
ATH434 Reverses Colorectal Dysfunction in the A53T Mouse Model of Parkinson’s Disease
Abstract
Background:
Gastrointestinal (GI) complications, that severely impact patient quality of life, are a common occurrence in patients with Parkinson’s disease (PD). Damage to enteric neurons and the accumulation of alpha-synuclein in the enteric nervous system (ENS) are thought to contribute to this phenotype. Copper or iron chelators, that bind excess or labile metal ions, can prevent aggregation of alpha-synuclein in the brain and alleviate motor-symptoms in preclinical models of PD.
Objective:
We investigated the effect of ATH434 (formally PBT434), a small molecule, orally bioavailable, moderate-affinity iron chelator, on colonic propulsion and whole gut transit in A53T alpha-synuclein transgenic mice.
Methods:
Mice were fed ATH434 (30 mg/kg/day) for either 4 months (beginning at ∼15 months of age), after the onset of slowed propulsion (“treatment group”), or for 3 months (beginning at ∼12 months of age), prior to slowed propulsion (“prevention group”).
Results:
ATH434, given after dysfunction was established, resulted in a reversal of slowed colonic propulsion and gut transit deficits in A53T mice to WT levels. In addition, ATH434 administered from 12 months prevented the slowed bead expulsion at 15 months but did not alter deficits in gut transit time when compared to vehicle-treated A53T mice. The proportion of neurons with nuclear Hu+ translocation, an indicator of neuronal stress in the ENS, was significantly greater in A53T than WT mice, and was reduced in both groups when ATH434 was administered.
Conclusion:
ATH434 can reverse some of the GI deficits and enteric neuropathy that occur in a mouse model of PD, and thus may have potential clinical benefit in alleviating the GI dysfunctions associated with PD.
INTRODUCTION
Parkinson’s disease (PD) is an increasingly prevalent neurodegenerative disorder, whose prominent symptoms are progressive movement dysfunction [1, 2]. However, the non-motor features of PD such as, dementia, apathy, anxiety, depression, hyposmia, sleep disorders, and gastrointestinal (GI) dysfunction are receiving more attention because of their impact on patient quality of life [3, 4]. Common GI dysfunctions associated with PD include swallowing difficulty (dysphagia), slowed stomach emptying (gastroparesis), slower nutrient absorption from the gut, and severe chronic constipation [5]. Chronic constipation affects ∼70% of PD patients and often precedes the onset of motor symptoms by decades [6, 7]. In PD, constipation frequently manifests with slow gut motility, decreased fecal water content [8], and dopaminergic deficits in the enteric nervous system (ENS) [9]. Current therapeutics commonly used for constipation are often ineffective in PD patients. Interestingly, aggregation of alpha-synuclein, a central nervous system (CNS) hallmark of PD, has also been found in the ENS of PD patients. It has been suggested that aggregation of alpha-synuclein in the ENS may contribute directly or indirectly to enteric neuron damage and subsequent GI dysfunction.
In the CNS, changes associated with the development of PD include an abnormal iron distribution in the brain. High levels of iron have been confirmed from postmortem studies, magnetic resonance imaging, and transcranial ultrasound studies [10–12]. Iron has been shown to promote the aggregation of alpha-synuclein, which is toxic to neurons and may cause their death [13, 14]. The iron chelators desferrioxamine, deferasirox, and deferiprone, which are currently in clinical use for treating iron overload in thalassemia major, have been shown to significantly attenuate dopaminergic neuronal loss in the substantia nigra pars compacta (SNpc) and striatum in the 6-hydroxydopamine model of PD, whilst reducing hydroxyl radical formation [15]. Moreover, small scale clinical trials have shown that reducing the iron burden can slow the progression of PD symptoms [16]. Thus, CNS data linking iron and alpha-synuclein have provided a rationale to interrogate the potential utility of iron chelation on enteric neuropathy and symptoms in the gut.
ATH434, previously referred to as PBT434, is an orally bioavailable 8-hydroxyquinazolinone, which binds to iron with an affinity that is intended not to disrupt physiological iron homeostasis [17]. In phase 1 trials in humans, the compound has been shown to be safe, although further large-scale testing is required [18]. ATH434 has been shown to significantly reduce loss of SNpc neurons and motor deficits in the MPTP mouse model of PD when administered for 20 day at a dose of 30 mg/kg/day [17]. Moreover, A53T transgenic mice treated for 4 months, prior to the onset of motor deficits, with a similar dose of ATH434 were found have preservation of dopamine-producing neurons in the SNpc, improved motor function, and reduced SN iron levels and nigral insoluble alpha-synuclein levels in the brain [17].
Whether or not ATH434 can improve non-motor symptoms, such as GI dysfunction, remains unknown. Given the promising results with ATH434 in the CNS, we hypothesized that ATH434 administration could alleviate enteric neuropathy and the GI complications associated with PD. In this study, we used the A53T mouse model of PD, which overexpresses human alpha-synuclein bearing the alanine to threonine mutation at position 53. This mouse model is known to develop decreased locomotion, hindlimb deficits, and a significant decrease in the number of nigral neurons by 8 months of age [19]. In addition, these mice develop GI deficits at ∼15 months of age, which progressively worsens throughout the course of disease [20]. In this study, our aim was to investigate whether ATH434 could alleviate or reverse the GI deficits observed in A53T mice. In different experiments we administered ATH434 before or after the onset of GI dysfunction. We measured colonic motility and whole gut transit and correlated functional changes with assessment of neuronal damage and alpha-synuclein aggregation in the ENS.
MATERIAL AND METHODS
Animals
All procedures involving mice conformed to the Australian National Health and Medical Research Council (NHMRC) code of practice for the care and use of animals for scientific purposes and were approved by the Florey Institute Animal Ethics Committee (approval #14-012). Mice that carry the human A53T mutation driven by the mouse prion promoter (Jax Stock No: 004479; B6;C3-Tg (Prnp-SNCA*A53T)83Vle/J; A53T alpha-synuclein transgenic line M83) were obtained from Jackson Laboratories (Bar Harbor, ME). A stable breeding colony was generated in house and the genotype of all mice used in the study were confirmed by qPCR. Two series of experiments were performed in this study: 1) A53T and wild-type (WT) (litter mate controls) mice were fed ATH434 (at 30 mg/kg/day) or vehicle that was mixed into rodent chow (Specialty Feeds, Western Australia) for a total of 4 months, beginning at ∼15 months of age (after the onset of GI dysfunction, ‘treatment’ paradigm; n = 9–24), and 2) A53T mice were fed ATH434 or vehicle for a total of 3 months, beginning at ∼12 months of age (prior to onset of GI dysfunction, ‘prevention’ paradigm; n = 8–10). The ‘treatment’ paradigm was designed to treat mice that already had GI dysfunction and determine if GI deficits could be reversed, while the ‘prevention’ paradigm was designed to treat mice before the onset of GI dysfunction and determine if onset of GI deficits could be prevented. Because we found there was no effect of ATH434 on WT mice in our treatment paradigm, we did not investigate WT mice in the prevention group. Mice were group-housed (2–5 animals/cage) in a temperature- and humidity-controlled room under a 12-h light/dark cycle. Food and water were available ad libitum. We and others have found that there are no significant differences in GI function with respect to gender [20, 21], therefore all results were combined for each genotype to increase power.
Bead expulsion
Deficits in colonic propulsion were measured using the bead expulsion test. For mice that commenced ATH434 treatment at ∼15 months of age, the bead expulsion test was performed at the beginning of each month (i.e., at ∼16, 17, 18, and 19 months of age), while mice that commenced feeding at ∼12 months of age (n = underwent the procedure at the end of the study (i.e., 3 months of feeding; ∼15 months of age). Mice were lightly anesthetized with 3% isoflurane inhalation to allow insertion of a bead (3 mm in diameter) into the distal colon 2 cm from the anus, as previously described [20]. Bead insertion was accomplished using a flexible plastic rod to avoid tissue damage. Following bead insertion, mice were placed in individual cages to recover from anesthesia. The time taken from insertion to expulsion was recorded to the nearest second.
Whole gut transit
For mice that commenced feeding at ∼15 months of age, assessment of whole-gut transit was performed at the beginning of each month (i.e., at ∼16, 17, 18, and 19 months of age), while mice that commenced feeding at ∼12 months of age were only tested at the end of the study (i.e., 3 months of feeding; ∼15 months of age). Briefly, transit time was assessed in mice after oral gavage of a 50% (v/v) cochineal extract solution (ethanol removed, Queen Fine Foods, Alderley, QLD), as previously described [20]. Mice were placed in individual cages containing food, tissue for nesting, and had free access to water. Post gavage, the mice were observed for up to 9 h until the time of excretion of the first red stool, which was recorded for each mouse. Mice that had not passed a red stool by 9 h were scored as > 9 h.
