
A Review on Response to Device-Aided Therapies Used in Monogenic Parkinsonism and GBA Variants Carriers: A Need for Guidelines and Comparative Studies
Abstract
Parkinson’s disease (PD) is in some cases predisposed-or-caused by genetic variants, contributing to the expression of different phenotypes. Regardless of etiology, as the disease progresses, motor fluctuations and/or levodopa-induced dyskinesias limit the benefit of pharmacotherapy. Device-aided therapies are good alternatives in advanced disease, including deep brain stimulation (DBS), levodopa-carbidopa intestinal gel, and continuous subcutaneous infusion of apomorphine. Candidate selection and timing are critical for the success of such therapies. Genetic screening in DBS cohorts has shown a higher proportion of mutation carriers than in general cohorts, suggesting that genetic factors may influence candidacy for advanced therapies. The response of monogenic PD to device therapies is not well established, and the contribution of genetic information to decision-making is still a matter of debate. The limited evidence regarding gene-dependent response to device-aided therapies is reviewed here. An accurate understanding of the adequacy and responses of different mutation carriers to device-aided therapies requires the development of specific studies with long-term monitoring.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative condition after Alzheimer’s disease and is the fastest-growing neurodegenerative disorder, with a projected prevalence of 12 million by 2040 [1]. The incidence ranges from 5 to 25 annual cases per 100,000, with the mean age of onset in the seventh decade [2].
There is still a knowledge gap in our understanding of the molecular basis for neurodegeneration in PD. Several environmental and genetic risk factors have been identified, including rare monogenic disorders [3].
A broad genotype-phenotype correlation can be recognized for certain variants [4]. Besides, monogenic PD may benefit from gene-specific treatment strategies (i.e., LRRK2 and GBA) [5]. However, it remains to be seen whether this applies to device-aided therapies such as deep brain stimulation (DBS), levodopa-carbidopa intestinal gel infusion (LCIG), or continuous subcutaneous apomorphine infusion (CSAI) [6, 7]. These therapies are potential options for individuals with PD with complications such as refractory “wearing-off” and levodopa-induced dyskinesias [8].
Surprisingly, genetic screening has demonstrated an overrepresentation of specific genetic variants in DBS cohorts (up to 29%) compared to the overall PD population (estimated at 5–10%). GBA, LRRK2, and PRKN variants are the most frequent [9].
This association raises whether individuals with certain genetic variants represent better candidates for specific device-aided therapies and whether these genetic factors affect the response to such therapies. We suggest reading a previous publication addressing the phenotype-genotype relationship in decision-making on device-aided therapies [10]. The latter issue, how genetic factors affect treatment response, is addressed below.
DEVICE-AIDED THERAPIES
The success of device-aided therapies depends on selecting the suitable device for the right patient. Key eligibility features include: (I) ≥1 hour of troublesome dyskinesia per day, or≥2 hours “off” symptoms per day and need to take levodopa≥5-times per day; (II) no more than mild dementia and absence of troublesome hallucinations; and (III) significant difficulty with activities of daily living. Patients demonstrating good levodopa response who are emotionally stable, physically healthy, cognitively intact, and younger (preferably < 70 years of age) are ideal candidates for CSAI, LCIG, or DBS [11]. While these therapies seem to provide a similar improvement in reducing “off” time by around 40–60%, their effects on dyskinesia and non-motor symptoms are heterogeneous, and their side effects and complications can be quite different [12]. Table 1 summarizes their main indications, advantages, disadvantages, and contraindications.
Table 1
Comparison between different device-aided therapies
| Ideal clinical indications | Advantages | Disadvantages | Relative contraindications |
| Deep Brain Stimulation | |||
| ⇒ PD>5 y (>4 y in EARLY Stim) | ⇒ 25–68% reduction in “off” periods | ⇒ Procedural: Invasive neurosurgery with uncommon but serious peri-operative complications (e.g., intracranial hemorrhage, postoperative wound or hardware infection, transient postoperative delirium) | ⇒ Poor cognition |
| ⇒ <75 y | ⇒ 40–60% reduction in dyskinesias | ⇒ Described AE: Balance and gait problems, freezing, depression, anxiety, apathy, suicide committed, psychosis, ICD, cognitive decline, dysarthria, swallowing problems, eyelid apraxia, dyskinesia, pain, weight gain. | ⇒ Troublesome hallucinations |
| ⇒ Motor improvement > 30% with Levodopa | ⇒ Significant benefit on tremor, bradykinesia, rigidity, dystonia, dyskinesia. | ⇒ Nonresponding symptoms: Autonomic dysfunction, dysphagia, dysphonia, cognition or verbal fluency, postural instability, RBD, excessive daytime sleepiness. | ⇒ Poor social support |
| ⇒ Significant motor fluctuations and dyskinesia | ⇒ Potential reduction on total LEDD (STN-DBS) | ⇒ Ref. [19, 111, 117–120] | ⇒ Target/most bothersome symptoms are not responsive to Levodopa (except tremor) |
| ⇒ Medication-resistant tremor | ⇒ Symptoms that may respond includes, truncal deviations, fatigue, inner restlessness, anxiety, depression, ICD, freezing of gait, urinary and gastrointestinal symptoms, weight loss | ⇒ Severe depression | |
| ⇒ Ref. [7, 13, 111–114] | ⇒ Ref. [19, 111, 115–118] | ⇒ Dopamine dysregulation or punding | |
| ⇒ Poor operative candidate, severe brain atrophy or intracranial lesions affecting surgical approach | |||
| ⇒ Severe systemic disease | |||
| ⇒ Lack of compliance at follow-up | |||
| ⇒ Ref. [7, 111–114] | |||
| Levodopa–carbidopa intestinal gel infusion | |||
| ⇒ Advanced PD | ⇒ 47–59% reduction in “off” periods | ⇒ Procedural: Invasive procedure with less common but serious peri-operative complications; PEG-J tube blockage or failure; PEG-J tube or skin infection | ⇒ Poor cognition |
| ⇒ with significant motor fluctuations | ⇒ 49–64% reduction in time with dyskinesias | ⇒ Infusion device thought large and cumbersome | ⇒ Troublesome hallucinations |
| ⇒ Troublesome “off” periods > 3 h per day | ⇒ Potential benefit on NMS: anxiety episodes, sleep, irritability, urinary and orthostatic symptoms, pain, constipation, attention and fatigue, delusions, mood disorders and ICD. | ⇒ Aesthetics, excessive granulation tissue | ⇒ Poor social support |
| ⇒ Ref. [7, 111, 121, 122] | ⇒ May be effective as monotherapy | ⇒ Described AE: Abdominal pain, nausea, flatulence, constipation, diarrhea, orthostatic hypotension, peak-dose dyskinesias, diphasic dyskinesias, insomnia, sleep attacks, anxiety. | ⇒ Poor fine motor skills |
| ⇒ Ref. [19, 111, 121–126] | ⇒ Rarely reported AE include peripheral neuropathy, weight loss | ⇒ Contraindications for PEG procedural | |
| ⇒ Nonresponding symptoms: Freezing and festination with no response to levodopa, postural instability, dysphagia, and dysarthria | ⇒ Conditions that interfere with kinetic of the drug | ||
| ⇒ Ref. [19, 111, 124, 126] | ⇒ pre-existing peripheral neuropathy | ||
| ⇒ Dopamine dysregulation syndrome or punding | |||
| ⇒ Severe systemic disease | |||
| ⇒ Ref. [7, 111] | |||
| Continuous subcutaneous infusion of apomorphine | |||
| ⇒ PD of > 3 y (TOLEDO study) | ⇒ 25–85% reduction in motor fluctuations | ⇒ Related to device: skin complications and nodules from subcutaneous injections | ⇒ Poor social support |
| ⇒ Advanced PD, with motor fluctuations, troublesome “off” time, and dyskinesias | ⇒ 43–64% reduction in time with dyskinesias | ⇒ Requirements for placement of subcutaneous line daily. | ⇒ Poor fine motor skills |
| ⇒ Good response to levodopa | ⇒ Older age, slight to moderate dementia and depression are not absolute contraindications | ⇒ Described AE: weight gain, neuropsychiatric side effects, daytime somnolence, nausea, orthostatic hypotension, coombs antiglobulin positive hemolytic anemia. | ⇒ Presence of dopamine dysregulation, punding or ICD |
| ⇒ Effective rescue doses of apomorphine but either needed too frequently or are associated with increasing dyskinesia | ⇒ Potential benefit on NMS, such as pain (related to dystonia), restless legs, panic attacks, depression, apathy, insomnia, slowness of thinking, swallowing, micturition disorders. | ⇒ Risk of ICD | ⇒ Severe hallucinations or dementia |
| ⇒ Ref. [7, 18, 111, 124, 127] | ⇒ Ref. [18, 19, 111, 128–134]. | ⇒ Potential benefit on dyskinesias may be delayed by weeks. | ⇒ Orthostatic hypotension |
| ⇒Ref. [19, 111] | ⇒ Severe fatigue syndrome | ||
| ⇒ Severe systemic disease | |||
| ⇒ Anticoagulation | |||
| ⇒ Ref. [7, 18, 111, 124] | |||
DBS, deep brain stimulation; LCIG, Levodopa-carbidopa intestinal gel; CASI, continual apomorphine subcutaneous infusion; iPD, idiopathic Parkinson’s disease; NMS, non motor symptoms; PEJ, percutaneous endoscopic Jejunostomy; AE, adverse effects; ICD, impulse control disorders; LEDD, Levodopa equivalent daily dose.
In terms of selecting which device therapy is most appropriate, DBS is favored with younger patients and minimal non-levodopa-responsive motor symptoms (except for tremor) [13, 14]. DBS may be contraindicated if there is dementia, hallucinations, uncontrolled depression, marked postural and gait problems, severe brain atrophy, or suspected atypical parkinsonism.
LCIG can still be considered for patients with mild-to-moderate dementia or age > 70 years, even with severe depression [15]. However, patients with dopamine dysregulation, punding, or pre-existent peripheral neuropathies may be less favorable candidates.
CSAI can be considered if there are mild hallucinations or moderate cognitive impairment. Moreover, it might improve neuropsychological performance [16, 17]. In addition, this device may ameliorate depression, apathy, “off” pain, and slowness of thinking [18]. However, it seems less favorable in patients with impulse control disorders (ICD), marked psychosis, daytime somnolence, and troublesome orthostatic hypotension [19].
We caution against dogmatism since there is also favorable evidence for device therapies in some of these reported contraindications. For example, there are cases where LCIG and CSAI improve ICD, and where LCIG decreases dopamine dysregulation syndrome [20–22]
A relevant factor in decision-making for device-aided therapies is the long-term outcome expectancy. Chronic STN-DBS can cause dysarthric speech, problems in verbal fluency, worsening freezing of gait, and axial symptoms. These are important determinants of quality of life [23–25]. Severe pre-operative gait difficulties might predict limited long-term DBS benefits [23, 24]. Moreover, motor outcomes one year after bilateral STN-DBS are inversely correlated with the rate of progression of motor symptoms [26].
The leading causes of discontinuation of LCIG therapy in long-term follow-up include worsening cognition, dyskinesias, chronic polyneuropathy, weight loss, and hallucinations. Eventually, LCIG may become ineffective [27, 28].
Like LCIG, long-term CSAI may worsen cognition, dyskinesias, postural instability, and hallucinations. CSAI also causes sedation and orthostatic hypotension. These factors may obscure the long-term benefits in some patients [29–32]. Furthermore, a decrease in therapeutic effect may become an important reason for discontinuation within the first four years [33].
In conclusion, it is essential to recognize patient heterogeneity and, if possible, identify biomarkers of short and long-term outcomes. Understanding phenotype-genotype relationships and how variants predict the risk of significant disease milestones [34] may affect the timing, appropriateness, expected outcome, and expectations for device-aided therapies [10].
CLINICAL FEATURES OF CAUSAL MUTATIONS AND GENETIC RISK FACTORS FOR PARKINSONISM
Most PD cases are sporadic, associated with genetic, epigenetic, and environmental risk factors [35]. The most frequent genetic risk factors for sporadic PD are GBA, SNCA, LRRK2, and MAPT. In addition, PD-causal monogenic variants account for 5 to 10% of cases [35, 37].
The severity and risk associated with GBA depend on the variant [38]. Overall, the GBA motor phenotype resembles idiopathic PD, possibly with faster progression, more bradykinesia, and levodopa-induced dyskinesias [39, 40]. Cognitive changes appear earlier and tend to be more prominent, particularly in memory and visuospatial domains [41–44]. The dementia risk with a severe GBA variant is 2.9 times higher than for mild variants and 5.6 times higher than for idiopathic PD [38]. Severe GBA variants may also have more neuropsychiatric and autonomic disturbances [45]. Bi-allelic carriers have faster disease progression and higher mortality than idiopathic PD [42, 46, 47].
