
Foslevodopa/Foscarbidopa Is Well Tolerated and Maintains Stable Levodopa and Carbidopa Exposure Following Subcutaneous Infusion
Abstract
Background:
Foslevodopa/foscarbidopa, formerly known as ABBV-951, is a formulation of levodopa/carbidopa prodrugs with solubility that allows for subcutaneous (SC) infusion and is in development for the treatment of motor complications for patients with advanced Parkinson’s disease (aPD).
Objective:
The current work characterizes the levodopa (LD) and carbidopa (CD) pharmacokinetics (PK) following SC infusions of foslevodopa/foscarbidopa delivered at four different infusion rates in PD patients.
Methods:
This was a Phase 1, single ascending dose, single-blind study conducted in 28 adult male and female subjects at seven sites in the United States. Foslevodopa/foscarbidopa was administered via abdominal SC infusion in PD patients over 72 hours. Patients were stratified in 4 groups and received a fixed dose of foslevodopa/foscarbidopa based on their oral daily LD intake. Serial plasma PK samples were collected to assay for LD and CD concentrations. Safety and tolerability were assessed throughout the study.
Results:
LD exposure quickly reached steady state and remained stable with minimal fluctuations. Foslevodopa/foscarbidopa infusion provides stable LD and CD exposures compared to oral LD/CD dosing with the average steady-state exposure ranging from 747-4660 ng/mL for the different groups.
Conclusion:
Foslevodopa/foscarbidopa was able to provide stable LD and CD exposures in PD patients over 72 hours via SC route of delivery with very low fluctuation in LD concentration level across a wide range of clinically relevant exposures. Foslevodopa/foscarbidopa had a favorable safety profile. The low PK fluctuation following foslevodopa/foscarbidopa infusion is expected to maintain LD exposure to treat aPD patients within a narrow therapeutic window.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disease and is characterized by progressive degeneration of the dopaminergic system resulting in bradykinesia, rigidity, resting tremor, and postural instability. Levodopa (LD), with a peripheral inhibitor of the aromatic L-amino acid decarboxylase such as carbidopa (CD), is the most effective symptomatic treatment for PD [1]; however, as PD progresses, oral medication inadequately controls symptoms due to an increasingly narrowing therapeutic window [2]. Duodopa (levodopa-carbidopa intestinal gel; LCIG), also known as Duopa (carbidopa-levodopa enteral suspension in the United States) provides superior efficacy compared to oral LD/CD by delivering a continuous infusion directly to the small intestine [3]; however, this delivery requires the surgical placement of a jejunal tube via percutaneous endoscopic gastrostomy.
Foslevodopa/foscarbidopa, formerly known as ABBV-951, is a formulation of LD/CD phosphate prodrugs. By generating prodrugs of LD and CD the solubility is increased by > 100-fold which allows for a highly concentrated foslevodopa/foscarbidopa solution and enables its administration through a minimally invasive subcutaneous (SC) infusion. Upon delivery to the body, foslevodopa/foscarbidopa is quickly and almost completely metabolized to LD/CD by alkaline phosphatases. Due to the high aqueous solubility of these monophosphate prodrugs, the continuous subcutaneous infusion (CSCI) of foslevodopa/foscarbidopa alone can achieve high sustained and steady-state therapeutic levels of plasma LD. Foslevodopa/foscarbidopa is in development for the treatment of motor complications for patients with advanced PD (aPD) and is intended to be delivered continuously for 24 h/day. The stability of LD exposure from foslevodopa/foscarbidopa CSCI has been already demonstrated in healthy volunteers [4]. The current work characterizes the LD and CD pharmacokinetics (PK) following SC infusions of foslevodopa/foscarbidopa delivered at four different infusion rates in PD patients.
METHODS
Study design and eligibility
This was a Phase 1, single ascending dose, single-blind study conducted in adult male and female subjects (45 to 85 years old) with a diagnosis of idiopathic PD, who were LD responsive, in general good health, and on a stable oral LD regimen receiving at least three doses per day. A total of 28 subjects (N = 28) were initially enrolled in the study (N =13 in Part 1 and N = 15 in Part 2) from seven sites in the United States. Part 1 of this study used a 4:1 foslevodopa:foscarbidopa ratio; however, a clinical study demonstrated that the bioavailability of CD was higher than expected following foslevodopa/foscarbidopa CSCI [4]. Following consultation with the United States Food and Drug Administration (FDA), the drug formulation was modified during the study to match the 4:1 exposure of oral LD/CD, which was obtained by reducing the amount of foscarbidopa relative to the amount of foslevodopa. The new ABBV-951 formulation, with a 20:1 foslevodopa:foscarbidopa ratio, was then administered in an additional 15 subjects (Part 2) and subsequently established as the formulation for the intended commercial product. Part 2 of this study, which is described in this manuscript, consisted of four treatment groups with four subjects in each group who received a 24 h/day dose of foslevodopa/foscarbidopa by CSCI via an infusion pump for 72 consecutive hours. Each dose was delivered as a foslevodopa/foscarbidopa SC bolus followed by a single CSCI to the abdomen at a constant rate.
The study protocol and informed consent form were approved by the Institutional Review Board at each participating site prior to the initiation of any screening or study-specific procedures. Written informed consent was obtained from each individual participating in the study. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines of 1975, as defined by the International Conference on Harmonization.