Ledged beam test
Motor coordination and balance in mice from each treatment group was assessed in the last month of feeding. As previously described [20], a beam, 1.5 m in total length, and comprising of six (10 cm length) sections that gradually decreased in width from 3.5 cm to 0.5 cm in 1 cm decrements was used. Animals were trained to traverse the beam (from widest to narrowest) directly into the animal’s home cage. Each mouse received training in the morning (4 trials each) followed by testing in the afternoon. During the testing phase, animals were recorded whilst traversing the beam over 4 trials, with a 15–30 s inter-trial period. Videos were analyzed in slow-motion by an investigator blinded to the genotype/treatment of the animals. The total number of foot faults per section was determined over the 4 trials and averaged.
Tissue collection and preparation
Mice were anesthetized by intraperitoneal injection with a mix of ketamine/xylazine (100 mg/kg ketamine and 10 mg/kg xylazine) and perfused transcardially with phosphate buffered saline (PBS). Mice from the “treatment paradigm” were euthanized and gut tissue was collected after 4 months of ATH434 treatment (when mice were ∼19 months of age). Mice from the “prevention paradigm” were euthanized and gut tissue was collected after 3 months of ATH434 treatment (when mice were ∼15 months of age). Gut tissue (colon and ileum) was flushed of fecal contents and opened along the mesenteric attachment, pinned flat onto balsa board with the lumen facing down, and fixed overnight in 2% formaldehyde, plus 0.2% picric acid in 0.1M sodium phosphate buffer, pH 7.2, at 4°C. Preparations were cleared of fixative by 3×10 min washes in dimethyl sulfoxide (DMSO) followed by 3×10 min washes in PBS. Fixed tissue was stored at 4°C in PBS containing sodium azide (0.1%).
Immunohistochemistry of colon and ileum tissue
The mucosa and circular muscle were removed from the fixed tissue and wholemounts of the myenteric plexus were prepared. Wholemount preparations were incubated with combinations of human anti-Hu (1:2000; a gift from Dr Miles Epstein [22]) a pan-neuronal marker that binds to RNA-binding proteins exclusively in neurons, sheep anti-neuronal nitric oxide synthase (nNOS, 1:2000; a gift from Dr. Piers Emson [23]), or rabbit anti-alpha-synuclein (1:1000, Abcam, VIC, Australia; for endogenous mouse alpha-synuclein) antibodies, overnight at 4°C. The wholemounts were then washed (3×10 min) in PBS before incubation with donkey anti-human Alexa 594, donkey anti-sheep 488, or donkey anti-rabbit Alexa 488 secondary antibodies, respectively (Molecular Probes, Eugene, OR, USA), for 1 h at room temperature. Preparations were washed once with PBS, followed by 3×5 min washes in distilled water and incubated with Hoeschst 33258 solution (10μg/ml Bisbenzimide-Blue in distilled water; Sigma-Aldrich, Sydney, NSW, Australia) for 5 min. Tissue was washed 3×10 min with distilled water before being mounted on glass slides using fluorescent mounting medium (Dako, Carpinteria, CA, USA). Images were captured using the Axio Imager.Z1 microscope (Carl Zeiss, Sydney, NSW, Australia) at 10×and 20×air objective. For Hu neurons per area studies, approximately 8–10 randomly captured images at 10×air objective were counted per preparation. Approximately 100–300 neurons per preparation were counted for neuron number, Hu translocation, and nNOS quantitative studies. ImageJ (1.52i, http://imagej.nih.gov/ij) [24] was used to quantify the area of alpha-synuclein coverage in a minimum of 50 ganglia per group in wholemount preparations. For all quantitative measurements, 5–6 wholemount preparations were used per cohort, which we have previously shown to be sufficient to reveal significant neuropathic changes [20].
Statistical analysis
Data are expressed as the mean±SEM. Comparisons between groups were performed using one- or two-way ANOVA with Sidak’s or Tukey’s multiple comparisons test. Analyses were performed using GraphPad Prism 8.3.0 (GraphPad software Inc., San Diego, CA, USA). p values of less than 0.05 were considered statistically significant.
RESULTS
ATH434 administered after the onset of GI symptoms (treatment group) improves deficits in colonic propulsion and whole gut transit, but not motor function in A53T mice
To determine the ability of ATH434 to reverse slowed colonic propulsion and whole gut transit, these parameters were assessed monthly in WT and A53T mice that were fed for a total of 4 months (from 15 to 19 months) with ATH434 after the onset of these GI deficits. WT mice served as a control to measure the effect of ATH434 in mice that lack Parkinsonian pathology and symptoms. ATH434 had no effect on fecal water content in either WT or A53T mice (data not shown) indicating that there were no constipation or diarrhea following long-term exposure to ATH434. Colonic propulsion, measured using the bead expulsion test, confirmed that vehicle-treated A53T mice had significant slowing when compared with vehicle-treated WT mice at all time points (p < 0.05; Fig. 1A). ATH434 significantly improved colonic propulsion in A53T mice by reducing the time taken to expel the bead when compared with vehicle-treated A53T mice (p < 0.05 at 17, 18, and 19 months; Fig. 1A). This was a progressive effect, seen more prominently at 19 months (after completion of 4 months treatment with ATH434) than at 16 months (after 1-month treatment with ATH434). ATH434 had no effect on bead expulsion in WT mice (Fig. 1B).
Fig. 1
Effect of ATH434 on bead expulsion and whole gut transit in mice with PD symptoms (treatment group). Vehicle-treated A53T mice had significant deficits in colonic motility, as assessed by the bead expulsion test, when compared to vehicle-treated WT mice at all timepoints (p < 0.05). Overall, ATH434, administered from the beginning of month 15 to the beginning of month 19, reduced the deficit in colonic motility, with a significant reduction in bead expulsion time observed at 2, 3, and 4 months after commencement of feeding in A53T mice (p < 0.05) (A). ATH434 had no effect on bead expulsion times in WT mice (B). Whole gut transit time was significantly delayed in A53T vehicle-treated mice when compared with vehicle-treated WT mice at all time points (p < 0.05). ATH434 significantly reduced whole gut transit time in A53T mice following 3 and 4 months of treatment (p < 0.05) (C), but had no effect on WT mice (D). Data were analyzed by two-way ANOVA (mixed model) followed by Sidak’s multiple comparison post hoc test. Data represent the mean±SEM; *p < 0.05 for WT (vehicle) versus A53T (vehicle); #p < 0.05 for A53T (vehicle) versus A53T (ATH434); n = 17–24 for WT vehicle and WT ATH434 mice and n = 9–18 for A53T vehicle and A53T ATH434 mice.
