
Safety, Pharmacokinetics, and Pharmacodynamics of Oral Venglustat in Patients with Parkinson’s Disease and a GBA Mutation: Results from Part 1 of the Randomized, Double-Blinded, Placebo-Controlled MOVES-PD Trial
Abstract
Background:
Glucocerebrosidase gene (GBA) mutations influence risk and prognosis of Parkinson’s disease (PD), possibly through accumulation of glycosphingolipids, including glucosylceramide (GL-1). Venglustat is a novel, brain penetrant glucosylceramide synthase inhibitor.
Objective:
Evaluate venglustat pharmacology, safety, and tolerability in patients with PD and GBA mutations (GBA-PD).
Methods:
Part 1 of the phase 2 MOVES-PD trial (NCT02906020) was a randomized, double-blinded, placebo-controlled, dose-escalation study performed in six countries. Eligible participants included Japanese and non-Japanese patients aged 18–80 years with PD diagnosis and heterozygous GBA mutation. Participants were randomized to three doses of once-daily oral venglustat or placebo and were followed up to 36 weeks (Japanese participants: 52 weeks). Primary endpoint was venglustat safety and tolerability versus placebo. Secondary and exploratory endpoints included venglustat pharmacokinetics and pharmacodynamics.
Results:
Participants (N = 29) received venglustat (Japanese, n = 9; non-Japanese, n = 13) or placebo (n = 3; n = 4). Eight (89%) Japanese and 12 (92%) non-Japanese venglustat-treated participants experienced at least one adverse event (AE) versus two (67%) and four (100%) participants from the respective placebo groups. Most AEs were mild or moderate; no serious AEs or deaths occurred. Two venglustat-treated non-Japanese participants discontinued due to AEs (confusional state and panic attack). Over 4 weeks, venglustat exposure in plasma and cerebrospinal fluid (CSF) increased, and GL-1 levels in plasma and CSF decreased, both in a dose-dependent manner. At the highest dose, CSF GL-1 decreased by 72.0% in Japanese and 74.3% in non-Japanese participants.
Conclusion:
Venglustat showed favorable safety and tolerability in MOVES-PD Part 1 and target engagement was achieved in CSF.
INTRODUCTION
Mutations in the GBA gene (glucosylceramidase beta; OMIM 606463), encoding the lysosomal enzyme glucocerebrosidase (GCase), increase the risk of developing Parkinson’s disease (PD) [1]. Although the penetrance of the gene is low and the majority of GBA mutation carriers will not develop synucleinopathies, an estimated 7–10% of patients with PD carry a GBA mutation, with prevalence varying depending on genotyping methods, ethnicity, and geography [1, 2]. These mutations are associated as a group with reduced enzyme activity, earlier disease onset, and more rapid cognitive decline [1, 3–6].
The exact mechanism by which GBA mutations contribute to PD pathogenesis is unclear, but one hypothesis posits that GBA loss of function causes an abnormal glycosphingolipid environment, leading to impaired lysosomal function, misprocessing of the neuronal protein α-synuclein, and neuronal dysfunction [7]. Deficient GCase leads to accumulation of its substrate, glucosylceramide (GL-1), affecting membrane fluidity of lysosomes [8–10]. Further evidence suggests GCase and α-synuclein form a bidirectional pathogenic loop in which excess GL-1 levels accelerate and stabilize formation of toxic α-synuclein oligomers, in turn blocking the endoplasmic reticulum–Golgi trafficking of GCase, leading to further GL-1 accumulation [11]. In addition, high plasma levels of lipids involved in ceramide metabolism, including GL-1, are associated with impaired cognition in patients with PD and may be markers of toxic α-synuclein deposition in the brain [12, 13]. Therefore, one of the proposed approaches to arrest the pathologic cross-talk between glucocerebrosidase and α-synuclein is modulation of the turnover of glycosphingolipids, such as GL-1 [7]. Indeed, reduction of GL-1 levels by glucosylceramide synthase (GCS) inhibitors reversed the stabilization of pathogenic α-synuclein species in neurons derived from induced pluripotent stem cells of patients with PD [14, 15], and treatment of synucleinopathy in animal models using a central nervous system (CNS)–penetrant GCS inhibitor reduced accumulation of pathologic α-synuclein aggregates in the hippocampus [16].
Venglustat is a potent, CNS-penetrant, small-mole-cule GCS inhibitor that reduces GL-1 production (Supplementary Figure 1). In healthy individuals, once-daily venglustat, administered orally for 2 weeks, was safe and well tolerated, and associated with a time- and dose-dependent reduction in plasma GL-1 levels [17]. The Multicenter pharmacOkinetics and interVEntional Study in Parkinson’s Disease (MOVES-PD) trial is a phase 2, randomized, double-blind, placebo-controlled study divided into two consecutive parts, aiming to evaluate the efficacy, safety, pharmacokinetics, and pharmacodynamics of orally administered venglustat in patients with PD and a GBA mutation. Here, we report results from Part 1 of the trial, a dose-escalation study with a sequential cohort design, including 29 heterozygous GBA mutation carriers with PD. The primary endpoint was safety and tolerability of venglustat treatment, the secondary endpoint evaluated pharmacokinetics in plasma and cerebrospinal fluid (CSF) of participants, and exploratory endpoints included plasma and CSF GL-1 level assessments through 4 weeks.
MATERIALS AND METHODS
Study design and participants
The MOVES-PD trial is a 3-year, phase 2, randomized, double-blind, placebo-controlled, multicenter study of venglustat in participants with early-stage PD carrying a GBA mutation. The trial consists of two consecutive parts. Part 1 was a placebo-controlled dose-escalation study of safety, pharmacokinetics, and pharmacodynamics of three venglustat doses using three sequential cohorts (Fig. 1 and Supplementary Figure 2). Part 2 is a placebo-controlled study of the efficacy and safety of venglustat and commenced after the appropriate dose was selected in Part 1.
Fig. 1
CONSORT diagram: disposition of participants enrolled in Part 1 of the MOVES-PD trial. Two non-Japanese participants permanently discontinued the study due to adverse events after receiving venglustat and post week 4 (one participant [low dose] discontinued due to confusional state, and one participant [high dose] due to a panic attack). aInclusion criterion: male or female patients with a diagnosis of PD (with at least two of the following signs: resting tremor, postural instability, akinesia/hypokinesia, and muscle rigidity) and who are heterozygous carriers of a GBA mutation. bInclusion criterion: patients carrying known sequence variants associated with GBA-PD, in addition to having a diagnosis of PD (with at least two of the following signs: resting tremor, postural instability, akinesia/hypokinesia, or muscle rigidity), must also have a diagnosis of RBD confirmed by historically documented polysomnography or by RBD screening questionnaire. GBA, glucocerebrosidase (glucosylceramidase beta) gene; PD, Parkinson’s disease; RBD, rapid eye movement sleep behavior disorder.
