
An Exploratory Study Using Electronic Medical Records to Assess the Feasibility of Establishing Cohorts of Patients with Genetic Causes of Parkinson’s Disease
Abstract
Background:
More efficient screening methods are needed to improve the ability to identify and follow genetic cohorts in Parkinson’s disease (PD).
Objective:
To explore the use of the electronic medical records (EMRs) to identify participants with PD.
Methods:
Using an algorithm previously developed in collaboration with Maccabi Healthcare Services (MHS), approximately 5,200 participants with PD were identified, more than 3,200 were screened, and 837 participants were enrolled and genotyped for leucine-rich repeat kinase 2 (LRRK2) and beta-glucocerebrosidase (GBA) variants. Questionnaires were completed to ascertain Ashkenazi Jewish (AJ) ancestry and family history of PD.
Results:
Among 837 participants with PD, 82% were 65 years and older and 72% had a family history of AJ ancestry. Among those with AJ ancestry, 15.6% reported having relatives with PD. The frequency of observed mutations for LRRK2 and GBA genes combined was approximately 15.4%. The frequency of observed LRRK2 mutation was 6.1% overall and 7.2% from those with AJ ancestry; and for GBA mutation was 9.3% overall and 11.2% from those with AJ ancestry.
Conclusion:
Although the frequency of observed mutations in this study was lower than anticipated, mutation carriers were enriched among those with a family history of AJ ancestry increasing nearly 2-3-fold, from 3% –7% (LRRK2) and 4% –11% (GBA). The identification (and selection) of PD patients through EMRs prior to genotyping is a viable approach, to establish a genetically defined cohort of patients with PD for clinical research.
INTRODUCTION
Parkinson’s disease (PD) is one of the most common age-related neurodegenerative disorders affecting about 1% of people over the age of 60 years [1, 2]. While most cases of PD are sporadic or idiopathic, human genetic studies, including a genome-wide association studies (GWAS) of large cohorts, have identified multiple genes that are associated with increased risk for PD and related disorders, including mutations of the beta-glucocerebrosidase (GBA) and leucine-rich repeat kinase 2 (LRRK2) genes [3]. Among the familial PD population of Ashkenazi Jews, the LRRK2 p.(Gly2019Ser); mutation was estimated at 28% compared to a worldwide estimate of about 4% [4]. Among Ashkenazi Jews with PD approximately 17.9% carry mutations in the GBA gene [5].
To better understand the mechanism of how LRRK2 and GBA contribute to the risk of developing PD, it is important to study this disease in PD patients who are carriers of selected genetic variants. PD is characterized by a wide spectrum of motor and non-motor symptoms, complicated by varying degrees of the neurodegenerative process (as it is likely to have begun 7–10 years before the appearance of the cardinal motor symptoms of bradykinesia, rigidity, and resting tremor) [6, 7].
However, recruitment of patients with PD currently focuses on clinical presentation, PD diagnoses generally from a healthcare providers’ database, and genetic testing (data) is rarely offered. Thus, to be able to include relevant mutation carriers with PD based on genetic predispositions, more effective methods for identifying and screening patients with PD are required.
Collectively, among people of AJ ancestry, it has been estimated that up to 30–40% of PD cases [4, 8–11] can be explained by mutations in LRRK2 and GBA. This makes the Israeli population uniquely suited to gain further insights into PD pathophysiology and to study the relevance of LRRK2 and GBA on PD manifestation and progression. Moreover, leveraging the second largest HMO in Israel (the Maccabi Healthcare Services (MHS) which serves approximately 2.5 million patients), can provide a unique opportunity to assess whether electronic medical records (EMR) can be an effective approach to identify mutation carriers for the design of future clinical studies, including potential interventional trials. The MHS databases integrate data from the central laboratory, medication prescriptions, hospitalizations and procedures, as well as socio-demographic data. Using an EMR database as a starting point to potentially cast a wider net (than traditional physician referrals), the goal of this study was to understand whether a genetically defined cohort of PD patients could be generated more efficiently leveraging the EMR within MHS, as a feasible method to identify, contact and recruit a patient population for clinical trials.
METHODS
Clinical study design
This was a non-randomized, multi-center, observational genetic study to assess whether EMRs can be a useful tool to establish, interrogate, and recall genetically defined mutation carrier cohorts among patients with PD. This study was conducted in conformance with Good Clinical Practice and with applicable country or local requirements regarding ethical committee review, informed consent, and other statutes or regulations regarding the protection of the rights and welfare of human participants participating in biomedical research. A method for communicating the genetic results to the participants was approved by the ethical committee review and the clinical study was approved by the Israel Ministry of Health (MOH).
Approximately 5,200 pre-screened participants with PD, who are active members in the health management organization (HMO), specifically MHS, were identified based on a preceding EMR-based study, “Feasibility and Demographic Analysis to Identify Patients with Parkinson’s Disease in the Maccabi Healthcare Database” (using an algorithm developed previously in collaboration with MHS). PD patients were identified using an in silico EMR review from the MHS database from January 1, 2000 to December 31, 2015. Data on membership in MHS was updated on December 4, 2016. The computerized databases of MHS included up to 20 years of data on 2.5 million members (an assumed representative sample comprising approximately 25% of the Israeli population). The MHS databases integrate data from the MHS central laboratory, medication prescriptions and purchases throughout the MHS pharmacy network, consultations, hospitalizations, procedures, and socio-demographic data. Physician diagnoses are coded using the International Classification of Disease, 9th Edition (ICD-9-CM) codes as well as internal MHS codes for sub-classification.