Study procedures
All study participants required ongoing LD/CD treatment for control of their PD symptoms to be eligible for participation in the study; thus, to maintain symptomatic control all subjects received active treatment with foslevodopa/foscarbidopa during the study treatment period. During treatment with foslevodopa/foscarbidopa no oral LD containing medications were allowed. Each subject had to be on a stable oral LD/CD dose for at least 4 weeks prior to the screening visit. Upon enrollment, the levodopa equivalent daily dose (LED) was calculated from the LD-containing medications taken by each subject prior to commencing foslevodopa/foscarbidopa. To keep subjects’ efficacy response to their previous treatments during the study, subjects were assigned to one of the four predefined foslevodopa/foscarbidopa dose groups that most closely matched their stable oral LD/CD therapy. The foslevodopa/foscarbidopa dose levels for each group were projected to deliver plasma LD/CD exposures that were comparable to those resulting from their usual oral treatment regimen LD/CD (Table 1).
Table 1
Summary of dose groups and steady state levodopa exposure
| Dosing | Daily oral | Foslevodopa | Average steady |
| group | LD intake range (mg) | dose (mg per 24 h period) | state LD exposure (mg/mL)* |
| 1 | ≤750 | 960 | 747 |
| 2 | 751–1250 | 2400 | 2380 |
| 3 | 1251–1750 | 3600 | 2920 |
| 4 | > 1750 | 4800 | 4660 |
*Steady state exposure estimated from hours 16–72.
To assess local tolerability and safety of the foslevodopa/foscarbidopa infusion, during the study treatment period, each subject was simultaneously administered a foslevodopa/foscarbidopa infusion and a saline placebo infusion, in parallel, at two infusion sites on the opposite sides of the abdomen. The side on which active foslevodopa/foscarbidopa was administered was randomly assigned, and subjects remained blinded to the treatment administered at each infusion site for the duration of the study.
Confinement at the study site began on Day –2. On Day –1 (1 day prior to beginning of foslevodopa/foscarbidopa infusion), subjects continued to take oral LD/CD according to their usual stable regimen, and timing of all meals and oral LD/CD doses were recorded. On the evening of Day –1, approximately 10 h prior to the initiation of study drug infusion, subjects discontinued their oral LD/CD regimen. On the morning of the following day (Study Day 1), after an approximate 10 h washout period from their oral LD/CD regimen, study drug infusion began (there was no Study Day 0). No sooner than 4 h after the end of the study drug infusion period, subjects resumed their habitual oral LD/CD regimen.
Subjects were monitored for adverse events; vital signs, electrocardiogram (ECG) and clinical laboratory tests were collected as well as medical device product complaint. Skin tolerability was assessed using the Infusion Site Evaluation Scale, a two-part numeric (0–7) and letter (A–G) grade scale, where 7 and G indicated the worst outcomes.
Pharmacokinetic sampling and assessments
Serial blood samples for determination of LD and CD concentrations were collected following initiation of foslevodopa/foscarbidopa doses. Over a 72 h period, blood samples were collected prior to priming of catheter, prior to infusion (0 h), and at 0.5, 1, 2, 4, 8, 12, 16, 24, 28, 32, 36, 40, 48, 52, 56, 60, 64, 72 (immediately prior to the end of infusion), 72.5, 73, 74, and 76 h after start of infusion on Day 1.
Plasma concentrations of LD and CD were determined using a validated liquid chromatography method with tandem mass spectrometric detection. The lower limit of quantitation (LLOQ) for LD and CD was established at 9.99 ng/mL and 9.27 ng/mL, respectively. For foslevodopa/foscarbidopa administration, values for the pharmacokinetic parameters of LD were determined using noncompartmental methods in Phoenix WinNonlin (Version 6.2, Pharsight Cor., Mountain View, CA). The maximum observed plasma concentration (Cmax), area under the plasma concentration-time curve (AUC), average plasma concentration (Cave) and minimum observed plasma concentration (Cmin). The degree of fluctuation for LD was determined from hours 16–72 as a transformation of the determined PK parameters (Cmax-Cmin)/Cave for each subject. As a comparator to the LD PK following the foslevodopa/foscarbidopa infusion, oral LD PK data was simulated using linear scaling from previously published oral LD/CD data in healthy volunteers [4]. The simulations represent 3 separate oral doses of 100/400 mg LD/CD administered every 5 h. The steady state LD exposure was determined for each subject as the AUC from hours 16–72 divided by 56 h (time difference over the AUC16–72 interval) following foslevodopa/foscarbidopa infusion. In order to address the dose proportionality of LD PK following foslevodopa/foscarbidopa infusion, the steady state LD exposure was fit using a linear regression model against the foslevodopa dose per 24-h period and the R2 value was determined.
Data availability statement
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial-level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
This clinical trial data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html.
RESULTS
Demographics
A total of 15 subjects (4 women and 11 men) were enrolled in Part 2 of this study. The mean age of the subjects was 66.4 years (range 52–84) and the mean weight was 82.5 kg (range 53–107). Subjects had a mean height of 171 cm (range 152–188). Eleven subjects were white (73%), three subjects were black (20%), and one subject was Native Hawaiian or Pacific Islander (7%).
