
A Practical Approach to Early-Onset Parkinsonism
Abstract
Early-onset parkinsonism (EO parkinsonism), defined as subjects with disease onset before the age of 40 or 50 years, can be the main clinical presentation of a variety of conditions that are important to differentiate. Although rarer than classical late-onset Parkinson’s disease (PD) and not infrequently overlapping with forms of juvenile onset PD, a correct diagnosis of the specific cause of EO parkinsonism is critical for offering appropriate counseling to patients, for family and work planning, and to select the most appropriate symptomatic or etiopathogenic treatments. Clinical features, radiological and laboratory findings are crucial for guiding the differential diagnosis. Here we summarize the most important conditions associated with primary and secondary EO parkinsonism. We also proposed a practical approach based on the current literature and expert opinion to help movement disorders specialists and neurologists navigate this complex and challenging landscape.
INTRODUCTION
The term “early-onset Parkinson’s disease” (EOPD, or young-onset PD - YOPD) refers to cases of PD with onset between the age of 21 and 40 years, as reported by Quinn et al. in their seminal paper from 1987, or between 21 and 50 years, according to other authors [1–4]. Compared with idiopathic cases of PD (iPD), patients with EOPD usually present a slower progression of the motor symptoms, a prevalence of bradykinesia over tremor, focal dystonia at onset or during off-state, satisfactory response even to low doses of levodopa, earlier motor complications (such as motor fluctuations and dyskinesias), a lower incidence of cognitive impairment and non-motor symptoms, while anxiety and depression are frequent [1, 5–16]. A positive family history can be often identified in these patients, suggesting an important role of genetics in the pathogenesis of several of these cases [15, 16].
Based on retrospective observational studies, classical EOPD accounts for about 3–7% of all cases of PD in the Western world and up to 10–14% in Japan [1, 3, 17–19], with an incidence between 0.29 and 3.3 per 100,000 persons-years in the current literature [8, 17, 20, 21].
In patients with EO parkinsonism, parkinsonian features can present as part of more complicated syndromes, with associated additional neurological manifestations (dystonia, pyramidal signs, cognitive impairment), psychiatric manifestations, systemic symptoms, alterations on brain imaging, and age of onset between 21 and 40 or 50 years.
Although rare, cases of EOPD or EO parkinsonism demand prompt diagnosis, as some forms can respond to treatments (i.e., PRKN-associated PD) and early interventions can prevent severe complications (i.e., Wilson’s disease or ataxia telangiectasia). Primary forms of EO parkinsonism are more frequent than secondary cases due, among the others, to drug exposures or vascular accidents [22]. A significant proportion of primary forms of EO parkinsonism are due to genetic causes. Given the impressive advancements in gene-based therapeutic approaches, some patients may qualify for these treatments in the near future [23].
Because of the broad range of the differential diagnosis, significant diagnostic delays are frequent with important implications for genetic counseling for these patients and their families.
In this review, we summarized the most important genetic and systemic causes of EOPD and EO parkinsonism (Table 1, Fig. 1). We also provide a practical approach for the clinical evaluation, examination, and genetic counseling of these patients to help guide the diagnostic process and the referral to appropriate available or future treatments.
Table 1
Demographic and radiological features of the spectrum of causes of EO parkinsonism
| Condition | Age of Onset | Heritability | Imaging | Response to L-dopa | Characteristic features |
| Primary Early Onset Parkinsonism | |||||
| Monogenic PD | |||||
| PARK-PRKN | Childhood to adulthood (mean 3rd-4th decade, median age 31) | AR | Brain MRI: normal | + | Dystonia (predominantly of the lower limbs, can be present at onset), symmetrical involvement, pyramidal signs, and psychiatric manifestations, excellent and sustained response to L-dopa with early motor fluctuations, sleep benefit |
| PARK-PINK1 | Childhood to adulthood (mean 32 years) | AR | Brain MRI: normal | + | Dystonia and psychiatric symptoms, excellent and sustained response to L-dopa |
| PARK-DJ1 | Adolescence to adulthood (3rd-4th decade, median age 27) | AR | Brain MRI: normal | + | Dystonia and psychiatric symptoms |
| PARK-ATP13A2 | 2nd-6th decade (mean 24) | AR | Diffuse atrophy; SWI, GRE, and T2* hypointensity of basal ganglia | –/+ | Vertical supranuclear gaze palsy, slow vertical saccades, pyramidal signs, facial-faucial-finger myoclonus, cognitive decline, optic atrophy |
| PARK-PLA2G6 | Childhood to adulthood (most common in the 3rd-4th decade) | AR | Cerebral and/or cerebellar atrophy; SWI, GRE, and T2* hypointensity of basal ganglia; claval hypertrophy in childhood onset | –/+ | Dystonia, rapid cognitive decline, psychiatric symptoms, pyramidal signs, bulbar involvement, limb ataxia, optic atrophy |
| PARK-FBXO7 | 2nd-6th decade (median age 17) | AR | Mild diffuse atrophy or normal brain MRI | + | Pyramidal signs, lower limb involvement, neuropsychiatric symptoms |
| RAB39B-related parkinsonism | 4th-7th decade (mean age 51) | XR | CT-hyperdensity and T2 hypointensity of the basal ganglia can be seen and correspond to calcifications | –/+ | Intellectual disability |
| PTRHD1-related parkinsonism | 3rd-4th decade | AR | Brain MRI: normal | + | Intellectual disability, pyramidal signs, psychiatric manifestations |
| PARK-SYNJ1 | 2nd-3rd decade (median age 21) | AR | Diffuse atrophy, T2 white matter hyperintensity, or normal brain MRI | + | Epilepsy with infantile onset, dystonia, cognitive decline |
| PARK-DNAJC6 | 1st-5th decade (median age 11) | AR | Mild diffuse atrophy or normal brain MRI | + | Epilepsy with childhood onset, pyramidal signs, rapid progression with cognitive decline |
| VPS13C-related parkinsonism | Childhood to late adulthood (median age 29) | AR | Brain MRI: normal | –/+ | Pyramidal signs, early cognitive impairment, autonomic dysfunction |
| PARK-LRRK2 | 4th-10th decade (mean age 58–61) | AD | Brain MRI: normal | + | Levodopa-responsive PD with slow progression and low incidence of dementia and psychiatric symptoms |
| PARK-SNCA | 2nd-7th decade | AD | Brain MRI: normal | –/+ | Cognitive decline and psychiatric features (p.Ala53Thr, p.Ala30Pro, p.Glu46Lys, dupl/tripl), pyramidal signs (p.Gly51Asp and p.Ala53Glu), myoclonus (p.Ala53Glu, dupl/tripl), cerebellar signs (p.Ala30Pro variant) |
| PARK-VPS35 | 4th-7th decade (mean age 51) | AD | Brain MRI: normal | + | tremor-predominant, slow progression, rare occurrence of learning disabilities, dementia, psychosis, depression |
| PARK-GBA | 4th-8th decade (mean age 56.8, median 58) | AD | Brain MRI: normal | + | higher incidence of dementia, dysautonomia, and psychiatric symptoms |
| Metals in the brain | |||||
| Wilson’s disease (ATP7B) | Childhood, adolescence, or early adulthood | AR | T2 hyperintensity of putamen and other deep brain nuclei (“giant panda” and “miniature panda” signs) | –/+ | Wing-beating tremor, bulbar signs, dystonia, Kayser-Fleischer ring, hepatic involvement |
| NBIA (PANK2, PLA2G6, WDR45, C19ORF12, ATP13A2, CP, FTL) | Childhood (PANK2, C19ORF12), adolescence and early adulthood (PLA2G6, ATP13A2, WDR45), or adulthood (CP, FTL) | AR (PANK2, PLA2G6, C19ORF12, ATP13A2, CP), AD (FTL, C19ORF12), XD (WDR45) | SWI, GRE, and T2* hypointensity of basal ganglia (eye-of-the-tiger sign in PKAN, hyperintense streak of medial medullary lamina in MPAN, T1-weighted signal hyperintensity surrounding substantia nigra in BPAN, cerebellar atrophy in PLAN) | –/+ | Dystonia is predominant, developmental delay, neuropsychiatric features, visual disturbances, choreoathetosis, pyramidal signs |
| Fahr’s disease (SLC20A2, PDGFRB, PDGFB, XPR1, MYORG, and secondary forms) | Adulthood | AD (except for MYORG ⟶ AR) | Symmetrical Hyperdense TC signal involving predominantly basal ganglia (pontine involvement ⟶ MYORG) | –/+ | Tremor, choreo-dystonic features and neuropsychiatric features, recurrent headaches |
| Manganese accumulation (SLC30A10, SLC39A14, and secondary forms) | Childhood (SLC39A14) or Early adulthood (SLC30A10) | AR | T1-hyperintensity of the globus pallidus and white matter, pathognomonic sparing of the ventral pons | - | hepatic involvement and polycythemia (SLC30A10), dystonia, “cock-walk” gait, tremor, bulbar signs |
| Mitochondrial disorders (mtDNA: tRNA(Lys), tRNA(Gln), ND1, ND2, ND3, ND6, Cytb; nDNA: \itPOLG1, TWINKLE, PEO, OPA1, multiple mtDNA deletions) | Childhood-late onset | AD, AR, matrilinear | Atrophy, white-matter lesions, calcifications, cysts | +/– | Possible associated symptoms: short stature, diabetes, ophthalmoplegia, deafness, cardiomyopathy, myoclonus, seizures, and stroke like episodes |
| Niemann-Pick disease type C (NPC1, NPC2) | >15 years (adult form) | AR | Brain MRI: normal (cortical and cerebellar atrophy at end-stage disease) | +/– | SNGP, hepatosplenomegaly, psychiatric manifestations |
| Chediak-Higashi syndrome (LYST) | 3rd-4th decade | AR | Brain MRI: normal or cortical atrophy | ++ | Immunodeficiency, bleeding tendency, hemophagocytic lymphohistiocytosis, and oculocutaneous albinism. |
| Peripheral blood smear: giant inclusions in leukocytes | |||||
| Ataxia Telangiectasia (ATM) | 2nd-3rd decade | AR | Brain MRI: cerebellar atrophy | +/– | Telangiectasia, immunodeficiency, increased risk of tumors, radiosensitivity, ataxia |
| Parkinsonism: mostly bradykinesia (less rigidity due to cerebellar hypotonia) | |||||
| Cerebrotendinous Xanthomatosis (CYP27A) | 4th-5th decade | AR | Brain MRI: cortical and cerebellar atrophy; dentate nuclei, cerebellar peduncles, and internal capsule, SN, GP, striatum T2 WM hyperintensity | Possible but limited | Systemic: Diarrhea, cataract, tendon xanthomas, osteopenia Neurological: cerebellar and pyramidal signs, cognitive impairment, polyneuropathy Increased serum cholestenol |
| Dopaminergic imaging: presynaptic damage | |||||
| Dystonia and Parkinsonism | |||||
| PARK2 (PRKN) (as reported above) | |||||
| PARK-GCH1 | Dystonia (childhood), parkinsonism 2nd-3rd decade | AD | Brain MRI: normal | Remarkable at low doses with less frequent motor complications compared to EOPD | Lower limbs dystonia, diurnal fluctuations, sleep benefit |
| DYT-PRKRA | 2nd-4th decade | AR | Brain MRI: normal | Limited | Young onset dystonia with opisthotonus and sardonic smile, mild parkinsonism |
| DYT-ATP1A3 (RODP) | 2nd-3rd decade | AD | Brain MRI: normal | Limited | Abrupt onset, limited progression, rostrocaudal progression |
| DYT-TAF1 | 2nd-7th decade (earlier onset in males compared to females) | X-linked recessive | Brain MRI: cortical and putaminal atrophy in advanced stages | +/– | Males> >females (Panay Island), Progressive and degenerative |
| Ataxia and Parkinsonism | |||||
| SCA1-2-3-6-7-8-10-12-17-19/ 22-21, 28 | 3rd-7th decade | AD | Brain MRI: cerebellar atrophy | + | Nystagmus; dysautonomia is possible (SCA1,2,3); cerebellar features and spastic paraplegia (SCA28) |
| FXTAS (FMR1) | 5th-6th decade | XR | Brain MRI: splenium CC, periventricular, pons, MCP WM hyperintensities (MCP sign), cortical atrophy | + | Affects males > females; parkinsonism reported in up to 28.9% cases, bradykinesia/rigidity > tremor |
| PEO and OA and Parkinsonism | |||||
| TWINKLE, POLG | 4th-6th decade | AD (incomplete penetrance) | Brain MRI: normal | + | Ptosis, PEO, +/–SANDO precede parkinsonism |
| SLC25A46 | 5th-6th decade | AR | Brain MRI: WM hyperintensities | + | Possible additional mitochondrial features (neuropathy and pes cavus, hearing loss) and non-motor symptoms |
| Spastic paraplegia and Parkinsonism | |||||
| SPG11 | 1st-2nd decade | AR | Brain MRI: thinning of CC | +/– | Large phenotypic variability: HSP, ALS, neuropathy, parkinsonism (5–30% cases) |
| SPG7 | 2nd-5th decade | AR | Brain MRI: reported cortical and cerebellar atrophy | + | Parkinsonism in up to 21% cases [25] |
| SPG15 | 2nd decade | AR | Brain MRI: thinning CC; sign of “the ear of the lynx” | + | Lower limbs spasticity +/–dysarthria, motor neuropathy, cognitive impairment, and retinal degeneration |
| Intellectual disability and Parkinsonism | |||||
| WDR45, RAB39B, PTHRD1 (as reported above) | |||||
| PPP2R5D | 2nd(-4th) decade | AD | Brain MRI: possible subcortical WM hyperintensities (FLAIR) / hydrocephalus and aqueductal stenosis (1 case) | + | Mild-moderate intellectual disability and developmental delay, development of motor fluctuations with levodopa |
| Secondary causes of EOPD | |||||
| Drug-induced parkinsonism | Any age | NA | Normal | +/– | Underlying symptoms for which the subjects was treated with the offending drug |
| Infectious diseases (HIV) | Adulthood | NA | Possible focal lesions (opportunistic infections) or basal ganglia atrophy | +/– | Risk factors, additional opportunistic conditions. Inconsistent results on the role of highly active antiretroviral therapy (HAART) in causing or treating parkinsonism in AIDS |
| Vascular parkinsonism | Any age | NA | Ischemic or hemorrhagic lesions or vascular malformations in strategic area / leukoaraiosis | +/– | Cardiovascular risk factors |
The table summarizes the different etiologies that can manifest with EO parkinsonism. Typical age of onset, mode of heritability, response to levodopa, characteristic imaging findings and distinctive clinical features are reported. AD, autosomal dominant; ALS, amyotrophic lateral sclerosis; AR, autosomal recessive; CC, corpus callosum; GRE, gradient echo; HSP, hereditary spastic paraplegia; MCP, medium cerebellar peduncle; NBIA, neurodegeneration with brain iron accumulation; OA, optic atrophy; PEO, Progressive external ophthalmoplegia; SANDO, sensory ataxic neuropathy, dysarthria and ophthalmoparesis; SNGP, supranuclear gaze palsy; SWI, susceptibility weighted imaging; XD, X-linked dominant; XR, X-linked recessive; WM, white matter; +, positive response; ++, robust response; +/–, inconsistent or limited response.
Fig. 1
Clinical presentation of the different forms of EO parkinsonism. Monogenic forms of EOPD and other forms of EO parkinsonism are listed on the left. For each condition the frequency of the listed clinical features (top row) is represented by the size and the color of the corresponding dot. Symptoms are described as frequent (if expected to be found in more than 75% of cases of a specific form of EO parkinsonism), present (expected to be found in 25–75% of cases), uncommon (expected to be found in < 25 of cases), or absent (never reported in the specific phenotype), based on data reported in the literature and authors’ experience. Conditions were ordered according to the frequency of parkinsonian features.
