
Infections and Changes in Commensal Bacteria and the Pathogenesis of Parkinson’s Disease
Abstract
The cause of Parkinson’s disease (PD) is unknown, but environmental factors are purported to influence risk. Interest in PD as a sequel of infection dates back to reports of parkinsonism arising from encephalitis lethargica. The objective of this paper is to review the literature as it relates to infections and changes in microbiome and the genesis of PD. There is evidence to support prior infection with Helicobacter pylori, hepatitis C virus, Malassezia, and Strep pneumonia in association with PD. A large number of studies support an association between changes in commensal bacteria, especially gut bacteria, and PD. Extant literature supports a role for some infections and changes in commensal bacteria in the genesis of PD. Studies support an inflammatory mechanism for this association, but additional research is required for translation of these findings to therapeutic options.
The most well-known association between infection and parkinsonism occurred in the early 20th century. There were thousands of cases of encephalitis in temporal proximity to the global influenza pandemic that assumed one of three phenotypes described by Constantin von Economo: somnolent-ophthalmoplegic, hyperkinetic, and amyostatic-akinetic (the predominant form). Post-encephalitic parkinsonism emerged in many survivors of the encephalitis, and even among asymptomatic persons months to years following the index infection. The syndrome distinguished itself from sporadic Parkinson’s disease (PD) by its epidemiology (earlier onset), and clinical (oculogyric crises, ocular motility disorders, autonomic instability, and behavioral changes) and pathological features (absence of Lewy bodies, extensive inflammatory changes), and the link to influenza was later characterized as tenuous [1].
The published literature was reviewed for citations reflecting a role for infectious agents and commensal bacteria (the microbiota) and PD.
INFLUENZA VIRUS
Some, but not all influenza strains are neurotropic, and influenza remains of interest among pathogens that might be causative in PD. Associations between the birth year of PD patients and pandemic flu years suggested a link to intrauterine exposure [2], and demonstration of influenza A virus labeling in substantia nigra melanocytes of encephalitis decedents buttressed this belief [3], though at least one case-control study in the United Kingdom was negative [4]. However, a more recent Danish case-control study that analyzed prospectively collected data over more than three decades in 10,271 newly diagnosed PD and 51,355 matched control subjects, found influenza infection was associated with PD diagnosis more than 10 years later (OR 1.73; 95% CI 1.11–2.71) [5].
HEPATITIS C VIRUS
Hepatitis C (HCV) is a neurotropic virus that replicates in the central nervous system (CNS) and may be associated with progressive dopaminergic neuronal demise [6]. HCV RNA has been detected in postmortem brain. In a population-based cohort in Taiwan, HCV infection was associated with an increased risk of developing PD (HR 1.29 (95% CI 1.06–1.56, adjusted for age, gender, and comorbidities)) [7, 8]. A meta-analysis showed a significant relationship between HCV and PD (pooled OR = 1.195 [9] 95% CI 1.012–1.410; p = 0.035) [10]. Treatment of HCV with interferon was associated with a decreased risk of developing PD over 6 years compared to persons who did not receive interferon, lending additional support to the concept that HCV increases risk of PD [8].
SARS-COV-2 VIRUS
SARS-CoV-2 is a respiratory pathogen implicated in a global pandemic that began in 2019 and continues at the time of this publication. The virus is a coronavirus, and the illness has killed nearly 6 million world citizens as of the end of February 2022. The clinical illness, COVID-19, has included many neurologic syndromes including stroke, meningitis, delirium, and gait disorder [11]. The frequency of anosmia in infections related to the SARS-CoV-2 virus has raised concerns that the global pandemic might give rise to an epidemic of PD [12]. It is not yet understood whether the virus is neurotropic, but it has been associated with robust cytokine response and CNS inflammation. A recent publication describes two patients who developed a rapidly-progressive atypical parkinsonism phenotype following SARS-CoV-2-related encephalopathy, including signs suggestive of encephalitis [13]. Features atypical for PD included gaze palsy, myoclonus, prominent dystonia, seizures, and limited levodopa response. These clinical features closely mirror those seen in the encephalitis lethargica epidemic of the early 20th century. One of the recent cases had CSF-restricted oligoclonal bands, but neither responded to immunotherapy. FDG PET-CT findings demonstrated brainstem, temporal lobe, and basal ganglia hypermetabolism and cortical hypometabolism [13]. No pathological studies in contemporary cases are yet available. However, some have been concerned that there will be cases of PD that arise from COVID-19, particularly in “long COVID” cases.