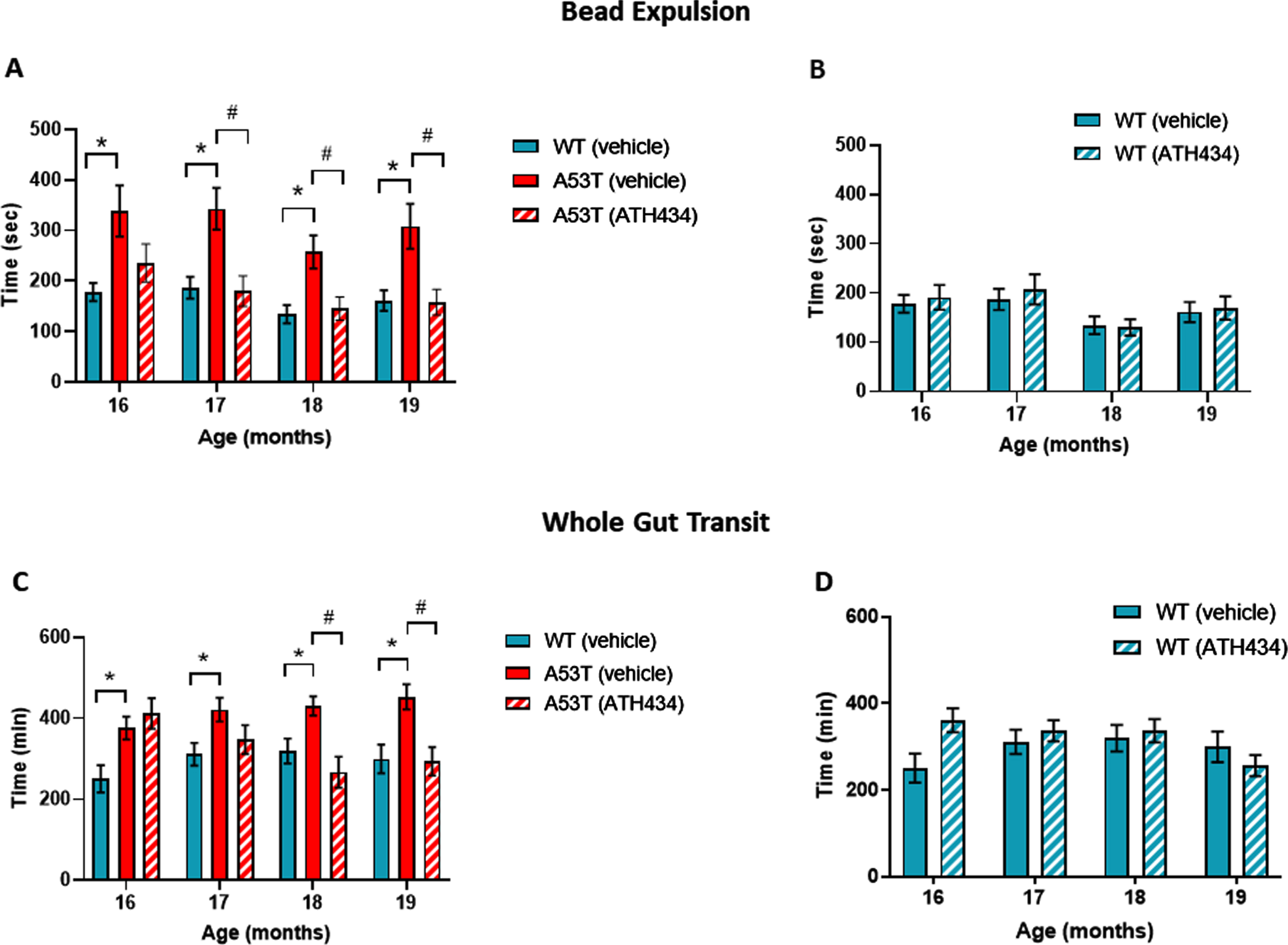
Whole gut transit in vehicle-treated A53T mice was significantly slower than in vehicle-treated WT mice between 16 and 19 months of age (p < 0.05; Fig. 1C). No significant effect was observed in WT mice treated with ATH434 when compared to vehicle-treated WT mice (Fig. 1C). ATH434 had no effect on whole gut transit after 1 and 2 months of treatment; however, ATH434 significantly improved whole gut transit time in A53T mice, compared to vehicle-treated A53T mice, after 3 and 4 months of treatment (p < 0.05) and reduced gut transit time to WT levels (Fig. 1D).
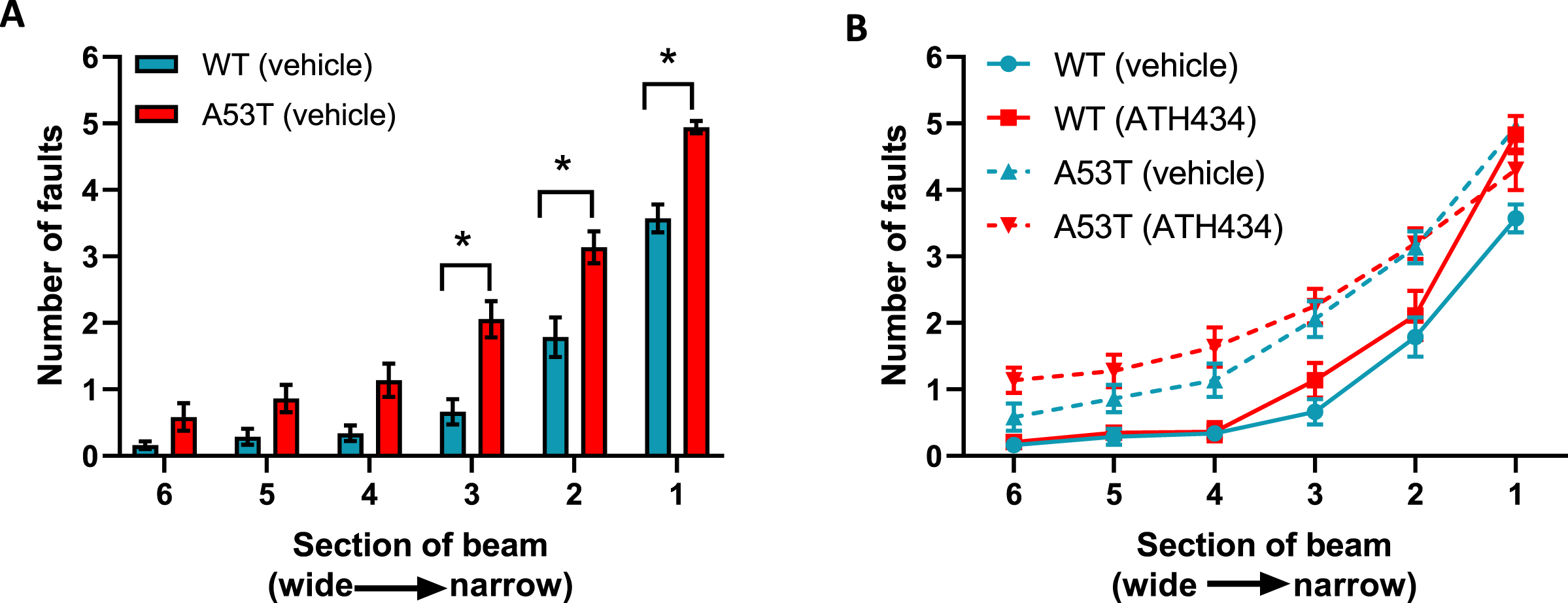
With respect to motor dysfunction, vehicle-treated A53T mice displayed a greater number of foot faults at narrower sections of the beam when compared to vehicle-treated WT mice (p < 0.05; Fig. 2A), indicating that A53T mice had motor impairments. Previously we have shown that ATH434 was able to prevent the onset of motor symptoms in mice treated from the age of 5 months [17]. In this study ATH434 was unable to reverse these established motor deficits when applied from 15 months (Fig. 2B). Taken together, these data indicate that the improvement in colonic propulsion and gut transit can be sustained over time and that orally administered ATH434 can reverse deficits in GI function.
Fig. 2
Lack of effect of ATH434 on motor deficits in mice with PD. When compared with vehicle-treated WT mice, vehicle-treated A53T mice had significant motor deficits (p < 0.05) (A), however the advanced motor dysfunction could not be reversed with ATH434 (B). Data were analyzed by two-way ANOVA (mixed model) followed by Sidak’s multiple comparison post hoc test. Data represent the mean±SEM; *p < 0.05, n = 17–24 for WT vehicle and WT ATH434 mice and n = 9–18 for A53T vehicle and A53T ATH434 mice.
