
Neuroinflammation and Immune Changes in Prodromal Parkinson’s Disease and Other Synucleinopathies
Abstract
Multiple lines of clinical and pre-clinical research support a pathogenic role for neuroinflammation and peripheral immune system dysfunction in Parkinson’s disease. In this paper, we have reviewed and summarised the published literature reporting evidence of neuroinflammation and peripheral immune changes in cohorts of patients with isolated REM sleep behaviour disorder and non-manifesting carriers of GBA or LRRK2 gene mutations, who have increased risk for Parkinsonism and synucleinopathies, and could be in the prodromal stage of these conditions. Taken together, the findings of these studies suggest that the early stages of pathology in Parkinsonism involve activation of both the central and peripheral immune systems with significant crosstalk. We consider these findings with respect to those found in patients with clinical Parkinson’s disease and discuss their possible pathological roles. Moreover, those factors possibly associated with the immune response, such as the immunomodulatory role of the affected neurotransmitters and the changes in the gut-brain axis, are also considered.
INTRODUCTION
Parkinson’s disease (PD) is a heterogeneous chronic neurodegenerative disease pathologically characterised by intracellular aggregates of α-synuclein (α-syn), in the Lewy bodies and neurites, and the loss of dopaminergic neurons in the substantia nigra (SN), which is responsible for the onset of the motor symptoms. However, in PD, α-syn pathology and neuronal degeneration also occur in non-dopaminergic pathways in the central and peripheral nervous systems (CNS and PNS). This might precede the degeneration in SN and is associated with a variety of non-motor symptoms, such as hyposmia, constipation, fatigue, depression, and sleep disorders [1, 2]. Based on the initial point of α-syn pathology and neurodegeneration, and its progression in the nervous system, Horsager and colleagues, have recently hypothesised the existence of two PD subtypes: the brain-first and the body-first, in which the neurodegeneration progresses from either the CNS or PNS respectively [3]. This new hypothesis might explain the diversity in the PD presentation and corroborates PD as a multisystem heterogeneous disease.
Although most PD cases are idiopathic (iPD), a small percentage of patients have genetic forms with several loci associated with PD [4]. Among these rare mutations, those in the leucine-rich repeat kinase 2 gene (LRRK2) or heterozygous mutations in the glucocerebrosidase gene (GBA) are the most commonly found. Within all these different PD-types, several common pathological mechanisms have been implicated, including defective protein clearance, mitochondrial dysfunction, and neuroinflammation [5–8]. Over the last 20 years, there has been a rapid growth in our knowledge of the role of neuroinflammation and peripheral immune changes in the pathophysiology of PD. Postmortem examinations and in vivo positron emission tomography (PET) studies have shown that the neurodegeneration in PD is accompanied by chronic widespread microgliosis and inflammation in the brain [7–10]. In addition, several studies support the involvement of peripheral immune cells in the PD immune response [11] with changes in peripheral blood immune cells in living patients [12–14] and postmortem studies showing infiltration of T-cells and natural killer cells in the patients’ brains [15–17]. These immune changes have been associated with the patients’ symptoms and/or with PD subtypes. Therefore, the central and peripheral immune systems are affected during PD. However, it is unclear whether the immune changes are a consequence of the initiated disease process or a causal factor. Supporting the latter, single nucleotide polymorphisms of the HLA-DR gene (MHCII system) have been genetically associated with PD risk [18]. Moreover, it is proposed that LRRK2— highly expressed in immune cells— has a function in the brain and peripheral immune system (see [19] for further reading). Furthermore, recent research supports a role for α-syn within the immune system as a chemoattractant and a factor involved in the maturation of dendritic cells in the gut immune responses [20, 21]. Therefore, immune dysregulation seems to be at the core of the disease, although it might occur in parallel to the above-mentioned pathological mechanisms in neurons.
Until recently, most of the studies addressing immune changes were performed in patients with established locomotor disability and, therefore, did not inform about the temporal relationship between the occurrence of immune changes and the clinical motor onset and progression. A better understanding of the role of the immune system in PD is now emerging from several studies in patients at high-risk of developing PD or a related synucleinopathy, who could be in a prodromal (pre-motor) stage of the disease. The International Parkinson and Movement Disorder Society (MDS) has recently proposed a division of early PD into three stages: preclinical PD, with neurodegenerative changes, but no clinical symptoms; prodromal PD, with symptoms not meeting the diagnostic criteria; and clinical PD, with the classical motor symptoms present [22]. There are currently no definitive diagnostic criteria for prodromal PD. However, for use in research settings the MDS has proposed criteria for “probable prodromal PD”, which correspond to a likelihood of ≥80% that a person has prodromal PD. The criteria present a threshold of multiplied likelihood ratios of risk markers. Such markers have been included based on their ability to predict PD onset in at least two independent prospective studies. Non-motor risk markers include polysomnography-proven isolated rapid eye movement sleep behaviour disorder (iRBD), hyposmia, constipation, excessive daytime somnolence, orthostatic hypotension, erectile dysfunction, urinary dysfunction, and depression [23, 24]. iRBD, a parasomnia characterized by the enactment of dreams due to a lack of muscle atonia, is currently considered the most important prodromal marker of PD and other synucleinopathies. Additionally, individuals carrying GBA or LRRK2 gene mutations are also at increased risk of developing PD during their lives and could be in the prodromal phase of PD. In this review, we will summarise the published literature focusing on the investigation of immune changes in these individuals who have a high risk for PD and discuss the findings with respect to those found in patients with clinical PD.
CENTRAL AND PERIPHERAL IMMUNE CELLS
Immune cells are mostly found in the bloodstream and the lymphoid tissue. White blood cells consist of myeloid cells including: polynuclear cells (basophils, neutrophils, and eosinophils), monocytes, and dendritic cells. These cells belong to the innate immune system and are the first responders during stress or infection. In addition, blood lymphoid cells include natural killer cells (also innate cells), and T-cells and B-cells that carry the load of the adaptive immunity and memory in the immune system. The antigen-presenting cells (APC) in the innate pool (mostly monocytes and dendritic cells) activate T-cells -through the MHC (HLA-DR) system, which in turn will activate B-cells, and a humoral response is mounted. Monocytes can infiltrate tissue and become macrophages, but, additionally, tissues possess the so-called resident macrophages, which arise from precursors of embryonic origin. Yolk sac primary erythro-myeloid progenitors— primitive macrophages— give rise to microglia [25]. While the major pool of the other tissue-resident macrophages is contributed later by foetal progenitors. Lastly, adult hematopoiesis occurs in the bone marrow.
Microglia, the brain’s resident innate immune cells (and APC), maintain homeostasis and act in a neuroprotective manner by removing unwanted synapses, dead cells, and dangerous molecules, and producing anti-inflammatory mediators to cease the inflammation. They may, however, also promote inflammation by secreting and reacting to pro-inflammatory cytokines and recruiting and activating other immune cells in response to, for example, misfolded proteins or pathogen molecular patterns [26]. The presence of higher microglia activation in Alzheimer’s disease patients with a slower disease progression suggests a neuroprotective function for activated microglia early in the disease [27]. Thus, it is speculated that chronic inflammation in neurodegenerative diseases results from an imbalance of the microglial activities in which a bias in the pro-inflammatory effectors leads to a progression of the pathology. However, the role of microglia could also be more dynamic, and evolve with the disease; being protective at initial points and later becoming detrimental.
In PD, α-syn plays a significant role not only in the neuronal dysfunction but also in the immune event, by activating innate immune cells such as microglia and monocytes/macrophages. α-Syn can act as a damage-associated molecular pattern and interact with several immune receptors widely expressed in innate cells leading to inflammatory activation and antigen presentation (MHCII expression), which in turn results in activation of adaptive immunity [28]. Accordingly, postmortem analysis associates microgliosis with areas of α-syn deposition and neuronal dysfunction or neurodegeneration. For example, PD brain areas with α-syn pathology show upregulation of HLA-DR [29, 30], the molecular pattern receptors TLR2 and TLR4 [31–33], and pro-inflammatory cytokines [34]. In vivo assessment of neuroinflammation is made possible by PET with radiopharmaceuticals targeting biomarkers of microglial activation, such as 11C-R-PK11195 (PK), a ligand for the 18-kDa translocator protein (TSPO). TSPO is mainly localised on the outer mitochondrial membrane in microglia, but also in other cells that partake in the neuroinflammatory process such as astrocytes [35]. Its exact function is still to be determined; however, it is markedly upregulated during microglial activation and inflammation, and thus increased TSPO signal is considered a sign of ongoing neuroinflammation [36].
EVIDENCE FROM RETROSPECTIVE STUDIES FOR IMMUNE CHANGES IN PRECLINICAL PD, AND ASSOCIATION TO FINDINGS IN ESTABLISHED PD
The PD-associated immune response is not only limited to changes in brain immune cells but also in the periphery and involves both innate and adaptive immune cells (Fig. 1). The early cellular peripheral immune response in preclinical PD has been corroborated by data from several recent retrospective population studies. The lymphocyte numbers are consistently found decreased in PD [37] and this seems to occur as soon as 10–20 years prior to diagnosis [38, 39], thus PD risk is associated with lower lymphocyte counts. However, the number of neutrophils, the most abundant innate cells in the blood, is increased in PD patients and this seems to occur years before diagnosis [39, 40] (Fig. 1). The lower lymphocyte count and higher neutrophils in prodromal and clinical PD have been also corroborated in a recent study using samples from the Parkinson’s Progression Marker Initiative (PPMI) cohort (https://www.ppmi-info.org), where higher expression of neutrophil enriched transcripts further validated the increased neutrophil population [41]. This increase was greatest during prodromal (high-risk) PD and once elevated it remained so during the disease. Accordingly, several groups have reported elevated neutrophil/lymphocyte ratio (NLR) in PD that correlated with symptoms or was associated with different symptomatic presentations [42, 43]. Interestingly, a significant negative correlation has been reported between NLR and white matter changes in areas associated with non-motor PD symptoms such as cognitive performance, REM sleep, and olfaction [44], suggesting that peripheral immunity is particularly relevant for non-motor symptoms.