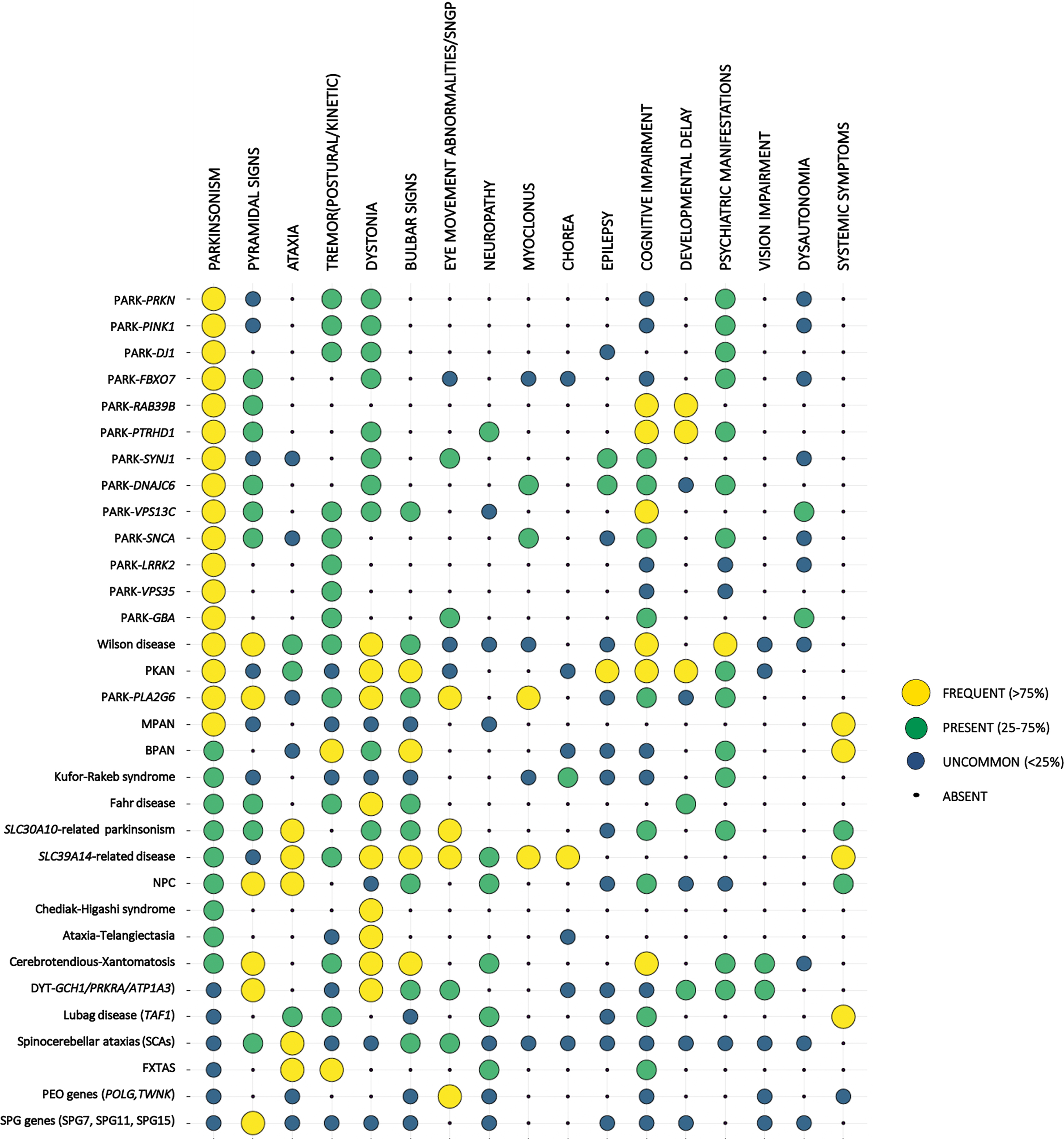
Although some of these forms can sometimes manifest before the age of 20 year, thus falling into the juvenile-onset PD (JOPD) category, here we will focus on the presentation of these various forms in the age range of 21 to 50 years.
The various etiologies of EO parkinsonism underlie distinct pathogenic mechanisms, particularly related to the genetic mutations. In general, earlier onset parkinsonism, whether in genetic or idiopathic cases, has been found to be associated with a more prominent degeneration of the basal ganglia, in certain cases lacking the classical Lewy bodies (such as in cases of PRKN-PD), with different biomarkers profiles (such as levels of p-Tau and t-Tau in the cerebrospinal fluid), molecular signatures, or with characteristic features (such as deposition of metals in the brain) due to the causative genetic mutations [24–27]. Although very interesting and certainly informative, a detailed description of the mechanistic aspects of the different forms of EO parkinsonism goes beyond the scope of this review.
METHODS
This work represents an expert opinion on this topic supported by the current literature. A narrative review of the literature was performed through a PubMed research using to the following key words: “early onset Parkinson’s disease”, “early onset parkinsonism”, “young onset Parkinson’s disease”, “young onset Parkinsonism”, “neurodegeneration with brain iron accumulation”, “secondary parkinsonism”, “parkinsonism” plus “lysosomal storage disease”, “mitochondrial disease”, “metabolic diseases”, “dystonia”, “ataxia”, “spasticity”, “spastic paraplegia”, “mental retardation”, “basal ganglia calcification”, “manganese”, “infectious disease”, “vascular diseases”, “Wilson’s disease”.
We focused only on the works referring to cases of EO parkinsonism. We included the more relevant and comprehensive cohort studies and reviews on the topics, as well as data from single case reports or small cohort studies for the rarer conditions.
Parkinson-predominant EO parkinsonism
Genetic forms
Autosomal recessive forms. Autosomal recessive mutations in genes associated with monogenic PD are a frequent cause of EOPD [28, 29]. Among these, mutations in Parkin (PRKN) (missense, nonsense, deletions, insertions, multiplications) are the most common, found in 50% of PD cases with onset before age 40 [30] and in up to 77% of patients with very young-onset parkinsonism before age 20 [31]. Disease onset spans from early childhood to after 50 years in rarer instances [32, 33], with the most common occurrence in the 3rd-4th decade and a median age at onset of 31 [33]. PTEN-induced kinase 1 (PINK1) mutations (point mutations and rarer large deletions) are the second most common cause of autosomal recessive PD [34], found in 4–7% of sporadic EOPD cases [35, 36], with a mean age at onset of 32 years [30]. Patients with mutations in PRKN or PINK1 share a similar phenotype, characterized by slow progression with robust and sustained response to levodopa. More peculiar features include onset with dystonia and symmetrical involvement, hyperreflexia, spasticity, psychiatric symptoms, and diurnal fluctuations with sleep benefit [30, 37]. Patients with PINK1 mutations have less pyramidal signs [30] and more commonly present with psychiatric symptoms [38, 39]. Despite young or very young-onset, disease progression is slow and the incidence of dementia and dysautonomia is low [31, 33, 40–43]. An increased risk for impulse control disorders and early motor fluctuations and dyskinesias in PRKN carriers has been described [44]. Mutations in DJ1 have been found in 0.4–1% of cases of EOPD [45, 46], with a median age of onset of 27 [30]. Despite their rare occurrence, DJ1 mutations are the third most common cause of autosomal recessive EOPD after PRKN and PINK1 mutations. The clinical presentation is similar to that of patients with PRKN or PINK1 mutations, however a higher risk of dementia and psychiatric symptoms (i.e., depression, anxiety, psychosis) has been reported [33, 46]. Brain MRI of EOPD associated with mutations in PRKN, PINK1, or DJ1 is usually normal.
Patients with mutations in FBXO7 present with juvenile parkinsonism typically associated with pyramidal tract signs, thus configuring a “pallido-pyramidal” or “parkinsonian-pyramidal” syndrome [47]. Pyramidal features, comprising spasticity and hyperreflexia predominantly in the lower extremities, Babinski sign, and equinovarus deformities, present in childhood and precede the onset of parkinsonism by 5–20 years [48]. Neuropsychiatric manifestations are often present, and slow vertical saccades have been described in ∼30% of patients [47, 49, 50]. Rare cases with limb myoclonus or chorea have also been reported [51]. Response to levodopa treatment is variable and often limited by dyskinesias and psychiatric complications [52, 53]. However, disease progression is usually slow and there is a low occurrence of dementia or dysautonomia [49, 52, 53]. Mild generalized atrophy can be seen, however brain MRI is often normal [48, 52, 53].
Very rare cases of rapidly progressive parkinsonism with median age at onset of 29 have been associated with mutations in VPS13C [54]. The course of this form is severe and characterized by early and dramatic cognitive decline, pyramidal signs, dysautonomia, and loss of ambulatory capacity within 15 years of onset [54].
Rarer autosomal recessive forms of PD present an early onset and are associated with atypical parkinsonism and complex phenotypes, often with a more severe course and are therefore discussed in the corresponding paragraphs below.
Autosomal dominant forms. Autosomal dominant forms of PD have a wide range of age at onset, from young adulthood to late adulthood, however they represent a rare cause of PD with early onset. Mutations in the LRRK2 gene are the most common variants found in all genetic PD cases [30]. Several missense mutations are considered pathogenic, each exhibiting a characteristic ethnic distribution. The frequency of the recurrent p.Gly2019Ser mutation ranges from 1–5% among European cases to 38–39% among North African populations and 10–28% among Ashkenazi Jews [38, 55]. Mutations at the 1441 site (p.Arg1441Gly, p.Arg1441Cys, and p.Arg1441His) are more common in Spain [56]. The p.Ile2020Thr has been reported in Japanese families [57–59], and the p.Gly2385Arg has been found in kindreds in China, Japan and Korea [60–62]. Although the onset of motor symptoms in carriers of mutations of this gene has been described as early as 28 years, the majority of mutations have been detected in late-onset cases (mean age 58–61 years) [55, 63–68]. The penetrance of LRRK2 mutation is largely incomplete and age-related, ranging from 14% to 90% according to various studies, and thus accounting for several apparently sporadic cases [69]. The clinical phenotype of LRRK2-related PD is that of typical levodopa-responsive PD with a milder disease course characterized by low occurrence of depression, hyposmia, RBD, and cognitive involvement [68, 70–74].
Mutations in SNCA are a highly penetrant and rare cause of PD, accounting for 0.2% of sporadic PD and 1-2% of familial PD cases [38, 75, 76]. The p.Ala53Thr, p.Gly51Asp, and p.Ala53Glu variants are associated with earlier onset PD before age 50 [33, 77, 78], whereas the p.Ala30Pro and p.Glu46Lys mutations cause a form of PD with typical onset in later adulthood [79, 80]. The p.Ala53Thr, p.Ala30Pro, and p.Glu46Lys variants yield a higher risk of cognitive decline, with behavioral changes, hallucinations, and progression to severe dementia and mutism in the later stages [81]. Atypical features can be seen in specific forms: pyramidal signs have been reported in patients with p.Gly51Asp and p.Ala53Glu [82–84], with the latter also presenting with myoclonus [85], while cerebellar signs have been described in patients with the p.Ala30Pro variant [81]. In addition to point mutations, heterozygous duplications and triplications of the SNCA gene have also been described in kindreds with early-onset PD [86]. SNCA multiplications are more common in European and Asian populations, and are linked to a more severe phenotype and a well-established gene dosage effect: triplications have been associated with an earlier onset, a more rapid progression, a higher risk of dementia and dysautonomia, and atypical signs (i.e., myoclonus) [87].
Mutations in VPS35 have a very low frequency (< 1%–1.5% in PD cohorts) [88–91] and represent a very rare cause of PD [123]. The mean age at onset is 51 years [89, 92–94]; however several cases with earlier onset in the 4th-5th decade have been described [90, 91, 95]. The phenotype associated with VPS35 mutations overlaps that of idiopathic PD of the tremor-predominant type with slow progression and good response to levodopa [30]. Atypical features are rare, however cases presenting with learning disabilities, dementia, psychosis, depression, and muscle spasms have been reported [91, 93, 96, 97].
GBA mutations are the most frequent genetic risk factor of PD, found in 5–20% of patients with PD across different populations [98–102]. A recent study screening a large cohort of PD patients identified an enrichment in GBA variants in EOPD [102]. Different variants of this gene have variable frequency across populations [103]. For example, the N370S, 84GG, E326K, and R496H variants are more frequent among Ashkenazi Jews, while the other common variant, L444P, is mostly found in non-Ashkenazi Jewish European subjects [99]. Compared with non-GBA carriers, PD patients with GBA mutations have an earlier onset of PD (median age 58) [98, 104], are more likely to develop PD before 50 years of age [98, 105], and have a higher risk and earlier presentation of non-motor features (i.e., dementia, dysautonomia, psychiatric manifestations) compared with idiopathic PD [99, 106, 107].
Secondary EO parkinsonism
Secondary causes of parkinsonism should be considered as well when assessing patients with EOPD, as they may represent treatable conditions, different from the majority of the degenerative processes. Drug-induced parkinsonism (secondary to the use of dopamine blocking agents, dopamine depletors, or, more rarely, associated with other drugs, such as calcium antagonists), and vascular parkinsonism are the most common forms of secondary parkinsonism [108]. Symmetric presentations and lack of the classical resting tremor are more frequent in these patients [109]. Although these conditions are more common among older subjects, they can be seen also in younger patients. When first evaluating a patient with EOPD, imaging studies of the brain should be considered to rule out rarer causes of secondary parkinsonism, such as vascular malformations (fistula, cavernoma, or arteriovenous malformations), single strategic lesions in the basal ganglia circuitry, supratentorial tumors or metastatic neoplastic lesions, as well as obstructive hydrocephalus [110]. Additional neurological symptoms can be frequently appreciated in secondary forms of EOPD and can help guide the diagnosis.
Systemic diseases that can present with parkinsonism in young age are represented by hepatic insufficiency (associated with bilateral hyperintensities of the basal ganglia on T1-weighted sequences or hyperintensities in the cerebellum and middle cerebellar peduncles on T2-weighted images), systemic lupus erythematosus (where characteristic serological biomarkers and systemic symptoms are crucial for the diagnosis), as well as infectious diseases (such as HIV and possible opportunistic infections) [109]. A high number of cases of secondary parkinsonism were observed during the pandemic of Spanish flu [111].
The use of street drugs (such as amphetamines derivatives or MPTP), as well as exposure to carbon monoxide, manganese - as described in the paragraph “Metals in the brain” -, cyanide and methanol can represent additional causes of secondary parkinsonism in young patients [112].
A recent work also showed a correlation with sport-related head trauma and EO parkinsonism, in subjects where the most common genetic causes of EOPD were excluded [113]. Although more studies will be necessary to further clarify this association, traumatic brain injuries have been already correlated with parkinsonism, thus this trigger may be taken into consideration among the other factors as a causative or precipitating event for EO parkinsonism in predisposed subjects [114].
EO parkinsonism associated with additional characteristic features
EO parkinsonism and metals in the brain
Brain metal accumulation can manifest clinically as early-onset parkinsonism in several neurological disorders. Various metals can be accumulated in the brain and other body tissues in these conditions (e.g., copper in Wilson’s disease, iron in neurodegeneration with brain iron accumulation (NBIA), calcium in Fahr’s disease, and manganese in hypermanganesemia). Some of these disorders are genetically inherited, while others are acquired [115–118].
Typically, the clinical suspect for these conditions emerges after brain imaging studies (i.e., brain MRI for Wilson’s disease, NBIA, and hypermanganesemia, and head CT for brain calcifications) [119]. However, associated systemic findings, such as hepatic involvement in Wilson’s disease and hypermanganesemia, can be the first clue to suspect metal accumulation in the brain [115, 118, 120].
Wilson’s disease
Wilson’s disease is distributed worldwide, with an estimated prevalence of 1 case per 30,000 live births in most populations [146]. However, in some isolated populations, such as a small mountain village on the island of Crete, the prevalence is much higher (1 in 15 births), due to high rates of consanguinity [121].
Wilson’s disease is an autosomal recessive disorder caused by loss-of-function mutations of ATP7B gene, which encodes for a copper-transporting ATPase which exports copper out of the cells, with a prominent role in the efflux of hepatic copper into the bile [122, 123]. The defective excretion of copper in the bile leads to hepatic copper overload and consequent damage, up to cirrhosis. Moreover, an impairment of copper incorporation into ceruloplasmin is also observed, resulting in low circulating levels of ceruloplasmin and elevated levels of unbound copper in plasma and urine. This altered copper metabolism leads to copper accumulation also in extrahepatic tissues, particularly in the basal ganglia [124]. In addition to the liver and brain, the cornea is also a characteristic site of copper deposition in Wilson’s disease. Indeed, the pathognomonic sign of this disease is the Kayser-Fleischer ring, an annular deposition of copper in the periphery of the cornea [125].
Copper accumulation is associated with a progressive neurodegeneration of basal ganglia leading to a complex movement disorder often associated with bulbar symptoms. The movement disorder is usually characterized by tremor, dystonia, and/or parkinsonism. Bulbar symptoms consist of dysarthria, drooling and/or dysphagia. In addition, a range of additional neurologic features may be present, including cerebellar ataxia, chorea, hyperreflexia, seizures, cognitive impairment, and psychiatric features [115].
While parkinsonism is rarely found in isolation or as the predominant feature in Wilson’s disease, this is commonly seen in combination with dystonia and/or tremor at presentation. Indeed, isolated parkinsonism occurs in less than 2% of neurologic presentations but parkinsonism in combination with other movement disorders is seen in the majority of the cases [115].
On brain MRI, T2 hyperintensity of the putamina and other deep brain nuclei is the most common abnormality. Involvement of the midbrain tegmentum can appear as a face of the “giant panda” on axial images. Axial T2 at the pons level may also show the face of a “miniature panda”. On T1-weighted images, these same areas show hypointensity. However, patients with severe hepatic dysfunction show areas of T1 hyperintensity, especially in the globus pallidus, similar to that seen in acquired hepatocerebral degeneration attributed to manganese deposition [126].
Neurodegeneration with brain iron accumulation
NBIA is a heterogeneous group of inherited movement disorders characterized by abnormal iron accumulation in basal ganglia and extrapyramidal movement disorders. The clinical features of NBIA range from global neurodevelopmental disorder in infancy to mild parkinsonism associated with minimal cognitive impairment in adulthood [116]. To date, ten NBIA genes have been identified, each associated with a different clinical syndrome and specific brain MRI features [118]. All forms of NBIA combined are considered to be ultra-rare with less than 1/1,000,000 affected world-wide, with the most common form being pantothenate kinase-associated neurodegeneration (PKAN), which accounts for approximately 50% of cases [127].
NBIA disorders presenting with parkinsonism as part of their clinical presentation include PKAN, PLA2G6-associated neurodegeneration (PLAN), beta-propeller protein-associated neurodegeneration (BPAN), mitochondrial-membrane protein-associated neurodegeneration (MPAN), Kufor-Rakeb Syndrome (KRS), Aceruloplasminemia, and Neuroferritinopathy [116]. EOPD is a frequent presentation of PLAN, BPAN, and KRS and typically shows atypical features, such as a poor levodopa response, lack of asymmetry, and the association with dystonia and pyramidal signs [116]. PLAN and KRS present brain iron deposition only in a subgroup of cases [128–130] and, therefore, are also classified in autosomal recessive forms of parkinsonism.