ATH434 administered before the onset of prominent GI symptoms (prevention paradigm) reduces the slowing of colonic propulsion, but does not affect whole gut transit or motor function in A53T mice
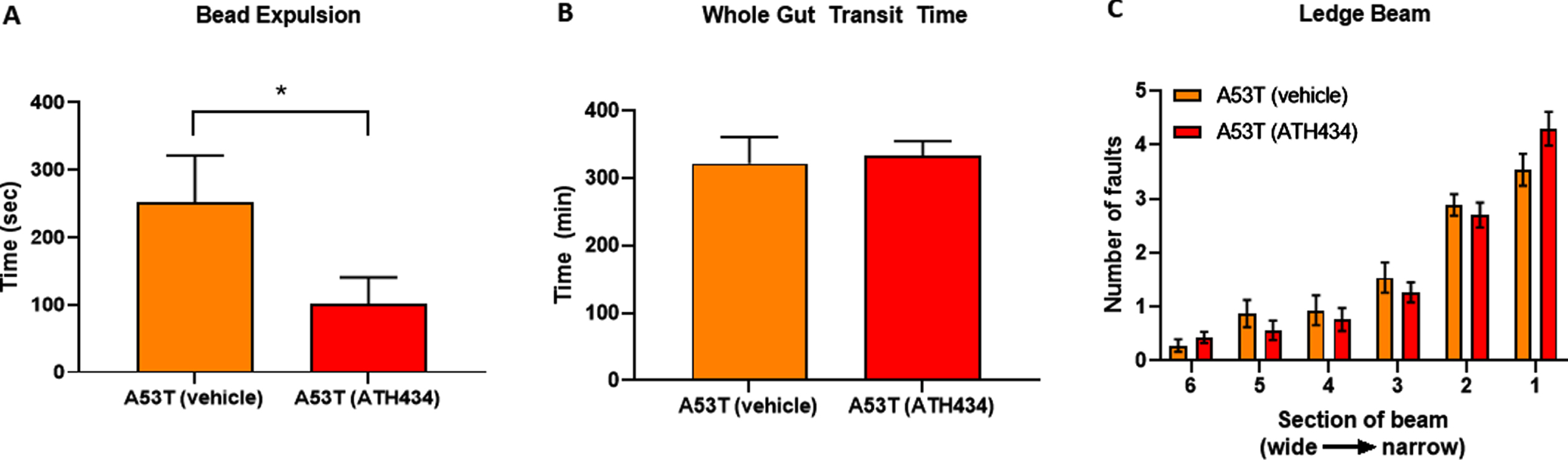
Given that ATH434 was able to reverse the deficits in colonic motility and whole-gut transit in A53T mice after the onset of GI symptoms (treatment paradigm), we aimed to determine if ATH434 could prevent or reduce the GI symptoms if administered before they were fully established in A53T mice. For this study, only A53T mice were used as no effects were observed in WT mice in the first series of experiments with ATH434. A53T mice were fed ATH434 for a total of 3 months, starting at ∼12 months of age, an age when GI symptoms are not prominent, to ∼15 months of age, an age when GI deficits are clearly observed [20]. Colonic motility and whole-gut transit were assessed at the end of the 3-month treatment period. ATH434 significantly reduced the time taken to expel the bead in A53T mice at 15 months of age when compared with vehicle-treated mice (p < 0.05; Fig. 3A) but had no effect on whole-gut transit (Fig. 3B) or reversal of motor dysfunction (Fig. 3C).
Fig. 3
Effects of ATH434 when administration before the development of GI symptoms (preventative paradigm). ATH434 had a significant effect on bead expulsion time (A). A53T mice treated for 3 months with ATH434 prior to the onset of GI dysfunction, had reduced bead expulsion times when compared with vehicle-treated A53T mice at 15 months of age (p < 0.05). ATH434 had no effect on whole-gut transit time at 15 months (B) or motor dysfunction as assessed by the ledge beam test (C). Data were analyzed by unpaired t-test. Data represent the mean±SEM; *p < 0.05. n = 8 A53T mice in vehicle group and n = 10 mice in ATH434 group.
ATH434 did not alter the number of Hu-positive neurons or the proportion of inhibitory motor neurons in the enteric nervous system
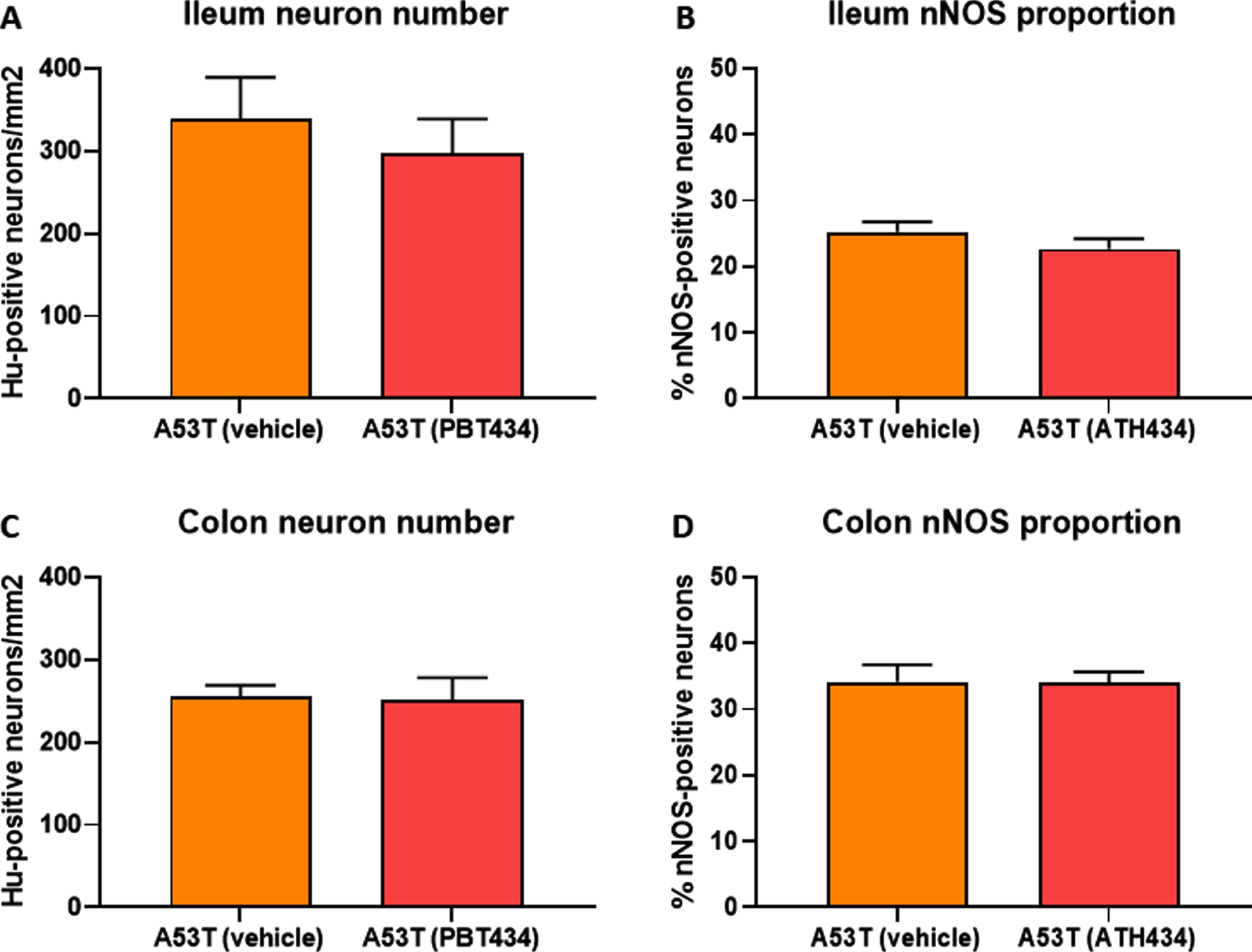
No change in the number of Hu-immunoreactive (IR) neurons in the ileum were observed between WT- and A53T-vehicle treated mice, and as expected, numbers did not change when mice were treated with ATH434 (Fig. 4A). Although there was a reduction in the number of Hu-IR neurons in the colon between vehicle-treated A53T and WT mice, ATH434 had no effect on the number of Hu-IR neurons in the colon (p < 0.05; Fig. 4B). No change in the proportion of nNOS-positive neurons was observed in the ileum (Fig. 4C) or colon (Fig. 4D) between vehicle-treated A53T and WT mice, treatment with ATH434 for 4 months (treatment paradigm) had no effect. Similar to mice in the treatment paradigm, no change in the number of Hu-IR neurons or proportion of nNOS-positive neurons was observed in mice treated for 3 months prior to the onset of GI symptoms in the ileum (Fig. 5A, C) or colon (Fig. 5B, D) of 15-month-old mice (preventative paradigm).
Fig. 4
Numbers of neurons in the A53T colon were reduced, but treatment with ATH434 for 4 months had no effect on neuron number or proportion of nNOS-positive neurons in PD mice with GI dysfunction. No change in the number of Hu+ neurons was observed in the ileum of A53T vehicle-treated mice when compared to WT mice (A), however, A53T mice had significantly fewer Hu+ neurons in the colon (p < 0.05) (B). ATH434 treatment had no effect on the number of Hu+ neurons/mm2 in either the ileum (A) or colon (B). The proportion of nNOS-positive neurons in vehicle-treated WT and A53T mice remained unchanged in both the ileum (C) and colon (D), and ATH434 had no effect on either genotype. Data were analyzed by one-way ANOVA followed by Tukey’s multiple comparison post hoc test. Data represent the mean±SEM (100–300 neurons per preparation were counted for neuron number and nNOS quantitation studies. All studies used 5–6 wholemount preparations per cohort); *p < 0.05.
