
Neuronal Presentation of Antigen and Its Possible Role in Parkinson’s Disease
Abstract
Patients with Parkinson’s disease (PD) and other synucleinopathies often exhibit autoimmune features, including CD4+ and some CD8+ T lymphocytes that recognize epitopes derived from alpha-synuclein. While neurons have long been considered to not present antigens, recent data indicate that they can be induced to do so, particularly in response to interferons and other forms of stress. Here, we review literature on neuronal antigen presentation and its potential role in PD. Although direct evidence for CD8+ T cell-mediated neuronal death is lacking in PD, neuronal antigen presentation appears central to the pathology of Rasmussen’s encephalitis, a pediatric neurological disorder driven by cytotoxic T cell infiltration and neuroinflammation. Emerging data suggest that T cells enter the brain in PD and other synucleinopathies, where the majority of neuromelanin-containing substantia nigra and locus coeruleus neurons express MHC Class I molecules. In cell culture, CD8+ T cell recognition of antigen:MHC Class I complexes on neuronal membranes leads to cytotoxic responses and neuronal cell death. Recent animal models suggest the possibility of T cell autoreactivity to mitochondrial antigens in PD. It remains unclear if neuronal antigen presentation plays a role in PD or other neurodegenerative disorders, and efforts are underway to better elucidate the potential impact of autoimmune responses on neurodegeneration.
OVERVIEW OF ANTIGEN PRESENTATION
Antigen presentation is central to adaptive immune system function, allowing host cells to display foreign and self-peptides on cell surface major histocompatibility complex (MHC) molecules for epitope recognition by T lymphocytes.
In one of two major arms of antigen presentation, degradation of viral and self-proteins by cytoplasmic proteasomes produces peptides of 8–10 amino acids in length; these peptides are then transported into the endoplasmic reticulum for loading onto MHC Class I (MHC-I). After peptide binding, the MHC-I alpha chain and light chain subunit β2-microglobulin (β2 m) are trafficked to the cell surface for antigen presentation to CD8+ T cells [1]. T-cell receptor (TCR) engagement by specific antigens presented on MHC-I causes cytotoxic responses by CD8+ T cells, also known as cytotoxic T lymphocytes (CTLs), leading to destruction of the antigen presenting cell. CTL-mediated destruction of target cells is beneficial in the context of viral infection or cancer, but recognition of native self-antigens can lead to autoimmunity [2].
Alternatively, extracellular foreign and self-proteins are imported via endocytosis and processed into peptides within the endolysosomal system. After degradation of the MHC Class II (MHC-II) invariant chain, MHC-II binds to peptides of 13–25 amino acids in length and is trafficked to the cell surface for presentation to CD4+ T cells [1]. Activation of CD4+ T cells leads to cell-cell interactions and secretion of cytokines that enhance the effector functions of other immune cells such as B cells, macrophages, and CTLs.
In contrast to MHC-I, which is expressed in most nucleated cells, MHC-II expression is limited to professional antigen presenting cells [1, 3]. Within the central nervous system (CNS), MHC-II expression may be limited to microglia [4]. Current evidence suggests that neurons can express MHC-I but not MHC-II. Thus, the primary focus of this review will be on neuronal antigen presentation via MHC-I.
NATIVE NEURONAL EXPRESSION OF MHC-I
Although MHC-I is constitutively expressed on most nucleated cells, neurons provide an important exception to this rule. Neuronal MHC-I regulates activity-dependent synapse refinement during cortical development [5, 6], although this activity is likely mediated by the MHC-I receptor PirB [7] and may be unrelated to antigen presentation. Consistent with the role of neuronal MHC-I in developmental synaptic plasticity [8, 9], neuronal MHC-I expression in humans declines rapidly after birth and is typically undetectable in the normal adult brain [10–12]. In contrast to the transient expression during early development, under basal conditions in adult animals, MHC-I expression is virtually undetectable in neurons [13, 14]. Postmortem investigations of ‘histologically normal’ human brain revealed a complete lack of parenchymal MHC-I immunoreactivity [15], leading to the general conclusion thought that adult CNS neurons do not express MHC-I [16].
Early studies indicated that even when infected by virus, neurons lack MHC-I expression [17, 18]. However, it has more recently become clear that many neurons can express MHC-I following viral infection or exposure to inflammatory cytokines (see below). Of particular relevance, we found MHC-I immunoreactivity in adult human dopaminergic and noradrenergic neurons within the ventral midbrain and locus coeruleus, respectively [19]. Using mass spectrometry, we also found MHC-I associated with neuromelanin [19], a product of oxidized catecholamines that accumulates in autophagocytic lysosomes within catecholamine neurons [20]. Other studies have reported that midbrain dopamine neurons in adult mice and rats express MHC-I under basal conditions [21, 22]. Indeed, evidence below suggests that oxyradical-stress from high cytosolic dopamine may produce neuronal MHC-I even in the absence of interferon-gamma (IFN-γ) or other cytokines.
In summary, neurons during early development are found to express MHC-I but may not present antigen, and this early expression is strongly implicated in developmental synaptic plasticity [8]. In the healthy adult, little neuronal MHC-I is found with the apparent exception of pigmented catecholamine neurons in the substantia nigra and locus coeruleus, which may result from constitutively high cytosolic dopamine levels. Thus, most evidence supports the idea that mature neurons normally suppress MHC-I expression, and specific stimuli or conditions are required to induce MHC-I [16, 23]. The lack of basal MHC-I expression may be related to neuron-specific differences in transcription factors or repressors [23, 24].
NEURONAL MHC-I IN HUMAN NEUROPATHOLOGY
Despite the general lack of detectable neuronal MHC-I in normal adult human brain tissue [15], absence of immunoreactivity does not definitively exclude the presence of low levels of neuronal MHC-I, especially given that neuronal membranes are highly damaged in autopsy material. However, MHC-I was observed in neurons in the setting of measles infection in patients with subacute sclerosing panencephalitis [25]. In postmortem histology studies of paraneoplastic disease directed towards intracellular antigens, MHC-I+ neurons are found closely opposed to granzyme B+ CTLs [26].
The most consistent pathological observation of neuronal MHC-I expression is in Rasmussen’s encephalitis, a rare pediatric neurological disease characterized by MHC-I restricted destruction of neurons and astrocytes by clonally-expanded CD8+ T cells [27–29]. Although the engagement of MHC-I+ cells by clonally expanded CTLs suggests an antigen-specific response [30], the presumptive viral or self-protein antigens are unknown [29]. On a related note, there has long been speculation that post-encephalitic parkinsonism associated with the 1918 Spanish flu and Von Economo’s encephalitis could be mediated by an autoantigen response directed towards dopamine neurons: there is presently no evidence for a similar role of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection in the initiation of parkinsonism, although there are reports of exacerbation of symptoms [31]. However, at this time it is still not clear if any viral, bacterial, or self-proteins are presented as antigens by CNS dopamine neurons [32] with the exception of neurons in culture (see below).
