unknown | Melatonin | MPTP/BV2 cell+mouse primary microglia treated with MPP++ATP | Launched | [100] |
| β-Arrestin2 mechanism | Unknown | Novel DRD2 agonist (UNC9995) DRD2 agonist (LY171555) | MPTP/mouse primary cultured astrocyte treated with LPS+ATP | Preclinical | [101, 102] |
| Nrf2 activation | Unknown | DDO-7263 | MPTP/THP-1 cell treated with LPS+ATP | Preclinical | [103] |
| Unknown | Unknown | Dimethylaminomylide (ACT001) | MPTP | Preclinical | [104] |
| Unknown | Unknown | Edaravone (MT-1186) | LPS/BV2 cell treated with LPS | Phase III for ALS | (NCT04577404) [105] |
| Unknown | Unknown | Ulinastatin | PC12 and SH-SY5Y cell treated with MPP+ | Launched | [106] |
NEK7, NIMA-related kinase 7; NLRP3, NOD-like receptor family-pyrin domain-containing 3; TLRs, toll-like receptors; NF-κB, nuclear factor kappa B; P2X7, P2X purinoceptor 7; ROS, reactive oxygen species; ASC, apoptosis-associated speck-like protein containing a CARD; LPS, lipopolysaccharide; 6-OHDA, 6-hydroxydopamine hydrobromide; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine.; MPP+, 1-methyl-4-phenylpyridinium (ion); PBMCs, peripheral blood mononuclear cells, ATP, adenosine triphosphate; DRD2, dopamine receptor D2; SIRT1, silence information regulator 1; Nrf2, nuclear factor erythroid 2-related factor 2; ALS, amyotrophic lateral sclerosis.
Preventing NLRP3 assembly by targeting each component protein
To fully activate the NLRP3 inflammasome, all inflammasome compositions need to be assembled. Therefore, current research aims to inhibit this assembling by targeting each of the constituent proteins: (1) NEK7; (2) NLRP3; (3) ASC adaptor protein; and (4) effector proteins, including caspase-1, (5) IL-1β, and IL-18.
In terms of NEK7, it is thought to be an upstream regulator of NLRP3 activation by binding to the LRR domain [24]. Therefore, preventing this association can reduce inflammasome activation. Oridonin (ORIN1001), a bioactive compound extracted from Rabdosia rubescens, has been shown to directly inhibit the NLRP3-NEK7 association by interacting with the NLRP3 NOD [107]. This compound recently demonstrated neuroprotective activity in traumatic brain injury in vivo models by inhibiting NLRP3 [108]. In an inflammatory bowel disease in vivo model, Chen et al. discovered the ability to suppress NEK7 expression of 4-methyl-N1-(3-phenylpropyl)-1,2-benzenediamine, JSH-23, an NF-κB transcriptional activity inhibitor, thereby downregulating the NLRP3 activity [109]. C1-27, a glutathione transferase omega-1 inhibitor, was proposed to inhibit NLRP3 via NEK7 deglutathionylation in multiple sclerosis in vivo model [110]. MCC950, a typical NLRP3 inhibitor, was recently shown to partially block NLRP3–NEK7 interaction in an in vivo lung ischemia–reperfusion model [111]. Ceritinib and lorlatinib, two anaplastic lymphoma kinase inhibitors, are thought to indirectly suppress the NLRP3–NEK7 interaction in ATP-stimulated macrophages [112]. However, only MCC950 was investigated in the PD model [6].
Some compounds directly target the NLRP3 subunit to inhibit this inflammasome. He et al. found that 3,4-methylenedioxy-β-nitrostyrene could bind to the LRR domain and NOD of NLRP3 and inhibit NLRP3 activation via its ATPase activity [113]. This hypothesis was recently validated by the finding that MCC950 interacts with the Walker B motif within the NLRP3 NOD to inhibit ATP hydrolysis and NLRP3 activation [114]. A number of acrylate derivative compounds, including IFN58, IFN39, and BOT-4-one, have a direct inhibitory effect on NLRP3 via its ATPase activity [115–117]. Two Inflazome candidates, IZD334 and Inzomelid, which have a direct inhibitory effect on NLRP3, have completed phase I trials in healthy volunteers and patients with CAPS (NCT04086602 and NCT04015076, respectively). OLT177 and CY-09 compounds were found to have an effect on osteoarthritis (OA) (NCT01768975) [118] and type 2 diabetes/CAPS in vivo models [119]. Similarly, Tranilast (Kissei) is thought to bind to the NOD of NLRP3 and prevent its oligomerization, and it was marketed in Japan and South Korea for its anti-allergic ability to CAPS (NCT03923140) [120]. However, with the exception of MCC950, no evaluation has yet been completed in PD [6].
The ASC adaptor protein may be a good target for inhibiting NLRP3 activation. The hydroxyl sulfonamide analog 5-chloro-N-[2-(4-hydroxysulfamoyl-phenyl)-ethyl]-2-methoxy-benzamide (JC-171) disrupts the NLRP3–ASC interaction in multiple sclerosis animal models [121]. Additionally, fenamate, a nonsteroidal anti-inflammatory drug, suppresses NLRP3 activation by inhibiting the ASC speck formation in Alzheimer’s disease (AD) rodent models [122]. Moreover, OLT1177, which suppresses NLRP3 via the NLRP3–ASC interaction, was found to be protective in an AD murine model [123]. Furthermore, beta-hydroxybutyrate inhibited NLRP3 activation by reducing ASC oligomerization to form specks in LPS-primed mouse macrophages [124]. However, this mechanism has not yet been evaluated in PD.
VX-765, one of the caspase-1 inhibitors, demonstrated neuroprotective effects in AD animal models by reducing neuropathology and cognitive impairment [125]. Another inhibitor, VX-740, was in phase II trials for rheumatoid arthritis but was discontinued because of hepatotoxicity in long-term treatment [126]. In a 6-OHDA-induced PD rat model, inhibiting caspase-1 activity by Ac-YVAD-CMK suppressed NLRP3 activation, rescued dopaminergic neurons in the substantia nigra region, and improved behavioral deficit [85]. Given that there are currently no clinical trials aimed at caspase-1 in PD, except for some animal data, targeting this is a real challenge.
In terms of inflammasome effector proteins, the use of IL-1β antibodies or IL-1 receptor antagonists such as anakinra, canakinumab, and rilonacept to treat NLRP3-related diseases has long been recognized [127]. However, its efficacy in targeting NLRP3 is questionable because suppressing these effectors may not prevent the release of other effectors, such as caspase-1, when NLRP3 is activated. Furthermore, because IL-1β and IL-18 are involved in not only the NLRP3 pathway but also other immune pathways, targeting this protein may accidentally increase infection risk in general.