Autosomal dominant (AD) PD includes SNCA, LRRK2, and VPS35. LRRK2 variants are the most frequent cause of monogenic PD, with p.G2019S, p.R1441C, and p.R1441G being the most common. The phenotype resembles idiopathic PD, with atypical signs rarely reported [48]. A more uniform disease course regardless of the age of onset [51] and a higher likelihood to develop dystonia and dyskinesias earlier have been reported [49]. Like iPD, LRRK2-PD most likely manifests PIGD phenotype with disease progression [50]. Remarkably, LRRK2 p.G2019S carriers with a PIGD phenotype have a lower risk of dementia than observed in non-carriers with this phenotype [51]. Overall, LRRK2-PD have a lower risk of dementia [48, 52–54], manifests less olfactory impairment [49, 55], and RBD [56, 57] than non-carriers with PD.
SNCA variants include duplications or triplications and missense mutations, with p.A53T as the most frequent. Some SNCA variants (p.A53T and p.E46K) are more likely to develop dementia [58]. Depression, psychosis, and autonomic compromise are also more common for certain SNCA variants compared to idiopathic PD [5, 48, 52, 59–62]. For VPS35, the most common point mutation p.D620N presents with classical PD features with minimal atypical signs, although postural instability and daytime sleepiness may be more common [48, 63, 64].
Autosomal recessive (AR) PD, such as PRKN, PINK1, and DJ1, present with a phenotype similar to idiopathic PD but with younger age of onset. Patients with PRKN variants can respond dramatically to low doses of dopaminergic agents [65]. In addition, it can present with exercise-induced lower extremity dystonia [66] and gait compromise associated with diphasic dyskinesias [67]. Variants of these three genes are uncommonly associated with atypical parkinsonian features [68].
On the other hand, ATP13A2, PLA2G6, FBXO7, DNAJC6, SYNJ1, and VPS13C represent AR forms that often manifest with juvenile parkinsonism, faster progression, and atypical features including supranuclear gaze palsy, oculomotor or eyelid apraxia, intellectual disability, facial-faucial-finger mini-myoclonus, ataxia, dysarthria, dysphagia, pyramidal signs, seizures, psychosis and dysautonomia [69–77].
Because of scarce evidence, how these causal or risk-modifying variants affect outcomes is debatable. Currently, decision-making for advanced therapies is based on clinical features, which are unreliable for inferring the underlying genetics. Incomplete penetrance (i.e., LRRK2, GBA), phenotype variability, and environmental factors affect clinical features [34]. At the same time, there is evidence that genetics affect outcomes in ways that would affect decision-making. This is reviewed in the next section.
CURRENT EVIDENCE FOR DEEP BRAIN STIMULATION IN MONOGENIC PARKINSONISM AND GBA VARIANTS CARRIERS
Pal et al. analyzed the Consortium On Risk for Early-onset PD (CORE-PD) cohort, emphasizing PRKN, LRRK2, and GBA. Ninety-nine individuals who received DBS, and 684 without DBS, were identified. Carriers of pathogenic (or “risk” variants for GBA) were more common in the DBS vs. non-DBS groups (26.5% vs. 16.8%, respectively) [78]. Performing genetic screening in a cohort of 94 DBS-treated PD patients, Angeli et al. found that 26% had PRKN, LRRK2 p.G2019S, or GBA variants. No pathogenic variants were found in SNCA [9]. Likewise, De Oliveira et al. reported that in addition to GBA variants, PRKN and LRRK2 were the most common monogenic forms in DBS cohorts [79]. Interestingly, the response to DBS seems to be related to the variants. Tables 2–5 summarize the evidence for outcomes obtained with device-aided therapies in monogenic parkinsonism and GBA carriers.
Table 2
Available evidence of outcomes for different device-aided therapies in GBA variants carriers
| Device | Best evidence available | Number of cases | Age at onset (y) | Disease duration till advanced therapy | Follow-up period | Outcome | Complications |
| LCIG | Case series (abstract) [105] | 11 | Mean about 54 | 13.54 (7.78) | Not specified (After titration and stabilization of LCIG treatment) | Higher UPDRS-III scores compared to idiopathic PD (44.3 vs. 29), possibly because of a more severe phenotype. | GBA carriers were treated on lower doses (1476 vs. 1702) due to higher rates of hallucinations (71.4% vs. 63.6%) and lower cognitive scores (MoCA 18 vs. 23.3), compared to idiopathic PD |
| DBS | Systematic Review | 50 (STN n = 33, GPi-DBS n = 4, Vim-DBS n = 1, NA n = 12) | 21 to 58 | 1.6 to 7.5 y | Favorable long term motor outcome in 18. Moderate benefit in 3. Poor outcome in 9. One study reported better outcomes with STN-DBS and Vim-DBS than with GPi-DBS. | Cognitive impairment faster than non-carriers | |
| STN-DBS | Systematic Review and Meta-analysis [81] | 33 31 heterozygous and 2 homozygous | Mean 41.4 –49-7 | 11.2 (5)–17.3 (5.5) | 24–90 mo | UPDRS-III score improved by 49% (20 points: 95% CI, 4.5–35.5; p = 0.01) (n = 33). LEDD was reduced by 22% (269.2 mg/d; 95% CI, 226.8–311.5 mg/d; p < 0.001) (n = 30). UPDRS-IV score improved by 37 to 80% (n = 16) | Progressive cognitive decline, in 3 studies with a mean follow-up of 72.2 [21.1] mo (n = 26) |
| DBS | Systematic Review [79] | 19 STN-DBS (n = 16) Gpi-DBS (n = 2) | 35.6–54 | 6.2–21 y | 1–10 y | STN-DBS at 2–6-y responses were marked in 1, satisfactory in 1 and unsatisfactory in 1. After > 6 y, two had satisfactory improvement. | All the studies showed cognitive decline in GBA patients who underwent STN or GPi-DBS |
| Vim-DBS (n = 1) | LEDD decreased. Motor complications improved from 37% to 100%. Two GPi-DBS cases had 24.8% improvement, and 1 Vim-DBS case showed 42.9% improvement (both at 1-y follow-up) |
LEDD, levodopa equivalent daily dose.
Table 3
Available evidence of outcomes for different device-aided therapies in autosomal dominant monogenic parkinsonism
| Gene | Device | Best evidence available | Number of cases | Age at onset (y) | Disease duration till advanced therapy | Follow-up period | Outcome | Complications |
| SNCA Duplication | DBS | Systematic Review | 2 STN-DBS | 35 and 41 | 5 and 6 y | One and 2 y | UPDRS III improved 43% and 52% LEDD decreased by 29.7% (n = 1). Motor complications improved by 87.5% (n = 1). Stable cognition. ICD resolved (n = 1) Depression scores improved or remained stable | Stimulation-induced right foot dystonia relieved by modulating stimulation parameters (n = 1) |
| Systematic Review [80] | 4 STN-DBS (n = 3) GPi-DBS (n = 1) | 18 to 40 (mean 33.5) | 5 to 8 y (mean 6.25) | From 1 mo to 2 y | Favorable motor outcome | Cognitive deterioration (n = 2) | ||
| Systematic Review [135] | 3 STN-DBS (n = 2) GPi-DBS (n = 1) | From 1 mo to 3 y | Improvement in motor features and reduction in LEDD | Foot dystonia, decline in verbal fluency and attention (n = 1). MMSE worsened from 26 to 23 (n = 1) | ||||
| Systematic Review and Meta-analysis [81] | 1 STN-DBS | 41 | 5 y | 12 mo | 43% motor improvement 63% LEDD reduction | MMSE worsened from 30 to 29 points. | ||
| SNCA missense | STN-DBS | Case report [136] | 1 p.A53E Heterozygous | 42 | 5 y | NR | Reduced fluctuations and increased “on” time. | NR |
| LRRK2 | LCIG | Case report [103] | 1 | 49 | 19 y | 2 y | Effective treatment up until his death | Deceased after 24 mo (colon cancer) |
| Case series (Abstract) [105] | 16 p.G2019S in Ashkenazi Jewish | NR | NR | NR | Similar motor outcomes to non-carriers LCIG dose 1622 (vs. 1702 in non-carriers) | Dyskinesia in 93.3% (vs. 90.6% in non-carriers) Hallucinations 42.8% (vs. 63.6% in non-carriers) | ||
| CSAI | Case report [108] | 1 | 42 | 14 y | NR | NR | NR | |
| DBS | Systematic Review [135] | 72 | NR | NR | 3 mo to 7 y | No significant differences compared to non-carriers | A single p.N1437H carrier with significant psychiatric comorbidity committed suicide after 6 mo | |
| Systematic Review[80] | 87 STN-DBS (n = 79) Target NR (n = 8) | 29 to 62 | 0.25 to 7 y | Outcome reported in 73 (83.9%) Poorer outcome in p.R1441G carriers compared to p.G2019S carriers | Two p.T2031S carriers developed neuropsychiatric problems after 5–7 y | |||
| Systematic Review [79] | 50 p.G2019S (n = 44) | 34 to 55 | 5 to 18 y | Intermediate follow-up (2–6 y) in 29 (58%) | At Intermediate follow-up all had marked or satisfactory improvement. Three of four p.R144G carriers had < 30% improvement. Motor complications improved from 33.3% to 75% LEDD decreased from 17.5% to 75% | Two p.T2031S carriers experienced behavioral disorders p.R1441G carriers had significantly less improvements in UPDRS II (22%) when compared with the LRRK2 negative group | ||
| Systematic Review and Meta-analysis [81] | 46 STN-DBS | 43.4 (3.7) | 13.4 (1.6) y | 6–24 mo | UPDRS-III improved by 46% in LRRK2 (n = 46) vs. 53% in iPD LEDD was reduced by 61% (n = 27) vs. 55% in iPD UPDRS-II improved 45.2% to 66.7% in p.G2019S carriers (n = 10). UPDRS-IV Improved by 50% to 75% (n = 15) | UPDRS-II deteriorated 10% in p.R1441G carriers (n = 4) Stable postsurgical Mattis dementia rating scale score (n = 9) | ||
| VPS35 | DBS | Systematic Review [79] | 3 p.D620N STN-DBS | 42, 45 and 49 | 13 and 19 y. NR in 1 case | 1, 5, and 8 y | LEDD was reduced 30 to 76.5% UPDRS-III improved 36 to 43.8% | One patient had episodes of FOG and falls after the drastic reduction of LEDD, which improved after medication adjustment and stimulation. |
| Systematic review [80] | 5 p.D620N STN-DBS (n = 3) NR (n = 2) | 42 to 49 (mean 45.75) (n = 4) | 10 to 13 y (mean 11.33) (n = 3) | 1–8 y (n = 3) | Favorable in 4, minor motor benefit in 1 UPDRS-III improvement of 69% (8 y), 36% (1 y) and 37% (n = 3) | FOG and falls after surgery, which improved with levodopa and stim (n = 1) Significant dysarthria (n = 1) | ||
| Systematic Review [135] | 6 p.D620N (n = 5) p.R524W (n = 1) | 37 to 49 (mean 46.6) | 7 to 21 y (mean = 12.8) (n = 4) | 1 to 8 y (n = 3) | Good outcome (n = 2) 76% improvement in UPDRS-III (8 y post-operative) 36% improvement in UPDRS-III (1 y) (n = 1) 37% improvement in UPDRS-III and decrease of peak-dose dyskinesia (n = 1) Modest effect (n = 1) | Dysarthria (n = 1) Increase frequency of falls and FOG (n = 1) |
NR, not reported; LEDD, levodopa equivalent daily dose.
