
Preventing Parkinson’s Disease: An Environmental Agenda
Abstract
Fueled by aging populations and continued environmental contamination, the global burden of Parkinson’s disease (PD) is increasing. The disease, or more appropriately diseases, have multiple environmental and genetic influences but no approved disease modifying therapy. Additionally, efforts to prevent this debilitating disease have been limited. As numerous environmental contaminants (e.g., pesticides, metals, industrial chemicals) are implicated in PD, disease prevention is possible. To reduce the burden of PD, we have compiled preclinical and clinical research priorities that highlight both disease prediction and primary prevention. Though not exhaustive, the “PD prevention agenda” builds upon many years of research by our colleagues and proposes next steps through the lens of modifiable risk factors. The agenda identifies ten specific areas of further inquiry and considers the funding and policy changes that will be necessary to help prevent the world’s fastest growing brain disease.
PREFACE
In 1992, Charlotte Haley, a 68-year-old woman was frustrated at the lack of scientific progress against breast cancer. The disease had affected both her sister and daughter, and she saw little progress. So in her dining room in a sprawling suburb of Los Angeles, Haley took action. She began making small loops out of peach-colored ribbons. She created packages of five and included a note that read, “The National Cancer Institute’s annual budget is $1.8 billion, only 5%goes for cancer prevention. Help us wake up legislators and America by wearing this ribbon.” She mailed them to famous women across the country and handed them out to less famous women in the parking lots of grocery stores. Thus began a grassroots campaign to prevent the second leading cause of cancer deaths among women [1].
Against Ms. Haley’s wishes, the peach ribbons were commercialized by a cosmetics company, changed to pink, and in the process, the message about prevention was subsumed by a focus on early diagnosis, treatment, and “survivors” [1]. Survival indeed increased, but the incidence of breast cancer has continued to rise [2].
We face the same situation for Parkinson’s disease. For decades, many public and private funders have devoted their resources toward seeking a cure for Parkinson’s disease, which has resulted in incremental therapeutic advances for those with the disease. The Michael J. Fox Foundation alone has invested more than a billion dollars in Parkinson’s disease research this century [3]. However, the number of Americans with the disease has increased 35%in the last decade [4]. The U.S. is far from alone. The rates of Parkinson’s disease, adjusted for age, continue to rise in almost every part of the world [5]. We need to stop and ask why.
THE ROLE OF THE ENVIRONMENT IN PARKINSON’S DISEASE
Parkinson’s disease (PD), once considered rare, is now the world’s fastest growing neurological disease and comes at a heavy societal and economic cost [6]. The estimated number of people with PD in 1990 was 2.5 million, which more than doubled to 6.2 million by 2015 [7]. Aging within populations alone is expected to double that number again to 12.9 million by 2040 [8]. Other factors (e.g., pesticides, chemicals, air pollution, decreased smoking) may drive that number even higher, as PD incidence currently outpaces the rate of aging and is disproportionately on the rise in newly industrialized areas of the globe [7].
In parallel, years of research focused on biology have deciphered crucial protein functions and cellular pathways involved in the primary PD pathology—the degeneration of dopaminergic neurons from the substantia nigra and their axonal projections to the striatum [9, 10]. In addition, molecular mechanisms relating to mitochondrial function, protein accumulation, or neuroinflammation show numerous critical pathways contribute to PD-related pathology, such as α-synuclein aggregation or lysosomal dysfunction [11]. Interest in linking genetic mutations to PD risk has been a strong focus of PD research, leading to the discovery of genes involved in both inherited and idiopathic PD, such as LRRK2 [12] and glucocerebrosidase (GBA) [13]. However, even the most liberal estimates place the narrow-sense heritability of PD around 27%[14], suggesting that PD risk is still largely influenced by exogenous factors. These non-genetic factors continue to be underappreciated and understudied.
Elucidating these factors is enormously difficult. The term “environment” encompasses any non-endogenous factor, such as pathogenic infection, head trauma, diet, pharmaceutical, supplement, drug use, other physiological stressors (e.g., PTSD), and of course, chemical toxicants that pollute our water, air, and soil. Some are clearly evident. For example, strong evidence shows that agricultural pesticide applicators are at increased PD risk [15]. Others represent a combination of insidious, variable exposures that extend over decades, such as pesticides, organic solvents [16], and metals [17], and air pollution. The relationship between exposure and disease is variable, implying that gene-environment and environment-environment interactions and other exposure dynamics contribute to disease phenotype [18–20]. Additionally, PD is extremely complex and still not fully understood, with a long prodromal period occurring over decades when exposures may influence phenotype and progression at multiple points within a lifespan. Because of this, predicting environmental risk for PD is often difficult and requires a circuitous process of evaluating contaminant exposure levels, animal and cell toxicity studies, biological markers, and epidemiological research. Despite these inherent challenges, understanding environmental contribution to PD is critical, as it may help prevent an incurable disease. We can change much of our environment. Even with gene-editing technology, our ability to modify human genes at a large scale remains science fiction.
To this end, we have outlined a research agenda to prevent PD that is anchored in the modification of environmental factors. In doing so, we recognize the remarkable work of our colleagues who have established links between environmental exposures and neurodegeneration and continue to strengthen this evidence today. In addition, we appreciate that no single approach is sufficient or all encompassing and that multiple interdisciplinary measures will be required to advance changes aimed at preventing PD.
AN ENVIRONMENTAL RESEARCH AGENDA FOR PREVENTING PD
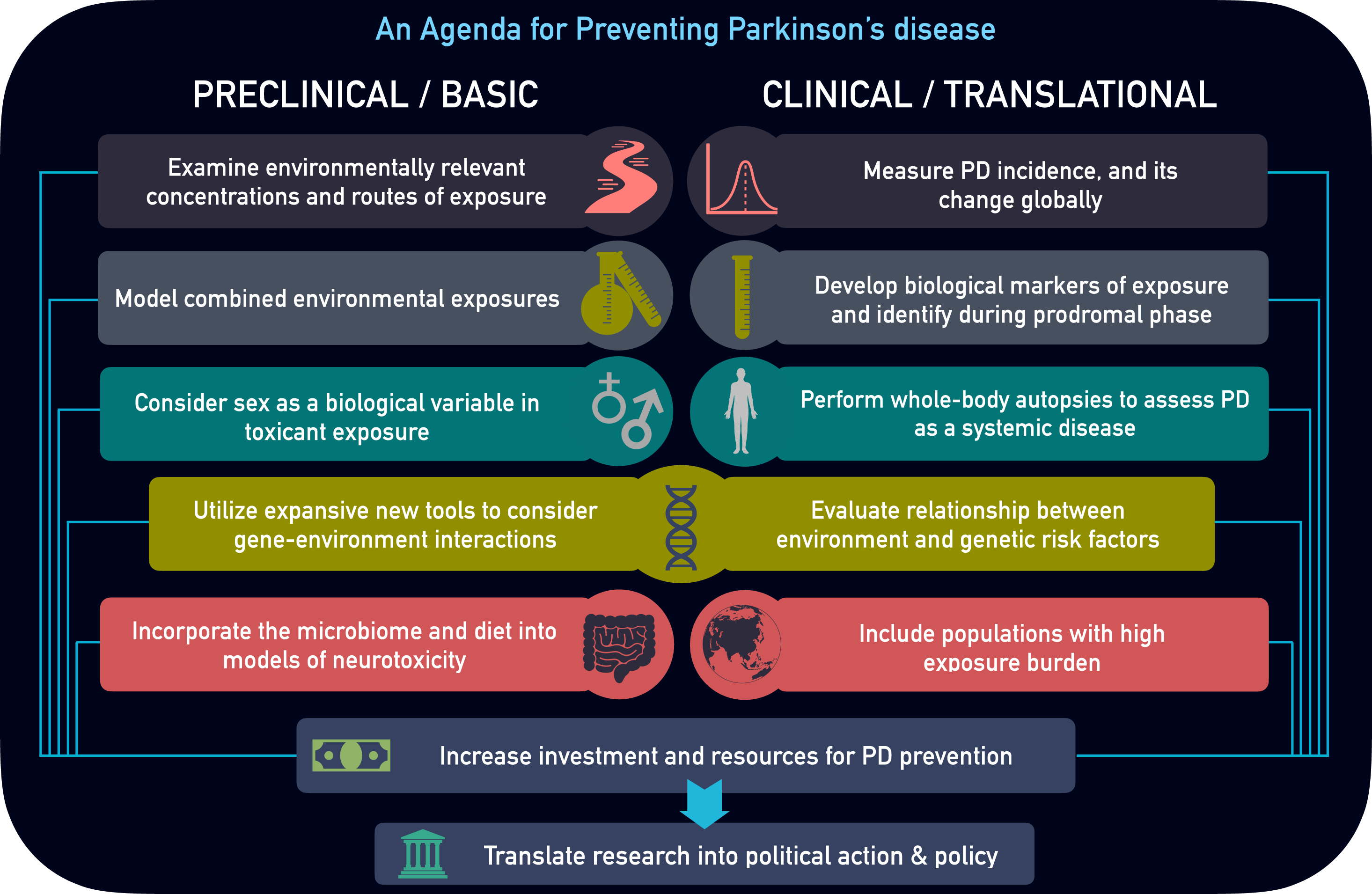
To prevent PD, our basic and clinical research activities must expand substantially. We present 10 key areas that could help accelerate disease prevention (Fig. 1).
Fig. 1
Parkinson’s disease Prevention Agenda. Preclinical and clinical research areas of focus to better characterize environmental influence and prevent Parkinson’s disease.
PRE-CLINICAL AND BASIC RESEARCH: FROM MOLECULAR BIOLOGY TO ANIMAL MODELS
Many key contributions to our understanding of environmental risk for PD in human populations have originated at the lab bench. After the curious observation of parkinsonism in heroin addicts reported by Langston and colleagues, scientists turned to the laboratory to study a range of environmental toxicants [21, 22]. The earliest exogenous toxicant models of PD, such as MPTP in mice and primates [23], ushered in a new era of pre-clinical research, providing a platform for biological mechanisms and experimental therapeutics development. Toxicant models also served as proof-of-principal concept that exogenous substances can produce selective degeneration of dopaminergic neurons from the substantia nigra (SN), thus generating evidence that other structurally or biochemically similar environmental contaminants may do the same. This was confirmed with rotenone and paraquat treatment in rodents, which when administered systemically, caused selective degeneration of dopaminergic neurons and other hallmark PD pathology [18, 24–28]. In parallel, epidemiological data linking rotenone or paraquat exposure to PD risk was published, providing complementary, empirical evidence for environmental risk related to these pesticides [15, 29, 30]. Multi-pronged approaches such as these are necessary to uncover both the risk and mechanisms associated with environmental factors for PD; however, as the link with pesticides such as paraquat remain strong, we need to expand this approach to numerous other toxicants implicated in the disease. In addition, a focus on improving translation of basic research will be required, which we discuss below.
Examine environmentally relevant concentrations and routes of exposure
In vivo toxicant models of PD historically fall into two categories: 1) models of neurodegeneration or 2) models of exposure. The former have been a major focus of basic PD research for the last several decades, where the significant loss of dopaminergic neurons from the SN and motor behavioral deficits are standard outcomes. For example, the toxin 6-hydroxydopamine (6-OHDA) causes severe dopaminergic cell death, which is ideal for unilateral lesioning of the SN or its projections to striatum [31–34]. Similarly, differential dosing with MPTP can cause acute dopaminergic neurodegeneration or can be administered repeatedly to produce a progressive SN lesion in rodents and non-human primates [35–37]. These models have been useful. Surgical treatments, such as deep brain stimulation (DBS), were developed based on animal models of PD [38], but this same success has not yet occurred for pharmacological treatments.
There is also room for improvement in models of exposure. Pesticide models of PD, such as rotenone and paraquat, emulate a relevant environmental risk, but often at much higher concentrations than encountered in environmental or even occupational settings. The reason for this is valid; paraquat or rotenone are used as tools to model systemic exposure that results in selective dopaminergic neurodegeneration or neuropathology in animals with a fraction of a human lifespan [24, 26, 39, 40]. And remarkably, pesticide models recapitulate features of PD in ways that are still poorly understood, such as endolysosomal impairment, disruptions to vesicular trafficking, calcium dysregulation, iron accumulation, and gastrointestinal dysfunction [25, 41, 42]. However, inferring risk from exposure via rodent models of PD is not equivalent to modeling environmental exposure, such as is commonly practiced to assess cancer risks. To do this, we must also include chronic, environmentally relevant concentrations via routes of exposure most applicable to putative risk factors. Dose or exposure extrapolation from human to animal is non-exact, however, the U.S. Food and Drug Administration (FDA) provides guidance for Human Equivalent Dose conversion based on body surface area using the equation: Human Equivalent Dose = animal dose in mg/kg x (animal weight in kg / human weight in kg)0.33 [43, 44]. An example for paraquat is provided in Table 1; however, such limits should be considered in context of the environment under investigation. In addition, age at exposure is key; developmental exposure (as discussed in the Clinical Agenda below) versus adult or aged adult exposure results in different neurotoxic, neurobehavioral, and neurodegenerative sequelae [45]. Lastly, the duration of exposure requires careful consideration for animal to human translation as environmental exposures associated with neurodegenerative disease often occur over a chronic time period. Mouse to human age conversion estimates suggest one mouse day is roughly equivalent to 30 human days, making one human year approximately 12 mouse days [46]. High doses of neurotoxicant treatment in rodents for a very short time period, even when accounting for age conversion, should be carefully considered as it may limit relevance to human exposure.
| Box 1: Types of Mixtures to Consider in Neurodegenerative Research Models |
| Simple Mixture: Small number of chemicals (less than 10) with known composition. |
| Examples: pesticide cocktail (paraquat + maneb), metals in drinking water (lead + copper + nickel), |
| pharmaceutical treatments. The composition could be based on known human exposures but should also consider |
| related compounds in the same chemical class. |
| Complex Mixture: More than 10 (could be hundreds or thousands) of chemicals with unknown composition. |
| Examples: air pollution, welding fumes, oil spills, contaminated drinking water. The composition could be |
| equimolar or derived from human population data. |
Table 1
Pesticide exposure dose extrapolated from human to rodent (mouse and rat)
| Risk Factor | Primary Route of Exposure | Exposure Limit/Maximum Contaminat Level | Extrapolated Daily Dose in Mice | Extrapolated Daily Dose in Rats |
| Paraquat | Inhalation | 0.1 mg/m3 † | 1.22 mg/m3 | 0.6 mg/m3 |
| Drinking Water | 20 ppb | 245μg/kg | 123μg/kg |
†Time Weighted Average: 8-hour total weight average, particulate size dependent. Inhalation dose calculated using the fixed allometric exponent: Xh = Xa (Ma/Mh)1 - .67 without Uncertainty Factors [47], however dosage is dependent on particulate size and duration of exposure. Oral dose calculated using the U.S. FDA conversion guidelines, Human Equivalent Dose = animal dose in mg/kg x (animal weight in kg / human weight in kg)0.33 [46].