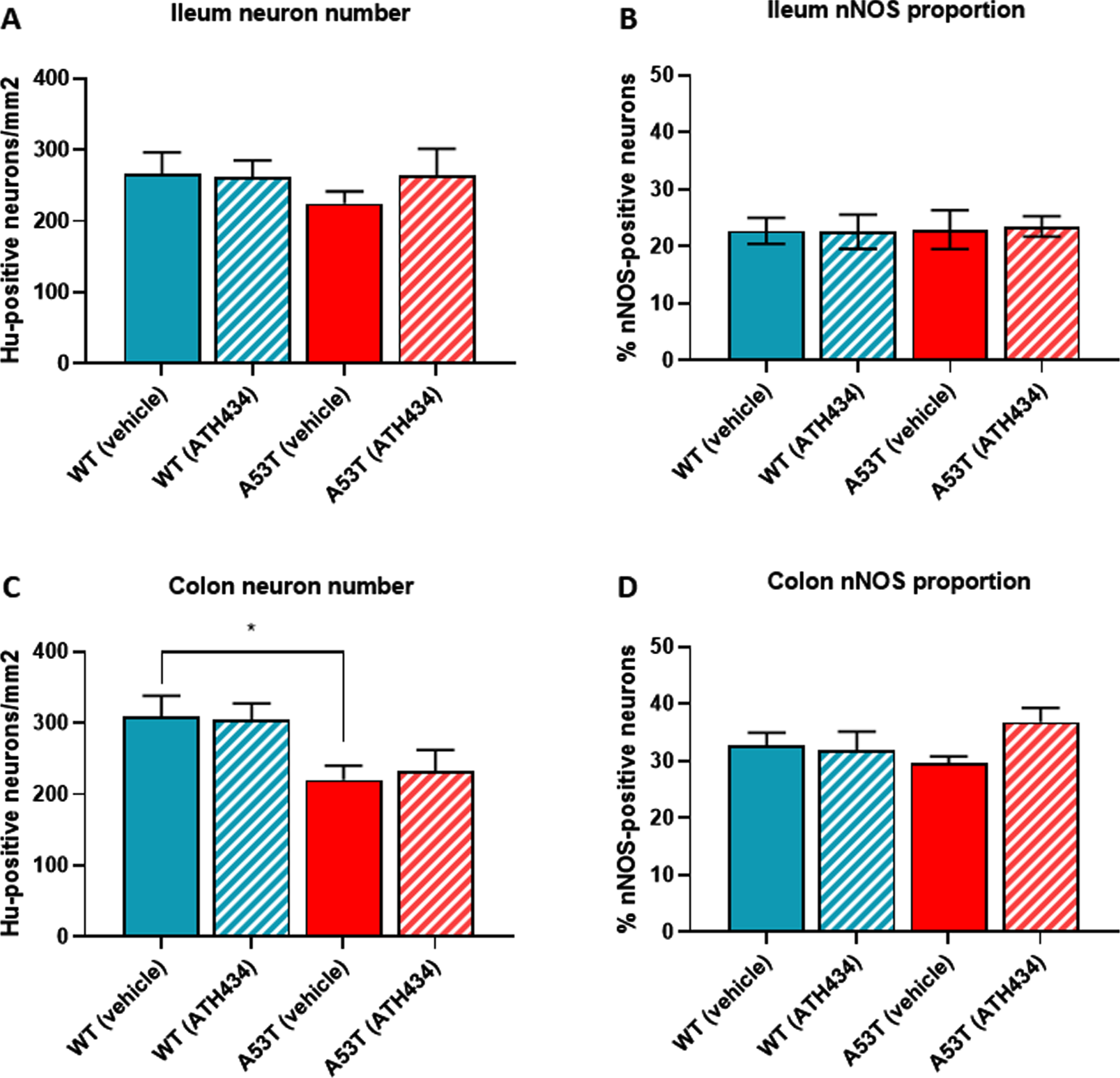
Fig. 5
ATH434 administered prior to the onset of GI dysfunction (at ∼12 months of age, continuing to 15 months) had no effect on the number of Hu+ neurons or the proportion of nNOS-positive neurons. Treatment with ATH434 had no effect on the number of Hu-positive neurons in either the ileum (A) or the colon (B), nor on the proportion of nNOS-positive neurons in the ileum (C) or the colon (D). Data were analyzed by one-way ANOVA followed by Tukey’s multiple comparison post hoc test. Data represent the mean±SEM (100–300 neurons per preparation were counted for neuron number and nNOS quantitation. All studies used 5–6 wholemount preparations per cohort).
Treatment with ATH434 reversed nuclear Hu+ translocation in mice with established GI dysfunction, but had no effect on alpha-synuclein accumulation
To determine if ATH434 treatment was associated with changes in pathological hallmarks at the level of the ENS, ileum and colon tissue was isolated from mice at the end of the feeding regime and processed immunohistochemically to assess nuclear Hu+ translocation and the proportion of alpha-synuclein accumulation per ganglion. No difference in nuclear Hu+ translocation was found in the ileum of vehicle-treated A53T mice when compared to WT, and as expected, treatment with ATH434 had no impact on nuclear Hu+ translocation in either WT or A53T mice (Fig. 6A). In the colon, A53T vehicle-treated mice displayed significantly greater nuclear Hu+ translocation in the colon when compared to WT (p < 0.05), and ATH434 significantly reduced this (p < 0.05) (Fig. 6B). Although there was a trend for increased alpha synuclein accumulation per ganglion in vehicle-treated A53T mice when compared to WT mice in both the ileum (Fig. 6C) and colon (Fig. 6D), the difference was not significant. However, there was a trend for a reduction in the proportion of alpha-synuclein accumulation per ganglion in the colon of A53T mice following ATH434 treatment (Fig. 6D; p = 0.059). In A53T mice who began ATH434 treatment prior to the onset of GI dysfunction, ATH434 significantly reduced the number of neurons with nuclear Hu+ translocation in the ileum (p < 0.05; Fig. 7A) but had no effect in the colon (Fig. 7B). ATH434 had no effect on the proportion of alpha-synuclein accumulation per ganglion in either the ileum (Fig. 7C) or the colon (Fig. 7D).
Fig. 6
ATH434 reduced the level of Hu translocation and showed a trend for reduced alpha-synuclein accumulation per ganglion in the colon of mice from the treatment paradigm. No significant difference in the proportion of neurons with nuclear Hu+ translocation was found in the ileum when comparing vehicle-treated A53T and WT mice (A). However, colons from vehicle treated A53T mice had a significantly greater proportion of neurons with nuclear Hu+ translocation than WT mice (p < 0.05) (B). Treatment with ATH434 had no effect in the ileum (A), but reduced the proportion of cells with nuclear Hu+ translocation in the colon (p < 0.05) (B). A trend for increased alpha-synuclein accumulation per ganglion was observed in the ileum of vehicle-treated A53T mice when compared to WT mice, but no effect was observed with ATH434 (C). Although not significant, ATH434 reduced alpha-synuclein accumulation per ganglion in the colon of A53T mice (p < 0.059) (D). Data were analyzed by one-way ANOVA followed by Tukey’s multiple comparison post hoc test. Data represent the mean±SEM (100–300 cells were counted for Hu translocation studies and n = 50 ganglia/group were quantified for alpha-synuclein immunoreactivity. All studies used 5–6 wholemount preparations per cohort); *p < 0.05.