OTHER VIRUSES
There have been reports of associations of PD with various other viruses including varicella zoster, measles, mumps, hepatitis B, Coxsackie, Japanese encephalitis B, western equine encephalitis, west Nile encephalitis, St. Louis encephalitis, human immunodeficiency virus, enteroviruses, dengue, and herpes viruses, but these reports are isolated or did not survive meta-analysis [10].
HELICOBACTER PYLORI
A Danish registry study demonstrated increased risk of PD in subjects prescribed Helicobacter pylori eradicating therapy or proton pump inhibitors five or more years before the onset of the disease [14] A meta-analysis of 10 studies (comprising 5,043 PD and 23,449 healthy subjects) showed an increased risk of PD in patients associated with Helicobacter pylori infection (pooled OR 1.47 (95% CI 1.25–1.70)) [15].
STREP PNEUMONIAE
A meta-analysis of 2 studies suggested a relationship between PD and prior infection with strep pneumonia (one with scarlet fever and the other with rheumatic fever) (OR 1.595 (95% CI 1.020–2.493)) [10].
OTHER BACTERIA
Reports of associations between Bordatella pertussis, Mycoplasma tuberculosis, and PD were not confirmed in a meta-analysis [10].
OTHER INFECTIOUS AGENTS AND PD
Toxoplama gondii
Toxoplasma gondii is an intracellular parasite. Between 10 and 80% of people have antibody evidence of exposure to the organism. A number of studies have suggested an increased risk of PD in persons with evidence of Toxoplasma gondii infection [16, 17]. However, a meta-analysis failed to confirm an association [18].
Malassezia
Malassezia is a lipophilic yeast that contributes to normal skin flora but can be associated with skin diseases such as dermatitis and folliculitis. While there have been some case-control studies that suggest an association with PD [19], seborrheic dermatitis is common in PD, which itself is associated with increased sebum production that may predispose to skin conditions [10]. Thus, Malassezia may be comorbid rather than contributory in PD etiopathogenesis.
OVERALL RISK OF PD AFTER INFECTION
The meta-analysis published by Wang et al. comprised an analysis of literature published from 1965 to 2019 in PubMed and EMBASE databases. Of more than 6,000 studies, 23 were included (6 retrospective cohorts and 17 case-control studies). The methods included data extraction, pooling of data with random/fixed effects models followed by sensitivity analysis and meta-regression. They included infection detection methods as well as PD diagnostic criteria. In all, 21 pathogenic organisms were included (8 bacteria, 12 viruses, one fungus). After limiting the analyses to pathogens covered in more than one study, analyses were limited to herpes virus, hepatitis B virus, pertussis, scarlet fever, influenza, mumps, chicken pox, measles, Malassezia, hepatitis C virus, Helicobacter pylori, pneumonia, and rubeola infections [10]. Those pathogens that survived this meta-analysis are shown in Table 1.
Table 1
Infectious agents associated with PD, meta-analysis
| Infectious agent | Pooled OR (95% CI) | p |
| Helicobacter pylori | 1.653 (1.426–1.915) | < 0.001 |
| Hepatitis C virus | 1.195 (1.012–1.410) | 0.035 |
| Malassezia | 1.694 (1.367–2.100) | < 0.001 |
| S. pneumoniae | 1.595 (1.020–2.493) | 0.041 |
Data from Wang et al. [10].
To answer a more global question on the role of infections in the etiology of PD, Meng et al. performed a quantitative assessment of case-control and cohort studies. They included 13 studies in their meta-analysis (8 case-control studies and 5 cohort studies) published between 1995–2016. They found that the development of PD was associated with prior infection (OR 1.20, 95% CI 10.7–1.32). However, viral infection showed no statistically significant association with subsequent PD diagnosis, while bacterial infection increased risk of PD by 40% (OR 1.40, 95% CI 1.32–1.48) [20].
