
Epidemiological Evidence for an Immune Component of Parkinson’s Disease
Abstract
There is a growing interest in the role the immune system and inflammatory response play on the pathophysiology of Parkinson’s disease (PD). Epidemiological evidence lends support for the hypothesis that PD is an immune-mediated condition. An association between inflammatory bowel disease, including Crohn’s and Ulcerative colitis, and the risk of PD has been described and replicated in several population-based cohorts. Other autoimmune conditions, such as Sjogren syndrome, ankylosing spondylitis, and rheumatoid arthritis also seem to be associated with an increased risk of PD. Immunosuppressant medications seem to be associated with a decreased risk of PD. Finally, variants in genes involved in immune system regulation are also shared between PD and autoimmune conditions. In this review, we will provide an overview of epidemiological evidence from population-based cohort studies, meta-analyses, and genome-wide association studies that analyze the association between the immune system and PD, discuss current gaps in the literature and future research directions in this field.
INTRODUCTION
There is growing evidence that inflammation plays a part in the pathophysiology of some neurodegenerative conditions, including Parkinson’s disease (PD) [1]. Neuroinflammation is a fundamental immune response to protect neurons from harm and compensate for neuronal damage. Importantly, this response can be considered as a double-edged sword, where it can be neuroprotective in the short term but can be neurotoxic when chronic. Aberrant functioning of the immune system, including changes in microglia, astrocytes, innate immune cells, and infiltrating peripheral immune cells, has been proposed as a critical component of susceptibility to and progression of PD [2]. There are several mechanisms by which the immune system may contribute to PD pathophysiology. Oxidative stress, inflammatory response, abnormal aggregation of proteins, and dysfunction of protein degradation systems are considered to participate in the cascade of events leading to the death of dopaminergic neurons. Inflammatory cytokines are reported as increased in PD patients and have been associated with worsened symptoms, including cognition, depression, anxiety, and sleep [3]. A meta-analysis including 25 studies compared blood cytokine concentrations in PD patients and controls and found higher peripheral concentrations of interleukin-6, 1B, 2 and 10, tumor necrosis factor and C-reactive protein in PD patients [4]. Systemic inflammation and chronic immune cell activation in the periphery may amplify microglial activation, a phenomenon known as microglia priming [5]. In this same manner, conditions associated with inflammation, such as autoimmune disorders, and medications that affect inflammation (e.g., immunosuppressants and anti-inflammatory medications) may influence risk and progression of PD.
The present manuscript is not intended to represent a systematic review, but rather an overview of the current epidemiological evidence for an association between the immune system and PD. For this purpose, we performed a literature search in National Center for Biotechnology Information’s PubMed database (https://www.ncbi.nlm.nih.gov/pubmed) utilizing the search terms “Parkinson Disease” AND “immune system”, “autoimmune”, “immunosuppressant”, “cardiovascular”, “infection”. We aim to summarize the state of the art of the field, as such we restricted our search to research published in the last 5 years. Additionally, we have only included population-based cohort, meta-analyses and genome wide association studies (GWAS) (Table 1). We have excluded preclinical and animal model studies since this is out of scope for the present review. We will focus on autoimmune conditions and the use of immunosuppressant drug, as well as the role of infection in PD. We also include type 2 diabetes mellitus (T2DM) as an example of a condition with known inflammatory derangements, that has been recently described as a risk factor for PD. Finally, we discuss current gaps in the literature and future research directions in this field.
Table 1
Summary of epidemiological studies investigating associations between Parkinson’s disease and immune-mediated diseases or immune-modulating drugs
| Study | Population/Database | n | Summary |
| Autoimmune conditions | |||
| Bacelis et al. (2021) [12] | Swedish medical registries | Inclusive branch (all PD patients): 8,256 PD subjects and 82, 452 HC. | OR 0.65 (CI 95% 0.46–0.89, p = 0.006) for PD diagnosis > 5 y after RA diagnosis. |
| Conservative branch (only patients where PD was assigned as main diagnosis): 4,738 PD and 47,269 HC. | OR 0.47 (CI 95% 0.28–0.75, p = 0.006) for PD diagnosis > 5 y after RA diagnosis. | ||
| Yeh et al. (2020) [13] | National Health Insurance Research Database of Taiwan | 6,440 AS patients and 25, 760 non-AS patients. | aHR 1.75 (95% CI 1.38–2.22) in subjects with AS. |
| Chang et al. (2018) [11] | National Health Insurance Research Database of Taiwan | 34,606 cases of autoimmune rheumatic diseases and 13,824 matched control cases. | aHR 1.14; 95% CI 1.03–1.2 in subjects with RA. |
| aHR 1.56; 95% CI 1.35–1.79 in subjects with Sjogren syndrome. | |||
| Witoelar et al. (2017) [8] | GWAS data from a selection of autoimmune diseases. NeuroX data for replication | 138,511 individuals of European ancestry. | Genes associated with both PD and autoimmune conditions: |
| CASZ1, FCGR2A, MROH3P, CXCR4, IL12A, GAK, GUCY1A3, TRIM10, BTNL2, HLA-DRB5, HLA-DQB1, CCNY, SLC2A13, LRRK2, BOLA2 SETD1A, MAP3K14, MAPT, KANSL1, WNT, RSPH6A, SYMPK | |||
| Wu et al. (2017) [10] | National Health Insurance Research Database of Taiwan | 7,716 subjects with newly diagnosed PD and 7,128 matched control subjects. | aOR 1.37 (95% CI 1.15–1.65) among subjects with Sjogren syndrome. |
| Inflammatory bowel disease (IBD) | |||
| Weimers et al. (2019) [27] | Swedish Patient Register | 39,652 individuals with IBD and 396,520 controls. | OR 1.4 (95% CI 1.2–1.8) for all patients. |
| OR 1.4 (95% CI 1.1–1.9) for UC and OR 1.6 (95% CI 1.1–2.3) for CD. | |||
| Villumsen et al. (2019) [28] | Danish National Patient Register | 76,477 individuals with IBD and 7,548,259 non-IBD individuals. | HR 1.22 (95% CI 1.09–1.35) in individuals with IBD. |
| Park et al. (2019) [29] | Korean National Health Care Insurance Service | 38,861 individuals with IBD. | aHR 2.23 (95% CI 1.12 – 4.45) for individuals with CD. |
| aHR 1.85 (95% CI 1.38 – 2.48) for individuals with UC. | |||
| Camacho-Soto et al. (2018) [30] | Medicare beneficiaries and Medicare base file | 89,790 newly diagnosed PD cases and 118,095 controls. | OR 0.85 (95% CI 0.80–0.91) in subjects with IBD. |
| Peter et al. (2018) [26] | Truven Health MarketScan and Medicare Supplemental Database | 144,018 individuals with IBD and 720,090 controls. | aIRR 1.26 (95% CI 1.03–1.53) for subjects with CD. |
| aIRR 1.31 (95% CI 1.14–1.51) for subjects with UC. | |||
| Gut microbiome | |||
| Toh et al. (2021) [38] | Meta-analysis of ten studies | 1,703 subjects (969 PD patients and 734 non-PD controls). | Differentially abundant bacteria taxa between PD and controls. |
| Liu et al. (2017) [39] | Swedish Patient Register | 9,439 vagotomized patients and 377, 200 reference individuals. | HR 0.96 (95% CI 0.78–1.17) in individuals who had received a vagotomy. |
| HR 0.59 (95% CI 0.37 – 0.93)>5 y after truncal vagotomy. | |||
| Irritable bowel syndrome (IBS) | |||