Table 4
Available evidence of outcomes for different device-aided therapies in monogenic autosomal recessive parkinsonism
| PRKN | LCIG | Case report | 1 heterozygous PRKN p.T240M | 10 | 14 y | 83 mo | 88% improvement in UPDRS III 74% improvement in NMSS 79% improvement in PDQ-8 | NR |
| Case series [103] | 2 | 28 and 43 | 35 and 22 y | 3 mo and 5 mo | NR | One case deceased 3 mo after | ||
| CSAI | Case series [109] | 2 | 7 and 29 | 25 and 18 y | Case 1: NR Case 2:2 y | Case 1: NR; Case 2: “with motor benefit” | Case 1: NR (severe motor and psychiatric features before starting CSAI). Case 2: Psychotic symptoms resolved after stopping CSAI. | |
| DBS | Systematic Review [80] | 67 STN-DBS (n = 51) GPi-DBS (n = 5) Zona inserta (n = 1) | 7 to 52 | 0.5 to 7 y | Favorable long-term motor outcome in 76.1% Modest improvement in 4 patients (6.0%) Poor benefit in 2 cases (3.0%) | Like non-carriers | ||
| Systematic Review [135] | 27 Homozygous/ compound heterozygous | 3 mo to 8 y | No difference compared to non-carriers. DBS efficacy in cases with disease duration up to 45 y, with sustained response for many years. | |||||
| Systematic Review [79] | 25 STN-DBS (n = 22) GPi-DBS (n = 3) (homozygotes/ compound heterozygotes | STN-DBS 18/22 had < 2 y follow-up 7/22 had a 2–6 y follow-up outcomes GPi-DBS cases had 12 mo follow-up | At 2–6 y of STN-DBS follow-up 2/7 had marked responses, 4/7 Satisfactory responses, and 1/7 Unsatisfactory responses The mean LEDD change range from 2.1% to 91.7% Improvement in UPDRS IV raged from 20% to 100% GPi Unsatisfactory motor response (21% of improvement) UPDRS-IV improved by 70% | 3 of 4 homozygous STN-DBS with slightly worsened of UPDRS-I after surgery worsening of parkinsonian hypophonia was observed in 1 PRKN patient 3 patients experienced ballistic dyskinesias after surgery | ||||
| Systematic Review and Meta-analysis STN-DBS [81] | 36 18 homozygotes, and 18 heterozygotes | 10 to 45 | 4 to 45 y. Mean = 23.9 (3.6) y | 3 mo to 6 y | UPDRS-III improved by 43 % LEDD was reduced by 61% (n = 23) UPDRS-II improved by 62% to 81.8% (n = 4) UPDRS-IV improved by 20% to 100% (n = 13) | PRKN carriers (n = 8) showed no or minimal undesired effects | ||
| PINK-1 | LCIG | Case report | 1 homozygous | 29 | 26 y | 4 y | Excellent response to low-moderate infusion rates (30–60 mg/h, LEDD 600–900). After 3 y, she required higher infusion rates plus 3–4 bolus of 40 mg/day (LEDD *1,200 mg/24 h). She developed dyskinesias and the hourly rate was reduced. | Sensory axonal polyneuropathy. Vitamin B6 deficit. 4 y after starting LCIG therapy she developed dopamine dysregulation syndrome, marked dyskinesias, ICD, punding behavior and psychosis |
| DBS | Case reported in a series [96] | 1 Homozygous STN-DBS | 30 | 30 y | 3–6 y | Favorable motor outcome comparable to patients without mutations. A 43.7% change in UPDRS-III in the long term | NR | |
| Case report [97] | 1 Homozygous GPi-DBS | 30 | 19 y | 2 mo | Improvement of gait, dystonia, and dyskinesia No difference in UPDRS-III score on-medication, but reduction in UPDRS IV-score and LEDD. | NR |
NR, not reported; LEDD, levodopa equivalent daily dose.
Table 5
Available evidence of outcomes for different device-aided therapies in monogenic autosomal recessive parkinsonism presenting with atypical feature
| ATP13A2 | STN-DBS | Case report | 1 Heterozygous ATP13A2 p.R449Q, and two Parkin variants—a deletion of exons 3 and 4, and duplications of exons 7 to 12 | 36 | 14 y | NR | Favorable | Postural instability and depression |
| PLA2G6 | CSAI | Case report [110] | 1 | 27 | 3 y | 1 y | Motor benefit with gait recovery | Intermittent visual hallucinations |
| DBS | Case report [98] | 1 GPi-DBS and Vim-DBS Atypical neuroaxonal dystrophy (NAD) phenotype. | Childhood | About 10 y | 9 mo | Good control of dystonic storm, oculogyric crises, and tremors | ||
| FBXO7 | CSAI | Case report [99] | 1 | 16 | 4 y | 6 mo | 50% reduction of daily “off” time | Severe dyskinesias after 6 mo |
| GPi-DBS | Case report [99] | 1 | 16 | 5 y | 6 mo | Good response (Not detailed) | Permanent anarthria | |
| DNAJC6 | STN-DBS | Case report homozygous [100] | 1 DNAJC6 p.T741= | 31 | 15 y | Not reported | Marked improvement | Not reported |
| VPS13C | STN-DBS | Case report [101] | 1 VPS13C compound heterozygous canonical splice-site | 39 | 2.5 y | Severe dysarthria and mild aphasia. |
NR, not reported; LEDD, levodopa equivalent daily dose.
Various authors have proposed different categories for motor outcomes. In their systematic review, de Oliveira et al. defined a mean UPDRS-III change of 50% or more as a marked response, a mean change of 30% to 50% as a satisfactory response, and less than 30% change as an unsatisfactory response [79]; on the other hand, Kuusimäki et al., defined an improvement of 30% or more in the UPDRS-III score as a favorable outcome; 20–30% a moderate outcome; and < 20% a poor/mild result [80].
DBS in carriers of genetic variants that modify the risk for developing PD or influence PD-related outcomes (GBA)
GBA carriers have DBS earlier in the disease course compared to LRRK2, PRKN, or non-mutation carriers [9, 78]. Most GBA carriers have marked or satisfactory short-term (<2 years) outcomes to STN-DBS [79]. Data for longer-term follow up are scarce, but outcomes tend to worsen over time. The authors hypothesized that because STN-DBS carries additional cognitive risk over GPi-DBS, the latter target may be preferable for GBA carriers, who are already at increased risk for cognitive impairment [79]. A separate study showed GBA carriers developed cognitive impairment and stimulation-resistant symptoms within 2 to 7 years after surgical treatment [81] (see Table 2). Thus, the overall benefit of DBS may be compromised due to the rapid progression of cognitive and neuropsychiatric symptoms [80].
A recent study screening for LRRK2, GBA, and PRKN mutations evaluating cognition at baseline and one-year post-DBS showed that high-risk or severe GBA variant was associated with pronounced postoperative cognitive decline [82]. The motor benefit was similar among groups.
Modeling different datasets, Pal et al. examined global cognition using the Mattis Dementia Rating Scale to compare the rate of change between GBA variant carriers and non-carriers with and without STN-DBS in PD. GBA carriers with DBS declined on average 2 points/year more than non-carriers with no DBS, 1.7 points/year more than GBA carriers with no DBS, and 1.5 points/year more than non-carriers with DBS [83]. Authors proposed that although non-randomized, this study suggests that GBA variants and STN-DBS’s combined effect negatively impact cognition, advising that GBA variant carriers should be counseled regarding potential risks associated with STN-DBS and alternative options may be considered [83].
Finally, the GPi target may be preferable for GBA carriers with dystonia and dyskinesia [79].
Both GPi-DBS and STN-DBS have similar outcomes on motor function measured by the UPDRS-III in the “on” and “off” medication state [84–86], and both targets have a beneficial effect on levodopa-induced dyskinesias [87]. STN-DBS achieves this goal mainly by a greater reduction in medication dosages [87, 88]; but also, stimulation of the area above the STN can directly suppress levodopa-induced “on”-dyskinesia [86]. In contrast, GPi-DBS may provide greater anti-dyskinetic effects possibly by a direct mechanism [84, 85, 87]. Hence, clinical guidelines recommend GPi as the target, especially when reduction of medication is not anticipated, and there is a goal to reduce the severity of “on” medication dyskinesias [84, 89].
On the other hand, although it seems relatively safe concerning cognitive function, chronic stimulation of STN has been associated with a subtle decline in cognitive domains, exceptionally verbal fluency, and executive function [90, 91]. Despite little data is supporting that STN-DBS has a worse cognitive outcome than GPi-DBS [92], more published information is required for validation [93]; if there is significant concern about cognitive decline, particularly regarding verbal fluency, processing speed, and working memory in a patient undergoing DBS, GPi has been recommended [84, 89].
DBS in autosomal dominant PD (SNCA, LRRK2, VPS35)
A systematic review showed that LRRK2 p.R1441G had poorer outcomes than other LRRK2 variants [79]. Overall, the response of LRRK2 p.G2019S carriers to STN-DBS was comparable to idiopathic PD [81]. There are reports of another variant, p.T2031S developing neuropsychiatric problems 5–7 years after DBS [80].
Thus far in the literature, five individuals carrying a VPS35 p.D620N variant have undergone DBS (STN = 3, unreported target = 2) and were followed for 1 to 8 years. The motor outcome was favorable in 3, moderate in 1, and poor in 1 who developed gait impairment, dysarthria, behavioral changes, and cognitive decline a few years later [80].
In a meta-analysis of SNCA duplications, three individuals had bilateral STN-DBS with good results. Two did not have cognitive decline at four-year follow-up. However, the third individual developed dementia [94]. Another individual with mosaicism of SNCA duplication, with motor complications, mild cognitive impairment, hallucinations, and an impulse control disorder, had bilateral GPi-DBS eight years after symptom onset. Good motor benefit was reported 1 month after surgery [95] (Table 3).
In summary, outcomes appear favorable for the most common LRRK2 pathogenic variant, p.G2019S, but may be poor for p.R1441G due to rapid cognitive decline and worsening of neuropsychiatric symptoms. The evidence remains very limited for SNCA and VPS35, with heterogeneous outcomes.
DBS in autosomal recessive PD (PRKN, PINK-1, DJ1)
PRKN carriers tend to have earlier disease onset yet longer disease duration at DBS surgery [9, 78]. After DBS, most of them have sustained motor improvement and in activities of daily living comparable to idiopathic PD [81]. Data for GPi-DBS are scarce. One PINK1 homozygous patient had satisfactory motor improvement after STN-DBS, but long-term results are not available, and nonmotor outcomes were not described [96, 97]. We found no published data on DJ1 variants undergoing DBS (see Table 4).
DBS in autosomal recessive parkinsonism with atypical features
Bilateral GPi-DBS and ventralis intermediate nucleus (Vim)-DBS has been successfully utilized for dystonic storm treatment in a 15-year-old girl with atypical neuroaxonal dystrophy (NAD) phenotype, a subgroup of PLA2G6-associated neurodegeneration (PLAN). She had a complex clinical picture characterized by progressive generalized dystonia, spasticity, myoclonus, intentional tremor, oculogyric crises, seizures, and poor cognition. She achieved good control of dystonic storm symptoms, oculogyric crises, and tremors at a 9-month follow-up [98]. The use of DBS for the PLA2G6-associated dystonia-parkinsonism phenotype has not been reported.
A female carrier of a FBXO7 homozygous variant with juvenile parkinsonism associated with postural instability, dysarthria, hypophonia, marked motor fluctuations, and levodopa-induced dyskinesias was reported to have satisfactory motor control with multiple device-aided therapies. At age 21, five years after symptoms onset, CSAI reduced daily “off” time by 50%. However, within 6 months, the patient developed severe “on”-period generalized chorea-dystonic dyskinesias. Bilateral GPi-DBS was implanted and achieved good control of those symptoms at a 6-month follow-up. However, severe dysarthria progressed to permanent anarthria [99].
In a cohort of early-onset sporadic or familial PD, a 46-year-old homozygous DNAJC6 p.T741 = female carrier with levodopa-responsive parkinsonism since age 31, who developed severe motor complications, underwent bilateral STN-DBS with marked improvement. However, the follow-up time and details were not reported [100].
A Caucasian woman with parkinsonism since age 39 had severe dyskinesias under dopaminergic treatment, dysarthria, tremor, mild dementia, hallucinations, dystonia, gait, and gastrointestinal tract problems. She had compound heterozygous canonical splice-site variants in VPS13C (p.Lys639AspfsTer14, p.Leu678GlufsTer26, p.Ala1072GlufsTer14, p.Ala1072_Gln1110del, p.Ser1076ArgfsTer4). She had initial benefit from STN-DBS but developed severe dysarthria and mild aphasia after 2.5 years. Rapid progression of symptoms was reported at a 4-year follow-up [101].
A Persian male bearing a p.R449Q heterozygous mutation in ATP13A2, who was also known to carry two Parkin mutations—a deletion of exons 3 and 4 and duplications of exons 7 to 12, was reported to have parkinsonian symptoms, including rest tremor and a good response to levodopa, since the age of 36. At 50, a favorable response to STN-DBS stimulation was reported, with mild postural instability and depression but no atypical neurological signs [102].
EVIDENCE ON THE USE OF LEVODOPA CARBIDOPA INTESTINAL GEL
Autosomal dominant PD and GBA mutations
In a cohort of 12 PD patients on LCIG in the UK, the authors reported one patient with LRRK2. This carrier had a 19-year history of PD and died 24 months after LCIG initiation because of colon cancer [103]. In a study in Tel Aviv, where 44 PD patients underwent LCIG, five were LRRK2 carriers, four were GBA heterozygotes, two were GBA homozygotes, and another was a carrier of both GBA and LRRK2. No significant differences were found between the carrier versus non-carrier group [104]. The same study group published an abstract of the data from 69 Ashkenazi Jewish patients with known GBA (11 cases) or LRRK2 p.G2019S mutations (16 cases) and 42 idiopathic PD. Motor UPDRS scores were significantly higher, and levodopa equivalent daily doses (LEDD) were lower among GBA-PD than in the two other groups. Although not statistically significant, GBA-PD had a higher rate of hallucinations and lower cognitive scores. The latter could explain the lower LEDD in this group [105] (see Tables 2 and 3).
Autosomal recessive PD with homogeneous presentations
A juvenile PD patient carrying a PRKN p.T240M variant in a heterozygous state had a marked improvement in motor and non-motor scores on LCIG in a long follow-up period [106]. A 63-year-old PRKN carrier with a history of 35 years of parkinsonism died 3 months after introducing LCIG from unspecified causes [103].