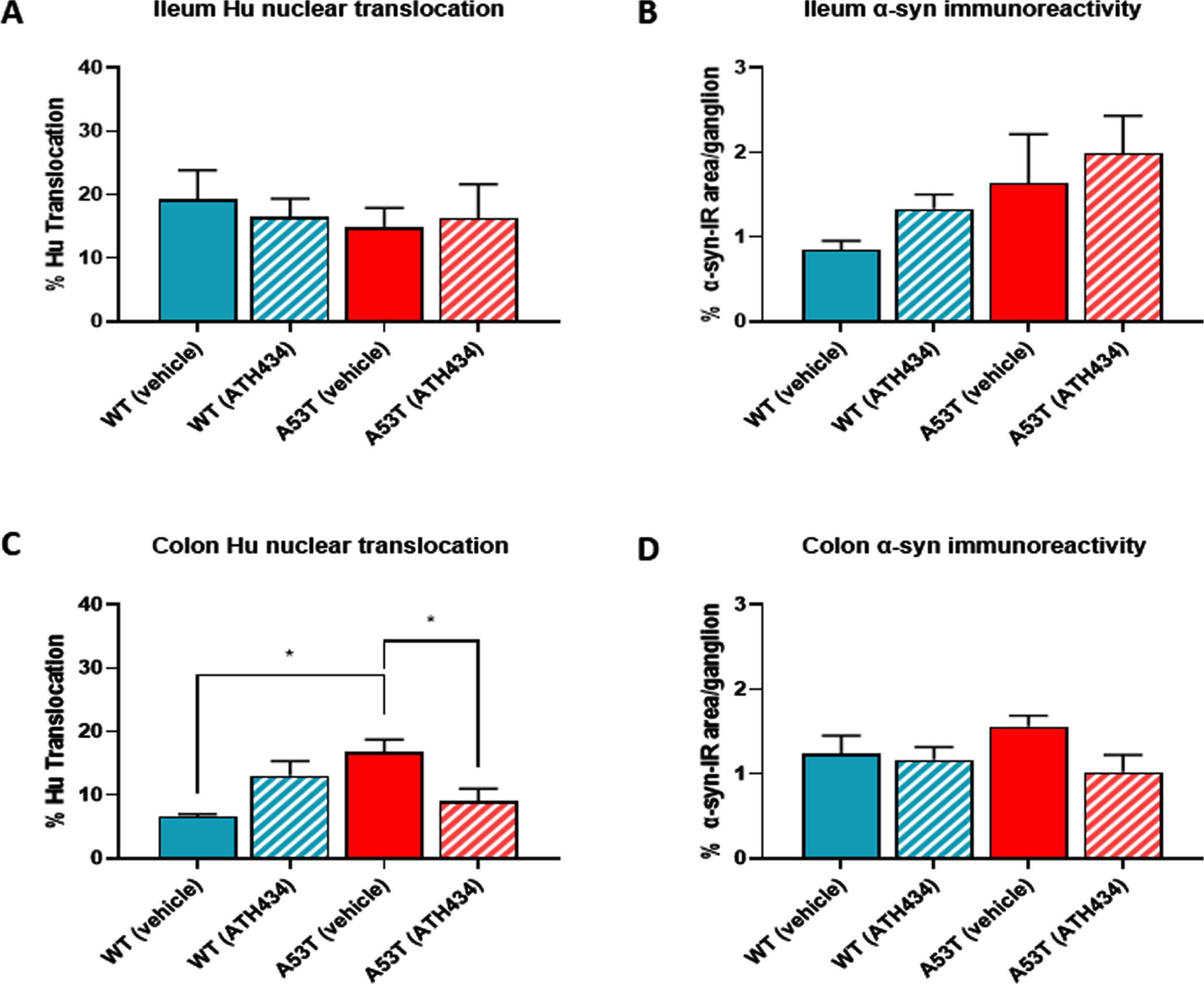
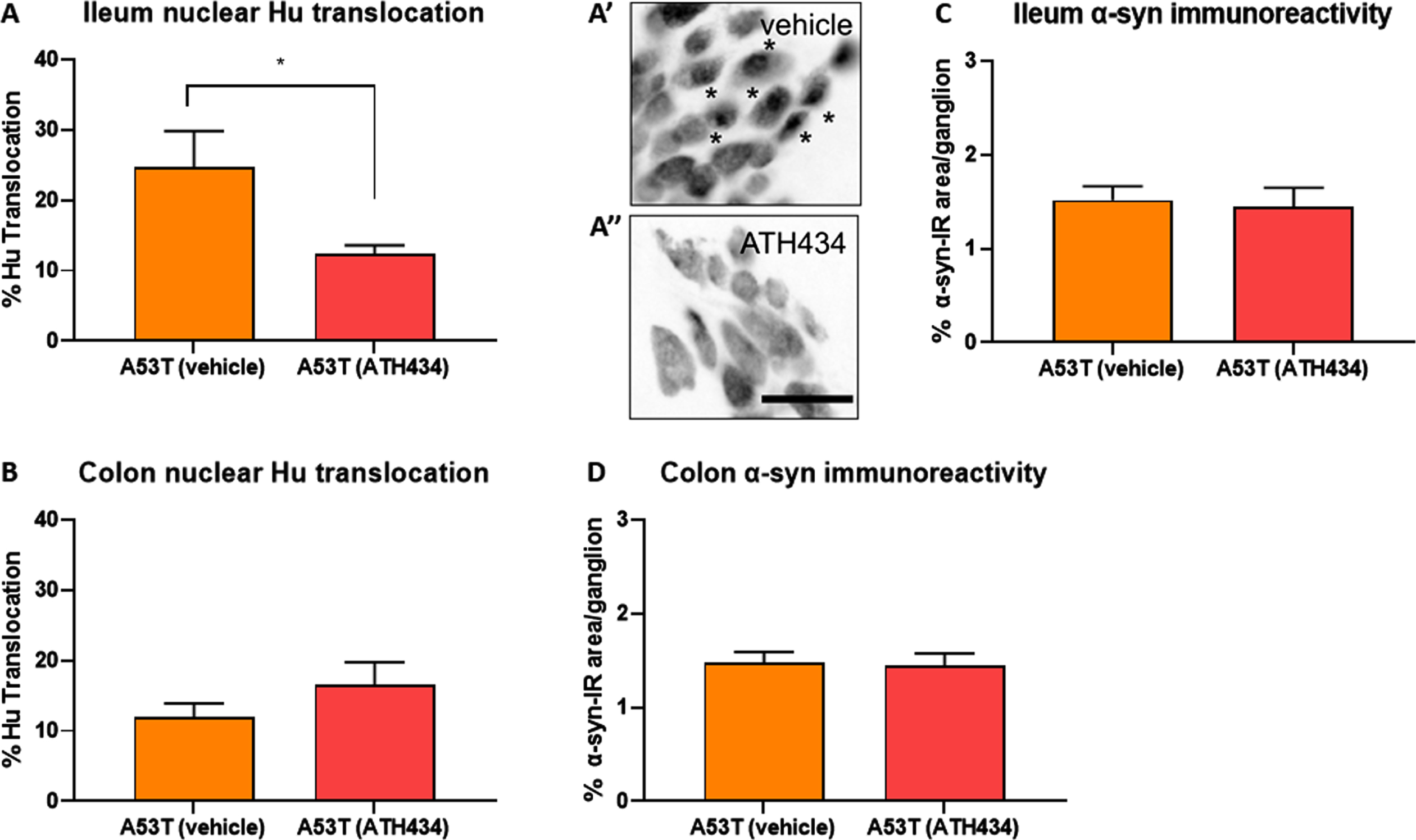
Fig. 7
ATH434 reduced the proportion of neurons with nuclear Hu+ translocation in the ileum but had no effect on alpha-synuclein accumulation in the prevention paradigm. Treatment with ATH434 reduced the proportion of neurons with nuclear translocation of Hu+ in the ileum (A, A’, and A”), but not the colon (B) in A53T mice. *indicate neurons with nuclear Hu translocation. Scale bar = 50μm. ATH434 had no effect on alpha-synuclein accumulation in the ileum (C) or colon (D). Data were analyzed by unpaired t-test. Data represent the mean±SEM (100–300 cells were counted for Hu translocation studies and n = 50 ganglia/group were quantified for alpha-synuclein immunoreactivity. All studies used 5–6 wholemount preparations per cohort); *p < 0.05.
DISCUSSION
Mice that carry the human A53T mutation of the alpha-synuclein gene develop a slowed colorectal transit, that is first seen from 3–15 months in different studies and progressively worsens [20, 25, 26]. Differences in onset times may be due to different levels of expression of the transgene or differences in methods of assessment. To investigate the effects of ATH434 in treating and preventing GI dysfunction, we used 2 experimental protocols. One group of A53T mice and their WT counterparts were treated with ATH434 for 4 months, beginning at 15 months of age, a time when GI dysfunction is well established (treatment paradigm), and a second cohort of A53T mice were treated for 3 months, starting at 12 months of age, which is prior to the onset of a prominent colorectal disorder in our colony (preventative paradigm).
The slowing of colorectal bead expulsion in A53T mice was improved by ATH434 treatment from month 16, but the reversal was only significant at month 17 (Fig. 1B). By months 18 and 19, bead expulsion in A53T mice was not different to that seen in WT mice. The effect of treatment on whole gut transit time was also slow to develop. Moreover, if the treatment commenced prior to constipation onset, at the beginning of 12 months of age, expulsion time was half that of vehicle-treated controls after 3 months of ATH434 feeding, indicating that prior ATH434 treatment inhibits progression of colonic dysfunction in A53T mice. It thus appears that ATH434 has effects that are slow to develop, but which are persistent. ATH434 did not accelerate transit in WT mice, indicating that the effect was dependent on there being an abnormality.