COMMENSAL BACTERIA AND PD: MICROBIOTA STUDIES
The microbiota is the population of microbes that dwell on epithelial and other surfaces, and the microbiome is the genetic material housed within these bacteria. Studies of microbiota and microbiome in man have garnered immense interest as it has become clear changes in bacterial populations may be important in the genesis of many illnesses. The analysis of the microbiome is very complex, and largely based on interrogation of the hypervariable genetic sequences in ribosomal RNA [21].
Nasal microbiota
There have been two negative studies of nasal microbiota associations with PD [22, 23]. Adequate sampling of the nasal mucosa can be uncomfortable for patients, and it is necessary to use a deep sampling approach to obtain optimal samples. Using one such approach, Pal et al. found significant differences between PD and non-household controls. Putative opportunistic bacteria were identified in PD, including Moraxella catarrhalis, a proinflammatory bacterium [24].
Oral microbiota
A study in 20 PD and 20 controls found bacteria associated with oral pathologies (Atinomyces, Veillonella, Kingella oralis, and Streptococcus mutans) were more prevalent in PD. PD subjects had gingival inflammation, exemplified by increased gingival crevice cytokines (pro-inflammatory cytokine IL1-β and the anti-inflammatory cytokine IL-1RA with a trend towards increase TNF-α levels) [25]. Another study by Pereira et al. in Finnish patents showed differences between PD and controls, but opportunistic oral pathogens were more common in men than women irrespective of PD diagnosis [23]. Poor oral hygiene, dysphagia, and drooling are common in PD and can independently contribute to changes in oral microbiota [26].
Gut microbiota
In healthy human gut, about 95% of bacteria belong to the phyla Bacteroidetes and Firmicutes, while those in a diseased state may harbor large numbers of other phyla (Proteobacteria, Verrucomicrobia, Actinobacteria, Fusobacteria). Variation in microbiota is the norm and there is no universally normal gut microbiota [27]. Moreover, gut microbial populations are dynamic, with frequent changes in patients with fluctuating primary bowel disease and slower changes related to diet, weight loss or other changes [21].
The most popular approach to the gut microbiome is amplicon analysis after amplification of hypervariable regions of 16S ribosomal RNA genes. Next generation sequencing allows placing the bacteria on a phylogenetic tree. Another approach uses shotgun genomics to analyze all DNA in a sample. The analysis of the resulting datasets is quite complex.
There have been nearly two dozen studies of gut flora in PD. A recent meta-analysis analyzed 22 studies using 16S ribosomal RNA-gene amplicon sequencing, for which raw sequencing data were available in 10 studies in 9 cohorts from 6 countries. One conclusion of this study was that most differences among the studies related to methodologic differences, including geographic region, participant selection, sample processing, sequencing methods, and which hypervariable region of the 16S rRNA gene was sequenced. However, the authors did conclude that there were robust differences in gut microbiota between PD subjects and control subjects. Representation of normally dominant bacteria was decreased in favor of microbes that are not normally abundant in stool. Roseburia, Fusicatenibacter, Blautia, Anaerostipes, and Faealibacterium were less abundant in PD feces [28]. There is a general agreement that the gut microbiota in PD is populated with fewer anti-inflammatory and more pro-inflammatory bacteria. One proposed consequence of this change in microbiota is reduced short-chain fatty acid (SCFA) production, particularly butyrate [28]. SCFA are the product of anaerobic fermentation of dietary polysaccharides and play critical roles in maintenance of the gut barrier, production of protective mucus, and anti-inflammatory activities. SCFA are also important in the microbiota-gut-brain axis. They have systemic anti-inflammatory effects and also a role in maintenance of the blood-brain barrier as well as neuronal function [29]. Parkinsonian phenotypes in animal PD models are enhanced by population of the animal gut with bacteria that predominate in PD subjects and reduced in the presence of bacteria associated with anti-inflammatory effects [30–33]. Predictive functional genetics studies suggest an over-representation of genes in metabolic pathways that favor pathogenicity and secretion of lipopolysaccharide, a bacterial endotoxin [34]. Localized and systemic inflammation is a direct result of these changes in commensal bacteria. Increased cytokine levels can be measured at the interfaces of the environment with epithelial surfaces and there are increases in systemic cytokines, including interleukins (IL) and others. Multiple studies support increased concentrations of IL-6, IL-8, IL-12, IL-18, IL-1-beta, and tumor necrosis factor-alpha [35].