![CONSORT diagram: disposition of participants enrolled in Part 1 of the MOVES-PD trial. Two non-Japanese participants permanently discontinued the study due to adverse events after receiving venglustat and post week 4 (one participant [low dose] discontinued due to confusional state, and one participant [high dose] due to a panic attack). aInclusion criterion: male or female patients with a diagnosis of PD (with at least two of the following signs: resting tremor, postural instability, akinesia/hypokinesia, and muscle rigidity) and who are heterozygous carriers of a GBA mutation. bInclusion criterion: patients carrying known sequence variants associated with GBA-PD, in addition to having a diagnosis of PD (with at least two of the following signs: resting tremor, postural instability, akinesia/hypokinesia, or muscle rigidity), must also have a diagnosis of RBD confirmed by historically documented polysomnography or by RBD screening questionnaire. GBA, glucocerebrosidase (glucosylceramidase beta) gene; PD, Parkinson’s disease; RBD, rapid eye movement sleep behavior disorder.](https://content.iospress.com:443/media/jpd/2022/12-2/jpd-12-2-jpd212714/jpd-12-jpd212714-g001.jpg)
In Part 1, we enrolled 29 participants from 13 sites, including four Japanese sites and nine sites in five countries outside Japan (Germany, Portugal, Spain, Sweden, and the United States), between January 26, 2017, and October 17, 2018. Enrolment in Japan was initiated after the trial had commenced in sites outside Japan. Japanese participants (n = 12) were evaluated separately as requested by the Japanese Pharmaceuticals and Medical Devices Agency to conduct a descriptive comparison of pharmacokinetics and pharmacodynamics of venglustat and efficacy and safety outcomes in this population versus non-Japanese participants (n = 17). Eligible participants were aged 18–80 years. The upper age limit was increased from 70 to 80 years after trial commenced, based on feedback from investigators, and the age range for Japanese participants (20–80 years) was defined after trial commenced. Also, participants had a diagnosis of PD based on established diagnostic criteria and PD symptoms for ≥2 years, were at stage 2 or less at baseline on the Hoehn and Yahr scale (after trial commenced, the state was inputted as ON for participants receiving a stable dose of PD medication) [18, 19], and were heterozygous carriers of a GBA mutation. All participants who were screened had GBA mutations that cause Gaucher disease (GD); two participants had additional variant GBA mutations. The GBA mutations assessed (Supplementary Table 1) included those known to cause mild (type 1) and severe GD (types 2 and 3) [20]. Per protocol, patients who had variants of unknown significance (e.g., E326K, T369M) were required to have documented history of rapid eye movement sleep behavior disorder (RBD) confirmed by historically documented polysomnography or by RBD screening questionnaire. All participants enrolled in MOVES-PD Part 1 carried GBA mutations that were pre-defined in the protocol. Participants could continue concomitant symptomatic PD medications if the regimens were stable for ≥ 30 days (≥ 60 days for rasagiline) before randomization. Key exclusion criteria included secondary or atypical parkinsonism, the presence of leucine-rich repeat kinase 2 gene (LRRK2) p.G2019S (p.Gly2019Ser) mutation, biallelic GBA mutations (i.e., GD diagnosis), Montreal Cognitive Assessment (MoCA) [21] score < 20 at baseline, history of deep brain stimulation, and presence of any medical disorders and/or clinically relevant findings that could interfere with study-related procedures (e.g., conditions that preclude the safe performance of routine lumbar puncture, bleeding diathesis, or clinically significant coagulopathy or thrombocytopenia). A complete list of inclusion and exclusion criteria is shown in Supplementary Table 2.
This study was conducted in accordance with international ethics guidelines, including the Declaration of Helsinki and the International Council for Harmonisation guidelines for good clinical practice, and all applicable laws, rules, and regulations. All study procedures were approved by local institutional ethics review boards of participating sites, and participants provided written informed consent.
Randomization and masking
In MOVES-PD Part 1, participants were randomized (3 : 1 for Japanese participants; 4 : 1 for non-Japanese participants) to receive venglustat or placebo based on permuted blocks of four (Japan) or five (outside Japan) and using an interactive voice-response system. Randomization was stratified by region (Japan or outside Japan), and by dose cohort. The sponsor provided a randomized treatment kit number list, and the study biostatistician provided the randomization scheme to the centralized treatment allocation system that generated the participant randomization list. Participants, investigators, and study site personnel were blinded to treatment allocation by use of visually indistinguishable capsules provided in identical packaging. The investigators, study site personnel, and sponsor were blinded to all pharmacokinetic and pharmacodynamic data throughout the study.
Procedures
Participants were randomized within 60 days from the screening visit. Baseline data, including complete GBA gene sequencing and LRRK2 p.G2019S (p.Gly2019Ser) genotyping results, were collected within the 60-day screening period. MoCA and Movement Disorder Society-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) [22, 23] scores were collected on the day of randomization. After the baseline visit on day 1, participants were seen at days 2 and 3, week 2 (one visit), week 4 (two visits), and week 8 (one visit), and were then followed every 4 weeks until the completion of Part 1 (up to a maximum of 36 weeks for non-Japanese participants and 52 weeks for Japanese participants). Venglustat was administered orally once daily, and dose was escalated in three sequential cohorts (low, mid, or high dose). Once participants completed the first 4-week course of therapy, safety and tolerability data were reviewed in a blinded manner by an internal review committee before dose escalation in the subsequent cohort could occur. A data monitoring committee was responsible for overseeing the safety of participants and the benefit/risk ratio throughout the study. Duration of exposure to venglustat was different among the doses, as per the sequential cohort design, with the low- and high-dose venglustat cohorts commencing the study first and last, respectively. Based on this sequential cohort design, participants randomized to low-, mid-, and high-dose venglustat had variable total follow-up, for a maximum of 36 weeks for non-Japanese participants and 52 weeks for Japanese participants. Randomized participants remained on their respective dosages throughout Part 1. The MOVES-PD trial allowed for participants to continue or receive as-needed concurrent symptomatic PD medications, recorded at the time of the screening visit and during the study.
This trial is registered with ClinicalTrials.gov, number NCT02906020.
Outcomes
The primary outcome in MOVES-PD Part 1 was safety and tolerability of venglustat versus placebo. Safety assessments were recorded after treatment initiation, at days 1, 2, and 3, weeks 2 and 4, and for every 4 weeks thereafter (up to a maximum of 36 weeks for non-Japanese participants and 52 weeks for Japanese participants). Safety was assessed by means of physical examination; neurologic examination; clinical laboratory evaluations conducted at a central laboratory, including hematology, biochemistry, urinalysis, and serology tests; vital signs; adverse events (AEs) and concomitant medication; ophthalmologic examination; and electrocardiogram. AEs, serious AEs, and AEs of special interest (AESIs) were recorded throughout the treatment-emergent period, defined as the period from first to last intake of treatment, with an additional 6-week post-treatment period. Serious AEs were defined as fatal or life-threatening events, events requiring or prolonging inpatient hospitalization, or disabling/incapacitating events, congenital anomalies, or medically important events (e.g., malignancies or chronic neurodegenerative diseases). Based on preclinical toxicology data, AESIs included new or worsening lens opacities and cataracts (as assessed by ophthalmologic examination at screening and at weeks 4, 12, 28, and 36 [weeks 4, 12, 36, 52 for Japanese participants]), pregnancy in participant or partner, increase of alanine aminotransferase, or symptomatic overdose with study drug.
The secondary outcome was pharmacokinetics of venglustat in plasma and CSF. Plasma samples were collected prior to the first dose and at 1, 2, 4, 8, and 24 h post dose (day 1 and week 4), 48 h post dose (day 1 only), and before taking the daily tablet for weeks 2 and 8. Venglustat concentration in plasma was determined using a validated liquid chromatography tandem mass spectrometry method with a lower limit of quantification of 0.500 ng/mL. Venglustat is stable in plasma samples at –20°C for up to 369 days and at –70°C for up to 735 days. CSF samples were collected by lumbar puncture during the screening period (within 14 days prior to randomization) or on the day of randomization, and at week 4, 2–4 h after taking the daily tablet. Venglustat stability in CSF samples has been established at –20°C and –70°C for 731 days. Venglustat concentration in CSF was determined using a validated liquid chromatography tandem mass spectrometry method with a lower limit of quantification of 0.100 ng/mL.
For plasma pharmacokinetics, the following para-meters were calculated using noncompartmental methods: maximum plasma concentration (Cmax), time to Cmax (tmax), area under the plasma concentration versus time curve from 0 to 24 h (AUC0–24; at day 1 and week 4), area under the plasma concentration versus time curve from 0 to 48 h (AUC0–48; at day 1 only), and apparent total systemic clearance at steady state (CLSS/F; at week 4 only). Trough plasma concentration observed just before treatment administration during repeated dosing (Ctrough) was also assessed at weeks 2, 4, and 8. For CSF pharmacokinetics, observed concentrations at week 4 were summarized using descriptive statistics.
