
The Compound ATH434 Prevents Alpha-Synuclein Toxicity in a Murine Model of Multiple System Atrophy
Abstract
Background:
An elevation in iron levels, together with an accumulation of α-synuclein within the oligodendrocytes, are features of the rare atypical parkinsonian disorder, Multiple System Atrophy (MSA). We have previously tested the novel compound ATH434 (formally called PBT434) in preclinical models of Parkinson’s disease and shown that it is brain-penetrant, reduces iron accumulation and iron-mediated redox activity, provides neuroprotection, inhibits alpha synuclein aggregation and lowers the tissue levels of alpha synuclein. The compound was also well-tolerated in a first-in-human oral dosing study in healthy and older volunteers with a favorable, dose-dependent pharmacokinetic profile.
Objective:
To evaluate the efficacy of ATH434 in a mouse MSA model.
Methods:
The PLP-α-syn transgenic mouse overexpresses α-synuclein, demonstrates oligodendroglial pathology, and manifests motor and non-motor aspects of MSA. Animals were provided ATH434 (3, 10, or 30 mg/kg/day spiked into their food) or control food for 4 months starting at 12 months of age and were culled at 16 months. Western blot was used to assess oligomeric and urea soluble α-synuclein levels in brain homogenates, whilst stereology was used to quantitate the number of nigral neurons and glial cell inclusions (GCIs) present in the substantia nigra pars compacta.
Results:
ATH434 reduced oligomeric and urea soluble α-synuclein aggregation, reduced the number of GCIs, and preserved SNpc neurons. In vitro experiments suggest that ATH434 prevents the formation of toxic oligomeric “species of synuclein”.
Conclusion:
ATH434 is a promising small molecule drug candidate that has potential to move forward to trial for treating MSA.
INTRODUCTION
Synucleinopathies are a group of conditions characterized by the pathological accumulation of intracellular deposits of α-synuclein (α-syn) [1]. This includes Parkinson’s disease (PD), Multiple System Atrophy (MSA), Dementia with Lewy bodies (DLB), and other rarer conditions such as pure autonomic failure [2]. Each of these conditions is distinguished by the anatomical location of the α-syn deposits, which can be within CNS neurons, glial cells or in peripheral nerves. Furthermore, recent evidence has suggested that different “strains” or “types” of α-syn may be involved in different diseases [3].
It has been shown that iron is elevated in synucleinopathies such as PD, MSA, and DLB, and the unique iron profile, more recently magnetic resonance imaging (MRI) is being developed as an aid in distinguishing the different conditions [4–8]. The significance of a localized accumulation of iron has been the subject of much speculation, but iron could initiate and/or potentiate oxidative stress [9, 10], influence the aggregation state and type of α-syn [11–14], and also regulate the amount of α-syn, as the mRNA contains an untranslated iron response element that is involved in the post-transcriptional regulation of the protein level [15, 16].
MSA is a rare, fatal neurodegenerative brain disorder that presents with predominant autonomic dysfunction, parkinsonism, cerebellar impairments, or a variable combination of these manifestations. MSA patients are generally not responsive to typical PD therapies, and there are no disease modifying treatments for this condition. As such, MSA represents an unmet medical need. Pathologically, MSA is characterized by glial cytoplasmic inclusions (GCIs), structures that are composed of aggregated, urea soluble α-syn that are found in the areas of degeneration of affected brain regions including the substantia nigra pars compacta, putamen, pontine nuclei, cerebellum, and spinal cord. Evidence suggests that the deposition of α-syn and the toxic effects of soluble α-syn aggregates in oligodendrocytes are key events in the hypothesized spreading of pathology from the oligodendrocytes to result in neuronal cell death. MSA is also associated with elevated iron concentrations in the pons and putamen, and this may be a critical factor in the pathogenesis of MSA given the potential role of iron in the aggregation of α-syn in GCIs [6].
ATH434 (formally called PBT434), which has successfully completed a Phase 1 study [17], is a brain-penetrant, small molecule inhibitor of α-syn aggregation. In several rodent models of PD, ATH434 reduced iron accumulation, α-syn aggregation, CSF α-syn levels, and oxidative stress, preserved neurons in the SN and improved motor function [18]. Importantly, one of the models in which ATH434 showed neuroprotection was the hA53T mutant α-syn transgenic mouse, which provided a rationale for exploring the effects of the compound in other synucleinopathies. The PLP-α-syn transgenic mouse is an animal model of MSA in as much as it overexpresses human α-syn in the oligodendrocytes (the synuclein transgene is driven by the myelin proteolipid protein (PLP) promoter), develops GCI pathology and manifests motor and non-motor aspects characteristic of the disease [19–21]. The aim of this study was to determine whether the effects of ATH434 would translate into therapeutic benefits in a genetic model of MSA.
METHODS
Mice
The MSA transgenic mice used in this study were derived from a colony initially established from breeding animals that were a generous gift from the laboratory of Dr Nadia Stefanova, Clinical Department of Neurology, Innsbruck Medical University, Innsbruck, Austria. These mice express human wild-type α-syn within the oligodendrocytes, driven by the proteolipid protein (PLP) promoter, they show that α-syn is localized only within cells immunopositive for cyclic nucleotide phosphodiesterase oligodendroglia [19]. The phenotype of these mice and pathology has been extensively characterized [20, 21]. The PLP-α-syn mice are maintained on a C57BL/6 background (Animal Resources Centre, Western Australia) and littermate wild types were bred from heterozygous mating at the Florey Neuroscience Institute animal facility for more than 10 generations. Mice were housed in Techniplast brand individually ventilated cages. Mice had ad libitum access to food and water. All procedures involving mice conformed to the Australian National Health and Medical Research Council Code of Practice for the care and use of animals for scientific purposes and were approved by the Florey Institute Animal Ethics Committee. All experiments were designed to minimize the number of animals used, whilst still providing sufficiently powered group sizes to observe effects. Both male (M) and female (F) mice were used in this study. In the 12 MO treated group and control groups –6 F and 3 M were used. In the 16 MO group; Control 9 F 6 M; 3 mg/kg 10 F 5 M; 10 mg/kg 9 F 6 M and 30 mg/kg 9 F 6 M.
Treatment
The mice were treated with either standard, non-irradiated rodent food or the same food but with the addition of ATH434 (provided by Alterity Therapeutics). The compound was incorporated into the food by Specialty Feeds (Glen Forrest, WA) at nominal doses of 0.025 g/kg food (equating to ∼3 mg/kg/day of ATH434 in the mice), 0.083 g/kg food (∼10 mg/kg/day of ATH434 in the mice), and 0.25 g/kg food (∼30 mg/kg/day of ATH434 in the mice). The mice and food were weighed weekly throughout the four months of the experiment. The group that was culled at 12 months of age were treated from 8 months of age; the group culled at 16 months of age were treated from 12 months of age. No mice in any of the treatment groups exhibited any adverse reaction to ATH434. One mouse in the control group was culled for human reasons.
Pole test of motor performance
The pole test was used to assess the speed of a complex motor skill and coordination. In the week before culling, the mice were placed (nose up) on top of a pole (wooden rod; 300 mm long and 5 mm in diameter; attached to a stable weighted base) and the time required for the mouse to rotate 180 degrees (nose down), and to return to the home cage (“Time Complete”) were recorded. The tests were performed on 2 different days (habituation day and test day). Each mouse attempted 5 trials and the fastest time was analyzed [18].
Transcardial perfusion
Following the treatments and the pole motor test was performed, the mice were anaesthetized with 80 mg/kg sodium pentobarbital (Lethabarb, Clifford Hallam, Australia) diluted in 0.9%saline. Mice were perfused and exsanguinated with 50 ml 0.1 M phosphate-buffered saline (PBS). The left-hand side of the brains were post-fixed in 4%paraformaldehyde (Sigma Aldrich, St. Louis, MO, USA) in PBS at 4°C overnight and then immersed in 30%sucrose in 0.1 M PBS at 4°C in preparation for histology. The right-hand side was micro-dissected and stored at –80°C for biochemical analysis and Inductively Coupled Plasma Mass Spectrometry (ICPMS).