Fig. 1
Proposed landscape on immune changes in preclinical and clinical Parkinson’s disease (PD). Despite some inconsistencies across studies data so far suggest the following. Based on retrospective studies and analysis in PD cohorts, the numbers of neutrophils are increased while lymphocytes are decreased years before diagnosis and remain so through the disease. Imaging studies suggest inflammation in the brain of iRBD patients and non-manifesting LRRK2 or GBA carriers, which remains at clinical PD stages. Data in serum during prodromal stages are inconsistent, with some studies suggesting the increase of pro-inflammatory cytokines that remains after diagnosis. The limited data published on iRBD patients show modified innate and adaptive cells, with classical monocytes enriched and expressing higher levels of CCR2 and CD11b, proteins associated with infiltration and transmigration. This seems to remain in iPD patients during the first few years after diagnosis. Later intermediate monocytes are increased and they show higher expression of HLA-DR, CD163. Clinical PD is also associated with increased expression of TLRs in monocytes. T-cells transcriptome is altered in iRBD patients and T-cells react to α-synuclein peptides years before and after PD diagnosis, and this seems to be more relevant early in the disease. Several studies suggest a Th1 and/or Th17 bias in the T-cell population in iPD patients. Microbiota might be altered at preclinical stages, as suggested by studies in iRBD cohorts. This dysbiosis remains through disease when signs of leaky gut are observed. See the text for more details. cMos, classical monocytes; iMos, intermediate monocytes; α-syn, α-synuclein. Figure created with bioRender.com.
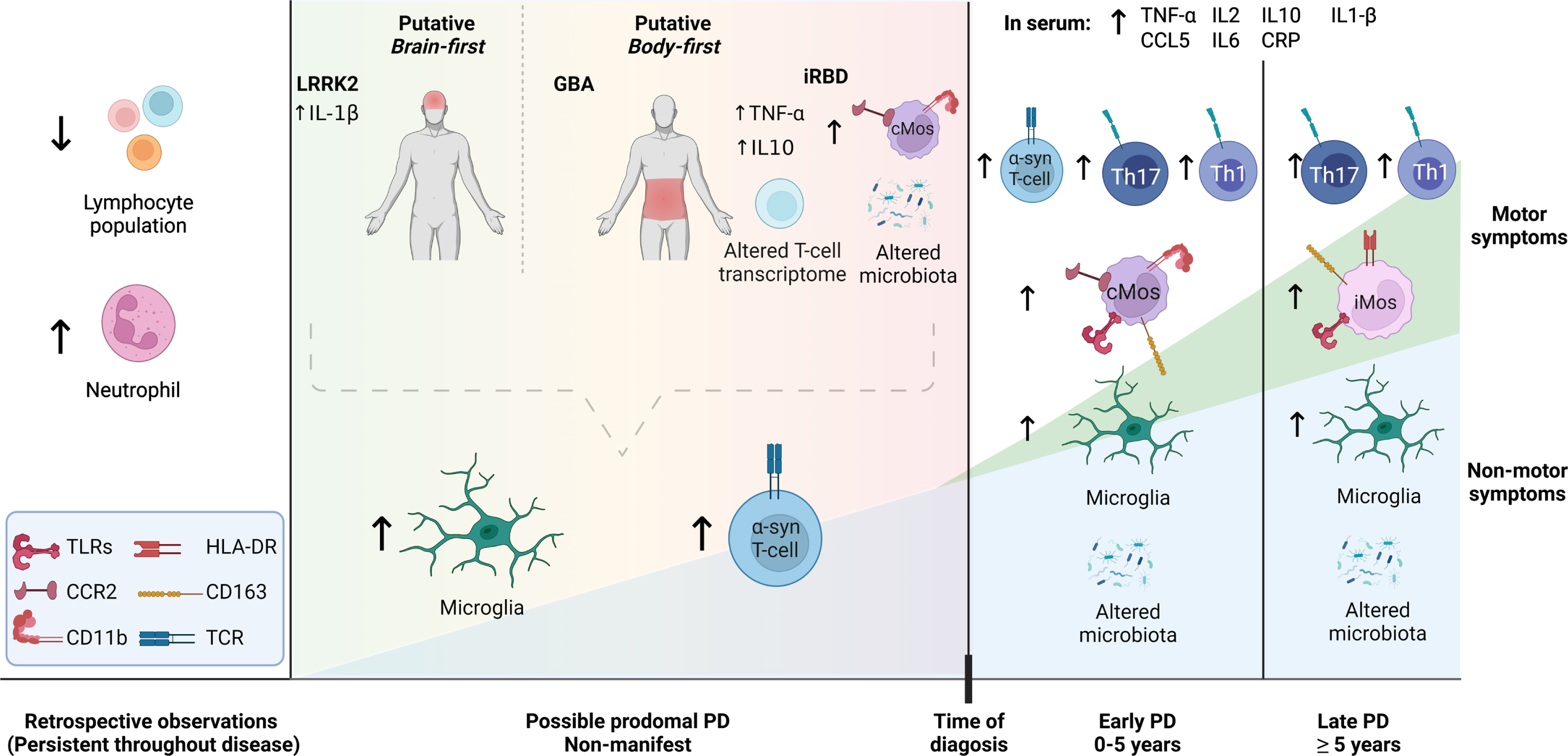
T-cells, both CD8 [15] and CD4 [16], have been proposed as key mediators of neuronal death in PD. PD patient-derived C8 T-cells show a profile suggesting a lack of immunosenescence, which could render them more prone to a reactive pro-inflammatory response [45, 46]. Interestingly, although the total T-cell numbers are decreased before and after PD diagnosis [47], in iPD patients, the effector CD4 T-cells have been reported overrepresented vs. the naïve population, with groups finding a bias towards the pro-inflammatory Th17 [13, 48, 49] or Th1 populations [13, 50]. The T-cell response seems to be more relevant in the early stage of the disease, with T-cells (CD8 and CD4) responding to α-syn-derived peptides found in a preclinical case and more prevalent in early-stage vs. late-stage PD [51, 52] (Fig. 1). PD patients have Th1 and cytotoxic CD4 T cell and terminal effector CD8 T cell populations clonally expanded by T-cell receptor (TCR)-dependent activation [53]. Further supporting a T- and B-cell early involvement, auto-anti-α-syn antibodies seemed elevated particularly in early PD, decreasing with time and disease progression [54]. Thus, adaptive immune cells and humoral response at prodromal stages of PD deserve further studies.
The decreased lymphocyte number might be related to the reported impaired lymphoproliferative response in clinical PD [55]. This was observed together with elevated signs of oxidative stress in blood cells [55], which could be a consequence of the elevated neutrophil population, since increased marker of oxidative stress, mitochondrial changes, and increased NO production have been reported in PD patients’ neutrophils [56, 57]. However, detailed changes in the neutrophil population in prodromal or clinical PD remain to be studied, and few are the groups that have addressed them. This is probably because most labs work with frozen isolated peripheral blood mononuclear cells when analysing immune cells, thus discarding granulocytes within the process. In conclusion, these studies support strongly early immune changes in PD years before diagnosis, with an unbalanced innate vs. adaptive cell pool.
NEUROINFLAMMATION IN PROBABLE PRODROMAL PD
iRBD is strongly associated with progression to a neurodegenerative disorder (primarily PD and dementia with Lewy bodies and less frequently multiple system atrophy), which develops in 75% of the patients [23]. Imaging studies have shown that most iRBD patients show neuronal changes resembling parkinsonian disorders despite the lack of motor symptoms [2, 58, 59]. iRBD patients who progress to PD are likely to have a more malignant PD course with autonomic dysfunction, cognitive loss, gait problems, and mortality. Non-iRBD prodromal PD is more likely to present with tremors and thus a faster PD diagnosis [2]. Prodromal iRBD is a strong marker of the body-first subtype hypothesized by Borghammer’s team, reflecting a neuropathologic brainstem predominant PD type (for further reading see [2]), a pattern that is also seen in GBA-associated PD, which shows denervation of PNS and RBD [2, 60].
In a cohort of 20 polysomnography-confirmed iRBD patients, Stockholm et al. found signs of neuroinflammation, as measured by PK-PET, in the SN compared to controls [58]. Smaller, however not significant, increased PK signals in the putamen and caudate nuclei were also found. A positive correlation was observed between PK binding values in SN and the putamen and the (right) caudate nucleus, possibly reflecting the dysfunction of somas and pre-synaptic terminals in the nigrostriatal pathway. This study also included 18F-DOPA PET, a marker of dopamine function, that showed nigrostriatal dopaminergic dysfunction in iRBD patients with microglial activation. However, a significant negative correlation between PK and 18F-DOPA PET uptake was only found in the caudate but not in the putamen. In the same cohort of iRBD patients, using a voxel-based analysis, increased PK binding was also found in the visual associative cortex of the occipital lobe [59]. Neuroinflammation in these brain areas could indicate early signs of cognitive changes and explain the poor performance of iRBD patients in visuospatial testing.
A longitudinal 18F-DOPA PET follow-up in a subgroup of these patients showed that the severity and extent of nigrostriatal terminal dysfunction increased significantly in iRBD patients over a 3-year period, and the greatest reductions were observed in subjects who had increased baseline microglial activation in their nigrostriatal structures. These findings would suggest a detrimental rather than a protective effect of neuroinflammation in the early stages of synucleinopathy [61]. This, however, needs to be confirmed in larger iRBD cohorts.
Stær and colleagues reported that in iRBD patients an increased PK binding in the substantia innominata, the major source of cholinergic input to the cortex [62], was associated with cortical cholinergic dysfunction, measured by 11C-Donepezil PET [63]. Interestingly, cholinergic anti-inflammatory neuroimmune function via the α7nChR has been related to memory and cognition ([64] see below). Longitudinal clinical and imaging follow-up studies in iRBD patients are needed to clarify if the changes observed in this cohort contribute to the development of cognitive impairment in iRBD patients and can be used to predict the onset of dementia.
