
Intraperitoneal Administration of Forskolin Reverses Motor Symptoms and Loss of Midbrain Dopamine Neurons in PINK1 Knockout Rats
Abstract
Background:
Parkinson’s disease (PD) is a relentless, chronic neurodegenerative disease characterized by the progressive loss of substantia nigra (SN) neurons that leads to the onset of motor and non-motor symptoms. Standard of care for PD consists of replenishing the loss of dopamine through oral administration of Levodopa; however, this treatment is not disease-modifying and often induces intolerable side effects. While the etiology that contributes to PD is largely unknown, emerging evidence in animal models suggests that a significant reduction in neuroprotective Protein Kinase A (PKA) signaling in the SN contributes to PD pathogenesis, suggesting that restoring PKA signaling in the midbrain may be a new anti-PD therapeutic alternative.
Objective:
We surmised that pharmacological activation of PKA via intraperitoneal administration of Forskolin exerts anti-PD effects in symptomatic PTEN-induced kinase 1 knockout (PINK1-KO), a bona fide in vivo model of PD.
Methods:
By using a beam balance and a grip strength analyzer, we show that Forskolin reverses motor symptoms and loss of hindlimb strength with long-lasting therapeutic effects (> 5 weeks) following the last dose.
Results:
In comparison, intraperitoneal treatment with Levodopa temporarily (24 h) reduces motor symptoms but unable to restore hindlimb strength in PINK1-KO rats. By using immunohistochemistry and an XF24e BioAnalyzer, Forskolin treatment reverses SN neurons loss, elevates brain energy production and restores PKA activity in SN in symptomatic PINK1-KO rats.
Conclusion:
Overall, our collective in vivo data suggest that Forskolin is a promising disease-modifying therapeutic alternative for PD and is superior to Levodopa because it confers long-lasting therapeutic effects.
INTRODUCTION
Among all the neurodegenerative diseases, Parkinson’s disease (PD) is the second-highest in terms of worldwide prevalence [1]. Its incidence and prevalence are increasing with a frequency expected to double by 2040 [2]. The economic, emotional, and physical toll from the individual and the societal perspective has proven immense. Economically speaking, it is estimated that the total financial burden of PD treatment and management will exceed 79 billion by 2037 [3]. Motor symptoms typically manifest in PD patients when more than 90 % of dopaminergic neurons die, leading to a significant reduction in the production of the neurotransmitter dopamine. A diagnosis of PD is made clinically, usually presenting with a unique set of motor symptoms (MS), including resting tremor, postural instability, slow gait with muscle stiffness [4], and early emotional impairment [5]. In addition, the accompanying non-motor symptoms (NMS) are substantial and include features such as anxiety, chronic fatigue, massive constipation, decreased REM sleep, apathy, and major clinical depression with bouts of anxiety as the primary emotional manifestations [6]. The pathogenesis of PD involves gene mutations that contribute to approximately 10% of all PD cases and environmental factors, including exposure to mitochondrial-specific toxins [7]. Indeed, genetic mutations in over more than 18 PD-associated genes encode for proteins that regulate mitochondrial function, lysosomal function, antioxidant sensing pathways, mitochondrial turnover, and the ubiquitin-proteasome pathway [7]. The discovery that mutations in PTEN-induced kinase 1 (PINK1), an atypical serine/threonine (ser/thr) kinase that modulates mitochondrial function/structure, leads to the second most common, autosomal-recessive, juvenile form of PD revealed that mitochondrial dysfunction and disruption in kinase signaling underlies the etiology of PD [8, 9]. Mitochondrial dysfunction and oxidative stress contribute to ubiquitin-proteasome system failure, which contributes to the misfolding of α-synuclein and protein aggregation [10–12]. It is this impairment of protein clearance (associated with dysfunction of the ubiquitin-proteasome and autophagy-lysosomal systems) that leads to the accumulation of intracellular protein aggregates termed Lewy bodies which can contribute to dopaminergic neuronal death [10, 11]. A reduction in neuroprotective PKA and neurotrophic signaling in the brain may contribute to etiology in PD [13]. Recently, our research group discovered that a decrease in the level of brain-derived neurotrophic factor (BNDF) and in the activity of Protein Kinase A (PKA)—a well-characterized ser/thr kinase that modulates critical neuronal functions, including neuronal plasticity, survival, and development- contributes to mitochondrial dysfunction, oxidative stress and loss of dendritic arbors in midbrain dopamine neurons in PINK1-deficient rodents [13, 14]. In aggregate, these observations in experimental animal models of PD suggest that restoring PKA signaling in the midbrain may be a therapeutic alternative to reverse the loss of SN neurons in PD.
Unfortunately, standard of care for PD does not alter the course of the disease [15]. In addition to the non-disease modifying nature of PD therapies, which consists of replenishing the loss in dopamine through oral or intramuscular administration of Levodopa, as well as providing other pharmaceutical alternatives to PD patients who are resistant to Levodopa treatment, including anticholinergics agents, dopaminergic agonists, catechol o-methyl transferase (COMT) inhibitors, and inhibitors of MAO-B, induce significant side effect profiles resulting from the delivery of dopamine to extra-striatal regions [16, 17]. Recent developments in the treatment of PD have explored a host of therapeutic alternatives, ranging from providing neurotrophic factors, gene-mediated regenerative therapies to drug repurposing existing FDA-approved drugs [18]. However, many of these therapeutic pursuits are invasive and carry their risks. Meanwhile, the development of more effective, non-invasive, and disease-modifying agents has never been more urgent.
Forskolin is a labdane-type diterpenoid made within the plant Coleus forskohlii, native to India and Southeast Asia. The compound itself is synthesized within the cells that contain structures known as oil bodies which are themselves stored inside cells within the bark of the root [19, 20 1981]. Interestingly, as an efficient adenylate cyclase activator that elevates the intracellular levels of cyclic AMP [21], Forskolin has shown great potential as a neuroprotective agent with the capacity for neurogenesis by enhancing PKA activity. In a recent study, Forskolin was shown to prevent the death of rat cerebellar neurons in vitro from damage induced by the long-term neurotoxic action of glutamate [22]. Forskolin is currently a safe, commercially available oral weight loss supplement worldwide with reported benefits [23, 24]. While some research studies have suggested that Forskolin may confer neuroprotection of neurons against a different toxic insults, its neuroprotective potential in the treatment of PD remains to be explored.
The objective of the present study was to compare the efficacy of a natural compound, Forskolin, to the current standard of care therapy, Levodopa, in the treatment of PD in the PINK-1 Knockout (PINK1-KO) [14, 25] rat model, a bone fide model of PD that faithfully recapitulates robust loss of SN neurons, exhibits impaired motor performance, loss of brain energy production, and corporeal fatigue as evident by a significant reduction in hindlimb strength [14]. By restoring the level of PKA activity in the brain, we hypothesized that Forskolin could improve motor symptoms, as measured by grip strength and motor coordination by using beam balance, to a greater extent than the Levodopa treatments. We also hypothesized that Forskolin has the capacity to restore energy production in the SN [26] in PINK1 KO rats and could restore the loss of dopaminergic neurons. Lastly, we hypothesized that Forskolin injection treatment could restore the loss of PKA activity observed in the SN in PINK1-KO rats.
METHODS
Animal procedures
All in vivo studies were performed following the National Institutes of Health Office of Laboratory Animal Welfare Policy and Institutional Animal Care and Use Committee (IACUC) at the University of Nevada, Reno (protocol #20-09-1086). In brief, male and female WT (Charles River Laboratories International, Inc.) and PINK1-KO (Horizon Discovery) Long-Evans rats at 10 months old were used for assessing for motor function and immunohistochemistry (IHC) studies. The following cohorts of animalsgroups were used for this study: 1) 14-month-old female PINK1-KO rats were used for the Forskolin vs. Levodopa study, and 2) female WT and PINK1-KO 11-month-old rats were used for the pharmacological “washout” studies to analyze the extent that Forskolin confers anti-PD effects following the intraperitoneal (I.P.) administration of the last dose. The animals were maintained under controlled temperature 25-26°C, 12 h light/dark cycle, with food and water ad libitum. Importantly, all animals were humanly treated per ARRIVE guidelines and individually caged three days before the behavioral assays.
Motor coordination and grip strength test
Every motor coordination and grip strength trial were recorded before (basal –“day 0”) and after the treatment with drugs (treated –“day 10”), and every week after for the “washout” assay (week 2 to 7) and Levodopa assay (week 2 to 4).
Grip strength assays
The grip strength test is a well-validated behavioral test used to measure rodent muscle strength of the hindlimbs (HL) and forelimbs (FL) [27]. This in vivo assay estimates the rat grip force on FL and HL. By employing a dynamometer, the rat grip strength (g) was normalized to the weight of each animal (kg) under baseline conditions (saline treated) and in pharmacologically-treated rats. The highest strength value for each rat was recorded and considered as the maximum muscular force.
The beam balance (BB) test measures the equilibrium and fine motor coordination in animals by allowing rats to cross through an inclined, 1-meter long beam from an open space to an enclosed dark box at varying widths (2–6 cm) [28]. Briefly, a beam balance apparatus (Panlab Harvard Apparatus) composed of a dark box and two ledges tapered beams of 2 cm (6 cm for training) mounted on two tripods at an angle of 15° from the floor (modified from Tung et al. [29]), with the highest point set at 90 cm from the level of the dark box. The setup included a safety ledge mounted below the beam and a light source at the end of the beam to motivate the animals to climb the beam up to the dark box as described in Sweis et al. [30] with minor modifications.
During the training phase, animals were trained once on two beams of gradually decreasing widths, followed by three trials at the 2 cm beam with 30 s of rest between attempts upon reaching the dark box the next day as previously published [30]. The cross-time of each beam (s) and the number of slips were measured by using the JWatcher™ software (version 0.9). To adhere to the guideline of rigor and reduce bias, it is worth nothing that the number of foot slips and falls were quantified blind to the observer recording and analyzing the videos (genotype or treatments were not known for each animal). Additionally, only the number of foot slips and falls were scored for each animal only for the time frames in which the animal was advancing towards the dark box at the end of the beam balance. To assess the rat’s behavioral performance on the BB, we assigned a value of 1 point to each foot slip, whereas a value of 2 points was assigned for each fall (value of 2). Both metrics were compiled as one score termed the Total Score (TS) value.
Pharmacological treatments of rats
The animals were subjected to intraperitoneal (I.P.) administration of Forskolin (1.6 mg/kg body weight) at days 1, 3, 5, 7, and 9 or with saline solution (vehicle control). As an additional control, some animals were left untreated to analyze any placebo effects caused by the saline solution. Overall, the following control and experimental groups were assigned: 1) WT rats intraperitoneally injected with vehicle (PBS 1x 10 % DMSO, WT), PINK1-KO rats injected with Forskolin (Fsk), and the PINK1-KO rats injected with the vehicle (Veh).
Washout studies with Forskolin
To measure the long-term effects of Forskolin, we designed a “washout” study using three groups of animals (WT, Veh, and Fsk). Eleven-month-old female rats were used for this study; the weight, crossing time, and motor coordination tests were recorded for baseline performance prior to the pharmacological treatments and analyzed again following I.P. injections with Forskolin (1.6 mg/kg) or Levodopa (12 mg/kg) and analyzed described above. The Forskolin treatment consisted of an I.P. injection of Forskolin or vehicle at days 1, 3, 5, 7, and 9. The behavioral tests were recorded weekly until the effects of Forskolin treatment wore off (week 2–6).
To evaluate the performance of Forskolin vs. Levodopa in terms of reducing motor symptoms and corporeal fatigue in PINK1-KO rats, we performed additional beam balance and grip strength studies in a select cohort of symptomatic PINK1-KO rats that were intraperitoneally treated with vehicle control, Forskolin, or with Levodopa. This study consists of two groups of 14-month-old female PINK1-KO rats; the Forskolin treated and the Levodopa-treated group.
The Levodopa group consisted of PINK1-KO rats that were treated with an I.P. injection of Levodopa (10 mg/kg body weight) and Carbidopa (12 mg/kg body weight) one hour before performing the beam balance test ten days following the last dose as previously published [31]. On day 11, animals were subjected to another dose of Levodopa/Carbidopa for another week before performing another assessment of motor coordination using the beam balance (week 2). Finally, the motor coordination test was performed as described above for an additional two weeks (weeks 3 and 4). The animals in the Forskolin group (Fsk) were dosed as described previously, with the sequential I.P. administration of Forskolin on days 1, 3, 5, 7, and 9 and analyzed for motor coordination for four weeks in a similar manner as the Levodopa-treated animals.