Model combined environmental exposures
Environmental contaminants are rarely, if ever, encountered in isolation. Toward this end, modeling combined exposures is critical for assessing PD risk in a basic research setting. Such an approach should also consider lifestyle factors, much the way epidemiology studies stratify PD risk based on smoking [48, 49]. This is a difficult task; regulatory agencies have struggled with how to deal with chemical mixtures. Besides the challenges of interpreting the scientific findings, the regulations are not designed to address mixtures. However, from a scientific perspective we must try to study the real-life norm of combined exposures.
An example of this strategy was employed to measure the combined toxicity of trichloroethylene (TCE) with traumatic brain injury (TBI), both of which are independently implicated in PD risk, on mitochondrial function in the nigrostriatial tract [50–53]. In an acute treatment model in male Fisher 344 rats, neither TCE nor TBI alone caused significant reduction in striatal mitochondrial complex I activity, but together produced approximately a 50%reduction in this brain region [54]. As TCE was heavily used by the military until 2007 [55], exposure to the organic solvent plus combat or training injury (e.g., concussion) are realistic combined exposures that may contribute to PD risk in military personnel [20, 56]. In addition, TCE is substantially metabolized in the body resulting in numerous byproducts that may contribute to toxicity of combined phyiological stressors [57].
Combined exposure to solvents, pesticides, metals, and other industrial byproducts also must be considered, as additive or synergistic effects of these compounds influence their individual toxicokinetics, and ultimately their combined neurotoxicity. This type of environment-environment interaction better models real life risk assessment, requires careful experimental design as mixtures are inherently complicated (Box 1), and may result in pathology specific to the type and complexity of the mixture. For example, simple mixitures, such as the two neurotoxicants manganese and copper, produced a synergistic interaction with dopamine which modulated DNA oxidation in neurons [58]. Likewise, a mixture designed to emulate Gulf War toxicant exposure (pyridostigmine bromide, chlorpyrifos, and N,N-diethyl-m-toluamide [DEET]) caused differential gene expression in the hippocampus of mice after two weeks of treatment, profiling one type of pathology underlying the many neurological symptoms of Gulf War veterans [59]. In contrast, diesel exhaust, a complex mixture and component of air pollution, disrupted autophagy, caused protein accumulation, and resulted in neurodegeneration in a zebrafish model [60]. Characterizing mixtures, and their resultant neurotoxicities, may help target intervention strategies based on relative environmental burden, such as metal exposure in drinking water, military service-related exposures, and individuals who live in areas with high air pollution.
Another exposure intersection that is poorly understood, but highly relevant for PD, is the interaction between environmental factors and pharmaceutical treatments. Such interactions may both increase risk of disease and influence the progression of extant disease. For example, most individuals with PD are eventually treated with levodopa (L-dopa) or dopamine agonists, which significantly impact metabolic pathways [61]. Increased cellular dopamine concentrations and changes in metabolism may readily modify toxicity from environmental contaminants. In this context, in vitro experiments with neuroblastoma cells exposed to arsenic showed synergistic toxicity when treated in combination with exogenous dopamine [62]. Thus, though minimally studied, the impact of environmental exposure combined with pharmaceutical use requires more focused attention, as pharmaceutical-environment interactions may modulate disease risk and progression [63].
Consider sex as a biological variable in toxicant exposure
Just as all biomedical research requires an equitable inclusion of women, measuring environmental risk for PD must also consider sex as a biological variable. While PD appears to be more prevalent in men (approximately 1.5 : 1), sexual dimorphisms are also apparent in PD symptoms [64]. An initial hypothesis that men were more likely to be employed in occupations that elevated exposure risk, such as pesticide applicators and factory workers, potentially explains some, but not all of the gender disparity in PD. In fact, sex differences in PD prevalence may be geographically or culturally specific. One study from Japan found that PD prevalence in women was significantly higher than men [65]; however, it is unclear whether this increased female-to-male PD ratio is due specifically to the environment. In Japan, women historically outnumbered men as farmers [66] suggesting exposure to agricultural pesticides could underlie this observation.
In the preclinical research setting, in vivo models using both male and female animals are the most direct way to assess sexual dimorphism in neurotoxicity, as they include both genetic and hormonal, primary and secondary sex characteristics of mammalian development [67]. This was recently demonstrated in mice exposed developmentally to the organochlorine pesticide dieldrin followed by stereotaxic α-synuclein fibril injection at twelve weeks of age, which produced male-specific behavioral deficits and increased dopamine turnover in the striatum [68]. Other studies support elevated sensitivity of the dopaminergic system to the pesticides paraquat and rotenone in male rodents [69, 70]. Underlining the value of modeling exposures in vivo, a similar observation was uncovered in previously collected (but unanalyzed) human epidemiological data, suggesting that women may indeed be less susceptible the neurotoxicity of rotenone [71].
Translation from combined studies such as these will help define the degree to which sexual dimorphism in PD is driven by environmental exposures, but they are not without challenges. Incorporating the stage of estrus into in vivo studies is key, as fluctuations in brain chemistry change during the estrus cycle, such as neuromodulatory signaling driven by adenosine levels [72]. In addition, investigating exposure in animal studies modeling pre-and-post menopausal life stages, including the use of hormone replacement, are needed to understand neurotoxicity relative to hormone levels that are important in human disease etiology. Nonetheless, reporting results of sex-specific toxicity in laboratory animals should be considered a requirement to fully understand environmental influence on chronic neurologic disease.
Utilize expansive new tools to consider gene-environment interaction
All disease phenotype is a result of our genes (intrinsic factors) and our environment (extrinsic factors). As we predict that gene-environment interaction drives the majority of idiopathic PD cases (further discussed in the Clinical Agenda below), preclinical research platforms provide a crucial resource for testing these associations. Transgenic and knock-in animal models, induced pluripotent stem cells (iPSCs) and other human derived cells, CRISPR-edited cell lines, zebrafish, drosophila, C. elegans, and yeast, are well-suited to manipulate genetic targets that interact with environmental contaminants. In addition, high-throughput content assays, such as transcriptomics (RNA-Seq, single-cell Seq, ATAC-Seq, spatial transcriptomics) and epigenetics, provide ideal measurements to evaluate toxicity of environmental contaminants linked to PD, and to screen for novel chemicals of concern. In combination, these assays are even more powerful. For example, Parmalee and colleagues have characterized a robust method to perform RNA-Seq in C. elegans exposed to manganese, which could be easily adaptable for any environmental exposure [73]. Combined with the relative ease of genetic manipulation in C. elegans, platforms such as these are ideal for amassing gene-environment information relative to disease mechanisms in PD, like mitochondria [74, 75] and monoamine function [76].
Genome-wide interaction studies that incorporate one or more environmental factor associated with PD can uncover novel risk loci, as was demonstrated by Hill-Burns et al. [77] that found the snaptic vesicle glycoprotein 2C (SV2C) interacted with nicotine in Drosophila and humans, providing a genetic basis for smoking and PD risk. Following this, studies in mice showed that SV2C mediated dopamine homeostasis in the basal ganglia and altered expression of SV2C was observed in postmortem brain tissue from PD cases [78]. SV2C was then identified in a GWAS study as a novel risk locus for PD [79], showing that the inclusion of environmental risk factors in GWAS studies combined with preclinical modeling may help discover other intrinsic vulnerabilities with relatively low penetrance.
With relevance to human-derived samples, iPSCs, organoids, and fibroblasts provide a distinct opportunity to assess intrinsic vulnerability to common environmental exposures, both for individuals with inherited PD, as well as sporadic cases that do not express common PD mutations [80]. These interactions can also uncover important biological similarities of PD pathogenesis between genetic mutations and environmental exposures, as was shown in iPSC-derived dopaminergic neurons expressing the LRRK2-G2019S mutation, which showed a genetic expression profile similar to that of control iPSCs treated with the pesticide rotenone [81]. As high-throughput and automated platforms continue to develop, a key factor for PD is to include relevant exposure conditions to ensure the biological basis of disease is not solely translated from an isolated laboratory environment.
Incorporate the microbiome and diet
The gut-brain axis has emerged in the last decade as an interesting target to understand PD origins and phenotype, as data from population-based cohort studies showed that individuals who underwent truncal vagotomy were at lower risk for PD [82]. A decade earlier, the gut-brain connection had been postulated as a potential route for toxicants, pathogens, or pathogenic processes to access to the brain via the vagus nerve [83]. In addition, animal studies confirmed the transmission of α-synuclein from the gut to the brain [84], which was limited by vagotomy in mice [85], while intestinal α-synuclein expression was influenced by bacterial lipopolysaccharide exposure [86]. The gastrointestinal system also represents a central target for environmental contaminants, as ingestion is a common route of exposure for systemic toxicants associated with PD (e.g., drinking water contaminants; [87]). A proof-of-prinicple example of this was shown in mice treated with rotenone via intragastric gavage, that displayed elevated levels of α-synuclein in the enteric nervous system as well as the brain [88].
Gastrointestinal microbiome heterogeneity or dysbiosis is a also major factor in xenobiotic metabolism [89], possibly driving differential disease risk among similarly exposed populations. Individuals with PD have distinct gut signatures and microbiome dysbiosis, some of which are caused PD medications [90], which could further influence chemical metabolism. Alterations in gastrointestinal microbiota are likewise driven by environmental PD toxicants, as was recently shown in a rat model of rotenone exposure [91] as well as chlorpyrifos [92], suggesting that environmental influence upon gastrointestinal homeostasis is likely bidirectional. As the aforementioned studies and others have already shown, the gastrointestinal system plays a central role in PD and is a vulnerable target for peripheral exposures that ultimately influence brain pathophysiology.
There are some natural limitations of microbiome research in animals as the gut microbiome differs between species and is heavily influenced by diet [93]. However, these limitations are also opportunities to better translate diet heterogenity into PD risk from the preclinical research stage, as human data already confirms that certain diets may influence PD risk from pesticide exposure, such as paraquat and rotenone [94]. Likewise, specific diets can potentiate neurotoxicity of environmental contaminants, as was observed in offspring from high fat diet-fed female mice combined with TCE exposure [95], which caused epigenetic and redox alterations in the cerebellum. More studies are needed to examine the connection between widespread environmental contaminants and diet in mechansims of parkinsonism that carefully controlled basic or laboratory research studies can provide in support of human population data.
BASIC SCIENCE: CONCLUSIONS AND A PATH FORWARD
The etiology of PD is influenced by environmental exposures [15, 16, 20, 96–98]. Thus, while not exhaustive, incorporating one or several environmentally focused components into basic PD research will ultimately improve our ability to prevent PD. Many of these efforts will require collaborative, cross-disciplinary groups and emerging technology combined with basic toxicological principles such as those that have been applied in other fields (e.g., cancer; [99]), but less frequently for PD.
One tool that has been specifically designed to do just this, is the Human Health Exposure Analysis Resource (HHEAR; https://hhearprogram.org/), a program sponsored by multiple NIH entities (NIEHS, NCI, NHLBI, and ECHO). HHEAR uses an eligibility and feasibility-based application for access to data analysis services that will incorporate exposure data into human health studies. With both laboratory and data analysis support, the incorporation of biomarkers, transcriptomics or metabolomics, and gene-environment interaction for a specific project would be an ideal mechanism for PD studies, both in the pre-clinical and clinical areas. However, one of the strengths of the HHEAR program is also a limitation. The very rigorous analysis approach may reduce the discovery of unknown associations as the bulk of the program targets already known environmental factors.
While a primary goal of an environmental research agenda for PD is to limit the negative impact of exposure on neurological health, there is also evidence for a benefit to therapeutic development. A recent example is the gene-environment interaction between the leuceine-rich repeat kinase 2 (LRRK2) protein and environmental toxicants, such as rotenone [100, 101], TCE [102], paraquat [103, 104], and manganese [105, 106]. Because the pathological elevation in LRRK2 kinase activity induced by environmental toxicants is similar to LRRK2 gain-of-function mutations, pharmaceutical inhibiton of LRRK2 by small molecule kinase inhibitors currently under clinical review [107], could be appropriate for individuals following certain exposures [63].
CLINICAL RESEARCH: EVALUATING THE ORIGINS OF PD
To prevent PD, we also need to study the origins and consequences of the disease beginning with the epidemiology of the disease and ending with whole-body autopsies.
Measure PD incidence and its change globally
Some of the basic facts about PD remain a mystery, beginning with how many individuals have parkinsonisms. For example, U.S. estimates vary by almost 50%. According to the Global Burden of Disease Study, the prevalence of PD in the U.S. in 2016 was 710,000 [5]. By contrast, a recent study estimated that in 2017 1,040,000 Americans were diagnosed with the disease [6]. In Europe, a review of “high-quality” studies found a more than two-fold difference in prevalence estimates [108].
The variability in these estimates highlights the dearth of rigorous studies on the prevalence and incidence of the disease. The most recent door-to-door study conducted in the U.S. occurred in the 1970s [4, 109]. Such studies are critical because the rates of undiagnosed PD can be enormous and result in underestimates of the disease’s true burden. The proportion of individuals with PD who are undiagnosed, and thus missed by claims-based estimates, ranges from 12%in Rotterdam, to 48%in Beijing, to 100%in rural Bolivia [110]. Similarly, temporal studies on the incidence of PD are few [111]. The absence of high-quality, accurate data on the epidemiology of PD limits our ability to understand its root causes. Estimated PD prevalence and percent change in age-stanzardized rates from 1990–2016 are presented in Table 2.
Table 2
Estimated Parkinson’s disease prevalence 1990–2016 with limited data on contaminant emissions
| Country | Prevalence of PD, 2016 | Percent change in age-standardized rates, 1990–2016 | Pesticide use 1990 (in tons) | Pesticide use 2000 (in tons) | Pesticide use 2016 (in tons) | Percent change in pesticide use 1990–2016 | SO2 Emissions 1970 (in thousand tons) | SO2 Emissions 1990 (in thousand tons) | SO2 Emissions 2000 (in thousand tons) | Percent Change in SO2 Emissions, 1970–2000 |
| Global | 6,063,000 | 22% | 2,304,000 | 3,082,000 | 4,161,000 | 81% | 140,480 | 141,980 | 104,560 | –26% |
| China | 1,408,000 | 116% | 775,000 | 1,280,000 | 1,773,000 | 129% | 7,330 | 171,90 | 21,390 | 192% |
| U.S. | 707,000 | 10% | 401,000 | 430,000 | 408,000 | 2% | 29,830 | 20,990 | 14,820 | –50% |
| India | 576,000 | 30% | 75,000 | 45,000 | 59,000 | –21% | 1,110 | 3,300 | 5,360 | 381% |
| Germany | 162,000 | 12% | 31,000 | 35,000 | 47,000 | 52% | 8,120 | 5,350 | 637 | –92% |
| Brazil | 129,000 | 16% | 50,000 | 140,000 | 377,000 | 654% | 777 | 1,640 | 1,730 | 123% |
| France | 120,000 | –2% | 98,000 | 98,000 | 72,000 | –27% | 3,360 | 1,370 | 670 | –80% |
| Canada | 104,000 | 43% | 30,000 | 40,000 | 91,000 | 203% | 5,150 | 3,080 | 2,230 | –57% |
| Argentina | 68,000 | 3% | 26,000 | 84,000 | 200,000 | 669% | 166 | 113 | 137 | –18% |
| Australia | 41,000 | 8% | 18,000 | 33,000 | 63,000 | 250% | 1,450 | 1,590 | 2,370 | 64% |
| Kenya | 7,000 | 22% | 3,000 | 2,000 | 2,000 | –33% | 21 | 42 | 67 | 215% |
| New Zealand | 6,000 | 14% | 3,000 | 4,000 | 5,000 | 67% | 102 | 54 | 64 | –38% |
| Ghana | 4,000 | 14% | 66 | 82 | 9,000 | 13,536% | 17 | 20 | 33 | 94% |
Pesticide use and sulfur dioxide (SO2; a component of air pollution) emission trends reported globally and in select countries. The lack of detailed environmental emission data highlights the need for studies assessing PD prevalence globally with quantifiable exposure assessment [5, 112–114].