Neuropathological studies of PD, multiple system atrophy, and Lewy body disease demonstrate enhanced numbers of both CD4+ and CD8+ T cells in the brain parenchyma [33–37]. A recent longitudinal, prospective case study of a PD patient found that alpha-synuclein-reactive T cells were most abundant in peripheral blood prior to the appearance of motor symptoms [38]. In a larger survey of peripheral blood from healthy controls and PD patients, alpha-synuclein-reactive T cells were more abundant in earlier stage patients and mostly absent by ten years after diagnosis [38]. Further support for the involvement of CD8+ T cells in early PD pathogenesis comes from a recent study demonstrating CTL infiltration into the substantia nigra (SN) of PD patients prior to the presence of alpha-synuclein aggregates [34].
The subcellular localization of CD8:MHC-I interactions and the downstream mechanisms underlying neuronal damage seem to vary based on context (reviewed in [39]). Most human pathology studies identify CD8+ T cells interacting with the neuronal cell body (perikarya), while CTL interactions with MHC-I+ neurites have been observed mostly in cell culture (see below). In Rasmussen’s encephalitis, granzyme B+ CD8+ T cells are frequently observed near neuronal perikarya [27]. CD8+ T cells are often identified near dopaminergic perikarya in the SN [19, 33, 34], and also near pigmented, MHC-I+ neuronal perikarya in the locus coeruleus [19]. It is difficult to establish direct CTL killing of neurons in post-mortem studies, but microglial engulfment of dying neurons (‘neuronophagia’) has been observed near CD8+ T cells in paraneoplastic encephalitis [40].
It should be noted that T cell interactions with neurites would be challenging to detect within the dense neuropil of brain tissue. The bias towards neuronal perikarya may also reflect the tissue chosen for study: for example, most studies of PD focus on the SN rather than the striatum, which contains the massive axonal arbors that comprise the majority of the dopamine neuronal volume, surface area, and protein [41, 42]. Thus, future post-mortem histology studies of neuronal antigen presentation and CD8+ T cells in PD should carefully consider the brain regions and disease stages studied.
NEURONAL ANTIGEN PRESENTATION BY MHC-I IN RODENT MODELS
Most studies of neuronal antigen presentation in rodent models have focused on infectious disease. In a rat model of Borna disease encephalitis, Borna disease virus-reactive CTLs isolated from the brain exhibited robust lysis of infected cells [43]. It was later shown that neurons infected with Borna disease virus expressed MHC-I and were directly recognized by CD8+ T cells, leading to morphological changes, but not immediate neuronal destruction [44]. More recently, neuronal antigen presentation via MHC-I was shown to be the key determinant of the murine immune response to intracerebral parasitic infection by Toxoplasmosis Gondii [45]. Conditional deletion of the H2-Ld allele using the neuron-specific CamKII-promoter resulted in poor parasite control, demonstrating that MHC-I presentation by neurons was critical for pathogen clearance in this model [45].
Peripheral sensory neurons are known to upregulate MHC-I after peripheral nerve injury [46] or acute infection with Herpes Simplex Virus [47]. Similarly, a majority of CNS neurons upregulated MHC-I, β2 m, and TAP1 mRNA following infection with neurovirulent Sindbis virus [48]. However, immunoreactivity for MHC-I was not observed, consistent with a lack of mRNA translation or rapid protein degradation. Nonetheless, CD8+ T cells can interact with neurons infected with Theiler’s murine encephalomyelitis virus (TMEV) and polarize CD8, T cell receptors, and granzyme B near the neuronal surface despite negligible immunoreactivity for neuronal MHC-I [49]. Similarly, CTL killing of dorsal root ganglion neurons occurred in the absence of overt MHC-I immunoreactivity [50]. In these examples, it is possible that physiologically relevant levels of neuronal MHC-I are too low for detection by available antibodies [49, 50].
There is also anecdotal evidence of neuronal antigen presentation in mouse models of multiple sclerosis based on viral infection. CNS infection with murine hepatitis virus, which causes a chronic inflammatory demyelinating disease in mice, drives expression of MHC-I in endothelial cells, microglia, oligodendrocytes, and neurons [4]. Although both oligodendrocytes and axons are damaged in this model, neuronal MHC-I was confined to the cell body and did not extend into axons or dendrites [4]. However, β2 m–/– mice lacking functional surface MHC-I were spared from axonal damage in a model of TMEV-induced demyelination, suggesting that CD8+ T cells may directly damage axons in this model of multiple sclerosis [51].
A well-studied mouse model of neuronal antigen presentation involves the infection of neonatal mice with neurotropic lymphocytic choriomeningitis virus (LCMV) [52]. Neonatal infection results in viral persistence within multiple organs including the CNS, where LCMV infection persists in neurons [53–55]. Adoptive transfer of LCMV-specific CTLs into persistently infected mice resulted in viral clearance, although this clearance from neurons did not appear to be driven by neuronal lysis [56, 57]. However, the same adoptive transfer of LCMV-specific CD8+ T cells resulted in in severe encephalitis following neuron-specific expression of a murine MHC Class I molecule (H2-Db), consistent with neuronal presentation of LCMV antigens in this model [58].
A related model based on neonatal LCMV infection, known as the “viral déjá vu model”, uses an attenuated LCMV that persists in neurons and does not initially generate robust CD8+ T cell responses [54]. Following infection of adult LCMV carrier mice with WT LCMV, a robust CD8+ T cell response against LCMV antigens leads to CTL invasion of the brain parenchyma and damage to persistently infected neurons presenting LCMV epitopes on MHC-I [54, 59]. In this in vivo mouse model, CD8+ T cell interactions with antigen presenting neurons lead to secretion of IFN-γ, upregulation of neuronal nuclear pSTAT1, and loss of synaptic inputs [59]. More recent work demonstrated that viral infection and nuclear pSTAT1 lead to neuronal expression of CCL2, recruitment of CD8+ T cells, and phagocyte-mediated synapse engulfment [60]. The fulminant encephalitis and pathological features observed in this mouse model are strikingly similar to Rasmussen’s encephalitis [27, 28].