Preparation of brain slice
The rats were anesthetized with isoflurane (5%) and perfused transcardially with 120 mL (10 mL/min) of artificial cerebrospinal fluid solution (120 mM NaCl, 3.5 mM KCl, 1.3 mM CaCl2, 1 mM MgCl2, 0.4 mM KH2PO4, 5 mM HEPES, 10 mM Glucose, pH 7.4) at 37°C. The right hemisphere was preserved for assessment of brain energy production by using the XF24e Seahorse Analyzer (see below), whereas the left hemisphere was saved for performing downstream IHC-mediated analysis of loss of dopamine neuron in the SN as further described below. The specific brain coordinates used to obtain slices from the prefrontal cortex (PFC) and the SN were localized using the stereotaxic atlas for rats from Paxinos and Watson [32].
Assessment of energy production in the brain
To assess the effects of Forskolin on brain energy production, an XF24e Extracellular Flux Analyzer was employed to measure the real-time oxygen consumption rates (OCRs) and extracellular acidification rates (ECARs), as proxies of mitochondrial function and glycolysis, respectively. Coronal sections of 150μm thickness of the right hemispheres were obtained by employing a vibratome (VT 1200S, Leica Microsystems, Germany). For this assay, we analyzed the bioenergetic profile in five brain slices per animal that were placed into the wells of a 24 well islet capture microplate (XF24 islet capture microplate, Seahorse Bioscience). Subsequently, OCRs and ECARs were measured by following a protocol previously published [14]. In brief, biopsy punches of 1.2 mm diameter from each brain region were isolated and mounted on the bottom of each well from the plate. Mesh capture screens, previously submerged in XF Base Medium were carefully mounted immediately over the tissue, and 700μL of Agilent Seahorse XF Base Medium (supplemented with 2 mM L-glutamine, 1 mM Na pyruvate, 10 mM glucose and 4 mg/ml BSA, pH 7.4) was added into each well.
Three dilutions of pharmacological reagents/toxins were prepared in XF Medium as follows: 25μM oligomycin from Streptomyces diastatochromogenes (1404-19-9, Sigma Aldrich), 15μM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP, 370-86-86, Sigma Aldrich) and a mixture of 20μM antimycin-A from Streptomyces sp. (1397-94-0, Sigma Aldrich) containing 20μM rotenone (R8875, Sigma Aldrich). Briefly, 75μL of each drug were injected successively in a final volume of 925μL in each well. Four wells, one per row, were employed as blank wells for each 24 well Seahorse microplate. To perform the mitochondrial stress assay using the XF24e Analyzer, we followed a well-validated protocol [14]. Each measuring cycle was programmed to consist of 3 min of mixing, 3 min of waiting, and OCRs and ECARs were measured for 2 min. In addition, five cycles were employed to analyze the baseline OCRs, seven cycles were used to evaluate ATP-dependent OCRs (oligomycin), five measurements were used to measure maximal OCRs (FCCP), and six cycles to evaluate mitochondrial-dependent OCRs.
Finally, the bioenergetics parameters in brain slices were recorded in accordance with the manufacturer’s recommendation (XF24e Extracellular Flux Analyzer Agilent Technologies): the non-mitochondrial OCRs, basal respiration, the maximal respiration, the proton (H+) leak, ATP production, and spare respiratory capacity. Also, the basal glycolysis and the glycolytic capacity as ECARs parameters were analyzed for each brain slice.
Immunofluorescence assay
Indirect immunolabeling of brain slices was performed as previously published [14] with the following minor modifications. Shortly, the left cerebral hemispheres from treated rats were fixed with 4 % paraformaldehyde (PFA, w/v), dehydrated by using a gradient of sucrose solution (10%, 20%, and 30%). Then, the brain hemisphere was embedded in a mixture of Optimial Cutting Temperature (OCT) media containing 30% sucrose, frozen with liquid nitrogen snap frozen, then stored at –80°C and sliced in a cryostat (Leica CM1510-S). The SN was sectioned in 12μm brain slices (stereotaxic coordinates were: 2.88 mm to 4.32 mm from the interaural line and –6.12 mm to –4.64 mm from bregma according to Paxinos and Watson [32]), collected on pre-coated slides (Superfrost Plus Gold Microscope Slides, Fisher Scientific) and stored at –20°C until used.
The tissue was blocked and permeabilized 1 h by using a 3 % bovine serum albumin (BSA) with 0.05 % Triton X-100 in PBS (BSA –1x PBS-T), then incubated for 2 h with tyrosine hydroxylase (TH) antibody (Thermo Fisher Scientific P21962, 1:400) in primary antibody solution (1x PBS 1.5% BSA), CREB (1:500 in 2.5% BSA & TBST; 9197S; Cell Signaling Technology, Denver, MA), or with Phospho-CREB (Ser133) (1:100 in 2.5% BSA & TBST; 9198S; Cell Signaling Technology, Denver, MA). Next, the brain slices were washed extensively in 1X PBS-T, 10 min per wash, three times, and then incubated with the secondary antibody (Alexa Fluor 647, goat anti-rabbit IgG, 1:1000). Finally, nuclei were labeled by exposing the tissue to nucleic acid stain 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) mixed in a solution of 70% glycerol at a final concentration of 1.25μg/ml to visualize nuclei. The brain slices were then cover-slipped and imaged by using an EVOS-FL Cell Imaging System (Life Technologies) equipped with EVOS Light cubes specific for GFP (excitation/emission of 470/510 nm), RFP (excitation/emission of 531/593 nm), and Cy5 (excitation/emission of 628/692 nm), at magnifications of 20× (numeric aperture 0.45) or 40×(numeric aperture 0.60). Epifluorescence images of TH-stained neurons were captured with a 4X objective. The collected images were analyzed using ImageJ 1.50i software.
In order to assess the loss of SN dopamine neurons as a consequence of a loss of endogenous PINK1 in PINK1-KO rats or assess for a rescue of neurodegeneration by Forskolin treatment, the integrated fluorescence intensity, mean fluorescence density over the area of the region of interest (ROI), was measured over the TH-specific area of each immunostained brains slice as quantified by using NIH ImageJ (Bethesda, MD, version 1.50i). The integrated density was then normalized to the total number of cells in the ROI based on DAPI staining. The ratio of the integrated density of TH by DAPI counts is an accurate and well accepted metric to assess for neurodegeneration of midbrain dopamine neurons as previously published [14].
PKA activity assay
The rats were anesthetized by using 5% isoflurane and transcardially perfused with saline solution as described above (Preparation of Brain Slice section). The brain was then harvested, collected, and washed in ice-cold 1x PBS solution. The midbrain samples were obtained as described before by employing the following brain coordinates: 2.88 mm to 4.32 mm from interaural line and –6.12 mm to –4.64 mm from bregma. 100 mg of tissue from the midbrain was then homogenized in 1 mL ice-cold lysis buffer (HEPES 25 mM, NaCl 150 mM, EDTA 5 mM, 10% glycerol, and 1% Triton-x100) followed by performing 20–25 strokes by using the tissue glass homogenizer. The 1 mL processed tissue was subsequently centrifuged (14,500 rpm, 30 min at 4°C), and the supernatant was collected and transferred to a new 1.5 mL tube. The protein quantification was performed by using the BCA reagent kit, and the sample was stored at –80°C until used.
The PKA assay was then performed per the manufacturer’s instructions with some modifications as previously published [13]. To assess for PKA activity, one duplicate sample of each tissue from each animal was treated with the pharmacological inhibitor of PKA N-{{(2E)-3-(4-Bromophenyl)prop-2-en-1-yl}amino}ethyl)isoquinoline-5-sulfonamide (H-89, 10μM) in order to calculate the PKA activity and subtract from the total activity in the sample.
Specifically, 1μl of lysate containing 0.25μg of total protein was added to the well with 29μl of kinase buffer assay, and 10μl of ATP was added to each well except the blank to start the reaction. The 96-well plate was then incubated for 25 min at 30°C, and the reaction was stopped by emptying the contents of each well. Then, 40μl of Phosphospecific Substrate Antibody to each well were added (except the blank) and incubated at room temperature for 60 min. The plate was washed by adding 100μl 1X Wash Buffer 4 times. After that, 40μl of diluted Anti-Rabbit HRP conjugated IgG secondary antibody was added to each well (except the blank) and incubated at room temperature for 30 min. The washing step was repeated 4x, 100μl per well. Finally, the 60μl of 3,3′,5,5′-Tetramethylbenzidine (TMB) substrate to each well was added and incubated at room temperature for 5 min. The colorimetric reaction was stopped by adding 20μl of stop solution to each well, and the absorbance was measured at 450 nm.
Statistical analyses
Unless otherwise indicated, all in vivo and in vitro data are expressed as mean±S.E.M. from at least three independent experiments or from at least 4-5 animals per experimental condition. Behavioral data from rats prior and after pharmacological administration to Forskolin, Levodopa, or saline solution (vehicle) were analyzed by employing a Student’s t-test (two-tailed) for pairwise comparisons or by Multiple group comparisons were done by performing one-way ANOVA followed by a Mann Whitney’s test for non-parametric data followed by Bonferroni-corrected Tukey’s test by using the GraphPad Prism software (version 6.0). p values less than 0.05 were considered statistically significant.
RESULTS
Unlike PINK1-KO mice, PINK1-KO rats recapitulate many symptoms of PD, including uncoordinated motor movement, loss of balance and gait, and decreased hindlimb muscle strength—an indicated of chronic fatigue—accompanied by overt loss of midbrain dopamine neurons and concomitant accumulation of phosphorylated α-synuclein [20, 25, 33–35]. Additionally, PINK1-KO rats show significant neurochemical and bioenergetic alterations in the midbrain and PFC and a significantly decreased level of brain-derived neurotrophic factor (BDNF) and various antioxidant proteins [14, 25, 33]. Given that pharmacological upregulation of PKA signaling or by applying molecular biology approaches (e.g., transient expression of PKA holoenzyme), can compensate for mitochondrial dysfunction, reverse the loss of ATP production and transmembrane potential, as well reverse the loss of dendrites induced by loss of PINK1 function in neurons [13, 36], we surmised that pharmacological activation of PKA via I.P. administration of Forskolin, a strong pharmacological activator of cyclic AMP-mediated signaling pathways [37], can exert neuroprotective effects in symptomatic PINK1-KO rats in a similar manner as observed in PINK1-deficient cells (PINK1-deficient SH-SY5Y neuroblastoma cells and primary cortical neurons) [13, 36]. To test this hypothesis, we evaluated the effects of intraperitoneal injection of Forskolin on motor symptoms, corporeal fatigue, loss of SN neurons, and brain metabolism by employing a battery of pre-validated behavioral and biochemical tests approaches as published [14] and as further described below.
Intraperitoneal administration of Forskolin reverses motor symptoms and hindlimb fatigue in PINK1-KO rats
Consistent with prior literature [14, 25], both male and female 10-month-old PINK1-KO rats show significantly impaired motor coordination and hind motor strength as assessed by the beam balance and grip strength analyzer, respectively. Indeed, PINK1-KO rats showed a significant two-fold increase in the number of foot slips and falls from the balance beam relative to 10-month-old WT rats (summarized as a total score, p≤0.0001, Fig. 1), a pathology that is concomitant with a significant 40 % decrease in hindlimb grip strength (p≤0.0001, Fig. 2). In aggregate, our behavioral data is consistent with prior reports that have observed significant onset of motor symptoms in middle-aged PINK1-KO rats [14, 25, 31, 33, 38, 39]. Interestingly, we observed gender-related differences in both motor deficits and hindlimb fatigue in PINK1-KO rats. Specifically, we observed significant motor deficits accentuated in 10-month-old male rats compared to age-matched WT rats, whereas a more modest effect was observed in female PINK1-KO rats (Figs. 1 and 2). Overall, our behavioral data suggest that PINK1-KO male rats show exacerbated motor deficits (p≤0.05) with respect to the PINK1-KO female rats, a pathology that is consistent with the reduced motor deficit previously observed in young 2-month-old PINK1-KO rats [14]. As in the beam balance test, we observed gender-related differences in hindlimb grip strength. Specifically, male 10-month-old PINK1-KO rats showed a more accentuated reduction in hindlimb grip strength (∼60% reduction) compared to WT rats, whereas female rats showed a more modest reduction in hindlimb grip strength (∼25% reduction) compared to WT rats (Fig. 2A, B). While there were no significant differences between WT males and females with respect to motor coordination as assessed by the balance beam test (p = 0.7425), there was a significant difference in hindlimb grip strength in age-matched WT males compared to age-matched WT females (Fig. 2, p≤0.01). Therefore, in light of these gender-based differences in hindlimb grip strength, we disaggregated all behavioral data based on gender and normalized value to its same-gender control group in subsequent in vivo assays.