A woman with homozygous PINK1 variants with parkinsonism since age 29 underwent LCIG at age 55 for motor fluctuations and dyskinesias. For three years, the motor response was satisfactory, but she required B6 vitamin replacement for sensory axonal polyneuropathy. Four years after LCIG, she developed marked dyskinesias, dopamine dysregulation syndrome, ICD, and punding [107].
EVIDENCE FOR CONTINUOUS APOMORPHINE SUBCUTANEOUS INFUSION
Autosomal dominant PD
In a case series of British LRRK2 patients, a 42-year-old man was reported to have a CSAI pump 14 years after disease onset for severe drug-induced dyskinesias. Before CSAI, he had depression, obsessive and hypomanic behavior, excessive alcohol drinking, and dopamine dysregulation syndrome. Outcomes were not specified [108].
Autosomal recessive PD with homogeneous presentations
Two PRKN patients have been reported with CSAI. The first case was a 32-year-old man with 25 years of progressive parkinsonism-dystonia syndrome, with deterioration of laryngeal dystonia on levodopa and severe neuropsychiatric symptoms. No outcomes were reported. The second case was a 57-year-old with a previous thalamotomy started on CSAI at age 47 with benefit. However, he developed psychosis with paranoid delusions that resolved after stopping apomorphine [109].
Autosomal recessive parkinsonism presenting with atypical features
A Turkish woman with homozygous PLA2G6 p.R747W and heterozygous LRRK2 p.W1295R variants developed parkinsonism at age 27. By age 29, she had severe parkinsonism with bilateral tremors, hypophonia, dysarthria, postural instability, and cognitive impairment. She had a good response to combined antiparkinsonian medications but developed irritability, restlessness, and ICD. By age 30, she was unable to stand or walk independently. After CSAI (5 mg/hour), she could walk for at least a 1-year follow-up. She developed intermittent visual hallucinations [110].
As mentioned above, a female FBXO7 homozygous variant carrier with juvenile parkinsonism was reported to have a transient motor benefit with CSAI. After 6 months, the patient developed severe “on” dyskinesias, requiring bilateral GPi-DBS [99].
DISCUSSION
The cumulative evidence for device-aided therapies in monogenic-PD and GBA carriers is still scarce.
Along with a regional difference in the prevalence of specific variants, the availability of advanced therapies is critical. Device-aided therapies offered in different countries may vary through healthcare systems, local experience, and center preferences. For instance, we have observed that publications on infusion therapies (i.e., LCIG and CSAI) in monogenic parkinsonism come predominantly from the UK and Middle Eastern countries (Israel, Saudi Arabia, and Turkey), and LCIG in GBA-PD from Israel. On the other hand, DBS-related publications are more widely distributed (i.e., North America, Europe, Middle East, Asia, South America, Australia), possibly because of increasing access to this therapy. Unfortunately, decision-making on device selection is not explicit in most reports from countries where more than one device-aided therapy is available (e.g., Italy, UK, Israel, Turkey, USA). Future reports should explain the selection of a specific device-aided therapy, especially when other alternatives are available.
Systematic reviews and a meta-analysis constitute the best evidence for DBS in monogenic PD. However, these are limited by small sample size, short follow-up, and incomplete data.
Moreover, several investigators have used different categories and cut-off values when defining DBS responses in gene-related PD populations; because of this heterogeneity, the same percentage of change in UPDRS-III would be qualified differently by distinct authors. An explicit limitation of this approach is the lack of consensus, adding difficulties when interpreting the literature. In addition to arbitrariness in establishing cut-off values, the effectiveness of these therapies has been firmly focused on the change in UPDRS-III scores, in our opinion lacking adequate emphasis on non-motor symptoms or changes in quality of life, which can be decisive in decision-making and in establishing the benefits of these therapies. Further, with some exceptions, reports on LCIG or CSAI lack objective and detailed results making a similar analysis difficult.
Mutation carriers seem to be overrepresented in DBS-cohorts compared to non-carrier PD populations. LRRK2 p.G2019S, homozygous or compound heterozygous PRKN, and GBA were the most frequent variants. This may be attributed to an overlap of the phenotype with criteria for device eligibility. However, this is not necessarily equivalent to suitableness in the selection. For instance, these all had motor outcomes comparable to patients with idiopathic PD, at least in the short term, with STN-DBS as the most frequent target. However, the motor benefit of STN-DBS in GBA-PD may be countered by a faster cognitive decline and axial symptoms following DBS than non-carriers. On the other hand, even if carriers may have poorer outcomes than non-carriers, this is not equal to absent benefit (e.g., motor benefit vs. cognitive decline). Future randomized studies should consider the quality of life as a primary outcome to better understand the risk-benefit ratio in GBA-PD.
This should be kept in mind when discussing prognosis, timing, and expectations for DBS.
Publications on DBS in autosomal recessive variants with atypical features are mainly limited to individual cases. Some patients have reported benefits, but outcomes are incompletely reported, and long-term data is scarce. Dysarthric speech, swallowing disturbances, freezing of gait, and balance problems are frequent features of atypical autosomal recessive parkinsonism (e.g., ATP13A2, PLA2G6, FBXO7, DNAJC6, SYNJ1, and VPS13C). On the other hand, these symptoms can occur using DBS parameters optimal for improving tremor, rigidity, and bradykinesia. Therefore, data is insufficient to differentiate device therapies outcomes (i.e., adverse effects) from disease progression or therapy non-responsiveness. At this point, in select cases, DBS seems to be a reasonable palliative therapy or a rescue treatment in emergencies such as dystonic storms.
Small genetic screening studies are the primary source of evidence for LCIG. There is no significant difference in motor outcomes between LRRK2 p.G2019S or GBA-carriers and non-carriers. GBA carriers in the LCIG studies had lower cognitive scores and higher scores for hallucinations.
CSAI has the most limited evidence of the three therapies in monogenic PD and GBA carriers. As expected, available cases tend to include individuals with very advanced diseases, given the typical patient selection criteria. CSAI may be a helpful alternative in recessive parkinsonism with atypical features, with some efficacy, as shown in the FBOX7 and PLA2G6 case reports.
The available information regarding individual monogenic variants and device-aided therapies is far from comprehensive. The data are limited to small numbers of patients, short follow-ups, and observational reports. Multicenter prospective cohort studies are needed to guide our knowledge and improve decision-making for device-aided therapies and PD-related variants.
In addition, we recommend that several key elements be included when reporting outcomes from device-aided therapy amongst genetic PD populations (Fig. 1).
Fig. 1
Key elements to consider when reporting the response to device-aided therapies in patients with monogenic parkinsonism or GBA variants carriers.
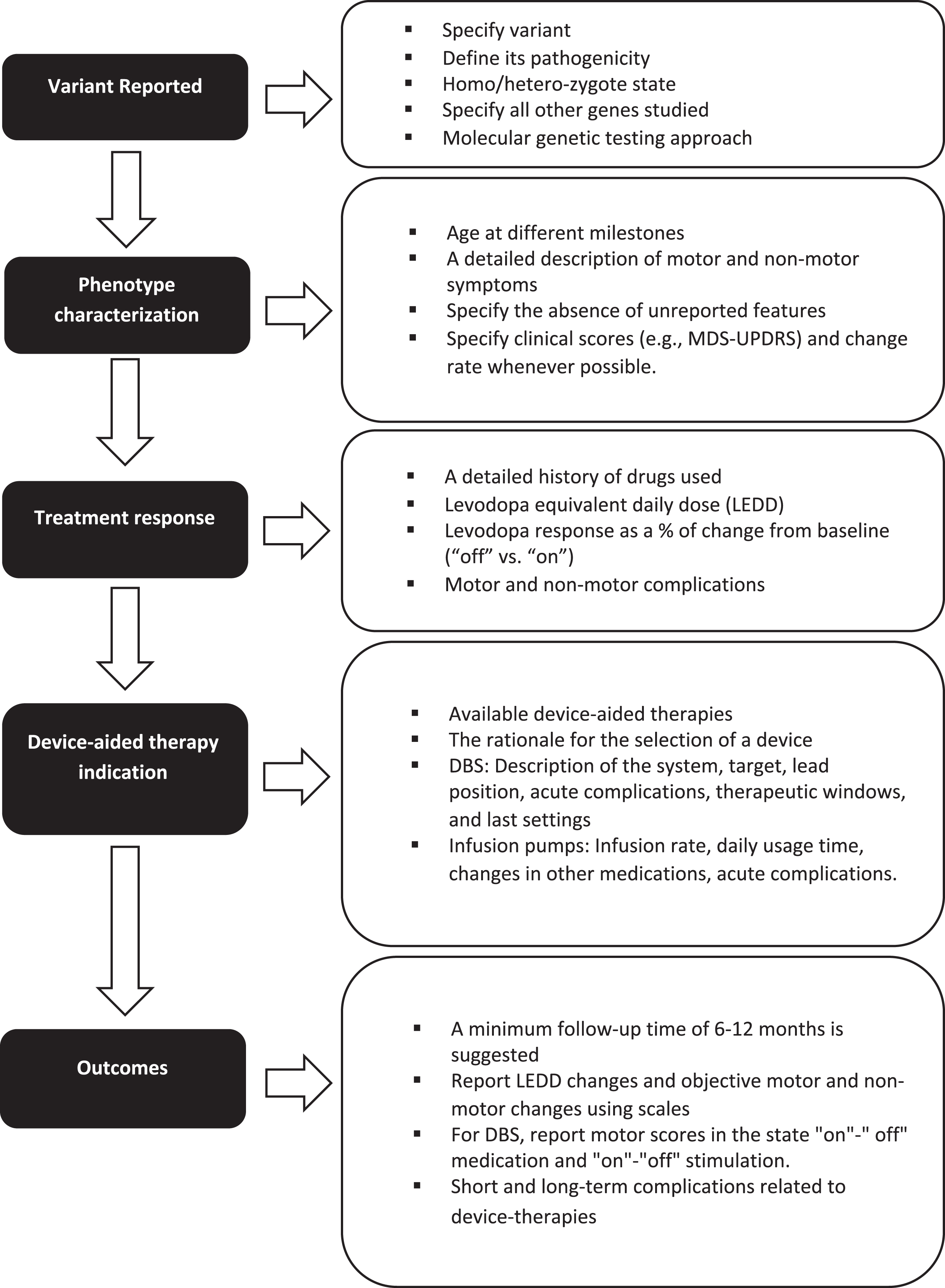
First, when discussing the genetic variant, the type of variant, its pathogenicity, and its homozygote or heterozygote state should be included. When using panels, all the genes studied should be mentioned, especially in patients belonging to ethnicities at risk for more than one type of variant (for example, LRRK2 p.G2019S and GBA variants in Ashkenazi Jews).
Second, when discussing phenotype, characterization must be rigorous, including the age at symptom onset, age at diagnosis, disease duration, initial clinical manifestation, presence of falls, freezing of gait, cognitive profile, neuropsychiatric manifestations, and other non-motor symptoms. The absence of unreported features should be specified. Individuals may be classified according to the MDS-UPDRS-III score (i.e., tremor dominant, intermediate, or postural instability/gait difficulty). Levodopa response should be described as % of change from baseline. Whenever possible, the rate of progression of motor and non-motor symptoms (i.e., cognitive decline) in the pre- and post-device-aided therapy stage should be included. A detailed history of the drugs used, related side effects and levodopa equivalent daily dose should also be included.
Third, the indication and rationale for each specific advanced device-aided therapy should be documented. In addition, for DBS, it is essential to define whether the surgery is uni- or bilateral, which commercial device was implanted, the target, lead position information, therapeutic window, and final stimulation parameters. The infusion rate, daily usage time, and changes in other medications should be indicated for infusion pumps.
Finally, long-term motor and non-motor outcomes should be measured objectively using, for example, the MDS-UPDRS scale administered at multiple time points. In the case of DBS, it is essential to report motor scores in the state “on"-” off” medication and “on"-“off” stimulation. Follow-up time should be sufficient for the device settings to reach a steady-state and assess disease progression, treatment efficacy, and long-term adverse effects. While there is no specific time, a reasonable minimum follow-up time would be greater than 6–12 months.
CONCLUSION
Based on current studies, it is unfeasible to establish evidence-based decision-making guidelines for device-aided therapies in monogenic parkinsonism. So far, an added prognostic value of genetic testing beyond a careful clinical assessment when patients are evaluated for device-aided therapies is yet to be demonstrated for monogenic parkinsonism. Large prospective cohorts combining genetic profiling with deep phenotyping, and randomized studies, can provide relevant data to address this question.
Although no randomized trials are available, based on accumulated evidence on the natural history and probable deleterious cognitive outcomes after STN-DBS in carriers of pathogenic variants in GBA, several authors have proposed that candidates for this device therapy should be screened for GBA mutations as part of the pre-surgical assessment. Carriers should be counseled regarding potential risks associated with STN-DBS, considering alternative options [83]. Comparative studies of different device-aided therapies in this population are pending.
