
Gene Therapy for Parkinson’s Disease Associated with GBA1 Mutations
Abstract
Human genetic studies as well as studies in animal models indicate that lysosomal dysfunction plays a key role in the pathogenesis of Parkinson’s disease. Among the lysosomal genes involved, GBA1 has the largest impact on Parkinson’s disease risk. Deficiency in the GBA1 encoded enzyme glucocerebrosidase (GCase) leads to the accumulation of the GCase glycolipid substrates glucosylceramide and glucosylsphingosine and ultimately results in toxicity and inflammation and negatively affect many clinical aspects of Parkinson’s disease, including disease risk, the severity of presentation, age of onset, and likelihood of progression to dementia. These findings support the view that re-establishing normal levels of GCase enzyme activity may reduce the progression of Parkinson’s disease in patients carrying GBA1 mutations. Studies in mouse models indicate that PR001, a AAV9 vector-based gene therapy designed to deliver a functional GBA1 gene to the brain, suggest that this therapeutic approach may slow or stop disease progression. PR001 is currently being evaluated in clinical trials with Parkinson’s disease patients carrying GBA1 mutations.
INTRODUCTION
Parkinson’s disease (PD) is a disease affecting the peripheral and central nervous systems that results in the dysfunction and loss of certain neuronal populations and clinically manifests with a plethora of motor and a non-motor symptoms. The lysosome is responsible for degradation and recycling of various proteins and other cellular components, and it is now widely recognized that lysosomal dysfunction plays a role in the pathogenesis of PD. Lysosomal dysfunction underlies the pathological accumulation of α-synuclein, a major component of the Lewy body intracellular aggregates, and leads to toxicity and inflammation. Human genetic studies have identified over 40 potentially causative and risk associated genes for Parkinson’s disease, and many of these genes are implicated in lysosomal function or lysosomal trafficking, indicating that lysosome dysfunction is a common denominator underlying Parkinson’s disease pathology. Of these, GBA1, which encodes the lysosomal enzyme glucocerebrosidase (GCase), stands out due to the relatively high prevalence of pathological mutations, the effect such mutations on the risk of developing PD, and their aggravating effect on clinical course of the disease. Animal model studies support the hypothesis that increasing GCase levels in PD patients’ CNS is a promising therapeutic approach to slow or stop disease progression.
PARKINSON’S DISEASE ASSOCIATED WITH THE GBA1 GENE
Glucosylceramidase (GCase) is a lysosomal enzyme encoded by the GBA1 gene and catalyzes the conversion of glucosylceramide (GluCer) into glucose and ceramide. GCase deficiency leads to the accumulation of GluCer, glucosylsphingosine (GluSph), and other glycolipid derangements, resulting in toxicity and inflammation [1–3]. GCase may play additional roles in lysosome function and protein homeostasis, and has been hypothesized to directly or indirectly impact α-synuclein aggregation [4–7]. Approximately 300 GBA1 mutations have been reported [8]. These mutations lead to various degrees of loss of function of GCase activity, and this is reflected in the variable extent and severity of clinical manifestations ranging from asymptomatic carriage to perinatal hydrops fatalis caused by Gaucher disease [2, 9, 10]. Gaucher disease has traditionally been subdivided into three phenotypes, herein referred to as GD1, GD2, and GD3. GD1 is considered to be the non-neuronopathic form, classically manifesting with hepatosplenomegaly and attendant sequelae, although GD1 patients are at high risk of developing PD as described below. GD2 and GD3 are the neuronopathic forms, having neurological in additional to the peripheral manifestations that accompany GD1 [11]. Of the two neuronopathic forms, GD2 is the more severe form, typically presenting in infanthood with multiple bulbar signs and symptoms including swallowing difficulty and supranuclear gaze palsy, opisthotonos, spasticity, epilepsy, and failure to thrive. Death typically occurs before age 2 [12]. GD3 usually presents in early childhood or adolescence with horizontal supranuclear gaze palsy and is often accompanied with other neurological manifestations including ataxia, myoclonic epilepsy, and learning disabilities [13, 14]. The clinical course tends to be slow. The distinction between GD2 and GD3 can be challenging, suggesting that the various forms of GD represents a phenotypic continuum [15].
