
Microglia in Parkinson’s Disease
Abstract
Microglia are the primary resident immune cells of the central nervous system. Neuropathological reports have identified augmented microglial activation in brains of patients with neurodegenerative disorders including Parkinson’s disease (PD). Extensive research over the years has strengthened the current view on microglia as a player in the pathogenesis of PD and other α-synucleinopathies. In this review, we summarize key findings of the recent three years on microglia in PD with specific relevance to understanding its heterogeneity, dual nature, and specific interactions with pathological α-synuclein strains to mediate its clearance and spreading. This review provides evidence on the relevance of microglia as a putative biomarker and therapeutic target in PD and related disorders.
MICROGLIA: MILESTONES
A century ago, the Spanish neuroscientist Pio Del Rio Hortega identified and described histologically microglia in silver carbonate staining as a part of “the third element” of the nervous system [1]. He provided the initial description of the morphological heterogeneity and suggested the transformation of microglia to amoeboid phenotype in brain diseases [2]. Importantly, in contrast to other types of neuroglia, which have ectodermal origin, microglia originate from yolk-sac myeloid cells that migrate within a restricted time-window during the embryonic development to populate the neuroepithelium and further the central nervous system (CNS) parenchyma [3]. Similarly, in boundary regions, non-parenchymal perivascular and meningeal macrophages have shown restricted origin from hematopoietic precursors during embryonic development. It is now accepted that parenchymal microglia as well as perivascular and meningeal macrophages possess longevity and self-renewal ability dictating their life-long persistence. In contrast, choroid macrophages may originate from blood-born myeloid cells during both embryonic development and adulthood and represent a more dynamic sub-population [4]. In addition to their different developmental origins, these cells have distinct transcriptome signatures and functions [4–6].
Focusing on microglia, the resident macrophages of the CNS provide environmental surveillance to support the brain homeostasis by a specific activation repertoire in aging, brain damage, or disease. Microglial activation is associated with changes in the morphology, the phagocytic function, and the inflammatory signaling, playing a major role in the neuroinflammatory response in pathological conditions. Recent methodological advances have allowed the determination of computational morphological patterns [7], while with the application of single cell RNA sequencing (scRNAseq) we have started gaining information on detailed functional profile changes of microglia in aging and disease [8–10].
MICROGLIA IN PARKINSON’S DISEASE: INCREASING EVIDENCE
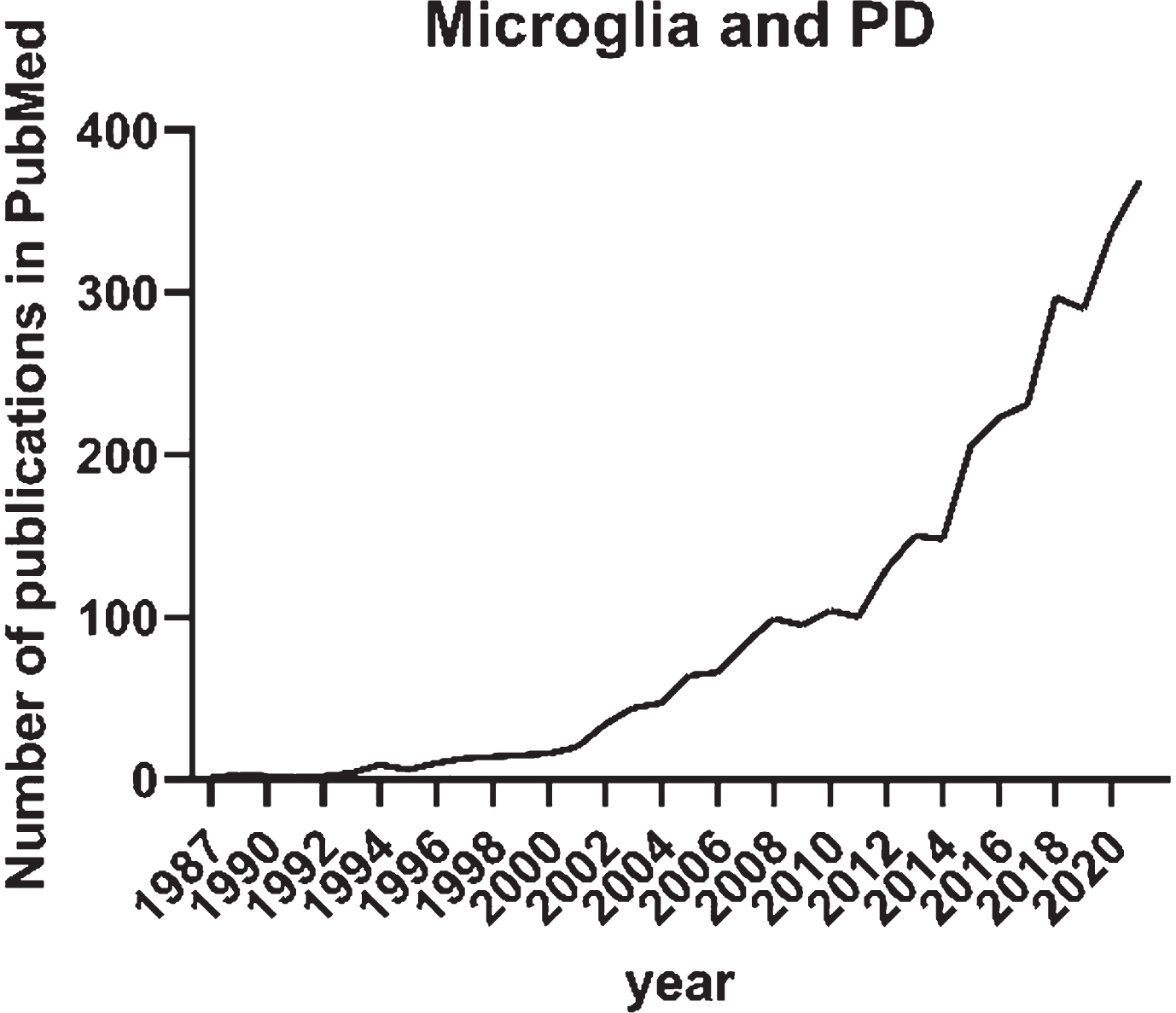
The initial interest in microglia and its role in Parkinson’s disease (PD) arose in the 1980s with the first publication on HLA-DR expression in neurological disorders including PD [11]. In a PubMed search with search terms “microglia” and “Parkinson’s disease” published before 2022, there are 2,831 publication records found (Fig. 1). Until 2011, the annual number of publications recorded in PubMed on the topic was less than 100. This was possibly a reflection of the old-school vision on microglia as a bystander in neurodegenerative disorders. Not until 2015, the annual publications on microglia and PD doubled and continuously increased up to 369 in 2021 as recorded in PubMed. The increasing interest in the involvement of microglia in PD in the recent 5 years is certainly influenced by the technological advancements