LRRK2-associated PD is an autosomal dominant condition with a penetrance of up to 74% by 80 years of age [65]. Homozygote mutations in GBA are the cause of the autosomal recessive disorder Gaucher’s disease, but, in heterozygous form, GBA mutations are associated with autosomal dominant PD with a penetrance of 30% by 80 years of age [66]. Non-manifesting carriers of these mutations could therefore represent a high-risk PD group. In a cohort of 286 mutation carriers with no motor symptoms, 18% of LRRK2-carriers and 3% of GBA-carriers had reduced tracer uptake on 123I-Ioflupane dopamine transporter imaging [67]. The low prevalence could be explained by the asymptomatic state of this population [2].
There is some initial evidence suggesting that neuroinflammation might occur in non-manifesting LRRK2 and GBA carriers. A recent study found putaminal, dopaminergic dysfunction by 18F-DOPA imaging in five out of eight non-manifesting LRRK2. Three of them had significantly raised PK-binding bilaterally in the SN [68], suggesting neuroinflammation along with subclinical nigrostriatal dysfunction. In 2021, Mullin and colleagues found a significant increase in PK binding in the SN of nine non-manifesting GBA compared to controls [69]. Additionally, a voxel-based analysis also localized increased neuroinflammation in several brain regions, including the occipital and temporal lobes, hippocampus, cerebellum, and mesencephalon. Interestingly, the degree of hyposmia correlated with nigral PK binding [69]. Even though these studies have small sample sizes, they strongly indicate the need for further investigation of neuroinflammation in the premotor stage of LRRK2- and GBA-associated PD.
Regarding immune biomarkers in CSF, one study showed no differences in levels of mostly pro-inflammatory cytokines (IL2, IL4, IL8, IL6, 1ILβ, IL10, and IFNγ and TNF) between iPD, and manifesting and non-manifesting GBA-PD and LRRK2-PD [70]. It should be noted, that the same study found no differences between PD and controls either, which is in disagreement with the blood increased IL6, IL10, IL1β, and TNF reported by many [71]. No differences in CSF cytokines and chemokines were observed in non-manifesting LRRK2 by another team, but once PD was manifested the differential markers (vs. iPD) were related to chemotaxis and transmigration: IL8 and VEGF, suggesting a brain permissive to the entrance of peripheral immune cells [72].
PERIPHERAL IMMUNE CHANGES IN PROBABLE PRODROMAL PD
Supporting an early immune peripheral involvement, Farmen and colleagues have recently described that in the iRBD cohort examined by Stockholm and colleagues [58], monocytic changes already occur in blood in parallel to the described inflammatory and neurodegenerative events in the brain revealed by PET [73]. The iRBD patients showed increased classical monocytes, a highly phagocytic population able to migrate to inflamed tissue [74]. Accordingly, they found an increased percentage of cells expressing CCR2 or/and CD11b, both receptors involved in transmigration [73]. CCL2, the CCR2 ligand, has been associated with PD [75, 76] and the CCR2-CCL2 axis seems crucial for the recruitment of immune cells to the brain in PD-like degeneration. CD11b binds CD18 to form the functional integrin heterodimer Mac-1 (or CR3). CD11b interaction with ICAM, which is also increased in PD [10], mediates leukocyte adhesion, and cellular extravasation [77]. The relevance of chemotaxis is further corroborated by longitudinal data in PD patients showing CCL3 and CCL2 as the serum biomarkers contributing the most to the predictive models of severity [78]. Interestingly, elevated expression of CCR2 and CD11b on monocytes is also seen early in PD (<5 years from diagnosis) [12] (Fig. 1). These and other changes in monocytic immune markers have been associated with cognition and dementia in clinical PD corroborating a significant role of the immune system in the cognitive component of the disease [12, 14, 79].
Importantly, Farmen et al. also observed a significant correlation of monocyte markers with inflammatory and neuronal markers in the brain in the iRBD cohort. The expression of TLR4 on monocytes increased with inflammation in the brain, i.e., PK binding, while dopaminergic neurotransmission -18F-DOPA PET- decreased [73]. Thus, suggesting a deleterious role for TLR4 expression in monocytes. TLR4 is one of the receptors associated with α-syn inflammatory activation [28] and also the endotoxin receptor. Interestingly, increased endotoxin levels in patients’ blood have been reported [14], which could result in a pro-inflammatory environment and could contribute to disease, since chronic TLR4 activation can promote neurodegeneration [80]. On the other hand, in the iRBD patients the number of cells expressing the CD163 scavenger receptor, normally associated with anti-inflammatory monocytes, was correlated to lower inflammation and better neuronal health in the brain [73]. This data strongly supports a role for peripheral monocytes in brain events in prodromal (iRBD+) PD stages and an active communication between the brain and periphery. It should be noted that this communication does not imply that immune proteins, cytokines, or chemokines changed in the periphery and brain, should be the same, but rather that the events are occurring under active communication and in close relation to neuronal changes.
Interestingly, data suggest that the monocytes changes seem to differ as time/PD-stages progress since the enriched classical monocytes observed in iRBD is also seen in PD patients with <5 years of disease duration, but it later evolves to enrichment on intermediate monocytes [12] (Fig. 1). This might explain the inconsistency in the observation between labs, with some showing increased classical [14] and other increased intermediates monocytes in clinical PD [13]. Moreover, the enrichment of intermediate monocytes becomes more relevant as the severity of the disease increases (H&Y stage) [13]. However, longitudinal studies are necessary to confirm this.
The early peripheral changes in immune cells in iRBD are not limited to the innate system. iRBD patients’ CD4 T cells show a transcription factor expression profile closer to those observed in T-cells from iPD patients than healthy individuals, although not identical, supporting again stage associated changes [81]. Finally, further supporting an early immune peripheral response, analysis of sera from iRBD patients showed enrichment of proteins related to inflammation and immunity processes [82]. Moreover, although both pro-inflammatory TNF and anti-inflammatory IL10 cytokines have been found increased in iRBD blood, those patients showing elevated TNF/IL10 ratio, i.e., a more pronounced pro-inflammatory environment, had a higher risk to develop a synucleinopathies later in life [83]. In conclusion, iRBD patients showed significant signs of immune activation in the periphery with monocytes of migratory capacity, T-cells with an altered transcriptomic profile, and a pro-inflammatory cytokine environment.
The studies addressing (peripheral) immune changes in non-manifesting GBA or LRRK2 mutation carriers are few and mostly refer to soluble biomarkers. LRRK2 is highly expressed in immune cells [84], and its expression is increased in peripheral blood immune cells, T and B cells, and CD16+ monocytes from iPD patients [85]. Regular use of NSAIDs has been associated with reduced penetrance in LRRK2-PD [86]. And numerous studies suggest a role for LRRK2 in the immune function and microglial activation [87]. However, most of the studies addressing non-manifesting LRRK2 have found none or few changes in immune biomarkers in blood [70, 88]. Brockmann et al. showed no differences between non-manifesting LRRK2 and controls, although in manifested LRRK2-PD and iPD certain inflammatory markers differed suggesting a differential immune response in these two PD types [88]. In a follow-up study, the team showed that indeed once PD is diagnosed the immune response— and particularly recruitment— is relevant, since malignant LRRK2-PD subtypes showed higher levels of biomarkers involved in chemotaxis and infiltration IL8, CCL2, and CCL4 [75]. Damzco et al. found higher IL1β in sera of non-manifesting LRRK2, supporting a relevance for NLRP3 in PD. Indeed, NLRP3 is upregulated in immune cells in PD patients’ blood [89] and IL1β is increased in sera (and CSF) from PD patients [71]. NLRP3 genetic polymorphisms associated with lower PD risk show downregulation of NLRP3 activity [90]. Interestingly, once LRRK2-PD was manifested LRRK2-PD patients showed elevated PDGF, another marker associated with transmigration [72, 78].
Glucocerebrosidase activity is reduced in monocytes from iPD patients, particularly in classical monocytes, and this was inversely correlated to motor symptom severity [91]. α-Syn levels in blood mononuclear cells were higher in non-manifesting GBA [92] and manifesting GBA-PD patients than those from iPD [93]. Gaucher’s disease iPSC-derived macrophages showed increased pro-inflammatory cytokine expression and lower phagocytosis [94]. Transcriptome analysis of monocyte-derived macrophages from a small number of controls, manifesting and non-manifesting GBA-PD, showed that pathways related to immune response, development, differentiation, and axonal growth were enriched in the non-manifesting group [95]. Thaler et al. found no differences in blood cytokines between iPD, controls, and manifesting and non-manifesting GBA-PD [70]. Although biallelic GBA mutation has been related to an increase of chemokines CCL18 and CCL3 in serum. Moreover, these chemokines levels were associated with motor and/or cognition defects in all PD patients (iPD and GBA-PD) [96]. One study has, however, reported differences in manifesting GBA-PD with increased IL8, CCL2, CCL3, and CCL4 corroborating the relevance of chemotaxis [97]. Therefore, the data so far does not show a particular pro-inflammatory cytokine environment in the blood of prodromal GBA or LRRK2 PD, but they suggest an environment with enhanced chemotaxis and transmigration. Studies addressing cellular change on blood immune cells in these groups are needed to better define any possible involvement of peripheral immune cells.
NEUROTRANSMITTERS WITH IMMUNOMODULATORY CAPACITY
Neurotransmitters possess immunomodulatory abilities; and not only do most immune cells (including microglia) express neurotransmitter receptors, but some can synthesize them to act in an auto- and paracrine manner modulating locally the immune process. In addition, several neuronal groups and circuits are involved in the regulation of the immune system and response to stress, namely: the enteric system, the vagus nerve (VN), and the hypothalamic-pituitary-adrenal (HPA) axis [98–100]. Although beyond the scope of this review, we will shortly discuss the potential relevance of the neuroimmunological aspect in prodromal PD.