GBA1 mutation(s) also increases one’s risk of developing PD, from 5× for mono-allelic carriers to up to 20× for bi-allelic carriers, as well as of related neurodegenerative disorders such as Lewy Body Dementia or Parkinson’s Disease Dementia [16–18]. It is estimated that 5% to 25% of patients with PD carry GBA1 mutation(s), and up to 30% of carriers will develop PD by age 80 [19, 20]. In light of the relatively large effect of GBA1 mutations on PD risk and the relatively high prevalence of these mutations, GBA1 mutations are considered to be the most important genetic cause of PD.
The extent and severity of the diseases caused by GBA1 mutations relate to the extent of the GCase deficiency. For example, GCase activity is reduced by approximately 20% to 50% in PD-GBA, often greater than 90% in neuropathic forms of GD, to virtually 100% in hydrops fetalis [2, 21–23]. The risk of developing PD follows the same “dose effect” principle: those with more severe GBA1 mutations (e.g., “neuronopathic” mutations) and/or biallelic carriage generally have lower GCase activity and are at higher risk of PD relative to patients with less severe mutations or mutations in only 1 chromosomal copy [22, 24–27].
The spectrum of the clinical manifestations of PD-GBA is similar to idiopathic PD, but PD-GBA tends to manifest at an earlier age, progress more rapidly, and be accompanied by cognitive impairment, and therefore is considered a more aggressive form of PD [19, 28–31]. Even in the absence of specific GBA1 mutations, reduced GCase activity in patients with idiopathic PD has been reported by multiple groups, further lending support to the hypothesis that reduced GCase is central to the development of PD [22, 32–34]. The observation of a genetic “dose-effect” of the level of GCase reduction correlating with the severity of disease, strongly supports the notion that an increased level of GCase would be beneficial in these individuals.
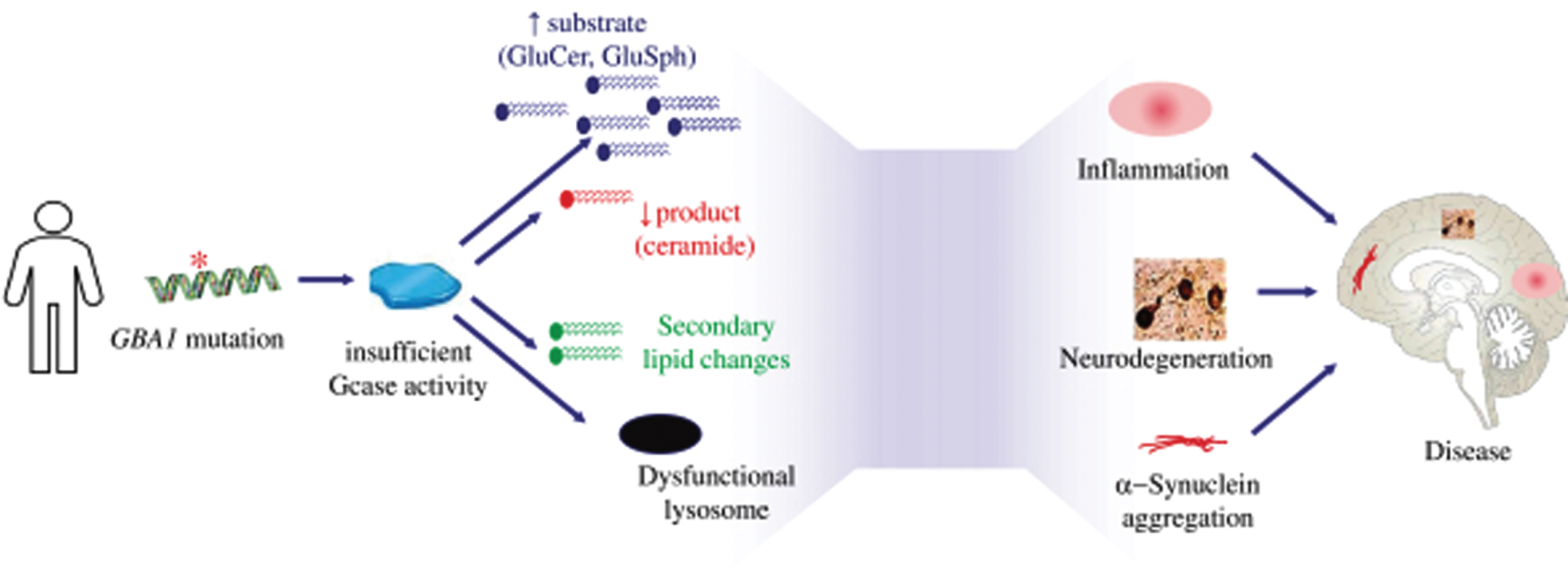
The pathophysiology of PD-GBA is partially understood. Reduced level of GCase activity in GBA-associated Parkinson (PD-GBA) patients is believed to lead to accumulation of glycolipid substrates including GluCer and GluSph, as well as altered production of ceramide and secondary changes in other lipids. The glycolipid substrate accumulation is toxic and pro-inflammatory, leading to lysosomal dysfunction. There is also evidence that accumulation of these glycolipids may impact the structure and aggregation of α-synuclein in cells. The reduction in ceramide has also been linked to neurodegeneration and α-synuclein pathology in model systems and has been observed in Parkinson’s disease brain. In animal models, GluCer accumulation correlates with increased aggregation of phosphorylated α-synuclein protein and aggregates [35, 36]. The pathophysiology of PD associated with GBA mutations is illustrated in Fig. 1.