To fill in these large knowledge gaps, we need to conduct door-to-door studies of the disease in multiple parts of the world to generate accurate point estimates of the prevalence of the disease. We also will need to address how PD is defined and separated from other movement disorders such as essential tremor or corticobasal degeneration. As a set of heterogenous diseases consisting of a long prodromal and progressive phase [115, 116], motor and non-motor symptoms [117, 118], variable cognitive features [119], and differing ages of onset [120], parkinsomisms are often misdiagnosed and diagnoses differ between nations, ethnic groups, ages, and sexes. Next, we need prospective studies of populations to understand temporal trends in the incidence of the disease and to assess these trends alongside data on known and potential environmental factors linked to the disease. Such data should include assessments of the air individuals breathe (indoor and ambient), the water they drink, and the food they eat. And the records of environmental testing of air, water, soil, and foods should be made freely available to the research community. As demonstrated in Table 2, global data for specific environmental factors correlated to PD prevalence is limited.
Prospective cohorts are also valuable because they allow for the assessment of prodromal features of the disease, such as hyposmia, constipation, and REM sleep behavioral disorder, which can be early harbingers of the disease [121–123]. These studies are especially important in areas of the world where prevalence rates have been historically low, such as sub-Saharan Africa, but where use of pesticides and industrial solvents are increasing and air pollution is rising [5, 124, 125]. Leveraging geospatial data to monitor disease as it relates to environmental health may present an opportunity to track PD at a global population level [126].
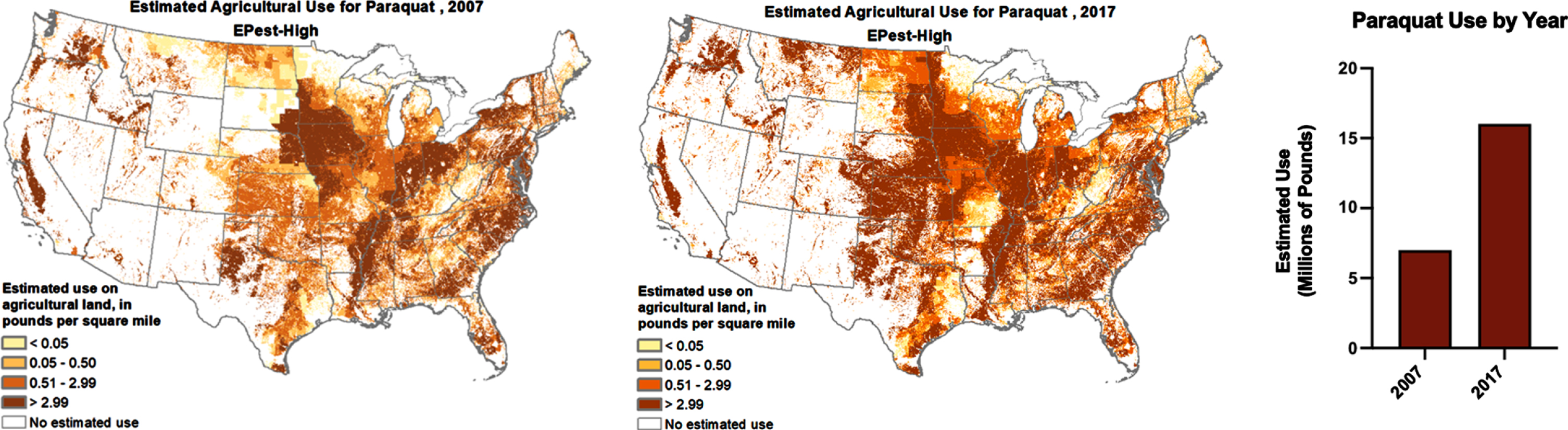
In addition to acquiring population-based data, we need to investigate clusters of PD as they can provide additional clues and evidence of preventable risk factors of the disease. Such clusters have been reported in both the lay and academic press [110, 127–130], though many more have likely gone unrecognized. A number of studies have documented high rates of PD among farmers, rural residents, and drinkers of water from private wells who may have increased exposure to pesticides associated with PD [131–133]. While numerous pesticides are linked to PD risk, the widely used herbicide paraquat dichloride has accumulated the strongest quantitative data for individuals living near its application, as was originally described in conjunction with the fungicide maneb in California’s Central Valley (OR 4.17, 95%CI 1.15–15.16) [134]. Despite this well-established link, paraquat usage has increased in U.S. agricultural areas over the last decade (Fig. 2), potentially exposing many more individuals to this herbicide. U.S. geospatial PD prevalence has been investigated by [4, 135, 136], but a comparison with detailed areas of pesticide application, such as paraquat, has not been conducted on a national scale.
Fig. 2
Paraquat usage in the U.S. has increased over the last decade. Maps and graph generated from the U.S. Geological Survey (USGS) Pesticide National Synthesis Project URL: http://water.usgs.gov/nawqa/pnsp/usage/maps/show_map.php?year=2017&map=PARAQUAT&hilo=L.
PD incidence is not limited to rural or agricultural areas, and in fact, appears to be more prevalent in urban counties [135, 137], as well as areas with high concentration of air pollution [138]. Investigations describing clusters of PD tied to industrial byproducts that might be found in urban areas are available, but far fewer than pesticides. Among the best studied of these is by Gash and colleagues who investigated 30 individuals who worked in a small industrial plant manufacturing small instruments like metal gauges in Berea, Kentucky [139]. The individuals worked with the organic solvent TCE in a degreasing area. The three individuals who worked nearest the large vats of TCE without any protective equipment like gloves, masks, or aprons all developed PD. When surveyed, fourteen others reported at least three parkinsonian signs. The remaining 13 reported no symptoms but when evaluated in clinic, the speed of their hand movements was significantly slower than age-matched controls [139]. Highlighting the value of identifying disease clusters, the Gash report led others to conduct an analytic epidemiologic twin study that confirmed the association with TCE (OR 6.1, 95%CI 1.2–33, p = 0.034) [140]. Given that TCE, which was ubiquitous in the 1970s [141], contaminates thousands of industrial sites in the U.S. and abroad and that up to 30%of U.S. groundwater is contaminated with the chemical [142], additional clusters are likely and worthy of investigation [140]. The challenge is that the time lag between exposure to TCE and related chemicals and development of parkinsonian symptoms can be a decade or longer making evaluation of populations difficult [140].
The TCE clusters are not isolated incidents. In 2004, Kumar and colleagues described three clusters of PD in Canada associated with variable work environments: a television crew, a college, and a garment factory [127]. Like the TCE incident in Kentucky, some may be attributed to direct chemical exposure in an occupational setting (a garment factory, for example, may use solvents widely). However, not all PD clusters are clearly defined by an individual’s occupation; some may be instead related to the environment in which they lived and worked. In fact, one of the largest organic solvent contamination events in U.S. history occurred at the Marine Corps Base Camp Lejeune in North Carolina. Over 30 years, Marines, their families, and civilians on the base were exposed to TCE and related chemicals at levels up to 3000 times above safety thresholds in the water they drank, cooked with, and bathed in. Consequently, up to one million individuals are at increased risk for several chronic diseases, including PD (Hazard Ratio [HR] 3.13, 95%CI 0.76–12.81) [87, 143–145]. Similar exposures that increase PD risk may be prevented in the future if we investigate and learn.
The flip side of investigating clusters of PD is to systematically assess sites with known contamination of or highly exposed populations. Several studies have been done for pesticides (e.g., the Agricultural Health Study; [29, 146]), but again far fewer have been conducted for other environmental comtaminants linked to PD risk, such as organic solvents like TCE and methylene chloride, or air pollution and metals. Unfortunately, such sites are not in short supply. Almost half of the sites on the U.S. National Priorities List (Superfund sites designated for clean up by the federal government) are contaminated with chemicals that are potentially neurotoxic [147]. In addition, there are thousands of these and similar sites across the U.S. and around the world. Evaluating such sites will help determine the neurodegenerative risk associated with such chemicals [140], understand the relationship between likely dose and response, help determine the scope of diseases associated with many of these known carcinogens, and inform our understanding of the extent to which these chemicals contribute to PD.
In conjunction with investigating clusters and contaminated sites, additional studies of occupational exposure can help evaluate potential risk factors for the disease. While some of these studies have been done, the results have been mixed [146, 148, 149]. Studies of individuals who commonly use industrial chemicals, such as mechanics, embalmers, computer chip manufacturers, and dry cleaners, are rare, but insightful. For example, a recent study examining PD risk from occupational exposure to solvents in a Finnish population concluded that PD risk was significantly associated with occupations that involved chlorinated hydrocarbon solvent use (OR 1.63, 95%CI 1.05–2.50 for electronic/telecommunications work; OR 1.40, 95%CI 0.98–1.99 for laboratory assistant; OR 1.23, 95%CI 0.99–1.52 for machine/engine mechanic) [150]. Beyond occupational exposure, where exposures are likely to be high, assessments of low-level, chronic exposure, such as eating pesticide-laden foods, are limited and may be best addressed in prospective studies. This information can inform policies, the public, and help reduce the risk of PD and potentially other chronic diseases.
Develop biological markers of exposure and identify during prodromal phase
Current epidemiological studies that assess environmental and thus preventable causes of PD are limited by the absence of robust biological markers of exposure. Such markers are needed because the time between exposure and diagnosis of the disease can be a decade or longer [140]. A few studies have found remnants of fat-soluble pesticides in the brains of individuals with PD [151–154], including polychlorinated biphenyls (PCBs) [155]. However, in vivo studies have been few [156] even though levels of some pesticides and their metabolites can be found in the body fat of humans [157, 158].
Markers of other industrial byproducts may be harder to find. For example, volatile compounds, such as solvents, are rapidly metabolized [159], and present in collectable samples for only weeks post exposure. Epigenetic modifications by environmental exposure represent a potential marker, as could changes in mitochondrial or other metabolites [160]. However, these studies are in early stages and need to be expanded. Biological markers for exposure to heavy metals and air pollutants are similarly needed.
One of the ongoing challenges in the field is the inherent bias investigators have with regard to which environmental factors to study. In laboratory and epidemiological studies, choices on what chemicals or exposures to measure must be made. Recently, the use of untargeted high-resolution mass spectrometry (HRMS) has been suggested to provide a more comprehensive and unbiased examination of environmental contributors to disease. The exposome represents the non-genetic drivers of disease [161] and could provide an important addition to PD research. By examining all of the detected peaks by HRMS, the effort becomes more analogous to GWAS studies [162]. HRMS-based exposomic studies build off of the field of metabolomics, but focus on the exogenous features in human samples. By combining those with the endogenous small molecules, which is the focus of metabolomics, it becomes possible to compare thousands of environmental features with thousands of endogenous molecules and hundreds of biological pathways. This sort of unbiased and systematic approach could provide the PD field important insight into the wide range of environmental factors that may play a role in disease etiology, progression, and response to therapy.
Idiopathic PD develops over decades and environmental factors may influence multiple stages of the disease such as initiation, symptom phenotype, and progression. Detection of exposure biomarkers before PD motor or cognitive symptoms develop is key, as many of the prodromal symptoms of PD (e.g., anosmia, gut motility changes) align with points of entry for environmental toxicants (inhalation, ingestion). There is very minimal research on prodromal symptoms of PD correlated with specific exposures; however, technology to measure exposure at the time it occurs, such as metabolomics, may improve this connection. In one example of this approach, the presenece of phthalate metabolites in cerebrospinal fluid were discovered to be higher in individuals with dementia with Lewy bodies (DLB) than those diagnosed with Alzheimer’s disease [163]. This study included a small amount of individuals at the prodromal DLB stage (16), which could indicate phthalate exposure is involved in the initiation of DLB. As others have suggested [164], the prodromal phase represents a critical disease stage where intervention could occur, whether through environmental factor modification or early therapeutic intervention, and therefore warrants increased attention.
Evaluate relationship between environmental and genetic risk factors
“Gene-environment interaction” is often used to describe the majority of idiopathic PD etiology [18, 19, 165], but most human population studies assess genetic risk for PD without any context of exposure. A few rare highly penetrant genetic mutations such as in the α-synuclein (SNCA) gene clearly cause the PD phenotype [10, 166]. In contrast, mutations in the LRRK2 gene, responsible for perhaps 1–2%of PD, are only 30%penetrant [167]. The much more common genetic variants (single nucleotide polymorphisms, SNPs) identified in large GWASs are associated with only minimally increased risk and are not even thought of in terms of their penetrance. These observations clearly suggest and are supported by twin studies [168, 169] that except in very rare circumstances, interactions with the environment (both intrinsic and extrinsic) are necessary to produce the disease. PD is not unique in this respect. Highly penetrant mutations cause only a small proportion of virtually all late life disorders. The converse of course is also true—highly “penetrant” environmental exposures are rare causes of late life disorders, perhaps with the exclusion of smoking related disease.