as well as the ground-breaking discoveries on the role of microglia in the most common neurodegenerative disorder in the aging population— Alzheimer’s disease. Keren-Shaul and colleagues described markers, spatial localization, and pathways associated with a specific subtype of microglia in neurodegenerative diseases (DAM). The researchers demonstrated that DAM are activated in a two-step process— a Trem2-independent initiation linked to downregulation of microglia checkpoints, followed by Trem2-dependent activation— with a potential to restrict neurodegeneration [12].
The aim of the current work is to review specifically the studies on microglia in PD published in the last three years (2019–2021).
Fig. 1
Annual number of publications until the end of 2021 recorded in PubMed and identified with the search “Microglia” and “Parkinson’s disease”.
NEW EVIDENCE FOR THE ROLE OF MICROGLIA IN PD PATHOGENESIS
One of the main questions, which remains open to date, is associated with the specific role of microglia in the pathogenesis of PD. A number of experimental studies described morphological heterogeneity of microglia in human PD as well as in relevant models [13–16]. A major limitation of the morphology-based studies on microglial heterogeneity has been the lack of clear link between morphology and specific functional activity of the different phenotypes; however, the morphological studies have been important drivers of the follow-up studies. Single-cell laser capture microscopy is one of the approaches recently applied to demonstrate different transcriptional profiles of microglia in a region- and disease-specific manner in the substantia nigra and hippocampus CA1 of postmortem PD [17]. It was shown that despite residing in known neurodegenerative, proinflammatory microenvironments, the hippocampal microglia in AD and the nigral microglia in PD had very few overlapping transcripts [17]. Microglial transcriptional subpopulations were also shown by scRNAseq in human and animal brains [18]. The study identified important specificities of human microglia as compared to rodents including higher heterogeneity and significant differences in complement, phagocytic, and susceptibility genes to neurodegeneration. Alternatively, single-nuclei RNA sequencing (snRNAseq) was introduced to circumvent the need of separation of intact single cells from aging and frozen samples, the usual human postmortem material. Interestingly, this technology did not identify in the human substantia nigra association of microglia with PD risk patterns [19]. However, another snRNAseq searching for cell-type specific risk for PD in combination with validation by immunolabeling and laser microdissection reported increase in PD nigral microglia with predominant amoeboid morphology, and a pro-inflammatory profile characterized by higher levels of IL1β, GPNMB, and HSP90AA1 [20]. The reason for the discrepancies in the outcomes of the high throughput screening systems may relate to different sensitivity of the methods to identify microglial subtypes as well as differences of the studied patient populations [21]. Understanding the heterogeneity of microglia and its activation in PD and related synucleinopathies is of tremendous relevance for the interpretation of its role in the disease pathogenesis as well as its relevance as a biomarker and therapeutic target. Multiple reports have pointed towards the dual roles of microglia in PD (Fig. 2). Therefore, identifying the physiological balance and pathological shift between pro-inflammatory and anti-inflammatory signals is crucial to understand the pathogenesis of a-synucleinopathies. Novel tools like for instance spatial transcriptome analysis of microglia will further expand the understanding on the heterogeneity of microglia.