Since primary and secondary lymphoid tissues receive autonomic sympathetic innervation, both noradrenaline and dopamine would influence the immune system [101]. Dopamine signalling on myeloid cells modulates inflammatory responses: it decreases NLRP3 [102], and LPS induced inflammation and promotes chemokinesis and chemotaxis on macrophages and microglia [103, 104]. Dopamine decreases oxidative stress production, cell migration, and phagocytic activity on human neutrophils [103]. Dopamine actions on T-cells depend on the type of dopamine receptors expressed and their activation status. It activates resting T cells, while it suppresses the production of IL2, IL4, and IFNγ in already activated T cells [105, 106]. This would be relevant since T-cells are seen in PD midbrain, although it is unknown how early this infiltration occurs. Notably, PD patients’ T cells show changes in the expression of dopamine receptors, and this was associated with motor symptoms [107]. Moreover, dopamine receptor gene polymorphism has been related to different clinical PD presentations and T-cell function (for a review, see [108]). Therefore, a decrease in dopaminergic neurotransmission in prodromal PD will affect both innate and adaptive immune responses. Equally, dopaminergic drugs might affect immune cells as shown in several studies analysing T-cells in PD patients [109, 110].
Locus coeruleus (LC) and noradrenaline have been associated with feedback modulation of the HPA axis and immunomodulation in the brain (see [111] for further reading). The HPA axis, with glucocorticoids as the main effectors, involves several brain nuclei that contribute to the neuroendocrine modulation of the immune response during infection and stress [100]. Noradrenaline downmodulates LPS pro-inflammatory activation in human whole blood cells in vitro [112]. It inhibits oxidative metabolism and adhesion of neutrophils [113, 114]. Equally, noradrenergic activation on human monocytes [113] and (non-human) microglia [115] is usually anti-inflammatory. This might be of relevance in prodromal stages of PD since several PET imaging studies point to decreased thalamic and sensorimotor cortex uptake of 11C-methylreboxetine (MeNER), a marker of noradrenergic function, and signs of neurodegeneration in LC in RBD patients (neuromelanin MRI) [116–118]. Moreover, the celiac ganglia release noradrenaline in the spleen, upon VN activation, that acts on beta-adrenergic receptors on T-cells leading to their release of acetylcholine, which will also exert an anti-inflammatory effect [119]. Therefore, VN denervation in prodromal PD stages will disrupt this, influencing immune response (see more below).
Lastly, serotonin has also a relevant role in both innate and adaptive immune cells [120]. Serotonin signalling modulates T-cell activation and function, depending on the receptor subtype. It can promote proliferation [121] and anti-inflammatory phenotypes in T-cells [122]. Increased serotonin levels and serotonin receptor activation seem to exert an anti-inflammatory effect in different cells [120, 123]. Interestingly, hypercholinergic activity in the cortex, thalamus, and limbic areas [124], and increased serotonin in the striatum, brainstem, and hypothalamus [125] have been seen in non-manifesting LRRK2 individuals. This might be of special relevance and could explain a milder progression in these individuals.
GUT-BRAIN AXIS: MICROBIOTA AND VAGUS NERVE IN PRODROMAL STAGES
Gastrointestinal dysfunction is a potential early PD symptom, which seems associated with the α-syn pathology observed in the gut of PD patients [126–128], and in healthy controls who later develop PD [129, 130]. This α-syn pathological accumulation could be the result of gut microbiota changes, and indeed, a range of studies has reported altered gut microbiota in PD patients [131]. However, it is not clear if this plays a causative role or is a consequence of the pathogenesis. Analysis of faecal microbiota in iRBD, iPD, and healthy controls, revealed that 80% of the differentially enriched gut microbes in iPD showed similar trends in iRBD, and they correlated with non-motor symptoms [132]. Among other changes, the study reported a decreased abundance of Prevotella in iRBD. Interestingly in a follow-up longitudinal study, the team found that microbiota changes in iPD were persistent and that Prevotella in the gut tends to be even less abundant in iPD patients with faster disease progression [133]. Furthermore, in their most recent study, they report that the constipation severity was lowest and subthreshold parkinsonism least frequent in individuals with the Prevotella-enriched enterotype [134]. Thus, gut bacteria might associate with relevant changes in disease and these changes appear early and persistent. Microbiota can modulate the immune system by releasing neurotransmitters and other molecules, thus dysbiosis will affect immune response. Moreover, it could also affect the integrity of the intestinal epithelial barrier, with subsequent leaking of intestinal content into the blood and initiation of systemic inflammation [135]. Supporting this, newly diagnosed PD patients showed signs of increased intestinal permeability, which correlated with the accumulation of α-syn in the intestine [127] and PD patients had increased endotoxin in blood [14]. However, these factors have not yet been studied in prodromal PD.
The gut is innervated by VN, which degenerates early in some patients, as suggested by PET studies and the early constipation observed in PD. VN serves as the conduct or a bidirectional connection in the gut-brain axis and signals transmitted by the dorsal motor nucleus of the VN modulate the immune response through the inflammatory reflex [98]. VN exerts an anti-inflammatory effect on the immune system involving acetylcholine [98] in a mechanism exerted on the gut and, via the celiac ganglia, on the spleen [136]. Ultimately acetylcholine binds α7nChR on macrophages, and inhibits their release of cytokines, shifting them towards an anti-inflammatory phenotype [137]. Although this has not been studied in detail, we could speculate that the compromised VN in PD would result in a failure of the anti-inflammatory reflex, thus favouring inflammation early in the disease, which would promote further degeneration. According to the mentioned body- vs. brain-first theory, the body-first subtype would have VN affected early in the disease, which could explain the faster disease progression and severity of this proposed subtype [138]. Contrary, in the brain-first the VN would be affected later, thus the inflammatory reflex is preserved, maybe explaining the slower progression. However, as of today, this is speculative, and more research is needed regarding the inflammatory reflex in PD and its association with PD subtypes.
CONCLUSIONS
Taken together the imaging findings suggest that microglial activation is present in the early stage of the neurodegenerative process in iRBD and non-manifesting GBA and LRRK2. However, it is yet unclear if the microglia activation has a protective function at this prodromal stage rather than the detrimental one suggested in advanced stages. Currently, no PET tracers are available to distinguish microglia subtypes. Additionally, these findings do not clarify the temporal relationship between neuroinflammation and the degenerative process. Moreover, the studies are limited by the use of PK, which despite its high sensitivity, has limited specificity due to relatively low brain permeability and its binding affinity to platelets, monocytes, and plasma proteins. Thus, PK is unable to detect the most subtle neuroinflammatory changes [139]. Unfortunately, the second-generation TSPO radiotracers also have their own shortcomings, such as the mixed affinity due to rs6971 polymorphism in the TSPO gene. A third generation of TSPO tracers that aims to overcome this fluctuating affinity is yet to be validated for clinical use [140]. Moreover, TSPO is also expressed on reactive astrocytes [141] and the observed increase in binding in these patients could also reflect reactive astrocytes, although this is also indicative of immune activation.
Regarding changes in the periphery, evidence points towards an affected peripheral immune system with increased neutrophils and decreased lymphocytes years before PD diagnosis that seems to remain through the disease. In iRBD the data is limited, but it also supports both innate and adaptive responses, with a coordinated immune response in the brain and periphery as suggested by the associations between blood monocyte marker and brain PET data [73]. But these peripheral immune changes might also occur just as a response to the damage of the PNS including VN degeneration. This in turn will have significant consequences on the immune system due to changes in the gut-brain axis and the loss of the inflammatory reflex, all together promoting disease. However, based on the few studies published, non-manifesting LRRK2 showed little signs of inflammation and immune activation in blood, although this was based on cytokine/chemokine levels since no study so far has been conducted analysing immune cellular changes in the blood, which prevents us from concluding. However, we can speculate that not all prodromal stages involve the (peripheral) immune system equally, but once the disease is manifested, the neurodegenerative process might more robustly include a systemic immune response. Accordingly, the little affection of PNS and the increase in cholinergic and serotoninergic neurotransmission in non-manifesting LRRK2 might contribute to reducing the inflammatory environment thus protecting neurons. However, the data collected so far is insufficient and more research needs to be done. Longitudinal studies in larger cohorts that include cellular and functional analysis of blood immune cells and with improved PET microglia tracers are needed to clarify the role of the immune system in PD subtypes and to determine the temporal relationship between immune activation and neurodegeneration in synucleinopathies. This will allow us to rationalise the potential of immunomodulatory agents as neuroprotective strategies in these patients.