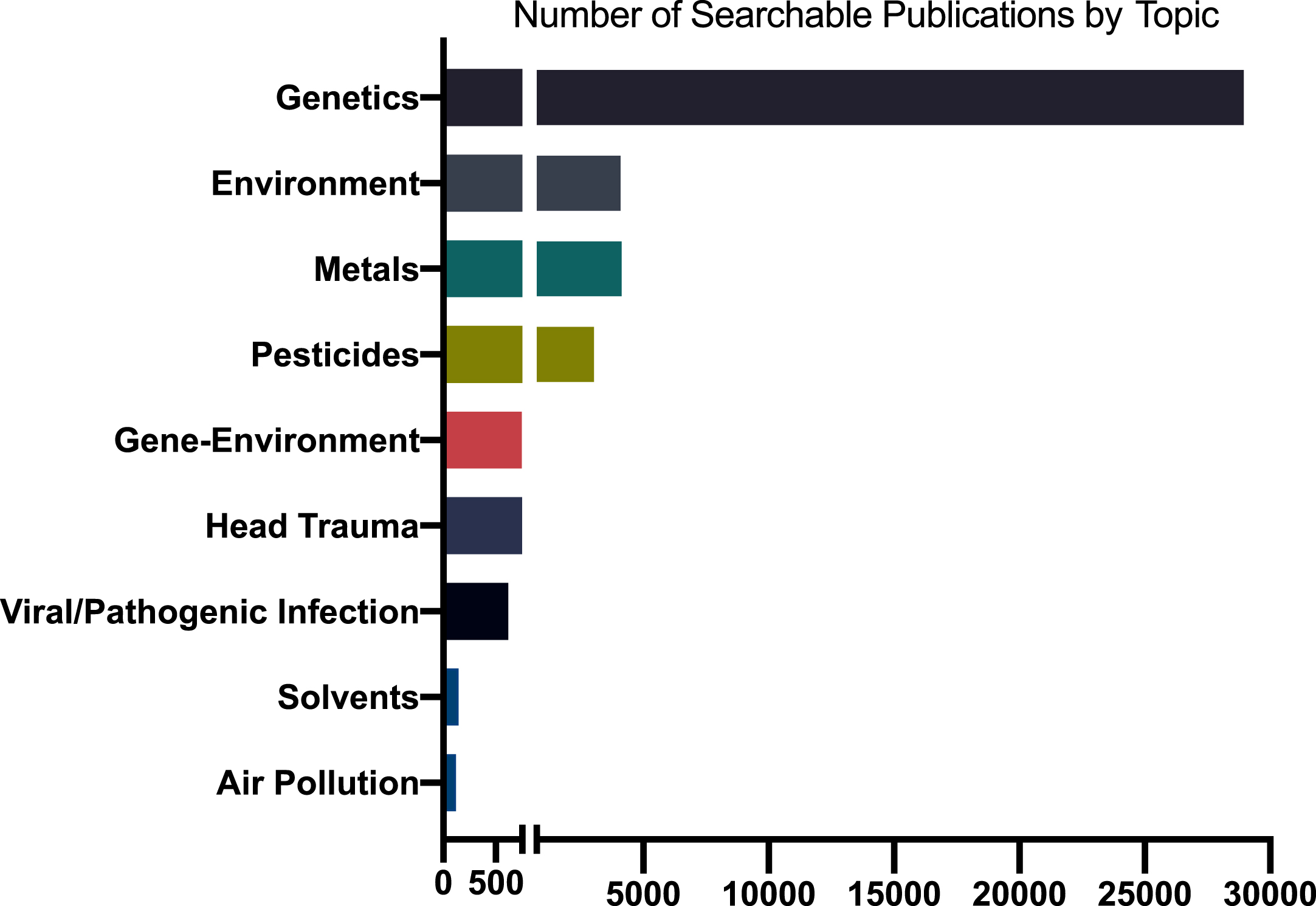
Notwithstanding the long-running arguments about their relative importance, there is substantial agreement in the scientific community that both genetic and environmental factors are important contributors to PD etiology. Despite this, shockingly little has been published on gene-environment interactions in human PD epidemiology or in animal or in vitro models (Fig. 3). Studying gene-environment interaction in human epidemiology poses major hurdles that can only be addressed through well-coordinated scientific policies. Except in very specialized populations, such as some occupational settings, determination of exposures is imprecise at best. A few prospective cohorts exist, but most of these were assembled to study other, more common diseases, have not collected much PD-relevant exposure data, and are generally underpowered for relatively rarer outcomes such as PD. Finally, genome-wide gene-environment interaction studies (GEWIS) analogous to GWASs require enormous sample sizes to identify statistically significant interactions. Thus, virtually all studies of gene-environment interaction have been “candidate-based,” with both gene and exposure pre-selected based on what we know, or think we know about their biological interactions.
Fig. 3
Number of publications on Parkinson’s disease and select topics, 1960-2021. Based on Medline search of Medical Subject Headings (MeSH) and PubMed for keywords or phrases in publications for PD-related topics. Estimates vary depending on search terms and database coverage.
Three general classes of genes have been most explored using this candidate-based approach: 1) those involved in xenobiotic metabolism and transport, and 2) in the protection from or 3) response to injury. Pesticides are the most consistently associated environmental risk factors for PD and studies of their interaction with these genes provide the most compelling examples to date. Several studies found that the risk associated with regular use of pesticides was modified by polymorphisms in CYP2D6 that reduce the activity of the Phase 1 xenobiotic metabolic enzyme cytochrome P450 2D6 (OR 8.41, 95%CI 1.01–69.76) [170, 171]. Notably, these same polymorphisms imparted no increased risk in the absence of pesticide use.
Other examples include interactions between organophosphorous insecticide exposure and the metabolic enzymes paraoxonase 1 (PON1) [19, 172, 173] and aldehyde dehydrogenase (ALDH2) [174]. One of the strongest reported interactions is the modification of risk associated with the common herbicide paraquat and the null variant of the Phase 2 conjugating enzyme glutathione-s-transferase-T1 (GSTT1) [175]. Although homozygous GSTT1 deletions (present in 20%of the population) imparted no increased risk in the absence of paraquat exposure, risk was increased 11-fold in those who were both GSTT1 null and exposed to paraquat (OR 11.1, 95%CI 3.0–44.6, p = 0.027).
Membrane transporters that extrude xenobiotics from cells comprise another class of genes that have consistently been found to modify risk associated with pesticide exposures. One of the best examples is ABCC1 (aka multidrug resistance protein 1, p-glycoprotein), which has been reported to interact with exposure to multiple pesticides including organochlorines such as dieldrin and DDT [175, 176], and the insecticide rotenone, a known substrate of ABCC1 [177, 178]. Lastly, numerous interactions with pesticides have been reported for genes that protect against oxidative stress. Among others, these include nitric oxide synthase (NOS1) [179, 180], manganese superoxide dismutase (SOD2) [181], and NAD(P)H quinone dehydrogenase (NQ01) [182].
Genetic interactions with other environmental risk factors have been less studied. One of particular note is the increased risk associated with exposure to solvents in persons lacking active glutathione transferase- M1 (GSTM1; OR 2.34, 95%CI 1.08–4.62, p = 0.03) [19], a phenotype that occurs in 40%of the population. A markedly increased risk of PD was also observed in solvent-exposed persons with the CYP2D6 slow metabolizer phenotype (OR 14.67, 95%CI 1.16–185.23) [183].
In addition, PD-related mutations and polymorphisms also play a role in gene-environment interaction, such as the SNCA REP1 263 genotype and paraquat [184] or traumatic brain injury (TBI) [185], and LRRK2 and NSAIDS [186]. Animal and in vitro studies support these observations, including interactions of LRRK2 with rotenone, paraquat, TCE, and manganese [100, 102, 104, 105]. Intersecting inflammatory pathways have been proposed as underlying many of these interactions [187]. As discussed in the basic research agenda, multiple exposures and environment-environment interactions are also important from a clinical perspective. For example, while both traumatic brain injury and paraquat increase the risk of PD independently, exposure to both factors almost triples the risk of the disease [53, 185]. Of note, each of these gene-environment interactions were studied in isolation, and it will be important to develop comprehensive and systematic approaches to these complex relationships.
Perform whole-body autopsies to assess PD as a systemic disease
PD is a multifactorial disease influenced by systemic factors as well as multiple types of daily exposures that gain entry through different portals (e.g., nose, gut, skin). As such, it should be studied holistically. And while numerous studies have evaluated the brains of individuals with PD, fewer have examined entire bodies. Those that have, such as the Arizona Study of Aging and Neurodegenerative Disorders, have found Lewy pathology in both those with and without PD [188]. Interestingly, this study did not find evidence of Lewy pathology in peripheral organs without it also being present in the brain, a contradictory finding to the ‘gut-first’ hypothesis. However, when present, Lewy pathology was universally found in the olfactory bulb [188]. As inhalation represents the greatest route of exposure for many of the environmental contaminants linked to PD (e.g., pesticides, air pollution, volatile organic compounds), Lewy pathology in the olfactory bulb could be directly linked to this exposure route. Moreover, smoking, which decreases the risks of PD, [48] alters the nasal epithelium [189, 190], which could influence the risk associated with inhaled exposures or pathogens.
Recent studies have also suggested that α-synuclein oligomers can be found throughout the body from the retina [191] to salivary glands [192] to the skin [193, 194]. Truncated forms of α-synuclein have also been found in the appendix, and appendectomy was associated with lower PD risk (OR 0.831, 95%CI, 0.756 to 0.907, p < 8.1×10–5), which was more profound for individuals living in rural areas (OR 0.769, 95%CI, 0.681 to 0.867, p < 1.5×10–5), suggesting that systemic exposures sustained in these environments influences Lewy pathology [195].
These efforts will require more whole body autopsies ideally from large population-based cohorts from around the world. By expanding our view beyond the brain for this systemic disease, we might better understand where and how the various parkinsonisms [196] begin and how they unfold [197]. In line with this, the distinction of parkinsonisms from other movement disorders (e.g., essential tremor) and the environmental influence on disease phenotype necessitates careful study design.
Include populations with high exposure burden
The current and projected global growth of PD in populous nations cannot alone be attributed to increased lifespan. Areas of the world that have seen the greatest growth of modern industrialization, such as China and India, have had the highest increase in age-adjusted prevalence estimates for PD [198]. Some of the increased prevalence in PD among these nations may be a result of better characterization and diagnosis of PD by neurologists or reductions in smoking, which is widely accepted to be protective against PD risk [199]. However, other critical factors are the products and by-products (e.g., pesticides, solvents, metals, air pollution) of industrialization. In the case of pesticides, China greatly outpaces the rest of the world in annual usage, estimated at over 1,800,000 tons, followed by the U.S. with approximately 55,000 tons [200]. In the last few decades, South American pesticide usage has risen sharply (30%sales growth between 2003 and 2004 alone; [200], including the increased usage of chemicals such as 2,4-D, paraquat, and chlorpyrifos, which are implicated in PD [201–203]). As regulations for chemical usage vary widely between nations, the risks for their harms may be especially high in certain parts of the world.
In relation, environmental exposures in any nation are not equally distributed. Almost all populations face challenges of environmental justice, where the most underprivileged areas also face the highest amount of environmental exposure risk [204]. For example, elevated blood-lead levels in children, which affects brain and cognitive development, can be predicted by socioeconomic factors in the United States, such as older housing and poverty [205]. In addition to socioeconomic status, race must also be better represented in the context of PD risk from exposure, as racial differences occur in PD prevalence, incidence, and medical treatment [206–209]. While some racial differences in PD may arise from biology [207], others may be based on exposure burden and gene-environment interaction. Therefore, while most of the PD field rightly urges a focus on inclusion of ethnically diverse populations, we cannot examine the full context of race without including the environment. Exposure to compounds that produce neurotoxicity are typically highest in industrialized (urban) or agricultural (rural) areas and associated with lower socioeconomic status, less accessability to green space (urban), and increased likihood to consume well water (rural; [210]). Combined with reduced access to healthcare, PD diagnoses related to sustained exposure may be underreported in these areas. As previously mentioned, using geospatial technology to monitor environmental health and its association with disease may provide an opportunity to better track areas with limited access [124].
Especially vulnerable populations such as children must also be considered in this context. Exposure to environmental toxicants during neurodevelopment, whether in utero, perinatal, or during childhood, may have a significant impact on future PD risk. As Goldman and colleagues observed, PD concordance in dizygotic twins is much higher than in typical siblings [168], which indicates the perinatal exposure period is a critical window for future disease development. There are also mechanistic data from experimental models to support “silent neurotoxicity,” such as the priming of microglia after a single dose of paraquat in mice, which predisposed animals to more severe dopaminergic neurodegeneration upon secondary insult [211]. Similarily, developmental exposure to organophosphate pesticides, such as diazinon and chlorpyrifos, casue gene transcriptional changes associated with PD [212]. As fat-soluable compounds implicated in neurodevelopmental toxicity, such as organochlorine pesticides and polybrominated diphenyl ethers (PBDEs), are often found in breast milk [213, 214], identifying these vulnerable populations may provide an avenue to prevent exposure at the earliest stages of life. Likewise, tools to accurately measure multifactorial exposures, such as the exposome approach, will be essential in characterizing populations of concern [162].
POLICY AGENDA
To prevent PD, research needs to be translated into action. Knowing or having evidence that smoking causes lung cancer meant little until policy (e.g., safety warnings, banning from restaurants, taxes) and other actions (e.g., media depictions) followed. The result has been plunging rates of lung cancer [215]. A similar result was achieved with the removal of lead from gasoline, decades after it was linked to neurotoxicity in adults and impaired cognitive development in children [216].
We have more than sufficient evidence to take some policy actions to prevent PD now. For certain pesticides and industrial chemicals, we have years of evidence from different investigators from around the world linking them to PD and other health concerns. Within the United States, we need to ban or further regulate substances based on their neurotoxicity, such as paraquat, chlorpyrifos, and TCE. Other countries around the world have already taken such action [110], as have some states within the U.S. Wisconsin banned TCE in 2020, followed by New York, which banned the “most harmful uses” of TCE later that year. In 2019, the bill H.R. 3817 –Protect Against Paraquat Act, which would ban the usage of paraquat in the U.S., was introduced by Rep. Nydia Velazquez to Congress, but as of this publication date has not been passed by the House of Representatives.
As we wait for legislation, paraquat usage in the U.S. has tripled in the last 25 years, doubled in the last decade, and increased 20%in the most recent year for which data are available [217]. Over 30 countries, including China, have already banned the herbicide. A unified voice from the individuals with PD, the research community, and their local and federal leaders can help drive policy changes to limit further exposure. Other simple steps that may have an immediate impact are policies that continue to educate and promote safe handling practices for toxicants, as studies confirm the use of protective gloves alone can reduce PD risk from pesticide exposure [218, 219], and could be reduced further with additional PPE.
FUNDING
The NIH, by far the world’s largest funder of research, spent $224 million on PD research in 2019 [220]. However, the economic burden of PD is $50 billion per year in the U.S. alone [6]. Spending less than 1%of that in research aimed at understanding, preventing, measuring, and treating the disease is not enough. Despite an overall growth in funding, when adjusted for inflation, NIH funding for PD research has actually decreased in the past decade at the same time the number of Americans with the disease has increased [4, 110, 220]. In relation, published research studies related to prevention as well as specific exposure risks lag behind other topics (Fig. 3). For example, the number of studies published on genetic risk for PD is approximately seven times higher than any extrinsic risk factors or gene-environment interaction.
Ultimately, if we want to change the course of PD, as we have for other diseases, we will have to increase our investment substantially. A much larger global investment in research and resources, including partnerships between private and public funding entities and data sharing initiatives that include an environmental focus, will pave the way for preventing PD. Recognizing the central role of exposure in disease—particularly for, but not limited to PD—may further promote cross disciplinary or cross-institution research funding that has typically fallen on one sector. However, a promising development in this area is the recent creation of the Office for Neural Exposome and Toxicology Research by the NINDS, which will collaborate with NIEHS to better understand the impact of environmental factors and neurological health [221]. In addition, an increase in research funding aimed at preventing PD has a synergistic benefit. Numerous chronic diseases, such as heart disease, cancer, Alzheimer’s disease, and amyotrophic lateral sclerosis, share one or more environmental risk factors with PD, potentially providing far-reaching impact on numerous chronic diseases.
CONCLUSIONS
The first response to any crisis whether from a fire or a virus is to contain it to prevent its spread. To date, we have failed to contain PD. Instead we are only fueling it. Prevention is the most important step to addressing the wave of PD.
We have prevented diseases in the past, and we can prevent them now. In the 1930s, a March of Dimes raised money for research that led to the development of vaccines that prevent polio. Prevention has been so effective for polio that the March of Dimes Foundation has had to change its mission. In the 1980s, a group of brave activists changed the course of HIV and in the process, our sexual practices [222]. The result is that likely millions, including many of us, have never been infected with the virus. At the same time, mothers whose children had been killed by drunk drivers said that 25,000 deaths a year in alcohol-related crashes was enough. Drunk driving is now no longer socially acceptable, and over 300,000 lives have been saved because of their efforts [223].