Fig. 2
The unresolved heterogeneity of microglia and DAMs in Parkinson’s disease. α-synuclein (α-syn) oligomers as well as pro-inflammatory signals released from the degenerating neuron may trigger activation of microglia in a disease-specific mode (DAMs). Importantly different α-syn polymorphs may induce different microglial responses. Microglia may get different morphological patterns of activation as well as execute different functions: i) the left lower panel shows pro-inflammatory signaling in microglia triggered by α-syn oligomers involving different membrane receptors, NLRP3 inflammasome activation, mitochondrial and oxidative stress, as well as release of toxic pro-inflammatory cytokines; ii). The right lower panel demonstrates a microglial cell which is involved in the uptake and clearance of α-syn either from the extracellular space or through putative nanotubes within the microglial network. Created with BioRender.com.
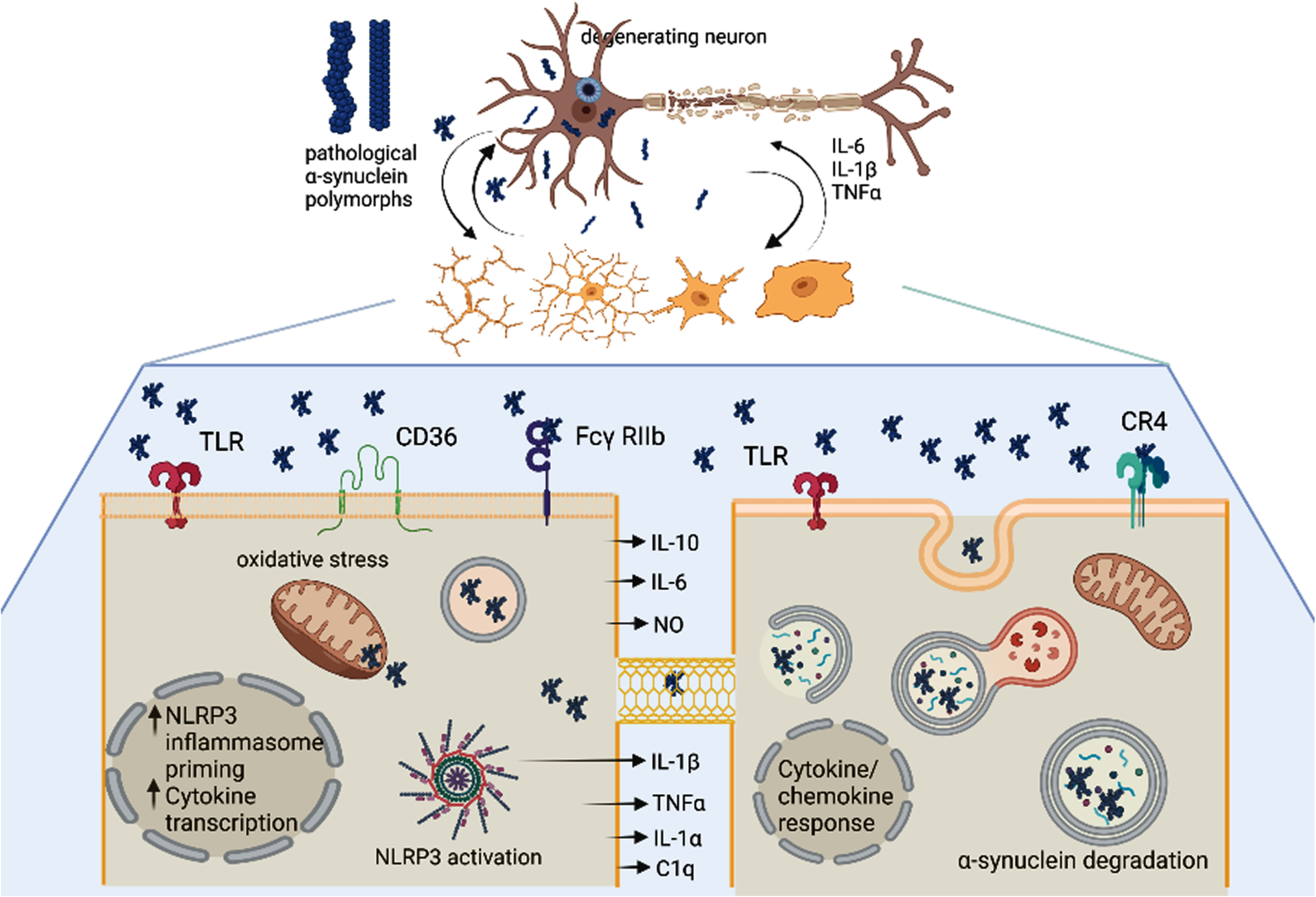
The evidence on the role of microglial pro-in-flammatory signaling in PD pathogenesis is strongly supported by a number of recent studies that show the involvement of the microglial NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome [22–26]. The NLRP3 inflammasome is an important component of the innate immune system that mediates caspase-1 activation and the secretion of proinflammatory cytokines IL-1β/IL-18 in response to infection, sterile inflammation or cellular damage including neurodegeneration [27, 28]. The NLRP3 inflammasome pathway may be triggered by fibrillary α-synuclein (α-syn) or directly by dopaminergic cell death (elicited by the neurotoxin 6-hydroxidopamine) in the absence of α-syn aggregates [22]. Interestingly, a recent study suggested Fyn kinase as the regulator of α-syn import in microglia, mediating in turn the priming and activation of microglial NLRP3 inflammasome [23]. In vitro analysis of the response of human microglia to fibrillar α-syn confirmed release of IL-1β dependent on the inflammasome assembly and recruitment of caspase-1 [24]. This finding was corroborated in a model of human iPSC-derived microglia and furthermore it was demonstrated that the process might involve dual stimulation by TLR2 engagement and mitochondrial damage [25]. Intriguingly, α-syn-antibody complexes were shown to exacerbate IL-1β secretion, indicating towards a possible aggravation of the pathology by anti-α-syn immunotherapies due to exacerbated microglial activation [25]. TLR2 was also implicated in the transmission of α-syn by reactive microglia. Both in vitro and in vivo, TLR2-dependent plasma exosome α-syn uptake by microglia was identified in PD. In addition, the authors demonstrated that neurons efficiently internalized human α-syn released by microglia previously exposed to PD plasma exosomes [29]. These observations have added an important feature to the profile of microglia in PD, namely a mediator of α-syn spreading.