Fig. 1
Forskolin treatment improves motor coordination in PINK1-KO rats. A) Quantification of motor coordination using the aggregate beam balance score in female WT and PINK1-KO rats that were untreated (basal), treated with vehicle (Veh) control, or treated I.P. with Forskolin solution (Fsk) at 1.6 mg/kg for 10 days. B) Quantification of motor coordination using the aggregate beam balance score assessed in male WT and PINK1-KO rats that were untreated (basal), treated with vehicle (Veh) control, or with Forskolin solution (Fsk) at 1.6 mg/kg for 10 days. C) Aggregate beam balance test scores in other females and males of WT and PINK1-KO rats that were untreated (basal), treated with vehicle (Veh) control, or treated I.P. with Forskolin solution (Fsk) at 1.6 mg/kg for 10 days. Note that the higher the score is a collective measure of both the mean number of slips and falls per crossing in the beam balance as previously published. The data shows that Forskolin completely reduced the impaired motor coordination in PINK1-KO rats to a similar level as in untreated or vehicle-treated WT rats. D) “Washout” studies showing the balance beam test score of female WT I.P. treated with vehicle (WT), and PINK1-KO rats, with vehicle (Veh) or treated with Forskolin solution (Fsk) at 1.6 mg/kg for 10 days. The magenta dashed arrow indicates the start of the indicated treatment, and the solid arrow shows the end of the treatment. For A, B, and C, six animals were employed per group, whereas three animals per group were used in D. Data are expressed as the total score per crossing. Mean±SEM. n = 8–11 animals per group, *p≤0.05, one-way ANOVA with Tukey’s multiple comparisons test. A #p < 0.05 vs. Fsk group; *p < 0.05 vs. WT group.
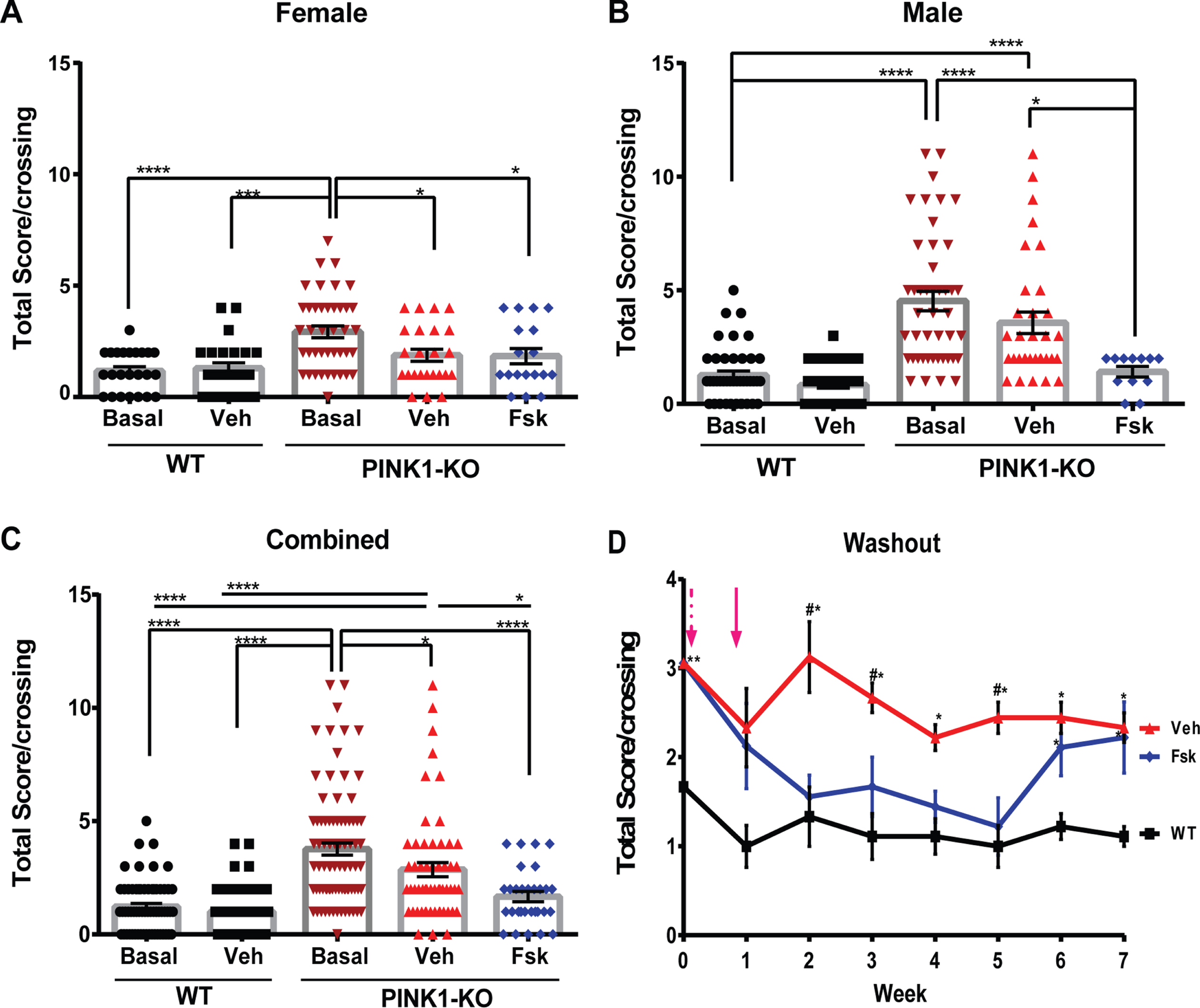
Fig. 2
Forskolin treatment reversed muscle fatigue in the hindlimbs of PINK1-KO rats. A) Compiled analysis of grip strength in the hind limbs as assessed by a grip strength analyzer in female WT and PINK1 KO rats that were left untreated (basal), or I.P. treated with vehicle (Veh), or with Forskolin solution (Fsk) at 1.6 mg/kg for 10 days or in B) male WT and PINK1-KO rats, or C) aggregated grip strength data for both male and female rats. D) Representative line graphs of a longitudinal washout study that assessed the mean grip strength in the hindlimbs of female WT treated or PINK1-KO rats that were left untreated (Basal) or I.P. treated with vehicle (Veh) or treated with Forskolin solution (Fsk). The magenta dashed arrow indicates the start of the treatment, whereas the solid arrow shows the end of the treatment. Data are expressed as the total score per crossing (Mean±SEM. *p≤0.05, One-way ANOVA with Tukey’s multiple comparisons test, n = 8–11 animals per group, A #=significant to Fsk group; *= significant to WT group).
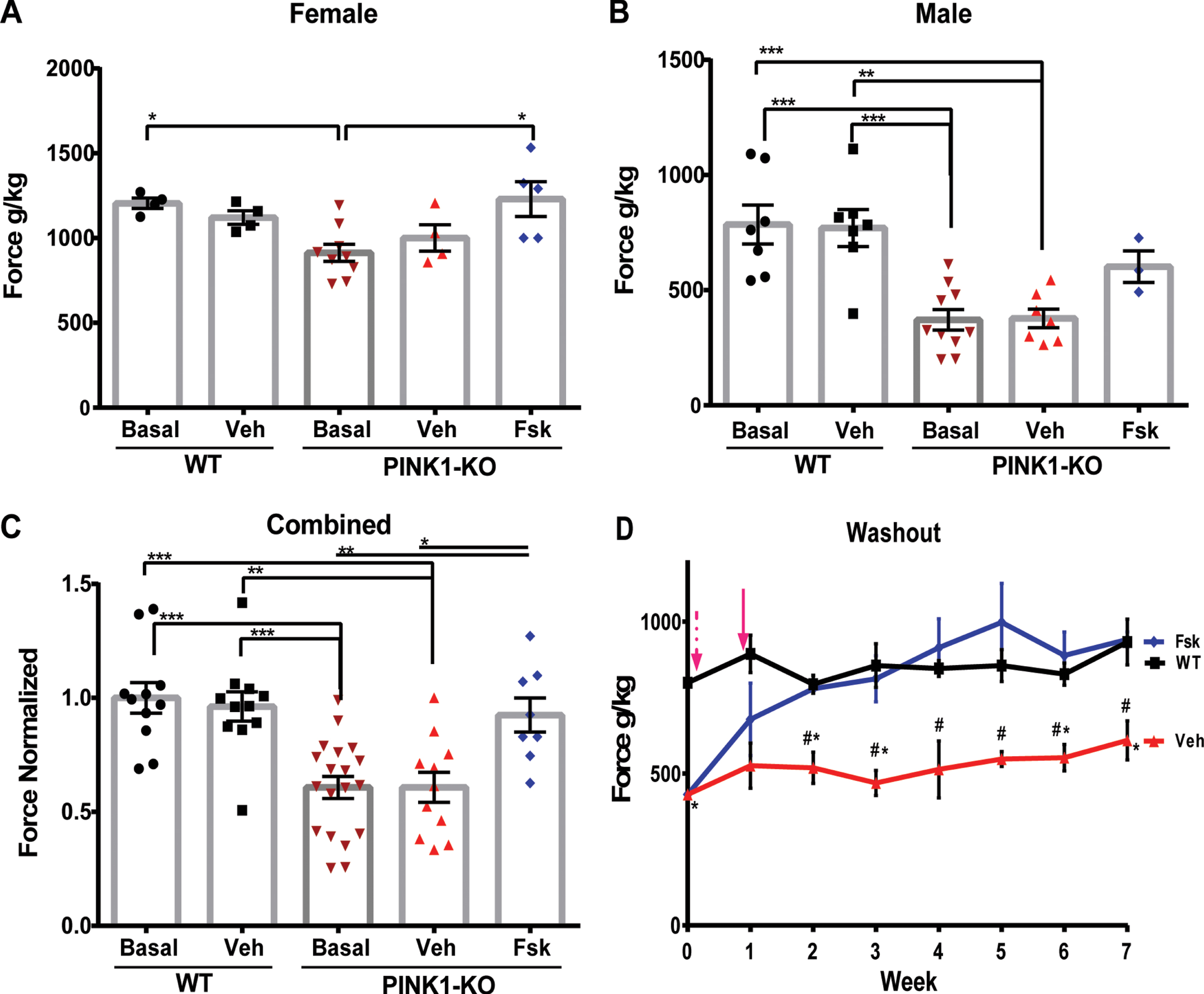
Next, we assessed the effects of pharmacological enhancement of cAMP-dependent increase of PKA signaling in the brain and PD symptoms in PINK1-KO rats. Specifically, treatment of PINK1-KO rats with I.P. injections of Forskolin (1.6 mg/kg body weight 5X for 10 days) significantly improved the balance beam performance of PINK1-KO rats as evident by a 55% decrease in the balance beam total score observed in Forskolin-treated PINK1-KO rats compared with untreated PINK1-KO basal values (p≤0.0001), and 41% with respect to the vehicle-treated group (Fig. 1A-C, p≤0.05). While it is clear that I.P. injections of Forskolin significantly improves motor performance in 10-month-old PINK1-KO rats, it is worth noting that vehicle-treated PINK1-KO rats showed a modest but significant improvement compared to untreated PINK1-KO rats (24% improvement, p≤0.05), which were not subjected to the beam balance test. This effect may be attributed to “intrinsic improvement”, a behavioral phenomenon observed in rats that show enhanced motor performance induced by practice and progressive learning in rats that are subjected to the repeated crossing of beam balance irrespective of any experimental conditions [40, 41]. Nevertheless, PINK1-KO rats treated with Forskolin showed a statistically significant improvement in motor performance control regardless of gender compared to those solely treated with the vehicle (Fig. 1A-C). With respect to hindlimb grip strength, we observed a 22% significant increase in hindlimb grip strength in Forskolin-treated PINK1-KO rats compared to untreated PINK1-KO rats and to a similar extent as vehicle-treated animals (Fig. 2C). In addition, I.P. administration of Forskolin was able to completely restore hindlimb grip strength in both females and males to a similar manner as in WT rats (Fig. 2A, B). It is worth noting that no statistical differences were observed between untreated/vehicle-treated WT rats and PINK1-KO rats treated with Forskolin, suggesting that pharmacological activation of the cAMP-PKA pathway reduces motor deficits and elevated hindlimb muscle strength in Parkinsonian rats in a similar manner as in WT rats.
Next, we evaluated the extent to which the in vivo neuroprotective effects of Forskolin persist following I.P. administration (1.6 mg/kg body weight 5X for 10 days) by performing “washout” studies in a cohort of severely symptomatic PINK1-KO rats. In an analogous manner to human clinical trials, the “washout” period in our animal studies is defined as the time by which PINK1-KO rats receive no additional “treatment” (Forskolin) and subsequently analyzed for motor performance and hindlimb muscle strength once a week for up to 6 weeks during the washout phase (immediately following the last dose) until the neuroprotective effects of Forskolin dissipated. Given that middle-aged male PINK1-KO rats gained excessive bodyweight compared to female rats (Supplementary Figure 1), we decided to perform the washout studies in symptomatic female 11-month-old PINK1-KO rats only as part of our study design and to rule out any undesired body mass effect and increase the rigor of our in vivo washout studies. Remarkably, Forskolin was able to efficiently reduce motor deficits and restore hindlimb muscle strength in a similar manner as in WT rats lasted at least three weeks (Fig. 1B). Interestingly, the anti-Parkinsonian effects of Forskolin in ameliorating motor coordination in symptomatic PINK1-KO rats persisted for three weeks before the animals “relapsed” following cessation of treatment, whereas the ability of Forskolin to restore hindlimb strength lasted more than five weeks with no indication of potential relapse at the observed time frame (Fig. 2D). In aggregate, our behavioral data suggest that I.P. administration of Forskolin can restore motor coordination and reverse muscle fatigue in symptomatic 10-month-old PINK1-KO rats.