The prevention agenda we have laid out here to investigate and address the environmental causes of PD is an imperfect and incomplete beginning. But the time has come to increase substantially our investment in slowing and eventually reversing the rise of PD. Our health and those of future generations depend on it.
ACKNOWLEDGMENTS
The authors would like to thank Maryam Zafar for her assistance in preparing this manuscript.
Research reported in this publication was supported in part by the National Institute of Neurological Disorders and Stroke under Award Number P50NS108676 (ERD) and the National Institute of Environmental Health Sciences under Award Numbers R00ES029986 (BRD) and R01ES023839 (GWM). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
CONFLICT OF INTEREST
Dr. Dorsey has received honoraria for speaking at American Academy of Neurology, American Neurological Association, Excellus BlueCross BlueShield, International Parkinson’s and Movement Disorders Society, National Multiple Sclerosis Society, Northwestern University, Physicians Education Resource, LLC, Stanford University, Texas Neurological Society, and Weill Cornell; received compensation for consulting services from Abbott, Abbvie, Acadia, Acorda, Alzheimer’s Drug Discovery Foundation, Ascension Health Alliance, Bial-Biotech Investments, Inc., Biogen, BluePrint Orphan, California Pacific Medical Center, Caraway Therapeutics, Clintrex, Curasen Therapeutics, DeciBio, Denali Therapeutics, Eli Lilly, Grand Rounds, Huntington Study Group, medical-legal services, Mediflix, Medopad, Medrhythms, Michael J. Fox Foundation, MJH Holding LLC, NACCME, Neurocrine, NeuroDerm, Olson Research Group, Origent Data Sciences, Otsuka, Pear Therapeutic, Praxis, Prilenia, Roche, Sanofi, Seminal Healthcare, Spark, Springer Healthcare, Sunovion Pharma, Sutter Bay Hospitals, Theravance, University of California Irvine, and WebMD; research support from Abbvie, Acadia Pharmaceuticals, Biogen, Biosensics, Burroughs Wellcome Fund, CuraSen, Greater Rochester Health Foundation, Huntington Study Group, Michael J. Fox Foundation, National Institutes of Health, Patient-Centered Outcomes Research Institute, Pfizer, PhotoPharmics, Safra Foundation, and Wave Life Sciences; editorial services for Karger Publications; and ownership interests with Grand Rounds (second opinion service). Dr. Miller receives royalties for his books The Exposome: A Primer and The Exposome: A New Paradigm for the Environment and Health. He also receives compensation as Editor of the journal Exposome, published by Oxford University Press. Dr. De Miranda has consulted for the Hartley Law Group. Drs Greenamyre and Goldman have no conflicts of interest to report.
REFERENCES
[1] | King S ((2004) ) Pink Ribbons Inc: Breast cancer activism and the politics of philanthropy. Int J Qual Stud Educ 17: , 473–492. |
[2] | Ji P , Gong Y , Jin M-L , Hu X , Di G-H , Shao Z-M ((2020) ) The burden and trends of breast cancer from 1990 to 2017 at the global, regional, and national levels: Results from the Global Burden of Disease Study 2017. Front Oncol 10: , 650. |
[3] | Ray F (2020) MJFF Recognized for Advocacy Role in Parkinson’s Research, Public Health Policy. Parkinson’s News Today. https://parkinsonsnewstoday.com/2020/12/11/mjff-recognized-advocacy-award-role-in-parkinsons-research-public-health-policy/ |
[4] | Marras C , Beck JC , Bower JH , Roberts E , Ritz B , Ross GW , Abbott RD , Savica R , Van Den Eeden SK , Willis AW , Tanner C ((2018) ) Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis 4: , 21. |
[5] | Dorsey ER , Elbaz A , Nichols E , Abd-Allah F , Abdelalim A , Adsuar JC , Ansha MG , Brayne C , Choi J-YJ , Collado-Mateo D , Dahodwala N , Do HP , Edessa D , Endres M , Fereshtehnejad S-M , Foreman KJ , Gankpe FG , Gupta R , Hankey GJ , Hay SI , Hegazy MI , Hibstu DT , Kasaeian A , Khader Y , Khalil I , Khang Y-H , Kim YJ , Kokubo Y , Logroscino G , Massano J , Mohamed Ibrahim N , Mohammed MA , Mohammadi A , Moradi-Lakeh M , Naghavi M , Nguyen BT , Nirayo YL , Ogbo FA , Owolabi MO , Pereira DM , Postma MJ , Qorbani M , Rahman MA , Roba KT , Safari H , Safiri S , Satpathy M , Sawhney M , Shafieesabet A , Shiferaw MS , Smith M , Szoeke CEI , Tabarés-Seisdedos R , Truong NT , Ukwaja KN , Venketasubramanian N , Villafaina S , Weldegwergs KG , Westerman R , Wijeratne T , Winkler AS , Xuan BT , Yonemoto N , Feigin VL , Vos T , Murray CJL ((2018) ) Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 17: , 939–953. |
[6] | Yang W , Hamilton JL , Kopil C , Beck JC , Tanner CM , Albin RL , Ray Dorsey E , Dahodwala N , Cintina I , Hogan P , Thompson T ((2020) ) Current and projected future economic burden of Parkinson’s disease in the U.S. NPJ Parkinsons Dis 6: , 15. |
[7] | Dorsey ER , Sherer T , Okun MS , Bloem BR ((2018) ) The emerging evidence of the Parkinson pandemic. J Parkinsons Dis 8: , S3–S8. |
[8] | Dorsey ER , Bloem BR ((2018) ) The Parkinson pandemic—a call to action. JAMA Neurology 75: , 9. |
[9] | Dickson DW ((2012) ) Parkinson’s disease and parkinsonism: Neuropathology. Cold Spring Harb Perspect Med 2: , a009258–a009258. |
[10] | Simon DK , Tanner CM , Brundin P ((2020) ) Parkinson disease epidemiology, pathology, genetics, and pathophysiology. Clin Geriatr Med 36: , 1–12. |
[11] | Maiti P , Manna J , Dunbar GL ((2017) ) Current understanding of the molecular mechanisms in Parkinson’s disease: Targets for potential treatments. Transl Neurodegener 6: , 28. |
[12] | Rui Q , Ni H , Li D , Gao R , Chen G ((2018) ) The role of LRRK2 in neurodegeneration of Parkinson disease. Curr Neuropharmacol 16: , 1348–1357. |
[13] | Lwin A , Orvisky E , Goker-Alpan O , LaMarca ME , Sidransky E ((2004) ) Glucocerebrosidase mutations in subjects with parkinsonism. Mol Genet Metab 81: , 70–73. |
[14] | Billingsley KJ , Bandres-Ciga S , Saez-Atienzar S , Singleton AB ((2018) ) Genetic risk factors in Parkinson’s disease. Cell Tissue Res 373: , 9–20. |
[15] | Narayan S , Liew Z , Bronstein JM , Ritz B ((2017) ) Occupational pesticide use and Parkinson’s disease in the Parkinson Environment Gene (PEG) study. Environ Int 107: , 266–273. |
[16] | Caudle WM , Guillot TS , Lazo CR , Miller GW ((2012) ) Industrial toxicants and Parkinson’s disease. Neurotoxicology 33: , 178–188. |
[17] | Aschner M , Erikson KM , Herrero Hernandez E , Tjalkens R ((2009) ) Manganese and its role in Parkinson’s disease: From transport to neuropathology. Neuromolecular Med 11: , 252–266. |
[18] | Cannon JR , Greenamyre JT ((2013) ) Gene-environment interactions in Parkinson’s disease: Specific evidence in humans and mammalian models. Neurobiol Dis 57: , 38–46. |
[19] | Dick FD , De Palma G , Ahmadi A , Osborne A , Scott NW , Prescott GJ , Bennett J , Semple S , Dick S , Mozzoni P , Haites N , Wettinger SB , Mutti A , Otelea M , Seaton A , Soderkvist P , Felice A , Geoparkinson Study Group ((2007) ) Gene-environment interactions in parkinsonism and Parkinson’s disease: The Geoparkinson study. Occup Environ Med 64: , 673–680. |
[20] | Tanner CM , Goldman SM , Ross GW , Grate SJ ((2014) ) The disease intersection of susceptibility and exposure: Chemical exposures and neurodegenerative disease risk. Alzheimers Dement 10: , S213–225. |
[21] | Langston JW , Langston EB , Irwin I ((1984) ) MPTP-induced parkinsonism in human and non-human primates–clinical and experimental aspects. Acta Neurol Scand Suppl 100: , 49–54. |
[22] | Langston JW ((2017) ) The MPTP story. J Parkinsons Dis 7: , S11–S19. |
[23] | Markey SP , Johannessen JN , Chiueh CC , Burns RS , Herkenham MA ((1984) ) Intraneuronal generation of a pyridinium metabolite may cause drug-induced parkinsonism. Nature 311: , 464–467. |
[24] | Cannon JR , Tapias V , Na HM , Honick AS , Drolet RE , Greenamyre JT ((2009) ) A highly reproducible rotenone model of Parkinson’s disease. Neurobiol Dis 34: , 279–290. |
[25] | Drolet RE , Cannon JR , Montero L , Greenamyre JT ((2009) ) Chronic rotenone exposure reproduces Parkinson’s disease gastrointestinal neuropathology. Neurobiol Dis 36: , 96–102. |
[26] | Brooks AI , Chadwick CA , Gelbard HA , Cory-Slechta DA , Federoff HJ ((1999) ) Paraquat elicited neurobehavioral syndrome caused by dopaminergic neuron loss. Brain Res 823: , 1–10. |
[27] | Smeyne RJ , Breckenridge CB , Beck M , Jiao Y , Butt MT , Wolf JC , Zadory D , Minnema DJ , Sturgess NC , Travis KZ , Cook AR , Smith LL , Botham PA ((2016) ) Assessment of the effects of MPTP and paraquat on dopaminergic neurons and microglia in the substantia nigra pars compacta of C57BL/6 mice. PLoS One 11: , e0164094. |
[28] | Sun J , Tian T , Wang Y , Yan W , Zhang B , Wang K , Yang H , Huang M ((2021) ) Paraquat-activated BV-2 microglia induces neuroinflammatory responses in the neuron model through NF-kappaB signaling pathway. Toxicol In Vitro 72: , 105076. |
[29] | Tanner CM , Kamel F , Ross GW , Hoppin JA , Goldman SM , Korell M , Marras C , Bhudhikanok GS , Kasten M , Chade AR , Comyns K , Richards MB , Meng C , Priestley B , Fernandez HH , Cambi F , Umbach DM , Blair A , Sandler DP , Langston JW ((2011) ) Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect 119: , 866–872. |
[30] | Gatto NM , Cockburn M , Bronstein J , Manthripragada AD , Ritz B ((2009) ) Well-water consumption and Parkinson’s disease in rural California. Environ Health Perspect 117: , 1912–1918. |
[31] | Apetz N , Paralikar K , Neumaier B , Drzezga A , Wiedermann D , Iyer R , Munns G , Scott E , Timmermann L , Endepols H (2021) Towards chronic deep brain stimulation in freely moving hemiparkinsonian rats: Applicability and functionality of a fully implantable stimulation system. J Neural Eng, doi: 10.1088/1741-2552/abe806. |
[32] | Glajch KE , Fleming SM , Surmeier DJ , Osten P ((2012) ) Sensorimotor assessment of the unilateral 6-hydroxydopamine mouse model of Parkinson’s disease. Behav Brain Res 230: , 309–316. |
[33] | Woodlee MT , Kane JR , Chang J , Cormack LK , Schallert T ((2008) ) Enhanced function in the good forelimb of hemi-parkinson rats: Compensatory adaptation for contralateral postural instability? Exp Neurol 211: , 511–517. |
[34] | Zigmond MJ , Hastings TG , Perez RG ((2002) ) Increased dopamine turnover after partial loss of dopaminergic neurons: Compensation or toxicity? Parkinsonism Relat Disord 8: , 389–393. |
[35] | Alvarez-Fischer D , Noelker C , Grunewald A , Vulinovic F , Guerreiro S , Fuchs J , Lu L , Lombes A , Hirsch EC , Oertel WH , Michel PP , Hartmann A ((2013) ) Probenecid potentiates MPTP/MPP+toxicity by interference with cellular energy metabolism. J Neurochem 127: , 782–792. |
[36] | Meredith GE , Totterdell S , Potashkin JA , Surmeier DJ ((2008) ) Modeling PD pathogenesis in mice: Advantages of a chronic MPTP protocol. Parkinsonism Relat Disord 14 Suppl 2: , S112–115. |
[37] | Singh A , Papa SM ((2020) ) Striatal oscillations in parkinsonian non-human primates. Neuroscience 449: , 116–122. |
[38] | Campbell BA , Cho H , Faulhammer RM , Hogue O , Tsai JP , Hussain MS , Machado AG , Baker KB (2021) Stability and effect of parkinsonian state on deep brain stimulation cortical evoked potentials. Neuromodulation, doi: 10.1111/ner.13508. |
[39] | Sherer TB , Richardson JR , Testa CM , Seo BB , Panov AV , Yagi T , Matsuno-Yagi A , Miller GW , Greenamyre JT ((2007) ) Mechanism oftoxicity of pesticides acting at complex I: Relevance toenvironmental etiologies of Parkinson’s disease. J Neurochem 100: , 1469–1479. |
[40] | Richardson JR , Quan Y , Sherer TB , Greenamyre JT , Miller GW ((2005) ) Paraquat neurotoxicity is distinct from that of MPTP and rotenone. Toxicol Sci 88: , 193–201. |
[41] | Martinez TN , Greenamyre JT ((2012) ) Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid Redox Signal 16: , 920–934. |
[42] | Chia SJ , Tan EK , Chao YX ((2020) ) Historical perspective: Models of Parkinson’s disease. Int J Mol Sci 21: , 2464. |
[43] | US Food and Drug Administration. (2005) Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials fot Therapeutics in Adult Healthy Volunteers. https://www.fda.gov/media/72309/download. Posted July 2005. Accessed August 12, 2021. |
[44] | Nair AB , Jacob S ((2016) ) A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 7: , 27–31. |
[45] | Moser VC , Chanda SM , Mortensen SR , Padilla S ((1998) ) Age- and gender-related differences in sensitivity to chlorpyrifos in the rat reflect developmental profiles of esterase activities. Toxicol Sci 46: , 211–222. |
[46] | Wang S , Lai X , Deng Y , Song Y ((2020) ) Correlation between mouse age and human age in anti-tumor research: Significance and method establishment. Life Sci 242: , 117242. |
[47] | Phillips JE ((2017) ) Inhaled efficacious dose translation from rodent to human: A retrospective analysis of clinical standards for respiratory diseases. Pharmacol Ther 178: , 141–147. |