In view of the role of different α-syn strains in α-synucleinopathies with variable rate of severity and progression [30, 31], it was important to address the role of the different α-syn species for the microglial activation profile. Scheiblich and co-workers [26] proposed that NLRP3 inflammasome and IL1β release were efficiently activated by α-syn monomers and oligomers via TLR2 and TLR5 ligation acting on different checkpoints, i.e., priming versus activation; however, other α-syn polymorphs— “fibrils”— needed microglial priming, while “ribbons” were ineffective to trigger NLRP3 inflammasome activation. Interestingly, α-syn monomers, oligomers, and fibrils induced TNF-α, IL-6, IL-10, NO, and CXCL2 release by microglia, while ribbons had no effect on the cytokine secretion. Furthermore, the activation of the NLRP3 inflammasome compromised α-syn phagocytosis and degradation of α-syn monomers and oligomers by microglia. This study has been the first to suggest the role of different high-molecular weight α-syn polymorphs as modulators of the type of neuroinflammatory response of microglia in α-synucleinopathies, while earlier studies have differentiated the inflammatory responses to monomers/dimers versus those to higher-ordered oligomeric α-syn [32, 33].
In addition to the pro-inflammatory signaling of microglia in PD and related disorders, microglia participate in the phagocytosis and clearance of α-syn. Recent study reported that complement receptor (CR) 4 selectively bound to fibrillary α-syn, triggered phagolysosome formation, and clearance of the pathological protein [34]. Earlier studies proposed that TLR4 initiated inflammatory response to α-syn, but also had important role for the phagocytic activity and the clearance of α-syn by microglia in vitro and in vivo [32, 35]. Recently, the role of microglial TLR4 was further assessed in vivo in a PD spreading model generated by an injection of α-syn preformed fibrils in the striatum. The accelerated progression of the pathology in this model in relation to TLR4 deficiency provided additional evidence for the role of TLR4 in α-syn processing by microglia. The study showed that TLR4 deficiency in vivo facilitated the α-syn seed spreading associated with reduced lysosomal activity of microglia and finally resulted in accelerated nigral neurodegeneration [36]. The role of extracellular α-syn to disrupt microglial autophagy activity was further reinforced by in vitro analysis suggesting that the autophagy inhibition by α-syn was processed via TLR4-dependent p38 and Akt-mTOR signaling cascades; however, in this study the authors could not exclude the possibility that the cytokines released from α-syn-activated microglia may also indirectly interfere and inhibit the microglial autophagy flux [37, 38].
A very intriguing dataset was presented by Scheiblich et al. [39] who proposed the presence of an “on-demand” functional network of microglia, which might improve pathogenic α-syn clearance. The authors observed the formation of F-actin-dependent intercellular connections, which were able to bridge and transfer α-syn from overloaded microglia to other/secondary naive microglia. These secondary microglial cells were then able to rapidly and effectively degrade the α-syn cargo.
In summary, the interaction between α-syn and microglia plays an important role in the pathogenesis of PD and related disorders as evidenced by multiple studies. However, this interaction is marked by a certain dualism, which may be linked to misbalance in the heterogeneous microglial response with aging and under pathological conditions.