Pharmacokinetic results
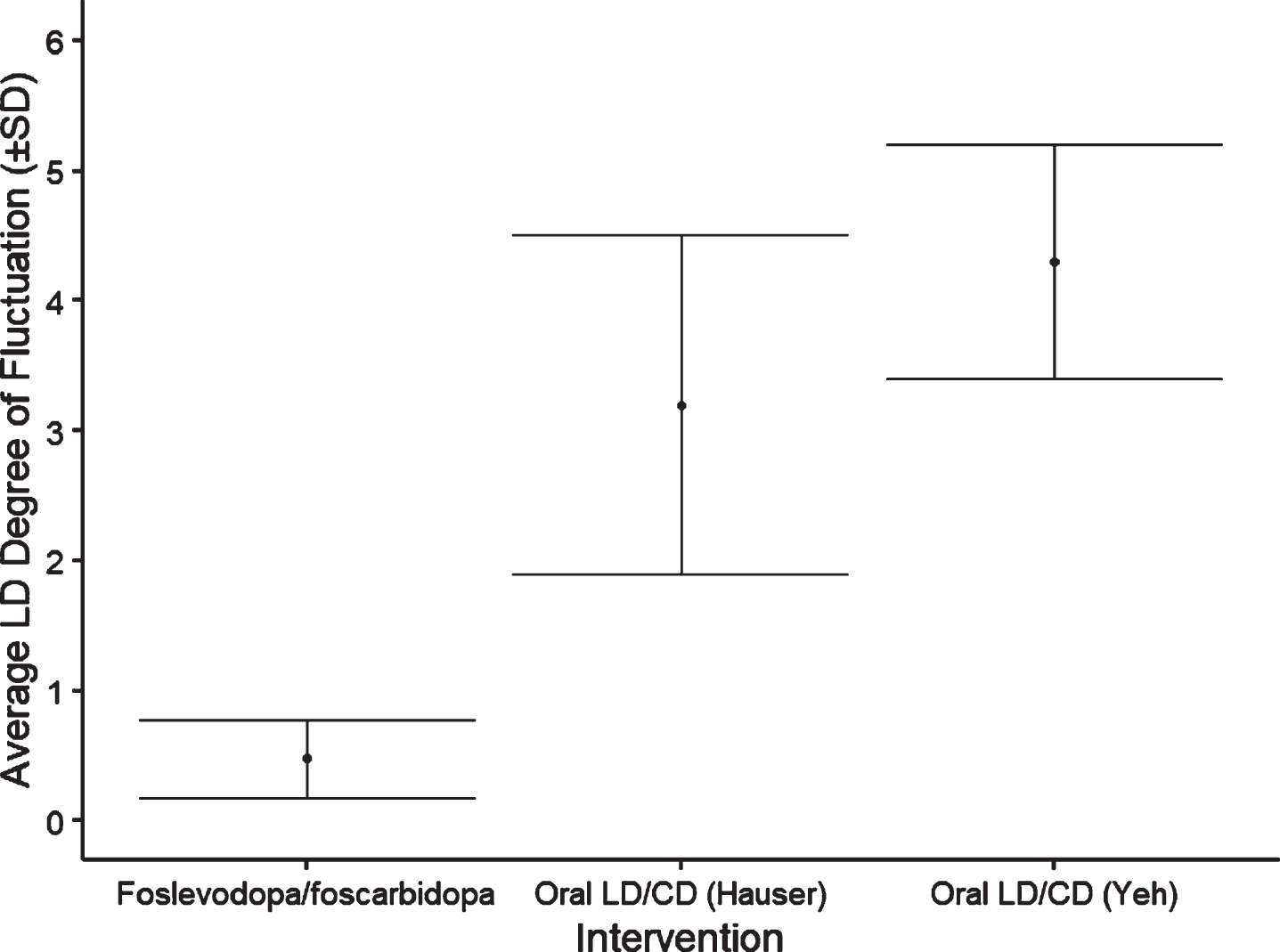
Results from 14/15 subjects who completed the study showed that following foslevodopa/foscarbidopa SC infusion, LD exposure quickly reached steady state and remained stable with minimal fluctuations (Fig. 1 and Table 1). The range of average steady-state exposure was 747–4660 ng/mL for the different groups. The LD PK appeared to be dose proportional following foslevodopa/foscarbidopa infusion as a linear regression model worked well to fit the foslevodopa/foscarbidopa dose vs LD exposure curve with an R2 value of 0.94 (Fig. 2). To assess PK fluctuation differences, the average LD and CD exposures following foslevodopa/foscarbidopa infusion were compared to simulated exposures from oral LD/CD (Fig. 3). Foslevodopa/foscarbidopa infusion provides stable LD and CD exposures compared to oral LD/CD dosing. Similar to the simulated oral LD/CD results, the degree of PK fluctuation in foslevodopa/foscarbidopa (0.31–0.66) is also significantly lower compared to historically reported values (3.2–4.3) for oral LD/CD degree of fluctuation [5, 6] (Fig. 4). The low PK fluctuation following foslevodopa/foscarbidopa infusion is expected to maintain exposure to treat aPD patients within a narrow therapeutic window.
Fig. 1
LD exposure (±SD) following foslevodopa/foscarbidopa infusion in PD patients.