We call for the development of guidelines that allow us to improve the quality and number of reports and randomized clinical studies that optimize our decision-making on device-aided therapies in monogenic parkinsonism and GBA carriers.
CONFLICT OF INTEREST
Philippe Salles MD did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
James Liao MD did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Umar Shuaib MD did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Ignacio Mata MD
Grants/Research Support: Dr. Mata has received research support from American Parkinson’s Disease Association, Parkinson’s Foundation, Michael J. Fox Foundation, and NIH/NINDS
Hubert Fernandez MD
Grants/Research Support: Dr. Fernandez has received research support from Acorda Therapeutics, Alkahest, Amneal, Biogen, Michael J. Fox Foundation, Movement Disorders Society, NIH/NINDS, Parkinson Study Group, and Sunovion but has no owner interest in any pharmaceutical company.
Honoraria: Dr. Fernandez has received honoraria from Cleveland Clinic and Boston University as a speaker in CME events. Dr. Fernandez has received honoraria from Bial Neurology, Biopas, Cerevel, CNS Ratings, Denali Therapeutics, Kyowa Hakko Kirin, Pfizer, Partners Healthcare System, Parkinson Study Group, Revance, Sun Pharmaceutical Industries, Sunovion Research, and Development Trust as a consultant, and from Elsevier as the Co-Editor-In-Chief of Parkinsonism and Related Disorders Journal.
Royalty: Dr. Fernandez has received royalty payments from Demos Publishing and Springer for serving as a book author/editor.
Contractual Services: The Cleveland Clinic has a contract with Teva for Dr. Fernandez’s role as a Co-Principal Investigator in Deutetrabenazine for Tardive Dyskinesia global studies.
REFERENCES
[1] | Dorsey ER , Sherer T , Okun MS , Bloemd BR ((2018) ) The emerging evidence of the Parkinson pandemic. J Parkinsons Dis 8: , S3–S8. |
[2] | Twelves D , Perkins KSM , Counsell C ((2003) ) Systematic review of incidence studies of Parkinson’s disease. Mov Disord 18: , 19–31. |
[3] | Simon DK , Tanner CM , Brundin P ((2020) ) Parkinson disease epidemiology, pathology, genetics, and pathophysiology. Clin Geriatr Med 36: , 1–12. |
[4] | Koros C , Simitsi A , Stefanis L ((2017) ) Genetics of Parkinson’s disease: Genotype-phenotype correlations. Int Rev Neurobiol 132: , 197–231. |
[5] | Puschmann A , Wszolek ZK ((2014) ) Genotype-phenotype correlations in Parkinson disease. InMovement Disorders: Genetics and Models, 2nd Edition, LeDoux MS, ed. Elsevier Academic Press, pp. 259–285. |
[6] | Antonini A , Moro E , Godeiro C , Reichmann H ((2018) ) Medical and surgical management of advanced Parkinson’s disease. Mov Disord 33: , 900–908. |
[7] | Siddiqui J , Aldaajani Z , Mehanna R , Changizi BK , Bhatti D , Al-Johani ZG , Shukla AW , Fernandez HH , Bajwa JA ((2018) ) Rationale and patient selection for interventional therapies in Parkinson’s disease. Expert Rev Neurother 18: , 811–823. |
[8] | Armstrong MJ , Okun MS ((2020) ) Diagnosis and treatment of Parkinson disease. JAMA 323: , 548. |
[9] | Angeli A , Mencacci NE , Duran R , Aviles-Olmos I , Kefalopoulou Z , Candelario J , Rusbridge S , Foley J , Pradhan P , Jahanshahi M , Zrinzo L , Hariz M , Wood NW , Hardy J , Limousin P , Foltynie T ((2013) ) Genotype and phenotype in Parkinson’s disease: Lessons in heterogeneity from deep brain stimulation. Mov Disord 28: , 1370–1375. |
[10] | Salles PA , Mata IF , Fernandez HH ((2021) ) Should we start integrating genetic data in decision-making on device-aided therapies in Parkinson disease? A Point of View. Parkinsonism Relat Disord 88: , 51–57. |
[11] | Antonini A , Stoessl AJ , Kleinman LS , Skalicky AM , Marshall TS , Sail KR , Onuk K , Odin PLA ((2018) ) Developing consensus among movement disorder specialists on clinical indicators for identification and management of advanced Parkinson’s disease: A multi-country Delphi-panel approach. Curr Med Res Opin 34: , 2063–2073. |
[12] | Timpka J , Nitu B , Datieva V , Odin P , Antonini A ((2017) ) Device-aided treatment strategies in advanced Parkinson’s disease. Int Rev Neurobiol 132: , 453–474. |
[13] | Bronstein JM , Tagliati M , Alterman RL , Lozano AM , Volkmann J , Stefani A , Horak FB , Okun MS , Foote KD , Krack P , Pahwa R , Henderson JM , Hariz MI , Bakay RA , Rezai A , Marks WJ , Moro E , Vitek JL , Weaver FM , Gross RE , DeLong MR ((2011) ) Deep brain stimulation for Parkinson disease an expert consensus and review of key issues. Arch Neurol 68: , 165–171. |
[14] | Lang AE , Houeto JL , Krack P , Kubu C , Lyons KE , Moro E , Ondo W , Pahwa R , Poewe W , Tröster AI , Uitti R , Voon V ((2006) ) Deep brain stimulation: Preoperative issues. Mov Disord 21: (Suppl 14), S171–96. |
[15] | Amjad F , Bhatti D , Davis TL , Oguh O , Pahwa R , Kukreja P , Zamudio J , Metman LV ((2019) ) Current practices for outpatient initiation of levodopa-carbidopa intestinal gel for management of advanced Parkinson’s disease in the United States. Adv Ther 36: , 2233–2246. |
[16] | de Gaspari D ((2006) ) Clinical and neuropsychological follow up at 12 months in patients with complicated Parkinson’s disease treated with subcutaneous apomorphine infusion or deep brain stimulation of the subthalamic nucleus. J Neurol Neurosurg Psychiatry 77: , 450–453. |
[17] | Alegret M , Valldeoriola F , Martí M , Pilleri M , Junqué C , Rumià J , Tolosa E ((2004) ) Comparative cognitive effects of bilateral subthalamic stimulation and subcutaneous continuous infusion of apomorphine in Parkinson’s disease. Mov Disord 19: , 1463–1469. |
[18] | Trenkwalder C , Chaudhuri KR , García Ruiz PJ , LeWitt P , Katzenschlager R , Sixel-Döring F , Henriksen T , Sesar Á , Poewe W , Baker M , Ceballos-Baumann A , Deuschl Günther , Drapier S , Ebersbach G , Evans A , Fernandez H , Isaacson S , van Laar T , Lees A , Lewis S , Castrillo JCM , Martinez-Martin P , Odin P , O’Sullivan J , Tagaris G , Wenzel K ((2015) ) Expert Consensus Group report on the use of apomorphine in the treatment of Parkinson’s disease - Clinical practice recommendations. Parkinsonism Relat Disord 21: , 1023–1030. |
[19] | Odin P , Ray Chaudhuri K , Slevin JT , Volkmann J , Dietrichs E , Martinez-Martin P , Krauss JK , Henriksen T , Katzenschlager R , Antonini A , Rascol O , Poewe W , Brücke T , Pirker W , Ransmayr G , Schwingenschuh P , Tomantschger V , Volc D , Jespersen H , Kamal A , Karlsborg M , Oppel L , Pedersen S , Avikainen S , Kaasinen V , Pekkonen E , Ruottinen H , Azulay JP , Corvol JC , Courbon CB , Defebvre L , Durif F , Houeto JL , Krack P , Tison F , Andrich J , Ehret R , Klostermann F , Krüger R , Lingor P , Liszka R , Schwarz J , Timmermann L , Warnecke T , Bostantjopoulou S , Konitsiotis S , Papageorgiou S , Stathishens P , Stefanis L , Zikos P , Browne P , Healy D , Lynch T , O’Riordan S , O’Sullivan S , Walsh R , Abbruzzese G , Lopiano L , Modugno N , Tamma F , Holmberg B , Linder J , Nyholm D , Pålhagen S , Matías Arbelo J , Bana RY , Castrillo JCM , Castro A , e Garcia Ruiz Espiga PJ , Kulisevsky J , Lezcano E , Luquin R , Mir P , Puente V , Valldeoriola F , Burn D , Clarke C , Foltynie T , Grosset D , Hindle J , Leake A , Lees A , Morris H , Stewart D , Walker R , Worth P ((2015) ) Collective physician perspectives on non-oral medication approaches for the management of clinically relevant unresolved issues in Parkinson’s disease: Consensus from an international survey and discussion program. Parkinsonism Relat Disord 21: , 1133–1144. |
[20] | Catalán MJ , de Pablo-Fernández E , Villanueva C , Fernández-Diez S , Lapeña-Montero T , García-Ramos R , López-Valdés E ((2013) ) Levodopa infusion improves impulsivity and dopamine dysregulation syndrome in Parkinson’s disease. Mov Disord 28: , 2007–2010. |
[21] | Todorova A , Samuel M , Brown RG , Chaudhuri KR ((2015) ) Infusion therapies and development of impulse control disorders in advanced Parkinson disease. Clin Neuropharmacol 38: , 132–134. |
[22] | Barbosa P , Lees AJ , Magee C , Djamshidian A , Warner TT ((2017) ) A retrospective evaluation of the frequency of impulsive compulsive behaviors in Parkinson’s disease patients treated with continuous waking day apomorphine pumps. Mov Disord Clin Pract 4: , 323–328. |
[23] | Castrioto A , Lozano AM , Poon Y-Y , Lang AE , Fallis M , Moro E ((2011) ) Ten-year outcome of subthalamic stimulation in Parkinson disease. Arch Neurol 68: , 1550. |
[24] | Fasano A , Romito LM , Daniele A , Piano C , Zinno M , Bentivoglio AR , Albanese A ((2010) ) Motor and cognitive outcome in patients with Parkinson’s disease 8 years after subthalamic implants. Brain 133: , 2664–2676. |
[25] | Hitti FL , Ramayya AG , McShane BJ , Yang AI , Vaughan KA , Baltuch GH ((2020) ) Long-Term outcomes following deep brain stimulation for Parkinson’s disease. J Neurosurg 132: , 205–210. |
[26] | Qi R , Geng X , Huang B , Chen Y , Jiang H , Zou Y , Wang W , Li Y , Li Y , Yin L , Liu A , Yang X , Li J , Yu H ((2020) ) Outcomes of STN-DBS in PD patients with different rates of disease progression over one year of follow-up. Front Neurol 11: , 600. |
[27] | Sensi M , Cossu G , Mancini F , Pilleri M , Zibetti M , Modugno N , Quatrale R , Tamma F , Antonini A , Aguggia M , Amboni M , Arca R , Bartolomei L , Bonetto N , Calandra-Buonaura G , Bove F , Calandrella D , Canesi M , Cannas A , Capecci M , Caputo E , Ceravolo MG , Ceravolo R , Cerrone G , Coletti Moja M , Comi C , Cortelli P , D’Antonio P , Dematteis F , di Lazzaro V , Eleopra R , Fabbrini G , Fichera M , Grassi E , Guido M , Gusmaroli G , Latorre A , Malaguti MC , Marano M , Marano P , Marconi R , Mazzucchi S , Meco G , Minafra B , Morgante F , Pacchetti C , Pierantozzi M , Pontieri FE , Riboldazzi G , Ricchi V , Ricchieri G , Rinaldo S , Rispoli V , Rossi S , Rubino A , Russo A , Saddi MV , Stefani A , Simoni S , Solla P , Tambasco N , Tamburin S , Tessitore A , Torre E , Ulivelli M , Vita MG , Volonté MA ((2017) ) Which patients discontinue? Issues on Levodopa/carbidopa intestinal gel treatment: Italian multicentre survey of 905 patients with long-term follow-up. Parkinsonism Relat Disord 38: , 90–92. |
[28] | Poewe W , Bergmann L , Kukreja P , Robieson WZ , Antonini A ((2019) ) Levodopa-carbidopa intestinal gel monotherapy: GLORIA registry demographics, efficacy, and safety. J Parkinsons Dis 9: , 531–541. |
[29] | Hughes AJ , Bishop S , Kleedorfer B , Turjanski N , Fernandez W , Lees AJ , Stern GM ((1993) ) Subcutaneous apomorphine in Parkinson’s disease: Response to chronic administration for up to five years. Mov Disord 8: , 165–170. |