Colorectal propulsion is dependent on intrinsic ENS reflexes and involves ascending excitatory pathways in which the final neurons are cholinergic, that evoke contractions above the bolus and descending inhibitory reflexes (final nitrergic neurons) that induce relaxation below the bolus, propelling contents anally. Interruption of the myenteric plexus is known to impact this excitatory and inhibitory conduction which is vital for colonic propulsion [27]. Our data demonstrates that ATH434 had a restorative effect on GI function, specifically colonic function, suggesting a potential action to reverse deficits at the level of the ENS.
In A53T mice, GI manifestations have been correlated with neuropathy in the ENS of the ileum and colon [20]. Data from the current study confirms that A53T mice suffer from significant neuronal loss and have significantly greater nuclear Hu+ translocation in the colon when compared to WT mice (Fig. 4B and Fig. 6B). In the ileum, the number of neurons was also reduced by ∼15% in A53T vehicle-treated mice when compared to WT mice, however this did not reach statistical significance (p < 0.2). When fed to mice with established GI dysfunction, ATH434 reduced the proportion of cells with nuclear Hu+ translocation in the colon of A53T mice, and when fed to mice prior to the onset of GI dysfunction, it significantly reduced nuclear Hu+ translocation in the ileum of A53T mice (Fig. 7A).
Intracellular localization of Hu+ is a useful tool in assessing the health status of enteric neurons, in conditions of high stress, such as those induced by hypoxia, ischemia and chemotherapy, Hu+ expression is predominantly localized to the nucleus [28–30]. Nuclear Hu+ translocation is often associated with markers of oxidative stress in enteric neurons, including accumulation of nitrosylated protein aggregates and the swelling and distortion of nitrergic neurons [29]. Oxidative stress is a feature of both aging and PD and is now known to hinder the performance of the iron trafficking apparatus resulting in atypical iron distribution [31, 32]. Recent findings have highlighted iron homeostasis as key in the pathological events involved in the neuronal toxicity and nigral degeneration associated with PD [33]. Cellular reactive oxygen species (ROS) production by alpha-synuclein oligomers has recently been shown to be dependent on the presence of metal ions, with the addition of metal chelators blocking oligomer-induced ROS production and preventing neuronal death [34]. In the current study, ATH434 reduced the proportion of cells with nuclear Hu+ translocation in A53T mice, however this was not correlated with enhanced survival of enteric neurons. This may in part be due to early loss of vulnerable neurons exhibiting Hu+ translocation, resulting in a proportional reduction of neurons with Hu+ translocation in the remaining population of neurons. Whether or not ATH434 alters markers of oxidative stress in the ENS is unknown and requires further investigation.
Alpha-synuclein aggregation is a hallmark pathology of PD. Accumulation of insoluble cytoplasmic protein inclusions, which contain alpha-synuclein, are thought to be key in the neurodegenerative processes associated with PD, contributing to neuronal impairment and degeneration [35]. Previously it has been found that ATH434 preserved SNpc neurons in a number of PD animal models, which translated to improved motor function accompanied by reduced levels of alpha-synuclein observed by western blot and markers of oxidative stress [17]. Whilst in the current study no significant difference in the proportion of alpha-synuclein accumulation per ganglion was observed when comparing vehicle-treated WT and A53T mice, there was a trend for reduction in the proportion of alpha-synuclein accumulation in neurons of the colon of A53T mice following treatment with ATH434. This reduction in alpha-synuclein accumulation in neurons was not associated with an increase in survival of enteric neurons, this may be due to early toxicity and loss of enteric neurons which was not prevented by subtle reductions in alpha-synuclein or may be due to downstream effects of ATH434 rather than a direct effect on alpha-synuclein. Whilst we have previously been able to examine the quantity of soluble and insoluble alpha-synuclein in the brain, this is not possible in GI tissues where the digestion of surrounding muscle and mucosa is difficult and the concentration of alpha-synuclein is low [36].
In the CNS, it is clear that the mechanism of action of ATH434 is dependent on the metal binding site, as shown in studies using the non-metal binding analog of ATH434 (formerly called PBT434-met), where no protective effect was observed when the metal binding site was blocked [17].
In the ENS, the precise mechanism of how ATH434 may restore gut function is unclear, and it is possible that ATH434 has off target effects that are independent of its metal chelator properties. The lack of robust alpha-synuclein accumulation in the ENS in this study suggests that ATH434 may exert its effects on the gut in an alpha-synuclein-independent manner. Several studies have shown that the gut microbiota of patients with PD differs to healthy individuals [37, 38]. The gut microbiome of A53T mice has also been shown to differ and has been used to model the human condition [39]. Therefore, it is possible that ATH434 may exert its effect by altering gut microbial health and potentially gut function, however further studies are needed to investigate this area.
As expected, data from this study indicate that ATH434 has no effect on motor phenotype in ageing A53T mice when treatment with ATH434 is started at either 12 or 15 months. Previous publications have shown that in A53T mice, significant neuronal loss in the SNpc occurs at 8 months of age [17, 19] and motor deficits emerge at ∼6 to 8 months of age [17, 20, 40]. As neuronal loss in the SNpc is already well established at 12 months we did not expect that ATH434 would prevent the motor deficit. However, it has been shown that when mice are fed from 4 months of age, prior to the onset of motor dysfunction, ATH434 can prevent the loss of neurons in the SNpc, and improve motor phenotype [17].
In the current study, the times of onset of specific deficits is of interest and should be noted. In humans it is well documented that GI dysfunction occurs prior to motor symptoms, however in these mice it is clear that motor symptoms and neuronal loss in the brain occur prior to GI symptoms and enteric neuron loss.
CONCLUSION
In conclusion, our data demonstrate that there is a distinct functional GI deficiency in aged A53T mice which is repaired with ATH434 treatment. ATH434 treatment halved colonic propulsion time in A53T mice, however this did not appear to be correlated with enhanced neuronal survival or alpha-synuclein pathology in the ENS. The strong effect of ATH434 on colonic propulsion regardless of its effects on enteric neurons suggests that it, and other compounds like it, may be clinically useful in other disorders of colonic motility. Further studies should investigate different doses and dosing regimes in other models of colonic dysfunction and/or constipation that are non-PD related.
ACKNOWLEDGMENTS
The Florey Institute of Neuroscience and Mental Health acknowledge the strong support from the Victorian Government and in particular the funding from the Operational Infrastructure Support Grant. This work was supported by funding from the National Health and Medical Research Council (grant APP1145686). ATH434 was generously supplied by Alterity Therapeutics.
CONFLICT OF INTEREST
DIF, PAA, RAC, and KJB have received personal compensation for consulting, serving on a scientific advisory board, speaking, or other activities with Alterity Therapeutics (previously Prana Biotechnology) and have received research support from Alterity Therapeutics.