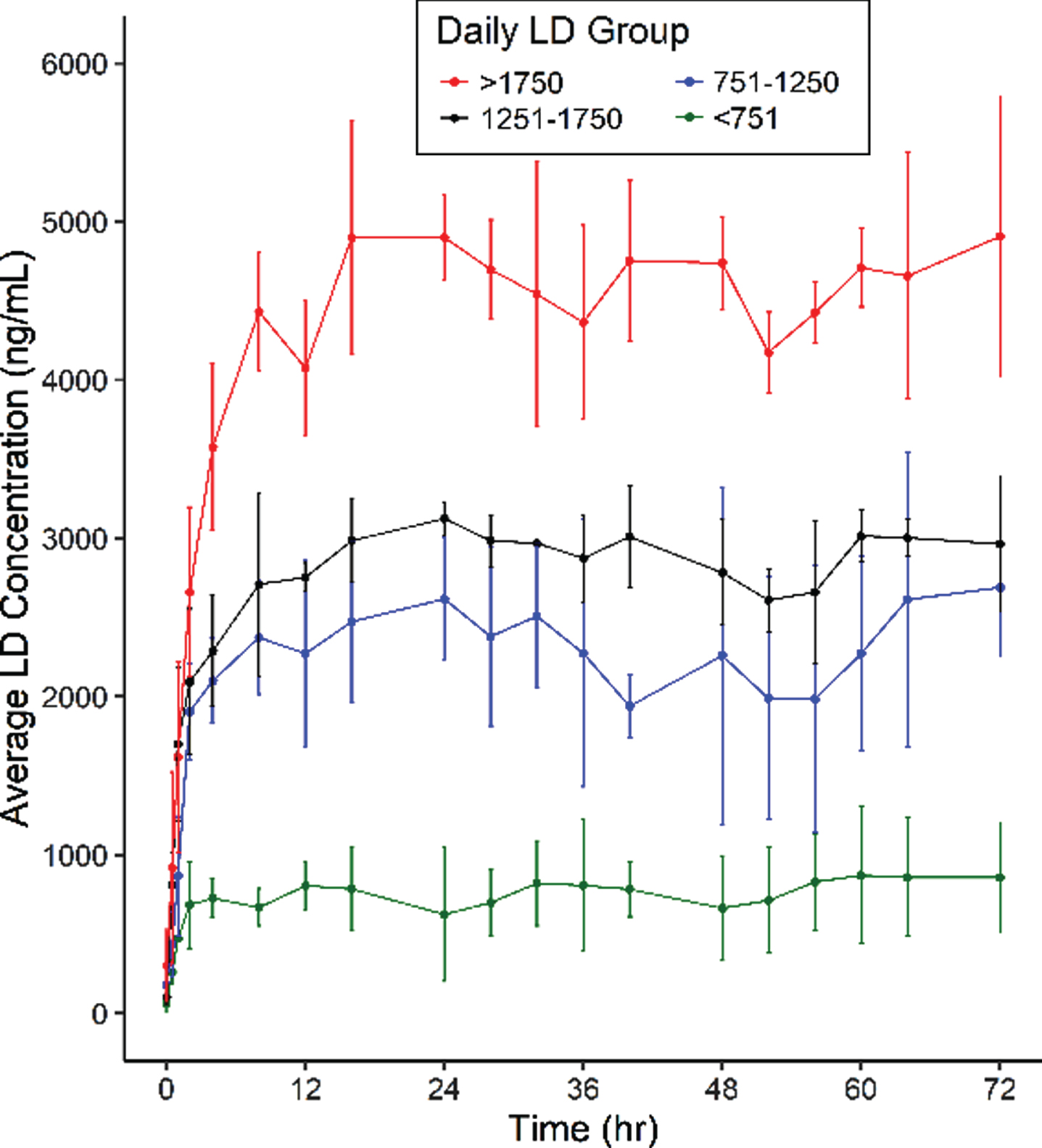
Fig. 2
Assessment of LD dose proportionality following foslevodopa/foscarbidopa infusion.
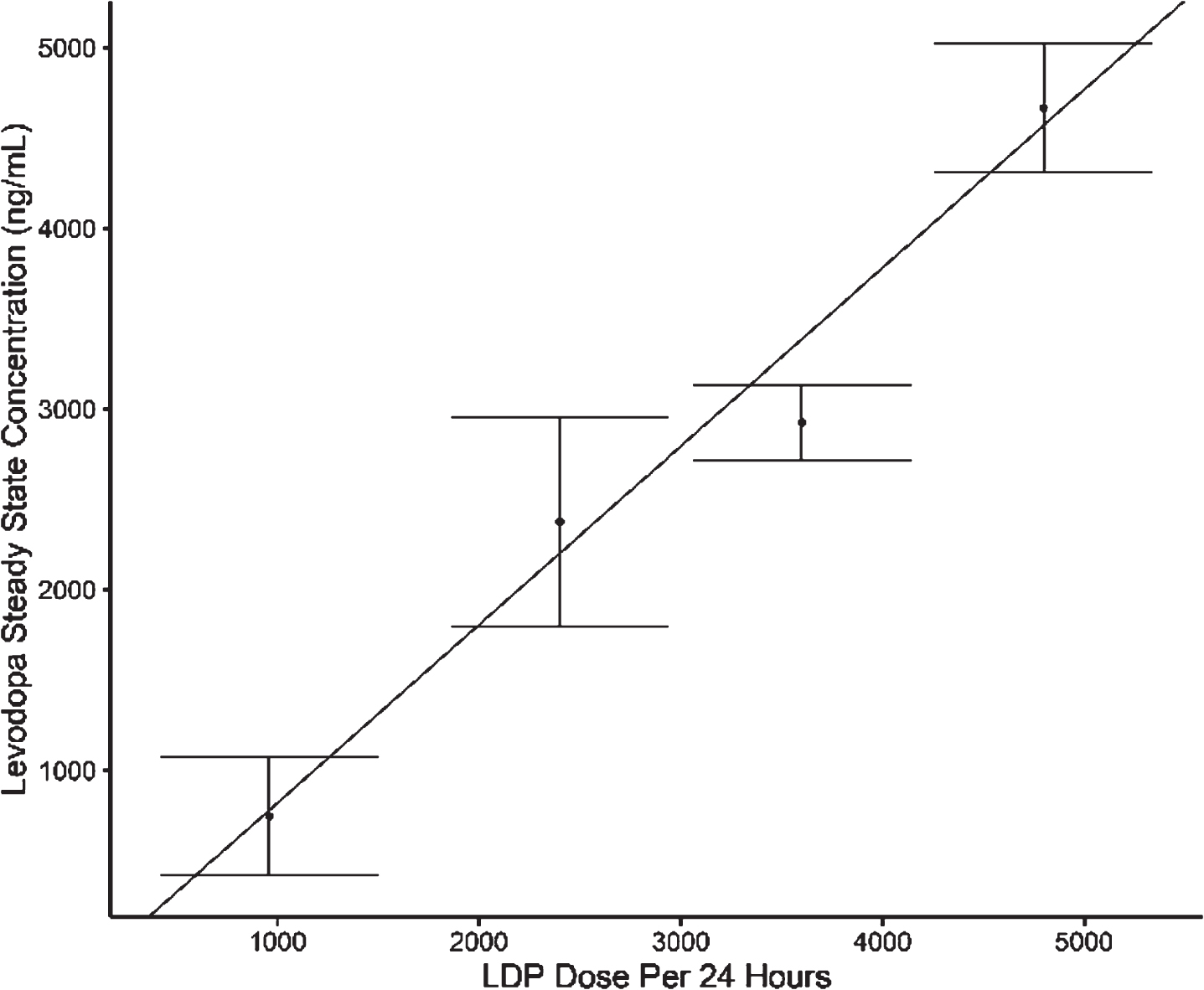
Fig. 3
LD and CD exposure following foslevodopa/foscarbidopa infusion in PD patients compared to oral LD/CD dosing. *Oral