Suspending NLRP3 activation by ceasing the priming process
Immunotherapy targeting TLR2/4, a ligand involved in the NLRP3 inflammasome transcription, can be applied to disrupt the priming process. Calycosin and cordycepin, two phytochemical compounds, showed neuroprotective effects in the MPTP model of PD by suppressing the TLR/NF-κB signaling pathway [86, 87]. Additionally, glaucocalyxin B, a diterpenoid, alleviated PD pathogenesis in the LPS model by inhibiting the same pathway [88]. Tomaralimab (NM-101), developed by Neuramedy and originally known as OPN-35 by Opsona Therapeutics, targets TLR2 and a phase II clinical trial in myelodysplastic syndrome (NCT02363491) was completed. Moreover, a small molecule, NPT520-34 (Neuropore), which has completed a phase I clinical trial (NCT03954600), reduced TLR2 mRNA expression in PD transgenic mice and thus improved neuropathology and motor deficits in these animals [89]. Furthermore, Ibudilast (MN-166) (MediciNova), which has completed phase II trials in multiple sclerosis (NCT01982942) and amyotrophic lateral sclerosis (NCT02714036), is another potential TLR4 antagonist. Notably, Ibudilast had a protective effect on the acute kidney injury model by inhibiting NLRP3 activation via the TLR4/NF-κB pathway [128]. Papaverine, a commercial drug, showed a protective effect on the PD animal model by acting on the downstream pathway of TLRs, the NF-κB-mediated NLRP3 activation [90].
Terminating the NLRP3 activation process by eliminating activation signals
In PD, the NLRP3 canonical activation pathway requires second signals from (1) ion flux in association with P2X7, (2) cathepsin B released in the ruptured lysosome, and (3) ROS. Therefore, it is currently thought that these mediators could be potential therapeutic targets for NLRP3-related diseases.
In terms of P2X7, P2X7 antagonists are novel for neurological diseases. For instance, JNJ54175446 and JNJ55308942, two candidates developed by Janssen, have completed phase I clinical trials (NCT02902601 and NCT03437590, respectively) and are expected to be used in major depressive disorder. However, CE-224535, another antagonist developed by Pfizer, has discontinued its clinical trial for OA due to a lack of efficacy (NCT00418782). Also, AZD9056 from AstraZeneca has completed its clinical trial in patients with rheumatoid arthritis but was clinically ineffective (NCT00520572). Conversely, GSK1482160 (GlaxoSmithKline), a CNS-penetrant antagonist, has completed a phase I clinical trial in inflammatory conditions (NCT00849134) and recently demonstrated a protective effect on the transgenic AD model [129]. Furthermore, Brilliant Blue G showed a protective effect on amyotrophic lateral sclerosis in vivo models and specifically PD animal models by reducing neuroinflammation caused by NLRP3 activation [130, 91]. In PD, common clinically used drugs, memantine and dopamine receptor agonists, changed P2X7-inflammasome activation in patients with de novo PD after 1 year of therapy (NCT03918616) [58].
Cathepsin B, which is released from the lysosome, can also be targeted to suppress NLRP3 activation. After treating the AD in vivo model with a prodrug CA074Me, which can inhibit cathepsin B after being converted into the active form, a protective effect for memory dysfunction and less amyloid-β plaque was observed [131]. Later, this inhibitor was found to inhibit ROS release in neuronal cells treated with aggregated αSyn [71]. Furthermore, aloxistatin (E64d), another cathepsin B inhibitor, is considered a promising candidate for traumatic brain injury [132]. In terms of the cathepsin B–inflammasome axis, the antimalaria drug hydroxychloroquine attenuated renal ischemia and reperfusion injury by preventing cathepsin release, resulting in NLRP3 inflammasome activation [133].
Preventing ROS released from damaged mitochondria and ruptured lysosomes could also be a potential treatment for neurodegenerative disease. In the 6-OHDA/MPTP-induced PD model, Antrodia camphorata polysaccharide, Bushen–Yizhi, and tenuigenin suppressed ROS-NLRP3 activation [78, 92, 93]. Notably, Dl-3-n-butylphthalide and fingolimod (FTY720), a sphingosine-1-phosphate receptor antagonist, suppressed mtROS, which further mediated NLRP3 inflammasome activation in the PD animal model [94, 96]. Furthermore, Kwon et al. proposed that GSK-650394, a serum/glucocorticoid-regulated kinase 1 inhibitor, could mitigate the pathogenesis of PD animal models by reducing mtROS and NLRP3 activation [95].
Abolishing NLRP3 activation via post-translational modification
Post-translational modifications such as phosphorylation, dephosphorylation, ubiquitination, deubiquitination, proteolytic processing, and others have been shown to positively and negatively regulate NLRP3 inflammasome activation. Each pathway uses a different mechanism to target the various NLRP3 inflammasome constituent domains, such as NLRP3 sensor protein, ASC adaptor protein, and caspase-1. This review will not go into detail but will instead suggest some therapeutic candidates based on evidence from previous PD research.
Yan et al. found that dopamine could suspend NLRP3 inflammasome activation by binding to dopamine receptor 1, mediating NLRP3 ubiquitination via E3 ubiquitin ligase MARCH7 [97]. This suggests a novel therapeutic approach for PD and other diseases involving NLRP3. Additionally, SP600125, a small molecule, is believed to have a neuroprotective effect on an MPTP-induced PD model [98]. Notably, this c-Jun N-terminal kinase inhibitor could prevent NLRP3 oligomerization via suppressing c-Jun N-terminal kinase 1-induced NLRP3 phosphorylation [134]. Furthermore, piceatannol, a natural compound that inhibits NLRP3 activation via the spleen tyrosine kinase Syk-induced ASC phosphorylation pathway, could reduce αSyn aggregation [99].
Other novel NLRP3 inhibitors for PD with known and unknown mechanisms
Zheng et al. discovered that melatonin could reduce neuroinflammation in PD animal models by reducing NLRP3 inflammasome activation [100], which was modulated by silence information regulator 1 (SIRT1), a histone deacetylase. However, whether SIRT1 inhibits NLRP3 by targeting its constituent proteins or through the priming or activation process remains unknown. Two dopamine receptor D2 agonists, UNC9995 and LY171555, were also shown to inhibit NLRP3 in the PD in vivo model [101, 102]. The beta arrestin 2 pathway (β-arrestin2) was suggested as the underlying mechanism of this process due to the β-arrestin2 and NLRP3 interaction and colocalization, but the direct mechanism of action of these compounds remains unknown. Another novel compound, 5-(3,4-difluorophenyl)-3-(6-methylpyridin-3-yl)-1,2,4-oxadiazole (DDO-7263), could reduce neuroinflammation in a PD in vivo model [103], by activating a transcription factor, nuclear factor E2-related factor 2. Nonetheless, the exact mechanism remains unknown. Furthermore, three other candidates, dimethylaminomylide (ACT001), edaravone (MT-1186), and ulinastatin, were recently found to suppress NLRP3 activation via unknown mechanisms [104–106].
DISCUSSION AND FUTURE PERSPECTIVES
To summarize, the NLRP3 inflammasome is activated in PD. PD pathology, such as misfolded αSyn, may activate the canonical pathway of NLRP3 via two processes: priming and activation. TLRs are the main sensor receptors in the priming pathway, and the activation pathway includes ion flux, mitochondrial damage, and lysosome rupture. After activation, the NLRP3 inflammasome releases caspase-1 and mature IL-1β/IL-18, which contribute significantly to PD pathogenesis by inducing neuroinflammation. Therefore, based on this mechanism, developing therapeutic trials for PD targeting NLRP3 inflammasome activation would be beneficial.