[48] | Hernan MA , Takkouche B , Caamano-Isorna F , Gestal-Otero JJ ((2002) ) A meta-analysis of coffee drinking, cigarette smoking, and the risk of Parkinson’s disease. Ann Neurol 52: , 276–284. |
[49] | Mentis AA , Dardiotis E , Efthymiou V , Chrousos GP ((2021) ) Non-genetic risk and protective factors and biomarkers for neurological disorders: A meta-umbrella systematic review of umbrella reviews. BMC Med 19: , 6. |
[50] | Cheng G , Kong RH , Zhang LM , Zhang JN ((2012) ) Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. Br J Pharmacol 167: , 699–719. |
[51] | Hiebert JB , Shen Q , Thimmesch AR , Pierce JD ((2015) ) Traumatic brain injury and mitochondrial dysfunction. Am J Med Sci 350: , 132–138. |
[52] | Hubbard WB , Joseph B , Spry M , Vekaria HJ , Saatman KE , Sullivan PG ((2019) ) Acute mitochondrial impairment underlies prolonged cellular dysfunction after repeated mild traumatic brain injuries. J Neurotrauma 36: , 1252–1263. |
[53] | Lee P-C , Bordelon Y , Bronstein J , Ritz B ((2012) ) Traumatic brain injury, paraquat exposure, and their relationship to Parkinson disease. Neurology 79: , 2061–2066. |
[54] | Sauerbeck A , Hunter R , Bing G , Sullivan PG ((2012) ) Traumatic brain injury and trichloroethylene exposure interact and produce functional, histological, and mitochondrial deficits. Exp Neurol 234: , 85–94. |
[55] | Environmental Protection Agency, Research Triangle Park, NC. Office of Air Quality Planning and Standards (2006) Economic Impact Analysis of the Proposed Halogenated Solvent Cleaners Residual Risk Standard. |
[56] | Snell FI , Halter MJ ((2010) ) A signature wound of war: Mild traumatic brain injury. J Psychosoc Nurs Ment Health Serv 48: , 22–28. |
[57] | Walker DI , Uppal K , Zhang L , Vermeulen R , Smith M , Hu W , Purdue MP , Tang X , Reiss B , Kim S , Li L , Huang H , Pennell KD , Jones DP , Rothman N , Lan Q ((2016) ) High-resolution metabolomics of occupational exposure to trichloroethylene. Int J Epidemiol 45: , 1517–1527. |
[58] | Ando M , Ueda K , Okamoto Y , Kojima N ((2011) ) Combined effects ofmanganese, iron, copper, and dopamine on oxidative DNA damage. Health Sci 57: , 204–209. |
[59] | Murray KE , Delic V , Ratliff WA , Beck KD , Citron BA ((2021) ) Acute gene expression changes in the mouse hippocampus following a combined Gulf War toxicant exposure.,. Life Sci 284: , 119845. |
[60] | Barnhill LM , Khuansuwan S , Juarez D , Murata H , Araujo JA , Bronstein JM ((2020) ) Diesel exhaust extract exposure induces neuronal toxicity by disrupting autophagy. Toxicol Sci 176: , 193–202. |
[61] | Branco RC , Ellsworth W , Niedzwiecki MM , Butkovich LM , Walker DI , Huddleston DE , Jones DP , Miller GW (2018) Levodopa and dopamine dynamics in Parkinson’s disease metabolomics. bioRxiv, 306266. |
[62] | Shavali S , Sens DA ((2008) ) Synergistic neurotoxic effects of arsenic and dopamine in human dopaminergic neuroblastoma SH-SY5Y cells. Toxicol Sci 102: , 254–261. |
[63] | Pristner M , Warth B ((2020) ) Drug-exposome interactions: The next frontier in precision medicine. Trends Pharmacol Sci 41: , 994–1005. |
[64] | Picillo M , Nicoletti A , Fetoni V , Garavaglia B , Barone P , Pellecchia MT ((2017) ) The relevance of gender in Parkinson’s disease: A review. J Neurol 264: , 1583–1607. |
[65] | Kimura H , Kurimura M , Wada M , Kawanami T , Kurita K , Suzuki Y , Katagiri T , Daimon M , Kayama T , Kato T ((2002) ) Female preponderance of Parkinson’s disease in Japan. Neuroepidemiology 21: , 292–296. |
[66] | Satake A (2020) Number ofWomen Farmers in Japan Continues to Decline. USDA Foreign Agricultural Service. https://gain.fas.usda.gov/. Posted May 19, 2020. Accessed June 18, 2021. |
[67] | Ngun TC , Ghahramani N , Sanchez FJ , Bocklandt S , Vilain E ((2011) ) The genetics of sex differences in brain and behavior. Front Neuroendocrinol 32: , 227–246. |
[68] | Gezer AO , Kochmanski J , VanOeveren SE , Cole-Strauss A , Kemp CJ , Patterson JR , Miller KM , Kuhn NC , Herman DE , McIntire A , Lipton JW , Luk KC , Fleming SM , Sortwell CE , Bernstein AI ((2020) ) Developmental exposure to the organochlorine pesticide dieldrin causes male-specific exacerbation of alpha-synuclein-preformed fibril-induced toxicity and motor deficits. Neurobiol Dis 141: , 104947. |
[69] | De Miranda BR , Fazzari M , Rocha EM , Castro S , Greenamyre JT ((2019) ) Sex differences in rotenone sensitivity reflect the male-to-female ratio in human Parkinson’s disease incidence. Toxicol Sci 170: , 133–143. |
[70] | Anderson T , Merrill AK , Eckard ML , Marvin E , Conrad K , Welle K , Oberdörster G , Sobolewski M , Cory-Slechta DA ((2020) ) Paraquat inhalation, a translationally relevant route of exposure: Disposition to the brain and male-specific olfactory impairment in mice. Toxicol Sci 180: , 175–185. |
[71] | Goldman SM , Tanner CM ((2019) ) Rotenone and Parkinson’s disease: Reduced sensitivity in females. Toxicol Sci 170: , 562–563. |
[72] | Borgus JR , Puthongkham P , Venton BJ ((2020) ) Complex sex and estrous cycle differences in spontaneous transient adenosine. J Neurochem 153: , 216–229. |
[73] | Parmalee NL , Maqbool SB , Ye B , Calder B , Bowman AB , Aschner M ((2015) ) RNASeq in C. elegans following manganese exposure. Curr Protoc Toxicol 65: , 11.20.1–11.20.17. |
[74] | Kim H , Perentis RJ , Caldwell GA , Caldwell KA ((2018) ) Gene-by-environment interactions that disrupt mitochondrial homeostasis cause neurodegeneration in C. elegans Parkinson’s models. Cell Death Dis 9: , 555. |
[75] | Mor DE , Sohrabi S , Kaletsky R , Keyes W , Tartici A , Kalia V , Miller GW , Murphy CT ((2020) ) Metformin rescues Parkinson’s disease phenotypes caused by hyperactive mitochondria. Proc Natl Acad Sci U S A 117: , 26438–26447. |
[76] | Bradner JM , Kalia V , Lau FK , Sharma M , Bucher ML , Johnson M , Chen M , Walker DI , Jones DP , Miller GW ((2021) ) Genetic or toxicant-induced disruption of vesicular monoamine storage and global metabolic profiling in Caenorhabditis elegans. Toxicol Sci 180: , 313–324. |
[77] | Hill-Burns EM , Singh N , Ganguly P , Hamza TH , Montimurro J , Kay DM , Yearout D , Sheehan P , Frodey K , McLear JA , Feany MB , Hanes SD , Wolfgang WJ , Zabetian CP , Factor SA , Payami H ((2013) ) A genetic basis for the variable effect of smoking/nicotine on Parkinson’s disease. Pharmacogenomics J 13: , 530–537. |
[78] | Dunn AR , Stout KA , Ozawa M , Lohr KM , Hoffman CA , Bernstein AI , Li Y , Wang M , Sgobio C , Sastry N , Cai H , Caudle WM , Miller GW ((2017) ) Synaptic vesicle glycoprotein 2C (SV2C) modulates dopamine release and is disrupted in Parkinson disease. Proc Natl Acad Sci U S A 114: , E2253–E2262. |
[79] | Foo JN , Chew EGY , Chung SJ , Peng R , Blauwendraat C , Nalls MA , Mok KY , Satake W , Toda T , Chao Y , Tan LCS , Tandiono M , Lian MM , Ng EY , Prakash KM , Au WL , Meah WY , Mok SQ , Annuar AA , Chan AYY , Chen L , Chen Y , Jeon BS , Jiang L , Lim JL , Lin JJ , Liu C , Mao C , Mok V , Pei Z , Shang HF , Shi CH , Song K , Tan AH , Wu YR , Xu YM , Xu R , Yan Y , Yang J , Zhang B , Koh WP , Lim SY , Khor CC , Liu J , Tan EK ((2020) ) Identification of risk loci for Parkinson disease in Asians and comparison of risk between Asians and Europeans: A genome-wideassociation study. JAMA Neurol 77: , 746–754. |
[80] | Avazzadeh S , Baena JM , Keighron C , Feller-Sanchez Y , Quinlan LR ((2021) ) Modelling Parkinson’s disease: iPSCs towards better understanding of human pathology. Brain Sci 11: , 373. |
[81] | Sandor C , Robertson P , Lang C , Heger A , Booth H , Vowles J , Witty L , Bowden R , Hu M , Cowley SA , Wade-Martins R , Webber C ((2017) ) Transcriptomic profiling of purified patient-derived dopamine neurons identifies convergent perturbations and therapeutics for Parkinson’s disease. Hum Mol Genet 26: , 552–566. |
[82] | Svensson E , Horvath-Puho E , Thomsen RW , Djurhuus JC , Pedersen L , Borghammer P , Sorensen HT ((2015) ) Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol 78: , 522–529. |
[83] | Braak H , Rub U , Gai WP , Del Tredici K ((2003) ) Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm (Vienna) 110: , 517–536. |
[84] | Challis C , Hori A , Sampson TR , Yoo BB , Challis RC , Hamilton AM , Mazmanian SK , Volpicelli-Daley LA , Gradinaru V ((2020) ) Gut-seeded alpha-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci 23: , 327–336. |
[85] | Kim S , Kwon SH , Kam TI , Panicker N , Karuppagounder SS , Lee S , Lee JH , Kim WR , Kook M , Foss CA , Shen C , Lee H , Kulkarni S , Pasricha PJ , Lee G , Pomper MG , Dawson VL , Dawson TM , Ko HS ((2019) ) Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103: , 627–641 e627. |
[86] | Kelly LP , Carvey PM , Keshavarzian A , Shannon KM , Shaikh M , Bakay RA , Kordower JH ((2014) ) Progression of intestinal permeability changes and alpha-synuclein expression in a mouse model of Parkinson’s disease. Mov Disord 29: , 999–1009. |
[87] | Bove FJ , Ruckart PZ , Maslia M , Larson TC ((2014) ) Evaluation of mortality among marines and navy personnel exposed to contaminated drinking water at USMC base Camp Lejeune: A retrospective cohort study. Environ Health 13: , 10. |
[88] | Pan-Montojo F , Anichtchik O , Dening Y , Knels L , Pursche S , Jung R , Jackson S , Gille G , Spillantini MG , Reichmann H , Funk RH ((2010) ) Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One 5: , e8762. |
[89] | Shakya R , Nepal MR , Kang MJ , Jeong TC ((2020) ) Effects of intestinal microbiota on pharmacokinetics of crocin and crocetin in male Sprague-Dawley rats. Metabolites 10: , 424. |
[90] | Hill-Burns EM , Debelius JW , Morton JT , Wissemann WT , Lewis MR , Wallen ZD , Peddada SD , Factor SA , Molho E , Zabetian CP , Knight R , Payami H ((2017) ) Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov Disord 32: , 739–749. |
[91] | Johnson ME , Stringer A , Bobrovskaya L ((2018) ) Rotenone induces gastrointestinal pathology and microbiota alterations in a rat model of Parkinson’s disease. Neurotoxicology 65: , 174–185. |
[92] | Liang Y , Zhan J , Liu D , Luo M , Han J , Liu X , Liu C , Cheng Z , Zhou Z , Wang P ((2019) ) Organophosphorus pesticide chlorpyrifos intake promotes obesity and insulin resistance through impacting gut and gut microbiota. Microbiome 7: , 19. |
[93] | Nagpal R , Wang S , Solberg Woods LC , Seshie O , Chung ST , Shively CA , Register TC , Craft S , McClain DA , Yadav H ((2018) ) Comparative microbiome signatures and short-chain fatty acids in mouse, rat, non-human primate, and human feces. Front Microbiol 9: , 2897. |
[94] | Kamel F , Goldman SM , Umbach DM , Chen H , Richardson G , Barber MR , Meng C , Marras C , Korell M , Kasten M , Hoppin JA , Comyns K , Chade A , Blair A , Bhudhikanok GS , Webster Ross G , William Langston J , Sandler DP , Tanner CM ((2014) ) Dietary fat intake, pesticide use, and Parkinson’s disease. Parkinsonism Relat Disord 20: , 82–87. |
[95] | Blossom SJ , Melnyk SB , Simmen FA ((2020) ) Complex epigenetic patterns in cerebellum generated after developmental exposure to trichloroethylene and/or high fat diet in autoimmune-prone mice. Environ Sci Process Impacts 22: , 583–594. |
[96] | Cannon JR , Greenamyre JT ((2011) ) The role of environmental exposures in neurodegeneration and neurodegenerative diseases. Toxicol Sci 124: , 225–250. |
[97] | Dick FD , De Palma G , Ahmadi A , Scott NW , Prescott GJ , Bennett J , Semple S , Dick S , Counsell C , Mozzoni P , Haites N , Wettinger SB , Mutti A , Otelea M , Seaton A , Soderkvist P , Felice A ((2007) ) Environmental risk factors for Parkinson’s disease and parkinsonism: The Geoparkinson study. Occup Environ Med 64: , 666–672. |
[98] | Marras C , Canning CG , Goldman SM ((2019) ) Environment, lifestyle, and Parkinson’s disease: Implications for prevention in the next decade. Mov Disord 34: , 801–811. |
[99] | Perera FP ((1997) ) Environment and cancer: Who are susceptible? Science 278: , 1068–1073. |
[100] | Rocha EM , De Miranda BR , Castro S , Drolet R , Hatcher NG , Yao L , Smith SM , Keeney MT , Di Maio R , Kofler J , Hastings TG , Greenamyre JT ((2020) ) LRRK2 inhibition prevents endolysosomal deficits seen in human Parkinson’s disease. Neurobiol Dis 134: , 104626. |
[101] | Di Maio R , Hoffman EK , Rocha EM , Keeney MT , Sanders LH , De Miranda BR , Zharikov A , Van Laar A , Stepan AF , Lanz TA , Kofler JK , Burton EA , Alessi DR , Hastings TG , Greenamyre JT ((2018) ) LRRK2 activation in idiopathic Parkinson’s disease. Sci Transl Med 10: , eaar5429. |
[102] | De Miranda BR , Castro SL , Rocha EM , Bodle CR , Johnson KE , Greenamyre JT ((2021) ) The industrial solvent trichloroethylene induces LRRK2 kinase activity and dopaminergic neurodegeneration in a rat model of Parkinson’s disease. Neurobiol Dis 153: , 105312. |