| Liu et al. (2021) [33] | Swedish Patient Register | Nested case-control study including 56,564 PD cases and 30 controls per case. | OR 1.44 (95% CI 1.27–1.63) |
| Swedish Twin Registry | Cohort study included 3046 individuals with self-reported IBS and 41,179 non-IBS individuals. | HR 1.25 (95% CI 0.87–1.81) | |
| Mertsalmi et al. (2021) [34] | Finnish Care Register | 28,150 individuals with IBS and 98,789 IBS-free. | aHR 1.70 (95% CI 1.27–2.26) |
| aHR 2.96 (95% CI 1.78–4.92) only during the first 2 y of follow up. | |||
| Zhang et al. (2021) [35] | Meta-analysis of five studies | 2,044,110 individuals. | HR 1.48 (85% CI 1.35–1.62) |
| Diabetes | |||
| Chohan et al. (2021) [49] | Meta-analysis including 28 articles | OR 1.21 (95% CI 1.07–1.36) for risk of PD. | |
| OR 1.08 (95% CI 1.02–1.14) for a causal effect using MR. | |||
| OR 1.10 (95% CI 1.01–1.20) for an effect on motor progression using MR. | |||
| De Pablo-Fernandez et al. (2018) [46] | Hospital Episode Statistics | 2,017,115 individuals with T2DM and 6,173,293 reference cohort. | HR 1.32 (95% CI 1.29–1.35) |
| De Pablo-Fernandez et al. (2017) [48] | NEDICES study | 79 PD patients, 4,919 controls. | OR 1.89 (95% CI 0.6–1.89) |
| Yang et al. (2017) [45] | National Health Insurance Research Database of Taiwan | 36,294 individuals with T2DM and 108,882 non-DM individuals. | aHR 1.19 (95% CI 1.08–1.32) |
| Kang et al. (2021) [58] | GWAS data from the IPDGC and 23andMe, Inc | 37,688 individuals with PD and 981,372 controls. | Mendelian randomization study with genetic variants in the vicinity of TNFRSF1A used as predictors of TNFR1 signaling blockade. |
| OR 0.99 (95% CI 0.91–1.08) for PD risk. | |||
| San Luciano et al. (2020) [54] | Parkinson Disease Genetic and Environmental Modifiers. Michael J. Fox Foundation LRRK2 Cohort Consortium | 577 participants (259 LRRK2-PD and 318 LRRK2-nonPD). | OR 0.34 (95% CI 0.21–0.57) for PD risk in subjects with regular NSAID use. |
| Yeh et al. (2020) [13] | National Health Insurance Research Database of Taiwan | 6,440 AS patients and 25, 760 non-AS patients. | aHR 0.69 (95% CI 0.5–0.96) in individuals using NSAIDs. |
| aHR 2.40 (95% CI 1.26 – 4.56) in individuals receiving immunosuppressant therapy. | |||
| Poly et al. (2019) [53] | Meta-analysis including 17 studies | 14,713 PD individuals and 2,498,258 controls. | RR 0.95 (95% CI 0.86 – 1.048) for use of NSAIDs. |
| Park et al. (2019) [29] | Korean National Health Care Insurance Service | 38,861 individuals with IBD. | aHR 0.08 (95% CI 0.02 – 0.33) in patients with CD who used corticosteroids. |
| Among 2,110 patients receiving anti-TNF, none experienced PD during 9950 person-years. | |||
| Fan et al. (2019) [55] | Medicare beneficiaries | 89,790 individuals with PD and 118,095 controls. | OR 0.63 (95% CI 0.53–0.75) for beneficiaries who had received a tissue transplant (kidney, heart, liver, lung, and bone marrow) at least 5 y prior to PD diagnosis. |
| Peter et al. (2018) [26] | Truven Health MarketScan and Medicare Supplemental Database | 144,018 individuals with IBD and 720,090 controls. | aIRR 0.22 (95% CI 0.05 – 0.88) for individuals with IBD who were exposed to anti-TNF therapy. |
| Racette et al. (2018) [56] | Medicare beneficiaries | 48,295 PD individuals and 52,324 controls. | RR 0.64 (95% CI 0.51–0.79) for IMDH inhibitors |
| RR 0.80 (95% CI 0.77–0.83) for corticosteroids | |||
| RR 0.84 (95% CI 0.74–0.95) for dihydrofolate reductase inhibitors | |||
| RR 0.91 (95% CI 0.49–1.70) for calcineurin inhibitors | |||
| RR 0.76 (95% CI 0.56–1.04) for any biologics (adalimumab, etanercept) | |||
| RR 0.77 (95% CI 0.65–0.90) for hydroxychloroquine | |||
| RR 0.88 (95% CI 0.69–1.13) for sulfasalazine | |||
| RR 0.79 (95% CI 0.64–0.96) for mesalamine | |||
| RR 0.85 (95% CI 0.54–1.34) for thalidomide/lenalidomide | |||
| Infection and PD | |||
| Smeyne et al. (2021)* [62] | Rochester Epidemiology Project | 464 individuals with parkinsonism and 464 matched controls. | OR 1.05 (95% CI 0.78–1.4) for infection-related hospitalization |
| OR 0.86 (95% CI 0.40–1.85) for sepsis | |||
| Cocoros el al. (2021) [66] | Danish National Patient Registry | 10,271 individuals with PD and 51,355 controls. | OR 1.73 (95% CI 1.11–2.71) for PD diagnosis more than 10 y after influenza. |
| Wang et al. (2020) [65] | Meta-analysis of 23 studies analyzing risk of 13 pathogenic microorganisms and risk of PD. | 23 articles (13,545 PD and 41,446 controls). | HP: pOR 1.65 (95% CI 1.42–1.91) |
| Malassezia pOR 1.69 (95% CI 1.36–2.1) | |||
| Chlamydophila pneumoniae pOR 1.5 (95% CI 1.02–2.49) | |||
| Antiviral treatment against HCV pOR 0.67 (95% CI 0.57–0.79) | |||
| Nerius et al. (2020) [72] | Health claims data from German health insurer. | 228,485 individuals aged 50 y and older. | HR 1.42 (95% CI 1.33–1.52) in individuals with a history of GII. |
| Tsai et al. (2016) [57] | Clalit Healthcare Service (healthcare provider in Israel) | 21,010 individuals with PD. | OR 1.08 (95% CI 1–1.16) in HBV-positive individuals. |
| OR 1.18 (95% CI 1.04–1.35) in HCV positive individuals. | |||
| OR 1.13 (95% CI 1.08–1.19) in patients diagnosed with NASH. | |||
aHR, adjusted hazard ratio; aIRR, adjusted incidence rate ratio; aOR, adjusted odds ratio; AS, Ankylosing spondylitis; CD, Crohn’s disease; GII, gastrointestinal infections; GWAS, genome wide association study; HBV, Hepatitis B virus; HCV, Hepatitis C virus; HP, Helicobacter pylori; HR, hazard ratio; IBD, inflammatory bowel disease; IBS, irritable bowel syndrome; IMDH, inosine monophosphate dehydrogenase inhibitors; IPDGC, International Parkinson’s Disease Genomics Consortium; MR, Mendelian randomization; NASH, nonalcoholic steatohepatitis; NSAID, non-steroidal anti-inflammatory drugs; OR, odds ratio; PD, Parkinson Disease; pOR, pooled OR; RA, rheumatoid arthritis; RR, risk ratio; SS, Sjogren syndrome; T2DM, Type 2 diabetes mellitus; TNFRSF1A, gene encoding TNF receptor 1; UC, ulcerative colitis. * Included MSA, DLB, PDD, and PD as cases.
IMMUNE SYSTEM AND GENETIC RISK IN PD
Genetic studies support an association between autoimmune conditions and PD. The most recent meta-analysis of GWAS identified more than 90 loci that are associated with idiopathic PD [6]. In this same study, PD genes were enriched in pathways involving a response to a stressor, including major histocompatibility complex, class II, DR Beta 5 (HLA-DRB5); which plays a central role in the immune system by presenting peptides derived from extracellular proteins [7].
LRRK2 also seems to be a common factor linking the immune system and PD. Variants in the LRRK2 gene are among the most frequent causes of familial PD. LRRK2 is expressed in both innate and adaptive immune cells and is a member of the receptor interacting protein kinase family, which are a group of proteins that detect and respond to cellular stress by regulating cell death and activation of the immune system [5]. It has been observed that LRRK2 expression increases in response to microbial pathogens [5]. Polymorphisms in the LRRK2 gene have been linked to inflammatory diseases such as leprosy and inflammatory bowel disease (IBD), highlighting its role in inflammation [5]. In a recent GWAS study, shared genetic risks were found between PD and Crohn’s disease (CD), with LRRK2 as one of the most significant genes shared by both [8]. For an in-depth review of the genetic basis of inflammation and PD, we refer the reader to the article “Immunogenetics in PD”, which is covered by others in this special issue of the Journal of Parkinson’s Disease.