MICROGLIA: A PUTATIVE BIOMARKER IN PD AND RELATED DISORDERS
Microglial activation is an important participant in the disease cascade of PD and for this reason it has been considered a putative biomarker for the diagnosis and progression of PD and related disorders. Microglial activation can be assessed by positron emission tomography (PET) imaging. Earlier studies widely relied on the [11C]-PK11195 radioligand, that binds to the translocator protein (TSPO), a protein that is upregulated in activated microglia and astrocytes. The initial studies with this radioligand in PD demonstrated increased binding in the midbrain and the basal ganglia [40, 41]. Due to several disadvantages of [11C]-PK11195 like low blood-brain barrier permeability, non-specific binding, relatively short life, a new generation of TSPO ligands has been developed and tested. Those have been also considered to account for TSPO polymorphism, which may interfere with the outcome of binding analysis. For example, [18F]-DPA714 binding was measured in PD compared to healthy controls and confirmed a region-specific pattern in the nigro-striatal region and the frontal cortex. However, the microglial activation in this specific study design did not correlate with disease severity or duration [42]. Recently, another radioligand, [18F]-FEMPA, was proposed and tested in Friedrich’s ataxia [43]. Intriguingly, despite its many disadvantages, [11C]-PK11195 PET was able to detect changes in the glial activation in the putamen of patients with multiple system atrophy (MSA) from baseline to 48 weeks after minocycline treatment supporting target engagement of the drug [44]. A phase 2a clinical trial applied [11C]-PBR28 PET to assess the effect of 8 weeks of treatment with AZD3241 (now BHV-3241, verdiperstat) on microglia in patients with PD. The community was very excited as the total distribution volume of [11C]-PBR28 binding to TSPO was significantly reduced compared to baseline both at 4 and 8 weeks after initiation of treatment with the drug [45]. This was in contrast to the lack of overall change in the placebo group. Recently, another clinical trial with observational and treatment phase on the effect of verdiperstat on microglial activation in MSA patients was announced (NCT04616456). One of the aims here will be to measure putative changes in [18F]-PBR06 PET signal and to assess the relationship between PET changes and clinical progression. Future development of the efforts to reliably measure changes of microglial activation is warranted. It is expected that the better definition of disease-specific DAM profiles may support the development of future selective microglia sub-type-specific ligands. Such imaging biomarkers may strongly support our understanding on the involvement of microglia in the progression of the neurodegenerative process. They will also be of high value for clinical therapeutic trials in PD and related disorders. Furthermore, they may support the evaluation of treatment efficacy even if the therapeutic target is not directly microglia. For example, multiple preclinical studies have demonstrated repeatedly that therapeutic targeting of α-syn is strongly associated with changes in microglial activation, which therefore may support the assessment of target engagement and efficacy [46–49]. Microglial imaging will be especially relevant in immunotherapy trials in PD and related disorders in view of the recent report that α-syn-antibody complexes may increase microglial activation/neuroinflammation in experimental conditions [25].
FUTURE PERSPECTIVES OR IS MICROGLIA A PUTATIVE THERAPEUTIC TARGET IN PD
Currently, α-syn is a major focus of the developers of disease modifying therapies (DMTs) for PD and related disorders. However, the role of microglial activation in PD is nowadays well acknowledged and accepted, therefore re-shaping the thinking on DMTs. Experimental data suggest that targeting microglia with DMTs may interfere with α-syn aggregation in addition to modifying the deleterious proinflammatory responses in α-synucleinopathies [50]. Certainly, a significant limitation at this stage is the incomplete understanding of microglial heterogeneity, its dual roles and their homeostatic balance and pathogenic dysbalance, and the dynamics of its shift during normal aging and in neurodegeneration. The advancement of the technologies applied for the characterization of microglia guarantees significant progress in the following years. Novel human and humanized models of PD and related disorders based on iPSC-technologies will step in. They will support outcomes from rodent models by addressing the species differences between human and rodent microglia, which may partly explain the poor preclinical predictive rate and translational success. Our experience has taught us of the importance of the early treatment initiation for the success of DMTs targeting microglial activation in PD and related disorders [50, 51]. The current knowledge postulates caution in the design of future clinical trials in association with careful interpretation of the preclinical data in all relevant aspects.
CONCLUSION
Microglia are a significant player in the pathogenesis of neurodegenerative disorders including PD. It may contribute to the disease process in multiple ways including pro- and anti-inflammatory signaling, α-syn clearance and α-syn spreading. It seems that the heterogeneity of microglia and its shift during aging or neurodegeneration may play a pivotal role in the initiation and progression of the disease. Because of its unequivocal participation in the disease process, microglia are a strong candidate for a diagnostic and progression biomarker as well as a clear target for DMTs in PD either alone or in combination with other putative targets.
ACKNOWLEDGMENTS
Prof. Stefanova receives support from the Austrian Science Fund and Grant of Alterity Therapeutics.
CONFLICT OF INTEREST
Prof. Stefanova is an employee of Medical University of Innsbruck.