ACKNOWLEDGMENTS
IHK is supported by a PhD Fellowship from the Health Faculty, Aarhus University. MHT and VSH are supported by PhD Fellowships from the Independent Research Fund, Denmark.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
REFERENCES
[1] | Postuma RB , Berg D , Stern M , Poewe W , Olanow CW , Oertel W , Obeso J , Marek K , Litvan I , Lang AE , Halliday G , Goetz CG , Gasser T , Dubois B , Chan P , Bloem BR , Adler CH , Deuschl G ((2015) ) MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 30: , 1591–1601. |
[2] | Berg D , Borghammer P , Fereshtehnejad SM , Heinzel S , Horsager J , Schaeffer E , Postuma RB ((2021) ) Prodromal Parkinson disease subtypes - key to understanding heterogeneity. Nat Rev Neurol 17: , 349–361. |
[3] | Horsager J , Andersen KB , Knudsen K , Skjaerbaek C , Fedorova TD , Okkels N , Schaeffer E , Bonkat SK , Geday J , Otto M , Sommerauer M , Danielsen EH , Bech E , Kraft J , Munk OL , Hansen SD , Pavese N , Goder R , Brooks DJ , Berg D , Borghammer P ((2020) ) Brain-first versus body-first Parkinson’s disease: A multimodal imaging case-control study. Brain 143: , 3077–3088. |
[4] | Blauwendraat C , Nalls MA , Singleton AB ((2020) ) The genetic architecture of Parkinson’s disease. Lancet Neurol 19: , 170–178. |
[5] | Lin KJ , Lin KL , Chen SD , Liou CW , Chuang YC , Lin HY , Lin TK ((2019) ) The overcrowded crossroads: Mitochondria, alpha-synuclein, and the endo-lysosomal system interaction in Parkinson’s disease. Int J Mol Sci 20: , 5312. |
[6] | Kalia LV , Lang AE ((2015) ) Parkinson’s disease. Lancet 386: , 896–912. |
[7] | Gerhard A , Pavese N , Hotton G , Turkheimer F , Es M , Hammers A , Eggert K , Oertel W , Banati RB , Brooks DJ ((2006) ) imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis 21: , 404–412. |
[8] | Ouchi Y , Yoshikawa E , Sekine Y , Futatsubashi M , Kanno T , Ogusu T , Torizuka T ((2005) ) Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol 57: , 168–175. |
[9] | McGeer PL , Itagaki S , Boyes BE , McGeer EG ((1988) ) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38: , 1285–1291. |
[10] | Imamura K , Hishikawa N , Sawada M , Nagatsu T , Yoshida M , Hashizume Y ((2003) ) Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol (Berl) 106: , 518–526. |
[11] | Harms AS , Ferreira SA , Romero-Ramos M ((2021) ) Periphery and brain, innate and adaptive immunity in Parkinson’s disease. Acta Neuropathol 141: , 527–545. |
[12] | Konstantin Nissen S , Farmen K , Carstensen M , Schulte C , Goldeck D , Brockmann K , Romero-Ramos M ((2022) ) Changes in CD163+, CD11b+, and CCR2+ peripheral monocytes relate to Parkinson’s disease and cognition. Brain Behav Immun 101: , 182–193. |
[13] | Thome AD , Atassi F , Wang J , Faridar A , Zhao W , Thonhoff JR , Beers DR , Lai EC , Appel SH ((2021) ) expansion of dysfunctional regulatory T lymphocytes restores suppressive function in Parkinson’s disease. NPJ Parkinsons Dis 7: , 41. |
[14] | Wijeyekoon RS , Kronenberg-Versteeg D , Scott KM , Hayat S , Kuan WL , Evans JR , Breen DP , Cummins G , Jones JL , Clatworthy MR , Andres Floto R , Barker RA , Williams-Gray CH ((2020) ) Peripheral innate immune and bacterial signals relate to clinical heterogeneity in Parkinson’s disease. Brain Behav Immun 87: , 473–488. |
[15] | Galiano-Landeira J , Torra A , Vila M , Bove J ((2020) ) CD8 T cell nigral infiltration precedes synucleinopathy in early stages of Parkinson’s disease. Brain 143: , 3717–3733. |
[16] | Brochard V , Combadiere B , Prigent A , Laouar Y , Perrin A , Beray-Berthat V , Bonduelle O , Alvarez-Fischer D , Callebert J , Launay JM , Duyckaerts C , Flavell RA , Hirsch EC , Hunot S ((2009) ) Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 119: , 182–192. |
[17] | Earls RH , Menees KB , Chung J , Gutekunst CA , Lee HJ , Hazim MG , Rada B , Wood LB , Lee JK ((2020) ) NK cells clear alpha-synuclein and the depletion of NK cells exacerbates synuclein pathology in a mouse model of alpha-synucleinopathy. Proc Natl Acad Sci U S A 117: , 1762–1771. |
[18] | Hamza TH , Zabetian CP , Tenesa A , Laederach A , Montimurro J , Yearout D , Kay DM , Doheny KF , Paschall J , Pugh E , Kusel VI , Collura R , Roberts J , Griffith A , Samii A , Scott WK , Nutt J , Factor SA , Payami H ((2010) ) Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet 42: , 781–785. |
[19] | Wallings RL , Herrick MK , Tansey MG ((2020) ) LRRK2 at the interface between peripheral and central immune function in Parkinson’s. Front Neurosci 14: , 443. |
[20] | Alam MM , Yang , Li XQ , Liu J , Back TC , Trivett A , Karim B , Barbut D , Zasloff M , Oppenheim JJ ((2022) ) Alpha synuclein, the culprit in Parkinson disease, is required for normal immune function. Cell Rep 38: , 110090. |
[21] | Stolzenberg E , Berry D , Yang , Lee EY , Kroemer A , Kaufman S , Wong GCL , Oppenheim JJ , Sen S , Fishbein T , Bax A , Harris B , Barbut D , Zasloff MA ((2017) ) A role for neuronal alpha-synuclein in gastrointestinal immunity. J Innate Immun 9: , 456–463. |
[22] | Berg D , Postuma RB , Bloem B , Chan P , Dubois B , Gasser T , Goetz CG , Halliday GM , Hardy J , Lang AE , Litvan I , Marek K , Obeso J , Oertel W , Olanow CW , Poewe W , Stern M , Deuschl G ((2014) ) Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson’s disease. Mov Disord 29: , 454–462. |
[23] | Berg D , Postuma RB , Adler CH , Bloem BR , Chan P , Dubois B , Gasser T , Goetz CG , Halliday G , Joseph L , Lang AE , Liepelt-Scarfone I , Litvan I , Marek K , Obeso J , Oertel W , Olanow CW , Poewe W , Stern M , Deuschl G ((2015) ) MDS research criteria for prodromal Parkinson’s disease. Mov Disord 30: , 1600–1611. |
[24] | Heinzel S , Berg D , Gasser T , Chen H , Yao C , Postuma RB ((2019) ) Update of the MDS research criteria for prodromal Parkinson’s disease. Mov Disord 34: , 1464–1470. |
[25] | Hoeffel G , Ginhoux F ((2018) ) Fetal monocytes and the origins of tissue-resident macrophages. Cell Immunol 330: , 5–15. |
[26] | Prinz M , Jung S , Priller J ((2019) ) Microglia biology: One century of evolving concepts. Cell 179: , 292–311. |
[27] | Hamelin L , Lagarde J , Dorothée G , Leroy C , Labit M , Comley RA , de Souza LC , Corne H , Dauphinot L , Bertoux M , Dubois B , Gervais P , Colliot O , Potier MC , Bottlaender M , Sarazin M ((2016) ) Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18F-DPA-714 PET imaging. Brain 139: , 1252–1264. |
[28] | Ferreira SA , Romero-Ramos M ((2018) ) Microglia response during Parkinson’s disease: Alpha-synuclein intervention. Front Cell Neurosci 12: , 247. |
[29] | Croisier E , Moran LB , Dexter DT , Pearce RK , Graeber MB ((2005) ) Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha-synuclein deposition. J Neuroinflammation 2: , 14. |
[30] | Kouli A , Camacho M , Allinson K , Williams-Gray CH ((2020) ) Neuroinflammation and protein pathology in Parkinson’s disease dementia. Acta Neuropathol Commun 8: , 211. |
[31] | Doorn KJ , Moors T , Drukarch B , van de Berg W , Lucassen PJ , van Dam AM ((2014) ) Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson’s disease patients. Acta Neuropathol Commun 2: , 90. |
[32] | Kim C , Ho DH , Suk JE , You S , Michael S , Kang J , Joong Lee S , Masliah E , Hwang D , Lee HJ , Lee SJ ((2013) ) Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 4: , 1562. |
[33] | Drouin-Ouellet J , St-Amour I , Saint-Pierre M , Lamontagne-Proulx J , Kriz J , Barker RA , Cicchetti F ((2014) ) Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson’s disease. Int J Neuropsychopharmacol 18: , pyu103. |
[34] | Imamura K , Hishikawa N , Sawada M , Nagatsu T , Yoshida M , Hashizume Y ((2003) ) Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol 106: , 518–526. |
[35] | Papadopoulos V , Baraldi M , Guilarte TR , Knudsen TB , Lacapère JJ , Lindemann P , Norenberg MD , Nutt D , Weizman A , Zhang MR , Gavish M ((2006) ) Translocator protein (18kDa): New nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci 27: , 402–409. |
[36] | Banati RB ((2002) ) Visualising microglial activation. Glia 40: , 206–217. |
[37] | Jiang S , Gao H , Luo Q , Wang P , Yang X ((2017) ) The correlation of lymphocyte subsets, natural killer cell, and Parkinson’s disease: A meta-analysis. Neurol Sci 38: , 1373–1380. |
[38] | Yazdani S , Mariosa D , Hammar N , Andersson J , Ingre C , Walldius G , Fang F ((2019) ) Peripheral immune biomarkers and neurodegenerative diseases: A prospective cohort study with 20 years of follow-up. Ann Neurol 86: , 913–926. |
[39] | Jensen MP , Jacobs BM , Dobson R , Bandres-Ciga S , Blauwendraat C , Schrag A , Noyce AJ , International Parkinson’s Disease Genomics Consortium (IPDGC) ((2021) ) Lower lymphocyte count is associated with increased risk of Parkinson’s disease. Ann Neurol 89: , 803–812. |