AUTOIMMUNE CONDITIONS AND PARKINSON DISEASE
An increased risk of PD has been associated with some autoimmune disorders, suggesting a role of autoimmunity in the pathogenesis of PD (Table 1). In one large epidemiological study conducted in Sweden, which involved more than 310,000 patients with different autoimmune disorders, patients with an autoimmune disease had a 33% excess risk of PD [9]. In another population-based cohort study conducted on the ethnic Chinese population from Taiwan, Sjogren syndrome was associated with an increased risk of PD [10]. In the same population, rheumatoid arthritis (RA) was also associated with an increased risk of PD, compared with age and sex-matched controls [11]. Nonetheless, a recent nested case-control study showed the risk of PD was decreased 30-50% in individuals who received an RA diagnosis compared to healthy controls [12]. Differences in ethnicity and lifestyle factors could contribute to these differences in outcomes.
A recent study also described a positive association between ankylosing spondylitis (AS) and the risk of PD regardless of sex or age [13]. In contrast, one study found that systemic lupus erythematous was associated with a decreased risk of PD, which the authors propose could be due to immunosuppressant treatment, although an unrecognized common risk factor is also provided as a possibility [14].
Fig. 1
Risk and protective factors associated with immune system function and PD development. Autoimmune conditions, except for systemic lupus erythematosus, are identified as risk factors for the development of Parkinson’s disease (PD). Genes identified through GWAS are highlighted as associated with Crohn’s disease, rheumatoid arthritis, and ulcerative colitis, as well as with PD. LRRK2 is specifically highlighted given its strong association with PD as well as Crohn’s disease. Among infections, Hepatitis B and hepatitis C viral infection, as well as H. pylori show the most consistent evidence and are highlighted. Type 2 diabetes and irritable bowel syndrome, which include immune system dysfunction have been described as risk factor for PD. Finally, immunosuppressant medications are used to treat autoimmune conditions and, along with NSAIDs, have an inverse association with PD. Risk factors are shown in orange and protective factors in green. Lighter shaded boxes represent lower level of evidence (e.g., only one study describing the association) while heavier shading represents robust evidence (e.g., those where several epidemiological studies are available and consistent).
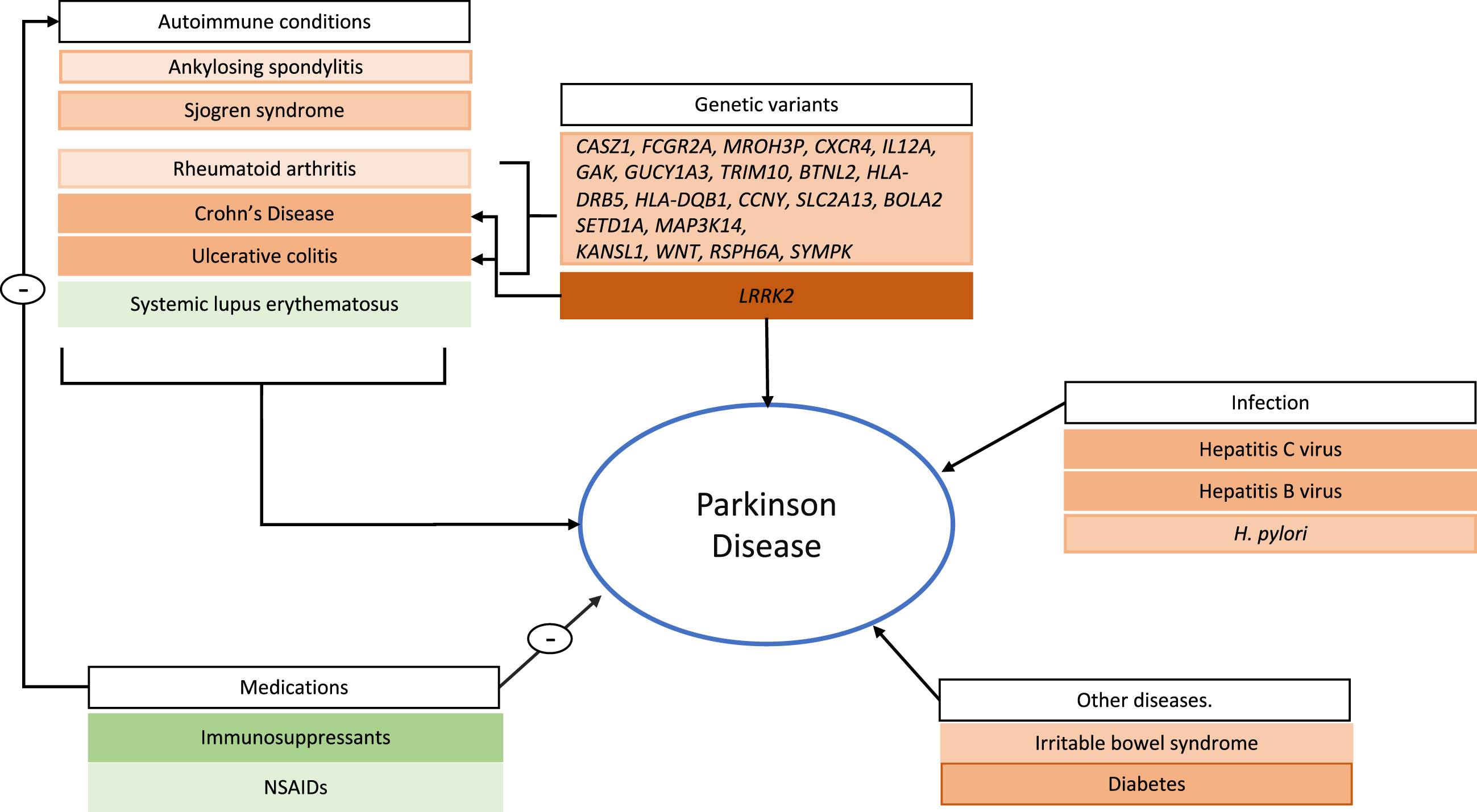
Secondary parkinsonism can be a feature of some of these disorders, which can confound interpretation of the existing literature [15]. Nonetheless, the consistency of these PD associations across multiple autoimmune diseases provides support for a true relationship mediated by the common feature of immune system activation.
This association is also supported by genetic data. Analysis of GWAS data on patients with PD or different autoimmune diseases, including CD, ulcerative colitis (UC), celiac disease, RA, psoriasis, and type 1 diabetes, identified several pleiotropic genes, with the strongest genetic overlap between PD and CD. Some of these genes include HLA-DRB5, HLA-DQB1, MAPT, and LRRK2 which were associated with CD and UC and GAK1 and CXCR4 which were associated with RA and type I diabetes [8].
INFLAMMATORY BOWEL DISEASE, IRRITABLE BOWEL SYNDROME, AND PARKINSON’S DISEASE
Gastrointestinal symptoms are frequently found in PD and may exist decades before motor symptom onset. Bidirectional communication between the central nervous system (CNS) and enteric nervous system (ENS) (“gut-brain axis”) has been proposed as a possible link between the intestinal environment and PD. Postmortem studies showing the presence of aggregated alpha-synuclein in the ENS, particularly the appendix [16]. In support of its role, patients with appendectomies have been reported to have a delayed age of PD onset [17]. Intestinal dysbiosis may also affect alpha-synuclein aggregation and lead to an excessive inflammatory response and potentially contribute to disease onset and progression. The gut microbiome and its effect on PD is covered by others in this special issue of the Journal of Parkinson’s Disease.