Mutations in ATP13A2 (missense, nonsense, and frameshift) have been found in patients presenting with a form of autosomal recessive hereditary parkinsonism with juvenile onset before age 20, known as Kufor-Rakeb syndrome [131–134]. Cases with onset in adulthood have been described, with a mean age at onset of 24 [129]. The clinical spectrum encompasses severe parkinsonism of the akinetic-rigid type and various atypical manifestations, most characteristically ocular movement abnormalities (i.e., vertical supranuclear gaze palsy, slow vertical saccades), pyramidal signs, and facial-faucial-finger myoclonus [131, 135–138]. Intellectual disability, cognitive decline, hallucinations, dystonia, ataxia, and axonal neuropathy are also common [139]. Parkinsonism in Kufor-Rakeb syndrome has a moderate response to levodopa treatment, however levodopa-induced motor fluctuations occur early in the disease course [49, 131, 135] and response to levodopa is lost with disease progression [133]. Brain MRI findings include diffuse atrophy and a hypointense signal in the striatum and pallidum on T2-weighted images, demonstrating the accumulation of paramagnetic substances [49, 137, 138].
Mutations in PLA2G6 have been associated with a form of parkinsonism-dystonia with onset between 10 and 30 years and with a rapid and severe progression to dementia (PLA2G6-associated Neurodegeneration or PLAN) [49, 140, 141]. Initial symptoms include psychiatric manifestations (i.e., depression, irritability, aggressive behavior, psychosis in some cases), gait changes with dragging of the foot and dysautonomia. Atypical features can include pyramidal signs, eye movement abnormalities (i.e., slow vertical saccades, nystagmus), optic atrophy, neuropathy, ataxia and cerebellar signs [135, 140–142]. Despite a modest response to dopaminergic agents, patients may develop early levodopa-induced dyskinesias. Brain MRI can show iron deposition in the cerebellum or in the pallidum, and cerebellar atrophy [49, 142]. Pontine atrophy with clival prominence can be seen in childhood-onset forms [143].
The clinical differential diagnosis among NBIA disorders is primarily based on the age at onset and on the different brain MRI features. PKAN, BPAN and MPAN typically have an early onset in childhood, PLAN and KRS begin in early adulthood, while neuroferritinopathy and aceruloplasminemia are adult-onset disorders [116]. Notably, BPAN-associated EOPD, due to mutations in WDR45, appears later in the course of the disease after intellectual disability and epilepsy which develop in childhood. MRI iron-sensitive sequences, such as susceptibility weighted imaging (SWI), gradient echo (GRE), and T2*, are needed to identify the characteristic hypointensity in specific NBIA disorders [144]. Concerning specific brain MRI changes of NBIA syndromes presenting with EOPD, the T1-weighted signal hyperintensity surrounding the substantia nigra is the hallmark of BPAN, the presence of associated cerebellar atrophy suggests a PLAN diagnosis, and marked atrophy and hypointensity of the globus pallidus in the initial phase is typical of KRS [144].
Fahr’s disease
The presence of neurological and psychiatric symptoms associated with bilateral basal ganglia calcifications identifies a clinical syndrome defined as Fahr’s disease [117]. This denomination mainly refers to idiopathic forms where no metabolic or other underlying causes are identified. Fahr’s disease commonly affects people after their 40s; therefore, this disorder is not a frequent cause of EOPD. Patients are usually in good health in their youth and tend to develop symptoms later in adulthood [145]. Calcifications are typically symmetrical and bilateral and involve the caudate, putamen, globus pallidus, thalamus, deep cortex, dentate nucleus, and, less frequently, the white matter [146]. CT scan is considered the gold standard for diagnosis identifying calcifications as hyperdense lesions [146]. Recently, mutations in five different genes (SLC20A2, PDGFRB, PDGFB, XPR1, and MYORG) were found to cause Fahr’s disease, or, more precisely, Familial idiopathic basal ganglia calcification (FBGC). MYORG-related FBGC is the only autosomal-recessively inherited form, while the other four types are transmitted with an autosomal dominant pattern of inheritance [147]. Patients with early onset FBGC present frequently with psychiatric/cognitive symptoms or migraine, while older patients exhibit mostly movement disorders, among which parkinsonism is the most frequent presentation [117]. Parkinsonism can present with both motor and non-motor symptoms [148]. The motor symptoms include the cardinal signs of idiopathic Parkinson’s disease (bradykinesia, rest tremor, rigidity, and postural and gait impairment) that tend to present symmetrically with the presence of coarse postural tremor. Levodopa response is variable from case to case. Parkinsonism can be associated with other movement disorders, such as choreoathetosis, non-parkinsonian tremor, dystonia, and orofacial dyskinesias. Associated neuropsychiatric symptoms consist of apathy, anxiety, depression, psychosis, and cognitive deterioration [117, 148].
Brain manganese accumulation
Manganese brain accumulation can be due to genetic or acquired causes. Genetically-determined brain manganese accumulations are ultra-rare conditions, with less than 100 cases reported world-wide so far [149]. There are two known genetic forms of brain manganese accumulation, associated with recessive mutations of SLC30A10 and SLC39A14 genes, but only the former can present as EOPD [118, 120]. Biallelic SLC30A10 mutations are associated with a clinical syndrome characterized by movement disorders (e.g., dystonia and/or parkinsonism) resulting from manganese accumulation in the basal ganglia, hypermanganesemia, polycythemia, and liver disease (e.g., hepatomegaly with variable degree of hepatic fibrosis). Parkinsonism develops in early adulthood as shuffling gait, rigidity, bradykinesia, hypomimia, and monotone speech, and it is unresponsive to levodopa treatment. Other neurologic findings may manifest in childhood and precede EOPD such as four-limb dystonia, leading to the characteristic “cock-walk gait”, dysarthria, and fine tremor. Hepatic failure, secondary complications of cirrhosis, and the neurologic disorder shorten life expectancy. Brain MRI T1-weighted images show characteristic hyperintensity of the basal ganglia, subthalamic nucleus, and dentate nucleus. Normalization of manganese blood levels with chelating therapies can improve brain MRI findings [120]. Differently, in SLC39A14 deficiency, affected individuals present with hypermanganesemia and rapidly progressive childhood-onset parkinsonism-dystonia due to manganese deposition in the basal ganglia. Brain MRI appearances are the same as for SLC30A10 disease. Distinguishing features are the absence of liver disease and polycythemia due to the lack of hepatic manganese deposition, which can be assessed by liver MRI [118].
Considering non-genetic forms, environmental exposure to high quantities of manganese is known to be neurotoxic and causes manganism, a clinical syndrome characterized by an extrapyramidal movement disorder (e.g., dystonia and/or parkinsonism) associated with T1 hyperintensity of the basal ganglia on brain MRI resulting from manganese accumulation in the basal ganglia. Manganism has been described in workers inhaling manganese-loaded dust or fumes (e.g., mining and welding industries), in individuals drinking manganese-contaminated water, and in drug addicts using intravenous drugs contaminated with potassium permanganate [150]. Brain accumulation of manganese is also observed in subjects with advanced hepatic cirrhosis or portosystemic shunts (acquired hepatocerebral degeneration) in which impaired biliary excretion of manganese results in manganese accumulation in the basal ganglia possibly causing debilitating movement disorders [151].
EO parkinsonism associated with metabolic conditions
A number of metabolic disorders can present with EO parkinsonism. Diseases caused by mutations in genes encoding for mitochondrial proteins are usually characterized by short stature, diabetes, ophthalmoplegia, deafness, cardiomyopathy, as well as neurological disorders such as myoclonus, seizures, and stroke like episodes [152]. The majority of the reports of parkinsonism and mitochondrial diseases consist of single case descriptions, as summarized in previous works [153, 154]. Although in most of these cases the neurological symptoms, including parkinsonism, manifest during childhood, onset after the age of 21 year is possible. Thus they should be considered in the differential diagnosis of EOPD. Some of these conditions are described below (paragraph “EO parkinsonism associated with other neurological symptoms”). Matrilineal heritability, due to mutations of genes in the mitochondrial DNA, can be a key factor for suspecting these diseases.
Parkinsonism has also been reported in a growing number of conditions caused by impairment of the lysosomal/lipid metabolisms, such as Gaucher disease (GD) (as described in paragraph “Monogenic forms of EOPD”). Parkinsonism has been described in single cases of Niemann Pick type C (NPC), although screenings of large cohorts of patients with PD failed to confirm the association between mutations of the NPC-causative genes, NPC1 and NPC2, and PD. On the contrary, variants of the SMPD1 gene, that is responsible for Niemann Pick types A and B, have been associated with PD across different populations [155–162]. NPC and GD are far more common among Ashkenazi Jews.
Chediak-Higashi syndrome (CHS) is a rare lysosomal disorder caused by mutations of the LYST gene and presenting with immunodeficiency, bleeding, lymphohistiocytosis, and oculocutaneous albinism. Few cases of EOPD with good response to levodopa have been found in carriers of homozygous mutations of this gene [151, 163–165]. Cerebrotendinous xanthomatosis (CTX) is due to mutation of the CYP27A gene which is involved in the cholesterol metabolism. CTX can frequently manifest with parkinsonism, in some cases as the presenting symptom, usually associated with other neurological manifestations, such as cerebellar ataxia and hyperreflexia (summarized in [166, 167]). CTX is more frequent in females, among Moroccan Jews and Druze in Israel (6/7,000 individuals) and in South and East Asia, compared with North America, Europe, and Africa [168].
Although the onset is more common during childhood, EOPD has also been reported in cases of gangliosidosis GM1, as well as late onset GM2 gangliosidosis (also called late onset Tay-Sachs disease, LOTS), and adult onset neuronal ceroid lipofuscinosis, usually in the context of additional neurological manifestations [15, 169–171]. Some of the conditions, such as LOTS, are more prevalent among Ashkenazi Jewish, although other clusters have been reported also within an Amish community in Pennsylvania and non-Jewish French Canadians in the area of St. Lawrence [172].
Parkinsonism can present in almost half of the cases of ataxia telangiectasia (A-T) (41% cases), usually in the second or third decade, when the other symptoms associated with this systemic disorder are already present (summarized in [173]). In a few cases levodopa has been reported to be beneficial in A-T.
Finally, young-onset parkinsonism can be present in the context of hemochromatosis, a hereditary condition caused by aberrant iron accumulation in the peripheral tissues causing, among the others, hepatic toxicity, anemia, diabetes, cardiac manifestations and hypogonadism [174–176]. In these cases, parkinsonism develops between the fourth and seventh decade and brain MRI usually shows hypointensity in the basal ganglia and substantia nigra [175, 176]. This condition is more frequent across people of Northern European descent, originally arising within the Celtic population [177].
EO parkinsonism associated with other neurological symptoms
Dystonia-parkinsonism syndromes
Parkinsonism may be part of the clinical spectrum of syndromes defined as “dystonia-parkinsonisms”. This term applies to rare conditions where the severity of dystonia usually equals that of parkinsonism. Unlike the parkinsonian syndromes with additional dystonia (i.e., PRKN-PARK), these conditions usually have their onset in childhood. The phenotype is often non-specific to a particular form and genetic investigations are required.
DYT-GCH1 typically presents with lower-limb dystonia, causing gait disturbance and a tendency to fall. Clinical features like diurnal fluctuation and sleep benefit are typically associated with this form and can be helpful in differentiating it from other forms of EOPD. The response to levodopa of both dystonic and parkinsonian signs is remarkable with low-doses (i.e., < 300–400 mg/daily) [178] with no development of the levodopa-induced dyskinesias.
Mutations in protein kinase, interferon-inducible double-stranded RNA-dependent activator (DYT-PRKRA) cause an autosomal recessive form of young onset, progressive generalized dystonia and mild parkinsonism [179]. A remarkable involvement of oromandibular and bulbar regions with a sardonic smile, tongue protrusion, significant speech and swallowing disturbances is typical of this form. The axial involvement of the neck and trunk can lead to opisthotonic postures. Parkinsonism is usually not a prominent feature, appears later than dystonia and is characterized by bradykinesia and rigidity, rather than resting tremor. The disorder was initially described in three Brazilian families [179]. In a few cases, bilateral striatal degeneration has been observed on brain MRI [180, 181]. However, the three described cases presented with a very young age of onset, before the age of 20 [180, 181].
Rapid-onset dystonia parkinsonism (RODP) due to mutations in the ATP1A3 gene (DYT-ATP1A3) is characterized by an abrupt onset of dystonia and parkinsonism over minutes to several weeks, usually triggered by febrile illness, physical or emotional stressors, alcoholic binges, or childbirth [182, 183]. The acute onset of symptoms and the autosomal dominant mode of inheritance are distinctive features of DYT-ATP1A3 that can help distinguish this form from other conditions. Age at onset is typically in the second to third decade of life. Initial symptoms rapidly evolve over weeks and then stabilize over time. A possible second abrupt worsening of symptoms after years is possible [184]. The rostrocaudal gradient of involvement with prominent dysarthria and hypophonia, and the asymmetric distribution of both dystonia and parkinsonian features is typical. Tremor is usually absent. Levodopa treatment is ineffective [185].
DYT-TAF1 is an adult-onset X-linked recessive neurodegenerative disorder, affecting predominantly men (male to female ratio of 93:1), described in patients from the island of Panay, Philippines, and very rarely in the general population [186, 187]. Dystonia is usually focal at onset, involving the cranio-cervical region with retrocollis, oromandibular dystonia, blepharospasm, and tongue protrusion, and it has a tendency to generalize as the disease progresses [188]. Deep brain stimulation of the internal pallidum (GPi-DBS) has proven effective with long-lasting benefit, especially when performed in the early stage of the disease [189, 190]. Ten years after the onset there is usually a decrease in the severity of dystonia and parkinsonism becomes gradually more prominent [188, 191]. This temporal pattern is quite unique and helps differentiate this condition from other disorders where there is a progression of both dystonic and parkinsonian features.
Parkinsonism-ataxia
Parkinsonism may be part of the clinical presentation of a range of spino-cerebellar ataxias (SCAs) associated with polyglutamine expansion (SCA1,2,3,6,7,8,10,12,17), other SCAs (19,21,22,28), and Fragile-X tremor ataxia syndrome (FXTAS) [192–194]. Characteristic features of the underlying disease (i.e. mental retardation, physiognomic features (long face, low set ears), action tremor in FXTAS; slow saccades in SCA2; pyramidal signs in SCA3; macular degeneration in SCA7; dementia, psychiatric features, chorea in SCA17; seizures in SCA10; cognitive impairment in SCA21; ophthalmoparesis, ptosis, spasticity in SCA28) can sometimes be overlooked by physicians but they are important to guide the diagnosis [195, 196]. SCA1,2,3 and 17 should be suspected in patients presenting with a phenotype suggestive of multiple system atrophy (MSA), as they can present with prominent parkinsonism and dysautonomia, although the classical “hot cross bun” sign on brain MRI, that is characteristic of MSA, is only rarely present in these forms [197].
Parkinsonism, PEO, or optic atrophy
Parkinsonism preceded by development of progressive external complete ophthalmoplegia (e.g., on both horizontal and vertical plane) has been reported in a few cases of subjects carrying mutations in the mitochondrial-related genes TWNK and POLG1. In these cases, parkinsonism may not be prominent and thus be overlooked. A variable phenotypic presentation and penetrance were present in the reported families. SANDO (sensory ataxic neuropathy, dysarthria, and ophthalmoparesis) syndrome has been reported as well in subjects with parkinsonism due to POLG1 variants [200]. POLG1 mutations can be found also in cases of parkinsonism associated with optic atrophy, manifesting with a gradual vision loss, as well mutations of the SLC25A46 [201].
Parkinsonism and spastic paraplegia
Mutations in the SPG11 gene have been identified to be a major cause of autosomal recessive hereditary spastic paraplegia (SPG) with thin corpus callosum. Juvenile-onset symmetric parkinsonism has been described, with variable response to dopamine intake [202, 203].
Other SPG syndromes may display parkinsonism as part of the clinical picture, in particular in SPG7 [204–206] and SPG15 [207, 208].
Parkinsonism and intellectual disability
Monogenic forms of early intellectual disability and early or juvenile onset parkinsonism have been described. The combination of early-onset parkinsonism and intellectual disability is a distinctive feature of patients with mutations in RAB39B [209] and PTRHD1 [210, 211]. Intellectual disability develops over a spectrum which encompasses delayed speech initiation, early learning difficulties, and obsessional and ritualistic behavior in childhood [212]. It is non-progressive and can be mild, and therefore is often neglected. Intellectual disability typically precedes the onset of parkinsonism, which has been described to occur as early as age 10 in males with RAB39B mutations [209] and in the 3rd-4th decade in patients with mutations in PTRHD1 [213]. RAB39B mutations (point mutations, deletions, and duplications) are the only known cause of PD with X-linked inheritance [30]. Affected females with mutations in RAB39B have a later onset of parkinsonism and a less severe phenotype [214]. Unaffected females with heterozygous RAB39B mutations have also been reported [214]. The clinical phenotype is that of typical parkinsonism with tremor at rest, bradykinesia and rigidity, and a good response to levodopa. Atypical features have been described in both RAB39B (i.e., postural tremor, seizures, macrocephaly, autism spectrum disorder and strabismus) [215–217] and PTRHD1 (i.e., pyramidal signs, distal muscular atrophy and hyposthenia, axonal sensorimotor polyneuropathy, hypersomnia, hypersexuality) [210, 213, 218]. Basal ganglia calcifications can be seen in RAB39B-related EOPD [209, 219], whereas brain MRI is normal in PTRHD1 mutations.