In exploratory analyses, venglustat pharmacodynamics in plasma and CSF was assessed by mean percent change in GL-1 levels from baseline through week 4. GL-1 levels were assessed in plasma and CSF samples collected at the day of randomization and week 4, with plasma GL-1 levels also assessed at week 2. GL-1 levels were normalized by volume, measured as μg/ml or ng/ml in plasma and CSF, respectively. Levels of glucosylsphingosine (lyso-GL-1), a metabolic product of GL-1, were also measured in plasma and CSF, but most values were found to be below the limit of quantification, reflecting the fact that enrolled participants are heterozygous carriers of GBA mutations, and they do not have a GD diagnosis. An additional exploratory outcome was the effect of venglustat on selected scales and questionnaires. In this paper, we report MDS-UPDRS (parts II [motor experiences of daily living; 13 items] and III [motor examination; 33 items]) [22, 23] scores through week 8. The MDS-UPDRS scale was assessed during the OFF state, with no PD medication taken for ≥12 h prior.
Statistical analysis
Analyses were based on all available data from participants who enrolled in Part 1 and were randomized to receive either venglustat or placebo for up to 52 weeks for Japanese participants and 36 weeks for non-Japanese participants. As Part 1 is an exploratory, dose-escalation study, the sample size was not based on a statistical power calculation, and there were no prespecified hypotheses. No formal statistical comparison between the Japanese and non-Japanese groups was performed. Although the study was not designed to analyze data from pooled Japanese and non-Japanese participants, post hoc analyses using pooled data were conducted (Supplementary Table 3). Participants included in the primary safety analyses were those who were randomized and received at least one dose of venglustat or placebo. Parameters for plasma pharmacokinetics were analyzed using noncompartmental methods and summarized using descriptive statistics. Observed CSF concentrations and plasma Ctrough were summarized using descriptive statistics at each time point. For pharmacodynamics, percent reduction of plasma or CSF GL-1 from baseline was calculated as (value –baseline) / baseline×100. Formal statistical testing was not conducted on MDS-UPDRS scale assessments.
Role of the funding source
The MOVES-PD study is funded by Sanofi, which oversaw the design and conduct of the trial, data analysis and interpretation, and review of the manuscript. Apart from the Sanofi employees listed as authors, the funders had no role in preparation, approval, and decision to submit the manuscript for publication. Although the clinical investigators were not directly involved in the design of the trial, some of the authors on this paper provided input as advisors/scientific advisory board members. The sponsor performed the statistical analyses. All authors had full access to data, participated in the analyses, wrote the manuscript, had final responsibility for the decision to submit for publication, and vouch for the accuracy and completeness of the findings herein, and the fidelity of the trial to the protocol.
RESULTS
Participants in Part 1 of the MOVES-PD trial were recruited between December 1, 2017, and October 17, 2018, in Japan and between January 26, 2017, and October 30, 2017, in sites outside Japan. A total of nine Japanese (low dose: n = 3; mid dose: n = 3; high dose: n = 3) and 13 non-Japanese (low dose: n = 4; mid dose: n = 5; high dose: n = 4) participants were randomized to receive venglustat, and three Japanese and four non-Japanese participants randomized to placebo, respectively (Fig. 1 and Supplementary Figure 2).
Demographics and baseline characteristics of the Japanese and non-Japanese groups are presented in Table 1. The mean time since symptom onset and mean time since diagnosis were similar between the Japanese and non-Japanese groups. Numerical differences were observed between groups in other baseline and disease characteristics (Table 1). Of note, 7/17 (41.2%) of the non-Japanese participants had a severe GBA mutation, including p.L444P (p.Leu483Pro); four participants, of whom one also had the p.A456P (p.Ala495Pro) mutation and was not diagnosed with GD (as displayed by similar GCase enzymatic activity in this patient to heterozygous individuals, suggesting these 2 mutations were present in the same allele) and 84GG (p.Leu29AlafsX18; three participants), and 10/17 (58.8%) carried other GBA mutations, including p.N370S (p.Asn409Ser; seven participants) and the variant p.E326K (p.Glu365Lys; three participants). The proportion of participants having a severe mutation was higher in the Japanese group (10/12 [83.3%], including p.L444P [p.Leu483Pro; five participants, of whom two also had the p.A456P (p.Ala495Pro) mutation and were determined not to have GD, based on similar GCase enzymatic activity to heterozygous individuals], p.R120W [p.Arg159Trp; three participants], p.D409H [p.Asp448His; one participant], and p.G193W [p.Gly232Trp; one participant]), whereas fewer Japanese participants had other mutations (2/12 [16.7% ]; both carrying the p.R496C (p.Arg535Cys) mutation). In all venglustat-treated participants, median (interquartile range [IQR]) duration of exposure at low, mid, and high doses was 35.7 (32.0–40.1) weeks, 20.3 (17.6–24.1) weeks, and 8.3 (8.0–12.1) weeks, respectively. All Japanese participants and 15/17 (88.2%) non-Japanese participants received concomitant symptomatic medications (Supplementary Table 4).
Table 1
Baseline characteristics in Part 1 of the MOVES-PD trial
| Japanese (n = 12) | Non-Japanese (n = 17) | |
| Age, y, mean (SD) | 54.3 (8.6) | 58.4 (7.9) |
| Sex, n (%) | ||
| Male | 7 (58.3) | 13 (76.5) |
| Female | 5 (41.7) | 4 (23.5) |
| Race, n (%) | ||
| White | 0 | 17 (100) |
| Asian | 12 (100) | 0 |
| Body mass index, kg/m2, mean (SD) | 23.1 (3.2) | 25.4 (3.1) |
| Time since symptoms onset, y | ||
| Mean (SD) | 6.7 (3.8) | 6.7 (4.0) |
| Median (min, max) | 5 (3, 14) | 6 (2, 18) |
| Time since diagnosis, years | ||
| Mean (SD) | 5.2 (4.0) | 5.2 (4.4) |
| Median (min, max) | 3.5 (1, 14) | 4.7 (0, 18) |
| Predominant symptoms at onset, n (%) | ||
| Rigidity/bradykinesia | 8 (66.7) | 5 (29.4) |
| Tremor | 4 (33.3) | 12 (70.6) |
| Family history of PD, n (%) | ||
| Yes | 3 (25.0) | 7 (41.2) |
| No | 9 (75.0) | 10 (58.8) |
| Hoehn and Yahr stage, n (%) | ||
| Stage 1 | 1 (8.3) | 0 |
| Stage 2 | 10 (83.3) | 17 (100) |
| Stage 3 | 1 (8.3) | 0 |
| MoCA total score, mean (SD) | 25.3 (4.0) | 27.4 (2.6) |
| MDS-UPDRS part II + part III score,a,b mean (SD) | 45.9 (12.7) | 44.6 (21.9) |
| MDS-UPDRS part II score,a mean (SD) | 10.8 (6.9) | 10.1 (7.0) |
| MDS-UPDRS part III score,b mean (SD) | 35.2 (9.9) | 34.6 (16.2) |
| Any severec GBA mutation, n (%) | 10 (83.3) | 7 (41.2) |
| p.L444P (p.Leu483Pro) | 5 (41.7) | 4 (23.5) |
| 84GG (p.Leu29AlafsX18) | 0 | 3 (17.6) |
| p.A456P (p.Ala495Pro) + p.L444P (p.Leu483Pro) | 2 (16.7)d | 1 (5.9)d |
| p.R120W (p.Arg159Trp) | 3 (25.0) | 0 |
| p.D409H (p.Asp448His) | 1 (8.3) | 0 |
| p.G193W (p.Gly232Trp) | 1 (8.3) | 0 |
| Any othere GBA mutation, n (%) | 2 (16.7) | 10 (58.8) |
| p.N370S (p.Asn409Ser) | 0 | 7 (41.2) |
| p.E326K (p.Glu365Lys) | 0 | 3 (17.6) |
| p.R496C (p.Arg535Cys) | 2 (16.7) | 0 |
aPart II (motor experiences of daily living, 13 items). bPart III (motor examination, 33 items) [22, 23]. cSevere GBA mutations are categorized as those that cause GD types 2 and 3 [20]. dAll participants with p.A456P (p.Ala495Pro) mutations also had p.L444P (p.Leu483Pro) mutations, and were determined not to have GD (based on similar GCase enzymatic activity in these patients to heterozygous individuals, suggesting these 2 mutations were present in the same allele). eOther, nonsevere GBA mutations included mild GBA mutations that have been associated with GD type 1 [20], and the p.E326K (p.Glu365Lys) variant (no participant carried the p.T369M [p.Thr408Met] variant). GBA, glucocerebrosidase (glucosylceramidase beta) gene; GD, Gaucher disease; max, maximum; MDS-UPDRS, Movement Disorder Society-Unified Parkinson’s Disease Rating Scale; min, minimum; MoCA, Montreal Cognitive Assessment; PD, Parkinson’s disease.