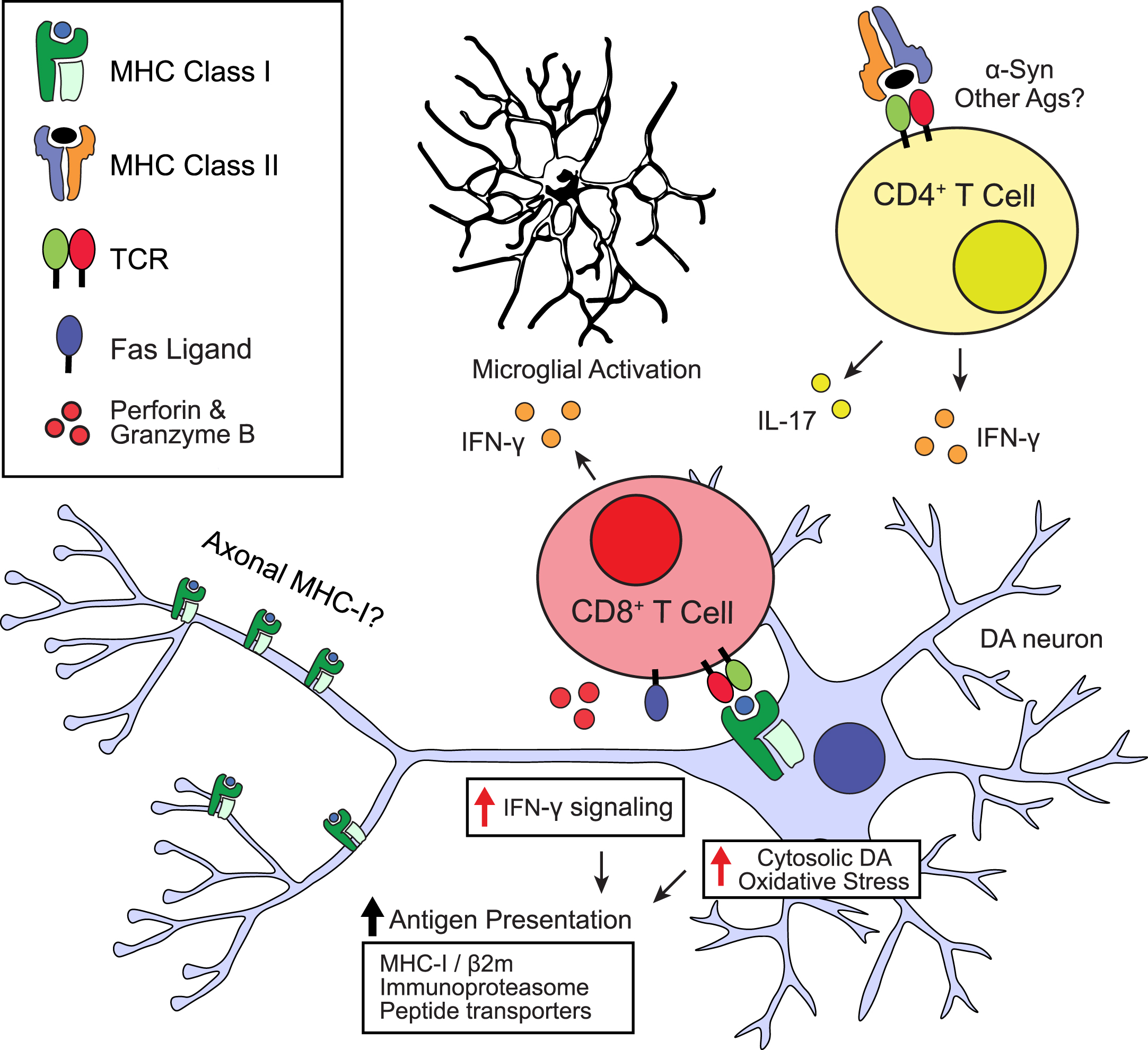
Fig. 1
Possible roles of neuronal antigen presentation in Parkinson’s disease. Schematic depicting potential stimuli leading to MHC-I upregulation and antigen presentation in dopamine neurons and possible downstream consequences. TCR, T cell receptor; DA, dopamine; Ags, antigens.
Beyond infectious disease, studies of neuronal antigen presentation in mouse models are more limited (Table 1). However, two recent studies demonstrated CD8+ T cell-mediated neuronal damage based on ectopic expression of influenza virus hemagglutinin (HA) as a neuronal ‘neo self-antigen’ [61, 62]. The first study introduced a model of paraneoplastic neurological disease based on expression of HA in both tumor cells and cerebellar Purkinje neurons [61]. Following CTLA4 blockade to induce anti-tumor T cell responses, HA-specific CD8+ T cells infiltrated the cerebellum and targeted HA:MHC-I+ Purkinje neurons, leading to neuroinflammation and cerebellar degeneration [61]. The second study developed a mouse model of narcolepsy based on the xpression of HA in orexin+ neurons in the lateral hypothalamus [62]. Adoptive transfer of HA-specific CD8+ T cells led to loss of orexinergic neurons and symptoms of narcolepsy [62]. Several features of this model are consistent with the detection of antigen-specific CD8+ T cells in the peripheral blood of Narcolepsy Type 1 patients [63].
Table 1
Key studies of neuronal MHC-I expression and/or antigen presentation in the adult brain
| Species | Region | Neuron | Context | Antigen | Ref. |
| Human | Locus coeruleus | Pigmented (Noradrenergic) | Normal aging &PD | Unknown | [19] |
| Human | Substantia nigra | Pigmented, (Dopaminergic) | Normal aging &PD | Unknown | [19] |
| Human | Grey matter | Unspecified | Measles infection (SSPE) | Unknown | [25] |
| Human | Brainstem, hippocampus, cerebellum, basal ganglia, medulla | Unspecified | Paraneoplastic encephalitis | Hu, Ma2 | [26] |
| Human | Temporal lobe | Unspecified | Rasmussen’s encephalitis | Unknown | [27] |
| Mouse | Substantia nigra | Dopaminergic | MPTP (PD model) | Unknown | [80] |
| Mouse | Ventral tegmental area | Dopaminergic | Normal aging &cocaine relapse model | Unknown | [21] |
| Mouse | Substantia nigra | Dopaminergic | Knockdown of alpha-synuclein | Unknown | [83] |
| Mouse | Hypothalamus | Orexinergic | Ectopic HA expr. (Narcolepsy model) | HA | [62] |
| Mouse | Cerebellum | Purkinje | Ectopic HA expr. (Paraneoplastic encephalitis model) | HA | [61] |
| Mouse | Cortex | Camk2a+ (Excitatory, Pyramidal) | T. Gondii infection | GRA6-OVA | [45] |
| Mouse | Cortex, hippocampus, basal ganglia, thalamus, hypothalamus, cerebellum, brain stem | Unspecified | Persistent LCMV infection | Db: NP396 | [54] |
| Mouse | Spinal cord | Medium to small (not α motor neurons) | Murine hepatitis virus infection | Unknown | [4] |
| Mouse | Thoracic ganglia | Sensory neurons | HSV infection | Unknown | |
| Mouse | Hippocampus, cortex, striatum, hypothalamus | Unspecified | TMEV infection | Db: VP2121 - 130 | [49] |
SSPE, subacute sclerosing panencephalitis; LCMV, lymphocytic choriomeningitis virus; Db, H2-Db allele of murine MHC Class I; TMEV, Theiler’s murine encephalomyelitis virus; HSV, Herpes simplex virus.
Recent studies have demonstrated a link between presentation of native mitochondria-derived antigens and the PD associated genes, PINK1 and parkin [64, 65]. In the absence of PINK1 and parkin, there is enhanced mitochondrial-derived vesicle cargo delivery to lysosomes and increased mitochondrial antigen presentation on MHC-I [64]. When Pink1–/– mice are infected with the intestinal pathogen Citrobacter rodentium, there is a clonal expansion of CD8+ T cells autoreactive to mitochondrial antigens derived from 2-oxoglutarate dehydrogenase (OGDH) [65]. Within 2–4 weeks of infection, OGDH-specific CD8+ T cells infiltrated the CNS, followed by a transient loss of dopaminergic axon terminals and motor symptoms at 6 months post-infection [65]. Future studies are required to determine whether neuronal presentation of OGDH antigens occurs in PD.