Fig. 1
Pathophysiology of Parkinson’s disease associated which GBA1 mutations.
GENE THERAPY TRANSDUCING GBA1 IN THE BRAIN
Delivering a normal GBA1 gene using an adeno-associated virus (AAV) vector to a variety of animal models of PD or PD-GBA, including various genetic models with α-synuclein or GBA1 gene mutations, has been shown to be efficacious by multiple independent investigators [37–41]. Most of the these in vivo studies delivered the AAV-GBA1 vector directly into the rodent CNS, although in at least two examples, intravenous delivery was used [42]. Increasing GCase levels and activity through the delivery of a GBA1 vector has been reported to reduce inflammation as well as the accumulation of aggregated α-synuclein in these multiple independent models of PD. Additional studies have used non-genetic approaches to increase GCase activity in rodent PD models and reported efficacy in the reduction of pathological features [43–45].
Based on the consideration above, we have pursued the development of a gene therapy to deliver a functional GBA1 gene to the CNS for PD-GBA patients. Our investigational drug, called PR001, represents the first clinic-staged experimental gene therapy for these patients. We selected the adeno-associated virus serotype 9 (AAV9) as the vector for a codon-optimized plasmid encoding a wild-type human GBA1 gene. AAV9 was chosen since it has demonstrated efficient brain transduction and multiple clinical experiences with this vector has shown a favorable safety profile in humans [46]. PR001 contains elements to constitutively express GBA1 under the control of the CMV enhancer and CBA promoter using a codon optimized coding sequence for GBA1.
PR001 efficacy was examined in established models of GCase deficiency that display phenotypic characteristics consistent with GD and PD-GBA (the preclinical findings obtained with PR001 were reported at scientific conferences [50, 51]). First, the CBE: conduritol-β-epoxide (CBE) mouse model is a chemical model in which a pharmacological inhibition of GCase activity is achieved using a selective and irreversible covalent competitive inhibitor of GCase, leading to glycolipid (GluCer and GluSph) accumulation, neuropathological changes including astrogliosis and microgliosis, and motor behavior eficits [47–49]. Second, the 4L/PS-NA mouse model is a genetic model that harbors mutations in Gba1 (V394L) and in the PSAP gene that encodes saposin C, an activator of GCase, and which displays a severe reduction in GCase activity, an accumulation of glycolipid substrates, and motor behavior deficits [52, 53]. In addition, the 4L/PS-NA mouse model displays accumulation of α-synuclein.