dosing simulated as 400/100 mg LD/CD TID.
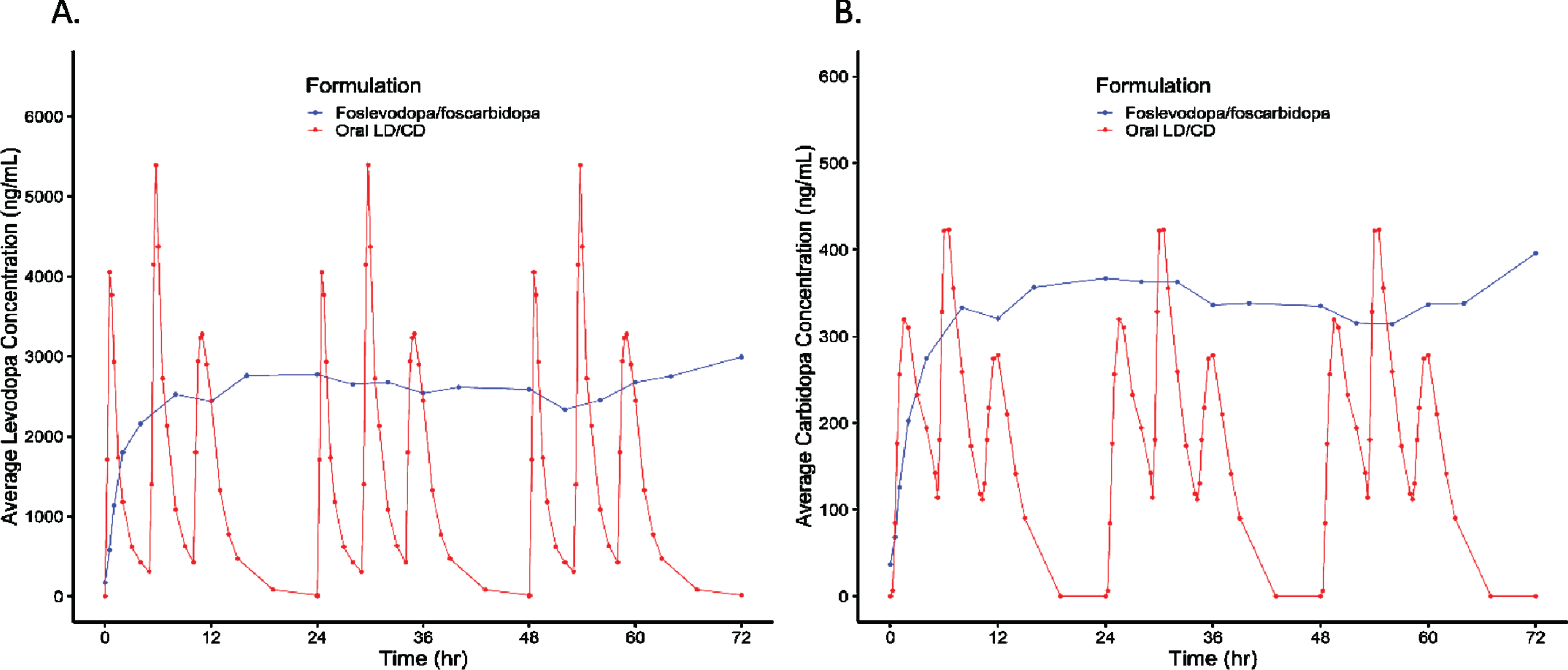
Fig. 4
Foslevodopa/foscarbidopa LD degree of fluctuation compared to oral immediate release LD/CD. *Degree of fluctuation determined as (Cmax-Cmin)/Cave from hours 16–72 for foslevodopa/foscarbidopa.