IBD, UC and CD, is also associated with intestinal dysbiosis [18]. IBD patients exhibit a decrease of bacteria with anti-inflammatory capacities and an increase in pro-inflammatory microbiota profile compared to healthy controls [19]. There are studies showing higher expression levels of alpha-synuclein in the colon of CD patients compared to healthy controls [20]. Intracellular alpha-synuclein staining in infiltrating monocytic cells from colonic biopsies of UC and CD patients was also described in a small study [21]. The co-occurrence of alpha-synuclein and tau deposits has been described in PD [22] and further supporting pathophysiological links between PD and CD, upregulation of two main human tau isoforms has been shown in the ENS of CD patients [23]. The relationship may be genetically mediated; as discussed above, there is evidence of genetic overlap between CD and PD, both from GWAS, as well as data from LRRK2 cohort studies [8, 24].
Several epidemiological studies have sought to determine whether IBD increases the risk of PD. To date, at least five population-based cohort studies have reported that the risk of PD is variably increased in IBD patients (Table 1). Specifically, IBD patients were found to have a 20%–90% higher risk of developing PD compared to individuals without IBD [25–29]. Although these studies can provide important insight into PD pathophysiology, some caution should be used in their interpretation, particularly since an inverse association has also been described. A population-based, case-control study of prodromal PD observed an inverse association between prodromal PD and IBD, IBD- associated surgical procedures and immunosuppressant use. The authors propose that the increased risk observed in other cohort studies may have been due to surveillance bias [30]. Similarly, in a population cohort from the Swedish National Patient Register, the effect of IBD on incident PD disappeared when adjusting for number of medical visits during follow-up, suggesting an important effect of surveillance bias [27]. By contrast, Danish and Korean cohort studies maintained a significant associated even after adjustment for number of healthcare visits [28, 29].
Some of the above associations seemed to be age dependent. In one meta-analysis patients with onset of IBD over age of 60 had a higher risk of PD [31]. By contrast, another study demonstrated a higher risk of PD in CD patients only in individuals with age at onset younger than 60 years [29]. The incidence of IBD among PD patients has also been controversial. A US study using Medicare data showed a lower risk of both CD and UC in PD patients compared to non-PD controls [30]. This discrepancy could be due to the gap in peak incidence age between diseases. It may also be that typical symptom of IBD may be masked by other gastrointestinal issues in PD, such as delayed gastric emptying and constipation.
Irritable bowel syndrome (IBS), while not an autoimmune condition, is associated with alterations in immune regulation and exposure to pathogens can be an important initiating event. IBS patients have increased expression of intestinal Toll-like receptors, which are important mediators of intestinal immune response to gut microbes. Increased serum levels of bacterial endotoxins (LPS) and anti-flagellin antibodies have also been demonstrated in IBS subjects [32]. IBS has been studied as a potential risk factor for PD, again because of the proposed gastrointestinal origin of PD (Table 1). A nested case-control study in the Swedish population estimated a higher risk of PD in individuals with a prior diagnosis of IBS. Nonetheless, the same group failed to confirm this finding in a cohort study using data from the Swedish Twin Registry [33]. Interestingly, a retrospective registry-based cohort study utilizing data from the Finnish Care Register for Health Care found that a diagnosis of IBS was associated with a higher hazard of PD; however, the ratio was not constant over time and the hazard was only significantly higher in the first two years of follow up. This suggests that this association may be better explained by reverse causation and detection bias [34]. Finally, a meta-analysis that included five studies did show a higher risk of PD among subjects with IBS although surveillance bias was identified as a potential explanation across most of the included studies [35].
A role of the gastrointestinal tract in the development of synucleinopathies could be mediated by bacterial activity of the gut microbiome. Endotoxins produced by some gut bacteria have been reported to have promote the aggregation of synuclein, generating toxic synuclein products that may participate in the cascade of events leading to PD [36]. Two recent meta-analysis included case-control studies in datasets from Finland, Russia, the United States, Germany, and Japan. 16S ribosomal ribonucleic acid (rRNA) gene sequencing data showed differentially abundant bacteria comparing PD and controls, including a decrease in short-fatty acid producing bacteria, which can lead to neuroinflammation [37, 38].
The association between gastrointestinal inflammation and CNS involvement in PD is hypothesized to be driven by alpha-synuclein deposition in the ENS, which is then transmitted to the CNS through the vagus nerve. While case-control and vagotomy animal models seem to support this, a nationwide Swedish register did not find an association with PD risk (HR 0.96; 95% CI 0.78–1.17). However, there was a suggestion of lower risk in those subjects that had received a truncal vagotomy at least 5 years before PD diagnosis (HR 0.59, 95% CI 0.37–0.93) [39].
Although gastrointestinal inflammatory conditions appear to be associated with PD, it is currently unclear whether the relationship is causal. Analysis of prodromal PD may aid in elucidating this. Recent advancements in in vitro seeding assays, such as real-time quaking induced conversion and protein misfolding cyclic amplification present the exciting possibility of molecular diagnosis of PD using readily accessible peripheral tissue samples [40]. Future studies could leverage these techniques to include the analyses of colon tissue samples from selected cohorts to help characterize this association.
DIABETES
Cardiovascular disease risk factors, including hypertension, obesity, and T2DM, share biological processes with PD, in particular inflammation and oxidative stress [41]. Hyperglycemia and insulin resistance in T2DM may also lead to mitochondrial dysfunction which in turn results in low-grade inflammation and overproduction of reactive oxygen species products and contribute to an elevated risk of PD [42, 43]. Hyperglycemia in diabetic patients also leads to protein glycation dysfunction and aggregation, which in turn may lead to alpha-synuclein aggregation, providing another potential link between T2DM and PD [44].
A retrospective, population-based study in Taiwan described a higher risk of PD in individuals diagnosed with T2DM compared to the non-T2DM cohort, with a larger magnitude in females, individuals aged 65 years and older and those with comorbidities including coronary artery disease, hyperlipidemia, asthma, and stroke. This increased risk remained during a mean follow-up of 7.3 years [45]. A similar risk was noted in T2DM patients from London England, with a greater magnitude in females and those with target organ damage. In contrast to the prior study, younger patients (less than 65 years of age) were at an increased risk of PD [46]. A retrospective cohort from Spain showed an overall higher risk of PD following diagnosis of not only in T2DM but also in prediabetic subjects; the higher risk was seen in both sexes in individuals younger than 65 years of age but was restricted to females among those over age 65 [47]. This study also described a higher rate of subsequent PD in overweight and obese subjects, independent of T2DM and prediabetes [47], suggesting that common metabolic risk factors may explain the associations observed between PD and T2DM. A cross-sectional population-based analysis in an elderly Spanish population study showed no association between T2DM and prevalence of PD, nonetheless, subgroup analysis revealed a positive association in those with long-duration diabetes (>10 years) [48]. This association is further supported in a recent meta-analysis [49].
T2DM has been associated with cognitive impairment in PD [50], which has been proposed to be mediated by inflammation. A study conducted in a large cohort examined the relationship between vascular, inflammatory, metabolic risk factors and dementia in PD subjects. Notably, PD patients with T2DM and dementia (PDD-T2DM) exhibited more vascular inflammatory factor derangements than PDD without T2DM. Derangements included elevated LDL cholesterol and fibrinogen. These factors were proposed to lead to greater fragmentation of capillaries and chronic inflammatory damage to capillary network in multiple brain regions, leading to cognitive changes [51].
IMMUNOSUPPRESSANT AND ANTI-INFLAMMATORY DRUGS AND PD
Given the association of PD and autoimmune conditions, it follows that medications used to treat these conditions may influence the risk of PD. Several epidemiological studies have been conducted to investigate the use of non-steroidal anti-inflammatory drugs (NSAIDs) before PD onset (Table 1). Initial observations suggested that NSAIDs protected against PD (for review, see [5]). However, meta-analyses published in 2018 and 2019 data did not identify a significant association between NSAID use and risk of PD, although a subset analysis in one study revealed a possible protective effect of non-aspirin NSAIDs [52, 53]. NSAID use was also associated with lower risk of PD in a cohort of individuals with AS (aHR 0.69) [13]. Finally, this association may be related to the underlying genetic variants. Regular use of NSAIDs (including aspirin and ibuprofen) was associated with a reduced odds of PD in a cohort of manifesting and non-manifesting LRRK2 carriers [54]. This may support the role of the LRRK2 protein in inflammatory pathways.