Tissue preparation
Substantia nigra samples (containing compacta and reticulata) were homogenized (sonication; Branson, CT, USA) according to their wet weight at 1 : 5 dilution in Dulbecco’s calcium- and magnesium-free PBS (Invitrogen, CA, USA) with a protease in-hibitor (Roche, Basel, Switzerland) and phosphatase inhibitor: phosStop (Sigma-Aldrich). Protein concentrations were determined with a Pierce BCA Protein Assay kit (Thermo Fisher Scientific, MA, USA). One of the duplicate samples was lyophilized for ICPMS analysis, the other frozen for α-syn analysis.
Iron content analysis by inductively coupled plasma mass spectrometry
Samples were weighed, lyophilized, and digested overnight at room temperature in nitric acid (HNO3) (65%Suprapur, Merck). The samples were further digested by heating at 90°C for 20 min. An equal volume of hydrogen peroxide (H2O2) (30%Aristar, BDH) was added to each sample and left for ∼30 min and then heated for 15 min at 70°C. Samples were then further diluted with 1%HNO3. Metal levels were measured using an Agilent 7700 series ICPMS instrument under routine multi-element operating conditions using a helium reaction gas cell. Results are expressed as microgram of metal per gram of protein weight (μg/g or ng/μg).
Western blot
The tissues were homogenized according to their wet weight at 1 : 5 dilution in Dulbecco’s calcium- and magnesium-free PBS (Invitrogen, #10010049) with a protease inhibitor (Roche, #05056489001) and phosphatase inhibitor: phosStop (Sigma-Aldrich, 4906837001) using a sonicator (Branson, stepped micro-tip). Protein concentrations were determined with a Protein Assay kit (Pierce, #23225). The fractionation procedure commenced with tissue homogenates of a set concentration (125μg in 500μl PBS). These were spun using an ultracentrifuge (Beckman Coulter, California, USA) at 100,000×g for 30 min at 4°C [22]. The supernatant (S1) comprised the soluble fraction; 10μg of protein was loaded on the S1 gels. The remaining pellet was washed 3 times in a PBS buffer and resuspended in 5%SDS (Sigma-Aldrich). The resuspended pellets were homogenized again using sonication and centrifuged at 100,000×g, 30 min at 4°C. The resulting supernatant (S2) was stored and not used. The remaining pellet was washed with 5%SDS/PBS solution and resuspended in 10μl 8%SDS/8M Urea (Sigma-Aldrich) in PBS to solubilize the fraction not soluble in SDS. This solution, soluble fraction 3 (S3), was re-homogenized and incubated at room temperature for 2 h. 5μl of the S3 fractions were reserved for the S3 gels. 10μg of S1 and 5μl S3 fractions were loaded into 4–12%Bis-Tris gels (Invitrogen) with 1X NuPAGE™ MES SDS Running Buffer (Invitrogen). The gel was run at 120V for approximately 90 min and transferred onto PVDF membrane (Invitrogen) using a semi-dry system (Iblot2, Invitrogen).
The membrane for the S1 fraction was stained with 0.1%Ponceau S Solution (Sigma-Aldrich) for at least 5 min to measure total protein levels (using digitized images, LAS-4000 FujiFilm). The stain was removed using Tris-Buffered Saline (TBST pH 8.0) and three washes with MilliQ water. Membranes were then blocked with 5%skim milk in TBST (pH 8.0) for 1 h at room temperature. After a rinse with TBST (pH 8.0), the membranes were incubated with primary antibody (1 : 10,000 LB509 Abcam, Cambridge, UK) followed by rocking overnight at 4°C. The membranes were then rinsed 3 times using TBST and then incubated in secondary antibody (1 : 10,0000, Rabbit-anti-mouse-HRP; Dako, Glostrup, Denmark), rocking for 1 hour at room temperature. Secondary antibody was then washed off and the membrane incubated with chemiluminescence reagents (1 : 1 ratio, ECL, GE Healthcare, Chicago, IL, USA) for at least 1 minute and imaged and analyzed using a LAS-4000 FujiFilm imager and Multi Gauge Software (GE Healthcare).
Immunohistochemistry
Brains were sectioned with a Leica Biosystem Cryostat at –24°C. The left-hand hemisphere was sectioned at 30μm through the SN and Pons, the right-hand hemisphere was used for the biochemistry. The sections were mounted to gelatinated microscope slides (0.5%gelatin in dH20) (Grale Scientific cat# S21102A). Sections were stored at –80°C until staining was performed. The slides were removed from –80°C then incubated in a blocking solution (containing 10%normal goat serum in 0.1 M PBS pH 7.4 for 30 min). Slides were then incubated in primary antibody overnight at room temperature (GCI: α-syn: MJFR1, Sapphire bioscience, AB138501 at 1 : 1000). The slides were then washed with PBS three times and incubated in secondary antibody (Goat Anti-Rabbit IgG Antibody, biotin-SP conjugate) for 3 h at room temperature. Slides were then washed with PBS three times and incubated in avidin peroxidase for 1 h at room temperature. The slides were washed again and incubated in 0.05%nickel sulphate and 0.05%cobalt chloride for 20 min and further developed by adding 0.001%H2O2 for a maximum of 5 min. Slides were then washed, counterstained with 2%Neutral Red solution for 2 min, dehydrated with increasing concentrations of ethanol (50%, 70%, 90%, and 100%), followed by xylene and mounted with Ultramount No.4 mounting medium (Trajan Scientific, Cat #II 065C). Neutral red is a versatile stain that is used to distinguish neuronal and glial cell membranes, lysosomes, and nuclei.
Stereology of substantia nigra pars compacta
The total number of neurons in the SN was estimated using an optical fractionator sampling design as published previously [23–25] using StereoInvestigator version 11 software (MBF Bioscience, Williston, VT, USA). The regions of interest were traced, and counts were made at regular predetermined grid intervals of x:140μm and y:140μm with an unbiased counting frame of 45μm×45μm and a dissector height of 12μm. Systematic samples of the area occupied by the Neutral Red stained nuclei were made from a random starting point. The average thickness of every section counted was also measured. All stereological analyses were undertaken in a blinded manner, with the data decoded after all the samples had been processed.
Glial cell inclusions stereology
Stereological estimates of GCIs were performed using a fractionator sampling design with the StereoInvestigator version 11 Software (MBF Bioscience, USA). Grid intervals of x: 100μm and y: 100μm with a counting frame of 50×50μm were systemically and randomly positioned by the software using a 63× objective lens (NA 1.63). All stereological analyses were undertaken in a blinded manner, with the data decoded after all of the samples had been processed.
In vitro α-syn assay
To test if ATH434 affected the rate of α-syn disaggregation, in vitro experiments were performed. Purified and lyophilized recombinant human α-syn (Monash Protein Production Unit, Monash University, Australia) was reconstituted with Tris Buffer Saline (TBS, pH 7.4). Pooled aliquots were spun at 100,000×g for 30 min at 4°C to remove pre-formed aggregates/seeds. The supernatant containing the monomeric form was collected and used in the assay. The protein concentration was determined using the BCA method. Iron nitrate was weighed and dissolved in TBS solution. ATH434 was dissolved in 100%DMSO, then diluted to a stock solution using MilliQ water. To each tube, TBS, iron, ATH434/ vehicle and then α-syn was added in sequence with equal concentrations. The final concentration of α-syn, Fe, and ATH434 was 140.0μM. Triplicates of the samples were assayed in the presence of ThT in a Perkin-Elmer Enspire multi-mode plate reader. ThT fluorescence intensity was measured over time at wavelengths of 450 nm emission and 485nm excitation. The relative fluorescent units (RFU) values were normalized to TBS + ThT control wells and were plotted over time. The method has been previously published [11, 18].