HOW MIGHT EXPOSURE TO INFECTIOUS AGENTS BE INVOLVED IN THE GENESIS OF PD?
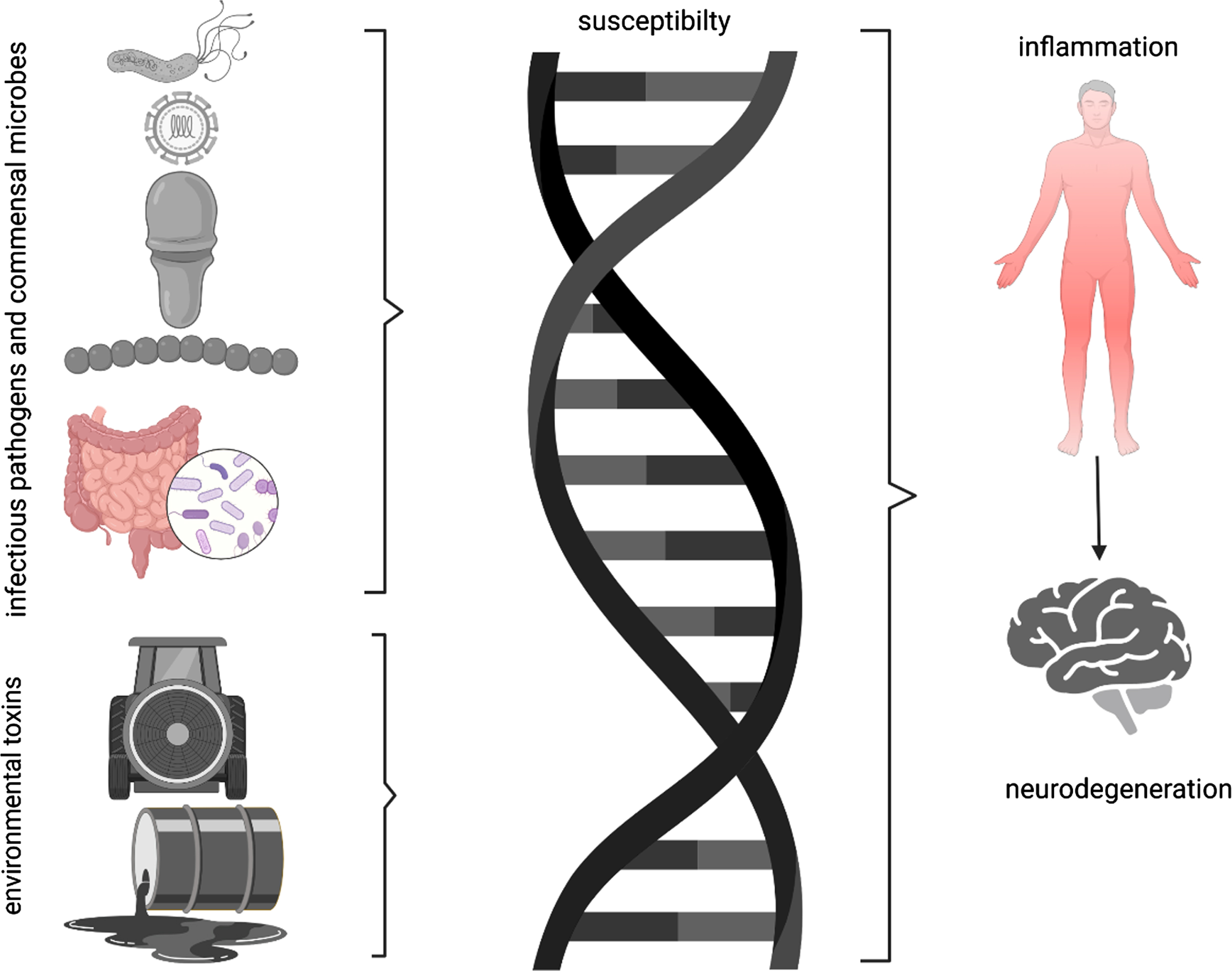
While associations among pathogens and PD are intriguing, it is clear we are not on a course to identify a single pathogen that triggers the cascade of events that results in PD. These agents should best be considered environmental agents, like environmental neurotoxins that conspire with genetic predisposition to incite the degenerative synucleinopathy that results in clinically manifest PD.