The association of immunosuppressant use and PD was also explored in a Medicare claims data study, which demonstrated a strong inverse association between tissue transplant and risk of PD; this was consistent across all types of tissue transplant, with the potential common link between patients being the use of immunosuppressants. One limitation of this study is the use of Medicare claims data, which restricted inclusion to subjects aged 65 and older and precluded the investigation of specific immunosuppressants due to insufficient prescription medication data [55]. Another population-based case-control study utilizing Medicare data described a lower risk of PD with the use of immunosuppressants. In this study, specific immunosuppressant categories that were associated with a particularly low risk included corticosteroids and inosine monophosphate dehydrogenase inhibitors (such as azathioprine, leflunomide, or mycophenolate) [56]. Interestingly, in a different study, the risk of PD was higher in patients with AS receiving immunosuppressants than in those not receiving immunosuppressants, this was attributed to a likely higher disease activity and stronger inflammatory response, rather than an effect from immunosuppressant treatment [13]. In a nationwide, population-based study using claims data from the Korean National Health care insurance, corticosteroid use was associated with an inverse association with development of PD in patients with CD but not with UC. In this same study, patients who received anti-tumor necrosis factor (anti-TNF) did not develop PD across 9950 person-years of follow-up [29].
In another cohort of subjects with IBD, anti-TNF treatment showed a 78% decrease in PD risk compared to those without anti-TNF treatment [26]. Two studies using the Taiwanese national health insurance database explored the role of interferon therapy for chronic Hepatitis C virus (HCV) infection and risk of PD, with a lower risk in the treated group. It should be noted that this same population had previously shown an increased risk of PD in those subjects diagnosed with HCV [57], further discussed below. A recent Mendelian randomization (MR) study did not support the notion that long-term blockade of TNF signaling affected the risk of PD in the general population. In this study, investigators focused on the association between single nucleotide polymorphisms in the vicinity of the gene TNFRSF1A (which encodes TNFR1, the principal effector of proinflammatory signaling following TNF agonism) and circulating markers of systemic inflammation. Data on select variants was then combined with corresponding association statistics from GWAS of PD traits, including age at disease onset and risk of PD (measured as self-reported or clinically ascertained disease). TNF-TNFR1 signaling inhibition was not estimated to affect PD risk or age at onset [58]. One important caveat is that TNF inhibition on direct measures of PD progression were not tested in this study, it still highlights the value of MR studies as an approach to help answer these questions. In MR, exposure is defined on the presence or absence of a specific allele, that influences the risk factor of interest. In this way, MR can avoid many of the typical biases that impact traditional epidemiological approaches. As such, tools such as MR can aid in causal inference related to many of these proposed immune risk factors [59, 60].
INFECTION AND PD
Infectious agents may contribute to PD by eliciting an inflammatory response. Peripheral infections may enhance neurodegeneration either via direct toxicity of bacteria or viral toxins or by circulating cytokines [61]. Neurotropic infectious agents could cause direct damage to brain regions affected by PD, increasing susceptibility [62]. It may also be that increased oxidative stress with age can render neurons vulnerable to the toxicity of infectious agents [62]. In addition, it is a commonly observed phenomenon that PD patients with viral or bacterial infections exhibit deterioration of both motor and cognitive function, suggesting that inflammation caused by infection may be a contributor to disease (for review, see [5]). Table 1 includes a summary of recent studies examining associations between PD and various infections.
There are a number of viral infections that have been studied for their association with PD. Pandemic outbreaks in the past century have been associated with encephalitis with parkinsonian features (for review, see [63]), and it has been noted that that the common feature is the induction of a systemic infection characterized by production of significantly high levels of cytokines and chemokines (for review, see [62]). However, evidence against high levels of cytokines and chemokines being sufficient to cause PD is provided by one study that did not show an association between severe infections including sepsis and future risk of PD [64]. The association between PD and influenza remains controversial. A meta-analysis combining data from four small case-control studies found no significant association between influenza infection and PD, with many of these studies including at least a decade of follow-up [65]. A case-control study from the Danish National Patient Registry ascertained influenza infection between 1977 and 2016 and showed that influenza diagnosed at any time during the calendar year was associated with PD more than 10 years later; interestingly no association was found for pneumonia or other respiratory infections [66]. While this study provides evidence for an association, as with any observational study design, causality remains to be established.
Serological evidence of prior infection among people with PD compared with those without has provided support for associations with several viruses. For example, a study showed that HSV1 peptides and alpha-synuclein peptides were cross recognized in PD patients. In this same study, PD patients had higher levels of antibodies against HSV-1 peptides compared to individuals without PD [67]. Data from epidemiological studies showed that seropositivity for EBV was higher in parkinsonian patients than in the general population [68]. This was explored in a recent study that reported the presence of antibodies against cytomegalovirus, Epstein Barr virus, herpex simplex virus type 1, Borrelia burgdorferi, Chlamydophila pneumoniae, and Helicobacter pylori (H. pylori) in serum was associated with PD diagnosis [69]; nonetheless further studies will be needed to clarify a causative role. Additionally, the potential role of medications used to treat infection, as well as the interaction between genetic predisposition and infectious exposure in the pathogenesis of PD remain to be studied.
Hepatitis B (HBV) and HCV have also been investigated for their association with PD. HCV infection includes a myriad of extrahepatic inflammatory and immune mediated reactions. The currently available epidemiologic evidence suggests a small, positive association between HCV and future development of PD. A large prospective study from the Taiwan national health insurance research database showed an association between prior diagnosis of HCV and an increased risk of subsequent PD [57]. Similarly, a UK-based study reported an association for both HCV and HBV. Finally, a population-study in Israel also reported a significantly increased risk for both HCV and for HBV [70].
Bacterial production of pro-inflammatory and neurotoxic factors could also play a role in the cascade of events leading to neurodegeneration. Nonetheless, there is less epidemiological evidence for associations between bacterial infection and PD compared to viral infections. Infection with H. pylori has been associated with PD. A large meta-analysis reported a 1.5- 2-fold increased risk of developing PD after H. pylori infection [71]. A prospective cohort study utilizing health claims data of the largest German health insurer, describes a significantly higher cumulative incidence of PD in those individuals with history of gastrointestinal infection, with the most frequent conditions being infectious gastroenteritis and colitis of unspecified origin [72]. This is of interest given the potential role of the gut microbiome, as mentioned in the preceding sections.
Given the above, there is some evidence that infection, particularly viral infection, may contribute to the etiology of PD in some individuals. This could align with the fact that symptoms of PD and other neurodegenerative diseases worsen in the context of infection and metabolic stress. The challenges in analyzing this association include the timing and relative impact of multiple infectious exposures over a lifetime, the wide variety of infectious agents and infection severity.
CONCLUSION AND FUTURE DIRECTIONS
Various lines of indirect epidemiological evidence lend support for a role of the immune system and inflammatory pathways in PD pathophysiology. As noted above, GWAS studies show evidence of shared genetic pathways between PD and different autoimmune conditions, including RA, CD, and UC. LRRK2, the most common genetic cause of PD, is also a significant risk factor for CD. Epidemiological studies also show an increased risk of developing PD in individuals with an underlying autoimmune condition. Conditions with a significant immune component, including T2DM and IBS have also been proposed to represent a higher risk of developing PD. Additionally, immunosuppressant medications show an inverse association with the risk of developing PD. Nonetheless, there are still many uncertainties that need to be addressed. In addition, for many of these studies there is a risk for surveillance bias, misclassification due to reliance on administrative diagnostic codes and unmeasured confounders (medications, comorbidities), asking for cautious interpretation of the published data.
The direction of association between PD and inflammation is yet to be determined. Epidemiological studies may be helpful to explore this question. Well-designed cohorts can provide additional data on temporal association between inflammatory response and the development of PD symptoms. In particular, the study of deeply phenotyped prodromal PD cohorts will be crucial to establish a sequence of events.
The role of immunomodulatory treatments of immune-mediated conditions as a possible confounder is understudied and requires further analysis. The role of immunosuppressant medications has been explored, mainly as part of cohort studies establishing a link between PD and other conditions (e.g., CD, UC, or HCV). In this area, further studies are needed to ascertain if the proposed effect on PD is limited to a specific immunosuppressant or if it is a class effect.