Data analysis
The data were plotted as Standard Error of the Mean (SEM). Data were analyzed with either a one-way ANOVA followed by Dunnett’s and Tukey multiple comparisons test, a two-way ANOVA, or unpaired t-tests as appropriate. The software used was GraphPad Prism (GraphPad Software, La Jolla, CA, USA).
RESULTS
Neuroprotective effects of ATH434
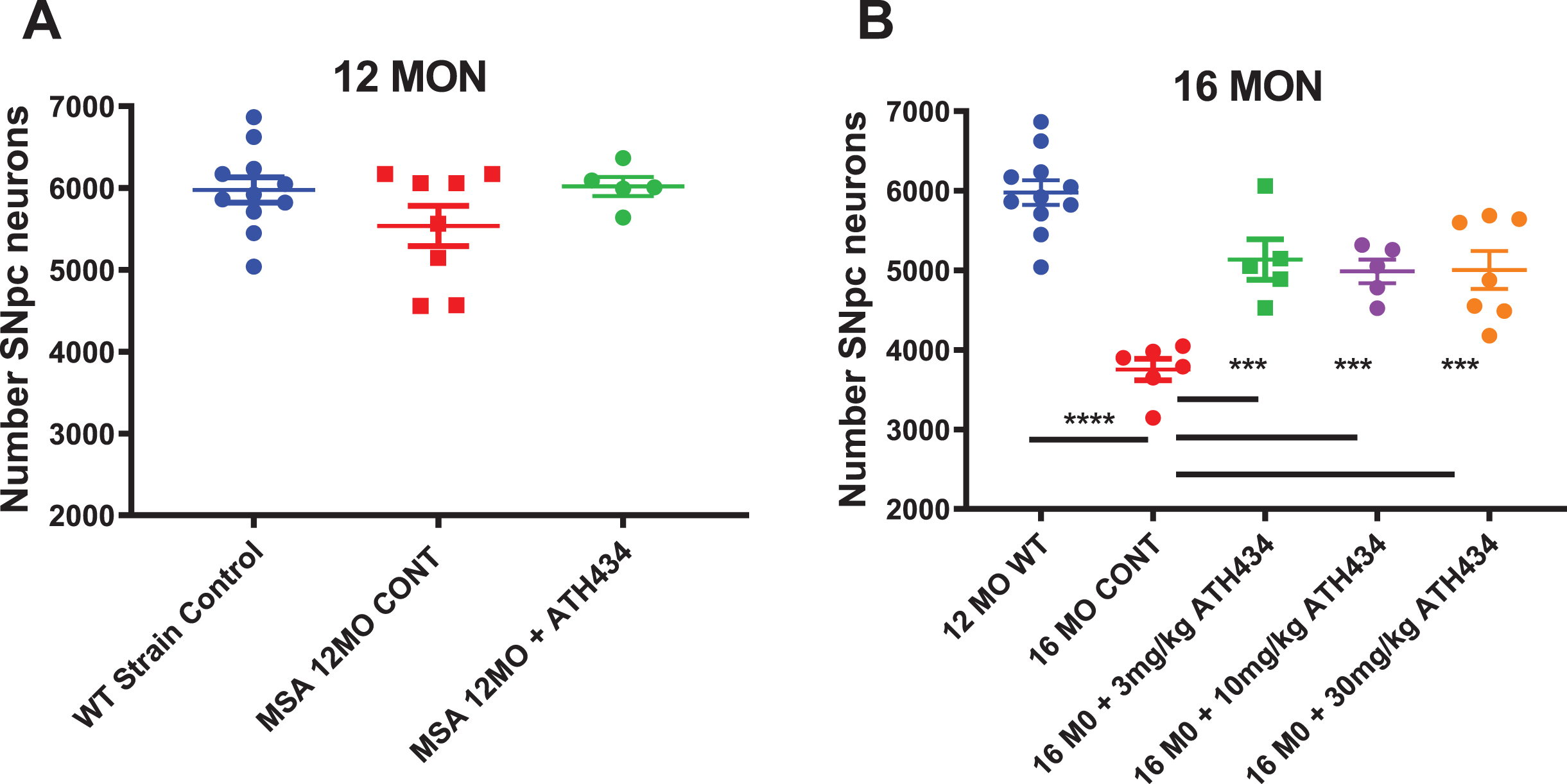
At 12 months of age the MSA mice did not show a significant decrease in SNpc neuron number compared to littermate controls, and treatment with ATH434 from 8 months of age did not alter nigral neuronal number (Fig. 1). As such, the rest of the studies focused on older mice. In contrast, the 16-month-old MSA mice had a 37%decrease in the number of nigral neurons compared to controls (thus providing the rationale for our selection of this timeframe as our primary analysis endpoint), and four months of treatment with ATH434 resulted in a significant protection against α-syn mediated nigral cell death (p < 0.001, One way ANOVA with Dunnett’s Post hoc test comparing to the 16-month untreated animals). Each of the doses of ATH434 trialed were equally effective suggesting a threshold effect below 3 mg/kg/day.
Fig. 1
Total Substantia Nigra pars compacta neuron counts in 12-month (Left) and 16-month (Right) PLP-α-syn mice after approximately 4 months of ATH434 or Control diet (Mean±SEM). A) No loss of neurons is observed in the 12-month group. B) At 16 months of age, there was a 37%loss in the number nigral neurons compared to the 12-month-old strain control. Treatment from the age of 12 months with ATH434 reduced the size of this loss to ∼16%at 16 months. One way ANOVA Dunnett’s Post hoc test comparing to the 16-month untreated animals ***p < 0.001, ****p < 0.0001. (12 MO WT vs. 16 MO CONT p≤0.0001; CONT vs. 3 mg/kg p = 0.0003; CONT vs. 10 mg/kg p = 0.0012; CONT vs. 30 mg/kg p = 0.0004)
Motor performance
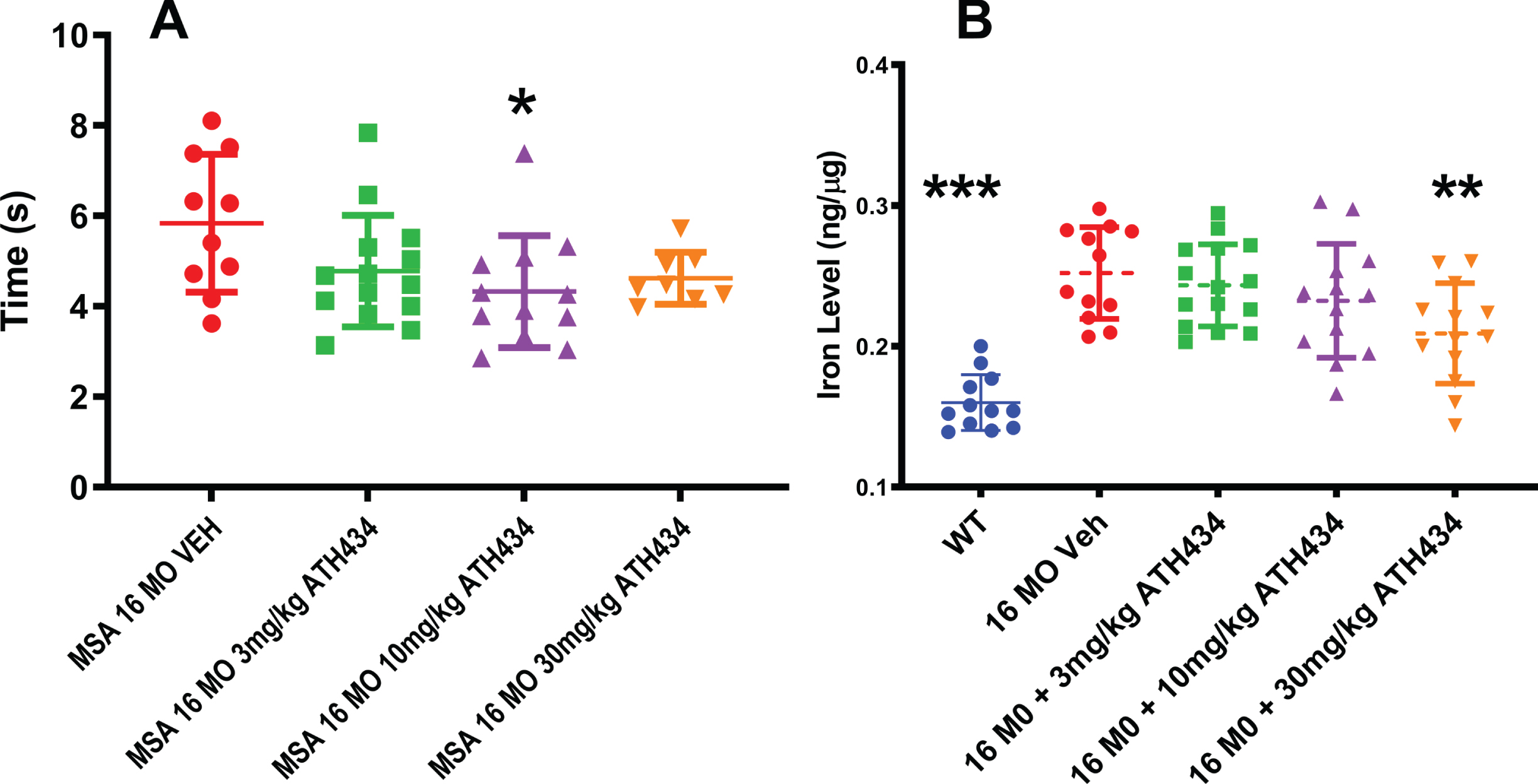
To investigate whether ATH434 altered nigral-mediated complex movement, mice were assessed for their performance on the pole test (Fig. 2A). The time to complete the pole test behavioral task was significantly improved (28%faster) in the MSA mice receiving the ATH434 food, as compared to those provided the control food (ANOVA, F = 3.01, p = 0.04). No dose response relationship was observed.