Consistent with other studies, we observed that PINK1-KO rats significantly gained an excessive amount of weight by 10 months of age compared with WT age-matched rats (Supplementary Figure 1) [14]. Given that Forskolin has been used over the years as an over-the-counter drug for weight loss, it is conceivable that long-term I.P. administration of Forskolin can induce significant weight loss and thereby improve motor coordination in PINK1-KO rats due to the potential thermogenic effects induced by consumption of Forskolin in animals and humans. However, this is not the case as we observed no significant differences in the mean weight of PINK1-KO rats treated with Forskolin compared to untreated or vehicle-treated rats for up to five weeks during the washout studies (Supplementary Figure 1).
Interestingly, we observed that 10-month-old PINK1-KO rats showed significantly reduced beam balance crossing times with respect to WT rats irrespective of the treatment (vehicle or Forskolin), presumably due to the hyperkinetic behavior obser-ved in middle-aged PINK1-KO rats as reported by other groups [42]. However, we observed no significant differences between vehicle-treated and Forskolin-treated PINK1-KO rats, suggesting that Forskolin does not enhance locomotion speed in PINK1-KO rats (Supplementary Figure 1).
Intraperitoneal administration of Forskolin can reverse the bioenergetic alterations in the SN of PINK1-KO rats
We have previously shown that 2-month-old PINK1-KO rats showed bioenergetic alterations in the brain [14]. Given that Forskolin, or enhancement of PKA-mediated signaling, can compensate for the loss in oxidative phosphorylation, reverse Drp1-dependent mitochondrial fission, and elevated oxidative stress in PINK1-deficient cortical neurons [13, 36], we wanted to evaluate the extent by Forskolin can reverse the deficit in mitochondrial respiration and glycolysis in SN dopamine neurons in PINK1-KO rats [14] by using the XF24e Extracellular Flux metabolic Bioanalyzer. Specifically, we evaluated the bioenergetic profile (oxidative phosphorylation and glycolysis) in 1 mm×1 mm, 10μm thick slices of the SN [26] and of the PFC derived from WT and PINK1-KO rats treated with either vehicle alone or with I.P. injection with Forskolin (1.6 mg/kg) for 10 days (Fig. 3). In brief, our aggregate bioenergetic data show that the lack of endogenous PINK1 induces a significant decrease in both the basal (p≤0.05, Fig. 3A, C) and maximal mitochondrial respiration (p≤0.01, Fig. 3E), decreased proton leak (p≤0.01, Fig. 3G) along with a modest decrease of the non-mitochondrial oxygen consumption (Fig. 3D) in the SN of PINK1-KO rats compared to WT rats, whereas no statistical changes were observed in PFC (data not shown). In addition, 10-month-old PINK1-KO rats showed a robust decrease in basal and maximal glycolytic rates (glycolysis) as measured by the basal and maximal ECARs (p≤0.05, Fig. 3B, I-J), whereas only a non-significant decrease in glycolytic capacity (also known as maximal glycolysis) was observed in PINK1-KO rats relative to WT rats (Fig. 3J). Thus far, our bioenergetic data indicate that PINK1-KO rats show a significant decrease in both glycolysis and oxidative phosphorylation in the SN.
Fig. 3
Forskolin restores total energy production in Substantia nigra (SN) of PINK1-KO rats. A) Representative trace for an oxygen consumption rate (OCR) assessment in midbrain slices derived from 10-month-old WT vs. PINK1-KO rats, B) representative trace for a Extracellular Acidification Rate (ECARs) assessment in midbrain slices derived from 10-month-old WT vs. PINK1-KO rats as measured from an XF24e Seahorse BioAnalyzer in SN slices treated with vehicle, untreated or with 1.6 mg/kg of Forskolin by I.P. administration for 10 days. The following bioenergetic parameters were calculated from at least three mitochondrial respiration and glycolysis assays, including: C) Basal respiration, D) Non-mitochondrial oxygen consumption rate. E) Maximal respiration. F) Spare respiratory capacity. G) Proton leak. H) ATP production, I) Basal glycolysis, and J) Glycolytic capacity. (Mean±SEM. *p≤0.05, one-way ANOVA with Tukey’s multiple comparisons test, n = 8–11 animals per treatment group).
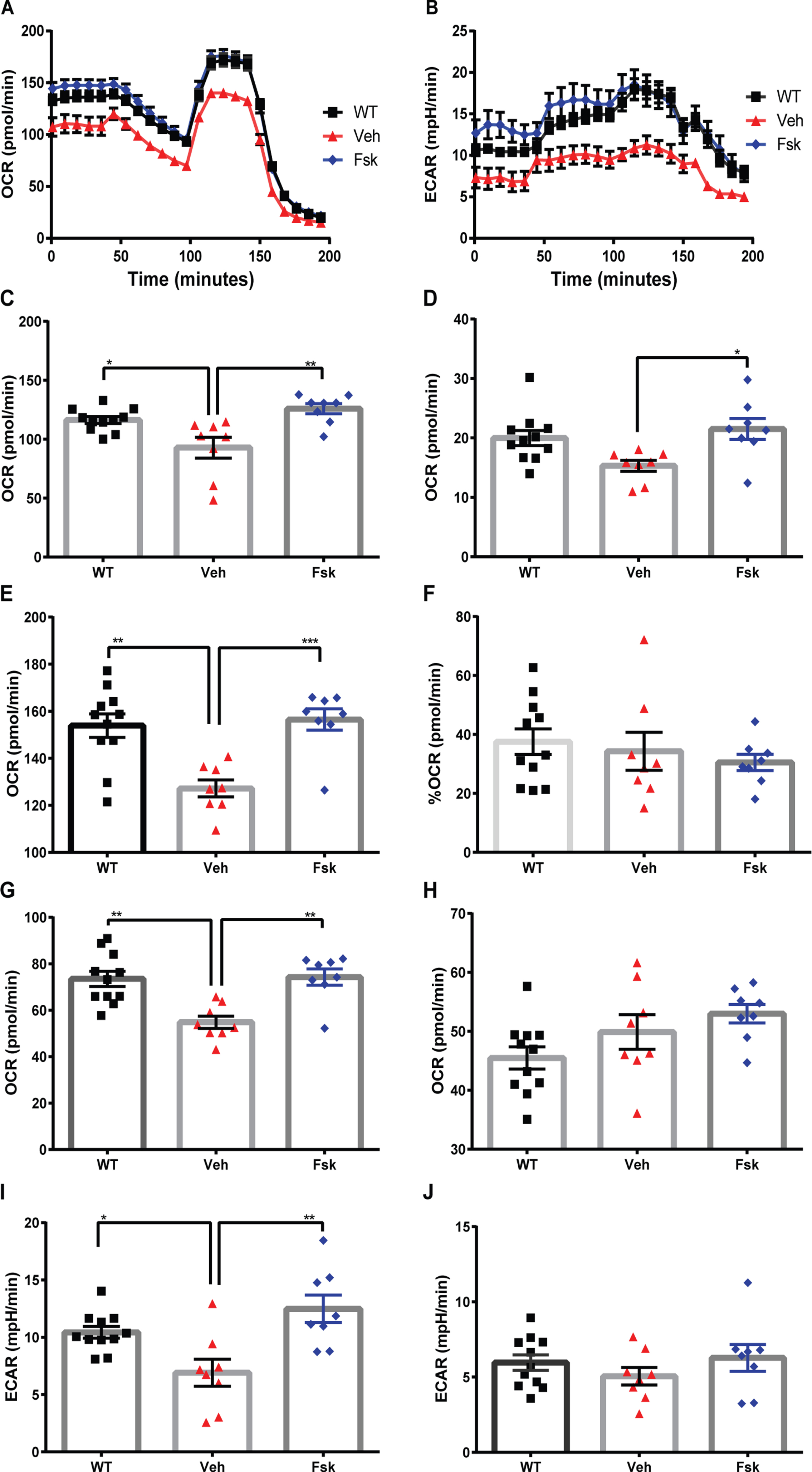
Conversely, I.P. administration of Forskolin for 10 days completely restored many parameters of OCRs and ECARs in the SN derived from 10-month-old PINK1-KO rats similarly as age-matched WT rats, including basal OCR (p≤0.01, Fig. 3C) and maximal (p≤0.001, Fig. 3E) OCRs, the non-mitochondrial OCRs (p≤0.05, Fig. 3D), proton leak (p≤0.01, Fig. 3G), and in basal glycolysis (p≤0.01, Fig. 3I) to a similar extent as SN slices derived from WT rats. Overall, our bioenergetic data suggest that activation of cyclic AMP-dependent signaling via I.P. administration of Forskolin can reverse the mitochondrial pathology and bioenergetic deficiencies in symptomatic PINK1-KO rats, presumably by elevating PKA signaling in the mitochondrion via the PKA scaffold Dual Specificity A-kinase anchoring protein 1 (D-AKAP1) [36].
Moreover, in order to understand the bioenergetic alterations induced by loss of PINK1 and the extent that Forskolin can reverse an imbalance in energy landscape in Parkinsonian rats, we analyzed the OCR/ECAR ratios for the baseline state (basal OCR/ECAR) in addition to the maximum bioenergetic potential as measured when the mitochondrial membrane potential is collapsed by the mitochondrial uncoupler FCCP (maximal OCR/ECAR) as previously published but with the minor modifications [14]. In brief, we observed that in SN slices, the lack of PINK1 elicits an unexpected increase in OCRs/ECARs ratio and as well as an increase in maximal OCRs/ECARs, as stimulated when brain slices are incubated in the presence of FCCP (Supplementary Figure 2), data that is consistent with our prior observations in PBMCs derived from young PINK1-KO rats [14]. Overall, the increase in the OCRs/ECARs ratio is likely a consequence of the more profound reduction in ECARs relative to the reduction in OCRs in PINK1-KO rats, given that both bioenergetic metrics are altered to some level (Fig. 3). However, we observed that PFC slices derived from PINK1-KO rats showed a metabolically switch from oxidative phosphorylation to glycolysis compared to WT rats, indicating a “pervasive” metabolic dysregulation in the brain that warrants further investigation. However, in both the SN and the PFC, Forskolin restored the OCR/ECAR ratio (bioenergetic landscape) to a similar extent as in WT rats (Supplementary Figure 2).