The primary outcome data are shown in Table 2. Overall, 8/9 (88.9%) Japanese participants and 12/13 (92.3%) non-Japanese participants in the venglustat treatment groups experienced at least one AE compared with 2/3 (66.7%) and 4/4 (100%) participants in the respective placebo groups. AE incidences were similar in low-, mid-, and high-dose venglustat-treated participants, with no unique safety signals observed in Japanese and non-Japanese groups exposed to increasing doses of venglustat. Most AEs were mild or moderate in severity and resolved during the study without corrective treatment. Neurologic events were reported in three cases each of placebo- and venglustat-treated participants, with no events reported in the high-dose groups. Psychiatric, gastrointestinal, and eye disorders were frequently reported AEs in both populations. Psychiatric disorders were only reported in the venglustat-treated participants, with higher frequencies for most events reported in the low- or mid-dose groups than in the high-dose groups, except anxiety and panic attacks (Supplementary Table 5). Eye disorders were only reported in venglustat-treated participants, with no events reported in the high-dose groups. No serious AEs or deaths were reported in either Japanese or non-Japanese participants. No Japanese participant discontinued the study due to AEs, and two non-Japanese participants had AEs leading to permanent discontinuation from the study after week 4 since venglustat initiation. One participant (low dose) discontinued due to confusional state, and one participant (high dose) due to a panic attack; both AEs were deemed related to venglustat by the reporting investigator.
Table 2
AEs throughout Part 1 of the MOVES-PD triala
| Participants with AEs by MedDRA Primary System Organ Class, n (%) | Japanese (n = 12) | Non-Japanese (n = 17) | ||||||
| Venglustat | Venglustat | |||||||
| Placebo (n = 3) | Low (n = 3) | Mid (n = 3) | High (n = 3) | Placebo (n = 4) | Low (n = 4) | Mid (n = 5) | High (n = 4) | |
| Any AE | 2 (66.7) | 3 (100) | 3 (100) | 2 (66.7) | 4 (100) | 4 (100) | 4 (80.0) | 4 (100) |
| Ear and labyrinth disorders | 0 | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 1 (25.0) |
| Eye disorders | 0 | 1 (33.3) | 1 (33.3) | 0 | 0 | 2 (50.0) | 1 (20.0) | 0 |
| Gastrointestinal disorders | 0 | 3 (100) | 0 | 1 (33.3) | 2 (50.0) | 2 (50.0) | 2 (40.0) | 0 |
| General disorders and administration site conditions | 0 | 0 | 0 | 0 | 0 | 0 | 1 (20.0) | 1 (25.0) |
| Infections and infestations | 1 (33.3) | 2 (66.7) | 0 | 1 (33.3) | 0 | 0 | 0 | 1 (25.0) |
| Injury, poisoning, and procedural complications | 0 | 0 | 1 (33.3) | 2 (66.7) | 0 | 1 (25.0) | 1 (20.0) | 1 (25.0) |
| Investigationsb | 1 (33.3) | 0 | 0 | 0 | 0 | 0 | 1 (20.0) | 0 |
| Musculoskeletal and connective tissue disorders | 0 | 0 | 0 | 0 | 0 | 2 (50.0) | 1 (20.0) | 0 |
| Nervous system disorders | 0 | 1 (33.3) | 0 | 0 | 3 (75.0) | 1 (25.0) | 1 (20.0) | 0 |
| Psychiatric disorders | 0 | 1 (33·3) | 2 (66.7) | 0 | 0 | 3 (75.0) | 3 (60.0) | 2 (50.0) |
| Renal and urinary disorders | 1 (33.3) | 0 | 0 | 1 (33.3) | 0 | 0 | 0 | 1 (25.0) |
| Respiratory, thoracic, and mediastinal disorders | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (25.0) |
| Skin and subcutaneous tissue disorders | 0 | 0 | 1 (33.3) | 0 | 0 | 0 | 0 | 1 (25.0) |
| Vascular disorders | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (25.0) |
| AEs leading to treatment discontinuationc,d | 0 | 0 | 0 | 0 | 0 | 1 (25.0) | 0 | 1 (25.0) |
| Confusional state | 0 | 0 | 0 | 0 | 0 | 1 (25.0) | 0 | 0 |
| Panic attack | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (25.0) |
aTable includes AEs that occurred during the treatment-emergent period (defined as the period from first intake of treatment to last intake of treatment, with an additional 6-week post-treatment period). bOne cardiac murmur in the venglustat mid-dose group (non-Japanese) and one event of increased hepatic enzyme in the placebo group (Japanese). cPresented by MedDRA Preferred Terms. dBoth patients discontinued the study after week 4 since venglustat initiation. AE, adverse event; MedDRA, Medical Dictionary for Regulatory Activities.
Secondary outcome data for pharmacokinetics at week 4 are shown in Table 3 (pharmacokinetic parameters at day 1 are shown in Supplementary Table 6). At week 4, venglustat was absorbed in plasma with median tmax values of 2.1–4.3 h in Japanese participants and 2.0–3.6 h in non-Japanese participants across all doses tested. Venglustat exposure in plasma and CSF increased in a close to dose-proportional manner in both Japanese and non-Japanese participants, as assessed by pharmacokinetic parameters at week 4. In participants treated with the high dose, the mean CSF concentration/mean plasma Cmax ratio of venglustat at week 4 was approximately 0.06 (determined from post hoc analysis of pooled data from Japanese and non-Japanese participants as shown in Supplementary Table 3). Graphically, as observed from the Ctrough versus time plots, steady state appeared to have been achieved on or before 2 weeks (Supplementary Figure 3).
Table 3
Plasma and CSF pharmacokinetic parameters of venglustat at week 4
| Japanese (n = 9) | Non-Japanese (n = 13) | |||||
| Venglustat | Venglustat | |||||
| Low (n = 3) | Mid (n = 3) | High (n = 3) | Low (n = 4) | Mid (n = 5) | High (n = 4) | |
| Plasma pharmacokinetic parameters at week 4a | ||||||
| Cmax, ng/mL, mean (SD) | 56.3 (3.6) | 83.7 (15.1) | 145 (17.6) | 41.6 (7.4) | 77.8 (20.1) | 136 (13.4) |
| Geometric mean (CV%) | 56.2 (6.5) | 82.8 (18.0) | 144 (12.2) | 41.0 (17.8) | 75.8 (25.8) | 135 (9.9) |
| tmax, h, median (range) | 4.33 (4.33–8.00) | 2.12 (1.92–3.58) | 2.10 (1.93–3.70) | 1.96 (1.00–24.00) | 3.55 (2.00–8.03) | 2.89 (2.00–3.98) |
| AUC0–24, ng·h/mL, mean (SD) | 1130 (NA)b | 1650 (382) | 3150 (NA)c | 766 (84) | 1510 (305) | 2510 (369) |
| Geometric mean (CV%) | 1130 (NA) | 1620 (23.1) | 3140 (NA) | 763 (11.0) | 1490 (20.2) | 2490 (14.7) |
| CLSS/F, mL/h, mean (SD) | 3540 (NA)b | 5010 (1070) | 4780 (NA)c | 5270 (626) | 5450 (1010) | 6100 (1050) |
| Geometric mean (CV%) | 3540 (NA) | 4930 (21.3) | 4770 (NA) | 5240 (11.9) | 5370 (18.5) | 6030 (17.2) |
| CSF pharmacokinetic concentrations at week 4 (2–4 h post dose) | ||||||
| CSF concentration, ng/mL, mean (CV%) | 2.11 (7.8) | 4.33 (37.3) | 10.10 (9.1) | 1.85 (6.2)d | 3.15 (32.1)d | 5.96 (15.2)e |
aPlasma samples were collected at 1 h predose and 1, 2, 4, 8, and 24 h post dose. bn = 1. cn = 2. dOne participant from the low-dose group and two from the mid-dose group were not included in the CSF pharmacokinetic analysis because week 4 CSF sample was collected before dose instead of at the protocol-specified postdose time. eOne participant was not included in week 4 CSF analysis because sample was not collected. AUC0–24, area under the plasma concentration versus time curve from 0 to 24 h; Cmax, maximum observed concentration; CLSS/F, total systemic clearance at steady state; CSF, cerebrospinal fluid; CV%, coefficient of variation; NA, not applicable; tmax, time to maximum concentration.