Current therapeutic attempts targeting inflammasome in PD are mostly in the preclinical stage, but this is changing because of the emerging mechanism involving inflammasome. However, because the inflammasome priming and activation pathways are diverse and the noncanonical pathway can replace the canonical pathway even in PD, targeting NLRP3 constituents or assembly may be a more efficient way than targeting one part of the various inflammasome cascades. For instance, direct targeting on NLRP3 constituents or the upstream transcription pathway via TLRs is more favorable. This is because inhibiting the effector proteins (caspases, IL-1β, IL-18, or GSDMD) may only hinder the downstream events but may not completely hinder the NLRP3 inflammasome activation pathway, which begins with pro-IL-1β/pro-IL-18 and NLRP3 upregulation (Fig. 2). Otherwise, we should look for the major stream among these alternatives or consider a drug with a multi-action mechanism on the inflammasome pathway or a drug with a general anti-inflammatory effect.
Reviewing previous and ongoing trials targeting inflammasomes in other diseases will help to find shortcuts for therapeutics in PD. Clinical trials targeting constituent proteins, NEK7, NLRP3, and ASC, and inflammasome assembly have already been conducted in other diseases, and the results of those trials allow us to select the candidate with the highest priority.
Previous research suggests that αSyn, like DAMPs or PAMPs, may activate the inflammasome pathway, but the relationship between αSyn and the inflammasome pathway needs to be investigated further in various aspects. It is unclear whether different αSyn species cause different patterns of NLRP3 inflammasome activation. In PD rodent models, αSyn fibril canonically activates the NLRP3 inflammasome [6]. Nonetheless, this pattern is not consistent in PD cell models. Although Panicker et al. suggested that fibrillar αSyn can prime and activate the NLRP3 inflammasome in mouse primary microglia in the absence of a priming signal (LPS), Scheiblich et al. later found that monomeric or oligomeric αSyn was sufficient to induce priming and activation [16, 18].
The pathomechanism of PD is based not only on the inflammasome but also on its interaction with other complex mechanisms. Therefore, even if all the inflammasome-related pathways are blocked, it is possible that misfolded αSyn accumulation, a dysfunctional protein clearance system, or other inflammation pathways and subsequent neurodegeneration will not be hindered. That is, understanding integrated mechanisms in PD is more important than focusing solely on peripheral issues. Therefore, clinical trials targeting inflammasome in PD may take a long time to observe, delaying the clinical progression of PD because of the impossibility of blocking all of these complex mechanisms. It implies that a strategic approach as well as reliable biomarkers to monitor disease progression is necessary for PD treatment.
Additionally, although the activation of the inflammasome and its related pathway in PD has been studied in recent years, the relationship between inflammasome and clinical characteristics of PD, including disease stage or motor and nonmotor features, has yet to be investigated. The differential manifestations of the inflammasome based on the clinical features of PD would provide a thorough understanding of the inflammasome’s role in PD.
Although the NLRP3 inflammasome is currently a promising target for PD treatment, there is a considerable knowledge gap in the complex interaction between the NLRP3 inflammasome and other PD mechanisms and finding promising therapeutic candidates and demonstrating efficacy even in complex mechanisms.
ACKNOWLEDGMENTS
This research was supported by Hallym University Research Fund and the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (2020R1F1A1076697).
CONFLICT OF INTEREST
The authors have no financial conflict of interest related to this study.
REFERENCES
[1] | Kalia LV , Lang AE ((2015) ) Parkinson’s disease. Lancet 386: , 896–912. |
[2] | Terkelsen MH , Klaestrup IH , Hvingelby V , Lauritsen J , Pavese N , Romero-Ramos M (2022) Neuroinflammation and immune changes in prodromal Parkinson’s disease and other synucleinopathies. JParkinsons Dis, doi: 10.3233/JPD-223245. |
[3] | Schroder K , Tschopp J ((2010) ) The inflammasomes. Cell 140: , 821–832. |
[4] | Voet S , Srinivasan S , Lamkanfi M , van Loo G ((2019) ) Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med 11: , 10248. |
[5] | von Herrmann KM , Salas LA , Martinez EM , Young AL , Howard JM , Feldman MS , Christensen BC , Wilkins OM , Lee SL , Hickey WF , Havrda MC ((2018) ) NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson’s disease. NPJ Parkinsons Dis 4: , 24. |
[6] | Gordon R , Albornoz EA , Christie DC , Langley MR , Kumar V , Mantovani S , Robertson AAB , Butler MS , Rowe DB , O’Neill LA , Kanthasamy AG , Schroder K , Cooper MA , Woodruff TM ((2018) ) Inflammasome inhibition prevents -synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med 10: , eaah4066. |
[7] | Fan Z , Pan YT , Zhang ZY , Yang H , Yu SY , Zheng Y , Ma JH , Wang XM ((2020) ) Systemic activation of NLRP3 inflammasome and plasma α-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J Neuroinflammation 17: , 11. |
[8] | Wang W , Nguyen LTT , Burlak C , Chegini F , Guo F , Chataway T , Ju S , Fisher OS , Miller DW , Datta D , Wu F , Wu CX , Landeru A , Wells JA , Cookson MR , Boxer MB , Thomas CJ , Gai WP , Ringe D , Petsko GA , Hoang QQ ((2016) ) Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein α-synuclein. Proc Natl Acad Sci U S A 113: , 9587–9592. |
[9] | Martinon F , Burns K , Tschopp J ((2002) ) The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10: , 417–426. |
[10] | Strowig T , Henao-Mejia J , Elinav E , Flavell R ((2012) ) Inflammasomes in health and disease. Nature 481: , 278–286. |