REFERENCES
[1] | Del Río-Hortega Bereciartu J ((2020) ) Pío del Río-Hortega: The revolution of glia. Anat Rec (Hoboken) 303: , 1232–1241. |
[2] | Sierra A , de Castro F , Del Río-Hortega J , Rafael Iglesias-Rozas J , Garrosa M , Kettenmann H ((2016) ) The “Big-Bang” for modern glial biology: Translation and comments on Pío del Río-Hortega 1919 series of papers on microglia. Glia 64: , 1801–1840. |
[3] | Ransohoff RM , Cardona AE ((2010) ) The myeloid cells of the central nervous system parenchyma. Nature 468: , 253–262. |
[4] | Goldmann T , Wieghofer P , Jordão MJ , Prutek F , Hagemeyer N , Frenzel K , Amann L , Staszewski O , Kierdorf K , Krueger M , Locatelli G , Hochgerner H , Zeiser R , Epelman S , Geissmann F , Priller J , Rossi FM , Bechmann I , Kerschensteiner M , Linnarsson S , Jung S , Prinz M ((2016) ) Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol 17: , 797–805. |
[5] | Jordão MJC , Sankowski R , Brendecke SM , Sagar , Locatelli G , Tai YH , Tay TL , Schramm E , Armbruster S , Hagemeyer N , Groß O , Mai D , çiçek Ö , Falk T , Kerschensteiner M , Grün D , Prinz M ((2019) ) Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363: , eaat7554. |
[6] | Van Hove H , Martens L , Scheyltjens I , De Vlaminck K , Pombo Antunes AR , De Prijck S , Vandamme N , De Schepper S , Van Isterdael G , Scott CL , Aerts J , Berx G , Boeckxstaens GE , Vandenbroucke RE , Vereecke L , Moechars D , Guilliams M , Van Ginderachter JA , Saeys Y , Movahedi K ((2019) ) A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci 22: , 1021–1035. |
[7] | Bennett ML , Bennett FC , Liddelow SA , Ajami B , Zamanian JL , Fernhoff NB , Mulinyawe SB , Bohlen CJ , Adil A , Tucker A , Weissman IL , Chang EF , Li G , Grant GA , Hayden Gephart MG , Barres BA ((2016) ) New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 113: , E1738–E1746. |
[8] | Safaiyan S , Besson-Girard S , Kaya T , Cantuti-Castelvetri L , Liu L , Ji H , Schifferer M , Gouna G , Usifo F , Kannaiyan N , Fitzner D , Xiang X , Rossner MJ , Brendel M , Gokce O , Simons M ((2021) ) White matter aging drives microglial diversity. Neuron 109: , 1100–1117.e1110. |
[9] | Provenzano F , Pérez MJ , Deleidi M ((2021) ) Redefining microglial identity in health and disease at single-cell resolution. Trends Mol Med 27: , 47–59. |
[10] | Masuda T , Sankowski R , Staszewski O , Prinz M ((2020) ) Microglia heterogeneity in the single-cell era. Cell Rep 30: , 1271–1281. |
[11] | McGeer PL , Itagaki S , McGeer EG ((1988) ) Expression of the histocompatibility glycoprotein HLA-DR in neurological disease. Acta Neuropathol 76: , 550–557. |
[12] | Keren-Shaul H , Spinrad A , Weiner A , Matcovitch-Natan O , Dvir-Szternfeld R , Ulland TK , David E , Baruch K , Lara-Astaiso D , Toth B , Itzkovitz S , Colonna M , Schwartz M , Amit I ((2017) ) A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169: , 1276–1290.e1217. |
[13] | Sanchez-Guajardo V , Febbraro F , Kirik D , Romero-Ramos M ((2010) ) Microglia acquire distinct activation profiles depending on the degree of alpha-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLoS One 5: , e8784. |
[14] | Barkholt P , Sanchez-Guajardo V , Kirik D , Romero-Ramos M ((2012) ) Long-term polarization of microglia upon alpha-synuclein overexpression in nonhuman primates. Neuroscience 208: , 85–96. |
[15] | Refolo V , Bez F , Polissidis A , Kuzdas-Wood D , Sturm E , Kamaratou M , Poewe W , Stefanis L , Angela CM , Romero-Ramos M , Wenning GK , Stefanova N ((2018) ) Progressive striatonigral degeneration in a transgenic mouse model of multiple system atrophy: Translational implications for interventional therapies. Acta Neuropathol Commun 6: , 2. |
[16] | Imamura K , Hishikawa N , Sawada M , Nagatsu T , Yoshida M , Hashizume Y ((2003) ) Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol (Berl) 106: , 518–526. |
[17] | Mastroeni D , Nolz J , Sekar S , Delvaux E , Serrano G , Cuyugan L , Liang WS , Beach TG , Rogers J , Coleman PD ((2018) ) Laser-captured microglia in the Alzheimer’s and Parkinson’s brain reveal unique regional expression profiles and suggest a potential role for hepatitis B in the Alzheimer’s brain. Neurobiol Aging 63: , 12–21. |
[18] | Geirsdottir L , David E , Keren-Shaul H , Weiner A , Bohlen SC , Neuber J , Balic A , Giladi A , Sheban F , Dutertre CA , Pfeifle C , Peri F , Raffo-Romero A , Vizioli J , Matiasek K , Scheiwe C , Meckel S , Mätz-Rensing K , van der Meer F , Thormodsson FR , Stadelmann C , Zilkha N , Kimchi T , Ginhoux F , Ulitsky I , Erny D , Amit I , Prinz M ((2019) ) Cross-species single-cell analysis reveals divergence of the primate microglia program. Cell 179: , 1609–1622.e1616. |