Forskolin reverses the loss of TH neurons in 10-month-old PINK1-KO rats
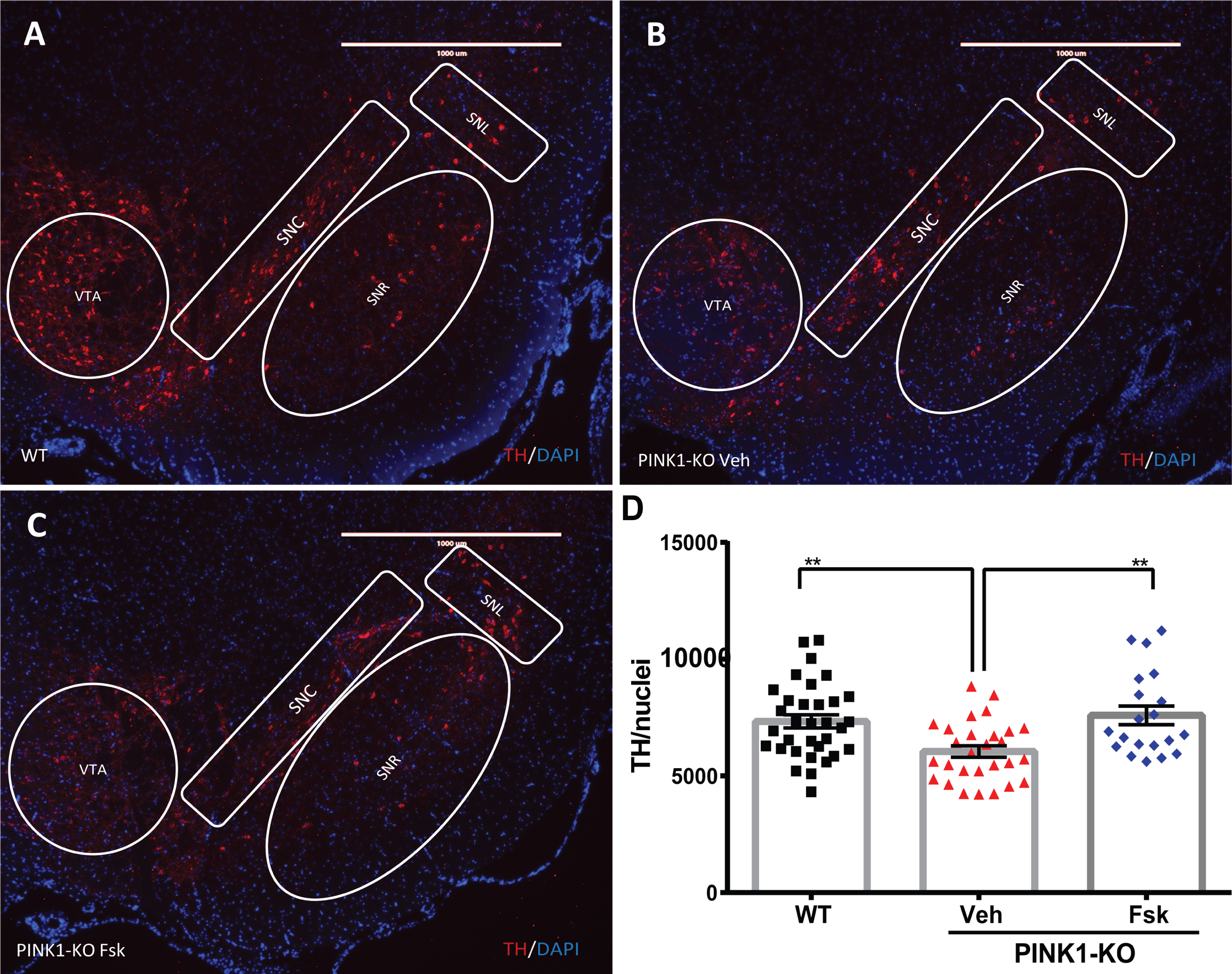
Consistent with other research groups, our IHC analysis of midbrain dopamine neurons in SN slices show that PINK1-KO rats experienced significant neurodegeneration in the SN as evident by a significant reduction in the TH-specific fluorescence in midbrain slices compared to age-matched WT rats at 10 months of age (p≤0.01, Fig. 4) [33, 38]. To assess the prevalence of TH-stained neurons in each SN slice, the integrated TH-specific fluorescence intensity for each representative epifluorescence micrograph was normalized the SN to the number of nuclei identified by DAPI staining. Given that I.P. administration of Forskolin elicits strong anti-Parkinsonian effects in 10-month-old PINK1-KO rats (Fig. 1), we then investigated whether the ability of Forskolin in reversing motor symptoms is associated with delaying/reversing the loss of SN neurons. Indeed, we observed that I.P. administration of Forskolin completely restores the TH-specific fluorescence in the SN from 10-month-old PINK1-KO rats to a similar extent as in age-matched WT rats (p = 0.58, WT/Veh vs. PINK1-KO/Fsk) and significantly elevated compared to PINK1-KO rats treated with vehicle control (p≤0.01, Fig. 4). Overall, our data show that I.P.-mediated administration of Forskolin can reverse the loss of SN midbrain dopamine neurons in middle-aged PINK1-KO rats.
Fig. 4
Forskolin treatment reverses the loss of SN neurons in PINK1-KO rats. A) Representative epifluorescence micrographs of midbrain slices immunostained for tyrosine hydroxylase (TH; red channel) to visualize midbrain dopamine neurons in the SN derived from A) 10-month-old WT rat treated with the vehicle solution or from B) 10-month-old PINK1-KO rat treated with the vehicle solution or from C) PINK1-KO rats I.P. treated with 1.6 mg/kg body weight of Forskolin. D) Compiled analysis on the mean fluorescence intensity of TH (red channel) in midbrain slices for the indicated treatment groups (WT, Veh, and FSK) in both male and female rats. The integrated density for the TH-specific fluorescence signal within the SN region was quantified and normalized to the number of DAPI-stained nuclei (blue channel) in the SN of WT rats treated with vehicle solution (WT) and PINK1-KO rats treated with vehicle solution (Veh) or Forskolin (Fsk) (Mean±SEM. *p≤0.05, One-way ANOVA with Tukey’s multiple comparisons test, n = 6 rats per experimental group). SNC, substantia nigra pars compacta; SNR, substantia nigra pars reticulata; SNL, substantia nigra pars lateralis. Scale bar: 100μm. DAPI, Blue; TH, Red.
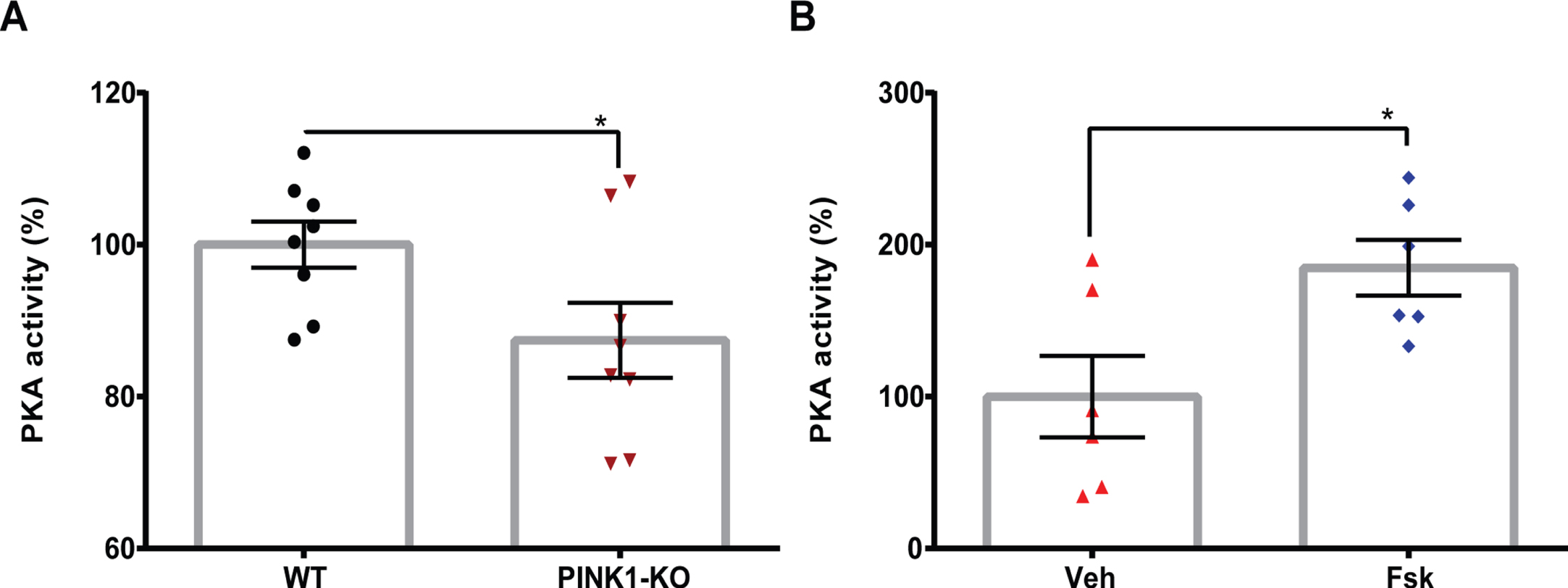
To determine whether the anti-Parkinsonian effects of Forskolin (Figs. 1–3) are associated with PKA activation in the brain, we performed PKA kinase activity assays by using a well-validated ELISA kit as previously published [13] but with minor modifications. Briefly, we evaluated the global PKA activity in the SN of WT and PINK1-KO rats treated with Forskolin or with vehicle solution. Consistent with a reduction of PKA activity in the SN observed in 10-month-old PINK1-KO mice [13], we observed a similar significant reduction in PKA activity in the SN derived from PINK1-KO rats (Fig. 5). On the other hand, the I.P. administration of Forskolin restored PKA activity in SN derived from 10-month-old PINK1-KO rats to a similar manner as age-matched WT rats. In further support of activation of PKA-related pathways by Forskolin, Western blot of lysates from the substantia nigra collected from WT or PINK1-KO rats treated with vehicle or I.P administration of Forskolin showed that Forskolin treatment was able to significantly reverse the loss in PKA-mediated phosphorylation of CREB (Serine 133) in PINK1-KO rats. Additionally, Forskolin in PINK1-KO rats achieved a modestly but significantly increase in p-CREB compared to WT rats treated with vehicle control (Supplementary Figure 3). In summary, our data suggest that the anti-Parkinsonian effects of Forskolin are causally linked to its ability to reverse the loss of PKA activity in the midbrain of Parkinsonian rats.
Fig. 5
Administration of Forskolin reverses the loss of PKA activity in the midbrain of PINK1-KO rats. A) Bar graph showing mean baseline global PKA activity assessed in the midbrain tissue derived from 3.5-month-old WT and PINK1-KO rats B) or in 3.5-month-old WT or PINK1-KO rats treated with vehicle (Veh), or with Forskolin at 1.6 mg/kg for 10 days (Fsk) by I.P. administration. The data was compiled from at least three animals per genotype. Data are expressed as the absorbance value normalized to the absorbance of WT group (A) or Veh group (B). (Mean±SEM. *p≤0.05, unpaired two-tailed t-test, 3–6 animals per group).
The ability of Forskolin to reverse motor symptoms of PD and corporeal fatigue are long-lasting
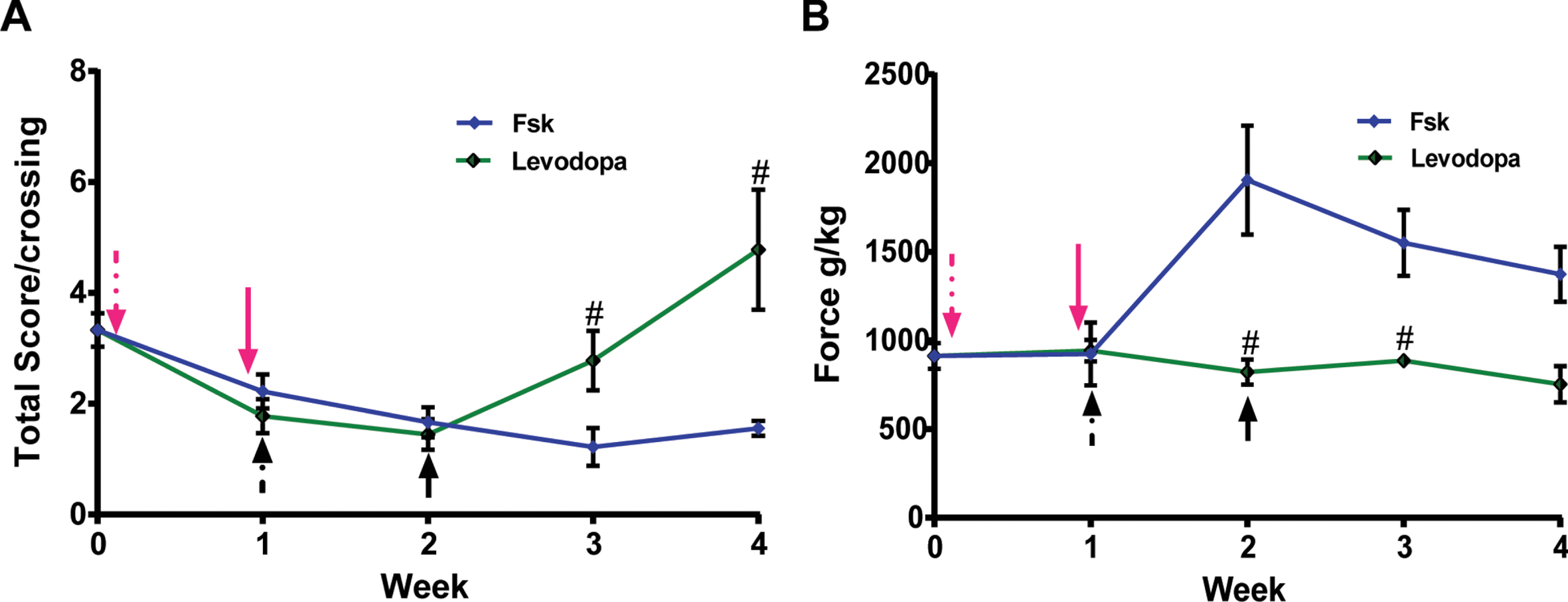
To date, there are no disease-modifying treatments for PD. Standard of care only provides symptomatic relief via oral or I.P. administration of Levodopa/Carbidopa in PD patients by replenishing the loss of dopamine in the central nervous system. Given the significant anti-Parkinsonian effects induced by I.P. administration of Forskolin in symptomatic PINK1-KO rats (Figs. 1–3), we then performed longitudinal in vivo studies to investigate the therapeutic efficacy of Forskolin in symptomatic PINK1-KO rats compared to standard of care (Levodopa/Carbidopa). To this end, another cohort of animals consisting of two groups of three symptomatic, female PINK1-KO rats with similar body weight and motor symptom scores were either I.P. injected with Levodopa/Carbidopa or with Forskolin for up to ten days, once a day, and assessed for motor symptoms and hindlimb strength by performing the beam balance assays and measuring muscle strength using the strength analyzer. As expected, I.P. administration with Levodopa/Carbidopa significant reverse motor symptoms as assessed by the beam balance assay in 14-month-old PINK1-KO rats as previously published Kelm-Nelson et al. [35], although that research study found a non-significant effect of Levodopa in reducing motor symptoms in 8-month-old PINK1-KO rats. However, the ability of Levodopa in reversing motor deficits in female PINK1-KO rats only lasted less than a week following the last dose, consistent with its short-term therapeutic effects in PD humans. Interestingly, no significant effects of I.P. administration of Levodopa on hindlimb strength were observed in 14-month-old PINK1-KO rats. However, unlike Levodopa/Carbidopa, and consistently with our previous findings, I.P. administration of Forskolin for five days significantly eliminated motor symptoms, and these anti-Parkinsonian effects persisted more than two weeks following cessation of treatments (Fig. 6A), data that is consistent with our prior washout studies (Fig. 1D and 2D). Additionally, unlike Levodopa/Carbidopa treatment, I.P. administration completely restored hindlimb grip strength which persisted more than two weeks following the last dose (Fig. 6B).