[40] | Kluter H , Vieregge P , Stolze H , Kirchner H ((1995) ) Defective production of interleukin-2 in patients with idiopathic Parkinson’s disease. J Neurol Sci 133: , 134–139. |
[41] | Craig DW , Hutchins E , Violich I , Alsop E , Gibbs JR , Levy S , Robison M , Prasad N , Foroud T , Crawford KL , Toga AW , Whitsett TG , Kim S , Casey B , Reimer A , Hutten SJ , Frasier M , Kern F , Fehlman T , Keller A , Cookson MR , Van Keuren-Jensen K , Hutten S , Van Keuren-Jensen K , Parkinson Progression Marker Initiative ((2021) ) RNA sequencing of whole blood reveals early alterations in immune cells and gene expression in Parkinson’s disease. Nature Aging 1: , 734–747. |
[42] | Sanjari Moghaddam H , Ghazi Sherbaf F , Mojtahed Zadeh M , Ashraf-Ganjouei A , Aarabi MH ((2018) ) Association between peripheral inflammation and DATSCAN data of the striatal nuclei in different motor subtypes of Parkinson disease. Front Neurol 9: , 234. |
[43] | Umehara T , Oka H , Nakahara A , Matsuno H , Murakami H ((2020) ) Differential leukocyte count is associated with clinical phenotype in Parkinson’s disease. J Neurol Sci 409: , 116638. |
[44] | Haghshomar M , Rahmani F , Hadi Aarabi M , Shahjouei S , Sobhani S , Rahmani M ((2019) ) White matter changes correlates of peripheral neuroinflammation in patients with Parkinson’s disease. Neuroscience 403: , 70–78. |
[45] | Kouli A , Jensen M , Papastavrou V , Scott KM , Kolenda C , Parker C , Solim IH , Camacho M , Martin-Ruiz C , Williams-Gray CH ((2021) ) T lymphocyte senescence is attenuated in Parkinson’s disease. J Neuroinflammation 18: , 228. |
[46] | Williams-Gray CH , Wijeyekoon RS , Scott KM , Hayat S , Barker RA , Jones JL ((2018) ) Abnormalities of age-related T cell senescence in Parkinson’s disease. J Neuroinflammation 15: , 166. |
[47] | Horvath S , Ritz BR ((2015) ) Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging (Albany NY) 7: , 1130–1142. |
[48] | Yan Z , Yang W , Wei H , Dean MN , Standaert DG , Cutter GR , Benveniste EN , Qin H ((2021) ) Dysregulation of the adaptive immune system in patients with early-stage Parkinson disease. Neurol Neuroimmunol Neuroinflamm 8: , e1036. |
[49] | Sommer A , Marxreiter F , Krach F , Fadler T , Grosch J , Maroni M , Graef D , Eberhardt E , Riemenschneider MJ , Yeo GW , Kohl Z , Xiang W , Gage FH , Winkler J , Prots I , Winner B ((2018) ) Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson’s disease. Cell Stem Cell 23: , 123–131 e126. |
[50] | Kustrimovic N , Comi C , Magistrelli L , Rasini E , Legnaro M , Bombelli R , Aleksic I , Blandini F , Minafra B , Riboldazzi G , Sturchio A , Mauri M , Bono G , Marino F , Cosentino M ((2018) ) Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: Cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naive and drug-treated patients. J Neuroinflammation 15: , 205. |
[51] | Lindestam Arlehamn CS , Dhanwani R , Pham J , Kuan R , Frazier A , Rezende Dutra J , Phillips E , Mallal S , Roederer M , Marder KS , Amara AW , Standaert DG , Goldman JG , Litvan I , Peters B , Sulzer D , Sette A ((2020) ) alpha-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat Commun 11: , 1875. |
[52] | Sulzer D , Alcalay RN , Garretti F , Cote L , Kanter E , Agin-Liebes J , Liong C , McMurtrey C , Hildebrand WH , Mao X , Dawson VL , Dawson TM , Oseroff C , Pham J , Sidney J , Dillon MB , Carpenter C , Weiskopf D , Phillips E , Mallal S , Peters B , Frazier A , Lindestam Arlehamn CS , Sette A ((2017) ) T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 546: , 656–661. |
[53] | Wang P , Yao L , Luo M , Zhou W , Jin X , Xu Z , Yan S , Li Y , Xu C , Cheng R , Huang Y , Lin X , Ma K , Cao H , Liu H , Xue G , Han F , Nie H , Jiang Q ((2021) ) Single-cell transcriptome and TCR profiling reveal activated and expanded T cell populations in Parkinson’s disease. Cell Discov 7: , 52. |
[54] | Scott KM , Kouli A , Yeoh SL , Clatworthy MR , Williams-Gray CH ((2018) ) A systematic review and meta-analysis of alpha synuclein auto-antibodies in Parkinson’s disease. Front Neurol 9: , 815. |
[55] | Vida C , Kobayashi H , Garrido A , Martinez de Toda I , Carro E , Molina JA , De la Fuente M ((2019) ) Lymphoproliferation impairment and oxidative stress in blood cells from early Parkinson’s disease patients. Int J Mol Sci 20: , 771. |
[56] | Vitte J , Michel BF , Bongrand P , Gastaut JL ((2004) ) Oxidative stress level in circulating neutrophils is linked to neurodegenerative diseases. J Clin Immunol 24: , 683–692. |
[57] | Gatto EM , Carreras MC , Pargament GA , Riobo NA , Reides C , Repetto M , Fernandez Pardal MM , Llesuy S , Poderoso JJ ((1996) ) Neutrophil function, nitric oxide, and blood oxidative stress in Parkinson’s disease. Mov Disord 11: , 261–267. |
[58] | Stokholm MG , Iranzo A , Østergaard K , Serradell M , Otto M , Svendsen KB , Garrido A , Vilas D , Borghammer P , Santamaria J , Møller A , Gaig C , Brooks DJ , Tolosa E , Pavese N ((2017) ) Assessment of neuroinflammation in patients with idiopathic rapid-eye-movement sleep behaviour disorder: A case-control study. Lancet Neurol 16: , 789–796. |
[59] | Stokholm MG , Iranzo A , Ostergaard K , Serradell M , Otto M , Bacher Svendsen K , Garrido A , Vilas D , Parbo P , Borghammer P , Santamaria J , Moller A , Gaig C , Brooks DJ , Tolosa E , Pavese N ((2018) ) Extrastriatal monoaminergic dysfunction and enhanced microglial activation in idiopathic rapid eye movement sleep behaviour disorder. Neurobiol Dis 115: , 9–16. |
[60] | Lebouvier T , Clairembault T , Devos D , Pallardy A , Coron E , Neunlist M , Derkinderen P ((2014) ) Peripheral autonomic nervous system involvement in Gaucher-related parkinsonism. J Parkinsons Dis 4: , 29–32. |
[61] | Staer K IA , Stokholm M , Ostergaard K , Serradell M , Otto M , Svendsen K , Pla A VD , Santamaria J , Moller A , Gaig C , Brooks D , Borghammer P , Tolosa E , Pavese N ((2020) ) Microglial activation associated with a faster progression of nigrostriatal dysfunction in patients with isolated REM sleep behavior disorder. Mov Disord 35: , 283. |
[62] | Mesulam MM , Geula C ((1988) ) Nucleus basalis (Ch4) and cortical cholinergic innervation in the human brain: Observations based on the distribution of acetylcholinesterase and choline acetyltransferase. J Comp Neurol 275: , 216–240. |
[63] | Stær K , Iranzo A , Stokholm MG , Østergaard K , Serradell M , Otto M , Svendsen KB , Garrido A , Vilas D , Santamaria J , Møller A , Gaig C , Brooks DJ , Borghammer P , Tolosa E , Pavese N ((2020) ) Cortical cholinergic dysfunction correlates with microglial activation in the substantia innominata in REM sleep behavior disorder. Parkinsonism Relat Disord 81: , 89–93. |
[64] | Maurer SV , Williams CL ((2017) ) The cholinergic system modulates memory and hippocampal plasticity via its interactions with non-neuronal cells. Front Immunol 8: , 1489. |
[65] | Healy DG , Falchi M , O’Sullivan SS , Bonifati V , Durr A , Bressman S , Brice A , Aasly J , Zabetian CP , Goldwurm S , Ferreira JJ , Tolosa E , Kay DM , Klein C , Williams DR , Marras C , Lang AE , Wszolek ZK , Berciano J , Schapira AH , Lynch T , Bhatia KP , Gasser T , Lees AJ , Wood NW ((2008) ) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol 7: , 583–590. |
[66] | Anheim M , Elbaz A , Lesage S , Durr A , Condroyer C , Viallet F , Pollak P , Bonaïti B , Bonaïti-Pellié C , Brice A ((2012) ) Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 78: , 417–420. |
[67] | Simuni T , Uribe L , Cho HR , Caspell-Garcia C , Coffey CS , Siderowf A , Trojanowski JQ , Shaw LM , Seibyl J , Singleton A , Toga AW , Galasko D , Foroud T , Tosun D , Poston K , Weintraub D , Mollenhauer B , Tanner CM , Kieburtz K , Chahine LM , Reimer A , Hutten SJ , Bressman S , Marek K ((2020) ) Clinical and dopamine transporter imaging characteristics of non-manifest LRRK2 and GBA mutation carriers in the Parkinson’s Progression Markers Initiative (PPMI): A cross-sectional study. Lancet Neurol 19: , 71–80. |
[68] | Gersel Stokholm M , Garrido A , Tolosa E , Serradell M , Iranzo A , Østergaard K , Borghammer P , Møller A , Parbo P , Stær K , Brooks DJ , Martí MJ , Pavese N ((2020) ) Imaging dopamine functionand microglia in asymptomatic LRRK2 mutation carriers. JNeurol 267: , 2296–2300. |
[69] | Mullin S , Stokholm MG , Hughes D , Mehta A , Parbo P , Hinz R , Pavese N , Brooks DJ , Schapira AHV ((2021) ) Brain microglial activation increased in glucocerebrosidase (GBA) mutation carriers without Parkinson’s disease. Mov Disord 36: , 774–779. |
[70] | Thaler A , Omer N , Giladi N , Gurevich T , Bar-Shira A , Gana-Weisz M , Goldstein O , Kestenbaum M , Shirvan JC , Cedarbaum JM , Orr-Urtreger A , Regev K , Shenhar-Tsarfaty S , Mirelman A ((2021) ) Mutations in GBA and LRRK2 are not associated with increased inflammatory markers. J Parkinsons Dis 11: , 1285–1296. |