Safety
Eleven subjects (11/15, 73.3%) experienced a total of 26 adverse events. Five subjects (33.3%) had adverse events that were considered to have a possible causal relationship with the study treatment (Table 2); six of these events occurred within the first 24 hours of dosing and included infusion site pain, infusion site swelling, dizziness, dyskinesia, paraesthesia, and euphoric mood. All adverse events were mild or moderate in severity, except for one severe adverse event of anxiety reported by one subject in dose group 3 (10/200 mg CDP/LDP loading dose followed by 180/3600 mg CDP/LDP per 24 h for 72 h) that led the subject to interrupt drug administration and prematurely discontinue from the study.
Table 2
Overview of treatment emergent adverse events
| ABBV-951 | ABBV-951 | ABBV-951 | ABBV-951 | Total | |
| 10/200 mg | 10/200 mg | 10/200 mg | 10/200 mg | (N = 15) | |
| CDP/LDP loading dose followed by 48/960 mg | CDP/LDP loading dose followed by 120/2400 mg | CDP/LDP loading dose followed by 180/3600 mg | CDP/LDP loading dose followed by 240/4800 mg | ||
| CDP/LDP per 24 h for 72 h | CDP/LDP per 24 h for 72 h | CDP/LDP per 24 h for 72 h | CDP/LDP per 24 h for 72 h | ||
| (N = 4) | (N = 4) | (N = 3) | (N = 4) | ||
| Any adverse event | 1 (25%) | 4 (100%) | 2 (66.7%) | 4 (100%) | 11 (73.3%) |
| Cardiac disorders | 1 (25%) | 0 | 0 | 0 | 1 (6.7%) |
| Bradycardia | 1 (25%) | 0 | 0 | 0 | 1 (6.7%) |
| Gastrointestinal disorders | 0 | 1 (25%) | 1 (33.3%) | 0 | 2 (13.3%) |
| Diarrhea | 0 | 1 (25%) | 0 | 0 | 1 (6.7%) |
| Dry mouth | 0 | 0 | 1 (33.3%) | 0 | 1 (6.7%) |
| Sensitivity of teeth | 0 | 0 | 1 (33.3%) | 0 | 1 (6.7%) |
| General disorders and administration site conditions | 0 | 2 (50%) | 1 (33.3%) | 3 (75%) | 6 (40%) |
| Application site dermatitis | 0 | 0 | 0 | 1 (25%) | 1 (6.7%) |
| Application site erythema | 0 | 1 (25%) | 0 | 0 | 1 (6.7%) |
| Infusion site erythema | 0 | 1 (25%) | 0 | 1 (25%) | 2 (13.3%) |
| Infusion site nodule | 0 | 1 (25%) | 0 | 0 | 1 (6.7%) |
| Infusion site pain | 0 | 0 | 1 (33.3%) | 1 (25%) | 2 (13.3%) |
| Infusion site swelling | 0 | 0 | 1 (33.3%) | 0 | 1 (6.7%) |
| Nodule | 0 | 1 (25%) | 0 | 0 | 1 (6.7%) |
| Oedema peripheral | 0 | 0 | 0 | 1 (25%) | 1 (6.7%) |
| Injury, poisoning and procedural complications | 0 | 0 | 1 (33.3%) | 0 | 1 (6.7%) |
| Muscle strain | 0 | 0 | 1 (33.3%) | 0 | 1 (6.7%) |
| Investigations | 0 | 0 | 1 (33.3%) | 0 | 1 (6.7%) |
| Blood creatine phosphokinase increased | 0 | 0 | 1 (33.3%) | 0 | 1 (6.7%) |
| Troponin increased | 0 | 0 | 1 (33.3%) | 0 | 1 (6.7%) |
| Musculoskeletal and connective tissue disorders | 0 | 1 (25%) | 0 | 0 | 1 (6.7%) |
| Musculoskeletal pain | 0 | 1 (25%) | 0 | 0 | 1 (6.7%) |
| Nervous system disorders | 0 | 1 (25%) | 2 (66.7%) | 1 (25%) | 4 (26.7%) |
| Dizziness | 0 | 0 | 2 (66.7%) | 0 | 2 (13.3%) |
| Dyskinesia | 0 | 1 (25%) | 0 | 0 | 1 (6.7%) |
| Headache | 0 | 0 | 0 | 1 (25%) | 1 (6.7%) |
| Paraesthesia | 0 | 0 | 1 (33.3%) | 0 | 1 (6.7%) |
| Psychiatric disorders | 0 | 1 (25%) | 1 (33.3%) | 0 | 2 (13.3%) |
| Anxiety | 0 | 0 | 1 (33.3%) | 0 | 1 (6.7%) |
| Euphoric mood | 0 | 0 | 1 (33.3%) | 0 | 1 (6.7%) |
| Sleep disorder | 0 | 1 (25%) | 0 | 0 | 1 (6.7%) |
| Skin and subcutaneous tissue disorders | 0 | 1 (25%) | 0 | 0 | 1 (6.7%) |
| Erythema | 0 | 1 (25%) | 0 | 0 | 1 (6.7%) |
| Vascular disorders | 0 | 1 (25%) | 0 | 1 (25%) | 2 (13.3%) |
| Hypertensive crisis | 0 | 1 (25%) | 0 | 0 | 1 (6.7%) |
| Orthostatic hypotension | 0 | 0 | 0 | 1 (25%) | 1 (6.7%) |
There were no notable skin reactions at the Infusion Site evaluation Scale (Table 3) for any subject in the study (all subjects had numeric Grades≤2 and letter Grades of A) (Table 4).