[11] | Tweedell RE , Kanneganti TD ((2020) ) Advances in inflammasome research: Recent breakthroughs and future hurdles. Trends Mol Med 26: , 969–971. |
[12] | Coll RC , Robertson AAB , Chae JJ , Higgins SC , Muñoz-Planillo R , Inserra MC , Vetter I , Dungan LS , Monks BG , Stutz A , Croker DE , Butler MS , Haneklaus M , Sutton CE , Núñez G , Latz E , Kastner DL , Mills KHG , Masters SL , Schroder K , Cooper MA , O’neill LAJ ((2015) ) A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 21: , 248–257. |
[13] | Li Y , Huang H , Liu B , Zhang Y , Pan X , Yu X-Y , Shen Z , Song Y-H ((2021) ) Inflammasomes as therapeutic targets in human diseases. Signal Transduct Target Ther 6: , 1–14. |
[14] | Han X , Sun S , Sun Y , Song Q , Zhu J , Song N , Chen M , Sun T , Xia M , Ding J , Lu M , Yao H , Hu G ((2019) ) Small molecule-driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: Implications for Parkinson disease. Autophagy 15: , 1860–1881. |
[15] | Zhang P , Shao XY , Qi GJ , Chen Q , Bu LL , Chen LJ , Shi J , Ming J , Tian B ((2016) ) Cdk5-dependent activation of neuronal inflammasomes in Parkinson’s disease. Mov Disord 31: , 366–376. |
[16] | Panicker N , Sarkar S , Harischandra DS , Neal M , Kam T-I , Jin H , Saminathan H , Langley M , Charli A , Samidurai M , Rokad D , Ghaisas S , Pletnikova O , Dawson VL , Dawson TM , Anantharam V , Kanthasamy AG , Kanthasamy A ((2019) ) Fyn kinase regulates misfolded α-synuclein uptake and NLRP3 inflammasome activation in microglia. J Exp Med 216: , 1411–1430. |
[17] | Pike AF , Varanita T , Herrebout MAC , Plug BC , Kole J , Musters RJP , Teunissen CE , Hoozemans JJM , Bubacco L , Veerhuis R ((2021) ) α-Synuclein evokes NLRP3 inflammasome-mediated IL-1β secretion from primary human microglia. Glia 69: , 1413–1428. |
[18] | Scheiblich H , Bousset L , Schwartz S , Griep A , Latz E , Melki R , Heneka MT ((2021) ) Microglial NLRP3 inflammasome activation upon TLR2 and TLR5 ligation by distinct α-synuclein assemblies. J Immunol 207: , 2143–2154. |
[19] | Lu M , Sun XL , Qiao C , Liu Y , Ding JH , Hu G ((2014) ) Uncoupling protein 2 deficiency aggravates astrocytic endoplasmic reticulum stress and nod-like receptor protein 3 inflammasome activation. Neurobiol Aging 35: , 421–430. |
[20] | Codolo G , Plotegher N , Pozzobon T , Brucale M , Tessari I , Bubacco L , de Bernard M ((2013) ) Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLos One 8: , e55375. |
[21] | Piancone F , Saresella M , la Rosa F , Marventano I , Meloni M , Navarro J , Clerici M ((2021) ) Inflammatory responses to monomeric and aggregated α-synuclein in peripheral blood of Parkinson disease patients. Front Neurosci 15: , 639646. |
[22] | Bauernfeind F , Ablasser A , Bartok E , Kim S , Schmid-Burgk J , Cavlar T , Hornung V ((2011) ) Inflammasomes: Current understanding and open questions. Cell Mol Life Sci 68: , 765–783. |
[23] | Anderson FL , von Herrmann KM , Andrew AS , Kuras YI , Young AL , Scherzer CR , Hickey WF , Lee SL , Havrda MC ((2021) ) Plasma-borne indicators of inflammasome activity in Parkinson’s disease patients. NPJ Parkinsons Dis 7: , 2. |
[24] | Sharif H , Wang li , li Wang W , Giri Magupalli V , Andreeva liudmila , Qiao Q , Hauenstein A v , Wu Z , Núñez G , Mao Y , Wu H , Wang L , Li Wang W ((2019) ) Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 570: , 338–343. |
[25] | Shi H , Wang Y , Li X , Zhan X , Tang M , Fina M , Su L , Pratt D , Hui Bu C , Hildebrand S , Lyon S , Scott L , Quan J , Sun Q , Russell J , Arnett S , Jurek P , Chen D , Kravchenko V v , Mathison JC , Moresco EMY , Monson NL , Ulevitch RJ , Beutler B ((2016) ) NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 17: , 250–258. |
[26] | Lu A , Magupalli V , Ruan J , Yin Q , Atianand MK , Vos M , Schröder GF , Fitzgerald KA , Wu H , Egelman EH ((2014) ) Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 156: , 1193–1206. |
[27] | Shi J , Zhao Y , Wang K , Shi X , Wang Y , Huang H , Zhuang Y , Cai T , Wang F , Shao F ((2015) ) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: , 660–665. |
[28] | Liu X , Zhang Z , Ruan J , Pan Y , Magupalli VG , Wu H , Lieberman J ((2016) ) Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535: , 153–158. |
[29] | Mangan MSJ , Olhava EJ , Roush WR , Seidel HM , Glick GD , Latz E ((2018) ) Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov 17: , 588–606. |
[30] | Franchi L , Eigenbrod T , Núñez G ((2009) ) Cutting Edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol 183: , 792–796. |
[31] | Bauernfeind FG , Horvath G , Stutz A , Alnemri ES , MacDonald K , Speert D , Fernandes-Alnemri T , Wu J , Monks BG , Fitzgerald KA , Hornung V , Latz E ((2009) ) Cutting Edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 183: , 787–791. |
[32] | Craven RR , Gao X , Allen IC , Gris D , Wardenburg JB , McElvania-TeKippe E , Ting JP , Duncan JA ((2009) ) Staphylococcus aureus α-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS One 4: , e7446. |
[33] | Gross O , Poeck H , Bscheider M , Dostert C , Hannesschläger N , Endres S , Hartmann G , Tardivel A , Schweighoffer E , Tybulewicz V , Mocsai A , Tschopp J , Ruland J ((2009) ) Syk kinase signalling couples to the Nlrp3 inflammasome for anti-fungal host defence. Nature 459: , 433–436. |
[34] | Allen IC , Scull MA , Moore CB , Holl EK , McElvania-TeKippe E , Taxman DJ , Guthrie EH , Pickles RJ , Ting JPY ((2009) ) The NLRP3 inflammasome mediates innate immunity to influenza A virus through recognition of viral RNA. in vivoImmunity 30: , 556–565. |
[35] | Kanneganti TD , Özören N , Body-Malapel M , Amer A , Park JH , Franchi L , Whitfield J , Barchet W , Colonna M , Vandenabeele P , Bertin J , Coyle A , Grant EP , Akira S , Núñez G ((2006) ) Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440: , 233–236. |
[36] | Halle A , Hornung V , Petzold GC , Stewart CR , Monks BG , Reinheckel T , Fitzgerald KA , Latz E , Moore KJ , Golenbock DT ((2008) ) The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol 9: , 857–865. |