Phenotypes associated with mutations in WDR45 have already been discussed (“Neurodegeneration with brain iron accumulation” paragraphs).
A dopamine-responsive parkinsonian syndrome has been reported in patients with mutations in the PPP2R5D gene associated with a rare autosomal-dominant disease with developmental delay and mild to severe intellectual disability [220–223].
Interestingly, a recent review found that impaired cognition or neuropsychiatric disorders are also an early presentation in 92% of patients with EOPD associated with mutations in ATP13A2, PLA2G6, or FBXO7, suggesting that these may occur early before parkinsonism develops [224].
Parkinsonism and seizures
Seizures with onset in infancy or childhood are seen in ∼50% [224] of patients with parkinsonism associated with mutations in DNAJC6 [88, 225, 226] and SYNJ1 [227–229]. Onset of parkinsonism, often associated with dystonia, ranges between 7 and 42 years in patients with DNAJC6 mutations [88, 226] and occurs between 20 and 40 years in SYNJ1 carriers [227]. Pyramidal signs and rapid progression to dementia are often seen in DNAJC6 [88, 225], whereas SYNJ1 patients can present with eyelid apraxia and supranuclear gaze palsy [227, 230]. Reports on brain imaging describe generalized atrophy in DNAJC6 and SYNJ1 cases and T2 hyperintensity in the posterior white matter and substantia nigra of few SYNJ1 patients [227, 228].
HOW TO APPROACH A PATIENT WITH EO PARKINSONISM
Clinical history
A detailed clinical history can provide important information to reach a diagnosis in patients presenting with EO parkinsonism. An initial presentation of pure parkinsonism within the 3rd decade, with no prominent additional clinical manifestations, is more characteristic of monogenic forms of PD (such as PARK-PRKN, followed by PARK-PINK1 and PARK-DJ1). Complicated forms where parkinsonism coexists with pyramidal symptoms, other movement disorders (mostly dystonia or cerebellar features), cognitive or psychiatric manifestations and an earlier age of onset are more suggestive of mutations of ATP13A2, PLA2G6, FBXO7, RAB39B, PTRHD1, GCH1, ATP1A3, Wilson’s disease, NPC and NBIA. Later age of onset, in the fourth or fifth decades, is more suggestive of mutation of the GBA gene, CTX, SCA (2, 3, 17), occasionally of mutations of the LRRK2 gene, as well as secondary forms such as drug-induced parkinsonism, or parkinsonism-related to systemic conditions.
Certain clinical features are very suggestive of specific forms of EO parkinsonism (Fig. 1). For example, epilepsy can be associated with SYNJ1 mutations or mitochondrial diseases. Cognitive impairment or developmental delay present early in Kufor-Rakeb syndrome, Fahr’s disease, PPP2R5D-related parkinsonism, while they can be a later manifestation of CTX and Wilson’s disease. Psychiatric features can be the initial manifestations of NPC, LOTS and Westphal syndrome, and they can complicate Fahr’s disease, Wilson’s disease, and SCA17 as the disease progresses, or can be consistent with a diagnosis of parkinsonism due to the use of dopamine blocking agents. Systemic symptoms are more characteristic of mitochondrial disorders (such as deafness, diabetes, lipomatosis), NBIA and hemochromatosis (diabetes and hepatotoxicity), NPC or GD (hepatosplenomegaly), history of diarrhea or early-onset cataracts in CTX, and the characteristic combination of hematological, immune and oculocutaneous manifestation in the CHS.
Because of the possibility of preventing severe complications when this condition is recognized and promptly treated, genetic analysis of ATP7B gene for the diagnosis of Wilson’s disease should be performed in any patient presenting with parkinsonism in childhood, adolescence or early adulthood, in particular when parkinsonism is subacute or rapidly progressive and associated with concomitant dystonia and/or dysarthria. In addition, the presence of the Kayser-Fleischer ring (observed in most neurologic cases) and evidences of liver disease (e.g., jaundice, unexplained cirrhosis, thrombocytopenia or coagulopathy) should immediately raise the diagnostic suspect of Wilson’s disease [231].
EOPD is usually slowly progressive. On the contrary, abrupt onset can be suggestive of RODP, viral, immune-mediated, or vascular (stroke, cavernomas or arteriovenous malformations) conditions. While acute forms usually have a more static presentation, the majority of the other forms of EO parkinsonism are progressive, especially the more complicated ones (such as NBIA, Fahr’s disease, and metabolic conditions) compared with pure monogenic forms of parkinsonism (such as PARK-PRKN, PARK-PINK1, PARK-DJ1) or DYT-GCH1. Very low doses of levodopa or dopamine agonists are able to provide a significant relief of symptoms in patients with PARK-PRKN, PARK-PINK1, PARK-DJ1 and DYT-GCH1. However, these patients, except for DYT-GCH1, will soon present motor fluctuations and levodopa-induced dyskinesias. On the contrary, the other forms of EO parkinsonism are poorly responsive to dopaminergic medications compared with iPD.
Non-motor symptoms can also help discriminating among different causes of EO parkinsonism. Symptoms such as dysautonomia, RBDs, hyposmia, which are typically found in patients with iPD, are much rarer, if not absent, in the majority of patients with EOPD. Their prominent presentation can be associated with mutations of the GBA gene. Dysautonomia and RBD can also be found in NPC and in some cases of SCA (2,3,17).
As part of the assessment of the clinical history, a detailed investigation of patients’ family history is mandatory, especially in the context of genetic forms. If family history suggests an autosomal dominant pattern of inheritance, genetic screening should include GBA (high prevalence, low penetrance), LRRK2 (medium-high prevalence, low penetrance), and SNCA (low prevalence, high penetrance). If consanguinity is present in the family and other family members (e.g., siblings) are affected by EOPD, then PRKN, PINK1, and DJ-1 are the most probable cause; if negative, other genes related to recessive forms can be considered (PLA2G6, FBXO7, ATP13A2, DNAJC6, SYNJ1, VPS13C, PTRHD1, ATP7B) [232].
Finally, patients’ ancestry should always be inquired, since some genetic mutations associated with EO parkinsonism present high prevalence in selected ethnic groups. For example, the LRRK2 G2019S mutation is a frequent cause of PD among North Africans, Ashkenazi Jews, and in the Mediterranean basin (e.g., Italy, Spain, and Portugal). Other population-specific LRRK2 mutations are the R1441G in Basques and the R1441C in Belgians and Southern Italians. Another example is the GBA N370S mutation, which is distributed worldwide, but presents a particular enrichment in Ashkenazi Jews [232].
Clinical exam
When approaching a patient with EOPD, a comprehensive clinical examination is essential to collect all elements that will aid in the diagnostic process. While the classic onset of iPD is in one body part or hemi-body, the initial presentation of EO parkinsonism observed in forms of NBIA and in Fahr’s disease is often symmetric and involves both halves of the body equally since the first stages. Similarly, acquired parkinsonisms due to exposure to dopamine-blocking agents and to vascular lesions in the basal ganglia typically have a symmetric presentation. Some forms of EO parkinsonism exhibit a characteristic distribution and preferential involvement of the limbs or the cranial district. DYT-GCH1, PARK-FBXO7, and vascular parkinsonism typically are more predominant in the lower limbs, while oromandibular involvement and bulbar signs are frequent in Wilson’s disease, DYT-PRKRA, DYT/PARK-ATP1A3, DYT-TAF. The classical distinction between tremor-dominant and akinetic-rigid phenotypes helps guide the clinical reasoning. Predominant rigidity and bradykinesia are frequently seen in PARK-ATP13A2, dystonia-parkinsonism (e.g., DYT-PRKRA, DYT/PARK-ATP1A3), as well as in drug-induced and vascular parkinsonism, whereas a tremor-dominant phenotype (rest, postural, or kinetic tremor) can point to a different etiology (Wilson’s disease, Fahr’s disease, PARK-LRRK2). A careful examination of ocular movements in all its components is critical, considering the velocity of the saccades (reduced in PARK-ATP13A2, NBIA/DYT/PARK-PLA2G6, PARK-FBXO7, and SCA2), gaze limitation (vertical gaze palsy in PARK-ATP13A2, NPC, A-T, PARK-SYNJ1), and ophthalmoplegia (mitochondrial disorders). Close inspection of the eyes can also show peculiar features, such as Kayser-Fleischer rings in Wilson’s disease and oculocutaneous albinism in CHS.
Additional movement disorders and neurological signs can accompany parkinsonism in several cases. Dystonia is characteristic of forms of monogenic EOPD (PRKN, PINK1, PLA2G6), dystonia-parkinsonism, and NBIA, as well as Wilson’s disease and hypermanganesemia. Chorea and choreoathetosis can be observed in Fahr’s disease, Wilsons’s disease, A-T, and most forms of NBIA. The association of parkinsonism and pyramidal signs may be indicative of specific phenotypes, most notably the ones associated with variants of FBXO7, ATP13A2, PLA2G6, DNAJC6, PRKN, PINK, RAB39B, PTRHD1, VPS13C, forms of NBIA, and CTX. Myoclonus can be seen in a minority of cases of EOPD, including PARK-ATP13A2 (facial-faucial-finger mini-myoclonus), PARK-SNCA, and mitochondrial cytopathies. Ataxia and cerebellar signs may be observed in PLA2G6-related parkinsonism, CTX, AT, NPC, SCAs, and FXTAS.
Imaging studies
Specific neuroradiological signs may strongly suggest the genetic diagnosis. For example, the T2*/SWI hypointensity of basal ganglia is a key feature of NBIA. The presence of basal ganglia calcifications on CT scan should address the genetic investigation to the FBGC forms. T2-hyperintensity of the putamina and other deep brain nuclei are the most common abnormality seen in Wilson’s disease. T2-hyperintensity of the white matter in the dentate nuclei, cerebellar peduncles, and internal capsule, substantia nigra, globus pallidus, striatum is found in CTX. Conversely, T1-hyperintensity of the basal ganglia can indicate manganese accumulation. Metabolic forms, such as NPC, CHS, and A-T can instead present cortical and cerebellar atrophy, particularly in advanced stages of the disease. A detailed description of specific MRI neuroradiological features associated with each form is reported in Table 1. All forms of Parkinson’s disease and the vast majority of degenerative parkinsonisms are associated with a reduction of the dopaminergic transporter (DaT) at the presynaptic dopaminergic terminal, as shown by dopaminergic functional imaging (e.g., Ioflupane I 123 SPECT) [233]. Fahr’s disease, hereditary spastic paraplegias, and spinocerebellar ataxias can be associated with abnormalities of the presynaptic dopaminergic terminal, more frequently if clinical parkinsonism is present. Differently, parkinsonian disorders associated with brain manganese accumulation (acquired or inherited) and non-degenerative dystonias display a normal Ioflupane I 123 SPECT imaging [234, 235]. A summary of functional imaging features of early-onset parkinsonisms is reported in Supplementary Table 1.
GENETIC COUNSELING
Next generation sequencing (NGS) panels or diagnostic exome sequencing represent the best diagnostic approach in the absence of strong candidate genes. With these techniques, all genes related to EOPD and EO parkinsonism are sequenced simultaneously, which is a cost and time effective method to identify possible mutations. Shortcomings related to this approach include a low sequence efficiency for difficult-to-sequence DNA regions and the risk of missing gross structural alterations. Each variant identified by gene panels should be confirmed by the Sanger approach. Moreover, dosage assay techniques (i.e., Multiplex Ligation-dependent Probe Amplification (MLPA) and qPCR) should always be implemented when analyzing SNCA, PRKN, PINK1, and DJ-1 genes in order to detect rearrangements that can be missed by NGS, especially heterozygous deletion or duplications. It is noteworthy that the interpretation of NGS results, which often leads to the detection of variants of unknown significance (VUS), requires the expertise of a neurogeneticist specialized in movement disorders. Additional studies are mandatory in case novel genetic variants are detected in order to confirm their pathogenetic role [232].
As an alternative approach, each single gene can also be analyzed by classic Sanger sequencing, a highly reliable but expensive technique, that can be used as a first approach only when few candidate genes are suspected as causative. Sanger sequencing should always be complemented with dosage assay techniques. Pathogenic mutations of LRRK2 gene have been found in exons 29-30-31-34-35-41-48, therefore only these exons should be analyzed for clinical purposes.
Genetic counseling of patients with EOPD involves the entire family. Whether genetic testing should be offered to asymptomatic family members is still very controversial. Except for genetic forms in which a disease-modifying therapeutic approach exists (e.g., Wilson’s disease) and an early genetic diagnosis can be useful to the patient, no defined guidelines are available for genetic counseling of family members of patients with EO parkinsonism.
The identification of causative genes of EOPD and EO parkinsonism contributed to increase our understanding of the genetic architecture and pathogenic mechanisms of these conditions. These discoveries led to the selection of targeted drugs for specific genetic forms (e.g., GBA- and LRRK2-related forms). At present, clinical trials testing these potential therapeutic approaches are ongoing [236].
CONCLUSIONS
EO parkinsonism can be the manifestation of a broad range of conditions. A prompt and accurate diagnosis is crucial for counseling of these patients, prognostic implications, and possibly early treatments to prevent further complications, such as in the case of Wilson’s disease. Specific genetic mutations can be more difficult to distinguish only based on phenotype in case of monogenic forms of EOPD, because of the extensive overlap of their clinical presentation. On the contrary, complicated, and rarer forms related to genetic and metabolic conditions, or metals deposition in the brain can be more easily differentiated, because of the characteristic clinical features, radiological and laboratory findings. Although rare, awareness of the different forms of EO parkinsonism by movement disorder specialists will certainly improve the management and care of these patients.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JPD-212815.