IN VITRO MODELS OF NEURONAL ANTIGEN PRESENTATION BY MHC-I
Early in vitro evidence that inflammatory conditions promote MHC-I expression in neurons came from treatment with IFN-γ and the sodium channel blocker tetrodotoxin [66, 67]. There appears to be a connection between MHC-I and electrical activity in some neurons, with multiple studies showed that electrical silencing by tetrodotoxin is required for MHC-I induction in neurons [66–68]. Dopamine neurons exhibit autonomous pacemaking activity [69] and would not typically be electrically silent in vivo, but disruption of pacemaking is often an early sign of dysfunction in models of PD [70, 71]. On the other hand, alterations in neuronal electrical activity may be a consequence of neuronal interactions with CTLs, rather than a step preceding MHC-I expression (reviewed in [39]). In hippocampal slices and neuronal cultures exposed to CD8+ T cells, release of perforin onto MHC-I+ neuronal membranes leads to shunting of membrane capacitance, electrical silencing, and elevated cytosolic Ca2 + [72].
Thus far, evidence for the direct interaction of CD8+ T cells with MHC-I+ axons and distal dendrites is largely limited to in vitro studies [68, 73]. In compartmentalized culture models, CTL interaction with MHC-I+ axons lead to axonal damage via a granzyme B-dependent mechanism [37] but did not cause degeneration of cell bodies [73, 74]. Interestingly, compartmentalized axonal exposure to IFN-γ led to retrograde signaling and upregulation of MHC-I [75]. Such retrograde signaling may be important for dopamine neurons, given that the majority of their surface area and volume lies in striatal axons [42]. In addition to cell bodies and dendrites, we found that MHC-I was localized to the axons of cultured mouse dopamine neurons [19]. Future studies are needed to investigate whether CTLs might interact with MHC-I on dopamine axons in vivo.
Dopamine neurons can be induced to express MHC-I by exposure to IFN-γ [19, 65]. After exposure to IFN-γ and either ovalbumin protein or the ovalbumin-derived peptide antigen, SIINFEKL, cultured postnatal mouse dopamine neurons are engaged and destroyed by SIINFEKL-specific CTLs from the OT-1 mouse [19, 76]. The presentation of MHC-I:SIINFEKL and neuronal cell death did not occur in neurons from β2 m–/– mice, which lack surface MHC-I expression [77]. Similar results were obtained when mouse dopamine neurons were loaded with OGDH peptide, stimulated with IFN-γ, and cultured with OGDH-specific CD8+ T cells [65]. CTL-mediated killing of dopamine neurons was blocked by inhibition of either the Fas/FasL or perforin/granzyme pathways [19]. Meanwhile, CTL-mediated damage occurred through the perforin pathway for cerebellar granule neurons [78], via a granzyme B-dependent mechanism for TMEV-infected neurons [49], and via Fas/FasL interactions for hippocampal neurons stimulated with IFN-γ/TTX [67]. Thus, multiple mechanisms of cytotoxic killing can be employed by CD8+ T cells that target neurons. Future studies are required to investigate the context-dependency for activation of these cytotoxic pathways.
All major proteins required for antigen processing and presentation harbor IFN-γ-responsive promoter elements, including MHC-I, β2 m, ER peptide transporters, and immunoproteasome subunits [79]. As noted above, neuronal MHC-I expression is most often observed following viral infection or by direct stimulation with IFN-γ. However, we found that cultured dopamine neurons challenged with high dose L-DOPA upregulate MHC-I, an effect likely due to elevated cytosolic dopamine and increased oxyradical stress [19] as well as neuromelanin synthesis (see above). Oxidative stress induced by MPP+/MPTP also induces dopamine neuronal MHC-I expression and parenchymal infiltration by CD8+ T cells [80]. Increased expression of the immunoproteasome subunit Lmp7 (Psmb8) has been observed in the SN of PD patients [81], and Lmp7 is upregulated in dopamine neurons in mouse models of dopaminergic toxicity (6-OHDA) [82] and alpha-synuclein aggregation [81]. Knockdown of endogenous alpha-synuclein in the midbrain of adult mice led to upregulation of MHC-I in dopamine neurons, recruitment of T cells, microglial activation, and loss of dopaminergic neurons [83]. Thus, upregulation of MHC-I antigen presentation machinery in dopamine neurons may be induced by a variety of cellular stress conditions in the absence of IFN-γ.
FUTURE DIRECTIONS: IS NEURONAL ANTIGEN PRESENTATION INVOLVED IN PARKINSON’S DISEASE?
With the exception of Rasmussen’s encephalitis, there is only indirect evidence for a role of neuronal antigen presentation in human disease, and there is much more to be done. An involvement of CD8+ T cells in diseases associated with older age is attractive, with recent studies showing an accumulation of clonally expanded CD8+ T cells in the aging mouse brain [84] and in the brain of patients with Alzheimer’s disease [85].
Nevertheless, multiple models of synucleinopathies show that CD4+ T cells that interact with MHC-II may be more responsible for neuronal death than CD8+ T cells, both in the central nervous system [33, 86–89], and enteric nervous system (Garretti et al., unpublished data). While some PD patients possess CD8+ T cells that recognize alpha-synuclein epitopes presented by MHC-I, most alpha-synuclein-reactive T cells are CD4+ and recognize epitopes presented by MHC-II [90–92]. Overexpression of beta-synuclein in neurons can lead to T cell-mediated gray matter damage in a rodent model of multiple sclerosis, but this damage appears to be mostly driven by CD4+ T cells, and only CD4+ T cells that recognize beta-synuclein were reported in MS patients and controls [92]. Consistent with these findings, genome wide association studies have identified PD risk variants linked to haplotypes of MHC-II genes [93, 94], including the DRB1*15:01 and DRB5*01:01 alleles that present specific alpha-synuclein epitopes [90].
Current evidence suggests that CD4+ T cell-mediated neuropathology is driven by interactions with microglia or monocytes [89]. A study on dopamine neurons derived from human induced pluripotent stem cells suggests that IL-17 producing CD4+ T cells may play an important role in neuronal death [95], with similar findings recently reported in postmortem studies of Lewy body disease [37]. Given that CD4+ T cells release IFN-γ or additional cytokines that may drive neuronal MHC-I expression, multiple steps may be involved in putative autoimmunity directed towards alpha-synuclein epitopes.
In contrast, the report that clonally expanded, mitochondrial antigen-specific CD8+ T cells can transiently damage substantia nigra neurons in PARK1 deficient mice [65] suggests that direct attack of CD8+ T cells on antigen presenting neurons may occur in some conditions. It is likely that many other unknown autoimmune and foreign antigens are presented by neurons during disease. Moreover, the aforementioned study suggests that axons may be particularly susceptible to such attack, and it will be important to study the timing and localization of MHC-I display, particularly for the long-range projection neurons that are the primary targets of many neuropsychiatric disorders and neurodegenerative diseases.
Much of the research on antigen presentation in neurodegeneration has been devoted to synucleinopathies, particularly PD. However, the detection of CD8+ T cell reactivity towards narcolepsy-associated antigens in Narcolepsy Type 1 patients [63] suggests a role of antigen presentation in multiple brain disorders. Indeed, as more autoimmune features of CNS disorders are identified [96], neuronal antigen presentation may be a key piece of the puzzle surrounding inflammation in neuronal disease.