[37] | Martinon F , Petrilli V , Mayor A , Tardivel A , Tschopp J ((2006) ) Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440: , 237–241. |
[38] | Kayagaki N , Warming S , Lamkanfi M , vande Walle L , Louie S , Dong J , Newton K , Qu Y , Liu J , Heldens S , Zhang J , Lee WP , Roose-Girma M , Dixit VM ((2011) ) Non-canonical inflammasome activation targets caspase-11. Nature 479: , 117–121. |
[39] | Baker PJ , Boucher D , Bierschenk D , Tebartz C , Whitney PG , D’Silva DB , Tanzer MC , Monteleone M , Robertson AAB , Cooper MA , Alvarez-Diaz S , Herold MJ , Bedoui S , Schroder K , Masters SL ((2015) ) NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol 45: , 2918–2926. |
[40] | Yang J , Zhao Y , Shao F ((2015) ) Non-canonical activation of inflammatory caspases by cytosolic LPS in innate immunity. Curr Opin Immunol 32: , 78–83. |
[41] | Rühl S , Broz P ((2015) ) Caspase-11 activates a canonical NLRP3 inflammasome by promoting+efflux. Eur J Immunol 45: , 2927–2936. |
[42] | Gaidt MM , Ebert TS , Chauhan D , Schmidt T , Schmid-Burgk JL , Rapino F , Robertson AAB , Cooper MA , Graf T , Hornung V ((2016) ) Human monocytes engage an alternative inflammasome pathway. Immunity 44: , 833–846. |
[43] | Doorn KJ , Moors T , Drukarch B , Dj Van De Berg W , Lucassen PJ , van Dam A-M ((2014) ) Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol Commun 2: , 90. |
[44] | Dzamko N , Gysbers A , Perera G , Bahar A , Shankar A , Gao J , Fu Y , Halliday GM ((2017) ) Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol 3: , 303–319. |
[45] | Fitz N , Maguire-Zeiss KA , Beraud D , Twomey M , Bloom B , Mittereder A , Ton V , Neitzke K , Chasovskikh S , Mhyre TR ((2011) ) α-Synuclein alters toll-like receptor expression. Front Neurosci 5: , 80. |
[46] | Fellner L , Irschick R , Schanda K , Reindl M , Klimaschewski L , Poewe W , Wenning GK , Stefanova N ((2013) ) Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 61: , 349–360. |
[47] | McCabe K , Concannon RM , McKernan DP , Dowd E ((2017) ) Time-course of striatal Toll-like receptor expression in neurotoxic, environmental and inflammatory rat models of Parkinson’s disease. J Neuroimmunol 310: , 103–106. |
[48] | Campolo M , Paterniti I , Siracusa R , Filippone A , Esposito E , Cuzzocrea S ((2019) ) TLR4 absence reduces neuroinflammation and inflammasome activation in Parkinson’s diseases in vivo model. Brain Behav Immun 76: , 236–247. |
[49] | Shao Q-H , Chen Y , Li F-F , Wang S , Zhang X-L , Yuan Y-H , Chen N-H ((2019) ) TLR4 deficiency has a protective effect in the MPTP/probenecid mouse model of Parkinson’s disease. Acta Pharmacol Sin 40: , 1503–1512. |
[50] | Muñoz-Planillo R , Kuffa P , Martínez-Colón G , Smith BL , Rajendiran TM , Núñez G ((2013) ) K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38: , 1142–1153. |
[51] | Lee GS , Subramanian N , Kim AI , Aksentijevich I , Goldbach-Mansky R , Sacks DB , Germain RN , Kastner DL , Chae JJ ((2012) ) The calcium-sensing receptor regulates the NLRP3 inflammasome through Ca2+ and cAMP. Nature 492: , 123–127. |
[52] | Coddou C , Yan Z , Obsil T , Huidobro-Toro JP , Stojilkovic SS ((2011) ) Activation and regulation of purinergic P2X receptor channels.. Pharmacol Rev 63: .A-AQ. |
[53] | Anke Di , Shiqin Xiong , Zhiming Ye , Thirumala-Devi Kanneganti , Jalees Rehman , Asrar Malik B. ((2018) ) The TWIK2 potassium efflux channel in macrophages mediates NLRP3 inflammasome-induced inflammation. Immunity 49: , 56–65. |
[54] | Liu H , Han X , Li Y , Zou H , Xie A ((2013) ) Association of P2X7 receptor gene polymorphisms with sporadic Parkinson’s disease in a Han Chinese population. Neurosci Lett 546: , 42–45. |
[55] | He Y , Taylor N , Fourgeaud L , Bhattacharya A ((2017) ) The role of microglial P2X7: Modulation of cell death and cytokine release. J Neuroinflammation 14: , 1–13. |
[56] | Van Weehaeghe D , Koole M , Schmidt ME , Deman S , Jacobs AH , Souche E , Serdons K , Sunaert S , Bormans G , Vandenberghe W , Van Laere K ((2019) ) [11C]JNJ54173717, a novel P2X7 receptor radioligand as marker for neuroinflammation: Human biodistribution, dosimetry, brain kinetic modelling and quantification of brain P2X7 receptors in patients with Parkinson’s disease and healthy volunteers. Eur J Nucl Med Mol Imaging 46: , 2051–2064. |
[57] | Pozo D , Xilouri M , Selvaraj S , Crabbe M , van der Perren A , Bollaerts I , Kounelis S , Baekelandt V , Bormans G , Casteels C , Moons L , van Laere K ((2019) ) Increased P2X7 receptor binding is associated with neuroinflammation in acute but not chronic rodent models for Parkinson’s disease. Front Neurosci 13: , 799. |
[58] | Solini A , Rossi C , Santini E , Giuntini M , Raggi F , Parolini F , Biancalana E , del Prete E , Bonuccelli U , Ceravolo R ((2021) ) P2X7 receptor/NLRP3 inflammasome complex and α-synuclein in peripheral blood mononuclear cells: A prospective study in neo-diagnosed, treatment-naïve Parkinson’s disease. Eur J Neurol 28: , 2648–2656. |
[59] | Chang D , Nalls MA , Hallgrímsdóttir IB , Hunkapiller J , van der Brug M , Cai F ; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team, Kerchner GA , Ayalon G , Bingol B , Sheng M , Hinds D , Behrens TW , Singleton AB , Bhangale TR , Graham RR ((2017) ) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49: , 1511–1516. |
[60] | Robak LA , Jansen IE , van Rooij J , Uitterlinden AG , Kraaij R , International Parkinson’s disease Genomics Consortium (IPDGC), Heutink P , Shulman JM ((2017) ) Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140: , 3191–3203. |
[61] | Keatinge M , Bui H , Menke A , Chen Y-C , Sokol AM , Bai Q , Ellett F , da Costa M , Burke D , Gegg M , Trollope L , Payne T , Mctighe A , Mortiboys H , de Jager S , Nuthall H , Kuo M-S , Fleming A , Schapira AH , Renshaw SA , Highley JR , Chacinska A , Panula P , Burton EA , O’neill MJ , Bandmann O ((2015) ) Glucocerebrosidase 1 deficient Danio rerio mirror key pathological aspects of human Gaucher disease and provide evidence of early microglial activation preceding alpha-synuclein-independent neuronal cell death. Hum Mol Genet 24: , 6640–6652. |