Table 3
Site Evaluation Scale
| Dermal response | Description |
| 0 | No evidence of irritation |
| 1 | Minimal erythema, barely perceptible |
| 2 | Definite erythema, readily visible, minimal edema or minimal popular response |
| 3 | Erythema and papules |
| 4 | Definite edema |
| 5 | Erythema, edema, and papules |
| 6 | Vesicular eruption |
| 7 | Strong reaction spreading beyond test site |
| Other effects | |
| A | Slight glazed appearance |
| B | Marked glazing |
| C | Glazing with peeling and cracking |
| F | Glazing with fissures |
| G | Film of dried serous exudate covering all or part of the patch site |
| H | Small petechial erosions and/or scabs |
Table 4
Infusion site evaluation
| Numeric grade | Letter grade | Hour 0 | Hour 12 | Hour 24 | Hour 48 | Hour 72 | Hour 78 | Highest grade during study | |
| (N = 15) | (N = 14) | (N = 15) | (N = 14) | (N = 14) | (N = 13) | (N = 15) | |||
| Foslevodopa/foscarbidopa | 0 | 15 (100%) | 12 (85.7%) | 15 (100%) | 14 (100%) | 13 (92.9%) | 13 (100%) | 13 (86.7%) | |
| 1 | 0 | 2 (14.3%) | 0 | 0 | 1 (7.1%) | 0 | 2 (13.3%) | ||
| Placebo | 0 | 15 (100%) | 13 (92.3%) | 15 (100%) | 15 (100%) | 13 (100%) | 13 (100%) | 14 (93.3%) | |
| 1 | 0 | 1 (7.1%) | 0 | 0 | 0 | 0 | 1 (6.7%) | ||
| Foslevodopa/foscarbidopa | A | 15 (100%) | 14 (100%) | 15 (100%) | 14 (100%) | 14 (100%) | 13 (100%) | 15 (100%) | |
| Placebo | A | 15 (100%) | 14 (100%) | 15 (100%) | 14 (100%) | 13 (100%) | 13 (100%) | 15 (100%) |
DISCUSSION
Previous work demonstrated that foslevodopa/foscarbidopa SC infusion was able to maintain stable LD exposure in healthy volunteers [4]; however, tolerable LD exposures in healthy volunteers are notably lower than what is reported in PD patients. High LD exposures in non-dopamine deficient healthy volunteers are associated with troublesome side effects making the exploration of higher foslevodopa/foscarbidopa doses in the healthy volunteer study population impractical.
In this study, four different doses (infusion rates) of foslevodopa/foscarbidopa were administered subcutaneously to the abdomen of PD patients assigned to the corresponding dose group based on their daily oral LED intake, continuously, 24 h/day, for 3 consecutive days. The foslevodopa/foscarbidopa infusion rates for Groups 2, 3, and 4 were all higher than previously administered in healthy volunteers.
The variability in LD plasma concentration that results from an unpredictable absorption of oral LD tablets, due to erratic gastric emptying [7], in combination with loss of storage capacity in remaining dopaminergic nerve terminals in the striatum [2] and the short (approximately 1.5–2 h) half-life of LD in the presence of an aromatic amino acid decarboxylase inhibitor (such as CD) [8, 9] result in progressively inadequate control of motor symptoms and lead to the onset of motor complications. Therefore, patients with aPD tend to increase the total amount and the frequency of intake of oral LD therapy. The advantage of continuous LD infusion compared to oral immediate release LD/CD has previously been demonstrated in aPD patients using LCIG [3]. The current work demonstrates that the formulation of LD and CD prodrugs was able to achieve stable LD exposures with all dose regimens through SC administration rather than via surgical delivery to the jejunum. The LD degree of fluctuation following foslevodopa/foscarbidopa administration (0.48±0.30) was similar to the LD degree of fluctuation following LCIG administration previously reported in a phase 1 study of 0.52±0.20 [10]. The degree of fluctuation following foslevodopa/foscarbidopa administration is also much lower than previously reported with oral LD/CD dosing [5, 6] (Fig. 4). Furthermore, since foslevodopa/foscarbidopa is administered subcutaneously, the gut is bypassed and there is no impact of food on the LD systemic absorption.
Although the LD therapeutic window narrows for many patients with aPD, the LD concentration needed to achieve optimal motor symptom control is highly variable from patient to patient. A previously published pharmacokinetic study showed a steady state LD concentration range from approximately 1500 to 4000 ng/mL in PD patients receiving therapeutic doses of LCIG [11] with an average of around 2910 ng/mL [10]. Results from the current study show that the average LD steady state exposure ranged from 747 to 4660 ng/mL following foslevodopa/foscarbidopa administration (Table 1). This suggests that SC infusion of foslevodopa/foscarbidopa, without any other LD-containing medications or catechol-o-methyl-transferase (COMT)-inhibitors, can achieve the LD exposure required to treat the majority of aPD patients. It is important to note that, when LD/CD is administered orally in patients with aPD, LD exposures have been recorded as high as 6000–9000 ng/mL [11] which indicates that LD infusion therapies can control motor symptoms and prevent motor fluctuations without excessive LD exposure, possibly reducing the risk of adverse events such as peak-dose dyskinesia. Finally, when steady state LD exposure was compared to foslevodopa/foscarbidopa dose, LD exposure appears to increase in a dose proportional manner (Fig. 2). Foslevodopa/foscarbidopa dose is intended to be modified by patients to optimally control their motor symptoms and the established dose proportionality suggests that modifications in dose will result in predictable exposure changes and symptomatic control.