Epidemiological evidence is most robust as it pertains to autoimmune conditions and PD; with most studies focusing on IBD. Future studies focusing on other autoimmune conditions may be beneficial to best understand the specific immune components driving this association. A clearer understanding of the specific genetic variants that overlap between PD and immune conditions and their effect on basal ganglia circuits is also necessary.
One salient observation emerging from this review is the fact that, while there is abundance of literature on animal and cell models exploring the role of the immune system on PD, epidemiological studies (i.e., population studies and genetic epidemiology studies) are less common and most focus on specific conditions such as IBD and immunosuppressant medication use. In this way, one of the biggest impediments to drawing conclusions is a lack of high-quality epidemiological studies.
We acknowledge that these studies may pose design difficulties, including exposure ascertainment, latency, duration, and confounding factors such as vaccination, treatment, and co-infection. Nonetheless, the use of well characterized cohorts such as the Parkinson Progression Marker Initiative (PPMI) [73] or the UK Biobank [74] may aid in conducting these analyses. These cohorts provide access to longitudinal clinical and biological data which can be leveraged to characterize immune system changes and their association not only with motor symptom onset (e.g., in Prodromal cohort) but also with disease progression. Genetic data can be used for novel epidemiological study designs such as MR.
We emphasize the need to study these associations in diverse populations. It is encouraging to see that many of these studies already include populations from Asia. Nonetheless, the inclusion of other ethnic and racial groups, such as Hispanic, African, and African American populations, is still lacking. This is an urgent need in our field which is necessary to better understand these associations and their possible contributions to disease onset and progression. This is of particular importance when considering autoimmune conditions, since there is previous data suggesting these are less common in non-European populations.
It is also worth considering that most studies have used PD as one condition. We now understand that there is considerable heterogeneity within “PD” and immune system changes may be a possible marker to distinguish those individuals with an important immune component and aid in tailoring future interventions. This will be of particular importance when designing and testing medications that target the immune system in PD. It has been proposed that recruitment of selective cohorts, in this case identified by markers of immune dysfunction, could ensure that putative therapies are tested only in those most likely to respond [75].
Future studies assessing the role of inflammation in the prodromal period vs after disease diagnosis are necessary, for example by studying non-manifesting LRRK2 or GBA carriers or REM Sleep Behavior disorders cohorts. Analyses of the association between the immune system and disease progression is also important, particularly to determine whether the immune system might be targeted with interventions aimed to modify established PD.
CONFLICT OF INTEREST
The authors have no conflict of interest related to this manuscript.
REFERENCES
[1] | Calabrese V , Santoro A , Monti D , Crupi R , Di Paola R , Latteri S , Cuzzocrea S , Zappia M , Giordano J , Calabrese EJ , Franceschi C ((2018) ) Aging and Parkinson’s disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic Biol Med 115: , 80–91. |
[2] | Tansey MG , Romero-Ramos M ((2019) ) Immune system responses in Parkinson’s disease: Early and dynamic. Eur J Neurosci 49: , 364–383. |
[3] | Rosen B , Kurtishi A , Vazquez-Jimenez GR , Moller SG ((2021) ) The intersection of Parkinson’s disease, viral infections, and COVID-19. Mol Neurobiol 58: , 4477–4486. |
[4] | Qin XY , Zhang SP , Cao C , Loh YP , Cheng Y ((2016) ) Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: A systematic review and meta-analysis. JAMA Neurol 73: , 1316–1324. |
[5] | Wallings RL , Herrick MK , Tansey MG ((2020) ) LRRK2 at the interface between peripheral and central immune function in Parkinson’s. Front Neurosci 14: , 443. |
[6] | Nalls MA , Blauwendraat C , Vallerga CL , Heilbron K , Bandres-Ciga S , Chang D , Tan M , Kia DA , Noyce AJ , Xue A , Bras J , Young E , von Coelln R , Simón-Sánchez J , Schulte C , Sharma M , Krohn L , Pihlstrøm L , Siitonen A , Iwaki H , Leonard H , Faghri F , Gibbs JR , Hernandez DG , Scholz SW , Botia JA , Martinez M , Corvol JC , Lesage S , Jankovic J , Shulman LM , Sutherland M , Tienari P , Majamaa K , Toft M , Andreassen OA , Bangale T , Brice A , Yang J , Gan-Or Z , Gasser T , Heutink P , Shulman JM , Wood NW , Hinds DA , Hardy JA , Morris HR , Gratten J , Visscher PM , Graham RR , Singleton AB ; 23andMe ResearchTeam; System Genomics of Parkinson’s Disease Consortium;International Parkinson’s Disease Genomics Consortium ((2019) ) Identification of novel risk loci, causal insights, and heritablerisk for Parkinson’s disease: A meta-analysis of genome-wideassociation studies. Lancet Neurol 18: , 1091–1102. |
[7] | Scholz EM , Marcilla M , Daura X , Arribas-Layton D , James EA , Alvarez I ((2017) ) Human leukocyte antigen (HLA)-DRB1*15:01 and HLA-DRB5*01:01 present complementary peptide repertoires. Front Immunol 8: , 984. |
[8] | Witoelar A , Jansen IE , Wang Y , Desikan RS , Gibbs JR , Blauwendraat C , Thompson WK , Hernandez DG , Djurovic S , Schork AJ , Bettella F , Ellinghaus D , Franke A , Lie BA , McEvoy LK , Karlsen TH , Lesage S , Morris HR , Brice A , Wood NW , Heutink P , Hardy J , Singleton AB , Dale AM , Gasser T , Andreassen OA , Sharma M , International Parkinson’s Disease Genomics Consortium (IPDGC), North American Brain Expression Consortium (NABEC), and United Kingdom Brain Expression Consortium (UKBEC) ((2017) ) Genome-wide pleiotropy between Parkinson disease and autoimmune diseases. JAMA Neurol 74: , 780–792. |
[9] | Li X , Sundquist J , Sundquist K ((2012) ) Subsequent risks of Parkinson disease in patients with autoimmune and related disorders: A nationwide epidemiological study from Sweden. Neurodegener Dis 10: , 277–284. |
[10] | Wu MC , Xu X , Chen SM , Tyan YS , Chiou JY , Wang YH , Lin LC , Chen CM , Wei JC ((2017) ) Impact of Sjogren’s syndrome on Parkinson’s disease: A nationwide case-control study. PLoS One 12: , e0175836. |
[11] | Chang CC , Lin TM , Chang YS , Chen WS , Sheu JJ , Chen YH , Chen JH ((2018) ) Autoimmune rheumatic diseases and the risk of Parkinson disease: A nationwide population-based cohort study in Taiwan. Ann Med 50: , 83–90. |
[12] | Bacelis J , Compagno M , George S , Pospisilik JA , Brundin P , Naluai AT , Brundin L ((2021) ) Decreased risk of Parkinson’s disease after rheumatoid arthritis diagnosis: A nested case-control study with matched cases and controls. J Parkinsons Dis 11: , 821–832. |