[62] | Soria FN , Engeln M , Martinez-Vicente M , Glangetas C , Opez-Gonz Alez J , Dovero S , Dehay B , Normand E , Vila M , Favereaux A , Georges F , lo Bianco C , Bezard E , Fernagut P-O ((2017) ) Glucocerebrosidase deficiency in dopaminergic neurons induces microglial activation without neurodegeneration. Hum Mol Genet 26: , 2603–2615. |
[63] | Kim C , Beilina A , Smith N , Li Y , Kim M , Kumaran R , Kaganovich A , Mamais A , Adame A , Iba M , Kwon S , Lee W-J , Shin S-J , Rissman R , You S , Lee S-J , Singleton AB , Cookson MR , Masliah E ((2020) ) LRRK2 mediates microglial neurotoxicity via NFATc2 in rodent models of synucleinopathies. Sci Transl Med 12: , eaay0399. |
[64] | McGlinchey RP , Lee JC ((2015) ) Cysteine cathepsins are essential in lysosomal degradation of α-synuclein. Proc Natl Acad Sci U S A 112: , 9322–9327. |
[65] | McGlinchey RP , Lacy SM , Huffer KE , Tayebi N , Sidransky E , Lee JC ((2019) ) C-terminal α-synuclein truncations are linked to cysteine cathepsin activity in Parkinson’s disease. J Biol Chem 294: , 9973–9984. |
[66] | Nakanishi H ((2020) ) Cathepsin regulation on microglial function. Biochim Biophys Acta Proteins Proteom 1868: , 140465. |
[67] | Campden RI , Zhang Y ((2019) ) The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch Biochem Biophys 670: , 32–42. |
[68] | Sun L , Wu Z , Hayashi Y , Peters C , Tsuda M , Inoue K , Nakanishi H ((2012) ) Neurobiology of disease microglial cathepsin B contributes to the initiation of peripheral inflammation-induced chronic pain. J Neurosci 32: , 11330–11342. |
[69] | Terada K , Yamada J , Hayashi Y , Wu Z , Uchiyama Y , Peters C , Nakanishi H ((2010) ) Involvement of cathepsin B in the processing and secretion of interleukin-1β in chromogranin a-stimulated microglia. Glia 58: , 114–124. |
[70] | Bogyo RM , Robertson SA , Gregory KL , Orlowski M , Colbert JD , Sharma S ((2015) ) Multiple cathepsins promote Pro-IL-1β synthesis and NLRP3-mediated IL-1β activation. J Immunol 195: , 1685–1679. |
[71] | Freeman D , Cedillos R , Choyke S , Lukic Z , Mcguire K ((2013) ) Alpha-synuclein induces lysosomal rupture and cathepsin dependent reactive oxygen species following endocytosis. PLoS One 8: , e62143. |
[72] | Zhou Y , Lu M , Du R-H , Qiao C , Jiang C-Y , Zhang K-Z , Ding J-H , Hu G ((2016) ) MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol Neurodegener 11: , 1–15. |
[73] | Bose A , Beal MF ((2016) ) Mitochondrial dysfunction in Parkinson’s disease. J Neurochem 139: , 216–231. |
[74] | Zhou R , Yazdi AS , Menu P , Tschopp J ((2011) ) A role for mitochondria in NLRP3 inflammasome activation. Nature 469: , 221–225. |
[75] | Mouton-Liger F , Rosazza T , Sepulveda-Diaz J , Amelie I , Hassoun S-M , Claire E , Mangone G , Brice A , Michel PP , Corvol J-C , Corti O ((2018) ) Parkin deficiency modulates NLRP3 inflammasome activation by attenuating an A20-dependent negative feedback loop. Glia 66: , 1763–1751. |
[76] | Park J , Min JS , Kim B , Chae UB , Yun JW , Choi MS , Kong IK , Chang KT , Lee DS ((2015) ) Mitochondrial ROS govern the LPS-induced pro-inflammatory response in microglia cells by regulating MAPK and NF-κB pathways. Neurosci Lett 584: , 191–196. |
[77] | Ganjam GK , Bolte K , Matschke LA , Neitemeier S , Dolga AM , Höllerhage M , Höglinger GU , Adamczyk A , Decher N , Oertel WH , Culmsee C ((2019) ) Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis 10: , 865. |
[78] | Han C , Shen H , Yang Y , Sheng Y , Wang J , Li W , Zhou X , Guo L , Zhai L , Guan Q , Liping Zhai C ((2020) ) Antrodia camphorata polysaccharide resists 6-OHDA-induced dopaminergic neuronal damage by inhibiting ROS-NLRP3 activation. Brain Behav 10: , 1–10. |
[79] | Wilkaniec A , Cieślik M , Murawska E , Babiec L , Gassowska-Dobrowolska M , Pałasz E , Jeśko H , Adamczyk A ((2020) ) P2X7 receptor is involved in mitochondrial dysfunction induced by extracellular alpha synuclein in neuroblastoma SH-SY5Y cells. Int J Mol Sci 21: , 3959. |
[80] | Shimada K , Crother TR , Karlin J , Dagvadorj J , Chiba N , Chen S , Krishnan Ramanujan V , Wolf AJ , Vergnes L , Ojcius DM , Rentsendorj A , Vargas M , Guerrero C , Wang Y , Fitzgerald KA , Underhill DM , Town T , Arditi M ((2012) ) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: , 401–414. |
[81] | Zhong Z , Liang S , Sanchez-Lopez E , He F , Shalapour S , Lin jia X , Wong J , Ding S , Seki E , Schnabl B , Hevener AL , Greenberg HB , Kisseleva T , Karin M ((2018) ) New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560: , 198–203. |
[82] | Borsche M , Kö IR , Delcambre S , Petrucci S , Balck A , Brü N , Zimprich A , Wasner K , Pereira SL , Avenali M , Deuschle C , Badanjak K , Ghelfi J , Gasser T , Kasten M , Rosenstiel P , Lohmann K , Brockmann K , Valente EM , Youle RJ , Grü Newald A , Klein C ((2020) ) Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/ PINK1 parkinsonism. Brain 143: , 3041–3051. |
[83] | Lowes H , Kurzawa-Akanbi M , Pyle A , Hudson G ((2020) ) Post-mortem ventricular cerebrospinal fluid cell-free-mtDNA in neurodegenerative disease. Sci Rep 10: , 15253. |
[84] | Furuya T , Hayakawa H , Yamada M , Yoshimi K , Hisahara S , Miura M , Mizuno Y , Mochizuki H ((2004) ) Neurobiology of disease caspase-11 mediates inflammatory dopaminergic cell death in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine mouse model of Parkinson’s disease. J Neurosci 24: , 1865–1872. |
[85] | Mao Z , Liu C , Ji S , Yang Q , Ye H , Han H , Xue Z ((2017) ) The NLRP3 inflammasome is involved in the pathogenesis of Parkinson’s disease in rats. Neurocheml Res 42: , 1104–1115. |
[86] | Yang J , Jia M , Zhang X , Wang P ((2019) ) Calycosin attenuates MPTP-induced Parkinson’s disease by suppressing the activation of TLR/NF-κB and MAPK pathways. Phytother Res 33: , 309–318. |
[87] | Cheng C , Zhu X ((2019) ) Cordycepin mitigates MPTP-induced Parkinson’s disease through inhibiting TLR/NF-κB signaling pathway. Life Sci 223: , 120–127. |