[19] | Agarwal D , Sandor C , Volpato V , Caffrey TM , Monzón-Sandoval J , Bowden R , Alegre-Abarrategui J , Wade-Martins R , Webber C ((2020) ) A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat Commun 11: , 4183. |
[20] | Smajić S , Prada-Medina CA , Landoulsi Z , Ghelfi J , Delcambre S , Dietrich C , Jarazo J , Henck J , Balachandran S , Pachchek S , Morris CM , Antony P , Timmermann B , Sauer S , Pereira SL , Schwamborn JC , May P , Grünewald A , Spielmann M ((2022) ) Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain 145: , 964–978. |
[21] | Thrupp N , Sala Frigerio C , Wolfs L , Skene NG , Fattorelli N , Poovathingal S , Fourne Y , Matthews PM , Theys T , Mancuso R , de Strooper B , Fiers M ((2020) ) Single-nucleus RNA-Seq is not suitable for detection of microglial activation genes in humans. Cell Rep 32: , 108189. |
[22] | Gordon R , Albornoz EA , Christie DC , Langley MR , Kumar V , Mantovani S , Robertson AAB , Butler MS , Rowe DB , O’Neill LA , Kanthasamy AG , Schroder K , Cooper MA , Woodruff TM ((2018) ) Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med 10: , eaah4066. |
[23] | Panicker N , Sarkar S , Harischandra DS , Neal M , Kam TI , Jin H , Saminathan H , Langley M , Charli A , Samidurai M , Rokad D , Ghaisas S , Pletnikova O , Dawson VL , Dawson TM , Anantharam V , Kanthasamy AG , Kanthasamy A ((2019) ) Fyn kinase regulates misfolded α-synuclein uptake and NLRP3 inflammasome activation in microglia. J Exp Med 216: , 1411–1430. |
[24] | Pike AF , Varanita T , Herrebout MAC , Plug BC , Kole J , Musters RJP , Teunissen CE , Hoozemans JJM , Bubacco L , Veerhuis R ((2021) ) α-Synuclein evokes NLRP3 inflammasome-mediated IL-1β secretion from primary human microglia. Glia 69: , 1413–1428. |
[25] | Trudler D , Nazor KL , Eisele YS , Grabauskas T , Dolatabadi N , Parker J , Sultan A , Zhong Z , Goodwin MS , Levites Y , Golde TE , Kelly JW , Sierks MR , Schork NJ , Karin M , Ambasudhan R , Lipton SA ((2021) ) Soluble α-synuclein-antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia. Proc Natl Acad Sci U S A 118: , e2025847118. |
[26] | Scheiblich H , Bousset L , Schwartz S , Griep A , Latz E , Melki R , Heneka MT ((2021) ) Microglial NLRP3 inflammasome activation upon TLR2 and TLR5 ligation by distinct α-synuclein assemblies. J Immunol 207: , 2143–2154. |
[27] | Heneka MT , McManus RM , Latz E ((2018) ) Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci 19: , 610–621. |
[28] | Swanson KV , Deng M , Ting JP ((2019) ) The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat Rev Immunol 19: , 477–489. |
[29] | Xia Y , Zhang G , Kou L , Yin S , Han C , Hu J , Wan F , Sun Y , Wu J , Li Y , Huang J , Xiong N , Zhang Z , Wang T ((2021) ) Reactive microglia enhance the transmission of exosomal α-synuclein via toll-like receptor 2.. Brain 144: , 2024–2037. |
[30] | Shahnawaz M , Mukherjee A , Pritzkow S , Mendez N , Rabadia P , Liu X , Hu B , Schmeichel A , Singer W , Wu G , Tsai AL , Shirani H , Nilsson KPR , Low PA , Soto C ((2020) ) Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 578: , 273–277. |
[31] | Schweighauser M , Shi Y , Tarutani A , Kametani F , Murzin AG , Ghetti B , Matsubara T , Tomita T , Ando T , Hasegawa K , Murayama S , Yoshida M , Hasegawa M , Scheres SHW , Goedert M ((2020) ) Structures of α-synuclein filaments from multiple system atrophy. Nature 585: , 464–469. |
[32] | Fellner L , Irschick R , Schanda K , Reindl M , Klimaschewski L , Poewe W , Wenning GK , Stefanova N ((2013) ) Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia 61: , 349–360. |
[33] | Daniele SG , Béraud D , Davenport C , Cheng K , Yin H , Maguire-Zeiss KA ((2015) ) Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci Signal 8: , ra45. |
[34] | Juul-Madsen K , Qvist P , Bendtsen KL , Langkilde AE , Vestergaard B , Howard KA , Dehesa-Etxebeste M , Paludan SR , Andersen GR , Jensen PH , Otzen DE , Romero-Ramos M , Vorup-Jensen T ((2020) ) Size-selective phagocytic clearance of fibrillar α-synuclein through conformational activation of complement receptor 4. J Immunol 204: , 1345–1361. |
[35] | Stefanova N , Fellner L , Reindl M , Masliah E , Poewe W , Wenning GK ((2011) ) Toll-like receptor 4 promotes alpha-synuclein clearance and survival of nigral dopaminergic neurons. Am J Pathol 179: , 954–963. |
[36] | Venezia S , Kaufmann WA , Wenning GK , Stefanova N ((2021) ) Toll-like receptor 4 deficiency facilitates α-synuclein propagation and neurodegeneration in a mouse model of prodromal Parkinson’s disease. Parkinsonism Relat Disord 91: , 59–65. |