[13] | Yeh FC , Chen HC , Chou YC , Lin CL , Kao CH , Lo HY , Liu FC , Yang TY ((2020) ) Positive association of Parkinson’s disease with ankylosing spondylitis: A nationwide population-based study. J Transl Med 18: , 455. |
[14] | Liu FC , Huang WY , Lin TY , Shen CH , Chou YC , Lin CL , Lin KT , Kao CH ((2015) ) Inverse Association of Parkinson disease with systemic lupus erythematosus: A nationwide population-based study. Medicine (Baltimore) 94: , e2097. |
[15] | Baizabal-Carvallo JF , Jankovic J ((2018) ) Autoimmune and paraneoplastic movement disorders: An update. J Neurol Sci 385: , 175–184. |
[16] | Beach TG , Adler CH , Sue LI , Vedders L , Lue L , White Iii CL , Akiyama H , Caviness JN , Shill HA , Sabbagh MN , Walker DG , Arizona Parkinson’s Disease Consortium ((2010) ) Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol 119: , 689–702. |
[17] | Killinger BA , Madaj Z , Sikora JW , Rey N , Haas AJ , Vepa Y , Lindqvist D , Chen H , Thomas PM , Brundin P , Brundin L , Labrie V ((2018) ) The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci Transl Med 10: , eaar5280. |
[18] | Zhang M , Sun K , Wu Y , Yang Y , Tso P , Wu Z ((2017) ) Interactions between intestinal microbiota and host immune response in inflammatory bowel disease. Front Immunol 8: , 942. |
[19] | Frank DN , St Amand AL , Feldman RA , Boedeker EC , Harpaz N , Pace NR ((2007) ) Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 104: , 13780–13785. |
[20] | Prigent A , Lionnet A , Durieu E , Chapelet G , Bourreille A , Neunlist M , Rolli-Derkinderen M , Derkinderen P ((2019) ) Enteric alpha-synuclein expression is increased in Crohn’s disease. Acta Neuropathol 137: , 359–361. |
[21] | Grathwohl S , Quansah E , Maroof N , Steiner JA , Spycher L , Benmansour F , Duran-Pacheco G , Siebourg-Polster J , Oroszlan-Szovik K , Remy H , Haenggi M , Stawiski M , Selhausen M , Mailver P , Wolfert A , Emrich T , Madaj Z , Su A , Escobar Galvis ML , Mueller C , Herrmann A , Brundin P , Britschgi M ((2021) ) Specific immune modulation of experimental colitis drives enteric alpha-synuclein accumulation and triggers age-related Parkinson-like brain pathology. Free Neuropathol 2: , 13. |
[22] | Torres-Garcia L , JM PD , Brandi E , Haikal C , Mudannayake JM , Bras IC , Gerhardt E , Li W , Svanbergsson A , Outeiro TF , Gouras GK , Li JY ((2022) ) Monitoring the interactions between alpha-synuclein and Tau in vitro and in vivo using bimolecular fluorescence complementation. Sci Rep 12: , 2987. |
[23] | Prigent A , Chapelet G , De Guilhem de Lataillade A , Oullier T , Durieu E , Bourreille A , Duchalais E , Hardonniere K , Neunlist M , Noble W , Kerdine-Romer S , Derkinderen P , Rolli-Derkinderen M ((2020) ) Tau accumulates in Crohn’s disease gut. FASEB J 34: , 9285–9296. |
[24] | Lee HS , Lobbestael E , Vermeire S , Sabino J , Cleynen I ((2021) ) Inflammatory bowel disease and Parkinson’s disease: Common pathophysiological links. Gut 70: , 408–417. |
[25] | Lin JC , Lin CS , Hsu CW , Lin CL , Kao CH ((2016) ) Association between Parkinson’s disease and inflammatory bowel disease: A nationwide Taiwanese retrospective cohort study. Inflamm Bowel Dis 22: , 1049–1055. |
[26] | Peter I , Dubinsky M , Bressman S , Park A , Lu C , Chen N , Wang A ((2018) ) Anti-tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol 75: , 939–946. |
[27] | Weimers P , Halfvarson J , Sachs MC , Saunders-Pullman R , Ludvigsson JF , Peter I , Burisch J , Olen O ((2019) ) Inflammatory bowel disease and Parkinson’s disease: A nationwide Swedish cohort study. Inflamm Bowel Dis 25: , 111–123. |
[28] | Villumsen M , Aznar S , Pakkenberg B , Jess T , Brudek T ((2019) ) Inflammatory bowel disease increases the risk of Parkinson’s disease: A Danish nationwide cohort study 1977-2014. Gut 68: , 18–24. |
[29] | Park S , Kim J , Chun J , Han K , Soh H , Kang EA , Lee HJ , Im JP , Kim JS ((2019) ) Patients with inflammatory bowel disease are at an increased risk of Parkinson’s disease: A South Korean nationwide population-based study. J Clin Med 8: , 1191. |
[30] | Camacho-Soto A , Gross A , Searles Nielsen S , Dey N , Racette BA ((2018) ) Inflammatory bowel disease and risk of Parkinson’s disease in Medicare beneficiaries. Parkinsonism Relat Disord 50: , 23–28. |
[31] | Wan QY , Zhao R , Wu XT ((2020) ) Older patients with IBD might have higher risk of Parkinson’s disease. Gut 69: , 193–194. |
[32] | Pimentel M , Lembo A ((2020) ) Microbiome and its role in irritable bowel syndrome. Dig Dis Sci 65: , 829–839. |
[33] | Liu B , Sjolander A , Pedersen NL , Ludvigsson JF , Chen H , Fang F , Wirdefeldt K ((2021) ) Irritable bowel syndrome and Parkinson’s disease risk: Register-based studies. NPJ Parkinsons Dis 7: , 5. |
[34] | Mertsalmi TH , But A , Pekkonen E , Scheperjans F ((2021) ) Irritable bowel syndrome and risk of Parkinson’s disease in Finland: A nationwide registry-based cohort study. J Parkinsons Dis 11: , 641–651. |
[35] | Zhang X , Svn Z , Liv M , Yang Y , Zeng R , Huang Q , Sun Q ((2021) ) Association between irritable bowel syndrome and risk of Parkinson’s disease: A systematic review and meta-analysis. Front Neurol 12: , 720958. |
[36] | Bhattacharyya D , Mohite GM , Krishnamoorthy J , Gayen N , Mehra S , Navalkar A , Kotler SA , Ratha BN , Ghosh A , Kumar R , Garai K , Mandal AK , Maji SK , Bhunia A ((2019) ) Lipopolysaccharide from gut microbiota modulates alpha-synuclein aggregation and alters its biological function. ACS Chem Neurosci 10: , 2229–2236. |
[37] | Nishiwaki H , Ito M , Ishida T , Hamaguchi T , Maeda T , Kashihara K , Tsuboi Y , Ueyama J , Shimamura T , Mori H , Kurokawa K , Katsuno M , Hirayama M , Ohno K ((2020) ) Meta-analysis of gut dysbiosis in Parkinson’s disease. Mov Disord 35: , 1626–1635. |
[38] | Toh TS , Chong CW , Lim SY , Bowman J , Cirstea M , Lin CH , Chen CC , Appel-Cresswell S , Finlay BB , Tan AH ((2021) ) Gut microbiome in Parkinson’s disease: New insights from meta-analysis. Parkinsonism Relat Disord 94: , 1–9. |
[39] | Liu B , Fang F , Pedersen NL , Tillander A , Ludvigsson JF , Ekbom A , Svenningsson P , Chen H , Wirdefeldt K ((2017) ) Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study. Neurology 88: , 1996–2002. |
[40] | Koga S , Sekiya H , Kondru N , Ross OA , Dickson DW ((2021) ) Neuropathology and molecular diagnosis of synucleinopathies. Mol Neurodegener 16: , 83. |
[41] | Tsalamandris S , Antonopoulos AS , Oikonomou E , Papamikroulis GA , Vogiatzi G , Papaioannou S , Deftereos S , Tousoulis D ((2019) ) The role of inflammation in diabetes: Current concepts and future perspectives. Eur Cardiol 14: , 50–59. |
[42] | Potashkin J , Huang X , Becker C , Chen H , Foltynie T , Marras C ((2020) ) Understanding the links between cardiovascular disease and Parkinson’s disease. Mov Disord 35: , 55–74. |
[43] | Khang R , Park C , Shin JH ((2015) ) Dysregulation of parkin in the substantia nigra of db/db and high-fat diet mice. Neuroscience 294: , 182–192. |