[88] | Xu W , Zheng D , Liu Y , Li J , Yang L , Shang X ((2017) ) Glaucocalyxin B alleviates lipopolysaccharide-induced Parkinson’s disease by inhibiting TLR/NF-κB and activating Nrf2/HO-1 pathway. Cell Physiol Biochem 44: , 2091–2104. |
[89] | Khan A , Johnson R , Wittmer C , Maile M , Tatsukawa K , Wong JL , Gill MB , Stocking EM , Natala SR , Paulino AD , Bowden-Verhoek JK , Wrasidlo W , Masliah E , Bonhaus DW , Price DL ((2021) ) NPT520-34 improves neuropathology and motor deficits in a transgenic mouse model of Parkinson’s disease. Brain 144: , 3692–3709. |
[90] | Leem YH , Park JS , Park JE , Kim DY , Kim HS ((2021) ) Papaverine exerts neuroprotective effect by inhibiting NLRP3 inflammasome activation in an MPTP-induced microglial priming mouse model challenged with LPS. Biomol Ther 29: , 295–302. |
[91] | Ren C , Li LX , Dong AQ , Zhang YT , Hu H , Mao CJ , Wang F , Liu CF ((2021) ) Depression induced by chronic unpredictable mild stress increases susceptibility to Parkinson’s disease in mice via neuroinflammation mediated by P2X7 receptor. ACS Chem Neurosci 12: , 1262–1272. |
[92] | Mo Y , Xu E , Wei R , Le B , Song L , Li D , Chen Y , Ji X , Fang S , Shen J , Yang C , Wang Q ((2018) ) Bushen-Yizhi formula alleviates neuroinflammation via inhibiting NLRP3 inflammasome activation in a mouse model of Parkinson’s disease. Evid Based Complementary Altern Med 2018: , 3571604. |
[93] | Fan Z , Liang Z , Yang H , Pan Y , Zheng Y , Wang X ((2017) ) Tenuigenin protects dopaminergic neurons from inflammation via suppressing NLRP3 inflammasome activation in microglia. J Neuroinflammation 14: , 256. |
[94] | Yao S , Li L , Sun X , Hua J , Zhang K , Hao L , Liu L , Shi D , Zhou H ((2019) ) FTY720 inhibits MPP+-induced microglial activation by affecting NLRP3 inflammasome activation. J Neuroimmune Pharmacol 14: , 478–492. |
[95] | Kwon O , Song J , Yang Y , Kim S , Kim JY , Seok M , Hwang I , Yu J , Karmacharya J , Maeng H , Kim J , Jho E , Ko SY , Son H , Chang M , Lee S ((2021) ) SGK1 inhibition in glia ameliorates pathologies and symptoms in Parkinson disease animal models. EMBO Mol Med 13: , e13076. |
[96] | Que R , Zheng J , Chang Z , Zhang W , Li H , Xie Z , Huang Z , Wang H-T , Xu J , Jin D , Yang W , Tan E-K , Wang Q ((2021) ) Dl-3-n-butylphthalide rescues dopaminergic neurons in Parkinson’s disease models by inhibiting the NLRP3 inflammasome and ameliorating mitochondrial impairment. Front Immunol 12: , 794770. |
[97] | Yan Y , Jiang W , Liu L , Wang X , Ding C , Tian Z , Zhou R ((2015) ) Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160: , 62–73. |
[98] | Wang W , Shi L , Xie Y , Ma C , Li W , Su X , Huang S , Chen R , Zhu Z , Mao Z , Han Y , Li M ((2004) ) SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Neurosci Res 48: , 195–202. |
[99] | Temsamani H , Krisa S , Decossas-Mendoza M , Lambert O , Merillon J-M , Richard T ((2016) ) Piceatannol and other wine stilbenes: A pool of inhibitors against α-synuclein aggregation and cytotoxicity. Nutrients 8: , 367. |
[100] | Zheng R , Ruan Y , Yan Y , Lin Z , Xue N , Yan Y , Tian J , Yin X , Pu J , Zhang B ((2021) ) Melatonin attenuates neuroinflammation by down-regulating NLRP3 inflammasome via a SIRT1-dependent pathway in MPTP-induced models of Parkinson’s disease. J Inflam Res 14: , 3063–3075. |
[101] | Zhu J , Sun T , Zhang J , Liu Y , Wang D , Zhu H , Yao H , Ding J , Hu G , Lu M ((2020) ) Drd2 biased agonist prevents neurodegeneration against NLRP3 inflammasome in Parkinson’s disease model via a β-arrestin2-biased mechanism. Brain Behav Immun 90: , 259–271. |
[102] | Zhu J , Hu Z , Han X , Wang D , Jiang Q , Ding J , Xiao M , Wang C , Lu M , Hu G ((2018) ) Dopamine D2 receptor restricts astrocytic NLRP3 inflammasome activation via enhancing the interaction of β-arrestin2 and NLRP3. Cell Death Differ 25: , 2037–2049. |
[103] | Xu LL , Wu YF , Yan F , Li CC , Dai Z , You QD , Jiang ZY , Di B ((2019) ) 5-(3,4-Difluorophenyl)-3-(6-methylpyridin-3-yl)-1,2,4-oxadiazole (DDO-7263), a novel Nrf2 activator targeting brain tissue, protects against MPTP-induced subacute Parkinson’s disease in mice by inhibiting the NLRP3 inflammasome and protects PC12 cells against oxidative stress. Free Radic Biol Med 134: , 288–303. |
[104] | Liu Q , Guo X , Huang Z , He Q , Zhu D , Zhang S , Peng Z , Che Y , Feng X ((2020) ) Anti-neuroinflammatory effects of dimethylaminomylide (DMAMCL, i.e., ACT001) are associated with attenuating the NLRP3 inflammasome in MPTP-induced Parkinson disease in mice. Behav Brain Res 383: , 1–9. |
[105] | Li J , Dai X , zhou L , Li X , Pan D ((2021) ) Edaravone Plays Protective Effects on LPS-Induced Microglia by Switching M1/M2 Phenotypes and Regulating NLRP3 Inflammasome Activation. Front Pharmacol 12: , 112539. |
[106] | Lin Y , Xu D , Gao F , Zheng X ((2021) ) Ulinastatin inhibits NLRP3-induced apoptosis in a PD cell model. Ann Transl Med 9: , 924. |
[107] | He H , Jiang H , Chen Y , Ye J , Wang A , Wang C , Liu Q , Liang G , Deng X , Jiang W , Zhou R ((2018) ) Oridonin is a covalent NLRP3 inhibitor with strong anti-inflammasome activity. Nat Commun 9: , 2550. |
[108] | Lason W , Tiwari V , Li K , Jin W , Yan C , Yan H , Mao J , Liu Y , Xu L , Zhao H , Shen J , Cao Y , Gao Y ((2020) ) Neuroprotective effect of oridonin on traumatic brain injury via inhibiting NLRP3 inflammasome in experimental mice. Front Neurosci 14: , 557170. |
[109] | Chen X , Liu G , Yuan Y , Wu G , Wang S , Yuan L ((2019) ) NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-κB signaling. Cell Death Dis 10: , 906. |
[110] | Hughes MM , Hooftman A , Angiari S , Tummala P , Zaslona Z , Runtsch MC , McGettrick AF , Sutton CE , Diskin C , Rooke M , Takahashi S , Sundararaj S , Casarotto MG , Dahlstrom JE , Palsson-McDermott EM , Corr SC , Mills KHG , Preston RJS , Neamati N , Xie Y , Baell JB , Board PG , O’Neill LAJ ((2019) ) Glutathione transferase omega-1 regulates NLRP3 inflammasome activation through NEK7 deglutathionylation. Cell 29: , 151–161. |