Several mechanisms (synuclein spread, neuroinflammation, metabolites) have been proposed to explain a relationship between infectious agents and PD. In 2006, Braak and colleagues [36] expanded on his studies of the distribution of alpha-synuclein (AS) pathology suggesting a spread of AS across contiguous neural pathways in CNS and suggested that AS pathology might begin consequent to a pathogen in the gastrointestinal tract, then spread in a prion-like fashion to the dorsal motor nucleus of the vagus, from which it ascends to the midbrain tegmentum and rostrally onwards. Further support for this mechanism comes from imaging studies suggesting that both brain-first and body-first pathways to PD are possible [37, 38]. Although support for spread of abnormal AS from the gut to the brain received some support from preclinical work [39], the failure to demonstrate any human decedents with isolated gastrointestinal tract synuclein aggregates has lessened enthusiasm for this potential mechanism from gastrointestinal sources [40]. Support for synuclein spread through olfactory pathways remains [41]. An alternative mechanistic explanation is that pathogens produce a systemic inflammatory response that accesses the brain and triggers a neuroinflammatory response, including microglial activation [9]. Indeed, the principle that unites these disparate agents to PD is the potential to cause systemic inflammation. Infectious agents activate pathogen-associated molecular pattern receptors and environmental toxins or the havoc they induce may activate damage-associated molecular pattern receptors, giving rise to cytokine production and release, with systemic inflammation. Systemic inflammation may gain access to the CNS via altered blood-brain barrier permeability or through migration of immunocompetent cells. Once the inflammatory process enters the CNS, cytokines can trigger synucleinopathy, which is itself self-sustaining [42]. In a series of studies, synuclein induced a robust microglial response in cell culture. Intracerebral injections of synuclein induced cytokine expression that was significantly greater after systemic lipopolysaccharide challenge [43]. Preclinical evidence suggests that bacterial lipopolysaccharide may induce a distinct strain of AS fibrils that is self-renewable and drives a mouse model of PD [44]. Another proposed mechanism is a direct effect the gut bacterial amyloid (Curli and others) on synuclein aggregation [45]. Figure 1 illustrates the potential mechanisms of PD genesis hypotheses.
Fig. 1
Parkinson’s disease is best thought as the end product of genetic susceptibility (supported by increased risk in first degree family members) and environmental exposure. Environmental exposures are generally thought to be chemical products in the external space. However, infectious pathogens and commensal microbes on surfaces or in tissues may also trigger inflammatory responses that ultimately provoke neuroinflammation and cell death.
THOUGHTS ON THERAPEUTIC APPROACHES
A number of anti-infective agents have been proposed as possible therapeutic agents in PD. These include ceftriaxone, doxycycline, minocycline, rifampicin, geldamycin, rapamycin, interferon, amantadine, and niclosamide [9]. However, in none of these cases is the potential neuroprotective effect mediated by eradicating an infectious organism. Rather, each is proposed to have an effect on neuroinflammation, microglial activation, oxidative stress, synuclein aggregation, or mitochondrial function. An accumulating body of evidence suggests that neuroinflammation is a promising target for therapy, but this has not yet yielded effective therapies. In the interim, approaches might include anti-inflammatory diet (Mediterranean diet), regular exercise, pre- and probiotic supplements, or fecal transplant.
CONFLICT OF INTEREST
The author has no conflict of interest to report.