ACKNOWLEDGMENTS
This work was funded in part by Aligning Science Across Parkinson’s [ASAP-000375] (DS) through the Michael J. Fox Foundation for Parkinson’s Research (MJFF). For the purpose of open access, the author has applied a CC BY public copyright license to all Author Accepted Manuscripts arising from this submission. This work was supported by the JPB Foundation (DS). This work was supported by NIH grants F30 DA047775-04 (BDH), R01 NS0954 (DS), R01 DA07418 (DS), and R01 MH122470 (DS).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Neefjes JJ , Momburg F ((1993) ) Cell biology of antigen presentation. Curr Opin Immunol 5: , 27–34. |
[2] | O’Garra A , Murphy K ((1993) ) T-cell subsets in autoimmunity. Curr Opin Immunol 5: , 880–886. |
[3] | Burgdorf S , Kurts C ((2008) ) Endocytosis mechanisms and the cell biology of antigen presentation. Curr Opin Immunol 20: , 89–95. |
[4] | Redwine JM , Buchmeier MJ , Evans CF ((2001) ) In vivo expression of major histocompatibility complex molecules on oligodendrocytes and neurons during viral infection. Am J Pathol 159: , 1219–1224. |
[5] | Corriveau RA , Huh GS , Shatz CJ ((1998) ) Regulation of class I MHC gene expression in the developing and mature CNS by neural activity. Neuron 21: , 505–520. |
[6] | Huh GS , Boulanger LM , Du H , Riquelme PA , Brotz TM , Shatz CJ ((2000) ) Functional requirement for class I MHC in CNS development and plasticity. Science 290: , 2155–2159. |
[7] | Djurisic M , Vidal GS , Mann M , Aharon A , Kim T , Santos AF , Zuo Y , Hübener M , Shatz CJ ((2013) ) PirB regulates a structural substrate for cortical plasticity. Proc Natl Acad Sci U S A 110: , 20771–20776. |
[8] | Goddard CA , Butts DA , Shatz CJ ((2007) ) Regulation of CNS synapses by neuronal MHC class I. Proc Natl Acad Sci U S A 104: , 6828–6833. |
[9] | Glynn MW , Elmer BM , Garay PA , Liu X-B , Needleman LA , El-Sabeawy F , McAllister AK ((2011) ) MHCI negatively regulates synapse density during the establishment of cortical connections. Nat Neurosci 14: , 442–451. |
[10] | Zhang A , Yu H , He Y , Shen Y , Zhang Y , Liu J , Fu B , Lv D , Miao F , Zhang J ((2015) ) Developmental expression and localization of MHC class I molecules in the human central nervous system. Exp Brain Res 233: , 2733–2743. |
[11] | Zhang A , Yu H , Shen Y , Liu J , He Y , Shi Q , Fu B , Miao F , Zhang J ((2013) ) The expression patterns of MHC class I molecules in the developmental human visual system. Neurochem Res 38: , 273–281. |
[12] | Zhang A , Yu H , He Y , Shen Y , Pan N , Liu J , Fu B , Miao F , Zhang J ((2013) ) The spatio-temporal expression of MHC class I molecules during human hippocampal formation development. Brain Res 1529: , 26–38. |
[13] | Wong GHW , Bartlett PF , Clark-Lewis I , Battye F , Schrader JW ((1984) ) Inducible expression of H–2 and Ia antigens on brain cells. Nature 310: , 688–691. |
[14] | Vass K , Lassmann H ((1990) ) Intrathecal application of interferon gamma. Progressive appearance of MHC antigens within the rat nervous system. Am J Pathol 137: , 789–800. |
[15] | Lampson LA , Hickey WF ((1986) ) Monoclonal antibody analysis of MHC expression in human brain biopsies: Tissue ranging from “histologically normal” to that showing different levels of glial tumor involvement. J Immunol 136: , 4054–4062. |
[16] | Rall GF ((1998) ) CNS neurons: The basis and benefits of low class I major histocompatibility complex expression. In Antigen Presentation, WhittonJL, ed. Springer, Berlin, Heidelberg, pp. 115–134. |
[17] | Joly E , Mucke L , Oldstone MB ((1991) ) Viral persistence in neurons explained by lack of major histocompatibility class I expression. Science 253: , 1283–1285. |
[18] | Joly E , Oldstone MBA ((1992) ) Neuronal cells are deficient in loading peptides onto MHC class I molecules. Neuron 8: , 1185–1190. |
[19] | Cebrián C , Zucca FA , Mauri P , Steinbeck JA , Studer L , Scherzer CR , Kanter E , Budhu S , Mandelbaum J , Vonsattel JP , Zecca L , Loike JD , Sulzer D ((2014) ) MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat Commun 5: , 3633. |
[20] | Sulzer D , Mosharov E , Talloczy Z , Zucca FA , Simon JD , Zecca L ((2008) ) Neuronal pigmented autophagic vacuoles: Lipofuscin, neuromelanin, and ceroid as macroautophagic responses during aging and disease. J Neurochem 106: , 24–36. |
[21] | Murakami G , Edamura M , Furukawa T , Kawasaki H , Kosugi I , Fukuda A , Iwashita T , Nakahara D ((2018) ) MHC class I in dopaminergic neurons suppresses relapse to reward seeking. Sci Adv 4: , eaap7388. |
[22] | Lindå H , Hammarberg H , Piehl F , Khademi M , Olsson T ((1999) ) Expression of MHC class I heavy chain and beta2-microglobulin in rat brainstem motoneurons and nigral dopaminergic neurons. J Neuroimmunol 101: , 76–86. |
[23] | Murphy C , Nikodem D , Howcroft K , Weissman JD , Singer DS ((1996) ) Active repression of major histocompatibility complex class I genes in a human neuroblastoma cell line. J Biol Chem 271: , 30992–30999. |
[24] | Massa PT , Ozato K , McFarlin DE ((1993) ) Cell type-specific regulation of major histocompatibility complex (MHC) class I gene expression in astrocytes, oligodendrocytes, and neurons. Glia 8: , 201–207. |
[25] | Gogate N , Swoveland P , Yamabe T , Verma L , Woyciechowska J , Tarnowska-Dziduszko E , Dymecki J , Dhib-Jalbut S ((1996) ) Major histocompatibility complex class I expression on neurons in subacute sclerosing panencephalitis and experimental subacute measles encephalitis. J Neuropathol Exp Neurol 55: , 435–443. |
[26] | Bien CG , Vincent A , Barnett MH , Becker AJ , Blümcke I , Graus F , Jellinger KA , Reuss DE , Ribalta T , Schlegel J , Sutton I , Lassmann H , Bauer J ((2012) ) Immunopathology of autoantibody-associated encephalitides: Clues for pathogenesis. Brain 135: , 1622–1638. |
[27] | Bien CG , Bauer J , Deckwerth TL , Wiendl H , Deckert M , Wiestler OD , Schramm J , Elger CE , Lassmann H ((2002) ) Destruction of neurons by cytotoxic T cells: A new pathogenic mechanism in Rasmussen’s encephalitis. Ann Neurol 51: , 311–318. |