In the CBE model, intraventricular PR001 treatment resulted in increased GCase expression, elevation of GCase enzyme activity, reduction of the accumulation of glycolipid substrates of GCase, correction of neuroinflammation, and behavioral improvements. Broad vector genome biodistribution of PR001 was seen in the CNS and peripheral organs, including the liver, spleen and lung. In the 4L/PS-NA model, PR001 treatment resulted in sustained GCase expression and suppression of glycolipid accumulation over 6 months and amelioration of the behavioral deficits. In our studies, insoluble α-synuclein levels in the cerebral cortex were nonsignificantly increased in the 4L/PS-NA relative to control mice, as reported in published studies and PR001 treatment reversed such accumulation, consistent with our in vitro analyses in cell cultures [6, 52, 54].
PR001 safety was evaluated in both mouse models and no adverse histopathologic findings or evidence of toxicity due to treatment was observed. Biodistribution and toxicology studies were performed in nonhuman primates. Broad distribution of PR001 vector and significant elevation of GCase protein levels were observed in the brain. There were no adverse findings or evidence of toxicity due to PR001.
PR001 is currently in early clinical trials in PD-GBA patients (PROPEL) and in children with GD2 (PROVIDE) [55, 56; http://www.clinicaltrials.gov]. PR001 is administered by a single injection into the cisterna magna of the intrathecal space. Animal studies have demonstrated that, by obviating the need to cross the blood brain barrier, intrathecal delivery results in efficient CNS gene transfer [57–60]. Lumbar puncture is the most common method for accessing CSF. However, delivery of an AAV9 vector into the CSF via a lumbar puncture was found to be at least 10-fold less effective at transducing cells of the brain and spinal cord compared to injection of the vector more superiorly at the level of the cisterna magna [55, 56] Adult rhesus macaques injected with the clinical candidate vector via suboccipital puncture into the cisterna magna exhibited motor neuron transduction at all levels of the spinal cord. In contrast, animals receiving vector injection via lumbar puncture showed substantially lower transduction at all levels of the spinal cord. This study illustrated the potential of delivering vector at the level of the cisterna magna, and supported the selection of suboccipital puncture as the clinical route of administration.
CONCLUSIONS
Human genetic studies strongly support a causative role of GCase deficiency in PD-GBA. Consistent with this, studies in animal models have demonstrated that increasing GCase activity by the delivery of the wild-type GBA1 gene can be efficacious. Clinical studies in PD-GBA and GD2 are now ongoing to investigate the hypothesis that increasing GCase slows or stops progression of the disease.
CONFLICT OF INTEREST
All authors are or have been employees of Prevail Therapeutics – a wholly owned subsidiary of Eli Lilly and Company.
REFERENCES
[1] | Do J , McKinney C , Sharma P , Sidransky E ((2019) ) Glucocerebrosidase and its relevance to Parkinson disease. Mol Neurodegener 14: , 1–16. |
[2] | Stirnemann J , Belmatoug N , Camou F , Serratrice C , Froissart R , Caillaud C , Levade T , Astudillo L , Serratrice J , Brassier A ((2017) ) A review of Gaucher disease pathophysiology, clinical presentation and treatments. Int J Mol Sci 18: , 441. |
[3] | Nguyen M , Wong YC , Ysselstein D , Severino A , Krainc D ((2019) ) Synaptic, mitochondrial, and lysosomal dysfunction in Parkinson’s disease. Trends Neurosci 42: , 140–149. |