REFERENCES
[1] | Cadar D , Jellinger KA , Riederer P , Strobel S , Monoranu CM , Tappe D ((2021) ) No metagenomic evidence of causative viral pathogens in postencephalitic parkinsonism following encephalitis lethargica. Microorganisms 9: , 1716. |
[2] | Mattock C , Marmot M , Stern G ((1988) ) Could Parkinson’s disease follow intra-uterine influenza? A speculative hypothesis. J Neurol Neurosurg Psychiatry 51: , 753–756. |
[3] | Rohn TT , Catlin LW ((2011) ) Immunolocalization of influenza A virus and markers of inflammation in the human Parkinson’s disease brain. PLoS One 6: , e20495. |
[4] | Toovey S , Jick SS , Meier CR ((2011) ) Parkinson’s disease or Parkinson symptoms following seasonal influenza. Influenza Other Respir Viruses 5: , 328–333. |
[5] | Cocoros NM , Svensson E , Szepligeti SK , Vestergaard SV , Szentkuti P , Thomsen RW , Borghammer P , Sorensen HT , Henderson VW ((2021) ) Long-term risk of Parkinson disease following influenza and other infections. JAMA Neurol 78: , 1461–1470. |
[6] | Wu WY , Kang KH , Chen SL , Chiu SY , Yen AM , Fann JC , Su CW , Liu HC , Lee CZ , Fu WM , Chen HH , Liou HH ((2015) ) Hepatitis C virus infection: A risk factor for Parkinson’s disease. J Viral Hepat 22: , 784–791. |
[7] | Tsai HH , Liou HH , Muo CH , Lee CZ , Yen RF , Kao CH ((2016) ) Hepatitis C virus infection as a risk factor for Parkinson disease: A nationwide cohort study. Neurology 86: , 840–846. |
[8] | Lin WY , Lin MS , Weng YH , Yeh TH , Lin YS , Fong PY , Wu YR , Lu CS , Chen RS , Huang YZ ((2019) ) Association of antiviral therapy with risk of Parkinson disease in patients with chronic hepatitis C virus infection. JAMA Neurol 76: , 1019–1027. |
[9] | Shen S , Zhang C , Xu YM , Shi CH ((2022) ) The role of pathogens and anti-infective agents in Parkinson’s disease, from etiology to therapeutic implications. J Parkinsons Dis 12: , 27–44. |
[10] | Wang H , Liu X , Tan C , Zhou W , Jiang J , Peng W , Zhou X , Mo L , Chen L ((2020) ) Bacterial, viral, and fungal infection-related risk of Parkinson’s disease: Meta-analysis of cohort and case-control studies.. Brain Behav 10: , e01549. |
[11] | Awogbindin IO , Ben-Azu B , Olusola BA , Akinluyi ET , Adeniyi PA , Di Paolo T , Tremblay ME ((2021) ) Microglial implications in SARS-CoV-2 infection and COVID-19: Lessons from viral RNA neurotropism and possible relevance to Parkinson’s disease. Front Cell Neurosci 15: , 670298. |
[12] | Ramirez-Zamora A , Hess CW , Nelson DR ((2019) ) Is interferon therapy for hepatitis C infection a treatable risk factor for Parkinson disease? . JAMA Neurol 76: , 1006–1007. |
[13] | Morassi M , Palmerini F , Nici S , Magni E , Savelli G , Guerra UP , Chieregato M , Morbelli S , Vogrig A ((2021) ) SARS-CoV-2-related encephalitis with prominent parkinsonism: Clinical and FDG-PET correlates in two patients. J Neurol 268: , 3980–3987. |
[14] | Nielsen HH , Qiu J , Friis S , Wermuth L , Ritz B ((2012) ) Treatment for Helicobacter pylori infection and risk of Parkinson’s disease in Denmark. Eur J Neurol 19: , 864–869. |
[15] | Dardiotis E , Tsouris Z , Mentis AA , Siokas V , Michalopoulou A , Sokratous M , Dastamani M , Bogdanos DP , Deretzi G , Kountouras J ((2018) ) H. pylori and Parkinson’s disease: Meta-analyses including clinical severity. Clin Neurol Neurosurg 175: , 16–24. |