[71] | King E , Thomas A ((2017) ) Systemic inflammation in Lewy body diseases: A systematic review. Alzheimer Dis Assoc Disord 31: , 346–356. |
[72] | Dzamko N , Rowe DB , Halliday GM ((2016) ) Increased peripheral inflammation in asymptomatic leucine-rich repeat kinase 2 mutation carriers. Mov Disord 31: , 889–897. |
[73] | Farmen K , Nissen SK , Stokholm MG , Iranzo A , Ostergaard K , Serradell M , Otto M , Svendsen KB , Garrido A , Vilas D , Borghammer P , Santamaria J , Moller A , Gaig C , Brooks DJ , Tolosa E , Pavese N , Romero-Ramos M ((2021) ) Monocyte markers correlate with immune and neuronal brain changes in REM sleep behavior disorder. Proc Natl Acad Sci U S A 118: , e2020858118. |
[74] | Geissmann F , Jung S , Littman DR ((2003) ) Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19: , 71–82. |
[75] | Brockmann K , Schulte C , Schneiderhan-Marra N , Apel A , Pont-Sunyer C , Vilas D , Ruiz-Martinez J , Langkamp M , Corvol JC , Cormier F , Knorpp T , Joos TO , Bernard A , Gasser T , Marras C , Schule B , Aasly JO , Foroud T , Marti-Masso JF , Brice A , Tolosa E , Berg D , Maetzler W ((2017) ) Inflammatory profile discriminates clinical subtypes in LRRK2-associated Parkinson’s disease. Eur J Neurol 24: , 427–e426. |
[76] | Hall S , Janelidze S , Surova Y , Widner H , Zetterberg H , Hansson O ((2018) ) Cerebrospinal fluid concentrations of inflammatory markers in Parkinson’s disease and atypical parkinsonian disorders. Sci Rep 8: , 13276. |
[77] | Fagerholm SC , Varis M , Stefanidakis M , Hilden TJ , Gahmberg CG ((2006) ) alpha-Chain phosphorylation of the human leukocyte CD11b/CD18 (Mac-1) integrin is pivotal for integrin activation to bind ICAMs and leukocyte extravasation. Blood 108: , 3379–3386. |
[78] | Ahmadi Rastegar D , Ho N , Halliday GM , Dzamko N ((2019) ) Parkinson’s progression prediction using machine learning and serum cytokines. NPJ Parkinsons Dis 5: , 14. |
[79] | Fan Z , Aman Y , Ahmed I , Chetelat G , Landeau B , Ray Chaudhuri K , Brooks DJ , Edison P ((2015) ) Influence of microglial activation on neuronal function in Alzheimer’s and Parkinson’s disease dementia. Alzheimers Dement 11: , 608–621 e607. |
[80] | Hughes CD , Choi ML , Ryten M , Hopkins L , Drews A , Botia JA , Iljina M , Rodrigues M , Gagliano SA , Gandhi S , Bryant C , Klenerman D ((2018) ) Picomolar concentrations of oligomeric alpha-synuclein sensitizes TLR4 to play an initiating role in Parkinson’s disease pathogenesis. Acta Neuropathol 137: , 103–120. |
[81] | De Francesco E , Terzaghi M , Storelli E , Magistrelli L , Comi C , Legnaro M , Mauri M , Marino F , Versino M , Cosentino M ((2020) ) CD4+ T-cell transcription factors in idiopathic REM sleep behavior disorder and Parkinson’s disease. Mov Disord 36: , 225–229. |
[82] | Mondello S , Kobeissy F , Mechref Y , Zhao J , Talih FR , Cosentino F , Antelmi E , Moresco M , Plazzi G , Ferri R ((2018) ) Novel biomarker signatures for idiopathic REM sleep behavior disorder: A proteomic and system biology approach. Neurology 91: , e1710–e1715. |
[83] | ZhangH, WangT, LiY, MaoW, HaoS, HuangZ, ChanP, CaiY ((2020) ) Plasma immune markers in an idiopathic REM sleep behavior disorder cohort. Parkinsonism Relat Disord 78: , 145–150. |
[84] | Hakimi M , Selvanantham T , Swinton E , Padmore RF , Tong Y , Kabbach G , Venderova K , Girardin SE , Bulman DE , Scherzer CR , LaVoie MJ , Gris D , Park DS , Angel JB , Shen J , Philpott DJ , Schlossmacher MG ((2011) ) Parkinson’s disease-linked LRRK2 is expressed in circulating and tissue immune cells and upregulated following recognition of microbial structures. J Neural Transm (Vienna) 118: , 795–808. |
[85] | Cook DA , Kannarkat GT , Cintron AF , Butkovich LM , Fraser KB , Chang J , Grigoryan N , Factor SA , West AB , Boss JM , Tansey MG ((2017) ) LRRK2 levels in immune cells are increased in Parkinson’s disease. NPJ Parkinsons Dis 3: , 11. |
[86] | San Luciano M , Tanner CM , Meng C , Marras C , Goldman SM , Lang AE , Tolosa E , Schule B , Langston JW , Brice A , Corvol JC , Goldwurm S , Klein C , Brockman S , Berg D , Brockmann K , Ferreira JJ , Tazir M , Mellick GD , Sue CM , Hasegawa K , Tan EK , Bressman S , Saunders-Pullman R , Michael J , Fox Foundation LRRK2 Cohort Consortium ((2020) ) Nonsteroidal anti-inflammatory use and LRRK2 Parkinson’s disease penetrance. Mov Disord 35: , 1755–1764. |
[87] | Magnusen AF , Hatton SL , Rani R , Pandey MK ((2021) ) Genetic defects and pro-inflammatory cytokines in Parkinson’s disease. Front Neurol 12: , 636139. |
[88] | Brockmann K , Apel A , Schulte C , Schneiderhan-Marra N , Pont-Sunyer C , Vilas D , Ruiz-Martinez J , Langkamp M , Corvol JC , Cormier F , Knorpp T , Joos TO , Gasser T , Schule B , Aasly JO , Foroud T , Marti-Masso JF , Brice A , Tolosa E , Marras C , Berg D , Maetzler W ((2016) ) Inflammatory profile in LRRK2-associated prodromal and clinical PD. J Neuroinflammation 13: , 122. |
[89] | Fan Z , Pan YT , Zhang ZY , Yang H , Yu SY , Zheng Y , Ma JH , Wang XM ((2020) ) Systemic activation of NLRP3 inflammasome and plasma alpha-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J Neuroinflammation 17: , 11. |
[90] | von Herrmann KM , Salas LA , Martinez EM , Young AL , Howard JM , Feldman MS , Christensen BC , Wilkins OM , Lee SL , Hickey WF , Havrda MC ((2018) ) NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson’s disease. NPJ Parkinsons Dis 4: , 24. |
[91] | Hughes LP , Pereira MMM , Hammond DA , Kwok JB , Halliday GM , Lewis SJG , Dzamko N ((2021) ) Glucocerebrosidase activity is reduced in cryopreserved Parkinson’s disease patient monocytes and inversely correlates with motor severity. J Parkinsons Dis 11: , 1157–1165. |
[92] | Emelyanov A , Usenko T , Nikolaev M , Senkevich K , Kulabukhova D , Lavrinova A , Andoskin P , Miliukhina I , Pchelina S ((2021) ) Increased alpha-synuclein level in CD45+ blood cells in asymptomatic carriers of GBA mutations. Mov Disord 36: , 1997–1998. |
[93] | Avenali M , Cerri S , Ongari G , Ghezzi C , Pacchetti C , Tassorelli C , Valente EM , Blandini F ((2021) ) Profiling the biochemical signature of GBA-related Parkinson’s disease in peripheral blood mononuclear cells. Mov Disord 36: , 1267–1272. |
[94] | Panicker LM , Miller D , Awad O , Bose V , Lun Y , Park TS , Zambidis ET , Sgambato JA , Feldman RA ((2014) ) Gaucher iPSC-derived macrophages produce elevated levels of inflammatory mediators and serve as a new platform for therapeutic development. Stem Cells 32: , 2338–2349. |
[95] | Usenko T , Bezrukova A , Basharova K , Panteleeva A , Nikolaev M , Kopytova A , Miliukhina I , Emelyanov A , Zakharova E , Pchelina S ((2021) ) Comparative transcriptome analysis in monocyte-derived macrophages of asymptomatic GBA mutation carriers and patients with GBA-associated Parkinson’s disease. Genes (Basel) 12: , 1545. |
[96] | Galper J , Balwani M , Fahn S , Waters C , Krohn L , Gan-Or Z , Dzamko N , Alcalay RN ((2021) ) Cytokines and Gaucher biomarkers in glucocerebrosidase carriers with and without Parkinson disease. Mov Disord 36: , 1451–1455. |
[97] | Chahine LM , Qiang J , Ashbridge E , Minger J , Yearout D , Horn S , Colcher A , Hurtig HI , Lee VM , Van Deerlin VM , Leverenz JB , Siderowf AD , Trojanowski JQ , Zabetian CP , Chen-Plotkin A ((2013) ) Clinical and biochemical differences in patients having Parkinson disease with vs without GBA mutations. JAMA Neurol 70: , 852–858. |
[98] | Chavan SS , Tracey KJ ((2017) ) Essential neuroscience in immunology. J Immunol 198: , 3389–3397. |
[99] | Yoo BB , Mazmanian SK ((2017) ) The enteric network: Interactions between the immune and nervous systems of the gut. Immunity 46: , 910–926. |
[100] | Mueller B , Figueroa A , Robinson-Papp J ((2022) ) Structural and functional connections between the autonomic nervous system, hypothalamic-pituitary-adrenal axis, and the immune system: A context and time dependent stress response network. Neurol Sci 43: , 951–960. |
[101] | Matt SM , Gaskill PJ ((2020) ) Where is dopamine and how do immune cells see it? Dopamine-mediated immune cell function in health and disease. J Neuroimmune Pharmacol 15: , 114–164. |
[102] | Yan Y , Jiang W , Liu L , Wang X , Ding C , Tian Z , Zhou R ((2015) ) Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 160: , 62–73. |
[103] | Pinoli M , Marino F , Cosentino M ((2017) ) Dopaminergic regulation of innate immunity: A review. J Neuroimmune Pharmacol 12: , 602–623. |
[104] | Mastroeni D , Grover A , Leonard B , Joyce JN , Coleman PD , Kozik B , Bellinger DL , Rogers J ((2009) ) Microglial responses to dopamine in a cell culture model of Parkinson’s disease. Neurobiol Aging 30: , 1805–1817. |