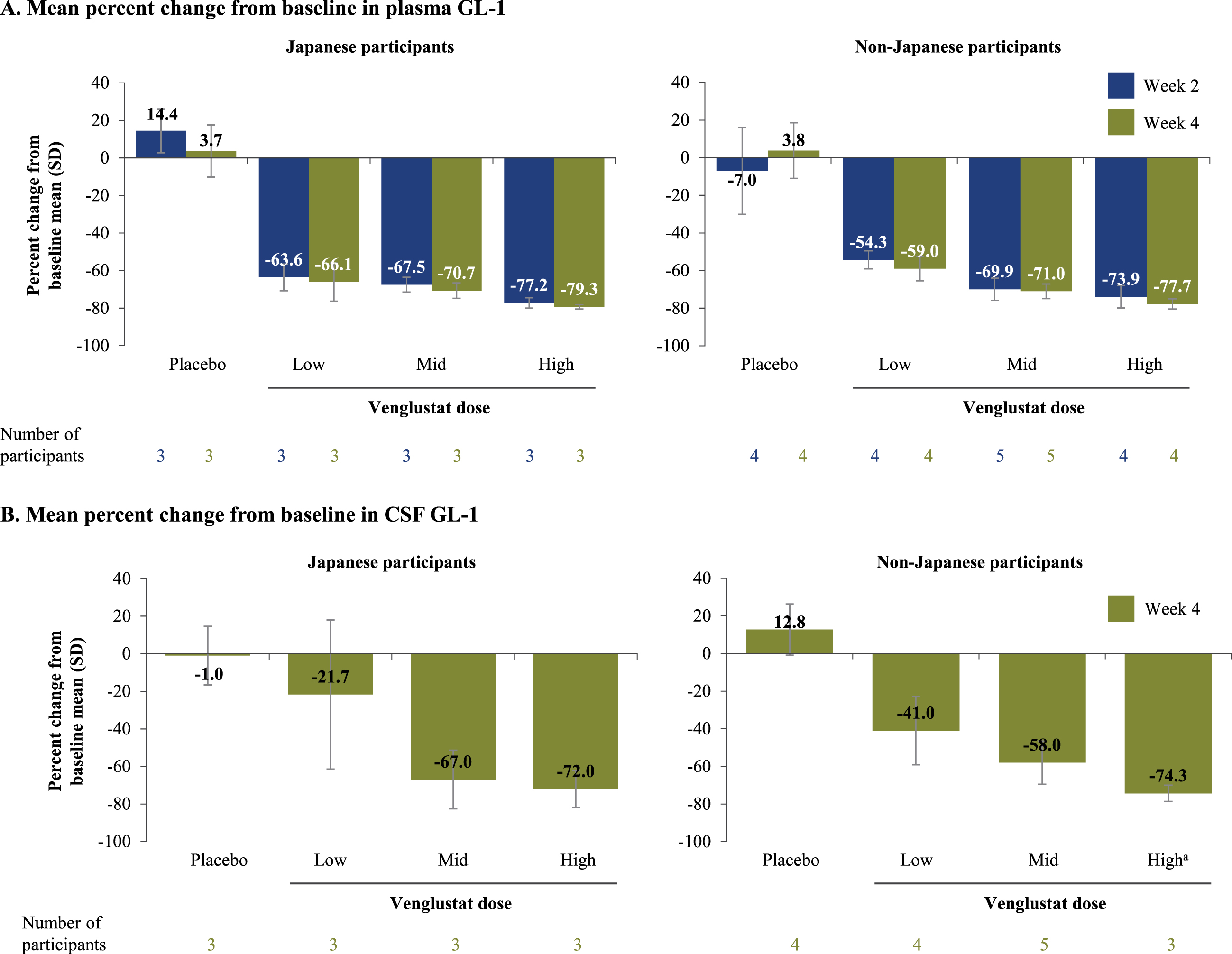
Plasma and CSF GL-1 levels of venglustat decreased from baseline in a dose-dependent manner over 4 weeks (Fig. 2 and Supplementary Figure 4). Mean GL-1 levels at baseline in both plasma and CSF were generally similar between venglustat- and placebo-treated participants and did not change over 4 weeks in the placebo groups (Supplementary Figure 4). For low-, mid-, and high-dose venglustat, the respective mean reductions in plasma GL-1 at week 4 were 66.1%, 70.7%, and 79.3% for Japanese participants and 59.0%, 71.0%, and 77.7% for non-Japanese participants; similar decreases in plasma GL-1 levels were observed at week 2 in both groups (Fig. 2). For low-, mid-, and high-dose venglustat, the respective mean reductions in CSF GL-1 at week 4 were 21.7%, 67.0%, and 72.0% for Japanese participants and 41.0%, 58.0%, and 74.3% for non-Japanese participants (Fig. 2). Post hoc analyses of pooled data from Japanese and non-Japanese participants showed mean reductions in CSF GL-1 levels from baseline at week 4 of 32.8%, 61.4%, and 73.2% for low-, mid-, and high-dose venglustat-treated participants, respectively (Supplementary Table 3).
Fig. 2
Percent reduction in plasma and CSF GL-1 levels after venglustat treatment. Mean percent change from baseline in plasma (A) and CSF (B) GL-1 levels in Japanese and non-Japanese participants who received placebo or were treated with venglustat (low, mid, or high dose) in Part 1 of the MOVES-PD trial. GL-1 levels were assessed in plasma samples collected at baseline, end of week 2, and end of week 4, and in CSF samples collected at baseline and end of week 4. aWeek 4 CSF sample was not collected for one participant from the non-Japanese population. CSF, cerebrospinal fluid; GL-1, glucosylceramide.
Through week 8, mean (SD) changes from baseline in MDS-UPDRS (parts II and III) scores performed in the OFF state in venglustat-treated Japanese and non-Japanese participants were –0.7 (10.7) and –4.9 (11.2) points, respectively, compared with 0.3 (2.5) and –3.0 (9.6) points observed in the respective placebo groups. There were no dose-proportional changes in MDS-UPDRS score assessments with venglustat (mean [SD] change from baseline to Week 8 with low-, mid-, and high-dose venglustat was –5.0 (11.0), –5.3 (9.5), and 8.3 (8.1) points for Japanese participants and –14.0 (10.7), 4.0 (7.7), and –2.5 (18.2) points for non-Japanese participants).
DISCUSSION
MOVES-PD is the first international, randomized, placebo-controlled trial of an oral, CNS-penetrant GCS inhibitor as a targeted treatment approach for a genetically defined patient population with PD. GCS inhibition by venglustat targets the accumulation of lipid substrates of mutant glucocerebrosidase, hence equilibrating biosynthesis of those substrates with the impaired catabolism as a result of GBA mutation [7]. The results from Part 1 of the trial demonstrated favorable safety and tolerability of venglustat and showed target engagement, justifying continuation into Part 2 to assess efficacy.