REFERENCES
[1] | Quinn N , Critchley P , Marsden CD ((1987) ) Young onset Parkinson’s disease. Mov Disord 2: , 73–91. |
[2] | Gibb WR , Lees AJ ((1988) ) A comparison of clinical and pathological features of young- and old-onset Parkinson’s disease. Neurology 38: , 1402–1406. |
[3] | Golbe LI ((1991) ) Young-onset Parkinson’s disease: A clinical review. Neurology 41: , 168–173. |
[4] | Butterfield PG , Valanis BG , Spencer PS , Lindeman CA , Nutt JG ((1993) ) Environmental antecedents of young-onset Parkinson’s disease. Neurology 43: , 1150–1158. |
[5] | Bostantjopoulou S , Logothetis J , Katsarou Z , Mentenopoulos G ((1991) ) Clinical observations in early and late onset Parkinson’s disease. Funct Neurol 6: , 145–149. |
[6] | Kostíc VS , Filipović SR , Lecić D , Momcilović D , Sokić D , Sternić N ((1994) ) Effect of age at onset on frequency of depression in Parkinson’s disease. J Neurol Neurosurg Psychiatry 57: , 1265–1267. |
[7] | Selikhova M , Williams DR , Kempster PA , Holton JL , Revesz T , Lees AJ ((2009) ) A clinico-pathological study of subtypes in Parkinson’s disease. Brain J Neurol 132: , 2947–2957. |
[8] | Alves G , Wentzel-Larsen T , Aarsland D , Larsen JP ((2005) ) Progression of motor impairment and disability in Parkinson disease: A population-based study. Neurology 65: , 1436–1441. |
[9] | Chung EJ , Ki C-S , Lee WY , Kim I-S , Kim J-Y ((2006) ) Clinical features and gene analysis in Korean patients with early-onset Parkinson disease. Arch Neurol 63: , 1170–1174. |
[10] | Ferguson LW , Rajput AH , Rajput A ((2016) ) Early-onset vs. late-onset Parkinson’s disease: A clinical-pathological study. Can J Neurol Sci 43: , 113–119. |
[11] | Wickremaratchi MM , Ben-Shlomo Y , Morris HR ((2009) ) The effect of onset age on the clinical features of Parkinson’s disease. Eur J Neurol 16: , 450–456. |
[12] | Marinus J , Zhu K , Marras C , Aarsland D , van Hilten JJ ((2018) ) Risk factors for non-motor symptoms in Parkinson’s disease. Lancet Neurol 17: , 559–568. |
[13] | Spica V , Pekmezović T , Svetel M , Kostić VS ((2013) ) Prevalence of non-motor symptoms in young-onset versus late-onset Parkinson’s disease. J Neurol 260: , 131–137. |
[14] | Schrag A , Schott JM ((2006) ) Epidemiological, clinical, and genetic characteristics of early-onset parkinsonism. Lancet Neurol 5: , 355–363. |
[15] | Inzelberg R , Korczyn AD ((1994) ) Parkinsonism in adult-onset GM2 gangliosidosis. Mov Disord 9: , 375–377. |
[16] | Diamond SG , Markham CH , Hoehn MM , McDowell FH , Muenter MD ((1989) ) Effect of age at onset on progression and mortality in Parkinson’s disease. Neurology 39: , 1187–1190. |
[17] | Ylikotila P , Tiirikka T , Moilanen JS , Kääriäinen H , Marttila R , Majamaa K ((2015) ) Epidemiology of early-onset Parkinson’s disease in Finland. Parkinsonism Relat Disord 21: , 938–942. |
[18] | Giovannini P , Piccolo I , Genitrini S , Soliveri P , Girotti F , Geminiani G , Scigliano G , Caraceni T ((1991) ) Early-onset Parkinson’s disease. Mov Disord 6: , 36–42. |
[19] | Rana AQ , Siddiqui I , Yousuf MS ((2012) ) Challenges in diagnosis of young onset Parkinson’s disease. J Neurol Sci 323: , 113–116. |
[20] | Winter Y , Bezdolnyy Y , Katunina E , Avakjan G , Reese JP , Klotsche J , Oertel WH , Dodel R , Gusev E ((2010) ) Incidence of Parkinson’s disease and atypical parkinsonism: Russian population-based study. Mov Disord 25: , 349–356. |
[21] | Camerucci E , Stang CD , Hajeb M , Turcano P , Mullan AF , Martin P , Ross OA , Bower JH , Mielke MM , Savica R ((2021) ) Early-onset parkinsonism and early-onset Parkinson’s disease: A population-based study (2010-2015). J Parkinsons Dis 11: , 1197–1207. |
[22] | Schrag A , Ben-Shlomo Y , Brown R , Marsden CD , Quinn N ((1998) ) Young-onset Parkinson’s disease revisited–clinical features, natural history, and mortality. Mov Disord 13: , 885–894. |
[23] | McFarthing K , Buff S , Rafaloff G , Dominey T , Wyse RK , Stott SRW ((2020) ) Parkinson’s disease drug therapies in the clinical trial pipeline: 2020. J Parkinsons Dis 10: , 757–774. |
[24] | Schneider SA , Alcalay RN ((2017) ) Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov Disord 32: , 1504–1523. |
[25] | Schirinzi T , Di Lazzaro G , Sancesario GM , Summa S , Petrucci S , Colona VL , Bernardini S , Pierantozzi M , Stefani A , Mercuri NB , Pisani A ((2020) ) Young-onset and late-onset Parkinson’s disease exhibit a different profile of fluid biomarkers and clinical features. Neurobiol Aging 90: , 119–124. |
[26] | Pagano G , Ferrara N , Brooks DJ , Pavese N ((2016) ) Age at onset and Parkinson disease phenotype. Neurology 86: , 1400–1407. |
[27] | Laperle AH , Sances S , Yucer N , Dardov VJ , Garcia VJ , Ho R , Fulton AN , Jones MR , Roxas KM , Avalos P , West D , Banuelos MG , Shu Z , Murali R , Maidment NT , Van Eyk JE , Tagliati M , Svendsen CN ((2020) ) iPSC modeling of young-onset Parkinson’s disease reveals a molecular signature of disease and novel therapeutic candidates. Nat Med 26: , 289–299. |
[28] | Bonifati V , Rizzu P , van Baren MJ , Schaap O , Breedveld GJ , Krieger E , Dekker MCJ , Squitieri F , Ibanez P , Joosse M , van Dongen JW , Vanacore N , van Swieten JC , Brice A , Meco G , van Duijn CM , Oostra BA , Heutink P ((2003) ) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299: , 256–259. |
[29] | Cherian A , Divya KP ((2020) ) Genetics of Parkinson’s disease. Acta Neurol Belg 120: , 1297–1305. |
[30] | Lunati A , Lesage S , Brice A ((2018) ) The genetic landscape of Parkinson’s disease. Rev Neurol (Paris) 174: , 628–643. |
[31] | Lücking CB , Dürr A , Bonifati V , Vaughan J , De Michele G , Gasser T , Harhangi BS , Meco G , Denèfle P , Wood NW , Agid Y , Brice A , French Parkinson’s Disease Genetics Study Group, European Consortium on Genetic Susceptibility in Parkinson’s Disease ((2000) ) Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med 342: , 1560–1567. |
[32] | Taghavi S , Chaouni R , Tafakhori A , Azcona LJ , Firouzabadi SG , Omrani MD , Jamshidi J , Emamalizadeh B , Shahidi GA , Ahmadi M , Habibi SAH , Ahmadifard A , Fazeli A , Motallebi M , Petramfar P , Askarpour S , Askarpour S , Shahmohammadibeni HA , Shahmohammadibeni N , Eftekhari H , Shafiei Zarneh AE , Mohammadihosseinabad S , Khorrami M , Najmi S , Chitsaz A , Shokraeian P , Ehsanbakhsh H , Rezaeidian J , Ebrahimi Rad R , Madadi F , Andarva M , Alehabib E , Atakhorrami M , Mortazavi SE , Azimzadeh Z , Bayat M , Besharati AM , Harati-Ghavi MA , Omidvari S , Dehghani-Tafti Z , Mohammadi F , Mohammad Hossein Pour B , Noorollahi Moghaddam H , Esmaili Shandiz E , Habibi A , Taherian-Esfahani Z , Darvish H , PaisÁn-Ruiz C ((2018) ) A clinical and molecular genetic study of 50 families with autosomal recessive parkinsonism revealed known and novel gene mutations. Mol Neurobiol 55: , 3477–3489. |
[33] | Kasten M , Hartmann C , Hampf J , Schaake S , Westenberger A , Vollstedt E-J , Balck A , Domingo A , Vulinovic F , Dulovic M , Zorn I , Madoev H , Zehnle H , Lembeck CM , Schawe L , Reginold J , Huang J , König IR , Bertram L , Marras C , Lohmann K , Lill CM , Klein C ((2018) ) Genotype-phenotype relations for the Parkinson’s disease genes Parkin, PINK1, DJ1: MDSGene systematic review. Mov Disord 33: , 730–741. |
[34] | Valente EM , Abou-Sleiman PM , Caputo V , Muqit MMK , Harvey K , Gispert S , Ali Z , Del Turco D , Bentivoglio AR , Healy DG , Albanese A , Nussbaum R , GonzÁlez-Maldonado R , Deller T , Salvi S , Cortelli P , Gilks WP , Latchman DS , Harvey RJ , Dallapiccola B , Auburger G , Wood NW ((2004) ) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304: , 1158–1160. |
[35] | Valente EM , Salvi S , Ialongo T , Marongiu R , Elia AE , Caputo V , Romito L , Albanese A , Dallapiccola B , Bentivoglio AR ((2004) ) PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol 56: , 336–341. |
[36] | Bonifati V , Rohé CF , Breedveld GJ , Fabrizio E , De Mari M , Tassorelli C , Tavella A , Marconi R , Nicholl DJ , Chien HF , Fincati E , Abbruzzese G , Marini P , De Gaetano A , Horstink MW , Maat-Kievit JA , Sampaio C , Antonini A , Stocchi F , Montagna P , Toni V , Guidi M , Dalla Libera A , Tinazzi M , De Pandis F , Fabbrini G , Goldwurm S , de Klein A , Barbosa E , Lopiano L , Martignoni E , Lamberti P , Vanacore N , Meco G , Oostra BA , Italian Parkinson Genetics Network ((2005) ) Early-onset parkinsonism associated with PINK1 mutations: Frequency, genotypes, and phenotypes. Neurology 65: , 87–95. |
[37] | Yamamura Y , Sobue I , Ando K , Iida M , Yanagi T ((1973) ) Paralysis agitans of early onset with marked diurnal fluctuation of symptoms. Neurology 23: , 239–244. |
[38] | Lesage S , Brice A ((2009) ) Parkinson’s disease: From monogenic forms to genetic susceptibility factors. Hum Mol Genet 18: , R48–59. |
[39] | Samaranch L , Lorenzo-Betancor O , Arbelo JM , Ferrer I , Lorenzo E , Irigoyen J , Pastor MA , Marrero C , Isla C , Herrera-Henriquez J , Pastor P ((2010) ) PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133: , 1128–1142. |
[40] | Lohmann E , Periquet M , Bonifati V , Wood NW , De Michele G , Bonnet A-M , Fraix V , Broussolle E , Horstink MWIM , Vidailhet M , Verpillat P , Gasser T , Nicholl D , Teive H , Raskin S , Rascol O , Destée A , Ruberg M , Gasparini F , Meco G , Agid Y , Durr A , Brice A , French Parkinson’s Disease Genetics Study Group, European Consortium on Genetic Susceptibility in Parkinson’s Disease ((2003) ) How much phenotypic variation can be attributed to parkin genotype? Ann Neurol 54: , 176–185. |
[41] | Khan NL , Graham E , Critchley P , Schrag AE , Wood NW , Lees AJ , Bhatia KP , Quinn N ((2003) ) Parkin disease: A phenotypic study of a large case series. Brain J Neurol 126: , 1279–1292. |
[42] | Doherty KM , Silveira-Moriyama L , Parkkinen L , Healy DG , Farrell M , Mencacci NE , Ahmed Z , Brett FM , Hardy J , Quinn N , Counihan TJ , Lynch T , Fox ZV , Revesz T , Lees AJ , Holton JL ((2013) ) Parkin disease: A clinicopathologic entity? JAMA Neurol 70: , 571–579. |
[43] | Ahlskog JE ((2009) ) Parkin and PINK1 parkinsonism may represent nigral mitochondrial cytopathies distinct from Lewy body Parkinson’s disease. Parkinsonism Relat Disord 15: , 721–727. |
[44] | Morgante F , Fasano A , Ginevrino M , Petrucci S , Ricciardi L , Bove F , Criscuolo C , Moccia M , De Rosa A , Sorbera C , Bentivoglio AR , Barone P , De Michele G , Pellecchia MT , Valente EM ((2016) ) Impulsive-compulsive behaviors in parkin-associated Parkinson disease. Neurology 87: , 1436–1441. |
[45] | Abou-Sleiman PM , Healy DG , Quinn N , Lees AJ , Wood NW ((2003) ) The role of pathogenic DJ-1 mutations in Parkinson’s disease. Ann Neurol 54: , 283–286. |
[46] | Kilarski LL , Pearson JP , Newsway V , Majounie E , Knipe MDW , Misbahuddin A , Chinnery PF , Burn DJ , Clarke CE , Marion M-H , Lewthwaite AJ , Nicholl DJ , Wood NW , Morrison KE , Williams-Gray CH , Evans JR , Sawcer SJ , Barker RA , Wickremaratchi MM , Ben-Shlomo Y , Williams NM , Morris HR ((2012) ) Systematic review and UK-based study of PARK2 (parkin), PINK1, PARK7 (DJ-1) and LRRK2 in early-onset Parkinson’s disease. Mov Disord 27: , 1522–1529. |
[47] | Di Fonzo A , Dekker MCJ , Montagna P , Baruzzi A , Yonova EH , Correia Guedes L , Szczerbinska A , Zhao T , Dubbel-Hulsman LOM , Wouters CH , de Graaff E , Oyen WJG , Simons EJ , Breedveld GJ , Oostra BA , Horstink MW , Bonifati V ((2009) ) FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 72: , 240–245. |
[48] | Shojaee S , Sina F , Banihosseini SS , Kazemi MH , Kalhor R , Shahidi G-A , Fakhrai-Rad H , Ronaghi M , Elahi E ((2008) ) Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am J Hum Genet 82: , 1375–1384. |
[49] | PaisÁn-Ruiz C , Guevara R , Federoff M , Hanagasi H , Sina F , Elahi E , Schneider SA , Schwingenschuh P , Bajaj N , Emre M , Singleton AB , Hardy J , Bhatia KP , Brandner S , Lees AJ , Houlden H ((2010) ) Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, FBXO7 and spatacsin mutations. Mov Disord 25: , 1791–1800. |
[50] | Conedera S , Apaydin H , Li Y , Yoshino H , Ikeda A , Matsushima T , Funayama M , Nishioka K , Hattori N ((2016) ) FBXO7 mutations in Parkinson’s disease and multiple system atrophy. Neurobiol Aging 40: , 192.e1–192.e5. |
[51] | Gündüz A , Eken AG , Bilgiç c B , Hanagasi HA , Bilgüvar K , Günel M , Başak AN , Ertan S ((2014) ) FBXO7-R498X mutation: Phenotypic variability from chorea to early onset parkinsonism within a family. Parkinsonism Relat Disord 20: , 1253–1256. |
[52] | Yalcin-Cakmakli G , Olgiati S , Quadri M , Breedveld GJ , Cortelli P , Bonifati V , Elibol B ((2014) ) A new Turkish family with homozygous FBXO7 truncating mutation and juvenile atypical parkinsonism. Parkinsonism Relat Disord 20: , 1248–1252. |
[53] | Wei L , Ding L , Li H , Lin Y , Dai Y , Xu X , Dong Q , Lin Y , Long L ((2018) ) Juvenile-onset parkinsonism with pyramidal signs due to compound heterozygous mutations in the F-Box only protein 7 gene. Parkinsonism Relat Disord 47: , 76–79. |
[54] | Lesage S , Drouet V , Majounie E , Deramecourt V , Jacoupy M , Nicolas A , Cormier-Dequaire F , Hassoun SM , Pujol C , Ciura S , Erpapazoglou Z , Usenko T , Maurage C-A , Sahbatou M , Liebau S , Ding J , Bilgic B , Emre M , Erginel-Unaltuna N , Guven G , Tison F , Tranchant C , Vidailhet M , Corvol J-C , Krack P , Leutenegger A-L , Nalls MA , Hernandez DG , Heutink P , Gibbs JR , Hardy J , Wood NW , Gasser T , Durr A , Deleuze J-F , Tazir M , Destée A , Lohmann E , Kabashi E , Singleton A , Corti O , Brice A , French Parkinson’s Disease Genetics Study (PDG), International Parkinson’s Disease Genomics Consortium (IPDGC) ((2016) ) Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/parkin-dependent mitophagy. Am J Hum Genet 98: , 500–513. |
[55] | Ishihara L , Warren L , Gibson R , Amouri R , Lesage S , Dürr A , Tazir M , Wszolek ZK , Uitti RJ , Nichols WC , Griffith A , Hattori N , Leppert D , Watts R , Zabetian CP , Foroud TM , Farrer MJ , Brice A , Middleton L , Hentati F ((2006) ) Clinical features of Parkinson disease patients with homozygous leucine-rich repeat kinase 2 G2019S mutations. Arch Neurol 63: , 1250–1254. |
[56] | Gorostidi A , Ruiz-Martínez J , Lopez de Munain A , Alzualde A , Martí Massó JF ((2009) ) LRRK2 G2019S and R1441G mutations associated with Parkinson’s disease are common in the Basque Country, but relative prevalence is determined by ethnicity. Neurogenetics 10: , 157–159. |
[57] | Funayama M , Hasegawa K , Ohta E , Kawashima N , Komiyama M , Kowa H , Tsuji S , Obata F ((2005) ) An LRRK2 mutation as a cause for the parkinsonism in the original PARK8 family. Ann Neurol 57: , 918–921. |
[58] | Hasegawa K , Stoessl AJ , Yokoyama T , Kowa H , Wszolek ZK , Yagishita S ((2009) ) Familial parkinsonism: Study of original Sagamihara PARK8 (I2020T) kindred with variable clinicopathologic outcomes. Parkinsonism Relat Disord 15: , 300–306. |