[103] | Dwyer Z , Rudyk C , Thompson A , Farmer K , Fenner B , Fortin T , Derksen A , Sun H , Hayley S , Clint ((2020) ) Leucine-rich repeat kinase-2 (LRRK2) modulates microglial phenotype and dopaminergic neurodegeneration. Neurobiol Aging 91: , 45–55. |
[104] | Rudyk C , Dwyer Z , Hayley S , membership C ((2019) ) Leucine-rich repeat kinase-2 (LRRK2) modulates paraquat-induced inflammatory sickness and stress phenotype. J Neuroinflammation 16: , 120. |
[105] | Chen J , Su P , Luo W , Chen J ((2018) ) Role of LRRK2 in manganese-induced neuroinflammation and microglial autophagy. Biochem Biophys Res Commun 498: , 171–177. |
[106] | Kim J , Pajarillo E , Rizor A , Son DS , Lee J , Aschner M , Lee E ((2019) ) LRRK2 kinase plays a critical role in manganese-induced inflammation and apoptosis in microglia. PLoS One 14: , e0210248. |
[107] | Zhao Y , Dzamko N ((2019) ) Recent developments in LRRK2-targeted therapy for Parkinson’s disease. Drugs 79: , 1037–1051. |
[108] | von Campenhausen S , Bornschein B , Wick R , Botzel K , Sampaio C , Poewe W , Oertel W , Siebert U , Berger K , Dodel R ((2005) ) Prevalence and incidence of Parkinson’s disease in Europe. Eur Neuropsychopharmacol 15: , 473–490. |
[109] | Schoenberg BS , Anderson DW , Haerer AF ((1985) ) Prevalence of Parkinson’s disease in the biracial population of Copiah County, Mississippi. Neurology 35: , 841–845. |
[110] | Dorsey R , Sherer T , Okun MS , Bloem BR (2020) Ending Parkinson’s Disease: A Prescription for Action, Public Affairs. |
[111] | Savica R , Grossardt BR , Bower JH , Ahlskog JE , Rocca WA ((2016) ) Time trends in the incidence of Parkinson disease. JAMA Neurol 73: , 981. |
[112] | Tubiello F (2018) FAOSTAT Statistical Database, Rome. http://www.fao.org/faostat/en/#data/RP/visualize |
[113] | Smith SJ , van Aardenne J , Klimont Z , Andres RJ , Volke A , Delgado Arias S ((2011) ) Anthropogenic sulfur dioxide emissions: 1850–2005. Atmos Chem Phys 11: , 1101–1116. |
[114] | Our World in Data. Sulphur dioxide (SO2) emissions, 1850-2000. https://ourworldindata.org/grapher/so-emissions-tonnes-per-year |
[115] | Berg D , Borghammer P , Fereshtehnejad SM , Heinzel S , Horsager J , Schaeffer E , Postuma RB ((2021) ) Prodromal Parkinson disease subtypes - key to understanding heterogeneity. Nat Rev Neurol 17: , 349–361. |
[116] | Venuto CS , Potter NB , Dorsey ER , Kieburtz K ((2016) ) A review of disease progression models of Parkinson’s disease and applications in clinical trials. Mov Disord 31: , 947–956. |
[117] | Ambar Akkaoui M , Geoffroy PA , Roze E , Degos B , Garcin B ((2020) ) Functional motor symptoms in Parkinson’s disease and functional parkinsonism: A systematic review. J Neuropsychiatry Clin Neurosci 32: , 4–13. |
[118] | Schapira AHV , Chaudhuri KR , Jenner P ((2017) ) Non-motor features of Parkinson disease. Nat Rev Neurosci 18: , 509. |
[119] | Aarsland D , Batzu L , Halliday GM , Geurtsen GJ , Ballard C , Ray Chaudhuri K , Weintraub D ((2021) ) Parkinson disease-associated cognitive impairment. Nat Rev Dis Primers 7: , 47. |
[120] | Pagano G , Ferrara N , Brooks DJ , Pavese N ((2016) ) Age at onset and Parkinson disease phenotype. Neurology 86: , 1400–1407. |
[121] | Liepelt-Scarfone I , Gauss K , Maetzler W , Muller K , Bormann C , Fruhmann Berger M , Timmers M , Streffer J , Berg D ((2013) ) Evaluation of progression markers in the premotor phase of Parkinson’s disease: The progression markers in the premotor phase study. Neuroepidemiology 41: , 174–182. |
[122] | Mahlknecht P , Seppi K , Poewe W ((2015) ) The concept of prodromal Parkinson’s disease. J Parkinsons Dis 5: , 681–697. |
[123] | Postuma RB ((2014) ) Prodromal Parkinson’s disease–using REM sleep behavior disorder as a window. Parkinsonism Relat Disord 20 Suppl 1: , S1–4. |
[124] | Shaddick G , Thomas ML , Mudu P , Ruggeri G , Gumy S ((2020) ) Half the world’s population are exposed to increasing air pollution. NPJ Clim Atmos Sci 3: , 23. |
[125] | Food and Agricultural Organizaiton of the United Nations (2018) FAOSTAT Database http://www.fao.org/faostat/en/#data/RP. Accessed June, 2021. |
[126] | National Academies of Science Engineering Medicine ((2021) ) Leveraging Advances in Remote Geospatial Technologies to Inform Precision Environmental Health Decisions. National Academic Press. |
[127] | Kumar A , Calne SM , Schulzer M , Mak E , Wszolek Z , Van Netten C , Tsui JKC , Stoessl AJ , Calne DB ((2004) ) Clustering of Parkinson disease. Arch Neurol 61: , 1057–1060. |
[128] | Hasan F (2002) Residents near old semiconductor plant worry about toxic exposure. My Voice. Embarcadero Publishing Company. https://www.mv-voice.com/morgue/2002/2002_11_22.resprknsn.html. Posted November 22, 2002. Accessed June 18, 2021. |
[129] | DeBolt D (2011) TCE causes cancer, other health woes, EPA says. Mountain View Voice. https://paloaltoonline.com/news/2011/10/07/tce-causes-cancer-other-health-problems-epa-says. Last Updated October 10, 2011. Accessed June 18, 2021. |
[130] | Duenwald M (2002) Parkinson’s ‘Clusters’ Getting a Closer Look. The New York Times. https://www.nytimes.com/2002/05/14/health/parkinson-s-clusters-getting-a-closer-look.html. Posted May 14, 2002. Accessed June 18, 2021. |
[131] | Gorell JM , Johnson CC , Rybicki BA , Peterson EL , Richardson RJ ((1998) ) The risk of Parkinson’s disease with exposure to pesticides, farming, well water, and rural living. Neurology 50: , 1346–1350. |
[132] | Ascherio A , Chen H , Weisskopf MG , O’Reilly E , McCullough ML , Calle EE , Schwarzschild MA , Thun MJ ((2006) ) Pesticide exposure and risk for Parkinson’s disease. Ann Neurol 60: , 197–203. |
[133] | Kab S , Spinosi J , Chaperon L , Dugravot A , Singh-Manoux A , Moisan F , Elbaz A ((2017) ) Agricultural activities and the incidence of Parkinson’s disease in the general French population. Eur J Epidemiol 32: , 203–216. |
[134] | Costello S , Cockburn M , Bronstein J , Zhang X , Ritz B ((2009) ) Parkinson’s disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am J Epidemiol 169: , 919–926. |
[135] | Wright Willis A , Evanoff BA , Lian M , Criswell SR , Racette BA ((2010) ) Geographic and ethnic variation in Parkinson disease: A population-based study of US Medicare beneficiaries. Neuroepidemiology 34: , 143–151. |
[136] | Mantri S , Fullard ME , Beck J , Willis AW ((2019) ) State-level prevalence, health service use, and spending vary widely among Medicare beneficiaries with Parkinson disease. NPJ Parkinsons Dis 5: , 1. |
[137] | Arguelles JL , Borroto Bermudez LA , Sosa Aguila LM , Rodriguez Carbajal A ((2019) ) Prevalence of Parkinson’s disease in an urban area of Cienfuegos city. Int J Fam Commun Med 3: , 12–14. |
[138] | Jo S , Kim YJ , Park KW , Hwang YS , Lee SH , Kim BJ , Chung SJ ((2021) ) Association of NO2 and other air pollution exposures with the risk of Parkinson disease. JAMA Neurol 78: , 800–808. |
[139] | Gash DM , Rutland K , Hudson NL , Sullivan PG , Bing G , Cass WA , Pandya JD , Liu M , Choi D-Y , Hunter RL , Gerhardt GA , Smith CD , Slevin JT , Prince TS ((2008) ) Trichloroethylene: Parkinsonism and complex 1 mitochondrial neurotoxicity. Ann Neurol 63: , 184–192. |
[140] | Goldman SM , Quinlan PJ , Ross GW , Marras C , Meng C , Bhudhikanok GS , Comyns K , Korell M , Chade AR , Kasten M , Priestley B , Chou KL , Fernandez HH , Cambi F , Langston JW , Tanner CM ((2012) ) Solvent exposures and Parkinson disease risk in twins. Ann Neurol 71: , 776–784. |
[141] | Barringer F (2011) E.P.A. Charts Risks of a Ubiquitous Chemical. The New York Times, https://green.blogs.nytimes.com/2011/09/30/e-p-a-quantifies-trichloroethylene-risks/. Accessed March 12, 2020. |
[142] | Environmental Working Group Tap Water Database - 2019 Update. https://www.ewg.org/tapwater/contaminant.php?contamcode=2984. Accessed April 18, 2021. |
[143] | Department of Veterans A ((2017) ) Diseases associated with exposure to contaminants in the water supply at Camp Lejeune. Final rule. Fed Regist 82: , 4173–4185. |
[144] | Ruckart PZ , Bove FJ , Maslia M ((2014) ) Evaluation of contaminated drinking water and preterm birth, small for gestational age, and birth weight at Marine Corps Base Camp Lejeune, North Carolina: A cross-sectional study. Environ Health 13: , 99. |
[145] | Sonnenfeld N , Hertz-Picciotto I , Kaye WE ((2001) ) Tetrachloroethylene in drinking water and birth outcomes at the US Marine Corps Base at Camp Lejeune, North Carolina. Am J Epidemiol 154: , 902–908. |
[146] | Kamel F , Tanner C , Umbach D , Hoppin J , Alavanja M , Blair A , Comyns K , Goldman S , Korell M , Langston J , Ross G , Sandler D ((2006) ) Pesticide exposure and self-reported Parkinson’s disease in the Agricultural Health Study. Am J Epidemiol 165: , 364–374. |
[147] | EPA (2021) Contaminants at Superfund Sites. United States Environmental Protection Agency. |
[148] | Firestone JA , Lundin JI , Powers KM , Smith-Weller T , Franklin GM , Swanson PD , Longstreth WT , Checkoway H ((2009) ) Occupational factors and risk of Parkinson’s disease: A population-based case-control study. Am J Ind Med 53: , 217–223. |
[149] | Tsui JKC , Calne DB , Wang Y , Schulzer M , Marion SA ((1999) ) Occupational risk factors in Parkinson’s disease. Can J Public Health 90: , 334–337. |
[150] | Nielsen SS , Warden MN , Sallmen M , Sainio M , Uuksulainen S , Checkoway H , Hublin C , Racette BA ((2021) ) Solvent exposed occupations and risk of Parkinson disease in Finland. Clin Park Relat Disord 4: , 100092. |
[151] | Abbott RD , Ross GW , Petrovitch H , Masaki KH , Launer LJ , Nelson JS , White LR , Tanner CM ((2016) ) Midlife milk consumption and substantia nigra neuron density at death. Neurology 86: , 512–519. |
[152] | Ross GW , Abbott RD , Petrovitch H , Duda JE , Tanner CM , Zarow C , Uyehara-Lock JH , Masaki KH , Launer LJ , Studabaker WB , White LR ((2019) ) Association of brain heptachlor epoxide and otherorganochlorine compounds with lewy pathology. Mov Disord 34: , 228–235. |
[153] | Fleming L , Mann JB , Bean J , Briggle T , Sanchez-Ramos JR ((1994) ) Parkinson’s disease and brain levels of organochlorine pesticides. Ann Neurol 36: , 100–103. |
[154] | Corrigan FM , Wienburg CL , Shore RF , Daniel SE , Mann D ((2000) ) Organochlorine insecticides in substantia nigra in Parkinson’s disease. J Toxicol Environ Health A 59: , 229–234. |
[155] | Hatcher-Martin JM , Gearing M , Steenland K , Levey AI , Miller GW , Pennell KD ((2012) ) Association between polychlorinated biphenyls and Parkinson’s disease neuropathology. Neurotoxicology 33: , 1298–1304. |
[156] | Dardiotis E , Aloizou A-M , Sakalakis E , Siokas V , Koureas M , Xiromerisiou G , Petinaki E , Wilks M , Tsatsakis A , Hadjichristodoulou C , Stefanis L , Hadjigeorgiou GM ((2020) ) Organochlorine pesticide levels in Greek patients with Parkinson’s disease. Toxicol Rep 7: , 596–601. |
[157] | Hashemy-Tonkabony SE , Soleimani-Amiri MJ ((1978) ) Chlorinated pesticide residues in the body fat of people in Iran. Environ Res 16: , 419–422. |
[158] | Jaga K , Dharmani C ((2003) ) Global surveillance of DDT and DDE levels in human tissues. Int J Occup Med Environ Health 16: , 7–20. |
[159] | Lash LH , Putt DA , Parker JC ((2006) ) Metabolism and tissue distribution of orally administered trichloroethylene in male and female rats: Identification of glutathione- and cytochrome P-450-derived metabolites in liver, kidney, blood, and urine. J Toxicol Environ Health A 69: , 1285–1309. |
[160] | Phillips RV , Rieswijk L , Hubbard AE , Vermeulen R , Zhang J , Hu W , Li L , Bassig BA , Wong JYY , Reiss B , Huang Y , Wen C , Purdue M , Tang X , Zhang L , Smith MT , Rothman N , Lan Q ((2019) ) Human exposure to trichloroethylene is associated with increased variability of blood DNA methylation that is enriched in genes and pathways related to autoimmune disease and cancer. Epigenetics 14: , 1112–1124. |
[161] | Wild CP ((2005) ) Complementing the genome with an “exposome”: Theoutstanding challenge of environmental exposure measurement inmolecular epidemiology. Cancer Epidemiol Biomarkers Prev 14: , 1847–1850. |
[162] | Vermeulen R , Schymanski EL , Barabasi AL , Miller GW ((2020) ) The exposome and health: Where chemistry meets biology. Science 367: , 392–396. |
[163] | Agin A , Blanc F , Bousiges O , Villette C , Philippi N , Demuynck C , Martin-Hunyadi C , Cretin B , Lang S , Zumsteg J , Namer IJ , Heintz D ((2020) ) Environmental exposure to phthalates and dementia with Lewy bodies: Contribution of metabolomics. J Neurol Neurosurg Psychiatry 91: , 968–974. |
[164] | Chen H , Ritz B ((2018) ) The search for environmental causes of Parkinson’s disease: Moving forward. J Parkinsons Dis 8: , S9–S17. |