Fig. 2
A) Time to complete the pole test motor task was improved by treatment with ATH434 in the PLP-α-syn mice after approximately 4 months of ATH434 compared to those eating Control diet (Mean±SEM, Ordinary ANOVA, F = 3.01, p = 0.04). One way ANOVA Dunnett’s Post hoc test comparing to the 16-month untreated animals indicated that the 10 mg/kg reached significance *p = 0.02. B) Substantia nigra iron was elevated was elevated in the 16-month PLP-α-syn mice compared to wild type litter mate controls. Four months of treatment with ATH434 in various doses reduced the iron within the SN (Mean±SEM, Ordinary ANOVA, F = 16.03, p < 0.0001). One way ANOVA with Dunnett’s Post hoc test comparing to the 16-month untreated animals indicated that the 30 mg/kg reached significance **p = 0.006; WT vs. VEH ***p≤0.0001
Substantia nigra iron levels
Utilizing inductively coupled plasma mass spectrometry, we demonstrated that iron levels were elevated by 57%in the SN of 16-month-old MSA mice, as compared to littermate wildtype controls. It should be noted that the microdissection included both the substantia nigra reticulata and pars compacta within the dissected tissue block. A primary question in this study was whether this iron elevation was tractable to therapy. As we have previously shown that ATH434 has a dissociation constant (Kd) for Fe(III) and Cu(II) of ∼10–10 M, and affinities for Fe(II) and Zn of ∼10–5 M and ∼10–7 M respectively [18], this is a likely candidate to target iron in the MSA mice. Four months of treatment with ATH434 in various doses reduced the iron within the SN (ANOVA, F = 16.03, p < 0.0001). A Dunnett’s post-hoc test revealed that only the 30 mg/kg treatment group reached significance (p = 0.006, –20%) (Fig. 2B).
Western blot data for α-syn
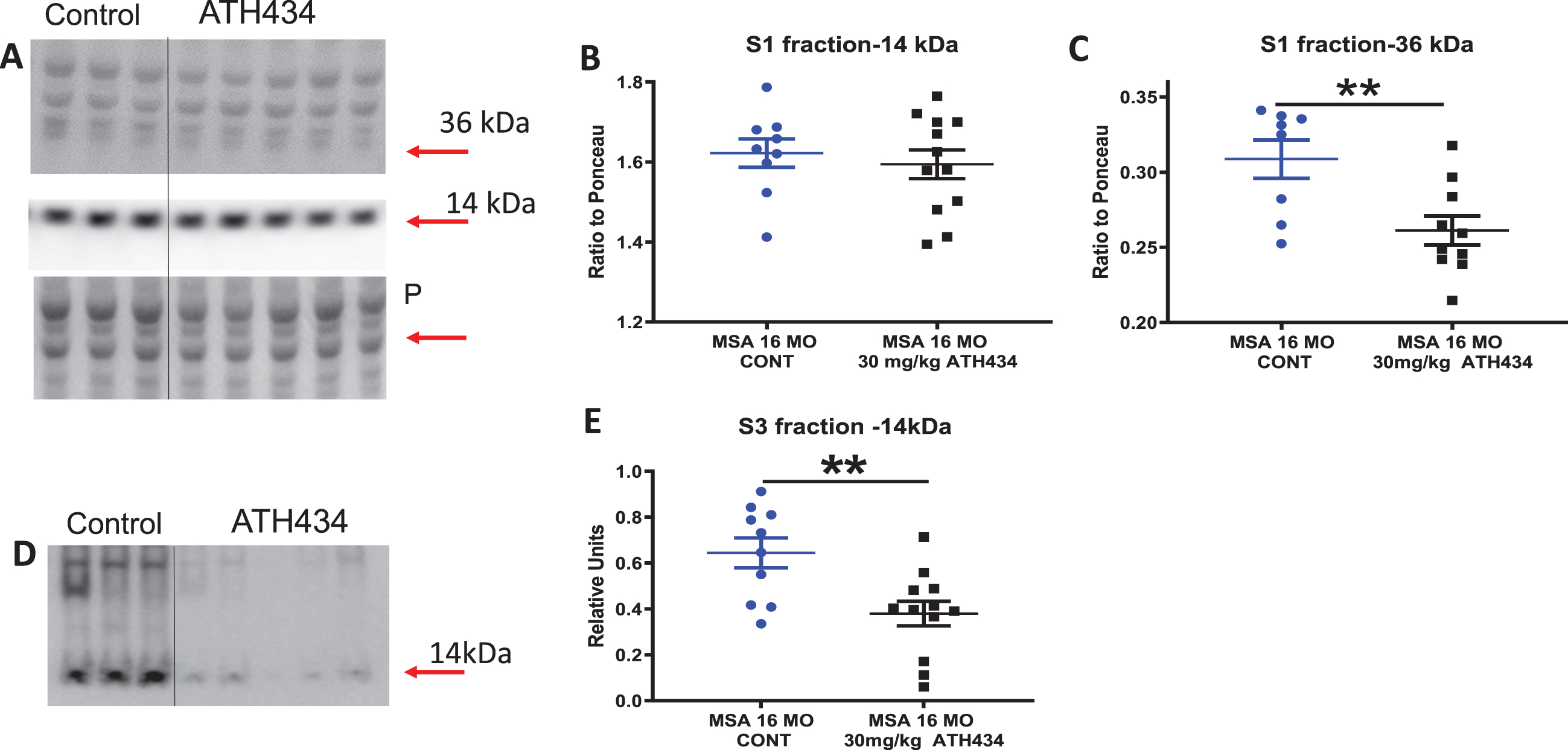
ATH434 treatment did not alter the levels of soluble monomeric α-syn (14 kDa) but significantly decreased both oligomeric (66 kDa, p < 0.01 by 18%, Fig. 3A–C) and insoluble (urea extracted, p < 0.01 by 69%) α- syn in the SN (Fig. 3D, E). Long term treatment of the mice with the compound appeared to either prevent the formation of higher order species or increased α- syn clearance from the nigra. It should be noted that the microdissection included both the substantia nigra reticulata and pars compacta within the dissected tissue block.