[28] | Schwab N , Bien CG , Waschbisch A , Becker A , Vince GH , Dornmair K , Wiendl H ((2009) ) CD8+T-cell clones dominate brain infiltrates in Rasmussen encephalitis and persist in the periphery. Brain 132: , 1236–1246. |
[29] | Bauer J , Elger CE , Hans VH , Schramm J , Urbach H , Lassmann H , Bien CG ((2007) ) Astrocytes are a specific immunological target in Rasmussen’s encephalitis. Ann Neurol 62: , 67–80. |
[30] | Schneider-Hohendorf T , Mohan H , Bien CG , Breuer J , Becker A , Görlich D , Kuhlmann T , Widman G , Herich S , Elpers C , Melzer N , Dornmair K , Kurlemann G , Wiendl H , Schwab N ((2016) ) CD8(+) T-cell pathogenicity in Rasmussen encephalitis elucidated by large-scale T-cell receptor sequencing. Nat Commun 7: , 11153. |
[31] | Sulzer D , Antonini A , Leta V , Nordvig A , Smeyne RJ , Goldman JE , Al-Dalahmah O , Zecca L , Sette A , Bubacco L , Meucci O , Moro E , Harms AS , Xu Y , Fahn S , Ray Chaudhuri K ((2020) ) COVID-19 and possible links with Parkinson’s disease and parkinsonism: From bench to bedside. NPJ Parkinsons Dis 6: , 18. |
[32] | Smeyne RJ , Noyce AJ , Byrne M , Savica R , Marras C ((2021) ) Infection and risk of Parkinson’s disease. J Parkinsons Dis 11: , 31–43. |
[33] | Brochard V , Combadière B , Prigent A , Laouar Y , Perrin A , Beray-Berthat V , Bonduelle O , Alvarez-Fischer D , Callebert J , Launay J-M , Duyckaerts C , Flavell RA , Hirsch EC , Hunot S ((2009) ) Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 119: , 182–192. |
[34] | Galiano-Landeira J , Torra A , Vila M , Bové J ((2020) ) CD8 T cell nigral infiltration precedes synucleinopathy in early stages of Parkinson’s disease. Brain 143: , 3717–3733. |
[35] | Iba M , Kim C , Sallin M , Kwon S , Verma A , Overk C , Rissman RA , Sen R , Sen JM , Masliah E ((2020) ) Neuroinflammation is associated with infiltration of T cells in Lewy body disease and α-synuclein transgenic models. J Neuroinflammation 17: , 214. |
[36] | Williams GP , Marmion DJ , Schonhoff AM , Jurkuvenaite A , Won W-J , Standaert DG , Kordower JH , Harms AS ((2020) ) T cell infiltration in both human multiple system atrophy and a novel mouse model of the disease. Acta Neuropathol 139: , 855–874. |
[37] | Gate D , Tapp E , Leventhal O , Shahid M , Nonninger TJ , Yang AC , Strempfl K , Unger MS , Fehlmann T , Oh H , Channappa D , Henderson VW , Keller A , Aigner L , Galasko DR , Davis MM , Poston KL , Wyss-Coray T ((2021) ) CD4+T cells contribute to neurodegeneration in Lewy body dementia. Science 374: , 868–874. |
[38] | Lindestam Arlehamn CS , Dhanwani R , Pham J , Kuan R , Frazier A , Rezende Dutra J , Phillips E , Mallal S , Roederer M , Marder KS , Amara AW , Standaert DG , Goldman JG , Litvan I , Peters B , Sulzer D , Sette A ((2020) ) α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat Commun 11: , 1875. |
[39] | Melzer N , Meuth SG , Wiendl H ((2009) ) CD8+T cells and neuronal damage: Direct and collateral mechanisms of cytotoxicity and impaired electrical excitability. FASEB J 23: , 3659–3673. |
[40] | Poh MQW , Simon NG , Buckland ME , Salisbury E , Watson S ((2014) ) Evidence of T-cell mediated neuronal injury in stiff-person syndrome with anti-amphiphysin antibodies. J Neurol Sci 337: , 235–237. |
[41] | Hobson BD , Choi SJ , Soni RK , Sulzer D , Sims PA ((2021) ) Subcellular proteomics of dopamine neurons in the mouse brain reveals axonal enrichment of proteins encoded by Parkinson’s disease-linked genes. bioRxiv 2021.06.01.446584. |
[42] | Matsuda W , Furuta T , Nakamura KC , Hioki H , Fujiyama F , Arai R , Kaneko T ((2009) ) Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci 29: , 444–453. |
[43] | Planz O , Bilzer T , Sobbe M , Stitz L ((1993) ) Lysis of major histocompatibility complex class I-bearing cells in Borna disease virus-induced degenerative encephalopathy. J Exp Med 178: , 163–174. |
[44] | Chevalier G , Suberbielle E , Monnet C , Duplan V , Martin-Blondel G , Farrugia F , Le Masson G , Liblau R , Gonzalez-Dunia D ((2011) ) Neurons are MHC class I-dependent targets for CD8 T cells upon neurotropic viral infection. PLoS Pathog 7: , e1002393. |
[45] | Salvioni A , Belloy M , Lebourg A , Bassot E , Cantaloube-Ferrieu V , Vasseur V , Blanié S , Liblau RS , Suberbielle E , Robey EA , Blanchard N ((2019) ) Robust control of a brain-persisting parasite through MHC I presentation by infected neurons. Cell Rep 27: , 3254–3268.e8. |
[46] | Maehlen J , Nennesmo I , Olsson AB , Olsson T , Schröder HD , Kristensson K ((1989) ) Peripheral nerve injury causes transient expression of MHC class I antigens in rat motor neurons and skeletal muscles. Brain Res 481: , 368–372. |
[47] | Pereira RA , Simmons A ((1999) ) Cell surface expression of H2 antigens on primary sensory neurons in response to acute but not latent herpes simplex virus infection in vivo. J Virol 73: , 6484–6489. |
[48] | Kimura T , Griffin DE ((2000) ) The role of CD8+T cells and major histocompatibility complex class I expression in the central nervous system of mice infected with neurovirulent Sindbis virus. J Virol 74: , 6117–6125. |
[49] | McDole JR , Danzer SC , Pun RYK , Chen Y , Johnson HL , Pirko I , Johnson AJ ((2010) ) Rapid formation of extended processes and engagement of Theiler’s virus-infected neurons by CNS-infiltrating CD8 T cells. Am J Pathol 177: , 1823–1833. |
[50] | Keane RW , Tallent MW , Podack ER ((1992) ) Resistance and susceptibility of neural cells to lysis by cytotoxic lymphocytes and by cytolytic granules. Transplantation 54: , 520–526. |
[51] | Rivera-Quiñones C , McGavern D , Schmelzer JD , Hunter SF , Low PA , Rodriguez M ((1998) ) Absence of neurological deficits following extensive demyelination in a class I-deficient murine model of multiple sclerosis. Nat Med 4: , 187–193. |
[52] | Bonthius DJ , Perlman S ((2007) ) Congenital viral infections of the brain: Lessons learned from lymhocytic choriomeningitis virus in the neonatal rat. PLoS Pathog 3: , e149. |