[59] | Ujiie S , Hatano T , Kubo S-I , Imai S , Sato S , Uchihara T , Yagishita S , Hasegawa K , Kowa H , Sakai F , Hattori N ((2012) ) LRRK2 I2020T mutation is associated with tau pathology. Parkinsonism Relat Disord 18: , 819–823. |
[60] | Funayama M , Li Y , Tomiyama H , Yoshino H , Imamichi Y , Yamamoto M , Murata M , Toda T , Mizuno Y , Hattori N ((2007) ) Leucine-rich repeat kinase 2 G2385R variant is a risk factor for Parkinson disease in Asian population. Neuroreport 18: , 273–275. |
[61] | Kim J-M , Lee J-Y , Kim HJ , Kim JS , Shin E-S , Cho J-H , Park SS , Jeon BS ((2010) ) The LRRK2 G2385R variant is a risk factor for sporadic Parkinson’s disease in the Korean population. Parkinsonism Relat Disord 16: , 85–88. |
[62] | Di Fonzo A , Wu-Chou Y-H , Lu C-S , van Doeselaar M , Simons EJ , Rohé CF , Chang H-C , Chen R-S , Weng Y-H , Vanacore N , Breedveld GJ , Oostra BA , Bonifati V ((2006) ) A common missense variant in the LRRK2 gene, Gly2385Arg, associated with Parkinson’s disease risk in Taiwan. Neurogenetics 7: , 133–138. |
[63] | Healy DG , Falchi M , O’Sullivan SS , Bonifati V , Durr A , Bressman S , Brice A , Aasly J , Zabetian CP , Goldwurm S , Ferreira JJ , Tolosa E , Kay DM , Klein C , Williams DR , Marras C , Lang AE , Wszolek ZK , Berciano J , Schapira AHV , Lynch T , Bhatia KP , Gasser T , Lees AJ , Wood NW , International LRRK2 Consortium ((2008) ) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol 7: , 583–590. |
[64] | San Luciano M , Lipton RB , Wang C , Katz M , Zimmerman ME , Sanders AE , Ozelius LJ , Bressman SB , Saunders-Pullman R ((2010) ) Clinical expression of LRRK2 G2019S mutations in the elderly. Mov Disord 25: , 2571–2576. |
[65] | Trinh J , Guella I , Farrer MJ ((2014) ) Disease penetrance of late-onset parkinsonism: A meta-analysis. JAMA Neurol 71: , 1535–1539. |
[66] | Marder K , Wang Y , Alcalay RN , Mejia-Santana H , Tang M-X , Lee A , Raymond D , Mirelman A , Saunders-Pullman R , Clark L , Ozelius L , Orr-Urtreger A , Giladi N , Bressman S , LRRK2 Ashkenazi Jewish Consortium ((2015) ) Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology 85: , 89–95. |
[67] | San Luciano M , Wang C , Ortega RA , Giladi N , Marder K , Bressman S , Saunders-Pullman R , Michael J Fox Foundation LRRK2 Consortium ((2017) ) Sex differences in LRRK2 G2019S and idiopathic Parkinson’s Disease. Ann Clin Transl Neurol 4: , 801–810. |
[68] | Aasly JO , Toft M , Fernandez-Mata I , Kachergus J , Hulihan M , White LR , Farrer M ((2005) ) Clinical features of LRRK2-associated Parkinson’s disease in central Norway. Ann Neurol 57: , 762–765. |
[69] | Lee AJ , Wang Y , Alcalay RN , Mejia-Santana H , Saunders-Pullman R , Bressman S , Corvol J-C , Brice A , Lesage S , Mangone G , Tolosa E , Pont-Sunyer C , Vilas D , Schüle B , Kausar F , Foroud T , Berg D , Brockmann K , Goldwurm S , Siri C , Asselta R , Ruiz-Martinez J , Mondragón E , Marras C , Ghate T , Giladi N , Mirelman A , Marder K , Michael J . Fox LRRK2 Cohort Consortium ((2017) ) Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov Disord 32: , 1432–1438. |
[70] | Marras C , Alcalay RN , Caspell-Garcia C , Coffey C , Chan P , Duda JE , Facheris MF , FernÁndez-Santiago R , Ruíz-Martínez J , Mestre T , Saunders-Pullman R , Pont-Sunyer C , Tolosa E , Waro B , LRRK2 Cohort Consortium ((2016) ) Motor and nonmotor heterogeneity of LRRK2-related and idiopathic Parkinson’s disease. Mov Disord 31: , 1192–1202. |
[71] | Saunders-Pullman R , Stanley K , San Luciano M , Barrett MJ , Shanker V , Raymond D , Ozelius LJ , Bressman SB ((2011) ) Gender differences in the risk of familial parkinsonism: Beyond LRRK2? Neurosci Lett 496: , 125–128. |
[72] | Saunders-Pullman R , Mirelman A , Wang C , Alcalay RN , San Luciano M , Ortega R , Raymond D , Mejia-Santana H , Ozelius L , Clark L , Orr-Utreger A , Marder K , Giladi N , Bressman SB ((2014) ) Olfactory identification in LRRK2 G2019S mutation carriers: A relevant marker? Ann Clin Transl Neurol 1: , 670–678. |
[73] | Somme JH , Molano Salazar A , Gonzalez A , Tijero B , Berganzo K , Lezcano E , Fernandez Martinez M , Zarranz JJ , Gómez-Esteban JC ((2015) ) Cognitive and behavioral symptoms in Parkinson’s disease patients with the G2019S and R1441G mutations of the LRRK2 gene. Parkinsonism Relat Disord 21: , 494–499. |
[74] | Srivatsal S , Cholerton B , Leverenz JB , Wszolek ZK , Uitti RJ , Dickson DW , Weintraub D , Trojanowski JQ , Van Deerlin VM , Quinn JF , Chung KA , Peterson AL , Factor SA , Wood-Siverio C , Goldman JG , Stebbins GT , Bernard B , Ritz B , Rausch R , Espay AJ , Revilla FJ , Devoto J , Rosenthal LS , Dawson TM , Albert MS , Mata IF , Hu S-C , Montine KS , Johnson C , Montine TJ , Edwards KL , Zhang J , Zabetian CP ((2015) ) Cognitive profile of LRRK2-related Parkinson’s disease. Mov Disord 30: , 728–733. |
[75] | Ahn T-B , Kim SY , Kim JY , Park S-S , Lee DS , Min HJ , Kim YK , Kim SE , Kim J-M , Kim H-J , Cho J , Jeon BS ((2008) ) alpha-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology 70: , 43–49. |
[76] | Brueggemann N , Odin P , Gruenewald A , Tadic V , Hagenah J , Seidel G , Lohmann K , Klein C , Djarmati A ((2008) ) Re: Alpha-synuclein gene duplication is present in sporadic Parkinson disease.1294; author reply. Neurology 71: , 1294. |
[77] | Kasten M , Klein C ((2013) ) The many faces of alpha-synuclein mutations. Mov Disord 28: , 697–701. |
[78] | Ricciardi L , Petrucci S , Di Giuda D , Serra L , Spanò B , Sensi M , Ginevrino M , Cocciolillo F , Bozzali M , Valente EM , Fasano A ((2016) ) The Contursi Family 20 years later: Intrafamilial phenotypic variability of the SNCA p.A53T mutation. Mov Disord 31: , 257–258. |
[79] | Krüger R , Kuhn W , Leenders KL , Sprengelmeyer R , Müller T , Woitalla D , Portman AT , Maguire RP , Veenma L , Schröder U , Schöls L , Epplen JT , Riess O , Przuntek H ((2001) ) Familial parkinsonism with synuclein pathology: Clinical and PET studies of A30P mutation carriers. Neurology 56: , 1355–1362. |
[80] | Zarranz JJ , Alegre J , Gómez-Esteban JC , Lezcano E , Ros R , Ampuero I , Vidal L , Hoenicka J , Rodriguez O , Atarés B , Llorens V , Gomez Tortosa E , del Ser T , Muñoz DG , de Yebenes JG ((2004) ) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55: , 164–173. |
[81] | Rosborough K , Patel N , Kalia LV ((2017) ) α-synuclein and parkinsonism: Updates and future perspectives. Curr Neurol Neurosci Rep 17: , 31. |
[82] | Kiely AP , Ling H , Asi YT , Kara E , Proukakis C , Schapira AH , Morris HR , Roberts HC , Lubbe S , Limousin P , Lewis PA , Lees AJ , Quinn N , Hardy J , Love S , Revesz T , Houlden H , Holton JL ((2015) ) Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation. Mol Neurodegener 10: , 41. |
[83] | Martikainen MH , Päivärinta M , Hietala M , Kaasinen V ((2015) ) Clinical and imaging findings in Parkinson disease associated with the A53E SNCA mutation. Neurol Genet 1: , e27. |
[84] | Lesage S , Anheim M , Letournel F , Bousset L , Honoré A , Rozas N , Pieri L , Madiona K , Dürr A , Melki R , Verny C , Brice A , French Parkinson’s Disease Genetics Study Group ((2013) ) G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol 73: , 459–471. |
[85] | Pasanen P , Myllykangas L , Siitonen M , Raunio A , Kaakkola S , Lyytinen J , Tienari PJ , Pöyhönen M , Paetau A ((2014) ) Novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging 35: , 2180.e1–5. |
[86] | Domingo A , Klein C ((2018) ) Genetics of Parkinson disease. Handb Clin Neurol 147: , 211–227. |
[87] | IbÁñez P , Lesage S , Janin S , Lohmann E , Durif F , Destée A , Bonnet A-M , Brefel-Courbon C , Heath S , Zelenika D , Agid Y , Dürr A , Brice A , French Parkinson’s Disease Genetics Study Group ((2009) ) Alpha-synuclein gene rearrangements in dominantly inherited parkinsonism: Frequency, phenotype, and mechanisms. Arch Neurol 66: , 102–108. |
[88] | Edvardson S , Cinnamon Y , Ta-Shma A , Shaag A , Yim Y-I , Zenvirt S , Jalas C , Lesage S , Brice A , Taraboulos A , Kaestner KH , Greene LE , Elpeleg O ((2012) ) A deleterious mutation in DNAJC6 encoding the neuronal-specific clathrin-uncoating co-chaperone auxilin, is associated with juvenile parkinsonism. PLoS One 7: , e36458. |
[89] | Lesage S , Condroyer C , Klebe S , Honoré A , Tison F , Brefel-Courbon C , Dürr A , Brice A , French Parkinson’s Disease Genetics Study Group ((2012) ) Identification of VPS35 mutations replicated in French families with Parkinson disease. Neurology 78: , 1449–1450. |
[90] | Ando M , Funayama M , Li Y , Kashihara K , Murakami Y , Ishizu N , Toyoda C , Noguchi K , Hashimoto T , Nakano N , Sasaki R , Kokubo Y , Kuzuhara S , Ogaki K , Yamashita C , Yoshino H , Hatano T , Tomiyama H , Hattori N ((2012) ) VPS35 mutation in Japanese patients with typical Parkinson’s disease. Mov Disord 27: , 1413–1417. |
[91] | Sheerin U-M , Charlesworth G , Bras J , Guerreiro R , Bhatia K , Foltynie T , Limousin P , Silveira-Moriyama L , Lees A , Wood N ((2012) ) Screening for VPS35 mutations in Parkinson’s disease. Neurobiol Aging 33: , 838.e1–5. |
[92] | Vilariño-Güell C , Wider C , Ross OA , Dachsel JC , Kachergus JM , Lincoln SJ , Soto-Ortolaza AI , Cobb SA , Wilhoite GJ , Bacon JA , Behrouz B , Melrose HL , Hentati E , Puschmann A , Evans DM , Conibear E , Wasserman WW , Aasly JO , Burkhard PR , Djaldetti R , Ghika J , Hentati F , Krygowska-Wajs A , Lynch T , Melamed E , Rajput A , Rajput AH , Solida A , Wu R-M , Uitti RJ , Wszolek ZK , Vingerhoets F , Farrer MJ ((2011) ) VPS35 mutations in Parkinson disease. Am J Hum Genet 89: , 162–167. |
[93] | Zimprich A , Benet-Pagès A , Struhal W , Graf E , Eck SH , Offman MN , Haubenberger D , Spielberger S , Schulte EC , Lichtner P , Rossle SC , Klopp N , Wolf E , Seppi K , Pirker W , Presslauer S , Mollenhauer B , Katzenschlager R , Foki T , Hotzy C , Reinthaler E , Harutyunyan A , Kralovics R , Peters A , Zimprich F , Brücke T , Poewe W , Auff E , Trenkwalder C , Rost B , Ransmayr G , Winkelmann J , Meitinger T , Strom TM ((2011) ) A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 89: , 168–175. |
[94] | Huang Y , Chen X , He X , Guo C , Sun X , Liang F , Long S , Lu X , Feng L , Guo W , Zeng Y , Pei Z ((2017) ) High expression levels of the D686N Parkinson’s disease mutation in VPS35 induces α-synuclein-dependent toxicity in yeast. Mol Med Rep 16: , 254–262. |
[95] | Chen Y-F , Chang Y-Y , Lan M-Y , Chen P-L , Lin C-H ((2017) ) Identification of VPS35 p.D620N mutation-related Parkinson’s disease in a Taiwanese family with successful bilateral subthalamic nucleus deep brain stimulation: A case report and literature review. BMC Neurol 17: , 191. |
[96] | Wider C , Skipper L , Solida A , Brown L , Farrer M , Dickson D , Wszolek ZK , Vingerhoets FJG ((2008) ) Autosomal dominant dopa-responsive parkinsonism in a multigenerational Swiss family. Parkinsonism Relat Disord 14: , 465–470. |
[97] | Deutschländer A , Ross OA , Wszolek ZK (1993) VPS35 related Parkinson disease. In GeneReviews®, Adam MP, Ardinger HH, Pagon RA,Wallace SE, Bean LJ, Mirzaa G, Amemiya A, eds. University of Washington, Seattle. |
[98] | Gan-Or Z , Giladi N , Rozovski U , Shifrin C , Rosner S , Gurevich T , Bar-Shira A , Orr-Urtreger A ((2008) ) Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 70: , 2277–2283. |
[99] | Sidransky E , Samaddar T , Tayebi N ((2009) ) Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset.1424-1425, author reply. Neurology 73: , 1425–1426. |
[100] | Ruskey JA , Greenbaum L , Roncière L , Alam A , Spiegelman D , Liong C , Levy OA , Waters C , Fahn S , Marder KS , Chung W , Yahalom G , Israeli-Korn S , Livneh V , Fay-Karmon T , Alcalay RN , Hassin-Baer S , Gan-Or Z ((2019) ) Increased yield of full GBA sequencing in Ashkenazi Jews with Parkinson’s disease. Eur J Med Genet 62: , 65–69. |
[101] | den Heijer JM , Cullen VC , Quadri M , Schmitz A , Hilt DC , Lansbury P , Berendse HW , van de Berg WDJ , de Bie RMA , Boertien JM , Boon AJW , Contarino MF , van Hilten JJ , Hoff JI , van Mierlo T , Munts AG , van der Plas AA , Ponsen MM , Baas F , Majoor-Krakauer D , Bonifati V , van Laar T , Groeneveld GJ ((2020) ) A large-scale full GBA1 gene screening in Parkinson’s disease in the Netherlands. Mov Disord 35: , 1667–1674. |
[102] | Petrucci S , Ginevrino M , Trezzi I , Monfrini E , Ricciardi L , Albanese A , Avenali M , Barone P , Bentivoglio AR , Bonifati V , Bove F , Bonanni L , Brusa L , Cereda C , Cossu G , Criscuolo C , Dati G , De Rosa A , Eleopra R , Fabbrini G , Fadda L , Garbellini M , Minafra B , Onofrj M , Pacchetti C , Palmieri I , Pellecchia MT , Petracca M , Picillo M , Pisani A , Vallelunga A , Zangaglia R , Di Fonzo A , Morgante F , Valente EM , ITA-GENE-PD Study Group ((2020) ) GBA-related Parkinson’s disease: Dissection of genotype-phenotype correlates in a large Italian cohort. Mov Disord 35: , 2106–2111. |
[103] | Riboldi GM , Di Fonzo AB ((2019) ) GBA, Gaucher disease, and Parkinson’s disease: From genetic to clinic to new therapeutic approaches. Cells 8: , 364. |
[104] | Oeda T , Umemura A , Mori Y , Tomita S , Kohsaka M , Park K , Inoue K , Fujimura H , Hasegawa H , Sugiyama H , Sawada H ((2015) ) Impact of glucocerebrosidase mutations on motor and nonmotor complications in Parkinson’s disease. Neurobiol Aging 36: , 3306–3313. |
[105] | Nichols WC , Pankratz N , Marek DK , Pauciulo MW , Elsaesser VE , Halter CA , Rudolph A , Wojcieszek J , Pfeiffer RF , Foroud T , Parkinson Study Group-PROGENI Investigators ((2009) ) Mutations in GBA are associated with familial Parkinson disease susceptibility and age at onset. Neurology 72: , 310–316. |
[106] | Brockmann K , Srulijes K , Pflederer S , Hauser A-K , Schulte C , Maetzler W , Gasser T , Berg D ((2015) ) GBA-associated Parkinson’s disease: Reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord 30: , 407–411. |
[107] | Brockmann K , Srulijes K , Hauser AK , Schulte C , Csoti I , Gasser T , Berg D ((2011) ) GBA-associated PD presents with nonmotor characteristics. Neurology 77: , 276–280. |
[108] | Fleury V , Brindel P , Nicastro N , Burkhard PR ((2018) ) Descriptive epidemiology of parkinsonism in the Canton of Geneva, Switzerland. Parkinsonism Relat Disord 54: , 30–39. |
[109] | Tse W , Cersosimo MG , Gracies J-M , Morgello S , Olanow CW , Koller W ((2004) ) Movement disorders and AIDS: A review. Parkinsonism Relat Disord 10: , 323–334. |
[110] | Cicarelli G , Pellecchia MT , Maiuri F , Barone P ((1999) ) Brain stem cystic astrocytoma presenting with “pure” parkinsonism. Mov Disord 14: , 364–366. |
[111] | Dickman MS ((2001) ) von Economo encephalitis. Arch Neurol 58: , 1696–1698. |
[112] | Dauer W , Przedborski S ((2003) ) Parkinson’s disease: Mechanisms and models. Neuron 39: , 889–909. |