Safety discussion
All reported adverse events were mild or moderate in severity, with the exception of one case of severe anxiety, which led to a premature discontinuation from the study. The most frequently reported skin event in subjects receiving foslevodopa/foscarbidopa was mild infusion site reaction; one subject reported moderate infusion site pain. No notable infusion site skin reactions were reported based on the rating scales. No clinically significant changes from baseline to study end in laboratory measurements, vital signs, suicidal behavior/ideation were observed. The reported adverse events did not show any trend or pattern, and no new safety issues were identified from this study.
The systemic and local (skin) safety profile of foslevodopa/foscarbidopa emerging from this study is consistent to that reported in a previous study, where healthy volunteers were exposed to foslevodopa/foscarbidopa for up to 72 h [4]. Although the duration of exposure is the same for the two studies (72 h), the doses of foslevodopa/foscarbidopa administered to PD patients in this study (Groups 2–4) are higher than those administered to healthy volunteers [4]. As healthy volunteers would not be dopamine deficient, they would not have tolerated therapeutic doses needed for PD patients. The higher doses did not seem to have any correlation with the reported systemic or local adverse events.
To minimize the risk of bias in reporting skin-related adverse events, each subject was concomitantly receiving an infusion of foslevodopa/foscarbidopa and an equal volume of saline in the opposite side of the abdomen. The attribution of the regimen to the side of the abdomen was randomly determined and both the subjects and raters were blinded to such determination. No differences were reported between infusion sites exposed to saline and those exposed to foslevodopa/foscarbidopa, confirming that even at high doses, foslevodopa/foscarbidopa has a benign safety profile for up to 72 consecutive hours.
This study showed that a 72 h CSCI of foslevodopa/foscarbidopa reaches a stable LD PK profile independent from the dose (infusion rate) administered and it is able to provide alone, in a dose-proportional manner, the wide LD exposures needed to control motor symptoms and manage motor fluctuations in the majority of patients with aPD. A long-term study to confirm the safety and tolerability of 24 h/daily continuous infusions of individualized therapeutic doses of foslevodopa/foscarbidopa in patients with aPD is warranted.
ACKNOWLEDGMENTS
AbbVie provided financial support for the study and participated in the design, study conduct, analysis and interpretation of data, as well as the writing, review, and approval of the publication. Medical writing support was provided by Wesley Wayman, PhD, an employee of AbbVie.
CONFLICTS OF INTEREST
MR, WL, MN, and MFF are AbbVie employees and may own stock.
REFERENCES
[1] | Olanow CW , Watts RL , Koller WC ((2001) ) An algorithm (decision tree) for the management of Parkinson’s disease (2001): Treatment guidelines. Neurology 56: , S1–S88. |
[2] | Olanow CW , Obeso JA , Stocchi F ((2006) ) Continuous dopamine-receptor treatment of Parkinson’s disease: Scientific rationale and clinical implications. Lancet Neurol 5: , 677–687. |
[3] | Olanow CW , Kieburtz K , Odin P , Espay AJ , Standaert DG , Fernandez HH , Vanagunas A , Othman AA , Widnell KL , Robieson WZ , Pritchett Y , Chatamra K , Benesh J , Lenz RA , Antonini A , Group LHS ((2014) ) Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson’s disease: A randomised, controlled, double-blind, double-dummy study. Lancet Neurol 13: , 141–149. |
[4] | Rosebraugh M , Voight EA , Moussa EM , Jameel F , Lou X , Zhang GGZ , Mayer PT , Stolarik D , Carr RA , Enright BP , Liu W , Facheris MF , Kym PR ((2021) ) Foslevodopa/foscarbidopa: A new subcutaneous treatment for Parkinson’s disease. Ann Neurol 90: , 52–61. |
[5] | Yeh KC , August TF , Bush DF , Lasseter KC , Musson DG , Schwartz S , Smith ME , Titus DC ((1989) ) Pharmacokinetics and bioavailability of Sinemet CR: A summary of human studies. Neurology 39: , 25–38. |
[6] | Hauser RA , Ellenbogen AL , Metman LV , Hsu A , O’Connell MJ , Modi NB , Yao HM , Kell SH , Gupta SK ((2011) ) Crossover comparison of IPX066 and a standard levodopa formulation in advanced Parkinson’s disease. Mov Disord 26: , 2246–2252. |
[7] | Nutt JG , Woodward WR , Hammerstad JP , Carter JH , Anderson JL ((1984) ) The “on-off” phenomenon in Parkinson’s disease. Relation to levodopa absorption and transport. N Engl J Med 310: , 483–488. |
[8] | Hardie RJ , Malcolm SL , Lees AJ , Stern GM , Allen JG ((1986) ) The pharmacokinetics of intravenous and oral levodopa in patients with Parkinson’s disease who exhibit on-off fluctuations. Br J Clin Pharmacol 22: , 429–436. |
[9] | Nutt JG , Woodward WR , Anderson JL ((1985) ) The effect of carbidopa on the pharmacokinetics of intravenously administered levodopa: The mechanism of action in the treatment of parkinsonism. Ann Neurol 18: , 537–543. |
[10] | Nyholm D , Odin P , Johansson A , Chatamra K , Locke C , Dutta S , Othman AA ((2013) ) Pharmacokinetics of levodopa, carbidopa, and 3-O-methyldopa following 16-hour jejunal infusion of levodopa-carbidopa intestinal gel in advanced Parkinson’s disease patients. AAPS J 15: , 316–323. |
[11] | Othman AA , Rosebraugh M , Chatamra K , Locke C , Dutta S ((2017) ) Levodopa-carbidopa intestinal gel pharmacokinetics: Lower variability than oral levodopa-carbidopa. J Parkinsons Dis 7: , 275–278. |