[30] | Borgemeester RWK , van Laar T ((2017) ) Continuous subcutaneous apomorphine infusion in Parkinson’s disease patients with cognitive dysfunction: A retrospective long-term follow-up study. Parkinsonism Relat Disord 45: , 33–38. |
[31] | Tyne HL , Parsons J , Sinnott A , Fox SH , Fletcher NA , Steiger MJ ((2004) ) A 10 year retrospective audit of long-term apomorphine use in Parkinson’s disease. J Neurol 251: , 1370–1374. |
[32] | García Ruiz PJ , Sesar Ignacio Á , Ares Pensado B , Castro García A , Alonso Frech F , Álvarez López M , Arbelo González J , Baiges Octavio J , Burguera Hernández JA , Calopa Garriga M , Campos Blanco D , Castaño García B , Carballo Cordero M , Chacón Peña J , Espino Ibáñez A , Gorospe Onisalde A , Giménez-Roldán S , Granés Ibáñez P , Hernández Vara J , Ibáñez Alonso R , Jiménez Jiménez FJ , Krupinski J , Kulisevsky Bojarsky J , Legarda Ramírez I , Lezcano García E , Martínez-Castrillo JC , Mateo González D , Miquel Rodríguez F , Mir P , Muñoz Fargas E , Obeso Inchausti J , Olivares Romero J , Olivé Plana J , Otermin Vallejo P , Pascual Sedano B , Pérez de Colosía Rama V , Pérez López-Fraile I , Planas Comes A , Puente Periz V , Rodríguez Oroz MC , Sevillano García D , Solís Pérez P , Suárez Muñoz J , Vaamonde Gamo J , Valero Merino C , Valldeoriola Serra F , Velázquez Pérez JM , Yáñez Baña R , Zamarbide Capdepon I ((2008) ) Efficacy of long-term continuous subcutaneous apomorphine infusion in advanced Parkinson’s disease with motor fluctuations: A multicenter study. Mov Disord 23: , 1130–1136. |
[33] | Borgemeester RWK , Drent M , van Laar T ((2016) ) Motor and non-motor outcomes of continuous apomorphine infusion in 125 Parkinson’s disease patients. Parkinsonism Relat Disord 23: , 17–22. |
[34] | Blauwendraat C , Nalls MA , Singleton AB ((2019) ) The genetic architecture of Parkinson’s disease. Lancet Neurol 4422: , 1–9. |
[35] | Deng H , Wang P , Jankovic J ((2018) ) The genetics of Parkinson disease. Ageing Res Rev 42: , 72–85. |
[36] | Kalinderi K , Bostantjopoulou S , Fidani L ((2016) ) The genetic background of Parkinson’s disease: Current progress and future prospects. Acta Neurol Scand 134: , 314–326. |
[37] | Lesage S , Brice A ((2009) ) Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum Mol Genet 18: , R48–R59. |
[38] | Avenali M , Blandini F , Cerri S ((2020) ) Glucocerebrosidase defects as a major risk factor for Parkinson’s disease. Front Aging Neurosci 12: , 97. |
[39] | Aharon-Peretz J , Badarny S , Rosenbaum H , Gershoni-Baruch R ((2005) ) Mutations in the glucocerebrosidase gene and Parkinson disease: Phenotype–genotype correlation: Neurology 65: , 1460–1461. |
[40] | Lesage S , Anheim M , Condroyer C , Pollak P , Durif F , Dupuits C , Viallet F , Lohmann E , Corvol J-C , Honoré A , Rivaud S , Vidailhet M , Dürr A , Brice A , Agid Y , Bonnet A-M , Borg M , Brice A , Broussolle E , Damier Ph , Destée A , Dürr A , Durif F , Lesage S , Lohmann E , Martinez M , Pollak P , Rascol O , Tison F , Tranchant C , Troiano A , Vérin M , Viallet F , Vidailhet M ((2011) ) Large-scale screening of the Gaucher’s disease-related glucocerebrosidase gene in Europeans with Parkinson’s disease. Hum Mol Genet 20: , 202–210. |
[41] | Sidransky E , Nalls MA , Aasly JO , Aharon-Peretz J , Annesi G , Barbosa ER , Bar-Shira A , Berg D , Bras J , Brice A , Chen C-M , Clark LN , Condroyer C , de Marco EV , Dürr A , Eblan MJ , Fahn S , Farrer MJ , Fung H-C , Gan-Or Z , Gasser T , Gershoni-Baruch R , Giladi N , Griffith A , Gurevich T , Januario C , Kropp P , Lang AE , Lee-Chen G-J , Lesage S , Marder K , Mata IF , Mirelman A , Mitsui J , Mizuta I , Nicoletti G , Oliveira C , Ottman R , Orr-Urtreger A , Pereira LV , Quattrone A , Rogaeva E , Rolfs A , Rosenbaum H , Rozenberg R , Samii A , Samaddar T , Schulte C , Sharma M , Singleton A , Spitz M , Tan E-K , Tayebi N , Toda T , Troiano AR , Tsuji S , Wittstock M , Wolfsberg TG , Wu Y-R , Zabetian CP , Zhao Y , Ziegler SG ((2009) ) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361: , 1651–1661. |
[42] | Brockmann K , Srulijes K , Pflederer S , Hauser AK , Schulte C , Maetzler W , Gasser T , Berg D ((2015) ) GBA-associated Parkinson’s disease: Reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord 30: , 407–411. |
[43] | Oeda T , Umemura A , Mori Y , Tomita S , Kohsaka M , Park K , Inoue K , Fujimura H , Hasegawa H , Sugiyama H , Sawada H ((2015) ) Impact of glucocerebrosidase mutations on motor and nonmotor complications in Parkinson’s disease. Neurobiol Aging 36: , 3306–3313. |
[44] | Alcalay RN , Caccappolo E , Mejia-Santana H , Tang MX , Rosado L , Reilly MO , Ruiz D , Ross B , Verbitsky M , Kisselev S , Louis E , Comella C , Colcher A , Jennings D , Nance M , Bressman S , Scott WK , Tanner C , Mickel S , Andrews H , Waters C , Fahn S , Cote L , Frucht S , Ford B , Rezak M , Novak K , Friedman JH , Pfeiffer R , Marsh L , Hiner B , Siderowf A , Payami H , Molho E , Factor S , Ottman R , Clark LN , Marder K ((2012) ) Cognitive performance of GBA mutation carriers with early-onset PD The CORE-PD study. Neurology 78: , 1434–1440. |
[45] | Brockmann K , Srulijes K , Hauser AK , Schulte C , Csoti I , Gasser T , Berg D ((2011) ) GBA-associated PD presents with nonmotor characteristics. Neurology 77: , 276–280. |
[46] | Thaler A , Gurevich T , Bar Shira A , Gana Weisz M , Ash E , Shiner T , Orr-Urtreger A , Giladi N , Mirelman A ((2017) ) A “dose” effect of mutations in the GBA gene on Parkinson’s disease phenotype. Parkinsonism Relat Disord 36: , 47–51. |
[47] | Cilia R , Tunesi S , Marotta G , Cereda E , Siri C , Tesei S , Zecchinelli AL , Canesi M , Mariani CB , Meucci N , Sacilotto G , Zini M , Barichella M , Magnani C , Duga S , Asselta R , Soldà G , Seresini A , Seia M , Pezzoli G , Goldwurm S ((2016) ) Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann Neurol 80: , 662–673. |
[48] | Trinh J , Zeldenrust FMJ , Huang J , Kasten M , Schaake S , Petkovic S , Madoev H , Grünewald A , Almuammar S , König IR , Lill CM , Lohmann K , Klein C , Marras C ((2018) ) Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov Disord 33: , 1857–1870. |
[49] | Healy DG , Falchi M , O’Sullivan SS , Bonifati V , Durr A , Bressman S , Brice A , Aasly J , Zabetian CP , Goldwurm S , Ferreira JJ , Tolosa E , Kay DM , Klein C , Williams DR , Marras C , Lang AE , Wszolek ZK , Berciano J , Schapira AH , Lynch T , Bhatia KP , Gasser T , Lees AJ , Wood NW ((2008) ) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol 7: , 583–590. |
[50] | Marras C , Alcalay RN , Caspell-Garcia C , Coffey C , Chan P , Duda JE , Facheris MF , Fernández-Santiago R , Ruíz-Martínez J , Mestre T , Saunders-Pullman R , Pont-Sunyer C , Tolosa E , Waro B ((2016) ) Motor and nonmotor heterogeneity of LRRK2 -related and idiopathic Parkinson’s disease. Mov Disord 31: , 1192–1202. |
[51] | Alcalay RN , Mirelman A , Saunders-Pullman R , Tang M-X , Mejia Santana H , Raymond D , Roos E , Orbe-Reilly M , Gurevich T , Bar Shira A , Gana Weisz M , Yasinovsky K , Zalis M , Thaler A , Deik A , Barrett MJ , Cabassa J , Groves M , Hunt AL , Lubarr N , San Luciano M , Miravite J , Palmese C , Sachdev R , Sarva H , Severt L , Shanker V , Swan MC , Soto-Valencia J , Johannes B , Ortega R , Fahn S , Cote L , Waters C , Mazzoni P , Ford B , Louis E , Levy O , Rosado L , Ruiz D , Dorovski T , Pauciulo M , Nichols W , Orr-Urtreger A , Ozelius L , Clark L , Giladi N , Bressman S , Marder KS ((2013) ) Parkinson disease phenotype in Ashkenazi jews with and without LRRK2 G2019S mutations. Mov Disord 28: , 1966–1971. |
[52] | Aarsland D , Kurz MW ((2010) ) The epidemiology of dementia associated with Parkinson disease. J Neurol Sci 289: , 18–22. |
[53] | Alcalay RN , Mejia-Santana H , Mirelman A , Saunders-Pullman R , Raymond D , Palmese C , Caccappolo E , Ozelius L , Orr-Urtreger A , Clark L , Giladi N , Bressman S , Marder K , Tang MX , Santana HM , Roos E , Orbe-Reilly M , Fahn S , Cote L , Waters C , Mazzoni P , Ford B , Louis E , Levy O , Rosado L , Ruiz D , Dorovski T , Greene P , Marder KS , Gurevich T , Shira AB , GanaWeisz M , Yasinovsky K , Zalis M , Thaler A , Balash Y , Hertzel S , Gan Z , Kobo H , Hillel A , Shkedy A , Deik A , Barrett MJ , Cabassa J , Groves M , Hunt AL , Lubarr N , Luciano MS , Miravite J , Sachdev R , Sarva H , Severt L , Shanker V , Swan MC , Soto-Valencia J , Johannes B , Ortega R ((2015) ) Neuropsychological performance in LRRK2 G2019S carriers with Parkinson’s disease. Parkinsonism Relat Disord 21: , 106–110. |
[54] | Srivatsal S , Cholerton B , Leverenz JB , Wszolek ZK , Uitti RJ , Dickson DW , Weintraub D , Trojanowski JQ , van Deerlin VM , Quinn JF , Chung KA , Peterson AL , Factor SA , Wood-Siverio C , Goldman JG , Stebbins GT , Bernard B , Ritz B , Rausch R , Espay AJ , Revilla FJ , Devoto J , Rosenthal LS , Dawson TM , Albert MS , Mata IF , Hu S-C , Montine KS , Johnson C , Montine TJ , Edwards KL , Zhang J , Zabetian CP ((2015) ) Cognitive profile of LRRK2-related Parkinson’s disease. Mov Disord 30: , 728–733. |
[55] | Ruiz-Martínez J , Gorostidi A , Goyenechea E , Alzualde A , Poza JJ , Rodríguez F , Bergareche A , Moreno F , López de Munain A , Martí Massó JF ((2011) ) Olfactory deficits and cardiac 123 I-MIBG in Parkinson’s disease related to the LRRK2 R1441G and G2019S mutations. Mov Disord 26: , 2026–2031. |
[56] | Ehrminger M , Leu-Semenescu S , Cormier F , Corvol J-C , Vidailhet M , Debellemaniere E , Brice A , Arnulf I ((2015) ) Sleep aspects on video-polysomnography in LRRK2 mutation carriers. Mov Disord 30: , 1839–1843. |
[57] | Vinagre-Aragón A , Campo-Caballero D , Mondragón-Rezola E , Pardina-Vilella L , Hernandez Eguiazu H , Gorostidi A , Croitoru I , Bergareche A , Ruiz-Martinez J ((2021) ) A more homogeneous phenotype in Parkinson’s disease related to R1441G mutation in the LRRK2 gene. Front Neurol 12: , 635396. |
[58] | Meade RM , Fairlie DP , Mason JM ((2019) ) Alpha-synuclein structure and Parkinson’s disease –lessons and emerging principles. Mol Neurodegener 14: , 29. |
[59] | Kasten M , Marras C , Klein C ((2017) ) Nonmotor signs in genetic forms of Parkinson’s disease. In Int Rev Neurobiol 133: , 129–178. |
[60] | Book A , Guella I , Candido T , Brice A , Hattori N , Jeon B , Farrer MJ ((2018) ) A meta-analysis of α-synuclein multiplication in familial parkinsonism. Front Neurol 9: , 1021. |
[61] | Meireles J , Massano J ((2012) ) Cognitive impairment and dementia in Parkinson’s disease: Clinical features, diagnosis, and management. Front Neurol 3: , 88. |