[113] | Schirinzi T , Grillo P , Di Lazzaro G , Zenuni H , Salimei C , Dams-O’Connor K , Sancesario GM , Mercuri NB , Pisani A ((2021) ) Effects of head trauma and sport participation in young-onset Parkinson’s disease. J Neural Transm 128: , 1185–1193. |
[114] | Ascherio A , Schwarzschild MA ((2016) ) The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol 15: , 1257–1272. |
[115] | Shribman S , Warner TT , Dooley JS ((2019) ) Clinical presentations of Wilson disease. Ann Transl Med 7: , S60. |
[116] | Gregory A , Hayflick S (1993) Neurodegeneration with brain iron accumulation disorders overview. In GeneReviews®, AdamMP, Ardinger HH, Pagon RA,Wallace SE, Bean LJ, Mirzaa G, Amemiya A, eds. University of Washington, Seattle. |
[117] | Donzuso G , Mostile G , Nicoletti A , Zappia M ((2019) ) Basal ganglia calcifications (Fahr’s syndrome): Related conditions and clinical features. Neurol Sci 40: , 2251–2263. |
[118] | Tuschl K , Gregory A , Meyer E , Clayton PT , Hayflick SJ , Mills PB , Kurian MA (1993) SLC39A14 Deficiency. In GeneReviews®, Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, Amemiya A, eds. University of Washington, Seattle. |
[119] | Dorman DC Metal imaging in the brain. In Biometals in Neurodegenerative Diseases Academic Press, pp. 349–362. |
[120] | Tuschl K , Clayton PT , Gospe SM , Mills PB (1993) Dystonia/parkinsonism, hypermanganesemia, polycythemia, and chronic liver disease. In GeneReviews®, Adam MP, Ardinger HH, Pagon RA,Wallace SE, Bean LJ, Mirzaa G, Amemiya A, eds. University of Washington, Seattle. |
[121] | Dedoussis GVZ , Genschel J , Sialvera T-E , Bochow B , Manolaki N , Manios Y , Tsafantakis E , Schmidt H ((2005) ) Wilson disease: High prevalence in a mountainous area of Crete. Ann Hum Genet 69: , 268–274. |
[122] | Bull PC , Thomas GR , Rommens JM , Forbes JR , Cox DW ((1993) ) The Wilson disease gene is a putative copper transporting P-type ATPase similar to the Menkes gene. Nat Genet 5: , 327–337. |
[123] | Harris ED ((2000) ) Cellular copper transport and metabolism. Annu Rev Nutr 20: , 291–310. |
[124] | Scheiber IF , Brůha R , Dušek P ((2017) ) Pathogenesis of Wilson disease. Handb Clin Neurol 142: , 43–55. |
[125] | Pandey N , John S ((2021) ) Kayser-Fleischer ring. In StatPearls StatPearls Publishing, Treasure Island (FL). |
[126] | Yu X-E , Gao S , Yang R-M , Han Y-Z ((2019) ) MR imaging of the brain in neurologic Wilson disease. AJNR Am J Neuroradiol 40: , 178–183. |
[127] | Hogarth P ((2015) ) Neurodegeneration with brain iron accumulation: Diagnosis and management. J Mov Disord 8: , 1–13. |
[128] | Chu Y-T , Lin H-Y , Chen P-L , Lin C-H ((2020) ) Genotype-phenotype correlations of adult-onset-associated Neurodegeneration: Case series and literature review. BMC Neurol 20: , 101. |
[129] | Yang X , Xu Y ((2014) ) Mutations in the ATP13A2 gene and Parkinsonism: A preliminary review. BioMed Res Int 2014: , 371256. |
[130] | Park J-S , Blair NF , Sue CM ((2015) ) The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms. Mov Disord 30: , 770–779. |
[131] | Di Fonzo A , Chien HF , Socal M , Giraudo S , Tassorelli C , Iliceto G , Fabbrini G , Marconi R , Fincati E , Abbruzzese G , Marini P , Squitieri F , Horstink MW , Montagna P , Libera AD , Stocchi F , Goldwurm S , Ferreira JJ , Meco G , Martignoni E , Lopiano L , Jardim LB , Oostra BA , Barbosa ER , Italian Parkinson Genetics Network, Bonifati V ((2007) ) ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 68: , 1557–1562. |
[132] | Najim al-Din AS , Wriekat A , Mubaidin A , Dasouki M , Hiari M ((1994) ) Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol Scand 89: , 347–352. |
[133] | Williams DR , Hadeed A , al-Din ASN , Wreikat A-L , Lees AJ ((2005) ) Kufor Rakeb disease: Autosomal recessive, levodopa-responsive parkinsonism with pyramidal degeneration, supranuclear gaze palsy, and dementia. Mov Disord 20: , 1264–1271. |
[134] | Ramirez A , Heimbach A , Gründemann J , Stiller B , Hampshire D , Cid LP , Goebel I , Mubaidin AF , Wriekat A-L , Roeper J , Al-Din A , Hillmer AM , Karsak M , Liss B , Woods CG , Behrens MI , Kubisch C ((2006) ) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 38: , 1184–1191. |
[135] | Schneider SA , Paisan-Ruiz C , Quinn NP , Lees AJ , Houlden H , Hardy J , Bhatia KP ((2010) ) ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord 25: , 979–984. |
[136] | Behrens MI , Brüggemann N , Chana P , Venegas P , Kägi M , Parrao T , Orellana P , Garrido C , Rojas CV , Hauke J , Hahnen E , GonzÁlez R , Seleme N , FernÁndez V , Schmidt A , Binkofski F , Kömpf D , Kubisch C , Hagenah J , Klein C , Ramirez A ((2010) ) Clinical spectrum of Kufor-Rakeb syndrome in the Chilean kindred with ATP13A2 mutations. Mov Disord 25: , 1929–1937. |
[137] | Malakouti-Nejad M , Shahidi G-A , Rohani M , Shojaee SM , Hashemi M , Klotzle B , Fan J-B , Elahi E ((2014) ) Identification of p.Gln858* in ATP13A2 in two EOPD patients and presentation of their clinical features. Neurosci Lett 577: , 106–111. |
[138] | Crosiers D , Ceulemans B , Meeus B , Nuytemans K , Pals P , Van Broeckhoven C , Cras P , Theuns J ((2011) ) Juvenile dystonia-parkinsonism and dementia caused by a novel ATP13A2 frameshift mutation. Parkinsonism Relat Disord 17: , 135–138. |
[139] | Eiberg H , Hansen L , Korbo L , Nielsen IM , Svenstrup K , Bech S , Pinborg LH , Friberg L , Hjermind LE , Olsen OR , Nielsen JE ((2012) ) Novel mutation in ATP13A2 widens the of spectrum Kufor-Rakeb syndrome (PARK9). Clin Genet 82: , 256–263. |
[140] | Paisan-Ruiz C , Bhatia KP , Li A , Hernandez D , Davis M , Wood NW , Hardy J , Houlden H , Singleton A , Schneider SA ((2009) ) Characterization ofas a locus for dystonia-parkinsonism. Ann Neurol 65: , 19–23. |
[141] | Sina F , Shojaee S , Elahi E , PaisÁn-Ruiz C ((2009) ) R632W mutation insegregates with dystonia-parkinsonism in a consanguineous Iranian family. Eur J Neurol 16: , 101–104. |
[142] | Yoshino H , Tomiyama H , Tachibana N , Ogaki K , Li Y , Funayama M , Hashimoto T , Takashima S , Hattori N ((2010) ) Phenotypic spectrum of patients withmutation and PARK14-linked parkinsonism. Neurology 75: , 1356–1361. |
[143] | Illingworth MA , Meyer E , Chong WK , Manzur AY , Carr LJ , Younis R , Hardy C , McDonald F , Childs AM , Stewart B , Warren D , Kneen R , King MD , Hayflick SJ , Kurian MA ((2014) ) -associated neurodegeneration (PLAN): Further expansion of the clinical, radiological and mutation spectrum associated with infantile and atypical childhood-onset disease. Mol Genet Metab 112: , 183–189. |
[144] | Lehéricy S , Roze E , Goizet C , Mochel F ((2020) ) MRI of neurodegeneration with brain iron accumulation. Curr Opin Neurol 33: , 462–473. |
[145] | Amisha F , Munakomi S ((2021) ) Fahr syndrome. In Stat Pearls StatPearls Publishing, Treasure Island (FL). |
[146] | Saade C , Najem E , Asmar K , Salman R , El Achkar B , Naffaa L ((2019) ) Intracranial calcifications on CT: An updated review. J Radiol Case Rep 13: , 1–18. |
[147] | Westenberger A , Balck A , Klein C ((2019) ) Primary familial brain calcifications: Genetic and clinical update. Curr Opin Neurol 32: , 571–578. |
[148] | Mufaddel AA , Al-Hassani GA ((2014) ) Familial idiopathic basal ganglia calcification (Fahr‘s disease). Neurosci Riyadh Saudi Arab 19: , 171–177. |
[149] | Anagianni S , Tuschl K ((2019) ) Genetic disorders of manganese metabolism. Curr Neurol Neurosci Rep 19: , 33. |
[150] | O’Neal SL , Zheng W ((2015) ) Manganese toxicity upon overexposure: A decade in review. Curr Environ Health Rep 2: , 315–328. |
[151] | Hauser RA , Zesiewicz TA , Rosemurgy AS , Martinez C , Olanow CW ((1994) ) Manganese intoxication and chronic liver failure. Ann Neurol 36: , 871–875. |
[152] | McFarland R , Taylor RW , Turnbull DM ((2010) ) A neurological perspective on mitochondrial disease. Lancet Neurol 9: , 829–840. |
[153] | Finsterer J ((2011) ) Parkinson’s syndrome and Parkinson’s disease in mitochondrial disorders. Mov Disord 26: , 784–791. |
[154] | Tranchant C , Anheim M ((2016) ) Movement disorders in mitochondrial diseases. Rev Neurol (Paris) 172: , 524–529. |
[155] | Ouled Amar Bencheikh B , Senkevich K , Rudakou U , Yu E , Mufti K , Ruskey JA , Asayesh F , Laurent SB , Spiegelman D , Fahn S , Waters C , Monchi O , Dauvilliers Y , Espay AJ , Dupré N , Greenbaum L , Hassin-Baer S , Rouleau GA , Alcalay RN , Fon EA , Gan-Or Z ((2020) ) Variants in the Niemann-Pick type C gene NPC1 are not associated with Parkinson’s disease. Neurobiol Aging 93: , 143.e1–143.e4. |
[156] | Clark LN , Chan R , Cheng R , Liu X , Park N , Parmalee N , Kisselev S , Cortes E , Torres PA , Pastores GM , Vonsattel JP , Alcalay R , Marder K , Honig LL , Fahn S , Mayeux R , Shelanski M , Di Paolo G , Lee JH ((2015) ) Gene-wise association of variants in four lysosomal storage disorder genes in neuropathologically confirmed Lewy body disease. PLoS One 10: , e0125204. |
[157] | Dagan E , Schlesinger I , Ayoub M , Mory A , Nassar M , Kurolap A , Peretz-Aharon J , Gershoni-Baruch R ((2015) ) The contribution of Niemann-Pick SMPD1 mutations to Parkinson disease in Ashkenazi Jews. Parkinsonism Relat Disord 21: , 1067–1071. |
[158] | Foo J-N , Liany H , Bei J-X , Yu X-Q , Liu J , Au W-L , Prakash KM , Tan LC , Tan E-K ((2013) ) Rare lysosomal enzyme gene SMPD1 variant (p.R591C) associates with Parkinson’s disease. Neurobiol Aging 34: , 2890.e13–15. |
[159] | Gan-Or Z , Ozelius LJ , Bar-Shira A , Saunders-Pullman R , Mirelman A , Kornreich R , Gana-Weisz M , Raymond D , Rozenkrantz L , Deik A , Gurevich T , Gross SJ , Schreiber-Agus N , Giladi N , Bressman SB , Orr-Urtreger A ((2013) ) The p.L302P mutation in the lysosomal enzyme gene SMPD1 is a risk factor for Parkinson disease. Neurology 80: , 1606–1610. |
[160] | Robak LA , Jansen IE , van Rooij J , Uitterlinden AG , Kraaij R , Jankovic J , International Parkinson’s Disease Genomics Consortium (IPDGC), Heutink P , Shulman JM ((2017) ) Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140: , 3191–3203. |
[161] | Ylönen S , Siitonen A , Nalls MA , Ylikotila P , Autere J , Eerola-Rautio J , Gibbs R , Hiltunen M , Tienari PJ , Soininen H , Singleton AB , Majamaa K ((2017) ) Genetic risk factors in Finnish patients with Parkinson’s disease. Parkinsonism Relat Disord 45: , 39–43. |
[162] | Alcalay RN , Mallett V , Vanderperre B , Tavassoly O , Dauvilliers Y , Wu RYJ , Ruskey JA , Leblond CS , Ambalavanan A , Laurent SB , Spiegelman D , Dionne-Laporte A , Liong C , Levy OA , Fahn S , Waters C , Kuo S-H , Chung WK , Ford B , Marder KS , Kang UJ , Hassin-Baer S , Greenbaum L , Trempe J-F , Wolf P , Oliva P , Zhang XK , Clark LN , Langlois M , Dion PA , Fon EA , Dupre N , Rouleau GA , Gan-Or Z ((2019) ) SMPD1 mutations, activity, and α-synuclein accumulation in Parkinson’s disease. Mov Disord 34: , 526–535. |
[163] | Silveira-Moriyama L , Moriyama TS , Gabbi TVB , Ranvaud R , Barbosa ER ((2004) ) Chediak-Higashi syndrome with parkinsonism. Mov Disord 19: , 472–475. |
[164] | Uyama E , Hirano T , Ito K , Nakashima H , Sugimoto M , Naito M , Uchino M , Ando M ((1994) ) Adult Chédiak-Higashi syndrome presenting as parkinsonism and dementia. Acta Neurol Scand 89: , 175–183. |
[165] | Bhambhani V , Introne WJ , Lungu C , Cullinane A , Toro C ((2013) ) Chediak-Higashi syndrome presenting as young-onset levodopa-responsive parkinsonism. Mov Disord 28: , 127–129. |
[166] | Stelten BML , van de Warrenburg BPC , Wevers RA , Verrips A ((2019) ) Movement disorders in cerebrotendinous xanthomatosis. Parkinsonism Relat Disord 58: , 12–16. |
[167] | Rubio-Agusti I , Kojovic M , Edwards MJ , Murphy E , Chandrashekar HS , Lachmann RH , Bhatia KP ((2012) ) Atypical parkinsonism and cerebrotendinous xanthomatosis: Report of a family with corticobasal syndrome and a literature review. Mov Disord 27: , 1769–1774. |
[168] | Salen G , Steiner RD ((2017) ) Epidemiology, diagnosis, and treatment of cerebrotendinous xanthomatosis (CTX). J Inherit Metab Dis 40: , 771–781. |
[169] | Roze E , Paschke E , Lopez N , Eck T , Yoshida K , Maurel-Ollivier A , Doummar D , Caillaud C , Galanaud D , Billette de Villemeur T , Vidailhet M , Roubergue A ((2005) ) Dystonia and parkinsonism in GM1 type 3 gangliosidosis. Mov Disord 20: , 1366–1369. |
[170] | Argov Z , Navon R ((1984) ) Clinical and genetic variations in the syndrome of adult GM2 gangliosidosis resulting from hexosaminidase A deficiency. Ann Neurol 16: , 14–20. |
[171] | van Diggelen OP , Thobois S , Tilikete C , Zabot MT , Keulemans JL , van Bunderen PA , Taschner PE , Losekoot M , Voznyi YV ((2001) ) Adult neuronal ceroid lipofuscinosis with palmitoyl-protein thioesterase deficiency: First adult-onset patients of a childhood disease. Ann Neurol 50: , 269–272. |
[172] | Toro C , Shirvan L , Tifft C (1993) HEXA disorders. In GeneReviews®, AdamMP, Ardinger HH, Pagon RA,Wallace SE, Bean LJ, Mirzaa G, Amemiya A, eds. University of Washington, Seattle. |
[173] | Levy A , Lang AE ((2018) ) Ataxia-telangiectasia: A review of movement disorders, clinical features, and genotype correlations. Mov Disord 33: , 1238–1247. |
[174] | Girotra T , Mahajan A , Sidiropoulos C ((2017) ) Levodopa responsive parkinsonism in patients with hemochromatosis: Case presentation and literature review. Case Rep Neurol Med 2017: , 5146723. |
[175] | Nielsen JE , Jensen LN , Krabbe K ((1995) ) Hereditary haemochromatosis: A case of iron accumulation in the basal ganglia associated with a parkinsonian syndrome. J Neurol Neurosurg Psychiatry 59: , 318–321. |
[176] | Costello DJ , Walsh SL , Harrington HJ , Walsh CH ((2004) ) Concurrent hereditary haemochromatosis and idiopathic Parkinson’s disease: A case report series. J Neurol Neurosurg Psychiatry 75: , 631–633. |
[177] | Pietrangelo A ((2004) ) Hereditary hemochromatosis–a new look at an old disease. N Engl J Med 350: , 2383–2397. |
[178] | Segawa M , Nomura Y , Nishiyama N ((2003) ) Autosomal dominant guanosine triphosphate cyclohydrolase I deficiency (Segawa disease). Ann Neurol 54: (Suppl 6), S32–45. |
[179] | Camargos S , Scholz S , Simón-SÁnchez J , PaisÁn-Ruiz C , Lewis P , Hernandez D , Ding J , Gibbs JR , Cookson MR , Bras J , Guerreiro R , Oliveira CR , Lees A , Hardy J , Cardoso F , Singleton AB ((2008) ) DYT16, a novel young-onset dystonia-parkinsonism disorder: Identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurol 7: , 207–215. |
[180] | Masnada S , Martinelli D , Correa-Vela M , Agolini E , Baide-Mairena H , Marcé-Grau A , Parazzini C , Veggiotti P , Perez-Duenas B , Tonduti D ((2021) ) PRKRA-related disorders: Bilateral striatal degeneration in addition to DYT16 spectrum. Mov Disord 36: , 1038–1040. |
[181] | Lemmon ME , Lavenstein B , Applegate CD , Hamosh A , Tekes A , Singer HS ((2013) ) A novel presentation of DYT 16: Acute onset in infancy and association with MRI abnormalities. Mov Disord 28: , 1937–1938. |