Fig. 3
A) Western blot data for soluble α-syn levels in the Substantia Nigra (SN) in 16-month PLP-α-syn mice after 4 months of ATH434 or Control diet, Soluble (S1) fraction of SN transferred to western blots and stained for α-syn with LB509 antibody. Arrows indicate the α-syn monomer and oligomeric bands. Loading control ponceau stain (P) is show under the labels. B) Plots of density (relative to ponceau) of 14 kDA. C) Plots of density of 36 kDA bands (relative to ponceau), Mean±SEM, T test **p = 0.0081. D) Western blot data for aggregated (urea soluble) α-syn levels in the Substantia Nigra in 16-month PLP-α-syn mice after approximately 4 months of ATH434 or Control diet. Urea soluble (S3) fraction of SN transferred to western blots and stained for α-syn with LB509 antibody. The arrow indicates the α-syn urea-soluble band. Laddering of higher order oligomeric bands can be observed in the control 16-month PLP-α-syn mice but are greatly reduced in the animals that have been treated with ATH434. Mean±SEM. T-test **p = 0.0049. The S3 plots were constructed using relative fluorescence units.
Glial cell inclusions in the substantia nigra pars compacta
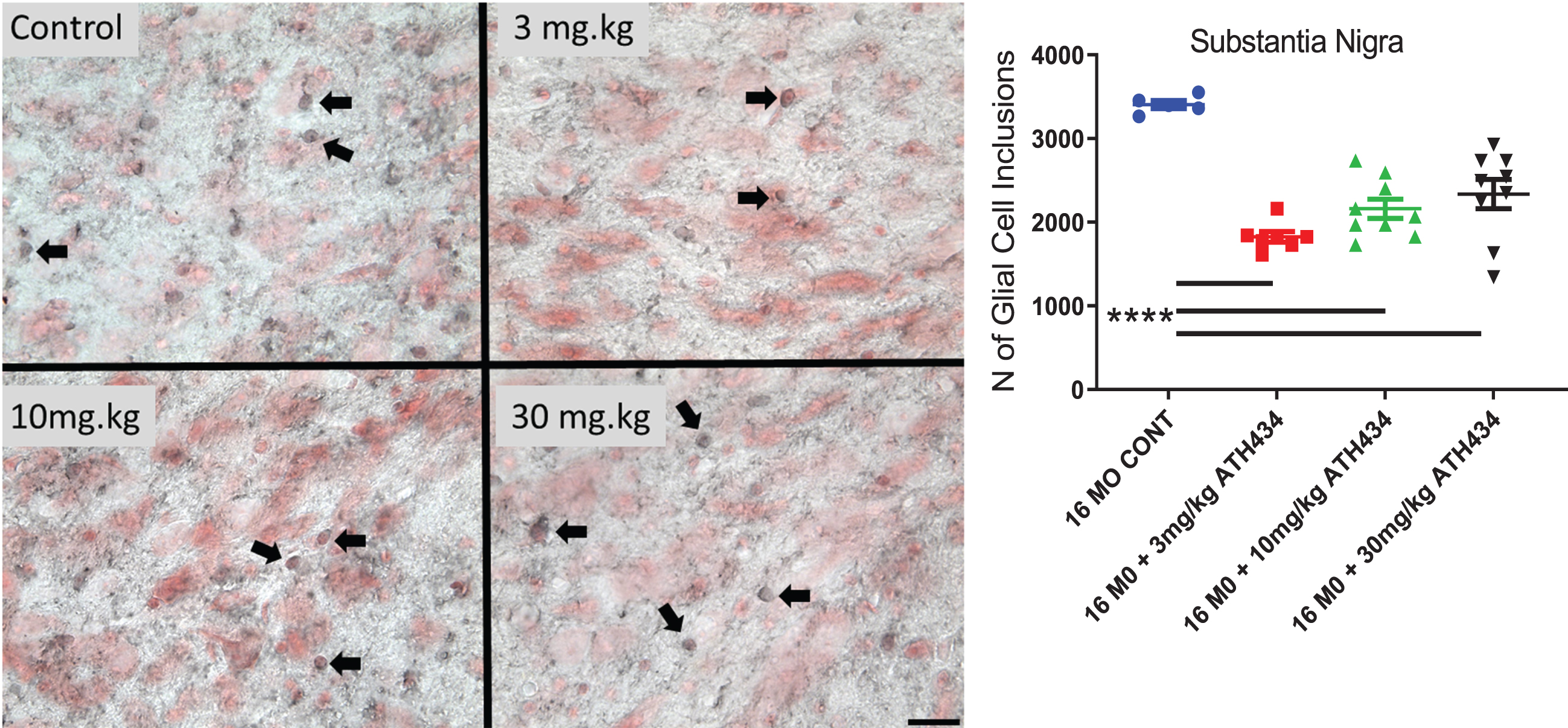
Four months of treatment with ATH434 significantly reduced the number of GCI within the SN (ANOVA, F = 19.98, p < 0.0001). A Dunnett’s post-hoc test demonstrated that each of the treatments significantly reduced the number of GCIs (3 mg/kg (46%p < 0.0001), 10 mg/kg (37%, p < 0.0001), 30 mg/kg (31%, p < 0.0001); Fig. 4).
Fig. 4
The number of Glial Cell Inclusions (dark staining, Arrows) in the Substantia Nigra pars compacta (neurons red staining) in 16-month PLP-α-syn mice. Four months of treatment with ATH434 in various doses reduced the number of GCI within the SN (Mean±SEM, Ordinary ANOVA, F = 19.98, p < 0.0001). One way ANOVA with Dunnett’s Post hoc test comparing to the 16-month untreated animals indicated that 3 mg/kg (46%decrease), 10 mg/kg (37%decrease), 30 mg/kg (31 %decrease) significantly reduced the number of GCI p < 0.0001. (16 MO CONT vs. 3 mg/kg p≤0.0001; 16 MO CONT vs. 10 mg/kg p≤0.0001; 16 MO CONT vs. 30 mg/kg p≤0.0001) Panels show photomicrographs showing GCIs within the SN (scalebar = 25 microns).