[16] | Alvarado-Esquivel C , Mendez-Hernandez EM , Salas-Pacheco JM , Ruano-Calderon LA , Hernandez-Tinoco J , Arias-Carrion O , Sanchez-Anguiano LF , Castellanos-Juarez FX , Sandoval-Carrillo AA , Liesenfeld O , Ramos-Nevarez A ((2017) ) Toxoplasma gondii exposure and Parkinson’s disease: A case-control study. BMJ Open 7: , e013019. |
[17] | Miman O , Kusbeci OY , Aktepe OC , Cetinkaya Z ((2010) ) The probable relation between Toxoplasma gondii and Parkinson’s disease. Neurosci Lett 475: , 129–131. |
[18] | Zhou Z , Zhou R , Li K , Wei W , Zhang Z , Zhu Y , Luan R ((2019) ) The association between Toxoplasma gondii infection and risk of Parkinson’s disease: A systematic review and meta-analysis. Biomed Res Int 2019: , 8186017. |
[19] | Laurence M , Benito-Leon J , Calon F ((2019) ) Malassezia and Parkinson’s disease. Front Neurol 10: , 758. |
[20] | Meng L , Shen L , Ji HF ((2019) ) Impact of infection on risk of Parkinson’s disease: A quantitative assessment of case-control and cohort studies. J Neurovirol 25: , 221–228. |
[21] | Keshavarzian A , Engen P , Bonvegna S , Cilia R ((2020) ) The gut microbiome in Parkinson’s disease: A culprit or a bystander? . Prog Brain Res 252: , 357–450. |
[22] | Heintz-Buschart A , Pandey U , Wicke T , Sixel-Doring F , Janzen A , Sittig-Wiegand E , Trenkwalder C , Oertel WH , Mollenhauer B , Wilmes P ((2018) ) The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov Disord 33: , 88–98. |
[23] | Pereira PAB , Aho VTE , Paulin L , Pekkonen E , Auvinen P , Scheperjans F ((2017) ) Oral and nasal microbiota in Parkinson’s disease. Parkinsonism Relat Disord 38: , 61–67. |
[24] | Pal G , Ramirez V , Engen PA , Naqib A , Forsyth CB , Green SJ , Mahdavinia M , Batra PS , Tajudeen BA , Keshavarzian A ((2021) ) Deep nasal sinus cavity microbiota dysbiosis in Parkinson’s disease. NPJ Parkinsons Dis 7: , 111. |
[25] | Fleury V , Zekeridou A , Lazarevic V , Gaia N , Giannopoulou C , Genton L , Cancela J , Girard M , Goldstein R , Bally JF , Mombelli A , Schrenzel J , Burkhard PR ((2021) ) Oral dysbiosis and inflammation in Parkinson’s disease. J Parkinsons Dis 11: , 619–631. |
[26] | Rozas NS , Tribble GD , Jeter CB ((2021) ) Oral factors that impact the oral microbiota in Parkinson’s disease. Microorganisms 9: , 1616. |
[27] | Allaband C , McDonald D , Vazquez-Baeza Y , Minich JJ , Tripathi A , Brenner DA , Loomba R , Smarr L , Sandborn WJ , Schnabl B , Dorrestein P , Zarrinpar A , Knight R ((2019) ) Microbiome 101: Studying, analyzing, and interpreting gut microbiome data for clinicians. Clin Gastroenterol Hepatol 17: , 218–230. |
[28] | Romano S , Savva GM , Bedarf JR , Charles IG , Hildebrand F , Narbad A ((2021) ) Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinsons Dis 7: , 27. |
[29] | Silva YP , Bernardi A , Frozza RL ((2020) ) The role of short-chain fatty acids from gut microbiota in gut-brain communication. Front Endocrinol (Lausanne) 11: , 25. |
[30] | Dodiya HB , Forsyth CB , Voigt RM , Engen PA , Patel J , Shaikh M , Green SJ , Naqib A , Roy A , Kordower JH , Pahan K , Shannon KM , Keshavarzian A ((2020) ) Chronic stress-induced gut dysfunction exacerbates Parkinson’s disease phenotype and pathology in a rotenone-induced mouse model of Parkinson’s disease. Neurobiol Dis 135: , 104352. |