In Part 1, most AEs were mild to moderate, and AE incidences were largely similar between venglu-stat- and placebo-treated participants, indicating the primary endpoint was reached. Nervous system disorders and gastrointestinal events were frequent in both placebo- and venglustat-treated participants, consistent with common PD symptoms [24, 25]. Psychiatric events (including anxiety with panic attack, depression, hallucinations, and insomnia) were observed in venglustat-treated participants (Supplementary Table 5) and not in placebo-treated participants. Data from the Part 2 study including a larger cohort of venglustat treated GBA-PD patients (n = 110) or placebo (n = 111), will be important in ascertaining any potential risk relating to psychiatric side effects. Overall, our data demonstrate venglustat was well tolerated in patients with GBA-PD, consistent with observations from the phase 1 trial of venglustat in healthy individuals [17].
The secondary objective of our study was met, demonstrating CNS penetration of venglustat and a close to dose-proportional increase in both plasma and CSF venglustat exposure. Our exploratory analyses of the pharmacodynamic response of venglustat also demonstrated a dose-dependent decrease of GL-1 levels in plasma and CSF samples over the 4-week period. Notably, at the highest venglustat dose, we observed a decrease of 72.0–74.3% in CSF GL-1 levels from baseline, suggesting venglustat at the achieved central compartment concentrations can modify the glycosphingolipid profile in the CSF. These findings are particularly relevant as glycosphingolipid homeostasis is reported to play a role in the process of α-synuclein oligomerization and aggregation [26]. Moreover, preclinical data have demonstrated that modulation of glycosphingolipid levels with a CNS-penetrant GCS inhibitor can ameliorate behavioral and pathologic abnormalities in animal models of disease [16]. We conclude that the reduction of GL-1 levels over 4 weeks in the CSF of participants in our study reflects sufficient target engagement of venglustat by inhibiting GCS, showing for the first time modulation of lipid profile in the CNS of patients with PD. Additional biomarkers, including α-synuclein, neurofilament light chain, tau, phospho-tau, and beta-amyloid were measured as exploratory endpoints over 4 weeks of treatment; however treatment duration may have been too short to observe any clinically meaningful changes in these parameters. It should also be mentioned that currently there are no validated biomarkers for PD. Future studies may explore the effect of venglustat on biomarkers and disease progression over longer treatment duration.
The present study has several limitations. First, the MOVES-PD Part 1 trial was a small study, with 3–4 participants per treatment group per region. Second, the participants were genetically heterogeneous, carrying a range of GBA mutations, and the study was not designed or powered to evaluate outcomes stratified by mutation severity (to counter this limitation, certain genetic risk confounders such as a LRRK2 p.G2019S (p.Gly2019Ser) mutation or GBA biallelic carriers were excluded). Third, the participants were also clinically heterogeneous, with varying degrees of cognitive and motor functionality at baseline. To address this limitation, we ensured that the enrolled participants had early-stage PD, with mild symptoms on one or both sides of the body and no impairment of balance [18, 19]. Fourth, participants were allowed to continue existing symptomatic PD treatment, or to initiate immediate-release levodopa as needed, with potential confounding effects on safety and efficacy outcomes. To counter this limitation, all participants treated with dopaminergic therapy were assessed in the defined medications OFF state. Finally, the study was not designed to detect any disease-modifying effect of venglustat, which was not expected, since the timeframe for assessment of MoCA and MDS-UPDRS parts II and III was only 8 weeks and the number of participants was small. As expected, we did not observe a short-term symptomatic effect of venglustat based on the mechanism of action. This observation is important for the design of the future efficacy studies.
The demonstration of a favorable safety/tolera-bility profile and sufficient CNS penetration to achieve target engagement highlight the potential of venglustat as a therapeutic approach offering precision medicine for a genetically defined population with PD. A major challenge in clinical trials on genetic forms of PD is that genetic testing must be made more accessible, allowing for recruitment of a large number of participants of various genotypes [27, 28]. The pharmacological profile, safety, and tolerability of venglustat are favorable in GBA-PD patients. First, a population with early-stage PD was enrolled, providing an opportunity to test benefits of early therapeutic intervention. Second, the results demonstrated that GL-1 is a reliable biomarker of target engagement in the CSF that can potentially be used to assess responses to venglustat therapy. Part 2 of the MOVES-PD study is evaluating whether the GL-1 level reduction in the CSF observed after venglustat treatment is associated with improved clinical outcomes in GBA-positive patients with PD. Although Sanofi has recently announced the discontinuation of the development of venglustat for GBA-PD [29, 30], the present results provide critical information to advance the clinical development of venglustat in related diseases of the nervous system (including GD type 3 and late-onset GM2 gangliosidosis [ClinicalTrials.gov Identifiers: NCT02843035 and NCT04221451, respectively]).
In conclusion, the present data from MOVES-PD Part 1 demonstrated the ability of venglustat to engage its target in the CNS displaying a robust pharmacokinetic-pharmacodynamic relationship and provided clear criteria for dose selection to enable the initiation of the clinical efficacy study which is currently being terminated. The lack of safety signals and the favorable tolerability allowed the continuation of clinical explorations in GBA-PD and lysosomal storage diseases. Importantly, the protocol described here, including the criteria for defining mutation severity and stratification criteria, will help enable the investigation of a range of molecules that are initiating clinical development as potential therapies for PD.
CONTRIBUTORS
MJP, PM, KM, GUH, CRS, AHVS, SPS, and TZF contributed to study design. HS, TH, TG, SHI, AHVS, and TS collected the data. PM and JS provided statistical support. All authors contributed to drafting and critical review of the manuscript and provided approval of the final submission.
ACKNOWLEDGMENTS
The authors and Sanofi thank the trial participants, as well as the MOVES-PD steering committee and investigators. Critical review of the manuscript was provided by Michael Yeakey, PharmD, and Sarah Strattman, MS, of Sanofi. Editorial and writing assistance was provided by Panos Xenopoulos, PhD, and Richard J Hogan, PhD, of Elevate Scientific Solutions, and was funded by Sanofi. The MOVES-PD study was funded by Sanofi.