[4] | Abeliovich A , Gitler AD ((2016) ) Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539: , 207–216. |
[5] | Stojkovska I , Krainc D , Mazzulli JR ((2018) ) Molecular mechanisms of α-synuclein and GBA1 in Parkinson’s disease. Cell Tissue Res 373: , 51–60. |
[6] | Mazzulli JR , Xu YH , Sun Y , Knight AL , McLean PJ , Caldwell GA , Sidransky E , Grabowski GA , Krainc D ((2011) ) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146: , 37–52. |
[7] | Taguchi YV , Liu J , Ruan J , Pacheco J , Zhang X , Abbasi J , Keutzer J , Mistry PK , Chandra SS ((2017) ) Glucosylsphingosine promotes alpha-synuclein pathology in mutant GBA-associated Parkinson’s disease. J Neurosci 37: , 9617–9631. |
[8] | Hruska KS , LaMarca ME , Scott CR , Sidransky E ((2008) ) Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 29: , 567–583. |
[9] | Roshan Lal T , Sidransky E ((2017) ) The spectrum of neurological manifestations associated with Gaucher disease. Diseases 5: , 10. |
[10] | Riboldi GM , Di Fonzo AB ((2019) ) GBA, Gaucher disease, and Parkinson’s disease: from genetic to clinic to new therapeutic approaches. Cells 8: , 364. |
[11] | Schiffmann R , Sevigny J , Rolfs A , Davies EH , Goker-Alpan O , Abdelwahab M , Vellodi A , Mengel E , Lukina E , Yoo HW ((2020) ) The definition of neuronopathic Gaucher disease. J Inherit Metab Dis 43: , 1056–1059. |
[12] | Lal TR , Seehra GK , Steward AM , Poffenberger CN , Ryan E , Tayebi N , Lopez G , Sidransky E ((2020) ) The natural history of type 2 Gaucher disease in the 21st century: A retrospective study. Neurology 95: , e2119–e2130. |
[13] | Goker-Alpan O , Schiffmann R , Park JK , Stubblefield BK , Tayebi N , Sidransky E ((2003) ) Phenotypic continuum in neuronopathic Gaucher disease: an intermediate phenotype between type 2 and type 3. J Pediatr 143: , 273–276. |
[14] | Steward AM , Wiggs E , Lindstrom T , Ukwuani S , Ryan E , Tayebi N , Lal TR , Lopez G , Schiffmann R , Sidransky E ((2019) ) Variation in cognitive function over time in Gaucher disease type 3. Neurology 93: , e2272–e2283. |
[15] | Schiffmann R , Sevigny J , Rolfs A , Davies EH , Goker-Alpan O , Abdelwahab M , Vellodi A , Mengel E , Lukina E , Yoo HW , Collin-Histed T , Narita A , Dinur T , Revel-Vilk S , Arkadir D , Szer J , Wajnrajch M , Ramaswami U , Sidransky E , Donald A , Zimran A ((2020) ) The definition of neuronopathic Gaucher disease. J Inherit Metab Dis 43: , 1056–1059. |
[16] | Sidransky E , Lopez G ((2012) ) The link between the GBA gene and parkinsonism. Lancet Neurol 11: , 986–998. |
[17] | Goker-Alpan O , Giasson BI , Eblan MJ , Nguyen J , Hurtig HI , Lee VM , Trojanowski JQ , Sidransky E ((2006) ) Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology 67: , 908–910. |
[18] | Nalls MA , Duran R , Lopez G , Kurzawa-Akanbi M , McKeith IG , Chinnery PF , Morris CM , Theuns J , Crosiers D , Cras P , Engelborghs S , De Deyn PP , Van Broeckhoven C , Mann DM , Snowden J , Pickering-Brown S , Halliwell N , Davidson Y , Gibbons L , Harris J , Sheerin UM , Bras J , Hardy J , Clark L , Marder K , Honig LS , Berg D , Maetzler W , Brockmann K , Gasser T , Novellino F , Quattrone A , Annesi G , De Marco EV , Rogaeva E , Masellis M , Black SE , Bilbao JM , Foroud T , Ghetti B , Nichols WC , Pankratz N , Halliday G , Lesage S , Klebe S , Durr A , Duyckaerts C , Brice A , Giasson BI , Trojanowski JQ , Hurtig HI , Tayebi N , Landazabal C , Knight MA , Keller M , Singleton AB , Wolfsberg TG , Sidransky E ((2013) ) A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol 70: , 727–735. |