Fig. 6
Intraperitoneal administration of Forskolin, but not Levodopa treatment, exhibits long-term anti-Parkinsonian effects in PINK1-KO rats. Representative longitudinal washout studies in 10-month-old WT and PINK1-KO rats that were I.P. treated with Levodopa (green trace) or with Forskolin (magenta trace) for ten days (dashed magenta arrow) prior to cessation of the treatment (solid red arrow) and assessed for motor coordination by using a beam balance (A) or assessed for grip strength by using a grip strength analyzer (B). In brief, while Levodopa can temporarily alleviate motor symptoms, only the anti-PD effects of Forskolin persisted more than two weeks following the cessation of treatment. Additionally, only I.P. administration of Forskolin but not Levodopa treatment was able to restore hindlimb muscle strength significantly in PINK1-KO rats, an anti-parkinsonian effect that persists following the cessation of treatment (solid arrow, magenta = Forskolin; Black = Levodopa).
In summary, our collective behavioral, metabolic, and immunofluorescence data show that I.P. injection of Forskolin induces strong anti-Parkinsonian effects, including reversing motor symptoms, bioenergetic alterations, corporeal fatigue, and loss of midbrain dopamine neurons, presumably by restoring PKA activity in the SN of symptomatic PINK1-KO rats. These anti-Parkinsonian effects induced by Forskolin, in terms of motor deficits and loss of grip strength, are long-lasting and superior to that conferred by Levodopa.
DISCUSSION
PD is the second most common neurodegenerative disorder that affects nearly 1% of the population over 50 years and 2.5% over 70 years [43, 44]. It is a progressive, debilitating neurodegenerative disorder characterized by the loss of dopaminergic neurons in the SN that compromises the patient quality of life [45]. PD is classified into subtypes based on age (early-onset or late-onset), genetic linkage (idiopathic or genetic), and phenotype, the presence of motor impairment or cognitive impairment [46]. The NMS include neuropsychiatric symptoms, autonomic impairment, cognitive impairment, major clinical depression, onset of bouts of anxiety, loss of olfaction, and taste [4, 44, 47]. Motor symptoms of PD include rigidity, bradykinesia, resting tremors, postural and gait instability. By the time motor symptoms become apparent, the progression of disease is already advanced in the patient as there is an approximate more than 90% loss of dopaminergic neurons [48]. Currently, no treatment can reverse the progression of PD.
The therapies for PD are classified based on their target of the action. While the primary focus of the PD treatments is to stabilize the dopaminergic and neurotransmitter system impairment of the brain, the alleviation of motor and NMS is a priority for neurologists as it severely impairs the quality of life of the PD patient, the patient family, and caregivers [6, 49]. PD treatment also incorporates counseling, exercise, deep brain stimulation, and various pharmacological agents focused on increasing neurotransmitter activity or reducing the breakdown of neuronal homeostasis [18, 50, 51]. Standard of care therapy include pharmacological agents like dopamine precursors and agonists and enzymatic inhibitors that decrease the neurotransmitter breakdown, monoamine oxidase (MAO) inhibitors, COMT inhibitors, aromatic amino acid decarboxylase (AAAD) inhibitors, cholinesterase inhibitors, etc. [52]. Among these drugs, Levodopa, a dopamine precursor, is the standard gold therapy for controlling motor symptoms of PD [51].
Levodopa, as the others PD treatments, is prescribed based the severity of the motor symptoms, comorbidities, safety, tolerability, and cost [1]. Levodopa can be administered orally, after which it is absorbed into the bloodstream and transported to the brain through the large neutral amino acid transporter. Once the Levodopa reaches the brain, it is converted to dopamine by the aromatic acid decarboxylase in the SN pars compacta and stored in the presynaptic neuron. When dopamine is needed, it is released into the synaptic cleft, where it can bind to the dopaminergic receptors (D1 and D2). In the intestine and blood, Levodopa is metabolized by the AAAD and the COMT, reducing its quantity and bioavailability to cross the brain-blood barrier and exert its therapeutic effect in the brain, co-administration with Carbidopa is necessary [53, 54].
Despite its wondrous ability to reduce the motor symptoms of PD, Levodopa treatment has a “wear-off” process, which diminishes its effect over time and requires co-administration of other drugs to fully address the motor symptoms and maintain the life quality of the patients at a later stage of PD [55, 56]. Furthermore, reports had shown that up to 40% of patients treated with Levodopa develop dyskinesia within five years of treatment initiation [53, 57–59], with 3% of the Levodopa treated patients presenting a severe form of dyskinesia. However, in early-onset PD patients showing non-motor symptoms or mild to moderate motor symptoms as the initial treatment is avoided, other drugs like dopamine agonists, cholinesterase inhibitors, etc., are prescribed to reduce the risk of Levodopa-induced dyskinesia [60]. Also, these drugs can be co-administered with the Carbidopa/Levodopa treatment to reduce the daily Levodopa dose up to 30%, reducing the onset of Levodopa-induced dyskinesia [57, 60]. Levodopa-induced dyskinesia is correlated with the age of onset of PD and the length and amount of the Levodopa treatment; therefore, early treatment with Levodopa increases the risk of Levodopa-induced dyskinesia [53]. Since the advent of Levodopa, the historical perspective on standard of care treatment for PD has shown little progress in developing proven disease modifying therapeutics that are safe, does not induce dyskinesias or resistance to therapy in advanced PD patients, and with long-lasting therapeutic effects. The present study, shows preclinical evidence in a Parkinsonian model that Forskolin, a pharmacological enhancer of cAMP-signaling, shows potent anti-PD effects in terms of reducing motor symptoms, alleviating chronic fatigue and in reversing loss of SN neurons in a feedforward loop that promotes long-lasting therapeutic effects (Figs. 1–3 and 5). Given that a reduction in global, neuroprotective PKA signaling in the brain contributes to PD pathology, the therapeutic effects of enhancers of cAMP signaling was previously explored by other research groups. Indeed, previous research groups have proposed that pharmacologically-active compounds that can “boost” cAMP production (adenylyl cyclase- AC activators), signaling (cAMP analogs or precursors), or that are able to reduce its breakdown (phosphodiesterase –PDE inhibitors) can have therapeutic use in PD by activating or potentiating the cAMP-PKA pathway while maintaining the dopamine agonists or precursors side effects to the minimum [61].
PDEs are enzymes that control cAMP and cGMP metabolism and act as modulators of cAMP-dependent signaling [62]. To date, only a few clinical studies have been conducted in a small population of PD patients. For instance, in the study published by Cassaccia et al. [63], PD patients were orally administered Rolipram, a PDE inhibitor, on a double-blind trial in conjunction with a Levodopa/Carbidopa and with Lisuride, a dopamine receptor agonist. The results of this small clinical study found that in a treatment of 3 mg of Rolipram daily, 50% of the patients presented modest, albeit non-significant, improvement of motor symptoms, without any adverse events noted when combined with Lisuride. Also, the lack of evidence of the therapeutic effect of caffeine (another PDE inhibitor) in PD, begs the question of whether or not the modulation of cAMP levels in the brain can be used as therapy for PD. The lack of therapeutic efficacy of the PDE inhibitors in PD can due to the fact that many isoforms of PDE in the brain have been identified (∼100 PDE enzymes), and from those, there are more than 9 that are expressed solely in the striatum [61, 64, 65].
Our study shows the first preclinical evidence that Forskolin exerts anti-Parkinsonian effects in vivo. Forskolin treatment restored the motor coordination and hindlimb grip strength of the PINK1-KO rats at 10-, 11-, and 14-month-old, therapeutic effects that lasted more than five weeks following the last day of treatment (Figs. 1 and 2). Given that female PINK1-KO rats showed moderate motor symptoms compared to male rats at 10-month-old (Fig. 1A, B), we performed “washout” studies in older female PINK1-KO rats. Our results showed that by 11 months of age, the female PINK1-KO rats demonstrated a Parkinsonian phenotype as indicated by the increased total score values in the balance beam and the hindlimb grip strength fatigue (Fig. 1D and 2D). Interestingly, the neuroprotective effects of I.P. administaion of Forskolin in ameliorating motor coordination in symptomatic PINK1-KO rats persisted for three weeks before the animals relapsed following cessation of treatment, whereas the ability of Forskolin to restore hindlimb strength lasted more than five weeks (Figs. 1 and 2). In aggregate, our behavioral data suggest that I.P. administration of Forskolin can restore motor coordination and reverse muscle fatigue in symptomatic 10-month-old PINK1-KO rats. When comparing Forskolin with Levodopa, both treatments improved the motor coordination in PINK1-KO rats; nevertheless Forskolin induced a long-lasting therapeutic effect in terms of reducing motor symptoms and reverse the loss of hindlimb strength compared to Levodopa. Previously, Kelm-Nelsen et al. [35] tested effects of Levodopa on motor coordination of 8-month-old PINK1-KO male rats on a tapered beam. However, they observed no significant improvement in the average number of foot sls of PINK1-KO rat group treated with Levodopa. Interestingly, we found that Forskolin induced a complete restoration of hindlimb grip strength whereas Levodopa did not have an effect (Fig. 6).
Our compiled bioenergetic data suggest that activation of cAMP-dependent signaling via I.P. administration of Forskolin can reverse bioenergetic deficiencies in symptomatic PINK1-KO rats (Fig. 3), presumably by elevating PKA signaling in the mitochondrion via the PKA scaffold Dual Specificity A kinase anchoring protein 1 (D-AKAP1) [36]. Previously, Villeneuve et al. [38] reported a significant increase in the OCRs in the striatum of PINK1-KO rats compared to WT rats, suggesting that the loss of PINK1 leads to substantial bioenergetic alterations in the striatum. Consistent with our data (Fig. 3), Zhi et al. [66] found a substantial decrease in the basal and maximal respiration in the SN of aged (10–14 months) PINK1-KO mice compared to WT mice.
Our bioergentic data in the SN of PINK1-KO rats shows that Forskolin likely potentiates neuroprotective effects of PKA activation when mitochondria are damaged by the lack of PINK1. The molecular mechanism through which Forskolin enhances mitochondrion function in vivo may be elicited via the activation of cAMP/PKA downstream of PINK1 [36]. Indeed, consistent with its ability to enhance oxidative phosphorylation and energy level in the brain, I.P. administration Forskolin in vivo enhances oxidative phosphorylationand glycolysis in the SN (Fig. 3), which can lead to an increase in OCRs by enhancing complex I activity [67] or the cytochrome c oxidase [68]. Previous studies have shown that Forskolin diminishes oxidative stress and neuroinflammation in rodents in different models of neurodegeneration [21, 69, 70]. Moreover, Forskolin has been demonstrated to enhance mitochondrial function and elicit mitochondrial fusion via PKA-mediated phosphorylation of Drp1, an enhancer of mitochondrial fission, and elevate neuronal bioenergetics, specifically by enhancing oxidative phosphorylation via PKA-mediated phosphorylation of complex I to increase electron flow through complex I activity, reduced level of ROS, and increase ATP production through the ATP synthase in neuronal cells [71], improved mitochondrial membrane potential in stressed neurons [22, 72], and restore complex I, II, IV and V activities in the brain, diminished the level of neuroinflammatory cytokines and improved behavior in a rat model of autism spectrum disorders [73]. Our in vivo bioenergetics data (Fig. 3) so far is consistent with Forskolin exerting an increase oxidative phosphorylation in the substantia nigra PINK1-KO rats which may be partly due to downstream enhancement of PKA-mediated phosphorylation of complex proteins or PKA-mediated fusion of mitochondria by phosphorylating Drp1. Therefore, additional future studies are warranted to determine whether PKA-mediated phosphorylation of the mitochondrial fission mediator Drp1 (Ser637) [74, 75], a proxy of PKA activity in mitochondria associated with neuroprotective effects against toxic insults, and increase electron flow of complexes I and IV are increased in mitochondria derived from the substantia nigra from PINK1-KO rats as a potential mechanism by which Forskolin elevates oxidative phosphorylation in vivo (Fig. 3).