[105] | Ghosh MC , Mondal AC , Basu S , Banerjee S , Majumder J , Bhattacharya D , Dasgupta PS ((2003) ) Dopamine inhibits cytokine release and expression of tyrosine kinases, Lck and Fyn in activated T cells. Int Immunopharmacol 3: , 1019–1026. |
[106] | Sarkar C , Basu B , Chakroborty D , Dasgupta PS , Basu S ((2010) ) The immunoregulatory role of dopamine: An update. Brain Behav Immun 24: , 525–528. |
[107] | Kustrimovic N , Rasini E , Legnaro M , Bombelli R , Aleksic I , Blandini F , Comi C , Mauri M , Minafra B , Riboldazzi G , Sanchez-Guajardo V , Marino F , Cosentino M ((2016) ) Dopaminergic receptors on CD4+ T naive and memory lymphocytes correlate with motor impairment in patients with Parkinson’s disease. Sci Rep 6: , 33738. |
[108] | Magistrelli L , Ferrari M , Furgiuele A , Milner AV , Contaldi E , Comi C , Cosentino M , Marino F ((2021) ) Polymorphisms of dopamine receptor genes and Parkinson’s disease: Clinical relevance and future perspectives. Int J Mol Sci 22: , 3781. |
[109] | Hurny A , Michalowska-Wender G , Wender M ((2013) ) Impact of L-DOPA treatment of patients with Parkinson’s disease on mononuclear subsets and phagocytosis in the peripheral blood. Folia Neuropathol 51: , 127–131. |
[110] | Hu ZX , Song WN , Lu XD , Zhou ML , Shao JH ((2018) ) Peripheral T lymphocyte immunity and l-dopamine in patients with Parkinson’s disease. J Biol Regul Homeost Agents 32: , 687–691. |
[111] | Butkovich LM , Houser MC , Tansey MG ((2018) ) alpha-synuclein and noradrenergic modulation of immune cells in Parkinson’s disease pathogenesis. Front Neurosci 12: , 626. |
[112] | Papandreou V , Kavrochorianou N , Katsoulas T , Myrianthefs P , Venetsanou K , Baltopoulos G ((2016) ) Adrenergic effect on cytokine release after healthy volunteers’ whole blood LPS stimulation. Inflammation 39: , 1069–1075. |
[113] | Scanzano A , Cosentino M ((2015) ) Adrenergic regulation of innate immunity: A review. Front Pharmacol 6: , 171. |
[114] | Trabold B , Lunz D , Gruber M , Fröhlich D , Graf B ((2010) ) Immunomodulation of neutrophil–endothelial interaction by inotropes. Injury 41: , 1079–1083. |
[115] | Liu H , Leak RK , Hu X ((2016) ) Neurotransmitter receptors on microglia. Stroke Vasc Neurol 1: , 52–58. |
[116] | Jiménez-Jiménez FJ , Alonso-Navarro H , García-Martín E , Agúndez JAG ((2021) ) Neurochemicalfeatures of Rem sleep behaviour disorder.. J Pers Med 11: , 880. |
[117] | Andersen KB , Hansen AK , Sommerauer M , Fedorova TD , Knudsen K , Vang K , Van Den Berge N , Kinnerup M , Nahimi A , Pavese N , Brooks DJ , Borghammer P ((2020) ) Altered sensorimotor cortex noradrenergic function in idiopathic REM sleep behaviour disorder - A PET study. Parkinsonism Relat Disord 75: , 63–69. |
[118] | Knudsen K , Fedorova TD , Hansen AK , Sommerauer M , Otto M , Svendsen KB , Nahimi A , Stokholm MG , Pavese N , Beier CP , Brooks DJ , Borghammer P ((2018) ) staging of pathology in REM sleep behaviour disorder: A multimodality imaging case-control study. Lancet Neurol 17: , 618–628. |
[119] | Wang H , Yu M , Ochani M , Amella CA , Tanovic M , Susarla S , Li JH , Wang H , Yang H , Ulloa L , Al-Abed Y , Czura CJ , Tracey KJ ((2003) ) Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 421: , 384–388. |
[120] | Hodo TW , de Aquino MTP , Shimamoto A , Shanker A ((2020) ) Critical neurotransmitters in the neuroimmune network. Front Immunol 11: , 1869. |
[121] | Ahern GP ((2011) ) 5-HT and the immune system. Curr Opin Pharmacol 11: , 29–33. |
[122] | Sacramento PM , Monteiro C , Dias ASO , Kasahara TM , Ferreira TB , Hygino J , Wing AC , Andrade RM , Rueda F , Sales MC , Vasconcelos CC , Bento CAM ((2018) ) Serotonin decreases the production of Th1/Th17 cytokines and elevates the frequency of regulatory CD4(+) T-cell subsets in multiple sclerosis patients. Eur J Immunol 48: , 1376–1388. |
[123] | Yu B , Becnel J , Zerfaoui M , Rohatgi R , Boulares AH , Nichols CD ((2008) ) Serotonin 5-hydroxytryptamine(2A) receptor activation suppresses tumor necrosis factor-alpha-induced inflammation with extraordinary potency. J Pharmacol Exp Ther 327: , 316–323. |
[124] | Liu SY , Wile DJ , Fu JF , Valerio J , Shahinfard E , McCormick S , Mabrouk R , Vafai N , McKenzie J , Neilson N , Perez-Soriano A , Arena JE , Cherkasova M , Chan P , Zhang J , Zabetian CP , Aasly JO , Wszolek ZK , McKeown MJ , Adam MJ , Ruth TJ , Schulzer M , Sossi V , Stoessl AJ ((2018) ) The effect of LRRK2 mutations on the cholinergic system in manifest and premanifest stages of Parkinson’s disease: A cross-sectional PET study. Lancet Neurol 17: , 309–316. |
[125] | Wile DJ , Agarwal PA , Schulzer M , Mak E , Dinelle K , Shahinfard E , Vafai N , Hasegawa K , Zhang J , McKenzie J , Neilson N , Strongosky A , Uitti RJ , Guttman M , Zabetian CP , Ding YS , Adam M , Aasly J , Wszolek ZK , Farrer M , Sossi V , Stoessl AJ ((2017) ) Serotonin and dopamine transporter PET changes in the premotor phase of LRRK2 parkinsonism: Cross-sectional studies. Lancet Neurol 16: , 351–359. |
[126] | Devos D , Lebouvier T , Lardeux B , Biraud M , Rouaud T , Pouclet H , Coron E , Bruley des Varannes S , Naveilhan P , Nguyen JM , Neunlist M , Derkinderen P ((2013) ) Colonic inflammation in Parkinson’s disease. Neurobiol Dis 50: , 42–48. |
[127] | Forsyth CB , Shannon KM , Kordower JH , Voigt RM , Shaikh M , Jaglin JA , Estes JD , Dodiya HB , Keshavarzian A ((2011) ) Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS One 6: , e28032. |
[128] | Gold A , Turkalp ZT , Munoz DG ((2013) ) Enteric alpha-synuclein expression is increased in Parkinson’s disease but not Alzheimer’s disease. Mov Disord 28: , 237–240. |
[129] | Shannon KM , Keshavarzian A , Dodiya HB , Jakate S , Kordower JH ((2012) ) Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov Disord 27: , 716–719. |
[130] | Hilton D , Stephens M , Kirk L , Edwards P , Potter R , Zajicek J , Broughton E , Hagan H , Carroll C ((2014) ) Accumulation of α-synuclein in the bowel of patients in the pre-clinical phase of Parkinson’s disease. Acta Neuropathol 127: , 235–241. |
[131] | Romano S , Savva GM , Bedarf JR , Charles IG , Hildebrand F , Narbad A ((2021) ) Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinsons Dis 7: , 27. |
[132] | Heintz-Buschart A , Pandey U , Wicke T , Sixel-Döring F , Janzen A , Sittig-Wiegand E , Trenkwalder C , Oertel WH , Mollenhauer B , Wilmes P ((2018) ) The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov Disord 33: , 88–98. |
[133] | Aho VTE , Pereira PAB , Voutilainen S , Paulin L , Pekkonen E , Auvinen P , Scheperjans F ((2019) ) Gut microbiota in Parkinson’s disease: Temporal stability and relations to disease progression. EBioMedicine 44: , 691–707. |
[134] | Heinzel S , Aho VTE , Suenkel U , von Thaler AK , Schulte C , Deuschle C , Paulin L , Hantunen S , Brockmann K , Eschweiler GW , Maetzler W , Berg D , Auvinen P , Scheperjans F ((2021) ) Gut microbiome signatures of risk and prodromal markers of Parkinson disease. Ann Neurol 90: , E1–E12. |
[135] | Campos-Acuña J , Elgueta D , Pacheco R ((2019) ) T-cell-driven inflammation as a mediator of the gut-brain axis involved in Parkinson’s disease. Front Immunol 10: , 239. |
[136] | Kressel AM , Tsaava T , Levine YA , Chang EH , Addorisio ME , Chang Q , Burbach BJ , Carnevale D , Lembo G , Zador AM , Andersson U , Pavlov VA , Chavan SS , Tracey KJ ((2020) ) Identification of a brainstem locus that inhibits tumor necrosis factor. Proc Natl Acad Sci U S A 117: , 29803–29810. |
[137] | de Jonge WJ , van der Zanden EP , The FO , Bijlsma MF , van Westerloo DJ , Bennink RJ , Berthoud HR , Uematsu S , Akira S , van den Wijngaard RM , Boeckxstaens GE ((2005) ) Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nat Immunol 6: , 844–851. |
[138] | Borghammer P , Van Den Berge N ((2019) ) Brain-first versus gut-first Parkinson’s disease: A hypothesis. J Parkinsons Dis 9: , S281–S295. |
[139] | Turkheimer FE , Rizzo G , Bloomfield PS , Howes O , Zanotti-Fregonara P , Bertoldo A , Veronese M ((2015) ) The methodology of TSPO imaging with positron emission tomography. Biochem Soc Trans 43: , 586–592. |
[140] | Best L , Ghadery C , Pavese N , Tai YF , Strafella AP ((2019) ) New and Old TSPO PET radioligands for imaging brain microglial activation in neurodegenerative disease. Curr Neurol Neurosci Rep 19: , 24. |
[141] | Lavisse S , Guillermier M , Hérard AS , Petit F , Delahaye M , Van Camp N , Ben Haim L , Lebon V , Remy P , Dollé F , Delzescaux T , Bonvento G , Hantraye P , Escartin C ((2012) ) Reactive astrocytes overexpress TSPO and are detected by TSPO positron emission tomography imaging. J Neurosci 32: , 10809–10818. |