[19] | Sidransky E , Nalls MA , Aasly JO , Aharon-Peretz J , Annesi G , Barbosa ER , Bar-Shira A , Berg D , Bras J , Brice A ((2009) ) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361: , 1651–1661. |
[20] | Anheim M , Elbaz A , Lesage S , Durr A , Condroyer C , Viallet F , Pollak P , Bonaiti B , Bonaïti-Pellié C , Brice A ((2012) ) Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology 78: , 417–420. |
[21] | Alcalay RN , Dinur T , Quinn T , Sakanaka K , Levy O , Waters C , Fahn S , Dorovski T , Chung WK , Pauciulo M ((2014) ) Comparison of Parkinson risk in Ashkenazi Jewish patients with Gaucher disease and GBA heterozygotes. JAMA Neurol 71: , 752–757. |
[22] | Alcalay RN , Levy OA , Waters CH , Fahn S , Ford B , Kuo S-H , Mazzoni P , Pauciulo MW , Nichols WC , Gan-Or Z ((2015) ) Glucocerebrosidase activity in Parkinson’s disease with and without GBA mutations. Brain 138: , 2648–2658. |
[23] | Grabowski GA , Zimran A , Ida H ((2015) ) Gaucher disease types 1 and 3: Phenotypic characterization of large populations from the ICGG Gaucher Registry. Am J Hematol 90 Suppl 1: , S12–18. |
[24] | Arkadir D , Dinur T , Mullin S , Mehta A , Baris HN , Alcalay RN , Zimran A ((2018) ) Trio approach reveals higher risk of PD in carriers of severe vs. mild GBA mutations. Blood Cells Mol Dis 68: , 115–116. |
[25] | Thaler A , Gurevich T , Shira AB , Weisz MG , Ash E , Shiner T , Orr-Urtreger A , Giladi N , Mirelman A ((2017) ) A “dose” effect of mutations in the GBA gene on Parkinson’s disease phenotype. Parkinsonism Relat Disord 36: , 47–51. |
[26] | Huh YE , Chiang MSR , Locascio JJ , Liao Z , Liu G , Choudhury K , Kuras YI , Tuncali I , Videnovic A , Hunt AL ((2020) ) β-Glucocerebrosidase activity in GBA-linked Parkinson disease: The type of mutation matters. Neurology 95: , e685–e696. |
[27] | Gan-Or Z , Amshalom I , Kilarski LL , Bar-Shira A , Gana-Weisz M , Mirelman A , Marder K , Bressman S , Giladi N , Orr-Urtreger A ((2015) ) Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology 84: , 880–887. |
[28] | Neumann J , Bras J , Deas E , O’Sullivan SS , Parkkinen L , Lachmann RH , Li A , Holton J , Guerreiro R , Paudel R ((2009) ) Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 132: , 1783–1794. |
[29] | Liu G , Boot B , Locascio JJ , Jansen IE , Winder-Rhodes S , Eberly S , Elbaz A , Brice A , Ravina B , van Hilten JJ ((2016) ) Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann Neurol 80: , 674–685. |
[30] | Alcalay R , Caccappolo E , Mejia-Santana H , Tang M-X , Rosado L , Reilly MO , Ruiz D , Ross B , Verbitsky M , Kisselev S ((2012) ) Cognitive performance of GBA mutation carriers with early-onset PD: the CORE-PD study. Neurology 78: , 1434–1440. |
[31] | Thaler A , Bregman N , Gurevich T , Shiner T , Dror Y , Zmira O , Gan-Or Z , Bar-Shira A , Gana-Weisz M , Orr-Urtreger A ((2018) ) Parkinson’s disease phenotype is influenced by the severity of the mutations in the GBA gene. Parkinsonism Relat Disord 55: , 45–49. |
[32] | Gegg ME , Burke D , Heales SJ , Cooper JM , Hardy J , Wood NW , Schapira AH ((2012) ) Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol 72: , 455–463. |
[33] | Murphy KE , Gysbers AM , Abbott SK , Tayebi N , Kim WS , Sidransky E , Cooper A , Garner B , Halliday GM ((2014) ) Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 137: , 834–848. |