In vitro α-syn assay
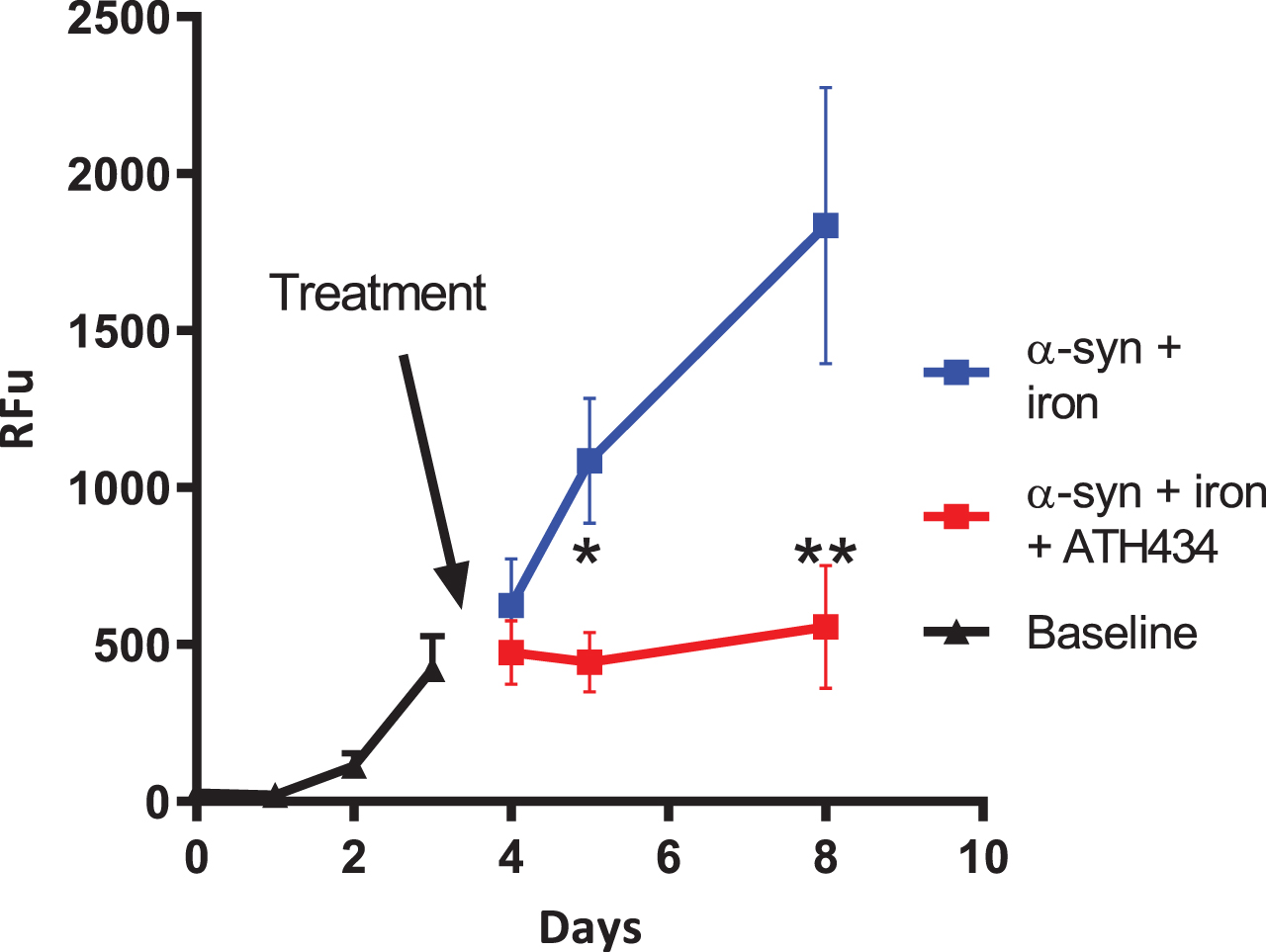
In an in vitro assay utilizing iron-mediated aggregation of α-syn, we previously demonstrated that the addition of ATH434 slowed the rate of aggregation [18]. An aim for some drug makers is to find compounds that break apart or disaggregate α-syn aggregates. In this study, we used the ThT fluorescence assay to assess if ATH434 could disaggregate already aggregated α-syn. After the day 3 measurements the samples were divided into two groups, control (α-syn + iron + water as vehicle control) or ATH434 (α-syn + iron + ATH434). Visual inspection of Fig. 5 suggests that the average rate of aggregation in the control group is the similar to baseline, whereas the addition of ATH434 the mixture did not allow new aggregates to form and importantly did not disaggregate the already formed fibrils. Analysis of data after treatments were introduced used a two-way ANOVA; samples were paired but the variance component for the pairing was zero. Pair wise multiple comparisons of treatment were performed for each day, as an interaction of treatment and day was detected. Pairwise analysis shows that at day 5 the differences reached a p = 0.013 (*) and at day 8 p < 0.001 (**) (Fig. 5).
Fig. 5
Inhibition of α-synuclein (α-syn) aggregation. Recombinant α-syn (140.0μM) was incubated in the presence of equimolar concentrations of iron nitrate (Fe = Fe(NO3)3) and or ATH434. The ATH434 was delivered at day 3 after the α-syn aggregation had commenced and prevented further iron-mediated α-syn aggregation. The presence of the ATH434 did not dis-aggregate the α-syn as the signal does not decrease. Aggregation was measured by using ThT fluorescence binding (RFU = relative fluorescent units) p = 0.013 (*) and at day 8 p < 0.001 (**)
DISCUSSION
Multiple System Atrophy is a rare atypical parkinsonian disorder in which affected individuals deteriorate quickly [26–28]. MSA is pathologically characterized both by the accumulation of α-syn within the oligodendrocytes and by an elevation in brain iron levels. As there is a known link between α-syn and iron, with oxidative stress and free iron shown to promote the aggregation of α-syn [11, 29] and to increase the toxicity to cells [30, 31], then there is a strong rationale for the hypothesis that iron-targeting compounds will be therapeutically efficacious in this disease.
To interrogate this hypothesis, we utilized a transgenic mouse model that expresses human wild-type α-syn within the oligodendrocytes, driven by the proteolipid protein (PLP) promoter. These MSA transgenic mice were originally imported from the laboratory of Dr. Nadia Stefanova, after which they were backcrossed onto a C57/BL6 background (Animal Resources Centre, Western Australia) for more than 10 generations. The phenotype of the mice appears to be very similar to the published data [19–21, 32], although as discussed later, there was some minor drift in the timing of the phenotype. To determine if iron was a tractable therapeutic target in these mice, we utilized an iron-targeting compound, ATH434, that we have previously shown to be efficacious in both PD toxin models and in the transgenic hA53T α-synuclein model of PD [18]. Further, this compound was well-tolerated in a first-in-human oral dosing study in healthy and older volunteers, and demonstrated a favorable, dose-dependent pharmacokinetic profile [17].
Mouse colony
Although originating from the same source, the minor drift in the timing of the phenotype observed in the Australian colony may be attributed to different genetic background of the mice or subtle variations in the experimental methods. Heras-Gavin et al. observed loss of TH immunoreactivity in six-month-old animals, consistent with data from Refollo et al. who also found behavioral differences at this time [20, 32]. The Australian colony showed a small but insignificant decrease in total SNpc neuron at 12 months of age, that became significant at 16 months. This finding of temporal differences in the results is not surprising as differences in colonies is known to occur [33]. Importantly GCIs are present in both colonies from an early age.
Alpha synuclein in MSA
It is believed that the pathogenesis of MSA results from spread of misfolded α-syn from neurons to oligodendroglia [34, 35]. The α-syn then accumulates in the oligodendroglia to form toxic fibrils and GCIs. How this accumulation within the glia subsequently causes death of neurons is not clear. Interestingly, it has been shown that the α-syn aggregates that form in the oligodendroglia micro-environment are more toxic and structurally distinct from the aggregates found within the neurons from individuals with PD [3, 36].
In the PLP promoter α-syn transgenic mice, the production of the α-syn is not under the natural promoter that has the iron response element within mRNA. In humans and wild type rodents the iron response element is within mRNA of α-syn [15, 16]. In this study we found that the amount of monomeric α-syn was not altered by ATH434, which is consistent with the α-syn being controlled by the PLP promoter. ATH434 reduced the amount of soluble aggregates and insoluble (urea-soluble) α-syn aggregates that are formed in the oligodendroglial microenvironment. The in vitro study indicated that ATH434 was not able to dis-aggregate α-syn but was able to prevent further aggregation. Therefore, it is deduced that the effects observed in vivo may have been achieved by increasing clearance of α-syn [37, 38] and/or slowing the formation of iron mediated α-syn aggregation. Importantly, this treatment culminated in a reduction in the number of glial cell inclusions which are constituted from aggregated α-syn. Given the data from this manuscript, ATH434 could act on several different pathways to prevent or slow the pathological process. These include potential effects on: 1) levels of synuclein protein, by removing access the iron-response element of the mRNA of α-syn; 2) preventing the formation of extracellular misfolded α-syn; 3) enhancing clearance of the α-syn burden; 4) prevent the formation intracellular α-syn aggregates.