[53] | Fazakerley JK , Southern P , Bloom F , Buchmeier MJ ((1991) ) High resolution in situ hybridization to determine the cellular distribution of lymphocytic choriomeningitis virus RNA in the tissues of persistently infected mice: Relevance to arenavirus disease and mechanisms of viral persistence. J Gen Virol 72 (Pt 7): , 1611–1625. |
[54] | Merkler D , Horvath E , Bruck W , Zinkernagel RM , Torre JC del la , Pinschewer DD ((2006) ) “Viral déjá vu” elicits organ-specific immune disease independent of reactivity to self. J Clin Invest 116: , 1254–1263. |
[55] | Rodriguez M , Buchmeier MJ , Oldstone MB , Lampert PW ((1983) ) Ultrastructural localization of viral antigens in the CNS of mice persistently infected with lymphocytic choriomeningitis virus (LCMV). Am J Pathol 110: , 95–100. |
[56] | Oldstone MB , Blount P , Southern PJ , Lampert PW ((1986) ) Cytoimmunotherapy for persistent virus infection reveals a unique clearance pattern from the central nervous system. Nature 321: , 239–243. |
[57] | Tishon A , Eddleston M , de la Torre JC , Oldstone MB ((1993) ) Cytotoxic T lymphocytes cleanse viral gene products from individually infected neurons and lymphocytes in mice persistently infected with lymphocytic choriomeningitis virus. Virology 197: , 463–467. |
[58] | Rall GF , Mucke L , Oldstone MB ((1995) ) Consequences of cytotoxic T lymphocyte interaction with major histocompatibility complex class I-expressing neurons in vivo. J Exp Med 182: , 1201–1212. |
[59] | Kreutzfeldt M , Bergthaler A , Fernandez M , Brück W , Steinbach K , Vorm M , Coras R , Blümcke I , Bonilla WV , Fleige A , Forman R , Müller W , Becher B , Misgeld T , Kerschensteiner M , Pinschewer DD , Merkler D ((2013) ) Neuroprotective intervention by interferon-γ blockade prevents CD8+T cell–mediated dendrite and synapse loss. J Exp Med 210: , 2087–2103. |
[60] | Di Liberto G , Pantelyushin S , Kreutzfeldt M , Page N , Musardo S , Coras R , Steinbach K , Vincenti I , Klimek B , Lingner T , Salinas G , Lin-Marq N , Staszewski O , Costa Jordão MJ , Wagner I , Egervari K , Mack M , Bellone C , Blümcke I , Prinz M , Pinschewer DD , Merkler D ((2018) ) Neurons under T cell attack coordinate phagocyte-mediated synaptic stripping. Cell 175: , 458–471.e19. |
[61] | Yshii LM , Gebauer CM , Pignolet B , Mauré E , Quériault C , Pierau M , Saito H , Suzuki N , Brunner-Weinzierl M , Bauer J , Liblau R ((2016) ) CTLA4 blockade elicits paraneoplastic neurological disease in a mouse model. Brain 139: , 2923–2934. |
[62] | Bernard-Valnet R , Yshii L , Quériault C , Nguyen X-H , Arthaud S , Rodrigues M , Canivet A , Morel A-L , Matthys A , Bauer J , Pignolet B , Dauvilliers Y , Peyron C , Liblau RS ((2016) ) CD8 T cell-mediated killing of orexinergic neurons induces a narcolepsy-like phenotype in mice. Proc Natl Acad Sci U S A 113: , 10956–10961. |
[63] | Pedersen NW , Holm A , Kristensen NP , Bjerregaard A-M , Bentzen AK , Marquard AM , Tamhane T , Burgdorf KS , Ullum H , Jennum P , Knudsen S , Hadrup SR , Kornum BR ((2019) ) CD8+T cells from patients with narcolepsy and healthy controls recognize hypocretin neuron-specific antigens. Nat Commun 10: , 837. |
[64] | Matheoud D , Sugiura A , Bellemare-Pelletier A , Laplante A , Rondeau C , Chemali M , Fazel A , Bergeron JJ , Trudeau L-E , Burelle Y , Gagnon E , McBride HM , Desjardins M ((2016) ) Parkinson’s disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell 166: , 314–327. |
[65] | Matheoud D , Cannon T , Voisin A , Penttinen A-M , Ramet L , Fahmy AM , Ducrot C , Laplante A , Bourque M-J , Zhu L , Cayrol R , Le Campion A , McBride HM , Gruenheid S , Trudeau L-E , Desjardins M ((2019) ) Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1–/– mice. Nature 571: , 565–569. |
[66] | Neumann H , Cavalié A , Jenne DE , Wekerle H ((1995) ) Induction of MHC class I genes in neurons. Science 269: , 549–552. |
[67] | Medana IM , Gallimore A , Oxenius A , Martinic MM , Wekerle H , Neumann H ((2000) ) MHC class I-restricted killing of neurons by virus-specific CD8+T lymphocytes is effected through the Fas/FasL, but not the perforin pathway. Eur J Immunol 30: , 3623–3633. |
[68] | Medana I , Martinic MA , Wekerle H , Neumann H ((2001) ) Transection of major histocompatibility complex class I-induced neurites by cytotoxic T lymphocytes. Am J Pathol 159: , 809–815. |
[69] | Grace AA , Bunney BS ((1984) ) The control of firing pattern in nigral dopamine neurons: Single spike firing. J Neurosci 4: , 2866–2876. |
[70] | Subramaniam M , Althof D , Gispert S , Schwenk J , Auburger G , Kulik A , Fakler B , Roeper J ((2014) ) Mutant α-synuclein enhances firing frequencies in dopamine substantia nigra neurons by oxidative impairment of A-type potassium channels. J Neurosci 34: , 13586–13599. |
[71] | Tozzi A , Sciaccaluga M , Loffredo V , Megaro A , Ledonne A , Cardinale A , Federici M , Bellingacci L , Paciotti S , Ferrari E , La Rocca A , Martini A , Mercuri NB , Gardoni F , Picconi B , Ghiglieri V , De Leonibus E , Calabresi P ((2021) ) Dopamine-dependent early synaptic and motor dysfunctions induced by α-synuclein in the nigrostriatal circuit. Brain 144: , 3477–3491. |
[72] | Meuth SG , Herrmann AM , Simon OJ , Siffrin V , Melzer N , Bittner S , Meuth P , Langer HF , Hallermann S , Boldakowa N , Herz J , Munsch T , Landgraf P , Aktas O , Heckmann M , Lessmann V , Budde T , Kieseier BC , Zipp F , Wiendl H ((2009) ) Cytotoxic CD8+T cell–neuron interactions: Perforin-dependent electrical silencing precedes but is not causally linked to neuronal cell death. J Neurosci 29: , 15397–15409. |
[73] | Sauer BM , Schmalstieg WF , Howe CL ((2013) ) Axons are injured by antigen-specific CD8(+) T cells through a MHC class I- and granzyme B-dependent mechanism. Neurobiol Dis 59: , 194–205. |
[74] | Manning PT , Johnson EM , Wilcox CL , Palmatier MA , Russell JH ((1987) ) MHC-specific cytotoxic T lymphocyte killing of dissociated sympathetic neuronal cultures. Am J Pathol 128: , 395–409. |