[34] | Parnetti L , Chiasserini D , Persichetti E , Eusebi P , Varghese S , Qureshi MM , Dardis A , Deganuto M , De Carlo C , Castrioto A , Balducci C , Paciotti S , Tambasco N , Bembi B , Bonanni L , Onofrj M , Rossi A , Beccari T , El-Agnaf O , Calabresi P ((2014) ) Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov Disord 29: , 1019–1027. |
[35] | Migdalska-Richards A , Schapira AH ((2016) ) The relationship between glucocerebrosidase mutations and Parkinson disease. J Neurochem 139: , 77–90. |
[36] | Sardi SP , Clarke J , Kinnecom C , Tamsett TJ , Li L , Stanek LM , Passini MA , Grabowski GA , Schlossmacher MG , Sidman RL ((2011) ) CNS expression of glucocerebrosidase corrects α-synuclein pathology and memory in a mouse model of Gaucher-related synucleinopathy. Proc Natl Acad Sci U S A 108: , 12101–12106. |
[37] | Jackson KL , Viel C , Clarke J , Bu J , Chan M , Wang B , Shihabuddin LS , Sardi SP ((2019) ) Viral delivery of a microRNA to Gba to the mouse central nervous system models neuronopathic Gaucher disease. Neurobiol Dis 130: , 104513. |
[38] | Rocha EM , Smith GA , Park E , Cao H , Brown E , Hayes MA , Beagan J , McLean JR , Izen SC , Perez-Torres E ((2015) ) Glucocerebrosidase gene therapy prevents α-synucleinopathy of midbrain dopamine neurons. Neurobiol Dis 82: , 495–503. |
[39] | Challis C , Hori A , Sampson TR , Yoo BB , Challis RC , Hamilton AM , Mazmanian SK , Volpicelli-Daley LA , Gradinaru V ((2020) ) Gut-seeded alpha-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci 23: , 327–336. |
[40] | Yun SP , Kim D , Kim S , Kim S , Karuppagounder SS , Kwon SH , Lee S , Kam TI , Lee S , Ham S , Park JH , Dawson VL , Dawson TM , Lee Y , Ko HS ((2018) ) alpha-Synuclein accumulation and GBA deficiency due to L444P GBA mutation contributes to MPTP-induced parkinsonism. Mol Neurodegener 13: , 1. |
[41] | Rockenstein E , Clarke J , Viel C , Panarello N , Treleaven CM , Kim C , Spencer B , Adame A , Park H , Dodge JC , Cheng SH , Shihabuddin LS , Masliah E , Sardi SP ((2016) ) Glucocerebrosidase modulates cognitive and motor activities in murine models of Parkinson’s disease. Hum Mol Genet 25: , 2645–2660. |
[42] | Morabito G , Giannelli SG , Ordazzo G , Bido S , Castoldi V , Indrigo M , Cabassi T , Cattaneo S , Luoni M , Cancellieri C , Sessa A , Bacigaluppi M , Taverna S , Leocani L , Lanciego JL , Broccoli V ((2017) ) AAV-PHP.B-mediated global-scale expression in the mouse nervous system enables GBA1 gene therapy for wide protection from synucleinopathy. Mol Ther 25: , 2727–2742. |
[43] | Migdalska-Richards A , Daly L , Bezard E , Schapira AH ((2016) ) Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann Neurol 80: , 766–775. |
[44] | Sun Y , Liou B , Xu YH , Quinn B , Zhang W , Hamler R , Setchell KD , Grabowski GA ((2012) ) Ex vivo and in vivo effects of isofagomine on acid beta-glucosidase variants and substrate levels in Gaucher disease. J Biol Chem 287: , 4275–4287. |
[45] | Sun Y , Zhang W , Xu YH , Quinn B , Dasgupta N , Liou B , Setchell KD , Grabowski GA ((2013) ) Substrate compositional variation with tissue/region and Gba1 mutations in mouse models–implications for Gaucher disease. PLoS One 8: , e57560. |
[46] | Mendell JR , Al-Zaidy S , Shell R , Arnold WD , Rodino-Klapac LR , Prior TW , Lowes L , Alfano L , Berry K , Church K , Kissel JT , Nagendran S , L’Italien J , Sproule DM , Wells C , Cardenas JA , Heitzer MD , Kaspar A , Corcoran S , Braun L , Likhite S , Miranda C , Meyer K , Foust KD , Burghes AHM , Kaspar BK ((2017) ) Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med 377: , 1713–1722. |
[47] | Farfel-Becker T , Vitner EB , Futerman AH ((2011) ) Animal models for Gaucher disease research. Dis Model Mech 4: , 746–752. |