Iron in MSA
Iron accumulates in a specific pattern in MSA[8]. Whilst studies have found that iron deposits in the putamen precede the clinical presentation of MSA by 2 years [39], most find that iron accumulation occurs during atrophy, and this is thought to be a secondary event of the neurodegeneration process [6]. Whilst the precise cellular and subcellular localization of this elevated iron has not been fully established, synchrotron microscopy technology is becoming available to address this question, although it does not yet appear to have been applied to PD or MSA [40].
We have been investigating the normal distribution of iron and the consequences of alterations in the metabolism and homeostasis of iron in parkinsonism [41–45]. We believed that this elevation in iron results in an increase in the weakly bound or labile iron pool [46, 47]. We have shown that this a tractable target and that a reduction in iron is associated with a reduction of damaging radicals which have a number of pathological consequences[18]. In the current MSA study we found that the elevation of iron caused by oligodendroglial α-syn expression could be reduced by the treatment with ATH434 in an apparent dose dependent way.
CONCLUSION
In 16-month-old MSA animals with a degenerating phenotype the compound ATH434; reduced oligomeric, aggregated and urea soluble α-synuclein, reduced the number of GCIs and preserved SN neurons. In vitro experiments suggest that ATH434 can prevent the formation of toxic oligomeric species of α-synuclein. The United Stated Food and Drug Administration and European Commission has granted Orphan drug status for ATH434 (formerly PBT434) as there are no approved treatments for MSA. Given this status, is concluded that ATH434 is a small molecule drug candidate with potential for treating MSA.
ACKNOWLEDGMENTS
The work was supported by funds from the Australian National Health and Medical Research Council, The Michael J. Fox Foundation for Parkinson’s Disease Research, Parkinsons UK and Alterity Ltd. The Florey Institute of Neuroscience and Mental Health acknowledge the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant. Joseph for making this project important and real. The independent statistical advice provided by Assoc Professor Sue Finch Deputy Director, Statistical Consulting Centre Melbourne Statistical Consulting Platform, Faculty of Science, The University of Melbourne.
This work is the culmination of many years of dedicated efforts and talents of: Irene Volitakis, Amelia Sedjahtera, Lisa Bray and Lydia Gunawan and Kali Perrones We thank Dr Margaret Bradbury for helpful suggestions to the manuscript.
To Suzie and Joannah Joseph for making this project important and real.
CONFLICT OF INTEREST
DIF, RAC, KJB, and PAA consulted for Alterity∖Prana.
REFERENCES
[1] | Erkkinen MG , Kim MO , Geschwind MD ((2018) ) Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb Perspect Biol 10: , a033118. |
[2] | Goedert M , Jakes R , Spillantini MG ((2017) ) The synucleinopathies: Twenty years on. J Parkinsons Dis 7: , S51–S69. |
[3] | Van der Perren A , Gelders G , Fenyi A , Bousset L , Brito F , Peelaerts W , Van den Haute C , Gentleman S , Melki R , Baekelandt V ((2020) ) The structural differences between patient-derived alpha-synuclein strains dictate characteristics of Parkinson’s disease, multiple system atrophy and dementia with Lewy bodies. Acta Neuropathol 139: , 977–1000. |
[4] | Kraft E , Schwarz J , Trenkwalder C , Vogl T , Pfluger T , Oertel WH ((1999) ) The combination of hypointense and hyperintense signal changes on T2-weighted magnetic resonance imaging sequences: A specific marker of multiple system atrophy? Arch Neurol 56: , 225–228. |
[5] | Dexter DT , Carayon A , Javoy-Agid F , Agid Y , Wells FR , Daniel SE , Lees AJ , Jenner P , Marsden CD ((1991) ) Alterations in the levels of iron, ferritin and other trace metals in Parkinson’s disease and other neurodegenerative diseases affecting the basal ganglia. Brain 114 (Pt 4): , 1953–1975. |
[6] | Kaindlstorfer C , Jellinger KA , Eschlbock S , Stefanova N , Weiss G , Wenning GK ((2018) ) The relevance of iron in the pathogenesis of multiple system atrophy: A viewpoint. J Alzheimers Dis 61: , 1253–1273. |
[7] | Mazzucchi S , Frosini D , Costagli M , Del Prete E , Donatelli G , Cecchi P , Migaleddu G , Bonuccelli U , Ceravolo R , Cosottini M ((2019) ) Quantitative susceptibility mapping in atypical Parkinsonisms. Neuroimage Clin 24: , 101999. |
[8] | Seki M , Seppi K , Mueller C , Potrusil T , Goebel G , Reiter E , Nocker M , Kremser C , Wildauer M , Schocke M , Gizewski ER , Wenning GK , Poewe W , Scherfler C ((2019) ) Diagnostic potential of multimodal MRI markers in atypical parkinsonian disorders. J Parkinsons Dis 9: , 681–691. |
[9] | Tsang AH , Chung KK ((2009) ) Oxidative and nitrosative stress in Parkinson’s disease. Biochim Biophys Acta 1792: , 643–650. |
[10] | Dias V , Junn E , Mouradian MM ((2013) ) The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis 3: , 461–491. |
[11] | Cappai R , Leck SL , Tew DJ , Williamson NA , Smith DP , Galatis D , Sharples RA , Curtain CC , Ali FE , Cherny RA , Culvenor JG , Bottomley SP , Masters CL , Barnham KJ , Hill AF ((2005) ) Dopamine promotes alpha-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J 19: , 1377–1379. |
[12] | Castellani RJ , Siedlak SL , Perry G , Smith MA ((2000) ) Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta Neuropathol 100: , 111–114. |
[13] | Febbraro F , Giorgi M , Caldarola S , Loreni F , Romero-Ramos M ((2012) ) alpha-Synuclein expression is modulated at the translational level by iron. Neuroreport 23: , 576–580. |
[14] | McFarland NR , Fan Z , Xu K , Schwarzschild MA , Feany MB , Hyman BT , McLean PJ ((2009) ) α-Synuclein S129 phosphorylation mutants do not alter nigrostriatal toxicity in a rat model of Parkinson disease. J Neuropathol Exp Neurol 68: , 515–524. |
[15] | Rogers JT , Mikkilineni S , Cantuti-Castelvetri I , Smith DH , Huang X , Bandyopadhyay S , Cahill CM , Maccecchini ML , Lahiri DK , Greig NH ((2011) ) The alpha-synuclein 5’untranslated region targeted translation blockers: Anti-alpha synuclein efficacy of cardiac glycosides and Posiphen. J Neural Transm 118: , 493–507. |
[16] | Friedlich AL , Tanzi RE , Rogers JT ((2007) ) The 5’-untranslated region of Parkinson’s disease alpha-synuclein messengerRNA contains a predicted iron responsive element. Mol Psychiatry 12: , 222–223. |
[17] | Stamler D , Bradbury M , Wong C , Offman E ((2019) ) A first in human study of PBT434, a novel small molecule inhibitor of α-synuclein aggregation (S4.001). Neurology 92: , S4.001. |
[18] | Finkelstein DI , Billings JL , Adlard PA , Ayton S , Sedjahtera A , Masters CL , Wilkins S , Shackleford DM , Charman SA , Bal W , Zawisza IA , Kurowska E , Gundlach AL , Ma S , Bush AI , Hare DJ , Doble PA , Crawford S , Gautier EC , Parsons J , Huggins P , Barnham KJ , Cherny RA ((2017) ) The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol Commun 5: , 53. |