[111] | Xu KY , Wu CY , Tong S , Xiong P , Wang SH ((2018) ) The selective NLRP3 inflammasome inhibitor Mcc950 attenuates lung ischemia-reperfusion injury. Biochem Biophy Res Commun 503: , 3031–3037. |
[112] | Zhang B , Wei W , Qiu J ((2018) ) ALK is required for NLRP3 inflammasome activation in macrophages. Biochem Biophy Res Commun 501: , 246–252. |
[113] | He Y , Varadarajan S , Muñoz-Planillo R , Burberry A , Nakamura Y , Núñez G ((2014) ) 3,4-methylenedioxy-β-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J Biol Chem 289: , 1142–1150. |
[114] | Coll RC , Hill JR , Day CJ , Zamoshnikova A , Boucher D , Massey NL , Chitty JL , Fraser JA , Jennings MP , Robertson AAB , Schroder K ((2019) ) MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol 15: , 556–559. |
[115] | Cocco M , Miglio G , Giorgis M , Garella D , Marini E , Costale A , Regazzoni L , Vistoli G , Orioli M , Massulaha-Ahmed R , Detraz-Durieux I , Groslambert M , Py BF , Bertinaria M ((2016) ) Design, synthesis, and evaluation of acrylamide derivatives as direct NLRP3 inflammasome inhibitors. Chem Med Chem 11: , 1790–1803. |
[116] | Cocco M , Pellegrini C , Martínez-Banaclocha H , Giorgis M , Marini E , Costale A , Miglio G , Fornai M , Antonioli L , López-Castejón G , Tapia-Abellan A , Angosto D , Hafner-Bratkovic I , Regazzoni L , Blandizzi C , Pelegrín P , Bertinaria M ((2017) ) Development of an acrylate derivative targeting the NLRP3 inflammasome for the treatment of inflammatory bowel disease. J Med Chem 60: , 3656–3671. |
[117] | Shim DW , Shin WY , Yu SH , Kim BH , Ye SK , Koppula S , Won HS , Kang TB , Lee KH ((2017) ) BOT-4-one attenuates NLRP3 inflammasome activation: NLRP3 alkylation leading to the regulation of its ATPase activity and ubiquitination. Sci Rep 7: , 15020. |
[118] | Marchetti C , Swartzwelter B , Koenders MI , Azam T , Tengesdal IW , Powers N , de Graaf DM , Dinarello CA , Joosten LAB ((2018) ) NLRP3 inflammasome inhibitor OLT1177 suppresses joint inflammation in murine models of acute arthritis. Arthritis Res Ther 20: , 169. |
[119] | Jiang H , He H , Chen Y , Huang W , Cheng J , Ye J , Wang A , Tao J , Wang C , Liu Q , Jin T , Jiang W , Deng X , Zhou R ((2017) ) Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med 214: , 3219–3238. |
[120] | Huang Y , Jiang H , Chen Y , Wang X , Yang Y , Tao J , Deng X , Liang G , Zhang H , Jiang W , Zhou R ((2018) ) Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med 10: , e8689. |
[121] | Guo C , Fulp JW , Jiang Y , Li X , Chojnacki JE , Wu J , Wang XY , Zhang S ((2017) ) Development and characterization of a hydroxyl-sulfonamide analogue, 5-Chloro-N-[2-(4-hydroxysulfamoyl-phenyl)-ethyl]-2-methoxy-benzamide, as a novel NLRP3 inflammasome inhibitor for potential treatment of multiple sclerosis. ACS Chem Neurosci 8: , 2194–2201. |
[122] | Daniels MJD , Rivers-Auty J , Schilling T , Spencer NG , Watremez W , Fasolino V , Booth SJ , White CS , Baldwin AG , Freeman S , Wong R , Latta C , Yu S , Jackson J , Fischer N , Koziel V , Pillot T , Bagnall J , Allan SM , Paszek P , Galea J , Harte MK , Eder C , Lawrence CB , Brough D ((2016) ) Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat Commun 7: , 12504. |
[123] | Lonnemann N , Hosseini S , Marchetti C , Skouras DB , Stefanoni D , Dinarello CA , Korte M ((2020) ) The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 117: , 32145–32154. |
[124] | Youm YH , Nguyen KY , Grant RW , Goldberg EL , Bodogai M , Kim D , D’Agostino D , Planavsky N , Lupfer C , Kanneganti TD , Kang S , Horvath TL , Fahmy TM , Crawford PA , Biragyn A , Alnemri E , Dixit VD ((2015) ) The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med 21: , 263–269. |
[125] | Flores J , Noël A , Foveau B , Lynham J , Lecrux C , LeBlanc AC ((2018) ) Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat Commun 9: , 3916. |
[126] | Mackenzie SH , Schipper JL , Clark AC ((2010) ) The potential for caspases in drug discovery. Curr Opin Drug Discov Devel 13: , 568–576. |
[127] | Jesus AA , Goldbach-Mansky R ((2014) ) IL-1 blockade in autoinflammatory syndromes. Annu Rev Med 65: , 223–244. |
[128] | Li X , Zou Y , Fu YY , Xing J , Wang KY , Wan PZ , Wang M , Zhai XY ((2021) ) Ibudilast attenuates folic acid–induced acute kidney injury by blocking pyroptosis through TLR4-mediated NF-κB and MAPK signaling pathways. Front Pharmacol 12: , 650283. |
[129] | Ruan Z , Delpech JC , Venkatesan Kalavai S , van Enoo AA , Hu J , Ikezu S , Ikezu T ((2020) ) P2RX7 inhibitor suppresses exosome secretion and disease phenotype in P301S tau transgenic mice. Mol Neurodegener 15: , 47. |
[130] | Apolloni S , Amadio S , Parisi C , Matteucci A , Potenza RL , Armida M , Popoli P , D’Ambrosi N , Volonte C ((2014) ) Spinal cord pathology is ameliorated by P2X7 antagonism in a SOD1-mutant mouse model of amyotrophic lateral sclerosis. Dis Models Mech 7: , 1101–1109. |
[131] | Hook VYH , Kindy M , Hook G ((2008) ) Inhibitors of cathepsin B improve memory and reduce beta-amyloid in transgenic Alzheimer disease mice expressing the wild-type, but not the Swedish mutant, beta-secretase site of the amyloid precursor protein. J Biol Chem 283: , 7745–7753. |
[132] | Hook G , Jacobsen JS , Grabstein K , Kindy M , Hook V ((2015) ) Cathepsin B is a new drug target for traumatic brain injury therapeutics: Evidence for E64d as a promising lead drug candidate. Front Neurol 6: , 178. |
[133] | Tang TT , Lv LL , Pan MM , Wen Y , Wang B , Li ZL , Wu M , Wang FM , Crowley SD , Liu BC ((2018) ) Hydroxychloroquine attenuates renal ischemia/reperfusion injury by inhibiting cathepsin mediated NLRP3 inflammasome activation. Cell Death Dis 9: , 351. |
[134] | Song N , Liu ZS , Xue W , Bai ZF , Wang QY , Dai J , Liu X , Huang YJ , Cai H , Zhan XY , Han QY , Wang H , Chen Y , Li HY , Li AL , Zhang XM , Zhou T , Li T ((2017) ) NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol Cell 68: , 185–197. |