REFERENCES
[1] | Alexander GE ((2004) ) Biology of Parkinson’s disease: Pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin Neurosci 6: , 259–280. |
[2] | Dorsey ER , Bloem BR ((2018) ) The Parkinson pandemic-a call to action. JAMA Neurol 75: , 9–10. |
[3] | Schapira AHV , Chaudhuri KR , Jenner P ((2017) ) Non-motor features of Parkinson disease. Nat Rev Neurosci 18: , 435–450. |
[4] | Weiss HD , Pontone GM ((2019) ) “Pseudo-syndromes” associated with Parkinson disease, dementia, apathy, anxiety, and depression. Neurol Clin Pract 9: , 354–359. |
[5] | Kim JS , Sung HY ((2015) ) Gastrointestinal autonomic dysfunction in patients with Parkinson’s disease. J Mov Disord 8: , 76–82. |
[6] | Fasano A , Visanji NP , Liu LW , Lang AE , Pfeiffer RF ((2015) ) Gastrointestinal dysfunction in Parkinson’s disease. Lancet Neurol 14: , 625–639. |
[7] | Savica R , Carlin JM , Grossardt BR , Bower JH , Ahlskog JE , Maraganore DM , Bharucha AE , Rocca WA ((2009) ) Medical records documentation of constipation preceding Parkinson disease: A case-control study. Neurology 73: , 1752–1758. |
[8] | Ashraf W , Pfeiffer RF , Park F , Lof J , Quigley EM ((1997) ) Constipation in Parkinson’s disease: Objective assessment and response to psyllium. Mov Disord 12: , 946–951. |
[9] | Singaram C , Ashraf W , Gaumnitz EA , Torbey C , Sengupta A , Pfeiffer R , Quigley EM ((1995) ) Dopaminergic defect of enteric nervous system in Parkinson’s disease patients with chronic constipation. Lancet 346: , 861–864. |
[10] | Gotz ME , Double K , Gerlach M , Youdim MB , Riederer P ((2004) ) The relevance of iron in the pathogenesis of Parkinson’s disease. Ann N Y Acad Sci 1012: , 193–208. |
[11] | Han YH , Lee JH , Kang BM , Mun CW , Baik SK , Shin YI , Park KH ((2012) ) Topographical differences of brain iron deposition between progressive supranuclear palsy and parkinsonian variant multiple system atrophy. J Neurol Sci 325: , 29–35. |
[12] | Walter U ((2010) ) Transcranial sonography in brain disorders with trace metal accumulation. Int Rev Neurobiol 90: , 166–178. |
[13] | Mandel S , Maor G , Youdim MB ((2004) ) Iron and alpha-synuclein in the substantia nigra of MPTP-treated mice: Effect of neuroprotective drugs R-apomorphine and green tea polyphenol (-)-epigallocatechin-3-gallate. J Mol Neurosci 24: , 401–416. |
[14] | Ostrerova-Golts N , Petrucelli L , Hardy J , Lee JM , Farer M , Wolozin B ((2000) ) The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci 20: , 6048–6054. |
[15] | Dexter DT , Statton SA , Whitmore C , Freinbichler W , Weinberger P , Tipton KF , Della Corte L , Ward RJ , Crichton RR ((2011) ) Clinically available iron chelators induce neuroprotection in the 6-OHDA model of Parkinson’s disease after peripheral administration. J Neural Transm (Vienna) 118: , 223–231. |
[16] | Devos D , Moreau C , Devedjian JC , Kluza J , Petrault M , Laloux C , Jonneaux A , Ryckewaert G , Garçon G , Rouaix N ((2014) ) Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid Redox Signal 21: , 195–210. |
[17] | Finkelstein DI , Billings JL , Adlard PA , Ayton S , Sedjahtera A , Masters CL , Wilkins S , Shackleford DM , Charman SA , Bal W ((2017) ) The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol Commun 5: , 53. |
[18] | Stamler D , Bradbury M , Wong C , Offman E ((2019) ) A first in human study of PBT434, a novel smallmolecule inhibitor of α-synuclein aggregation (S4. 001). AAN Enterprises. |
[19] | Oaks AW , Frankfurt M , Finkelstein DI , Sidhu A ((2013) ) Age-dependent effects of A53T alpha-synuclein on behavior and dopaminergic function. PLoS One 8: , e60378. |
[20] | Diwakarla S , Finkelstein DI , Constable R , Artaiz O , Di Natale M , McQuade RM , Lei E , Chai XY , Ringuet MT , Fothergill LJ , Lawson VA , Ellett LJ , Berger JP , Furness JB ((2019) ) Chronic isolation stress is associated with increased colonic and motor symptoms in the A53T mouse model of Parkinson’s disease. Neurogastroenterol Motil 32: , e13755. |
[21] | Rota L , Pellegrini C , Benvenuti L , Antonioli L , Fornai M , Blandizzi C , Cattaneo A , Colla E ((2019) ) Constipation, deficit in colon contractions and alpha-synuclein inclusions within the colon precede motor abnormalities and neurodegeneration in the central nervous system in a mouse model of alpha-synucleinopathy. Transl Neurodegener 8: , 5. |
[22] | Fairman CL , Clagett-Dame M , Lennon VA , Epstein ML ((1995) ) Appearance of neurons in the developing chick gut. Dev Dyn 204: , 192–201. |
[23] | Herbison AE , Simonian SX , Norris PJ , Emson PC ((1996) ) Relationship of neuronal nitric oxide synthase immunoreactivity to GnRH neurons in the ovariectomized and intact female rat. J Neuroendocrinol 8: , 73–82. |
[24] | Schindelin J , Arganda-Carreras I , Frise E , Kaynig V , Longair M , Pietzsch T , Preibisch S , Rueden C , Saalfeld S , Schmid B , Tinevez JY , White DJ , Hartenstein V , Eliceiri K , Tomancak P , Cardona A ((2012) ) Fiji: An open-source platform for biological-image analysis. Nat Methods 9: , 676–682. |
[25] | Kuo Y-M , Li Z , Jiao Y , Gaborit N , Pani AK , Orrison BM , Bruneau BG , Giasson BI , Smeyne RJ , Gershon MD ((2010) ) Extensive enteric nervous system abnormalities in mice transgenic for artificial chromosomes containing Parkinson disease-associated α-synuclein gene mutations precede central nervous system changes. Hum Mol Genet 19: , 1633–1650. |
[26] | Vidal-Martinez G , Vargas-Medrano J , Gil-Tommee C , Medina D , Garza NT , Yang B , Segura-Ulate I , Dominguez SJ , Perez RG ((2016) ) FTY720/fingolimod reduces synucleinopathy and improves gut motility in A53T mice: Contributions of pro-brain-derived neurotrophic factor (PRO-BDNF) and mature BDNF. J Biol Chem 291: , 20811–20821. |
[27] | Costa M , Furness J ((1976) ) The peristaltic reflex: An analysis of the nerve pathways and their pharmacology. Naunyn Schmiedebergs Arch Pharmacol 294: , 47–60. |
[28] | Desmet AS , Cirillo C , Vanden Berghe P ((2014) ) Distinct subcellular localization of the neuronal marker HuC/D reveals hypoxia-induced damage in enteric neurons. Neurogastroenterol Motil 26: , 1131–1143. |
[29] | Rivera LR , Thacker M , Pontell L , Cho H-J , Furness JB ((2011) ) Deleterious effects of intestinal ischemia/reperfusion injury in the mouse enteric nervous system are associated with protein nitrosylation. Cell Tissue Res 344: , 111–123. |
[30] | Carbone S , Jovanovska V , Brookes S , Nurgali K ((2016) ) Electrophysiological and morphological changes in colonic myenteric neurons from chemotherapy-treated patients: A pilot study. Neurogastroenterol Motil 28: , 975–984. |
[31] | Orth ES , Medeiros M , Bortolotto T , Terenzi Hn , Kirby AJ , Nome F ((2011) ) Dephosphorylation reactions with deferoxamine, a potential chemical nuclease. J Org Chem 76: , 10345–10348. |
[32] | Medeiros MS , Schumacher-Schuh A , Cardoso AM , Bochi GV , Baldissarelli J , Kegler A , Santana D , Chaves CMMBS , Schetinger MRC , Moresco RN ((2016) ) Iron and oxidative stress in Parkinson’s disease: An observational study of injury biomarkers. PLoS One 11: , e0146129. |
[33] | Ayton S , Lei P , Mclean C , Bush AI , Finkelstein DI ((2016) ) Transferrin protects against Parkinsonian neurotoxicity and is deficient in Parkinson’s substantia nigra. Signal Transduct Target Ther 1: , 16015. |
[34] | Deas E , Cremades N , Angelova PR , Ludtmann MH , Yao Z , Chen S , Horrocks MH , Banushi B , Little D , Devine MJ ((2016) ) Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson’s disease. Antioxid Redox Signal 24: , 376–391. |
[35] | Rocha EM , De Miranda B , Sanders LH ((2018) ) Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol Dis 109: , 249–257. |
[36] | Ellett LJ , Hung LW , Munckton R , Sherratt NA , Culvenor J , Grubman A , Furness JB , White AR , Finkelstein DI , Barnham KJ , Lawson VA ((2016) ) Restoration of intestinal function in an MPTP model of Parkinson’s disease. Sci Rep 6: , 30269. |
[37] | Scheperjans F , Aho V , Pereira PA , Koskinen K , Paulin L , Pekkonen E , Haapaniemi E , Kaakkola S , Eerola-Rautio J , Pohja M ((2015) ) Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord 30: , 350–358. |
[38] | Mulak A , Bonaz B ((2015) ) Brain-gut-microbiota axis in Parkinson’s disease. World J Gastroenterol 21: , 10609–10620. |
[39] | Vidal-Martinez G , Chin B , Camarillo C , Herrera GV , Yang B , Sarosiek I , Perez RG ((2020) ) A pilot microbiota study in Parkinson’s disease patients versus control subjects, and effects of FTY720 and FTY720-mitoxy therapies in parkinsonian and multiple system atrophy mouse models. J Parkinsons Dis 10: , 185–192. |
[40] | Paumier KL , Sukoff Rizzo SJ , Berger Z , Chen Y , Gonzales C , Kaftan E , Li L , Lotarski S , Monaghan M , Shen W , Stolyar P , Vasilyev D , Zaleska M , W DH, Dunlop J ((2013) ) Behavioral characterization of A53T mice reveals early and late stage deficits related to Parkinson’s disease. PLoS One 8: , e70274. |