[37] | Tu HY , Yuan BS , Hou XO , Zhang XJ , Pei CS , Ma YT , Yang YP , Fan Y , Qin ZH , Liu CF , Hu LF ((2021) ) α-synuclein suppresses microglial autophagy and promotes neurodegeneration in a mouse model of Parkinson’s disease. Aging Cell 20: , e13522. |
[38] | Jin MM , Wang F , Qi D , Liu WW , Gu C , Mao CJ , Yang YP , Zhao Z , Hu LF , Liu CF ((2018) ) A critical role of autophagy in regulating microglia polarization in neurodegeneration. Front Aging Neurosci 10: , 378. |
[39] | Scheiblich H , Dansokho C , Mercan D , Schmidt SV , Bousset L , Wischhof L , Eikens F , Odainic A , Spitzer J , Griep A , Schwartz S , Bano D , Latz E , Melki R , Heneka MT ((2021) ) Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 184: , 5089–5106.e5021. |
[40] | Gerhard A , Pavese N , Hotton G , Turkheimer F , Es M , Hammers A , Eggert K , Oertel W , Banati RB , Brooks DJ ((2006) ) imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. }. Neurobiol Dis 21: , 404–412. |
[41] | Ouchi Y , Yoshikawa E , Sekine Y , Futatsubashi M , Kanno T , Ogusu T , Torizuka T ((2005) ) Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol 57: , 168–175. |
[42] | Lavisse S , Goutal S , Wimberley C , Tonietto M , Bottlaender M , Gervais P , Kuhnast B , Peyronneau MA , Barret O , Lagarde J , Sarazin M , Hantraye P , Thiriez C , Remy P ((2021) ) Increased microglial activation in patients with Parkinson disease using [(18)F]-DPA714 TSPO PET imaging. Parkinsonism Relat Disord 82: , 29–36. |
[43] | Khan W , Corben LA , Bilal H , Vivash L , Delatycki MB , Egan GF , Harding IH ((2022) ) Neuroinflammation in the cerebellum and brainstem in Friedreich Ataxia: An [18F]-FEMPA PET study. Mov Disord 37: , 218–224. |
[44] | Dodel R , Spottke A , Gerhard A , Reuss A , Reinecker S , Schimke N , Trenkwalder C , Sixel-Doring F , Herting B , Kamm C , Gasser T , Sawires M , Geser F , Kollensperger M , Seppi K , Kloss M , Krause M , Daniels C , Deuschl G , Bottger S , Naumann M , Lipp A , Gruber D , Kupsch A , Du Y , Turkheimer F , Brooks DJ , Klockgether T , Poewe W , Wenning G , Schade-Brittinger C , Oertel WH , Eggert K ((2010) ) Minocycline 1-year therapy in multiple-system-atrophy: Effect on clinical symptoms and [(11)C] (R)-PK11195 PET (MEMSA-trial). Mov Disord 25: , 97–107. |
[45] | Jucaite A , Svenningsson P , Rinne J , Cselenyi Z , Varrone A , Karlsson P , Varnäs K , Johnström P , McLoughlin MJ , Halldin C , Posener J , Salter H , Kugler A , Budd S , Farde L ((2014) ) Effect of the myeloperoxidase inhibitior AZD3241 on translocator protein binding in brain - a positron emission tomography study in patients with Parkinson’s disease. Mov Disord 29: , S662–S662. |
[46] | Khan A , Johnson R , Wittmer C , Maile M , Tatsukawa K , Wong JL , Gill MB , Stocking EM , Natala SR , Paulino AD , Bowden-Verhoek JK , Wrasidlo W , Masliah E , Bonhaus DW , Price DL ((2021) ) NPT520-34 improves neuropathology and motor deficits in a transgenic mouse model of Parkinson’s disease. Brain 144: , 3692–3709. |
[47] | Heras-Garvin A , Weckbecker D , Ryazanov S , Leonov A , Griesinger C , Giese A , Wenning GK , Stefanova N ((2019) ) Anle138b modulates α-synuclein oligomerization and prevents motor decline and neurodegeneration in a mouse model of multiple system atrophy. Mov Disord 34: , 255–263. |
[48] | Lemos M , Venezia S , Refolo V , Heras-Garvin A , Schmidhuber S , Giese A , Leonov A , Ryazanov S , Griesinger C , Galabova G , Staffler G , Wenning GK , Stefanova N ((2020) ) Targeting α-synuclein by PD03 AFFITOPE® and Anle138b rescues neurodegenerative pathology in a model of multiple system atrophy: Clinical relevance. Transl Neurodegener 9: , 38. |
[49] | Heras-Garvin A , Refolo V , Schmidt C , Malfertheiner K , Wenning GK , Bradbury M , Stamler D , Stefanova N ((2021) ) ATH434 reduces α-synuclein-related neurodegeneration in a murine model of multiple system atrophy. Mov Disord 36: , 2605–2614. |
[50] | Stefanova N , Georgievska B , Eriksson H , Poewe W , Wenning GK ((2012) ) Myeloperoxidase inhibition ameliorates multiple system atrophy-like degeneration in a transgenic mouse model. Neurotox Res 21: , 393–404. |
[51] | Kaindlstorfer C , Sommer P , Georgievska B , Mather RJ , Kugler AR , Poewe W , Wenning GK , Stefanova N ((2015) ) Failure of neuroprotection despite microglial suppression by delayed-start myeloperoxidase inhibition in a model of advanced multiple system atrophy: Clinical implications. Neurotox Res 28: , 185–194. |