[75] | Clarkson BDS , Patel MS , LaFrance-Corey RG , Howe CL ((2018) ) Retrograde interferon-gamma signaling induces major histocompatibility class I expression in human-induced pluripotent stem cell-derived neurons. Ann Clin Transl Neurol 5: , 172–185. |
[76] | Moore MW , Carbone FR , Bevan MJ ((1988) ) Introduction of soluble protein into the class I pathway of antigen processing and presentation. Cell 54: , 777–785. |
[77] | Koller BH , Marrack P , Kappler JW , Smithies O ((1990) ) Normal development of mice deficient in beta 2M, MHC class I proteins, and CD8+T cells. Science 248: , 1227–1230. |
[78] | Rensing-Ehl A , Malipiero U , Irmler M , Tschopp J , Constam D , Fontana A ((1996) ) Neurons induced to express major histocompatibility complex class I antigen are killed via the perforin and not the Fas (APO-1/CD95) pathway. Eur J Immunol 26: , 2271–2274. |
[79] | van den Elsen PJ , Holling TM , Kuipers HF , van der Stoep N ((2004) ) Transcriptional regulation of antigen presentation. Curr Opin Immunol 16: , 67–75. |
[80] | Wang B-Y , Ye Y-Y , Qian C , Zhang H-B , Mao H-X , Yao L-P , Sun X , Lu G-H , Zhang S-Z ((2021) ) Stress increases MHC-I expression in dopaminergic neurons and induces autoimmune activation in Parkinson’s disease. Neural Regen Res 16: , 2521–2527. |
[81] | Ugras S , Daniels MJ , Fazelinia H , Gould NS , Yocum AK , Luk KC , Luna E , Ding H , McKennan C , Seeholzer S , Martinez D , Evans P , Brown D , Duda JE , Ischiropoulos H ((2018) ) Induction of the immunoproteasome subunit Lmp7 links proteostasis and immunity in α-synuclein aggregation disorders. EBioMedicine 31: , 307–319. |
[82] | Mo M-S , Li G-H , Sun C-C , Huang S-X , Wei L , Zhang L-M , Zhou M-M , Wu Z-H , Guo W-Y , Yang X-L , Chen C-J , Qu S-G , He J-X , Xu P-Y ((2018) ) Dopaminergic neurons show increased low-molecular-mass protein 7 activity induced by 6-hydroxydopamine in vitro and in vivo. Transl Neurodegener 7: , 19. |
[83] | Benskey MJ , Sellnow RC , Sandoval IM , Sortwell CE , Lipton JW , Manfredsson FP ((2018) ) Silencing alpha synuclein in mature nigral neurons results in rapid neuroinflammation and subsequent toxicity. Front Mol Neurosci 11: , 36. |
[84] | Dulken BW , Buckley MT , Navarro Negredo P , Saligrama N , Cayrol R , Leeman DS , George BM , Boutet SC , Hebestreit K , Pluvinage JV , Wyss-Coray T , Weissman IL , Vogel H , Davis MM , Brunet A ((2019) ) Single-cell analysis reveals T cell infiltration in old neurogenic niches. Nature 571: , 205–210. |
[85] | Gate D , Saligrama N , Leventhal O , Yang AC , Unger MS , Middeldorp J , Chen K , Lehallier B , Channappa D , De Los Santos MB , McBride A , Pluvinage J , Elahi F , Tam GK-Y , Kim Y , Greicius M , Wagner AD , Aigner L , Galasko DR , Davis MM , Wyss-Coray T ((2020) ) Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577: , 399–404. |
[86] | Williams GP , Schonhoff AM , Jurkuvenaite A , Gallups NJ , Standaert DG , Harms AS ((2021) ) CD4 T cells mediate brain inflammation and neurodegeneration in a mouse model of Parkinson’s disease. Brain 144: , 2047–2059. |
[87] | Qin H , Buckley JA , Li X , Liu Y , Fox TH , Meares GP , Yu H , Yan Z , Harms AS , Li Y , Standaert DG , Benveniste EN ((2016) ) Inhibition of the JAK/STAT pathway protects against α-synuclein-induced neuroinflammation and dopaminergic neurodegeneration. J Neurosci 36: , 5144–5159. |
[88] | Subbarayan MS , Hudson C , Moss LD , Nash KR , Bickford PC ((2020) ) T cell infiltration and upregulation of MHCII in microglia leads to accelerated neuronal loss in an α-synuclein rat model of Parkinson’s disease. J Neuroinflammation 17: , 242. |
[89] | Harms AS , Cao S , Rowse AL , Thome AD , Li X , Mangieri LR , Cron RQ , Shacka JJ , Raman C , Standaert DG ((2013) ) MHCII is required for α-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J Neurosci 33: , 9592–9600. |
[90] | Sulzer D , Alcalay RN , Garretti F , Cote L , Kanter E , Agin-Liebes J , Liong C , McMurtrey C , Hildebrand WH , Mao X , Dawson VL , Dawson TM , Oseroff C , Pham J , Sidney J , Dillon MB , Carpenter C , Weiskopf D , Phillips E , Mallal S , Peters B , Frazier A , Lindestam Arlehamn CS , Sette A ((2017) ) T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 546: , 656–661. |
[91] | Singhania A , Pham J , Dhanwani R , Frazier A , Rezende Dutra J , Marder KS , Phillips E , Mallal S , Amara AW , Standaert DG , Sulzer D , Peters B , Sette A , Lindestam Arlehamn CS ((2021) ) The TCR repertoire of α-synuclein-specific T cells in Parkinson’s disease is surprisingly diverse. Sci Rep 11: , 302. |
[92] | Lodygin D , Hermann M , Schweingruber N , Flügel-Koch C , Watanabe T , Schlosser C , Merlini A , Körner H , Chang H-F , Fischer HJ , Reichardt HM , Zagrebelsky M , Mollenhauer B , Kügler S , Fitzner D , Frahm J , Stadelmann C , Haberl M , Odoardi F , Flügel A ((2019) ) β-Synuclein-reactive T cells induce autoimmune CNS grey matter degeneration. Nature 566: , 503–508. |
[93] | Hamza TH , Zabetian CP , Tenesa A , Laederach A , Montimurro J , Yearout D , Kay DM , Doheny KF , Paschall J , Pugh E , Kusel VI , Collura R , Roberts J , Griffith A , Samii A , Scott WK , Nutt J , Factor SA , Payami H ((2010) ) Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet 42: , 781–785. |
[94] | Wissemann WT , Hill-Burns EM , Zabetian CP , Factor SA , Patsopoulos N , Hoglund B , Holcomb C , Donahue RJ , Thomson G , Erlich H , Payami H ((2013) ) Association of Parkinson disease with structural and regulatory variants in the HLA region. Am J Hum Genet 93: , 984–993. |
[95] | Sommer A , Marxreiter F , Krach F , Fadler T , Grosch J , Maroni M , Graef D , Eberhardt E , Riemenschneider MJ , Yeo GW , Kohl Z , Xiang W , Gage FH , Winkler J , Prots I , Winner B ((2018) ) Th17 lymphocytes induce neuronal cell death in a human iPSC-based model of Parkinson’s disease. Cell Stem Cell 23: , 123–131.e6. |
[96] | Geis C , Planagumá J , Carreño M , Graus F , Dalmau J ((2019) ) Autoimmune seizures and epilepsy. J Clin Invest 129: , 926–940. |