CONFLICT OF INTEREST
MJP, SJMG, PM, SS, JS, SW, and SPS report receiving personal compensation as employees of Sanofi, and may hold shares and/or stock options in the company. HS reports receiving grant support from Boehringer Japan, Dainippon Sumitomo, Kyowa Hakko-Kirin, and Otsuka, and honoraria from Dainippon Sumitomo, Kyowa Hakko-Kirin, Medtronic, Novartis Pharma KK, Otsuka FP, and Takeda. TH reports receiving speaker’s honoraria and research funding from Dainippon Sumitomo, Eisai Co, Ltd, Kyowa Hakko-Kirin, Novartis Pharma KK, Otsuka, Sanofi, and Takeda, and grant support from Dainippon Sumitomo, Eisai Co, Ltd, the Setsuro Fujii Memorial, the Osaka Foundation for Promotion of Fundamental Medical Research, and the Japan Agency for Medical Research and Development under grant number 19dm0107156, the Setsuro Fujii Memorial. TG reports receiving speaker’s honoraria from MedUpdate, Novartis, Teva, and UCB Pharma, and grant support from the German Research Foundation, the German Federal Ministry of Education and Research, the European Commission, the Helmholtz Association, and the Michael J Fox Foundation; he also serves as chairman of the Scientific Advisory Board of the “Joint Programming for Neurodegenerative Diseases” program, funded by the European Commission. SHI has received honoraria for CME services, consulting, research grants, and/or promotional speaking on behalf of AbbVie, Acadia, Acorda, Adamas, Addex, Affiris, Alexva, Allergan, Amarantus, Amneal, Aptinyx, Axial, Axovant, Benevolent, Biogen, Britannia, Cadent, Cala, Cerecor, Cerevel, Cipla, Eli Lilly, Enterin, GE Healthcare, Global Kinetics, Impax, Impel, Intec Pharma, Ipsen, Jazz, Kyowa, Lundbeck, Merz, the Michael J Fox Foundation, Mitsubishi Tanabe, Neuralys, Neurocrine, Neuroderm, Parkinson Study Group, Pharma2B, Prilenia, Promentis, Revance, Roche, Sanofi, Sunovion, Sun Pharma, Teva, Theravance, UCB, US WorldMeds and Zambon. RNA reports research support from the NIH, the Department of Defense, the Parkinson’s Foundation, and the Michael J Fox Foundation, and receives consultation fees from Janssen, Restorbio, Roche, and Sanofi. GC reports participating on Data and Safety Monitoring Boards for Astra-Zeneca, Avexis Pharmaceuticals, Biolinerx, Brainstorm Cell Therapeutics, Bristol Meyers Squibb/Celgene, CSL Behring, Galmed Pharmaceuticals, Green Valley Pharma, Mapi Pharmaceuticals LTD, Merck, Merck/Pfizer, Mitsubishi Tanabe Pharma Holdings, Neurim, Novartis, Ophazyme, Opko Biologics, Reata Pharmaceuticals, Sanofi-Aventis, Teva pharmaceuticals, VielaBio Inc, NHLBI (Protocol Review Committee), and NICHD (OPRU oversight committee); reports receiving consulting fees or participating on advisory boards from Alexion, Antisense Therapeutics, Biodelivery Sciences International, Biogen, Clinical Trial Solutions LLC, Genzyme, Genentech, GW Pharmaceuticals, Immunic, Klein-Buendel Incorporated, Medimmune/Viela Bio, Medday, Merck/Serono, Neurogenesis LTD, Novartis, Osmotica Pharmaceuticals, Perception Neurosciences, Reckover Pharmaceuticals, Recursion/Cerexis Pharmaceuticals, Regeneron, Roche, SAB Biotherapeutics, and TG Therapeutics; is employed by the University of Alabama at Birmingham; and is President of Pythagoras, Inc, a private consulting company located in Birmingham, AL, USA. NH reports receiving consulting fees for participating in advisory boards from Dainippon Sumitomo, Eisai, FP, Kyowa Hakko-Kirin, Ono, Otsuka, Tanabe-Mitsubishi, and grant support from Biogen, Boehringer Japan, Boston Scientific, Eisai, Medtronic, Meiji, Otsuka, and Takeda; and honoraria from Dainippon, FP, Novartis, Otsuka, and Takeda. GUH reports receiving consulting fees or participating on advisory boards from AbbVie, Alzprotect, Asceneuron, Biogen, Biohaven, Lundbeck, Novartis, Retrotope, Roche, Sanofi, UCB, and the Weston Brain Institute, and has received honoraria for scientific presentations from AbbVie, Biogen, Roche, Teva, UCB, and Zambon. KM reports receiving consulting fees from Ceraspir, GE Healthcare, Invicro, LTI, Lundbeck, the Michael J Fox Foundation, Neuroderm, Neuron23, Proclara, Roche, Takeda, and UBC. AHVS reports receiving consulting fees from Inflammazome, Kyowa, Prevail, and Sanofi. CRS is named as co-inventor on a US patent application on sphingolipids biomarkers that is jointly held by Brigham & Women’s Hospital and Sanofi; has consulted for Sanofi; has collaborated with Genzyme, Lysosomal Therapies, Opko, Pfizer, and Proteome Sciences; is on the Scientific Advisory Board of the American Parkinson Disease Association; has served as Advisor to the Michael J Fox Foundation, NIH, Department of Defense, and Google; and has received funding from the NIH, the US Department of Defense, the Michael J Fox Foundation, and the American Parkinson Disease Association. TS reports receiving consulting fees and honoraria from Acadia, Adamas, Teva, and UCB Pharma; consulting fees from AbbVie, Acorda, Anavex, Allergan, NeuroDerm, PhotoPharmics, Revance, Sanofi, Sunovion, Voyager, US WorldMeds, and the Michael J Fox Foundation; and research funding from Biogen, NeuroDerm, Roche, Sanofi, NINDS, the Michael J Fox Foundation, and the Parkinson Foundation. NG reports owning stock from BOL, Lysosomal Therapeutic Ltd and Vibrant; has served as a consultant for AbbVie, Biogen, BOL, Denali, NeuroDerm, Pharma2B, Sanofi Genzyme and Vibrant; has participated in advisory boards for Biogen, Denali, NeuroDerm, Sanofi Genzyme and Sionara; has received honoraria from AbbVie, Sanofi Genzyme and the Movement Disorder Society; and has received grants from Biogen, Ionis, the European Union, the Israel Science Foundation, the Michael J Fox Foundation and the National Parkinson Foundation. TZF received compensation as an employee of Sanofi at the time the study was conducted, and is currently employed by Alnylam Pharmaceuticals (Cambridge, MA, USA).
DATA SHARING
Qualified researchers may request access to patient-level data and related study documents in-cluding the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan, and data set specifications. Patient-level data will be anonymized and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi’s data sharing criteria, eligible studies, and process for requesting access can be found at: https://www.clinicalstudydatarequest.com.
PREVIOUS PRESENTATION OF DATA
Some of the data have been presented at the World Parkinson Congress (June 4–7, 2019), the 13th European Working Group on Gaucher Disease (July 4–6, 2019), the International Congress of Parkinson’s Disease and Movement Disorders (September 22–26, 2019, and September 12–16, 2020), the 2020 Virtual Annual Meeting of the American Academy of Neurology, the 16th Annual Research Meeting of WORLDSymposiumTM (February 10–13, 2020), and the 6th Congress of the European Academy of Neurology (May 23–26, 2020).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JPD-212714.