In addition, our in vivo data show that I.P. administration of Forskolin is disease modifying, at least in the PINK1-KO rat model. Indeed, I.P. administration of Forskolin can restore the dopaminergic loss in the SN of Parkinsonians rats (Fig. 4). Our data shows that PINK1-KO rats present significant neurodegeneration of the SN, as evidenced by a significant reduction in the integrated density of TH-specific fluorescence signal in midbrain slices compared to age-mated WT rats at 10 months. The significant reduction TH-specific fluorescence is consistent with other groups that reported a decrease in TH-positive neurons and a reduction of the area of the SN [33, 38]. In the PINK1-KO rats, the dopaminergic loss has been controversial, some groups reported no dopaminergic neurodegeneration [42] or decrease in dopamine concentration in the SN [76], however, it is worth noting that those studies have been conducted at an early age (8-month-old). As previously reported, the NMS can precede the dopaminergic neuron loss and the dopaminergic loss precedes the MS of PD [48]. PINK1-KO rats faithfully recapitulates NMS as previously reported. For instance, Marquis et al. [42] showed that PINK1-KO rats exhibited NMS including vocalization deficits, anhedonia and anxiety at 8 months old. In our study, we observed significant loss of SN neurons in symptomatic 10-month-old PINK1-KO rats which occurred with a significant loss of hindlimb strength, a NMS associated with chronic fatigue in PD patients. Given that Forskolin has been used extensively in cell culture protocols to stimulate differentiation of neuronal stem cells and neuropheres to midbrain dopamine neurons, the ability of Forskolin to restore the dopaminergic loss in the SN could be attributed to its ability to stimulate neurogenesis in the substantia nigra. This is consistent with previous in vitro studies that demonstrate cytoprotective effects of Forskolin. For example, Maruoka et al. (2011) found that Forskolin induces neurite outgrowth in PC12 cells via Nur77, an orphan nuclear receptor and immediate-early response gene; also, it has been described that Forskolin can induce neuronal differentiation of embryonal carcinoma stem cells Ntera2 [77]. However, given that neurogenesis occurs throughout the lifetime of the rat and a basal level of neurogenesis has been observed to occur following brain lesion with MPTP treatment [78–81], it is possible that an intrinsic, low baseline level of neurogenesis is further enhanced with I.P. treatment with Forskolin in middle aged, symptomatic PINK1-KO rats. However, future Western blot studies are needed to determine whether markers of neurogenesis are enhanced by Forskolin in I.P.-treated PINK1-KO rats or by performing BrdU content assays in the midbrain in BrdU-fed PINK1-KO rats to further functionally dissect the molecular mechanisms by which Forskolin reverses loss of midbrain dopanie neurons.
In this study, we show that Forskolin can exert its neuroprotective and neuromodulatory effect in PINK1-KO rats via the activation of the AC-cAMP-PKA pathway. The anti-Parkinsonian effects of Forskolin has been linked to its ability to reverse the loss of PKA activity in the midbrain of Parkinsonian rats as assessed by performing an ELISA-based assay to measure PKA activity [13]. On the other hand, I.P. administration of Forskolin completely restored PKA activity in SN derived from 10-month-old PINK1-KO rats to a similar manner as age matched WT rats (Fig. 5). This data is consistent with our previous research findings showing that enhancing downstream PKA activity can compensate for loss of PINK1 function in neuroblastoma cells and in PINK1-deficient primary cortical neurons [13, 36]. Consistent with the PKA activity assay data, Western blot analysis in substantia nigra from PINK1-KO rats I.P. treated with Forskolin was able to not only reverse the loss in the ratio of the level of phospho-CREB (Ser 133) to total CREB in PINK1-KO rats but also significantly increased the level of p-CREB relative to vehicle treated WT. Overall this Western blot data further demonstrates that I.P. treatment of symptomatic PINK1-KO rats with Forskolin elicits activation of neuroprotective PKA-related signaling pathways (Supplementary Figure 3). Therefore, based on our in vivo data, the neuroprotective effects of Forskolin is likely attributed to an increase in the synthesis of cAMP synthesis via adenylate cyclase, especially given the fact that cAMP-mediated signaling pathways modulates a myriad of neuronal functions. Specifically, in the striatum, cAMP participates in the regulation of glutamatergic excitatory synaptic transmission, facilitates the striatal neurotransmission [61, 82] and can trigger the PKA pathway phosphorylating the CREB which regulates the expression of immediate early genes [83].
In summary, our results show that Forskolin have an anti-Parkinsonian effect in vivo in a bone fide animal model for PD. Our studies also support that the therapeutic effect of Forskolin is mediated trough the AC-cAMP-PKA pathway; our preclinical data opens the possibility of using Forskolin and other drugs that elevate the cAMP levels. Finally, our results also highlight the need to further evaluate the therapeutic benefits of Forskolin in the setting of PD, either in isolation or in combination with current standard of care options such as Levodopa.
ACKNOWLEDGMENTS
This study was funded by the Pennington Foundation (Nevada), by National Institutes of Health (NIH) grant R01 NS105783, and MG Ph.D-fellowship (CVU 548217) by the National Council of Science and Technology (CONACYT).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JPD-213016.
REFERENCES
[1] | Seppi K , Ray Chaudhuri K , Coelho M , Fox SH , Katzenschlager R , Perez Lloret S , Weintraub D , Sampaio C , the collaborators of the Parkinson’s Disease Update on Non-Motor Symptoms Study Group on behalf of the Movement Disorders Society Evidence-Based Medicine C ((2019) ) Update on treatments for nonmotor symptoms of Parkinson’s disease-an evidence-based medicine review. Mov Disord 34: , 180–198. |
[2] | Mayeux R , Stern Y ((2012) ) Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006239. |
[3] | Yang W , Hamilton JL , Kopil C , Beck JC , Tanner CM , Albin RL , Ray Dorsey E , Dahodwala N , Cintina I , Hogan P , Thompson T ((2020) ) Current and projected future economic burden of Parkinson’s disease in the U.S. NPJ Parkinsons Dis 6: , 15. |
[4] | Gokcal E , Gur VE , Selvitop R , Babacan Yildiz G , Asil T ((2017) ) Motor and non-motor symptoms in Parkinson’s disease: Effects on quality of life. Noro Psikiyatr Ars 54: , 143–148. |
[5] | Pfeiffer RF ((2016) ) Non-motor symptoms in Parkinson’s disease. Parkinsonism Relat Disord 22: (Suppl 1), S119–122. |
[6] | Mendonca IP , Duarte-Silva E , Chaves-Filho AJM , Andrade da Costa B , Peixoto CA ((2020) ) Neurobiological findings underlying depressive behavior in Parkinson’s disease: A review. Int Immunopharmacol 83: , 106434. |
[7] | Klein C , Westenberger A ((2012) ) Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med 2: , a008888. |
[8] | Ammal Kaidery N , Thomas B ((2018) ) Current perspective of mitochondrial biology in Parkinson’s disease. Neurochem Int 117: , 91–113. |
[9] | Pickrell AM , Youle RJ ((2015) ) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85: , 257–273. |
[10] | Lansbury PT Jr , Brice A ((2002) ) Genetics of Parkinson’s disease and biochemical studies of implicated gene products. Curr Opin Genet Dev 12: , 299–306. |
[11] | Lotharius J , Brundin P ((2002) ) Pathogenesis of Parkinson’s disease: Dopamine, vesicles and alpha-synuclein. Nat Rev Neurosci 3: , 932–942. |
[12] | Sherer TB , Betarbet R , Greenamyre JT ((2001) ) Pathogenesis of Parkinson’s disease. Curr Opin Invest Drugs 2: , 657–662. |
[13] | Das Banerjee T , Dagda RY , Dagda M , Chu CT , Rice M , Vazquez-Mayorga E , Dagda RK ((2017) ) PINK1 regulates mitochondrial trafficking in dendrites of cortical neurons through mitochondrial PKA. J Neurochem 142: , 545–559. |
[14] | Grigoruta M , Martinez-Martinez A , Dagda RY , Dagda RK ((2020) ) Psychological stress phenocopies brain mitochondrial dysfunction and motor deficits as observed in a parkinsonian rat model. Mol Neurobiol 57: , 1781–1798. |
[15] | Cotzias GC , Van Woert MH , Schiffer LM ((1967) ) Aromatic amino acids and modification of parkinsonism. N Engl J Med 276: , 374–379. |
[16] | Rinne UK , Mölsä P ((1979) ) Levodopa with benserazide or carbidopa in Parkinson disease. Neurology 29: , 1584–1589. |
[17] | Tolosa E , Martí MJ , Valldeoriola F , Molinuevo JL ((1998) ) History of levodopa and dopamine agonists in Parkinson’s disease treatment. Neurology 50: (6 Suppl 6), S2–10; discussion S44-8. |
[18] | Stoker TB , Barker RA (2020) Recent developments in the treatment of Parkinson’s disease. F1000Res 9, F1000 Faculty Rev-862. |
[19] | Pateraki I , Andersen-Ranberg J , Hamberger B , Heskes AM , Martens HJ , Zerbe P , Bach SS , Moller BL , Bohlmann J , Hamberger B ((2014) ) Manoyl oxide (13R), the biosynthetic precursor of forskolin, is synthesized in specialized root cork cells in Coleus forskohlii. Plant Physiol 164: , 1222–1236. |
[20] | Seamon KB , Padgett W , Daly JW ((1981) ) Forskolin unique diterpene activator of adenylate cyclase in membranes and in intact cells. Proc Natl Acad Sci U S A 78: , 3363–3367. |
[21] | Owona BA , Zug C , Schluesener HJ , Zhang ZY ((2016) ) Protective effects of forskolin on behavioral deficits and neuropathological changes in a mouse model of cerebral amyloidosis. J Neuropathol Exp Neurol 75: , 618–627. |
[22] | Ivanova MA , Sitnikova LS , Stepanenko YD , Karelina TV , Sibarov DA , Abushik PA , Antonov SM ((2018) ) Forskolin as a neuroprotector and modulator of glutamate-evoked Ca2+entry into cerebellar neurons. Biochem (Mosc) Suppl Ser A Membr Cell Biol 12: , 390–393. |
[23] | Godard MP , Johnson BA , Richmond SR ((2005) ) Body composition and hormonal adaptations associated with forskolin consumption in overweight and obese men. Obes Res 13: , 1335–1343. |
[24] | Henderson S , Magu B , Rasmussen C , Lancaster S , Kerksick C , Smith P , Melton C , Cowan P , Greenwood M , Earnest C , Almada A , Milnor P , Magrans T , Bowden R , Ounpraseuth S , Thomas A , Kreider RB ((2005) ) Effects of coleus forskohlii supplementation on body composition and hematological profiles in mildly overweight women. J Int Soc Sports Nutr 2: , 54–62. |
[25] | Dave KD , De Silva S , Sheth NP , Ramboz S , Beck MJ , Quang C , Switzer RC , 3rd, Ahmad SO , Sunkin SM , Walker D , Cui X , Fisher DA , McCoy AM , Gamber K , Ding X , Goldberg MS , Benkovic SA , Haupt M , Baptista MA , Fiske BK , Sherer TB , Frasier MA ((2014) ) Phenotypic characterization of recessive gene knockout rat models of Parkinson’s disease. Neurobiol Dis 70: , 190–203. |
[26] | Dolle C , Flones I , Nido GS , Miletic H , Osuagwu N , Kristoffersen S , Lilleng PK , Larsen JP , Tysnes OB , Haugarvoll K , Bindoff LA , Tzoulis C ((2016) ) Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat Commun 7: , 13548. |
[27] | Maurissen JPJ , Marable BR , Andrus AK , Stebbins KE ((2003) ) Factors affecting grip strength testing. Neurotoxicol Teratol 25: , 543–553. |