[19] | Kahle PJ , Neumann M , Ozmen L , Muller V , Jacobsen H , Spooren W , Fuss B , Mallon B , Macklin WB , Fujiwara H , Hasegawa M , Iwatsubo T , Kretzschmar HA , Haass C ((2002) ) Hyperphosphorylation and insolubility of alpha-synuclein in transgenic mouse oligodendrocytes. EMBO Rep 3: , 583–588. |
[20] | Refolo V , Bez F , Polissidis A , Kuzdas-Wood D , Sturm E , Kamaratou M , Poewe W , Stefanis L , Angela Cenci M , Romero-Ramos M , Wenning GK , Stefanova N ((2018) ) Progressive striatonigral degeneration in a transgenic mouse model of multiple system atrophy: Translational implications for interventional therapies. Acta Neuropathol Commun 6: , 2. |
[21] | Boudes M , Uvin P , Pinto S , Voets T , Fowler CJ , Wenning GK , De Ridder D , Stefanova N ((2013) ) Bladder dysfunction in a transgenic mouse model of multiple system atrophy. Mov Disord 28: , 347–355. |
[22] | Zhou J , Broe M , Huang Y , Anderson JP , Gai WP , Milward EA , Porritt M , Howells D , Hughes AJ , Wang X , Halliday GM ((2011) ) Changes in the solubility and phosphorylation of alpha-synuclein over the course of Parkinson’s disease. Acta Neuropathol 121: , 695–704. |
[23] | West MJ , Gundersen HJ ((1990) ) Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol 296: , 1–22. |
[24] | Stanic D , Finkelstein DI , Bourke DW , Drago J , Horne MK ((2003) ) Timecourse of striatal re-innervation following lesions of dopaminergic SNpc neurons of the rat. Eur J Neurosci 18: , 1175–1188. |
[25] | Finkelstein DI , Stanic D , Parish CL , Tomas D , Dickson K , Horne MK ((2000) ) Axonal sprouting following lesions of the rat substantia nigra. Neuroscience 97: , 99–112. |
[26] | Lieto M , Roca A , Bruzzese D , Antenora A , Alfieri G , Sacca F , Bellofatto M , Bilo L , Barbato S , De Michele G , Filla A ((2019) ) Longitudinal study of a cohort of MSA-C patients in South Italy: Survival and clinical features. Neurol Sci 40: , 2105–2109. |
[27] | Foubert-Samier A , Pavy-Le Traon A , Guillet F , Le-Goff M , Helmer C , Tison F , Rascol O , Proust-Lima C , Meissner WG ((2020) ) Disease progression and prognostic factors in multiple system atrophy: A prospective cohort study. Neurobiol Dis 139: , 104813. |
[28] | Bjornsdottir A , Gudmundsson G , Blondal H , Olafsson E ((2013) ) Incidence and prevalence of multiple system atrophy: A nationwide study in Iceland. J Neurol Neurosurg Psychiatry 84: , 136–140. |
[29] | Deas E , Cremades N , Angelova PR , Ludtmann MH , Yao Z , Chen S , Horrocks MH , Banushi B , Little D , Devine MJ , Gissen P , Klenerman D , Dobson CM , Wood NW , Gandhi S , Abramov AY ((2016) ) Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson’s disease. Antioxid Redox Signal 24: , 376–391. |
[30] | Ostrerova-Golts N , Petrucelli L , Hardy J , Lee JM , Farer M , Wolozin B ((2000) ) The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci 20: , 6048–6054. |
[31] | Angelova PR , Choi ML , Berezhnov AV , Horrocks MH , Hughes CD , De S , Rodrigues M , Yapom R , Little D , Dolt KS , Kunath T , Devine MJ , Gissen P , Shchepinov MS , Sylantyev S , Pavlov EV , Klenerman D , Abramov AY , Gandhi S ((2020) ) Alpha synuclein aggregation drives ferroptosis: An interplay of iron, calcium and lipid peroxidation. Cell Death Differ 27: , 2781–2796. |
[32] | Heras-Garvin A , Weckbecker D , Ryazanov S , Leonov A , Griesinger C , Giese A , Wenning GK , Stefanova N ((2019) ) Anle138b modulates alpha-synuclein oligomerization and prevents motor decline and neurodegeneration in a mouse model of multiple system atrophy. Mov Disord 34: , 255–263. |
[33] | Lei P , Ayton S , Moon S , Zhang Q , Volitakis I , Finkelstein DI , Bush AI ((2014) ) Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol Neurodegener 9: , 29. |
[34] | Jellinger KA ((2018) ) Multiple system atrophy: An oligodendroglioneural synucleinopathy. J Alzheimers Dis 62: , 1141–1179. |
[35] | Prusiner SB , Woerman AL , Mordes DA , Watts JC , Rampersaud R , Berry DB , Patel S , Oehler A , Lowe JK , Kravitz SN , Geschwind DH , Glidden DV , Halliday GM , Middleton LT , Gentleman SM , Grinberg LT , Giles K ((2015) ) Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A 112: , E5308–5317. |
[36] | Peelaerts W , Bousset L , Van der Perren A , Moskalyuk A , Pulizzi R , Giugliano M , Van den Haute C , Melki R , Baekelandt V ((2015) ) alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522: , 340–344. |
[37] | Meredith GE , Totterdell S , Petroske E , Santa Cruz K , Callison RC Jr. , Lau YS ((2002) ) Lysosomal malfunction accompanies alpha-synuclein aggregation in a progressive mouse model of Parkinson’s disease. Brain Res 956: , 156–165. |
[38] | Ebrahimi-Fakhari D , McLean PJ , Unni VK ((2012) ) Alpha-synuclein’s degradation in vivo: Opening a new (cranial) window on the roles of degradation pathways in Parkinson disease. Autophagy 8: , 281–283. |
[39] | Kikuchi Y , Shibata M , Hirayanagi K , Nagashima K , Mihara B , Ikeda Y ((2018) ) Putaminal iron deposition precedes MSA-P onset by 2 years. Neurology 90: , 1071–1072. |
[40] | Brooks J , Everett J , Lermyte F , Tjendana Tjhin V , Sadler PJ , Telling N , Collingwood JF ((2020) ) Analysis of neuronal iron deposits in Parkinson’s disease brain tissue by synchrotron x-ray spectromicroscopy. J Trace Elem Med Biol 62: , 126555. |
[41] | Ayton S , Lei P , Duce JA , Wong BX , Sedjahtera A , Adlard PA , Bush AI , Finkelstein DI ((2013) ) Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease. Ann Neurol 73: , 554–559. |
[42] | Ayton S , Lei P , Hare DJ , Duce JA , George JL , Adlard PA , McLean C , Rogers JT , Cherny RA , Finkelstein DI , Bush AI ((2015) ) Parkinson’s disease iron deposition caused by nitric oxide-induced loss of beta-amyloid precursor protein. J Neurosci 35: , 3591–3597. |
[43] | Lei P , Ayton S , Finkelstein DI , Spoerri L , Ciccotosto GD , Wright DK , Wong BX , Adlard PA , Cherny RA , Lam LQ , Roberts BR , Volitakis I , Egan GF , McLean CA , Cappai R , Duce JA , Bush AI ((2012) ) Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med 18: , 291–295. |
[44] | Ayton S , Lei P , McLean C , Bush AI , Finkelstein DI ((2016) ) Transferrin protects against Parkinsonian neurotoxicity and is deficient in Parkinson’s substantia nigra. Signal Transduct Target Ther 1: , 16015. |
[45] | McAllum EJ , Hare DJ , Volitakis I , McLean CA , Bush AI , Finkelstein DI , Roberts BR ((2020) ) Regional iron distribution and soluble ferroprotein profiles in the healthy human brain. Prog Neurobiol 186: , 101744. |
[46] | Barnham KJ , Bush AI ((2008) ) Metals in Alzheimer’s and Parkinson’s diseases. Curr Opin Chem Biol 12: , 222–228. |
[47] | Barnham KJ , Bush AI ((2014) ) Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem Soc Rev 43: , 6727–6749. |