REFERENCES
[1] | Migdalska-Richards A , Schapira A ((2016) ) The relationship between glucocerebrosidase mutations and Parkinson disease. J Neurochem 139: , 77–90. |
[2] | Blandini F , Cilia R , Cerri S , Pezzoli G , Schapira AHV , Mullin S , Lanciego JL ((2019) ) Glucocerebrosidase mutations and synucleinopathies: toward a model of precision medicine. Mov Disord 34: , 9–21. |
[3] | Cilia R , Tunesi S , Marotta G , Cereda E , Siri C , Tesei S , Zecchinelli AL , Canesi M , Mariani CB , Meucci N , Sacilotto G , Zini M , Barichella M , Magnani C , Duga S , Asselta R , Solda G , Seresini A , Seia M , Pezzoli G , Goldwurm S ((2016) ) Survival and dementia in-associated Parkinson’s disease: the mutation matters. Ann Neurol 80: , 662–673. |
[4] | Liu G , Boot B , Locascio JJ , Jansen IE , Winder-Rhodes S , Eberly S , Elbaz A , Brice A , Ravina B , van Hilten JJ , Cormier-Dequaire F , Corvol JC , Barker RA , Heutink P , Marinus J , Williams-Gray CH , Scherzer CR , International Genetics of Parkinson Disease Progression (IGPP) Consortium ((2016) ) Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann Neurol 80: , 674–685. |
[5] | Davis MY , Johnson CO , Leverenz JB , Weintraub D , Trojanowski JQ , Chen-Plotkin A , Van Deerlin VM , Quinn JF , Chung KA , Peterson-Hiller AL , Rosenthal LS , Dawson TM , Albert MS , Goldman JG , Stebbins GT , Bernard B , Wszolek ZK , Ross OA , Dickson DW , Eidelberg D , Mattis PJ , Niethammer M , Yearout D , Hu SC , Cholerton BA , Smith M , Mata IF , Montine TJ , Edwards KL , Zabetian CP ((2016) ) Association of mutations and the E326K polymorphism with motor and cognitive progression in Parkinson disease. JAMA Neurol 73: , 1217–1224. |
[6] | Huh YE , Chiang MSR , Locascio JJ , Liao Z , Liu G , Choudhury K , Kuras YI , Tuncali I , Videnovic A , Hunt AL , Schwarzschild MA , Hung AY , Herrington TM , Hayes MT , Hyman BT , Wills AM , Gomperts SN , Growdon JH , Sardi SP , Scherzer CR ((2020) ) beta-Glucocerebrosidase activity in GBA-linked Parkinson disease: the type of mutation matters. Neurology 95: , e685–e696. |
[7] | Sardi SP , Cedarbaum JM , Brundin P ((2018) ) Targeted therapies for Parkinson’s disease: from genetics to the clinic. Mov Disord 33: , 684–696. |
[8] | Plotegher N , Bubacco L , Greggio E , Civiero L ((2019) ) Ceramides in Parkinson’s disease: from recent evidence to new hypotheses. Front Neurosci 13: , 330. |
[9] | Batta G , Soltész L , Kovács T , Bozó T , Mészár Z , Kellermayer M , Szöllősi J , Nagy P ((2018) ) Alterations in theproperties of the cell membrane due to glycosphingolipidaccumulation in a model of Gaucher disease. Sci Rep 8: , 157. |
[10] | Pavićević A , Lakočević M , Popović M , Popović-Bijelić A , Daković M , Mojović M ((2018) ) Changes of the peripheral blood mononuclear cells membrane fluidityfrom type 1 Gaucher disease patients: an electron paramagneticresonance study. Biol Chem 399: , 447–452. |
[11] | Mazzulli JR , Xu YH , Sun Y , Knight AL , McLean PJ , Caldwell GA , Sidransky E , Grabowski GA , Krainc D ((2011) ) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146: , 37–52. |
[12] | Mielke MM , Maetzler W , Haughey NJ , Bandaru VV , Savica R , Deuschle C , Gasser T , Hauser AK , Graber-Sultan S , Schleicher E , Berg D , Liepelt-Scarfone I ((2013) ) Plasma ceramide and glucosylceramide metabolism is altered in sporadic Parkinson’s disease and associated with cognitive impairment: a pilot study. PLoS One 8: , e73094. |
[13] | Lerche S , Schulte C , Wurster I , Machetanz G , Roeben B , Zimmermann M , Deuschle C , Hauser AK , Bohringer J , Krageloh-Mann I , Waniek K , Lachmann I , Petterson XT , Chiang R , Park H , Wang B , Liepelt-Scarfone I , Maetzler W , Galasko D , Scherzer CR , Gasser T , Mielke MM , Hutten SJ , Mollenhauer B , Sardi SP , Berg D , Brockmann K ((2021) ) The mutation matters: CSF profiles of GCase, sphingolipids, alpha-synuclein in PDGBA. Mov Disord 36: , 1216–1228. |
[14] | Zunke F , Moise AC , Belur NR , Gelyana E , Stojkovska I , Dzaferbegovic H , Toker NJ , Jeon S , Fredriksen K , Mazzulli JR ((2018) ) Reversible conformational conversion of alpha-synuclein into toxic assemblies by glucosylceramide. Neuron 97: , 92–107. |
[15] | Kim S , Yun SP , Lee S , Umanah GE , Bandaru VVR , Yin X , Rhee P , Karuppagounder SS , Kwon SH , Lee H , Mao X , Kim D , Pandey A , Lee G , Dawson VL , Dawson TM , Ko HS ((2018) ) GBA1 deficiency negatively affects physiological alpha-synuclein tetramers and related multimers. Proc Natl Acad Sci U S A 115: , 798–803. |
[16] | Sardi SP , Viel C , Clarke J , Treleaven CM , Richards AM , Park H , Olszewski MA , Dodge JC , Marshall J , Makino E , Wang B , Sidman RL , Cheng SH , Shihabuddin LS ((2017) ) Glucosylceramide synthase inhibition alleviates aberrations in synucleinopathy models. Proc Natl Acad Sci U S A 114: , 2699–2704. |
[17] | Peterschmitt MJ , Nigel PS , Crawford MB , Sebastiaan JM , Gaemers MD , Allena JJ , Sharma J , Pham TT ((2020) ) Pharmacokinetics, pharmacodynamics, safety, and tolerability of oral venglustat in healthy volunteers . Clin Pharmacol Drug Devel 0: , 1–13. |
[18] | Hoehn MM , Yahr MD ((1967) ) Parkinsonism: onset, progression and mortality. Neurology 17: , 427–442. |
[19] | Goetz CG , Poewe W , Rascol O , Sampaio C , Stebbins GT , Counsell C , Giladi N , Holloway RG , Moore CG , Wenning GK , Yahr MD , Seidl L , Movement Disorder Society Task Force on Rating Scales for Parkinson’s Disease ((2004) ) Movement Disorder Society Task Force report on the Hoehn and Yahr staging scale: status and recommendations. Mov Disord 19: , 1020–1028. |
[20] | Gan-Or Z , Amshalom I , Kilarski LL , Bar-Shira A , Gana-Weisz M , Mirelman A , Marder K , Bressman S , Giladi N , Orr-Urtreger A ((2015) ) Differential effects of severe vs mild mutations on Parkinson disease. Neurology 84: , 880–887. |
[21] | Nasreddine ZS , Phillips NA , Bedirian V , Charbonneau S , Whitehead V , Collin I , Cummings JL , Chertkow H ((2005) ) The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc 53: , 695–699. |
[22] | Goetz CG , Fahn S , Martinez-Martin P , Poewe W , Sampaio C , Stebbins GT , Stern MB , Tilley BC , Dodel R , Dubois B , Holloway R , Jankovic J , Kulisevsky J , Lang AE , Lees A , Leurgans S , LeWitt PA , Nyenhuis D , Olanow CW , Rascol O , Schrag A , Teresi JA , Van Hilten JJ , LaPelle N ((2007) ) Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): process, format, and clinimetric testing plan. Mov Disord 22: , 41–47. |
[23] | Goetz CG , Tilley BC , Shaftman SR , Stebbins GT , Fahn S , Martinez-Martin P , Poewe W , Sampaio C , Stern MB , Dodel R , Dubois B , Holloway R , Jankovic J , Kulisevsky J , Lang AE , Lees A , Leurgans S , LeWitt PA , Nyenhuis D , Olanow CW , Rascol O , Schrag A , Teresi JA , van Hilten JJ , LaPelle N , Movement Disorder Society UPDRS Revision Task Force ((2008) ) Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord 23: , 2129–2170. |
[24] | DeMaagd G , Philip A ((2015) ) Parkinson’s disease and its management: Part 1: disease entity, risk factors, pathophysiology, clinical presentation, and diagnosis. P T 40: , 504–532. |
[25] | Poirier AA , Aube B , Cote M , Morin N , Di Paolo T , Soulet D ((2016) ) Gastrointestinal dysfunctions in Parkinson’s disease: symptoms and treatments. Parkinsons Dis 2016: , 6762528. |
[26] | Suzuki M , Sango K , Wada K , Nagai Y ((2018) ) Pathological role of lipid interaction with alpha-synuclein in Parkinson’s disease. Neurochem Int 119: , 97–106. |
[27] | Schneider SA , Alcalay RN ((2020) ) Precision medicine in Parkinson’s disease: emerging treatments for genetic Parkinson’s disease. J Neurol 267: , 860–869. |
[28] | Giladi N , Mirelman A , Thaler A , Orr-Urtreger A ((2016) ) A personalized approach to Parkinson’s disease patients based on founder mutation analysis. Front Neurol 7: , 71. |
[29] | Sanofi press release, https://www.sanofi.com/en/media-room/press-releases/2021/2021-02-05-07-30-00, Accessed July 1, 2021. |
[30] | Peterschmitt MJ , Giladi N , Alcalay RN , Cutter G , Höglinger GU , Schapira AHV , Scherzer CR , Simuni T , Gurevich T , Gasser T , Pacchetti C , Marek K , Minini P , Sardi SP ; on behalf of the MOVES-PDInvestigators (2021) Venglustat in Parkinson’s disease patients with a GB Amutation: results from part 2 of the phase 2MOVES-PD trial. The 15th International Conference on Alzheimer’s & Parkinson’s diseases (AD/PD™). |