[48] | Manning-Bog AB , Schule B , Langston JW ((2009) ) Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: a biological link between Gaucher disease and parkinsonism. Neurotoxicology 30: , 1127–1132. |
[49] | Rocha EM , Smith GA , Park E , Cao H , Graham AR , Brown E , McLean JR , Hayes MA , Beagan J , Izen SC , Perez-Torres E , Hallett PJ , Isacson O ((2015) ) Sustained systemic glucocerebrosidase inhibition induces brain alpha-synuclein aggregation, microglia and complement C1q activation in mice. Antioxid Redox Signal 23: , 550–564. |
[50] | Heckman LD , Sheehan P , Feen T , Wong LC , Nelson S , Garimally S , Haller J , Daily J , Politi J , Day Y , Hefti F , Abeliovich A ((2020) ) PR001 gene therapy improves PD-GBA phenotypes in mouse models by increasing GCase activity. AD/PD Conference, abstract. |
[51] | Sheehan P , Heckman LD , Fenn T , Wong LC , Nelson S , Garimalla W , Haller J , Daily J , Politi J , Dai Y , Hefti F , Abeliovich A. ((2020) ) PR001 gene therapy improved phenotypes in models of Parkinson’s disease with GBA1 mutation. Alzheimers Dement 16: (S2), e043614. |
[52] | Xu YH , Sun Y , Ran H , Quinn B , Witte D , Grabowski GA ((2011) ) Accumulation and distribution of alpha-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol Genet Metab 102: , 436–447. |
[53] | Xu YH , Xu K , Sun Y , Liou B , Quinn B , Li RH , Xue L , Zhang W , Setchell KD , Witte D , Grabowski GA ((2014) ) Multiple pathogenic proteins implicated in neuronopathic Gaucher disease mice. Hum Mol Genet 23: , 3943–3957. |
[54] | Sun Y , Quinn B , Witte DP , Grabowski GA ((2005) ) Gaucher disease mouse models: point mutations at the acid beta-glucosidase locus combined with low-level prosaposin expression lead to disease variants. J Lipid Res 46: , 2102–2113. |
[55] | Youssef EA , Tamburi P , Beckerman Y , Lowrey M , Mahoney E , McNamara I , Sevigny J. ((2020) ) Design of a Phase 2/2 study to evaluate safety and efficacy of PR001, an AAV9-based gene therapy in infants with type 2 Gaucher Disease (PROVIDE Trial). ASGCT Conference, abstract. |
[56] | Uspenskaya O , Mahoney E , Verselis L , Lowrey M , Velaga J , Sevigny J ((2020) ) Design of the first-in-human trial of novel AAV9-based gene therapy for Parkinson’s disease with pathogenic GBA1 mutations. AD/PD Conference, abstract. |
[57] | Samaranch L , Salegio EA , San Sebastian W , Kells AP , Foust KD , Bringas JR , Lamarre C , Forsayeth J , Kaspar BK , Bankiewicz KS ((2012) ) Adeno-associated virus serotype 9 transduction in the central nervous system of nonhuman primates. Hum Gene Ther 23: , 382–389. |
[58] | Gray SJ , Nagabhushan Kalburgi S , McCown TJ , Jude Samulski R ((2013) ) Global CNS gene delivery and evasion of anti-AAV-neutralizing antibodies by intrathecal AAV administration in non-human primates. Gene Ther 20: , 450–459. |
[59] | Ohno K , Samaranch L , Hadaczek P , Bringas JR , Allen PC , Sudhakar V , Stockinger DE , Snieckus C , Campagna MV , San Sebastian W , Naidoo J , Chen H , Forsayeth J , Salegio EA , Hwa GGC , Bankiewicz KS ((2019) ) Kinetics and MR-based monitoring of AAV9 vector delivery into cerebrospinal fluid of nonhuman primates. Mol Ther Methods Clin Dev 13: , 47–54. |
[60] | Hinderer C , Bell P , Katz N , Vite CH , Louboutin JP , Bote E , Yu H , Zhu Y , Casal ML , Bagel J , O’Donnell P , Wang P , Haskins ME , Goode T , Wilson JM ((2018) ) Evaluation of intrathecal routes of administration for adeno-associated viral vectors in large animals. Hum Gene Ther 29: , 15–24. |