[165] | Fleming SM ((2017) ) Mechanisms of gene-environment interactions in Parkinson’s disease. Curr Environ Health Rep 4: , 192–199. |
[166] | Deng H , Wang P , Jankovic J ((2018) ) The genetics of Parkinson disease. Ageing Res Rev 42: , 72–85. |
[167] | Troiano AR , Elbaz A , Lohmann E , Belarbi S , Vidailhet M , Bonnet AM , Lesage S , Pollak P , Cazeneuve C , Borg M , Feingold J , Durr A , Tazir M , Brice A , French Parkinson Disease Genetic Study Group ((2010) ) Low disease risk in relatives of North African LRRK2 Parkinson disease patients. Neurology 75: , 1118–1119. |
[168] | Goldman SM , Marek K , Ottman R , Meng C , Comyns K , Chan P , Ma J , Marras C , Langston JW , Ross GW , Tanner CM ((2019) ) Concordance for Parkinson’s disease in twins: A 20-year update. Ann Neurol 85: , 600–605. |
[169] | Tanner CM , Ottman R , Goldman SM , Ellenberg J , Chan P , Mayeux R , Langston JW ((1999) ) Parkinson disease in twins: An etiologic study. JAMA 281: , 341–346. |
[170] | Deng Y , Newman B , Dunne MP , Silburn PA , Mellick GD ((2004) ) Further evidence that interactions between CYP2D6 and pesticide exposure increase risk for Parkinson’s disease. Ann Neurol 55: , 897. |
[171] | Elbaz A , Levecque C , Clavel J , Vidal JS , Richard F , Amouyel P , Alperovitch A , Chartier-Harlin MC , Tzourio C ((2004) ) CYP2D6 polymorphism, pesticide exposure, and Parkinson’s disease. Ann Neurol 55: , 430–434. |
[172] | Lee PC , Rhodes SL , Sinsheimer JS , Bronstein J , Ritz B ((2013) ) Functional paraoxonase 1 variants modify the risk of Parkinson’s disease due to organophosphate exposure. Environ Int 56: , 42–47. |
[173] | Manthripragada AD , Costello S , Cockburn MG , Bronstein JM , Ritz B ((2010) ) Paraoxonase 1, agricultural organophosphate exposure, and Parkinson disease. Epidemiology 21: , 87–94. |
[174] | Fitzmaurice AG , Rhodes SL , Cockburn M , Ritz B , Bronstein JM ((2014) ) Aldehyde dehydrogenase variation enhances effect of pesticides associated with Parkinson disease. Neurology 82: , 419–426. |
[175] | Goldman SM , Kamel F , Ross GW , Bhudhikanok GS , Hoppin JA , Korell M , Marras C , Meng C , Umbach DM , Kasten M , Chade AR , Comyns K , Richards MB , Sandler DP , Blair A , Langston JW , Tanner CM ((2012) ) Genetic modification of the association of paraquat and Parkinson’s disease. Mov Disord 27: , 1652–1658. |
[176] | Narayan S , Sinsheimer JS , Paul KC , Liew Z , Cockburn M , Bronstein JM , Ritz B ((2015) ) Genetic variability in ABCB1, occupational pesticide exposure, and Parkinson’s disease. Environ Res 143: , 98–106. |
[177] | Lacher SE , Skagen K , Veit J , Dalton R , Woodahl EL ((2015) ) P-Glycoprotein Transport of Neurotoxic Pesticides. J Pharmacol Exp Ther 355: , 99–107. |
[178] | Goldman SM , Kamel F , Meng C , Korell M , Umbach DM , Hoppin J , Ross GW , Marras C , Kasten M , Chade A , Comyns K , Sandler D , Blair A , Tanner CM (2016) Rotenone and Parkinson’s disease (PD): Effect modification by membrane transporter variants. International Parkinson and Movement Disorder Society, 2016 International Congress, Abstract 466. |
[179] | Hancock DB , Martin ER , Vance JM , Scott WK ((2008) ) Nitric oxide synthase genes and their interactions with environmental factors in Parkinson’s disease. Neurogenetics 9: , 249–262. |
[180] | Paul KC , Sinsheimer JS , Rhodes SL , Cockburn M , Bronstein J , Ritz B ((2016) ) Organophosphate pesticide exposures, nitric oxide synthase gene variants, and gene-pesticide interactions in a case-control study of Parkinson’s disease, California (USA). Environ Health Perspect 124: , 570–577. |
[181] | Fong CS , Wu RM , Shieh JC , Chao YT , Fu YP , Kuao CL , Cheng CW ((2007) ) Pesticide exposure on southwestern Taiwanese with MnSOD and NQO1 polymorphisms is associated with increased risk of Parkinson’s disease. Clin Chim Acta 378: , 136–141. |
[182] | Kaye J , Lima L , Tanner C , Finkbeiner S , Swanson R , Goldman S (2019) Exposed-only analysis of gene-pesticide interaction in Parkinson’s disease (PD). International Parkinson and Movement Disorder Society. |
[183] | De Palma G , Mozzoni P , Mutti A , Calzetti S , Negrotti A ((1998) ) Case-control study of interactions between genetic and environmental factors in Parkinson’s disease. Lancet 352: , 1986–1987. |
[184] | Gatto NM , Rhodes SL , Manthripragada AD , Bronstein J , Cockburn M , Farrer M , Ritz B ((2010) ) alpha-Synuclein gene may interact with environmental factors in increasing risk of Parkinson’s disease. Neuroepidemiology 35: , 191–195. |
[185] | Goldman SM , Kamel F , Ross GW , Jewell SA , Bhudhikanok GS , Umbach D , Marras C , Hauser RA , Jankovic J , Factor SA , Bressman S , Lyons KE , Meng C , Korell M , Roucoux DF , Hoppin JA , Sandler DP , Langston JW , Tanner CM ((2012) ) Head injury, alpha-synuclein Rep1, and Parkinson’s disease. Ann Neurol 71: , 40–48. |
[186] | San Luciano M , Tanner CM , Meng C , Marras C , Goldman SM , Lang AE , Tolosa E , Schule B , Langston JW , Brice A , Corvol JC , Goldwurm S , Klein C , Brockman S , Berg D , Brockmann K , Ferreira JJ , Tazir M , Mellick GD , Sue CM , Hasegawa K , Tan EK , Bressman S , Saunders-Pullman R , Michael JFFLCC ((2020) ) Nonsteroidal anti-inflammatory use and LRRK2 Parkinson’s disease penetrance. Mov Disord 35: , 1755–1764. |
[187] | Kline EM , Houser MC , Herrick MK , Seibler P , Klein C , West A , Tansey MG ((2021) ) Genetic and environmental factors in Parkinson’s disease converge on immune function and inflammation. Mov Disord 36: , 25–36. |
[188] | Adler CH , Beach TG ((2016) ) Neuropathological basis of nonmotor manifestations of Parkinson’s disease. Mov Disord 31: , 1114–1119. |
[189] | Zhang X , Sebastiani P , Liu G , Schembri F , Zhang X , Dumas YM , Langer EM , Alekseyev Y , O’Connor GT , Brooks DR , Lenburg ME , Spira A ((2010) ) Similarities and differences between smoking-related gene expression in nasal and bronchial epithelium. Physiol Genomics 41: , 1–8. |
[190] | Vachier I ((2004) ) Inflammatory features of nasal mucosa in smokers with and without COPD. Thorax 59: , 303–307. |
[191] | Ortuño-Lizarán I , Beach TG , Serrano GE , Walker DG , Adler CH , Cuenca N ((2018) ) Phosphorylated α-synuclein in theretina is a biomarker of Parkinson’s disease pathology severity. Mov Disord 33: , 1315–1324. |
[192] | Campo F , Carletti R , Fusconi M , Pellicano C , Pontieri FE , Di Gioia CR , de Vincentiis M ((2019) ) Alpha-synuclein in salivary gland as biomarker for Parkinson’s disease. Rev Neurosci 30: , 455–462. |
[193] | Kim JY , Illigens BM , McCormick MP , Wang N , Gibbons CH ((2019) ) Alpha-synuclein in skin nerve fibers as a biomarker for alpha-synucleinopathies. J Clin Neurol 15: , 135–142. |
[194] | Mazzetti S , Basellini MJ , Ferri V , Cassani E , Cereda E , Paolini M , Calogero AM , Bolliri C , De Leonardis M , Sacilotto G , Cilia R , Cappelletti G , Pezzoli G ((2020) ) α-Synuclein oligomers in skin biopsy of idiopathic and monozygotic twin patients with Parkinson’s disease. Brain 143: , 920–931. |
[195] | Killinger BA , Madaj Z , Sikora JW , Rey N , Haas AJ , Vepa Y , Lindqvist D , Chen H , Thomas PM , Brundin P , Brundin L , Labrie V ((2018) ) The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci Transl Med 10: , eaar5280. |
[196] | Weiner WJ ((2008) ) There is no Parkinson disease. Arch Neurol 65: , 705–708. |
[197] | Borghammer P , Van Den Berge N ((2019) ) Brain-first versus gut-first Parkinson’s disease: A hypothesis. J Parkinsons Dis 9: , S281–S295. |
[198] | Dorsey ER , Constantinescu R , Thompson JP , Biglan KM , Holloway RG , Kieburtz K , Marshall FJ , Ravina BM , Schifitto G , Siderowf A , Tanner CM ((2007) ) Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 68: , 384–386. |
[199] | Cheng Y , Wang Y-J ((2020) ) Tobacco smoking and the reduced risk of Parkinson disease. Neurology 94: , 860–861. |
[200] | Sharma A , Kumar V , Shahzad B , Tanveer M , Sidhu GPS , Handa N , Kohli SK , Yadav P , Bali AS , Parihar RD , Dar OI , Singh K , Jasrotia S , Bakshi P , Ramakrishnan M , Kumar S , Bhardwaj R , Thukral AK ((2019) ) Worldwide pesticide usage and its impacts on ecosystem. Sn Appl Sci 1: , 1446. |
[201] | Tanner CM , Ross GW , Jewell SA , Hauser RA , Jankovic J , Factor SA , Bressman S , Deligtisch A , Marras C , Lyons KE , Bhudhikanok GS , Roucoux DF , Meng C , Abbott RD , Langston JW ((2009) ) Occupation and risk of parkinsonism: A multicenter case-control study. Arch Neurol 66: , 1106–1113. |
[202] | Ali SJ , Ellur G , Patel K , Sharan K ((2019) ) Chlorpyrifos exposure induces parkinsonian symptoms and associated bone loss in adult Swiss albino mice. Neurotox Res 36: , 700–711. |
[203] | Freire C , Koifman S ((2012) ) Pesticide exposure and Parkinson’s disease: Epidemiological evidence of association. Neurotoxicology 33: , 947–971. |
[204] | Evans GW , Kim P ((2010) ) Multiple risk exposure as a potential explanatory mechanism for the socioeconomic status-health gradient. Ann N Y Acad Sci 1186: , 174–189. |
[205] | Gleason JA , Nanavaty JV , Fagliano JA ((2019) ) Drinking water lead and socioeconomic factors as predictors of blood lead levels in New Jersey’s children between two time periods. Environ Res 169: , 409–416. |
[206] | Hemming JP , Gruber-Baldini AL , Anderson KE , Fishman PS , Reich SG , Weiner WJ , Shulman LM ((2011) ) Racial and socioeconomic disparities in parkinsonism. Arch Neurol 68: , 498–503. |
[207] | Yacoubian TA , Howard G , Kissela B , Sands CD , Standaert DG ((2009) ) Racial differences in Parkinson’s disease medication use in the reasons for geographic and racial differences in stroke cohort: A cross-sectional study. Neuroepidemiology 33: , 329–334. |
[208] | Dahodwala N , Siderowf A , Xie M , Noll E , Stern M , Mandell DS ((2009) ) Racial differences in the diagnosis of Parkinson’s disease. Mov Disord 24: , 1200–1205. |
[209] | Cheng EM , Siderowf AD , Swarztrauber K , Lee M , Vassar S , Jacob E , Eisa MS , Vickrey BG ((2008) ) Disparities of care in veterans with Parkinson’s disease. Parkinsonism Relat Disord 14: , 8–14. |
[210] | Iqubal A , Ahmed M , Ahmad S , Sahoo CR , Iqubal MK , Haque SE ((2020) ) Environmental neurotoxic pollutants: Review. Environ Sci Pollut Res Int 27: , 41175–41198. |
[211] | Purisai MG , McCormack AL , Cumine S , Li J , Isla MZ , Di Monte DA ((2007) ) Microglial activation as a priming event leading to paraquat-induced dopaminergic cell degeneration. Neurobiol Dis 25: , 392–400. |
[212] | Slotkin TA , Seidler FJ ((2011) ) Developmental exposure to organophosphates triggers transcriptional changes in genes associated with Parkinson’s disease in vitro and in vivo. Brain Res Bull 86: , 340–347. |
[213] | Adgent MA , Hoffman K , Goldman BD , Sjodin A , Daniels JL ((2014) ) Brominated flame retardants in breast milk and behavioural and cognitive development at 36 months. Paediatr Perinat Epidemiol 28: , 48–57. |
[214] | Lu D , Wang D , Ni R , Lin Y , Feng C , Xu Q , Jia X , Wang G , Zhou Z ((2015) ) Organochlorine pesticides and their metabolites in human breast milk from Shanghai, China. Environ Sci Pollut Res Int 22: , 9293–9306. |
[215] | CDC, Lung Cancer Statistics, https://www.cdc.gov/cancer/lung/statistics/index.htm. Last Updated June 8, 2021. Accessed July 13, 2021. |
[216] | Needleman HL ((2000) ) The removal of lead from gasoline: Historical and personal reflections. Environ Res 84: , 20–35. |
[217] | USGS (2020) esticide National Synthesis Project. NationalWater-Quality Assessment Project, https://water.usgs.gov/nawqa/pnsp/usage/maps/show_map.php?year=2017&map=PARAQUAT&hilo=L&disp=Paraquat. Last Updated June 18, 2020. Accessed March 15, 2021. |
[218] | Furlong M , Tanner CM , Goldman SM , Bhudhikanok GS , Blair A , Chade A , Comyns K , Hoppin JA , Kasten M , Korell M , Langston JW , Marras C , Meng C , Richards M , Ross GW , Umbach DM , Sandler DP , Kamel F ((2015) ) Protective glove use and hygiene habits modify the associations of specific pesticides with Parkinson’s disease. Environ Int 75: , 144–150. |
[219] | Shrestha S , Parks CG , Umbach DM , Richards-Barber M , Hofmann JN , Chen H , Blair A , Beane Freeman LE , Sandler DP ((2020) ) Pesticide use and incident Parkinson’s disease in a cohort of farmers and their spouses. Environ Res 191: , 110186. |
[220] | NIH RePORT, Estimates of Funding for Various Research, Condition, and Disease Categories (RCDC), https://report.nih.gov/funding/categorical-spending#/. Accessed February 24, 2020. |
[221] | NIEHS Environmental Factor (2021) Environmental factor. Exposome research, anti-racism on tap at NIEHS council meeting. https://factor.niehs.nih.gov/2021/7/feature/3-feature-niehs-council/index.htm. |
[222] | Shilts R ((1987) ) And the Band Played On, St. Martinȁs Press. |
[223] | Mothers Against Drunk Driving (MADD). https://madd.org/history. Accessed May 21, 2021. |