[182] | de Carvalho Aguiar P , Sweadner KJ , Penniston JT , Zaremba J , Liu L , Caton M , Linazasoro G , Borg M , Tijssen MAJ , Bressman SB , Dobyns WB , Brashear A , Ozelius LJ ((2004) ) Mutations in the Na+/K+ -ATPase alpha3 geneare associated with rapid-onset dystonia parkinsonism. Neuron 43: , 169–175. |
[183] | Dobyns WB , Ozelius LJ , Kramer PL , Brashear A , Farlow MR , Perry TR , Walsh LE , Kasarskis EJ , Butler IJ , Breakefield XO ((1993) ) Rapid-onset dystonia-parkinsonism. Neurology 43: , 2596–2602. |
[184] | Sweney MT , Newcomb TM , Swoboda KJ ((2015) ) The expanding spectrum of neurological phenotypes in children withmutations, alternating hemiplegia of childhood, rapid-onset dystonia-parkinsonism, CAPOS and beyond. Pediatr Neurol 52: , 56–64. |
[185] | Brashear A , Dobyns WB , de Carvalho Aguiar P , Borg M , Frijns CJM , Gollamudi S , Green A , Guimaraes J , Haake BC , Klein C , Linazasoro G , Münchau A , Raymond D , Riley D , Saunders-Pullman R , Tijssen MAJ , Webb D , Zaremba J , Bressman SB , Ozelius LJ ((2007) ) The phenotypic spectrum of rapid-onset dystonia-parkinsonism (RDP) and mutations in thegene. Brain 130: , 828–835. |
[186] | Bragg DC , Mangkalaphiban K , Vaine CA , Kulkarni NJ , Shin D , Yadav R , Dhakal J , Ton M-L , Cheng A , Russo CT , Ang M , Acuña P , Go C , Franceour TN , Multhaupt-Buell T , Ito N , Müller U , Hendriks WT , Breakefield XO , Sharma N , Ozelius LJ ((2017) ) Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1.E11020-E. Proc Natl Acad Sci U S A 114: , 11028. |
[187] | Lee LV , Maranon E , Demaisip C , Peralta O , Borres-Icasiano R , Arancillo J , Rivera C , Munoz E , Tan K , Reyes MT ((2002) ) The natural history of sex-linked recessive dystonia parkinsonism of Panay, Philippines (XDP). Parkinsonism Relat Disord 9: , 29–38. |
[188] | Lee LV , Rivera C , Teleg RA , Dantes MB , Pasco PMD , Jamora RDG , Arancillo J , Villareal-Jordan RF , Rosales RL , Demaisip C , Maranon E , Peralta O , Borres R , Tolentino C , Monding MJ , Sarcia S ((2011) ) The unique phenomenology of sex-linked dystonia parkinsonism (XDP, DYT3, “Lubag”). Int J Neurosci 121: (Suppl 1), 3–11. |
[189] | Abejero JEE , Jamora RDG , Vesagas TS , Teleg RA , Rosales RL , Anlacan JP , Velasquez MS , Aguilar JA ((2019) ) Long-term outcomes of pallidal deep brain stimulation in X-linked dystonia parkinsonism (XDP): Up to 84 months follow-up and review of literature. Parkinsonism Relat Disord 60: , 81–86. |
[190] | Kilbane C , Witt J , Galifianakis NB , Glass GA , Volz M , Heath S , Starr PA , Ostrem JL ((2018) ) Long-term outcomes of bilateral pallidal deep brain stimulation for X-linked dystonia and parkinsonism. Stereotact Funct Neurosurg 96: , 320–326. |
[191] | Lee LV , Kupke KG , Caballar-Gonzaga F , Hebron-Ortiz M , Müller U ((1991) ) The phenotype of the X-linked dystonia-parkinsonism syndrome. An assessment of 42 cases in the Philippines. Medicine (Baltimore) 70: , 179–187. |
[192] | Niu Y-Q , Yang J-C , Hall DA , Leehey MA , Tassone F , Olichney JM , Hagerman RJ , Zhang L ((2014) ) Parkinsonism in fragile X-associated tremor/ataxia syndrome (FXTAS): Revisited. Parkinsonism Relat Disord 20: , 456–459. |
[193] | Salcedo-Arellano MJ , Wolf-Ochoa MW , Hong T , Amina S , Tassone F , Lechpammer M , Hagerman R , Martínez-Cerdeño V ((2020) ) Parkinsonism versus concomitant Parkinson’s disease in fragile X-associated tremor/ataxia syndrome. Mov Disord Clin Pract 7: , 413–418. |
[194] | Hall DA , Berry-Kravis E ((2018) ) Fragile X syndrome and fragile X-associated tremor ataxia syndrome. Handb Clin Neurol 147: , 377–391. |
[195] | Bird TD (1993) Hereditary ataxia overview. In GeneReviews®, Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, Amemiya A, eds. University of Washington, Seattle. |
[196] | Park H , Kim H-J , Jeon BS ((2015) ) Parkinsonism in spinocerebellar ataxia. BioMed Res Int 2015: , 125273. |
[197] | Kim H-J , Jeon BS , Shin J , Lee W-W , Park H , Jung YJ , Ehm G ((2014) ) Should genetic testing for SCAs be included in the diagnostic workup for MSA? Neurology 83: , 1733–1738. |
[198] | Baloh RH , Salavaggione E , Milbrandt J , Pestronk A ((2007) ) Familial parkinsonism and ophthalmoplegia from a mutation in the mitochondrial DNA helicase twinkle. Arch Neurol 64: , 998–1000. |
[199] | Miguel R , Gago MF , Martins J , Barros P , Vale J , Rosas MJ ((2014) ) -related levodopa-responsive parkinsonism. Clin Neurol Neurosurg 126: , 47–54. |
[200] | Bandettini di Poggio M , Nesti C , Bruno C , Meschini MC , Schenone A , Santorelli FM ((2013) ) Dopamine-agonist responsive Parkinsonism in a patient with the SANDO syndrome caused by POLG mutation. BMC Med Genet 14: , 105. |
[201] | Bitetto G , Malaguti MC , Ceravolo R , Monfrini E , Straniero L , Morini A , Di Giacopo R , Frosini D , Palermo G , Biella F , Ronchi D , Duga S , Taroni F , Corti S , Comi GP , Bresolin N , Giometto B , Di Fonzo A ((2020) ) mutations in patients with Parkinson’s disease and optic atrophy. Parkinsonism Relat Disord 74: , 1–5. |
[202] | Faber I , Martinez ARM , Martins CR , Maia ML , Souza JP , Lourenço CM , Marques W , Montecchiani C , Orlacchio A , Pedroso JL , Barsottini OGP , Ramos CD , Lopes-Cendes Í , Friedman JH , Amorim BJ , França MC ((2018) ) -related parkinsonism: Clinical profile, molecular imaging and l-dopa response. Mov Disord 33: , 1650–1656. |
[203] | Guidubaldi A , Piano C , Santorelli FM , Silvestri G , Petracca M , Tessa A , Bentivoglio AR ((2011) ) Novel mutations incause hereditary spastic paraplegia associated with early-onset levodopa-responsive Parkinsonism. Mov Disord 26: , 553–556. |
[204] | De la Casa-Fages B , FernÁndez-Eulate G , Gamez J , Barahona-Hernando R , Morís G , García-Barcina M , Infante J , Zulaica M , FernÁndez-Pelayo U , Muñoz-Oreja M , Urtasun M , Olaskoaga A , Zelaya V , Jericó I , Saez-Villaverde R , Catalina I , Sola E , Martínez-SÁez E , Pujol A , Ruiz M , Schlüter A , Spinazzola A , Muñoz-Blanco JL , Grandas F , Holt I , Álvarez V , López de Munaín A ((2019) ) Parkinsonism and spastic paraplegia type 7: Expanding the spectrum of mitochondrial Parkinsonism. Mov Disord 34: , 1547–1561. |
[205] | Pedroso JL , Vale TC , Bueno FL , Marussi VHR , Amaral do LLF , França MC , Barsottini OG ((2018) ) SPG7 with parkinsonism responsive to levodopa and dopaminergic deficit. Parkinsonism Relat Disord 47: , 88–90. |
[206] | Magri S , Fracasso V , Plumari M , Alfei E , Ghezzi D , Gellera C , Rusmini P , Poletti A , Di Bella D , Elia AE , Pantaleoni C , Taroni F ((2018) ) Concurrent AFG3L2 and SPG7 mutations associated with syndromic parkinsonism and optic atrophy with aberrant OPA1 processing and mitochondrial network fragmentation. Hum Mutat 39: , 2060–2071. |
[207] | Mallaret M , Lagha-Boukbiza O , Biskup S , Namer IJ , Rudolf G , Anheim M , Tranchant C ((2014) ) SPG15: A cause of juvenile atypical levodopa responsive parkinsonism. J Neurol 261: , 435–437. |
[208] | Araujo FMM , Junior WM , Tomaselli PJ , Pimentel ÂV , Macruz Brito MC , Tumas V ((2020) ) SPG15: A rare correlation with atypical juvenile parkinsonism responsive to Levodopa. Mov Disord Clin Pract 7: , 842–844. |
[209] | Shi C-H , Zhang S-Y , Yang Z-H , Yang J , Shang D-D , Mao C-Y , Liu H , Hou H-M , Shi M-M , Wu J , Xu Y-M ((2016) ) A novel RAB39B gene mutation in X-linked juvenile parkinsonism with basal ganglia calcification. Mov Disord 31: , 1905–1909. |
[210] | Jaberi E , Rohani M , Shahidi GA , Nafissi S , Arefian E , Soleimani M , Moghadam A , Arzenani MK , Keramatian F , Klotzle B , Fan J-B , Turk C , Steemers F , Elahi E ((2016) ) Mutation in ADORA1 identified as likely cause of early-onset parkinsonism and cognitive dysfunction. Mov Disord 31: , 1004–1011. |
[211] | Kuipers DJS , Carr J , Bardien S , Thomas P , Sebate B , Breedveld GJ , van Minkelen R , Brouwer RWW , van Ijcken WFJ , van Slegtenhorst MA , Bonifati V , Quadri M ((2018) ) PTRHD1 Loss-of-function mutation in an african family with juvenile-onset Parkinsonism and intellectual disability. Mov Disord 33: , 1814–1819. |
[212] | Wilson GR , Sim JCH , McLean C , Giannandrea M , Galea CA , Riseley JR , Stephenson SEM , Fitzpatrick E , Haas SA , Pope K , Hogan KJ , Gregg RG , Bromhead CJ , Wargowski DS , Lawrence CH , James PA , Churchyard A , Gao Y , Phelan DG , Gillies G , Salce N , Stanford L , Marsh APL , Mignogna ML , Hayflick SJ , Leventer RJ , Delatycki MB , Mellick GD , Kalscheuer VM , D’Adamo P , Bahlo M , Amor DJ , Lockhart PJ ((2014) ) Mutations in RAB39B cause X-linked intellectual disability and early-onset Parkinson disease with α-synuclein pathology. Am J Hum Genet 95: , 729–735. |
[213] | Khodadadi H , Azcona LJ , Aghamollaii V , Omrani MD , Garshasbi M , Taghavi S , Tafakhori A , Shahidi GA , Jamshidi J , Darvish H , PaisÁn-Ruiz C ((2017) ) PTRHD1 (C2orf79) mutations lead to autosomal-recessive intellectual disability and parkinsonism. Mov Disord 32: , 287–291. |
[214] | Mata IF , Jang Y , Kim C-H , Hanna DS , Dorschner MO , Samii A , Agarwal P , Roberts JW , Klepitskaya O , Shprecher DR , Chung KA , Factor SA , Espay AJ , Revilla FJ , Higgins DS , Litvan I , Leverenz JB , Yearout D , Inca-Martinez M , Martinez E , Thompson TR , Cholerton BA , Hu S-C , Edwards KL , Kim K-S , Zabetian CP ((2015) ) The RAB39B p.G192R mutation causes X-linked dominant Parkinson’s disease. Mol Neurodegener 10: , 50. |
[215] | Laxova R , Brown ES , Hogan K , Hecox K , Opitz JM ((1985) ) An X-linked recessive basal ganglia disorder with mental retardation. Am J Med Genet 21: , 681–689. |
[216] | Giannandrea M , Bianchi V , Mignogna ML , Sirri A , Carrabino S , D’Elia E , Vecellio M , Russo S , Cogliati F , Larizza L , Ropers H-H , Tzschach A , Kalscheuer V , Oehl-Jaschkowitz B , Skinner C , Schwartz CE , Gecz J , Van Esch H , Raynaud M , Chelly J , de Brouwer APM , Toniolo D , D’Adamo P ((2010) ) Mutations in the small GTPase gene RAB39B are responsible for X-linked mental retardation associated with autism, epilepsy, and macrocephaly. Am J Hum Genet 86: , 185–195. |
[217] | Woodbury-Smith M , Deneault E , Yuen RKC , Walker S , Zarrei M , Pellecchia G , Howe JL , Hoang N , Uddin M , Marshall CR , Chrysler C , Thompson A , Szatmari P , Scherer SW ((2017) ) Mutations in RAB39B in individuals with intellectual disability, autism spectrum disorder, and macrocephaly. Mol Autism 8: , 59. |
[218] | Puschmann A ((2017) ) New genes causing hereditary Parkinson’s disease or parkinsonism. Curr Neurol Neurosci Rep 17: , 66. |
[219] | Ciammola A , Carrera P , Di Fonzo A , Sassone J , Villa R , Poletti B , Ferrari M , Girotti F , Monfrini E , Buongarzone G , Silani V , Cinnante CM , Mignogna ML , D’Adamo P , Bonati MT ((2017) ) X-linked parkinsonism with intellectual disability caused by novel mutations and somatic mosaicism in RAB39B gene. Parkinsonism Relat Disord 44: , 142–146. |
[220] | Walker IM , Riboldi GM , Drummond P , Saade-Lemus S , Martin-Saavedra JS , Frucht S , Bardakjian TM , Gonzalez-Alegre P , Deik A ((2021) ) PPP2R5D genetic mutations and early-onset parkinsonism. Ann Neurol 89: , 194–195. |
[221] | Kim CY , Wirth T , Hubsch C , Németh AH , Okur V , Anheim M , Drouot N , Tranchant C , Rudolf G , Chelly J , Tatton-Brown K , Blauwendraat C , Vonsattel JPG , Cortes E , Alcalay RN , Chung WK ((2021) ) Reply to “PPP2R5D Genetic Mutations and Early Onset Parkinsonism.”. Ann Neurol 89: , 195–196. |
[222] | Hetzelt KLML , Kerling F , Kraus C , Rauch C , Thiel CT , Winterholler M , Reis A , Zweier C ((2021) ) Early-onset parkinsonism in PPP2R5D-related neurodevelopmental disorder. Eur J Med Genet 64: , 104123. |
[223] | Kim CY , Wirth T , Hubsch C , Németh AH , Okur V , Anheim M , Drouot N , Tranchant C , Rudolf G , Chelly J , Tatton-Brown K , Blauwendraat C , Vonsattel JPG , Cortes E , Alcalay RN , Chung WK ((2020) ) Early-onset parkinsonism is a manifestation of the PPP2R5D p.E200K mutation. Ann Neurol 88: , 1028–1033. |
[224] | Morales-Briceño H , Mohammad SS , Post B , Fois AF , Dale RC , Tchan M , Fung VSC ((2020) ) Clinical and neuroimaging phenotypes of genetic parkinsonism from infancy to adolescence. Brain 143: , 751–770. |
[225] | Köroğlu ç , Baysal L , Cetinkaya M , Karasoy H , Tolun A ((2013) ) DNAJC6 is responsible for juvenile parkinsonism with phenotypic variability. Parkinsonism Relat Disord 19: , 320–324. |
[226] | Olgiati S , Quadri M , Fang M , Rood JPMA , Saute JA , Chien HF , Bouwkamp CG , Graafland J , Minneboo M , Breedveld GJ , Zhang J , International Parkinsonism Genetics Network, Verheijen FW , Boon AJW , Kievit AJA , Jardim LB , Mandemakers W , Barbosa ER , Rieder CRM , Leenders KL , Wang J , Bonifati V ((2016) ) DNAJC6 mutations associated with early-onset Parkinson’s disease. Ann Neurol 79: , 244–256. |
[227] | Krebs CE , Karkheiran S , Powell JC , Cao M , Makarov V , Darvish H , Di Paolo G , Walker RH , Shahidi GA , Buxbaum JD , De Camilli P , Yue Z , PaisÁn-Ruiz C ((2013) ) The Sac1 domain of SYNJ1 identified mutated in a family with early-onset progressive Parkinsonism with generalized seizures. Hum Mutat 34: , 1200–1207. |
[228] | Kirola L , Behari M , Shishir C , Thelma BK ((2016) ) Identification of a novel homozygous mutation Arg459Pro in SYNJ1 gene of an Indian family with autosomal recessive juvenile Parkinsonism. Parkinsonism Relat Disord 31: , 124–128. |
[229] | Ben Romdhan S , Sakka S , Farhat N , Triki S , Dammak M , Mhiri C ((2018) ) A novel SYNJ1 mutation in a Tunisian family with juvenile Parkinson’s disease associated with epilepsy. J Mol Neurosci 66: , 273–278. |
[230] | Quadri M , Fang M , Picillo M , Olgiati S , Breedveld GJ , Graafland J , Wu B , Xu F , Erro R , Amboni M , Pappatà S , Quarantelli M , Annesi G , Quattrone A , Chien HF , Barbosa ER , International Parkinsonism Genetics Network, Oostra BA , Barone P , Wang J , Bonifati V ((2013) ) Mutation in the SYNJ1 gene associated with autosomal recessive, early-onset Parkinsonism. Hum Mutat 34: , 1208–1215. |
[231] | Chang IJ , Hahn SH ((2017) ) The genetics of Wilson disease. Handb Clin Neurol 142: , 19–34. |
[232] | Di Fonzo A , Monfrini E , Erro R ((2018) ) Genetics of movement disorders and the practicing clinician; who and what to test for? Curr Neurol Neurosci Rep 18: , 37. |
[233] | Langston JW , Wiley JC , Tagliati M ((2018) ) Optimizing Parkinson’s disease diagnosis: The role of a dual nuclear imaging algorithm. NPJ Parkinsons Dis 4: , 5. |
[234] | Banks KP , Peacock JG , Clemenshaw MN , Kuo PH ((2019) ) Optimizing the diagnosis of parkinsonian syndromes with 123I-ioflupane brain SPECT. AJR Am J Roentgenol 213: , 243–253. |
[235] | Sikk K , Haldre S , Aquilonius S-M , Taba P ((2011) ) Manganese-induced parkinsonism due to ephedrone abuse. Parkinsons Dis 2011: , 865319. |
[236] | Schneider SA , Alcalay RN ((2020) ) Precision medicine in Parkinson’s disease: Emerging treatments for genetic Parkinson’s disease. J Neurol 267: , 860–869. |