[31] | Sampson TR , Debelius JW , Thron T , Janssen S , Shastri GG , Ilhan ZE , Challis C , Schretter CE , Rocha S , Gradinaru V , Chesselet MF , Keshavarzian A , Shannon KM , Krajmalnik-Brown R , Wittung-Stafshede P , Knight R , Mazmanian SK ((2016) ) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167: , 1469–1480 e1412. |
[32] | Sun MF , Zhu YL , Zhou ZL , Jia XB , Xu YD , Yang Q , Cui C , Shen YQ ((2018) ) Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson’s disease mice: Gut microbiota, glial reaction and TLR4/TNF-alpha signaling pathway. Brain Behav Immun 70: , 48–60. |
[33] | Zhao Z , Ning J , Bao XQ , Shang M , Ma J , Li G , Zhang D ((2021) ) Fecal microbiota transplantation protects rotenone-induced Parkinson’s disease mice via suppressing inflammation mediated by the lipopolysaccharide-TLR4 signaling pathway through the microbiota-gut-brain axis. Microbiome 9: , 226. |
[34] | Keshavarzian A , Green SJ , Engen PA , Voigt RM , Naqib A , Forsyth CB , Mutlu E , Shannon KM ((2015) ) Colonic bacterial composition in Parkinson’s disease. Mov Disord 30: , 1351–1360. |
[35] | Qin XY , Zhang SP , Cao C , Loh YP , Cheng Y ((2016) ) Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: A systematic review and meta-analysis. JAMA Neurol 73: , 1316–1324. |
[36] | Braak H , de Vos RA , Bohl J , Del Tredici K ((2006) ) Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett 396: , 67–72. |
[37] | Borghammer P , Knudsen K , Fedorova TD , Brooks DJ ((2017) ) Imaging Parkinson’s disease below the neck. NPJ Parkinsons Dis 3: , 15. |
[38] | Borghammer P , Van Den Berge N ((2019) ) Brain-first versus gut-first Parkinson’s disease: A hypothesis. J Parkinsons Dis 9: , S281–S295. |
[39] | Uemura N , Yagi H , Uemura MT , Hatanaka Y , Yamakado H , Takahashi R ((2018) ) Inoculation of alpha-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol Neurodegener 13: , 21. |
[40] | Beach TG , Adler CH , Sue LI , Shill HA , Driver-Dunckley E , Mehta SH , Intorcia AJ , Glass MJ , Walker JE , Arce R , Nelson CM , Serrano GE ((2021) ) Vagus nerve and stomach synucleinopathy in Parkinson’s disease, incidental Lewy body disease, and normal elderly subjects: Evidence against the “body-first” hypothesis. J Parkinsons Dis 11: , 1833–1843. |
[41] | Rey NL , George S , Steiner JA , Madaj Z , Luk KC , Trojanowski JQ , Lee VM , Brundin P ((2018) ) Spread of aggregates after olfactory bulb injection of alpha-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol 135: , 65–83. |
[42] | Houser MC , Tansey MG ((2017) ) The gut-brain axis: Is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? . NPJ Parkinsons Dis 3: , 3. |
[43] | Couch Y , Alvarez-Erviti L , Sibson NR , Wood MJ , Anthony DC ((2011) ) The acute inflammatory response to intranigral alpha-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J Neuroinflammation 8: , 166. |
[44] | Kim C , Lv G , Lee JS , Jung BC , Masuda-Suzukake M , Hong CS , Valera E , Lee HJ , Paik SR , Hasegawa M , Masliah E , Eliezer D , Lee SJ ((2016) ) Exposure to bacterial endotoxin generates a distinct strain of alpha-synuclein fibril. Sci Rep 6: , 30891. |
[45] | Sampson TR , Challis C , Jain N , Moiseyenko A , Ladinsky MS , Shastri GG , Thron T , Needham BD , Horvath I , Debelius JW , Janssen S , Knight R , Wittung-Stafshede P , Gradinaru V , Chapman M , Mazmanian SK ((2020) ) A gut bacterial amyloid promotes alpha-synuclein aggregation and motor impairment in mice. Elife 9: , e53111. |