[62] | Factor SA , Steenland NK , Higgins DS , Molho ES , Kay DM , Montimurro J , Rosen AR , Zabetian CP , Payami H ((2011) ) Disease-related and genetic correlates of psychotic symptoms in Parkinson’s disease. Mov Disord 26: , 2190–2195. |
[63] | Deng H , Gao K , Jankovic J ((2013) ) The VPS35 gene and Parkinson’s disease. Mov Disord 28: , 569–575. |
[64] | Bianca B , Gerhard R , Alexander Z , Walter S ((2017) ) VPS35-linked Parkinson’s disease resembles the idiopathic disease: A review of clinical trials. J Alzheimers Dis Parkinsonism 7: , 6–8. |
[65] | Corvol JC , Poewe W ((2017) ) Pharmacogenetics of Parkinson’s disease in clinical practice. Mov Disord Clin Pract 4: , 173–180. |
[66] | Ruiz-Lopez M , Freitas ME , Oliveira LM , Munhoz RP , Fox SH , Rohani M , Rogaeva E , Lang AE , Fasano A ((2019) ) Diagnostic delay in Parkinson’s disease caused by PRKN mutations. Parkinsonism Relat Disord 63: , 217–220. |
[67] | Yamamura Y , Hattori N , Matsumine H , Kuzuhara S , Mizuno Y ((2000) ) Autosomal recessive early-onset parkinsonism with diurnal fluctuation: Clinicopathologic characteristics and molecular genetic identification. Brain Dev 22: , 87–91. |
[68] | Kasten M , Hartmann C , Hampf J , Schaake S , Westenberger A , Vollstedt EJ , Balck A , Domingo A , Vulinovic F , Dulovic M , Zorn I , Madoev H , Zehnle H , Lembeck CM , Schawe L , Reginold J , Huang J , König IR , Bertram L , Marras C , Lohmann K , Lill CM , Klein C ((2018) ) Genotype-phenotype relations for the Parkinson’s disease genes Parkin, PINK1, DJ1: MDSGene systematic review. Mov Disord 33: , 730–741. |
[69] | Benson DL , Huntley GW ((2019) ) Are we listening to everything the PARK genes are telling us? J Comp Neurol 527: , 1527–1540. |
[70] | Tranchant C , Koob M , Anheim M ((2017) ) Parkinsonian-Pyramidal syndromes: A systematic review. Parkinsonism Relat Disord 39: , 4–16. |
[71] | Park JS , Blair NF , Sue CM ((2015) ) The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms. Mov Disord 30: , 770–779. |
[72] | Schneider SA , Paisan-Ruiz C , Quinn NP , Lees AJ , Houlden H , Hardy J , Bhatia KP ((2010) ) ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord 25: , 979–984. |
[73] | di Fonzo A , Chien HF , Socal M , Giraudo S , Tassorelli C , Iliceto G , Fabbrini G , Marconi R , Fincati E , Abbruzzese G , Marini P , Squitieri F , Horstink MW , Montagna P , Libera AD , Stocchi F , Goldwurm S , Ferreira JJ , Meco G , Martignoni E , Lopiano L , Jardim LB , Oostra BA , Barbosa ER , Bonifati V , Bonifati V , Vanacore N , Meco G , Fabrizio E , Locuratolo N , Scoppetta C , Manfredi M , Berardelli A , Lopiano L , Giraudo S , Bergamasco B , Pacchetti C , Nappi G , Antonini A , Pezzoli G , Riboldazzi G , Bono G , Raudino F , Manfredi M , Fincati E , Tinazzi M , Bonizzato A , Ferracci C , Dalla Libera A , Abbruzzese G , Marchese R , Montagna P , Marini P , Massaro F , Guidi M , Minardi C , Rasi F , Onofrj M , Thomas A , Stocchi F , Vacca L , de Pandis F , de Mari M , Diroma C , Iliceto G , Lamberti P , Toni V , Trianni G , Mauro A , de Gaetano A , Rizzo M , Cossu G , Rieder CRM , Saraiva-Pereira ML ((2007) ) ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 68: , 1557–1562. |
[74] | Lu CS , Lai SC , Wu RM , Weng YH , Huang CL , Chen RS , Chang HC , Wu-Chou YH , Yeh TH ((2012) ) PLA2G6 mutations in PARK14-linked young-onset parkinsonism and sporadic Parkinson’s disease. Med Genet B Neuropsychiatr Genet 159 B (2): , 183–191. |
[75] | Guo Y , Tang B , Guo J ((2018) ) PLA2G6-Associated Neurodegeneration (PLAN): Review of clinical phenotypes and genotypes. Front Neurol 9: , 1100. |
[76] | Conedera S , Apaydin H , Li Y , Yoshino H , Ikeda A , Matsushima T , Funayama M , Nishioka K , Hattori N ((2016) ) FBXO7 mutations in Parkinson’s disease and multiple system atrophy. Neurobiol Aging 40: , 192.e1–192.e5. |
[77] | Lesage S , Drouet V , Majounie E , Deramecourt V , Jacoupy M , Nicolas A , Cormier-Dequaire F , Hassoun SM , Pujol C , Ciura S , et al. ((2016) ) Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am J Hum Genet 98: , 500–513. |
[78] | Pal GD , Hall D , Ouyang B , Phelps J , Alcalay R , Pauciulo MW , Nichols WC , Clark L , Mejia-Santana H , Blasucci L , Goetz CG , Comella C , Colcher A , Gan-Or Z , Rouleau GA , Marder K ((2016) ) Genetic and clinical predictors of deep brain stimulation in young-onset Parkinson’s disease. Mov Disord Clin Pract 3: , 465–471. |
[79] | de Oliveira LM , Barbosa ER , Aquino CC , Munhoz RP , Fasano A , Cury RG ((2019) ) Deep brain stimulation in patients with mutations in Parkinson’s disease–related genes: A systematic review. Mov Disord Clin Pract 6: , 359–368. |
[80] | Kuusimäki T , Korpela J , Pekkonen E , Martikainen MH , Antonini A , Kaasinen V ((2020) ) Deep brain stimulation for monogenic Parkinson’s disease: A systematic review. J Neurol 267: , 883–897. |
[81] | Artusi CA , Dwivedi AK , Romagnolo A , Pal G , Kauffman M , Mata I , Patel D , Vizcarra JA , Duker A , Marsili L , Cheeran B , Woo D , Contarino MF , Verhagen L , Lopiano L , Espay AJ , Fasano A , Merola A ((2019) ) Association of subthalamic deep brain stimulation with motor, functional, and pharmacologic outcomes in patients with monogenic Parkinson disease: A systematic review and meta-analysis. JAMA Netw Open 2: , e187800. |
[82] | Mangone G , Bekadar S , Cormier-Dequaire F , Tahiri K , Welaratne A , Czernecki V , Pineau F , Karachi C , Castrioto A , Durif F , Tranchant C , Devos D , Thobois S , Meissner WG , Navarro MS , Cornu P , Lesage S , Brice A , Welter ML , Corvol J-C , Benchetrit E , Delaby L , Berthet D , Danjou F , Vidaihlet M , Krack P , Pelissier P , Morand D , Delaigue C , Barun N , Anheim M , Pleuvret M , Destée A , Defebvre L , Moreau C , Simonin C , Ryckewaert G , Kreisler A , Mutez E , Carrière N , Hopes L , Tard C , Grolez G , Dujardin K , Pecheux N , Delliaux M , Rolland A-S , Broussolle E , Laurencin C , Tison F , Burbaud P ((2020) ) Early cognitive decline after bilateral subthalamic deep brain stimulation in Parkinson’s disease patients with GBA mutations. Parkinsonism Relat Disord 76: , 56–62. |
[83] | Pal G , Mangone G , Hill EJ , Ouyang B , Liu Y , Lythe V , Ehrlich D , Saunders-Pullman R , Shanker V , Bressman S , Alcalay RN , Garcia P , Marder KS , Aasly J , Mouradian MM , Link S , Rosenbaum M , Anderson S , Bernard B , Wilson R , Stebbins G , Nichols WC , Welter M-L , Sani S , Afshari M , Verhagen L , de Bie RMA , Foltynie T , Hall D , Corvol J-C , Goetz CG ((2022) ) Parkinson disease and STN-DBS: Cognitive effects in GBA mutation carriers. Ann Neurol 91: , 424–435. |
[84] | Williams NR , Foote KD , Okun MS ((2014) ) Subthalamic nucleus versus globus pallidus internus deep brain stimulation: Translating the rematch into clinical practice. Mov Disord Clin Pract 1: , 24–35. |
[85] | Zhang J , Li J , Chen F , Liu X , Jiang C , Hu X , Ma L , Xu Z ((2021) ) STN versus GPi deep brain stimulation for dyskinesia improvement in advanced Parkinson’s disease: A meta-analysis of randomized controlled trials. Clin Neurol Neurosurg 201: , 106450. |
[86] | Li J , Mei S , Jia X , Zhang Y ((2021) ) Evaluation of the direct effect of bilateral deep brain stimulation of the subthalamic nucleus on levodopa-induced on-dyskinesia in Parkinson’s disease. Front Neurol 12: , 595741. |
[87] | Fan S-Y , Wang K-L , Hu W , Eisinger RS , Han A , Han C-L , Wang Q , Michitomo S , Zhang J-G , Wang F , Ramirez-Zamora A , Meng F-G ((2020) ) Pallidal versus subthalamic nucleus deep brain stimulation for levodopa-induced dyskinesia. Ann Clin Transl Neurol 7: , 59–68. |
[88] | Liu Y , Li W , Tan C , Liu X , Wang X , Gui Y , Qin L , Deng F , Hu C , Chen L ((2014) ) Meta-analysis comparing deep brain stimulation of the globus pallidus and subthalamic nucleus to treat advanced Parkinson disease. J Neurosurg 121: , 709–18. |
[89] | Rughani A , Schwalb JM , Sidiropoulos C , Pilitsis J , Ramirez-Zamora A , Sweet JA , Mittal S , Espay AJ , Martinez JG , Abosch A , Eskandar E , Gross R , Alterman R , Hamani C ((2018) ) Congress of Neurological Surgeons systematic review and evidence-based guideline on subthalamic nucleus and globus pallidus internus deep brain stimulation for the treatment of patients with Parkinson’s disease: Executive summary. Neurosurgery 82: , 753–756. |
[90] | Wang J-W , Zhang Y-Q , Zhang X-H , Wang Y-P , Li J-P , Li Y-J ((2016) ) Cognitive and psychiatric effects of STN versus GPi deep brain stimulation in Parkinson’s disease: A meta-analysis of randomized controlled trials. PLoS One 11: , e0156721. |
[91] | Xie Y , Meng X , Xiao J , Zhang J , Zhang J ((2016) ) Cognitive changes following bilateral deep brain stimulation of subthalamic nucleus in Parkinson’s disease: A meta-analysis. Biomed Res Int 2016: , 3596415. |
[92] | Mehanna R , Bajwa JA , Fernandez H , Wagle Shukla AA ((2017) ) Cognitive impact of deep brain stimulation on Parkinson’s disease patients. Parkinsons Disease 2017: , 3085140. |
[93] | Lachenmayer ML , Mürset M , Antih N , Debove I , Muellner J , Bompart M , Schlaeppi J-A , Nowacki A , You H , Michelis JP , Dransart A , Pollo C , Deuschl G , Krack P ((2021) ) Subthalamic and pallidal deep brain stimulation for Parkinson’s disease—meta-analysis of outcomes. NPJ Parkinsons Dis 7: , 77. |
[94] | Konno T , Ross OA , Puschmann A , Dickson DW , Wszolek ZK ((2016) ) Autosomal dominant Parkinson’s disease caused by SNCA duplications. Parkinsonism Relat Disord 22: , S1–S6. |
[95] | Perandones C , Aráoz Olivos N , Raina GB , Pellene LA , Giugni JC , Calvo DS , Radrizzani M , Piedimonte F , Micheli FE ((2015) ) Successful GPi stimulation in genetic Parkinson’s disease caused by mosaicism of alpha-synuclein gene duplication: First description. J Neurol 262: , 222–223. |
[96] | Moro E , Volkmann J , König IR , Winkler S , Hiller A , Hassin-Baer S , Herzog J , Schnitzler A , Lohmann K , Pinsker MO , Voges J , Djarmatic A , Seibler P , Lozano AM , Rogaeva E , Lang AE , Deuschl G , Klein C ((2008) ) Bilateral subthalamic stimulation in Parkin and PINK1 parkinsonism. Neurology 70: , 1186–1191. |
[97] | Borellini L , Cogiamanian F , Carrabba G , Locatelli M , Rampini P , di Fonzo A , Bana C , Barbieri S , Ardolino G ((2017) ) Globus pallidus internus deep brain stimulation in PINK-1 related Parkinson’s disease: A case report. Parkinsonism Relat Disord 38: , 93–94. |
[98] | Cif L , Kurian MA , Gonzalez V , Garcia-Ptacek S , Roujeau T , Gelisse P , Moura de Ribeiro AM , Crespel A , MacPherson L , Coubes P ((2014) ) Atypical PLA2G6-associated neurodegeneration: Social communication impairment, dystonia and response to deep brain stimulation. Mov Disord Clin Pract 1: , 128–131. |