[28] | Carter RJ , Morton J , Dunnett BB ((2001) ) Motor coordination and balance in rodents. Curr Protoc Neurosci Chapter 8: , Unit 8.12. |
[29] | Tung VW , Burton TJ , Quail SL , Mathews MA , Camp AJ ((2016) ) Motor performance is impaired following vestibular stimulation in ageing mice. Front Aging Neurosci 8: , 12. |
[30] | Sweis BM , Bachour SP , Brekke JA , Gewirtz JC , Sadeghi-Bazargani H , Hevesi M , Divani AA ((2016) ) A modified beam-walking apparatus for assessment of anxiety in a rodent model of blast traumatic brain injury. Behav Brain Res 296: , 149–156. |
[31] | Kelm-Nelson CA , Brauer AFL , Barth KJ , Lake JM , Sinnen MLK , Stehula FJ , Muslu C , Marongiu R , Kaplitt MG , Ciucci MR ((2018) ) Characterization of early-onset motor deficits in the Pink1-/- mouse model of Parkinson disease. Brain Res 1680: , 1–12. |
[32] | Paxinos G , Watson C ((2008) ), The Rat Brain in Stereotaxic Coordinates. Elsevier. |
[33] | Grant LM , Kelm-Nelson CA , Hilby BL , Blue KV , Paul Rajamanickam ES , Pultorak JD , Fleming SM , Ciucci MR ((2015) ) Evidence for early and progressive ultrasonic vocalization and oromotor deficits in a PINK1 gene knockout rat model of Parkinson’s disease. J Neurosci Res 93: , 1713–1727. |
[34] | Grigoruta M , Dagda RK , Diaz-Sanchez AG , Martinez-Martinez A ((2020) ) Psychological distress and lack of PINK1 promote bioenergetics alterations in peripheral blood mononuclear cells. Sci Rep 10: , 9820. |
[35] | Kelm-Nelson CA , Trevino MA , Ciucci MR ((2018) ) Quantitative analysis of catecholamines in the Pink1 -/- rat model of early-onset Parkinson’s disease. Neuroscience 379: , 126–141. |
[36] | Dagda RK , Gusdon AM , Pien I , Strack S , Green S , Li C , Van Houten B , Cherra SJ , 3rd, Chu CT ((2011) ) Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell Death Differ 18: , 1914–1923. |
[37] | Insel PA , Ostrom RS ((2003) ) Forskolin as a tool for examining adenylyl cyclase expression, regulation, and G protein signaling. Cell Mol Neurobiol 23: , 305–314. |
[38] | Villeneuve LM , Purnell PR , Boska MD , Fox HS ((2016) ) Early expression of Parkinson’s disease-related mitochondrial abnormalities in PINK1 knockout rats. Mol Neurobiol 53: , 171–186. |
[39] | Ren X , Butterfield DA ((2021) ) Fidelity of the PINK1 knockout rat to oxidative stress and other characteristics of Parkinson disease. Free Radic Biol Med 163: , 88–101. |
[40] | Schaar KL , Brenneman MM , Savitz SI ((2010) ) Functional assessments in the rodent stroke model. Exp Transl Stroke Med 2: , 13. |
[41] | Schallert T ((2006) ) Behavioral tests for preclinical intervention assessment. NeuroRx 3: , 497–504. |
[42] | Marquis JM , Lettenberger SE , Kelm-Nelson CA ((2020) ) Early-onset Parkinsonian behaviors in female Pink1-/- rats. Behav Brain Res 377: , 112175. |
[43] | Rizek P , Kumar N , Jog MS ((2016) ) An update on the diagnosis and treatment of Parkinson disease. CMAJ 188: , 1157–1165. |
[44] | Tibar H , El Bayad K , Bouhouche A , Ait Ben Haddou EH , Benomar A , Yahyaoui M , Benazzouz A , Regragui W ((2018) ) Non-motor symptoms of Parkinson’s disease and their impact on quality of life in a cohort of Moroccan patients. Front Neurol 9: , 170. |
[45] | DeMaagd G , Philip A ((2015) ) Parkinson’s disease and its management 1: Disease entity, risk factors, pathophysiology, clinical presentation, and diagnosis. P T 40: , 504–532. |
[46] | Marras C , Chaudhuri KR , Titova N , Mestre TA ((2020) ) Therapy of Parkinson’s disease subtypes. Neurotherapeutics 17: , 1366–1377. |
[47] | Frucht SJ ((2004) ) Non-motor symptoms of Parkinson disease update on the diagnosis and treatment. Neurologist 10: , 185–194. |
[48] | Vermeiren Y , Hirschberg Y , Mertens I , De Deyn PP ((2020) ) Biofluid markers for prodromal Parkinson’s disease: Evidence from a catecholaminergic perspective. Front Neurol 11: , 595. |
[49] | Menon B , Nayar R , Kumar S , Cherkil S , Venkatachalam A , Surendran K , Deepak KS ((2015) ) Parkinson’s disease, depression, and quality-of-life. Indian J Psychol Med 37: , 144–148. |
[50] | Sun C , Armstrong MJ ((2021) ) Treatment of Parkinson’s disease with cognitive impairment: Current approaches and future directions. Behav Sci (Basel) 11: , 54. |
[51] | Connolly BS , Lang AE ((2014) ) Pharmacological treatment of Parkinson disease: A review. JAMA 311: , 1670–1683. |
[52] | Truong DD , Bhidayasiri R , Wolters E ((2008) ) Management of non-motor symptoms in advanced Parkinson disease. J Neurol Sci 266: , 216–228. |
[53] | Kumar N , Van Gerpen JA , Bower JH , Ahlskog JE ((2005) ) Levodopa-dyskinesia incidence by age of Parkinson’s disease onset. Mov Disord 20: , 342–344. |
[54] | DeMaagd G , Philip A ((2015) ) Parkinson’s disease and its management 2: Introduction to the pharmacotherapy of Parkinson’s disease, with a focus on the use of dopaminergic agents. P T 40: , 590–600. |
[55] | DeMaagd G , Philip A ((2015) ) Parkinson’s disease and its management 4: Treatment of motor complications. P T 40: , 747–773. |
[56] | Colombo D , Abbruzzese G , Antonini A , Barone P , Bellia G , Franconi F , Simoni L , Attar M , Zagni E , Haggiag S , Stocchi F ((2015) ) The “gender factor” in wearing-off among patients with Parkinson’s disease: A post hoc analysis of DEEP study. ScientificWorldJournal 2015: , 787451. |
[57] | Turcano P , Mielke MM , Bower JH , Parisi JE , Cutsforth-Gregory JK , Ahlskog JE , Savica R ((2018) ) Levodopa-induced dyskinesia in Parkinson disease: A population-based cohort study. Neurology 91: , e2238–e2243. |
[58] | Calabresi P , Ghiglieri V , Mazzocchetti P , Corbelli I , Picconi B ((2015) ) Levodopa-induced plasticity: A double-edged sword in Parkinson’s disease? Philos Trans R Soc Lond B Biol Sci 370: , 20140184. |
[59] | Ahlskog JE , Muenter MD ((2001) ) Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 16: , 448–458. |
[60] | DeMaagd G , Philip A ((2015) ) Parkinson’s disease and its management 3: Nondopaminergic and nonpharmacological treatment options. P T 40: , 668–679. |
[61] | Padovan-Neto FE , West AR ((2017) ) Regulation of striatal neuron activity by cyclic nucleotide signaling and phosphodiesterase inhibition: Implications for the treatment of Parkinson’s disease. In Phosphodiesterases: CNS Functions and Diseases, Zhang H-T, Xu Y, O’Donnell JM, eds. Springer International Publishing, pp. 257–283. |
[62] | Nthenge-Ngumbau DN , Mohanakumar KP ((2018) ) Can cyclic nucleotide phosphodiesterase inhibitors be drugs for Parkinson’s disease? Mol Neurobiol 55: , 822–834. |
[63] | Casacchia M , Meco G , Castellana F , Bedini L , Cusimano G , Agnoli A ((1983) ) Therapeutic use of a selective camp phosphodiesterase inhibitor (Rolipram) in Parkinson’s disease. Pharmacol Res Commun 15: , 329–334. |
[64] | Menniti FS , Faraci WS , Schmidt CJ ((2006) ) Phosphodiesterases in the CNS: Targets for drug development. Nat Rev Drug Discov 5: , 660–670. |
[65] | Polito M , Klarenbeek J , Jalink K , Paupardin-Tritsch D , Vincent P , Castro LR ((2013) ) The NO/cGMP pathway inhibits transient cAMP signals through the activation of PDE2 in striatal neurons. Front Cell Neurosci 7: , 211. |
[66] | Zhi L , Qin Q , Muqeem T , Seifert EL , Liu W , Zheng S , Li C , Zhang H ((2019) ) Loss of PINK1 causes age-dependent decrease of dopamine release and mitochondrial dysfunction. Neurobiol Aging 75: , 1–10. |
[67] | Papa S , Sardanelli AM , Cocco T , Speranza F , Scacco SC , Technikova-Dobrova Z ((1996) ) The nuclear-encoded 18 kDa (IP) AQDQ subunit of bovine heart complex I is phosphorylated by the mitochondrial cAMP-dependent protein kinase. FEBS Lett 379: , 299–301. |
[68] | Acin-Perez R , Salazar E , Kamenetsky M , Buck J , Levin LR , Manfredi G ((2009) ) Cyclic AMP produced inside mitochondria regulates oxidative phosphorylation. Cell Metab 9: , 265–276. |
[69] | Kumar A , Singh N ((2017) ) Pharmacological activation of protein kinase A improves memory loss and neuropathological changes in a mouse model of dementia of Alzheimer’s type. Behav Pharmacol 28: , 187–198. |
[70] | Mehan S , Parveen S , Kalra S ((2017) ) Adenyl cyclase activator forskolin protects against Huntington’s disease-like neurodegenerative disorders. Neural Regen Res 12: , 290–300. |
[71] | Palorini R , De Rasmo D , Gaviraghi M , Sala Danna L , Signorile A , Cirulli C , Chiaradonna F , Alberghina L , Papa S ((2013) ) Oncogenic K-ras expression is associated with derangement of the cAMP/PKA pathway and forskolin-reversible alterations of mitochondrial dynamics and respiration. Oncogene 32: , 352–362. |
[72] | Ivanova MA , Sitnikova LS , Karelina TV , Sibarov DA , Abushik PA , Antonov SM ((2019) ) The neuroprotective effect of forskolin and its influence of mitochondrial dysfunction in neurons in primary rat cerebellum cultures. Neurosci Behav Physiol 49: , 1096–1099. |
[73] | Mehan S , Rahi S , Tiwari A , Kapoor T , Rajdev K , Sharma R , Khera H , Kosey S , Kukkar U , Dudi R ((2020) ) Adenylate cyclase activator forskolin alleviates intracerebroventricular propionic acid-induced mitochondrial dysfunction of autistic rats. Neural Regen Res 15: , 1140–1149. |
[74] | Chang CR , Blackstone C ((2007) ) Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem 282: , 21583–21587. |
[75] | Zhang J , Feng J , Ma D , Wang F , Wang Y , Li C , Wang X , Yin X , Zhang M , Dagda RK , Zhang Y ((2019) ) Neuroprotective mitochondrial remodeling by AKAP121/PKA protects HT22 cell from glutamate-induced oxidative stress. Mol Neurobiol 56: , 5586–5607. |
[76] | de Haas R , Heltzel L , Tax D , van den Broek P , Steenbreker H , Verheij MMM , Russel FGM , Orr AL , Nakamura K , Smeitink JAM ((2019) ) To be or not to be pink(1): Contradictory findings in an animal model for Parkinson’s disease. Brain Commun 1: , fcz016. |
[77] | Tegenge MA , Roloff F , Bicker G ((2011) ) Rapid differentiation of human embryonal carcinoma stem cells (NT2) into neurons for neurite outgrowth analysis. Cell Mol Neurobiol 31: , 635–643. |
[78] | Arenas E , Denham M , Villaescusa JC ((2015) ) How to make a midbrain dopaminergic neuron. Development 142: , 1918–1936. |
[79] | Malmersjö S , Liste I , Dyachok O , Tengholm A , Arenas E , Uhlén P ((2010) ) Ca2+and cAMP signaling in human embryonic stem cell-derived dopamine neurons. Stem Cells Dev 19: , 1355–1364. |
[80] | Zhang X , Klueber KM , Guo Z , Cai J , Lu C , Winstead WI , Qiu M , Roisen FJ ((2006) ) Induction of neuronal differentiation of adult human olfactory neuroepithelial-derived progenitors. Brain Res 1073-1074: , 109–119. |
[81] | Zhao M , Momma S , Delfani K , Carlen M , Cassidy RM , Johansson CB , Brismar H , Shupliakov O , Frisen J , Janson AM ((2003) ) Evidence for neurogenesis in the adult mammalian substantia nigra. Proc Natl Acad Sci U S A 100: , 7925–7930. |
[82] | Colwell CS , Levine MS ((1995) ) Excitatory synaptic transmission in neostriatal neurons: Regulation by cyclic AMP-dependent mechanisms. J Neurosci 15: , 1704–1713. |
[83] | Pereira JB , Svenningsson P , Weintraub D , Brønnick K , Lebedev A , Westman E , Aarsland D ((2014) ) Initial cognitive decline is associated with cortical thinning in early Parkinson disease. Neurology 82: , 2017–2025. |