[44] | Vicente Miranda H , El-Agnaf OM , Outeiro TF ((2016) ) Glycation in Parkinson’s disease and Alzheimer’s disease. Mov Disord 31: , 782–790. |
[45] | Yang YW , Hsieh TF , Li CI , Liu CS , Lin WY , Chiang JH , Li TC , Lin CC ((2017) ) Increased risk of Parkinson disease with diabetes mellitus in a population-based study. Medicine (Baltimore) 96: , e5921. |
[46] | De Pablo-Fernandez E , Goldacre R , Pakpoor J , Noyce AJ , Warner TT ((2018) ) Association between diabetes and subsequent Parkinson disease: A record-linkage cohort study. Neurology 91: , e139–e142. |
[47] | Sanchez-Gomez A , Diaz Y , Duarte-Salles T , Compta Y , Marti MJ ((2021) ) Prediabetes, type 2 diabetes mellitus and risk of Parkinson’s disease: A population-based cohort study. Parkinsonism Relat Disord 89: , 22–27. |
[48] | De Pablo-Fernandez E , Sierra-Hidalgo F , Benito-Leon J , Bermejo-Pareja F ((2017) ) Association between Parkinson’s disease and diabetes: Data from NEDICES study. Acta Neurol Scand 136: , 732–736. |
[49] | Chohan H , Senkevich K , Patel RK , Bestwick JP , Jacobs BM , Bandres Ciga S , Gan-Or Z , Noyce AJ ((2021) ) Type 2 diabetes as a determinant of Parkinson’s disease risk and progression. Mov Disord 36: , 1420–1429. |
[50] | Ong M , Foo H , Chander RJ , Wen MC , Au WL , Sitoh YY , Tan L , Kandiah N ((2017) ) Influence of diabetes mellitus on longitudinal atrophy and cognition in Parkinson’s disease. J Neurol Sci 377: , 122–126. |
[51] | Wang T , Yuan F , Chen Z , Zhu S , Chang Z , Yang W , Deng B , Que R , Cao P , Chao Y , Chan L , Pan Y , Wang Y , Xu L , Lyu Q , Chan P , Yenari MA , Tan EK , Wang Q ((2020) ) Vascular, inflammatory and metabolic risk factors in relation to dementia in Parkinson’s disease patients with type 2 diabetes mellitus. Aging (Albany NY) 12: , 15682–15704. |
[52] | Ren L , Yi J , Yang J , Li P , Cheng X , Mao P ((2018) ) Nonsteroidal anti-inflammatory drugs use and risk of Parkinson disease: A dose-response meta-analysis. Medicine (Baltimore) 97: , e12172. |
[53] | Poly TN , Islam MMR , Yang HC , Li YJ ((2019) ) Non-steroidal anti-inflammatory drugs and risk of Parkinson’s disease in the elderly population: A meta-analysis. Eur J Clin Pharmacol 75: , 99–108. |
[54] | San Luciano M , Tanner CM , Meng C , Marras C , Goldman SM , Lang AE , Tolosa E , Schüle B , Langston JW , Brice A , Corvol JC , Goldwurm S , Klein C , Brockman S , Berg D , Brockmann K , Ferreira JJ , Tazir M , Mellick GD , Sue CM , Hasegawa K , Tan EK , Bressman S , Saunders-Pullman R ; Michael J. Fox Foundation LRRK2 Cohort Consortium ((2020) ) Nonsteroidal anti-inflammatory use and LRRK2 Parkinson’s disease penetrance. Mov Disord 35: , 1755–1764. |
[55] | Fan J , Searles Nielsen S , Faust IM , Racette BA ((2019) ) Transplant and risk of Parkinson disease. Parkinsonism Relat Disord 63: , 149–155. |
[56] | Racette BA , Gross A , Vouri SM , Camacho-Soto A , Willis AW , Searles Nielsen S ((2018) ) Immunosuppressants and risk of Parkinson disease. Ann Clin Transl Neurol 5: , 870–875. |
[57] | Tsai HH , Liou HH , Muo CH , Lee CZ , Yen RF , Kao CH ((2016) ) Hepatitis C virus infection as a risk factor for Parkinson disease: A nationwide cohort study. Neurology 86: , 840–846. |
[58] | Kang X , Ploner A , Pedersen NL , Bandres-Ciga S , Noyce AJ , Wirdefeldt K , Williams DM ((2021) ) Tumor necrosis factor inhibition and Parkinson disease: A Mendelian randomization study. Neurology 96: , e1672–e1679. |
[59] | Lawlor DA , Harbord RM , Sterne JA , Timpson N , Davey Smith G ((2008) ) Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat Med 27: , 1133–1163. |
[60] | Richmond RC , Davey Smith G ((2022) ) Mendelian randomization: Concepts and scope. Cold Spring Harb Perspect Med 12: , a040501. |
[61] | Brown GC ((2019) ) The endotoxin hypothesis of neurodegeneration. J Neuroinflammation 16: , 180. |
[62] | Smeyne RJ , Noyce AJ , Byrne M , Savica R , Marras C ((2021) ) Infection and risk of Parkinson’s disease. J Parkinsons Dis 11: , 31–43. |
[63] | Henry J , Smeyne RJ , Jang H , Miller B , Okun MS ((2010) ) Parkinsonism and neurological manifestations of influenza throughout the 20th and 21st centuries. Parkinsonism Relat Disord 16: , 566–571. |
[64] | Hasan S , Mielke MM , Ahlskog JE , Bower J , Turcano P , Savica R ((2020) ) Infections or sepsis preceding clinically diagnosed alpha-synucleinopathies: A case-control study. Mov Disord 35: , 1684–1689. |
[65] | Wang H , Liu X , Tan C , Zhou W , Jiang J , Peng W , Zhou X , Mo L , Chen L ((2020) ) Bacterial, viral, and fungal infection-related risk of Parkinson’s disease: Meta-analysis of cohort and case-control studies. Brain Behav 10: , e01549. |
[66] | Cocoros NM , Svensson E , Szepligeti SK , Vestergaard SV , Szentkuti P , Thomsen RW , Borghammer P , Sorensen HT , Henderson VW ((2021) ) Long-term risk of Parkinson disease following influenza and other infections. JAMA Neurol 78: , 1461–1470. |
[67] | Caggiu E , Paulus K , Arru G , Piredda R , Sechi GP , Sechi LA ((2016) ) Humoral cross reactivity between alpha-synuclein and herpes simplex-1 epitope in Parkinson’s disease, a triggering role in the disease? J Neuroimmunol 291: , 110–114. |
[68] | Woulfe JM , Gray MT , Gray DA , Munoz DG , Middeldorp JM ((2014) ) Hypothesis: A role for EBV-induced molecular mimicry in Parkinson’s disease. Parkinsonism Relat Disord 20: , 685–694. |
[69] | Patrick KL , Bell SL , Weindel CG , Watson RO ((2019) ) Exploring the “multiple-hit hypothesis” of neurodegenerative disease: Bacterial infection comes up to bat. Front Cell Infect Microbiol 9: , 138. |
[70] | Goldstein L , Fogel-Grinvald H , Steiner I ((2019) ) Hepatitis B and C virus infection as a risk factor for Parkinson’s disease in Israel-A nationwide cohort study. J Neurol Sci 398: , 138–141. |
[71] | Shen X , Yang H , Wu Y , Zhang D , Jiang H ((2017) ) Meta-analysis: Association of Helicobacter pylori infection with Parkinson’s diseases. Helicobacter 22: , e12398. |
[72] | Nerius M , Doblhammer G , Tamguney G ((2020) ) GI infections are associated with an increased risk of Parkinson’s disease. Gut 69: , 1154–1156. |
[73] | Parkinson Progression Marker I ((2011) ) The Parkinson Progression Marker Initiative (PPMI). Prog Neurobiol 95: , 629–635. |
[74] | Sudlow C , Gallacher J , Allen N , Beral V , Burton P , Danesh J , Downey P , Elliott P , Green J , Landray M , Liu B , Matthews P , Ong G , Pell J , Silman A , Young A , Sprosen T , Peakman T , Collins R ((2015) ) UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 12: , e1001779. |
[75] | Espay AJ , Kalia LV , Gan-Or Z , Williams-Gray CH , Bedard PL , Rowe SM , Morgante F , Fasano A , Stecher B , Kauffman MA , Farrer MJ , Coffey CS , Schwarzschild MA , Sherer T , Postuma RB , Strafella AP , Singleton AB , Barker RA , Kieburtz K , Olanow CW , Lozano A , Kordower JH , Cedarbaum JM , Brundin P , Standaert DG , Lang AE ((2020) ) Disease modification and biomarker development in Parkinson disease: Revision or reconstruction? Neurology 94: , 481–494. |