[99] | Spielberger S , Seppi K , Wolf E , Eisner W , Poewe W ((2015) ) Invasive treatment strategies in a patient with PARK 15–associated parkinsonism. Mov Disord Clin Pract 2: , 434–435. |
[100] | Olgiati S , Quadri M , Fang M , Rood JPMA , Saute JA , Chien HF , Bouwkamp CG , Graafland J , Minneboo M , Breedveld GJ , Zhang J , Verheijen FW , Boon AJW , Kievit AJA , Jardim LB , Mandemakers W , Barbosa ER , Rieder CRM , Leenders KL , Wang J , Bonifati V ((2016) ) DNAJC6 mutations associated with early-onset Parkinson’s disease. Ann Neurol 79: , 244–256. |
[101] | Schormair B , Kemlink D , Mollenhauer B , Fiala O , Machetanz G , Roth J , Berutti R , Strom TM , Haslinger B , Trenkwalder C , Zahorakova D , Martasek P , Ruzicka E , Winkelmann J ((2018) ) Diagnostic exome sequencing in early-onset Parkinson’s disease confirms VPS13C as a rare cause of autosomal-recessive Parkinson’s disease. Clin Genet 93: , 603–612. |
[102] | Djarmati A , Hagenah J , Reetz K , Winkler S , Behrens MI , Pawlack H , Lohmann K , Ramirez A , Tadić V , Brüggemann N , Berg D , Siebner HR , Lang AE , Pramstaller PP , Binkofski F , Kostić VS , Volkmann J , Gasser T , Klein C ((2009) ) ATP13A2 variants in early-onset Parkinson’s disease patients and controls. Mov Disord 24: , 2104–2111. |
[103] | Foltynie T , Magee C , James C , Webster GJM , Lees AJ , Limousin P ((2013) ) Impact of duodopa on quality of life in advanced Parkinson’s disease: A UK case series. Parkinsons Dis 2013: , 362908. |
[104] | Thaler A , Hillel A , Shabtai H , Giladi N , Gurevich T ((2017) ) Assessing the response to L-dopa/carbidopa intestinal gel infusion (Deudopa) based on genetic status. Mov Disord 32: , Abstract number 1015. |
[105] | Thaler A , Livneh V , Hillel A , Strauss H , Yahalom H , Shabtai H , Migirov Senderovich A , Israel-Korn S , Fay-Carmon T , Giladi N , Hassin Baer S , Gurevich T ((2019) ) Assessing the response to L-dopa/carbidopa intestinal gel infusion based on genetic status. Mov Disord 34: , Abstract number 434. |
[106] | Bohlega S , Abou Al-Shaar H , Alkhairallah T , Al-Ajlan F , Hasan N , Alkahtani K ((2015) ) Levodopa-carbidopa intestinal gel infusion therapy in advanced Parkinson’s disease: Single middle eastern center experience. Eur Neurol 74: , 227–236. |
[107] | Paviour DC , Marion M-H ((2012) ) PINK1: Pumps, paraesthesia, punding and psychosis. J Neurol 259: , 1241–1242. |
[108] | Khan NL , Jain S , Lynch JM , Pavese N , Abou-Sleiman P , Holton JL , Healy DG , Gilks WP , Sweeney MG , Ganguly M , Gibbons V , Gandhi S , Vaughan J , Eunson LH , Katzenschlager R , Gayton J , Lennox G , Revesz T , Nicholl D , Bhatia KP , Quinn N , Brooks D , Lees AJ , Davis MB , Piccini P , Singleton AB , Wood NW ((2005) ) Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: Clinical, pathological, olfactory and functional imaging and genetic data. Brain 128: , 2786–2796. |
[109] | Khan NL , Graham E , Critchley P , Schrag AE , Wood NW , Lees AJ , Bhatia KP , Quinn N ((2003) ) Parkin disease: A phenotypic study of a large case series. Brain 126: , 1279–1292. |
[110] | Giri A , Guven G , Hanagasi H , Hauser AK , Erginul-Unaltuna N , Bilgic B , Gurvit H , Heutink P , Gasser T , Lohmann E , Simón-Sánchez J ((2016) ) PLA2G6 mutations related to distinct phenotypes: A new case with early-onset parkinsonism. Tremor Other Hyperkinet Mov (N Y) 2016: , 1–6. |
[111] | Ryden LE , Lewis SJG ((2019) ) Parkinson’s disease in the era of personalised medicine: One size does not fit all. Drugs Aging 36: , 103–113. |
[112] | Broggi G , Franzini A , Marras C , Romito L , Albanese A ((2003) ) Surgery of Parkinson’s disease: Inclusion criteria and follow-up. Neurol Sci 24: , 38–40. |
[113] | Deuschl G , Schade-Brittinger C , Krack P , Volkmann J , Schäfer H , Bötzel K , Daniels C , Deutschländer A , Dillmann U , Eisner W , Gruber D , Hamel W , Herzog J , Hilker R , Klebe S , Kloß M , Koy J , Krause M , Kupsch A , Lorenz D , Lorenzl S , Mehdorn HM , Moringlane JR , Oertel W , Pinsker MO , Reichmann H , Reuß A , Schneider GH , Schnitzler A , Steude U , Sturm V , Timmermann L , Tronnier V , Trottenberg T , Wojtecki L , Wolf E , Poewe W , Voges J ((2006) ) A randomized trial of deep-brain stimulation for Parkinson’s disease. N Engl J Med 355: , 896–908. |
[114] | Schuepbach WMM , Rau J , Knudsen K , Volkmann J , Krack P , Timmermann L , Hälbig TD , Hesekamp H , Navarro SM , Meier N , Falk D , Mehdorn M , Paschen S , Maarouf M , Barbe MT , Fink GR , Kupsch A , Gruber D , Schneider GH , Seigneuret E , Kistner A , Chaynes P , Ory-Magne F , Brefel Courbon C , Vesper J , Schnitzler A , Wojtecki L , Houeto JL , Bataille B , Maltête D , Damier P , Raoul S , Sixel-Doering F , Hellwig D , Gharabaghi A , Krüger R , Pinsker MO , Amtage F , Régis JM , Witjas T , Thobois S , Mertens P , Kloss M , Hartmann A , Oertel WH , Post B , Speelman H , Agid Y , Schade-Brittinger C , Deuschl G ((2013) ) Neurostimulation for Parkinson’s disease with early motor complications. N Engl J Med 368: , 610–622. |
[115] | Deuschl G , Agid Y ((2013) ) Subthalamic neurostimulation for Parkinson’s disease with early fluctuations: Balancing the risks and benefits. Lancet Neurol 12: , 1025–1034. |
[116] | Almeida L , Deeb W , Spears C , Opri E , Molina R , Martinez-Ramirez D , Gunduz A , Hess CW , Okun MS ((2017) ) Current practice and the future of deep brain stimulation therapy in Parkinson’s disease. Semin Neurol 37: , 205–214. |
[117] | Yin Z , Cao Y , Zheng S , Duan J , Zhou D , Xu R , Hong T , Lu G ((2018) ) Persistent adverse effects following different targets and periods after bilateral deep brain stimulation in patients with Parkinson’s disease. J Neurol Sci 393: , 116–127. |
[118] | Kurtis MM , Rajah T , Delgado LF , Dafsari HS ((2017) ) The effect of deep brain stimulation on the non-motor symptoms of Parkinson’s disease: A critical review of the current evidence. NPJ Parkinsons Dis 3: , 16024. |
[119] | Eisinger RS , Ramirez-Zamora A , Carbunaru S , Ptak B , Peng-Chen Z , Okun MS , Gunduz A ((2019) ) Medications, deep brain stimulation, and other factors influencing impulse control disorders in Parkinson’s disease. Front Neurol 10: , 1–14. |
[120] | Limousin P , Foltynie T ((2019) ) Long-term outcomes of deep brain stimulation in Parkinson disease. Nat Rev Neurol 15: , 234–242. |
[121] | Olanow CW , Kieburtz K , Odin P , Espay AJ , Standaert DG , Fernandez HH , Vanagunas A , Othman AA , Widnell KL , Robieson WZ , Pritchett Y , Chatamra K , Benesh J , Lenz RA , Antonini A ((2014) ) Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson’s disease: A randomised, controlled, double-blind, double-dummy study. Lancet Neurol 13: , 141–149. |
[122] | Lopiano L , Modugno N , Marano P , Sensi M , Meco G , Solla P , Gusmaroli G , Tamma F , Mancini F , Quatrale R , Zangaglia R , Bentivoglio A , Eleopra R , Gualberti G , Melzi G , Antonini A ((2019) ) Motor and non-motor outcomes in patients with advanced Parkinson’s disease treated with levodopa/carbidopa intestinal gel: Final results of the GREENFIELD observational study. J Neurol 266: , 2164–2176. |
[123] | Fernandez HH , Standaert DG , Hauser RA , Lang AE , Fung VSC , Klostermann F , Lew MF , Odin P , Steiger M , Yakupov EZ , Chouinard S , Suchowersky O , Dubow J , Hall CM , Chatamra K , Robieson WZ , Benesh JA , Espay AJ ((2015) ) Levodopa-carbidopa intestinal gel in advanced Parkinson’s disease: Final 12-month, open-label results. Mov Disord 30: , 500–509. |
[124] | Krüger R , Hilker R , Winkler C , Lorrain M , Hahne M , Redecker C , Lingor P , Jost WH ((2016) ) Advanced stages of PD: Interventional therapies and related patient-centered care. J Neural Transm 123: , 31–43. |
[125] | Fasano A , Ricciardi L , Lena F , Bentivoglio AR , Modugno N ((2012) ) Intrajejunal levodopa infusion in advanced Parkinson’s disease: Long-term effects on motor and non-motor symptoms and impact on patient’s and caregiver’s quality of life. Eur Rev Med Pharmacol Sci 16: , 79–89. |
[126] | Buongiorno M , Antonelli F , Cámara A , Puente V , de Fabregues-Nebot O , Hernandez-Vara J , Calopa M , Pascual-Sedano B , Campolongo A , Valldeoriola F , Tolosa E , Kulisevsky J , Martí MJ ((2015) ) Long-term response to continuous duodenal infusion of levodopa/carbidopa gel in patients with advanced Parkinson disease: The Barcelona registry. Parkinsonism Relat Disord 21: , 871–876. |
[127] | Katzenschlager R , Poewe W , Rascol O , Trenkwalder C , Deuschl G , Chaudhuri KR , Henriksen T , van Laar T , Spivey K , Vel S , Staines H , Lees A ((2018) ) Apomorphine subcutaneous infusion in patients with Parkinson’s disease with persistent motor fluctuations (TOLEDO): A multicentre, double-blind, randomised, placebo-controlled trial. Lancet Neurol 17: , 749–759. |
[128] | Mundt-Petersen U , Odin P ((2017) ) Infusional therapies, continuous dopaminergic stimulation, and nonmotor symptoms. Int Rev Neurobiol 134: , 1019–1044. |
[129] | van Laar T , Postma AG , Drent M ((2010) ) Continuous subcutaneous infusion of apomorphine can be used safely in patients with Parkinson’s disease and pre-existing visual hallucinations. Parkinsonism Relat Disord 16: , 71–72. |
[130] | Ray Chaudhuri K , Critchley P , Abbott RJ , Pye IF , Millac PAH ((1988) ) Subcutaneous apomorphine for on-off oscillations in Parkinson’s disease. Lancet 332: , 1260. |
[131] | Antonini A , Isaias IU , Rodolfi G , Landi A , Natuzzi F , Siri C , Pezzoli G ((2011) ) A 5-year prospective assessment of advanced Parkinson disease patients treated with subcutaneous apomorphine infusion or deep brain stimulation. J Neurol 258: , 579–585. |
[132] | Pietz K , Hagell P , Odin P ((1998) ) Subcutaneous apomorphine in late stage Parkinson’s disease: A long term follow up. J Neurol Neurosurg Psychiatry 65: , 709–716. |
[133] | Katzenschlager R , Hughes A , Evans A , Manson AJ , Hoffmann M , Swinn L , Watt H , Bhatia K , Quinn N , Lees AJ ((2005) ) Continuous subcutaneous apomorphine therapy improves dyskinesias in Parkinson’s disease: A prospective study using single-dose challenges. Mov Disord 20: , 151–157. |
[134] | Kaňovský P , Kubová D , Bareš M , Hortová H , Streitová H , Rektor I , Znojil V ((2002) ) Levodopa-induced dyskinesias and continuous subcutaneous infusions of apomorphine: Results of a two-year, prospective follow-up. Mov Disord 17: , 188–191. |
[135] | Ligaard J , Sannæs J , Pihlstrøm L ((2019) ) Deep brain stimulation and genetic variability in Parkinson’s disease: A review of the literature. NPJ Parkinsons Dis 5: , 1–10. |
[136] | Martikainen MH , Päivärinta M , Hietala M , Kaasinen V ((2015) ) Clinical and imaging findings in Parkinson disease associated with the A53E SNCA mutation. Neurol Genet 1: , e27. |