Eligibility
Inclusion criteria
Participants were at least 18 years old; actively insured within the MHS for at least 1 year before index diagnosis; and the PD diagnoses were defined either by using the International Classification of Disease, 9th Edition (ICD-9-CM) codes as entered by a qualified physician or by using internal Maccabi codes for sub-classification for clinical symptoms that are strongly associated with PD and, therefore, are consistent with a highly probable diagnosis of PD.
After the initiation of the study, the inclusion criteria were amended to exclude patients in the category of highly probable or suspected PD; and only patients with clinically established or clinically probable PD, based on Movement Disorder Society (MDS) Clinical Diagnostic Criteria for PD, were eligible to enroll. However, 35 participants with suspected PD diagnoses, were included in the final analysis as genotyping was completed from these participants before the protocol amendment was finalized.
For exclusion criteria
Participants were excluded if the participants: 1) had a diagnosis of secondary parkinsonism, including but not limited to neuroleptic- or other drug-induced parkinsonism, traumatic brain injury, carbon monoxide or other chemical poisoning; 2) had a history of a basal ganglia, subcortical, or brainstem stroke with acute or residual movement disorder within 6 months prior to diagnosis of PD; 3) was taking, or had taken within the last 6 months, dopamine blocking agents (e.g., typical and atypical anti-psychotics); 4) had a prior or current history of schizophrenia or other psychotic disorder. However, psychosis symptoms considered related explicitly to PD and patients treated for psychosis symptoms related to PD were not exclusionary.
Qualifying individuals with PD (N = 3,218) after EMR pre-screening were invited to participate by telecommunication by protocol trained study coordinators and nurses. Participants (N = 837) consented to all study procedures, including genetic testing for LRRK2 and GBA variants. During the screening visit, participants completed a brief questionnaire (Supplementary Table 1) to identify their ethnic origin and family history of PD, their willingness to be re-contacted for possible participation in future studies and an option to receive their genotyping results. Medical history was reviewed to determine whether the participants met study criteria by the primary investigator (PI) and sub-investigators.
Based on the MOH approval of the clinical study, for those patients who provided consent to have results disclosed, the investigator notified them of their genetic status via letter, i.e., whether they tested positive or negative for mutations in the LRRK2 and/or the GBA gene (no specific genotyping data/results were referred in the letter). Genotype testing was performed in a CLIA accredited laboratory in the USA. Additionally, the letter invited those patients who tested positive for a mutation, to schedule a meeting with the PI (Neurologist) for additional information and genetic counseling was offered.
Statistical analysis
The analysis population consisted of all participants contacted from the MHS EMR database who provided consent for LRRK2 and GBA genetic testing and completed the study. No formal hypotheses were tested. The number of patients with PD, with LRRK2 and GBA mutations were estimated. Summaries of the number and percentage of patients contacted from the Maccabi EMR database who provided consent were generated. Among those providing consent and who had genetic testing, the number and percentage of patients with LRRK2 and GBA mutations were summarized. In addition, a summary of the number and percentage of patients contacted from the Maccabi EMR database who did not meet the inclusion/exclusion criteria at the screening visit(s) was generated.
Genotyping
One time collection of blood (8.5 ml) for LRRK2/GBA genotyping was conducted in the clinic following the signing of the informed consent (ICF). The ICF was available in English Hebrew and Russian languages and adhered to the IRB/IEC requirements, applicable laws and regulations and Sponsor requirements.
Mutations in LRRK2 and GBA were assessed using genetic assays in a CLIA (Clinical Laboratory Improvement Amendments) certified laboratory (Brooks Life sciences, Rutgers, NJ, USA). DNA was extracted from blood drawn into PAxGene® DNA tubes and standard DNA quality control was performed to ensure high quality DNA before genotyping. Genotyping was conducted to specifically measure, LRRK2 p.(Gly2019Ser); and 7 variants for GBA: p.(Leu29Alafs*18), c.(115 + 1G>A), p.(Glu326Lys), p.(Leu444Pro), p.(Asn370Ser), p.(Val389Leu), and p.(Arg496His) (see the Supplementary Material for detailed description of genotyping assays). Positive and negative control samples were obtained either as hapmap samples (Coriel, Camden, NJ) or synthesized synthetic String controls (Thermo Fisher) and were included in each run.
RESULTS
Initially, 5,326 potentially eligible participants with PD were identified using the algorithm, which was further refined by applying relevant exclusion criteria, evaluating a subset of samples for probable PD cases, and physician case adjudication, resulting in approximately 5,200 eligible, pre-screened participants with PD.
Out of 5,200, 3,218 (62%) participants who met the study criteria for PD or high probability of PD as identified from the HMO EMRs were contacted to schedule a screening visit. The remaining, eligible 1,982 participants (38%) were not contacted, as the study ended early at the discretion of the Sponsor. Most of the participants contacted for a screening visit (74%, N = 2,381) either declined (n = 838), failed to meet screening/study criteria (n = 369), were deceased (n = 204) or provided other reasons for not participating (n = 970), (see Fig. 1), resulting in enrollment of 837 (26%) participants with PD. Genotyping was performed on the 837 enrolled participants. The baseline characteristic and disposition are presented in Supplementary Table 2 and a summary of recruitment/participation based on the responses from the questionnaire is provided in the consort diagram in Fig. 1.
Fig. 1
Summary of recruitment to genetic results. Out of 5,200 participants who met the criteria for PD or high probability of PD as identified from the EMR review: 3,218 were contacted and 1,982 were not contacted due to early study completion. More than 2,380 participants out of 3,218 participants contacted either declined, did not meet the study criteria, or failed to meet screening criteria resulting in enrollment of 837 participants. The genetic dataset is shown within the dashed shaded box. Of the 837 participants, a total of 823 participants completed the study, and 14 participants discontinued primarily for not meeting study inclusion eligibility post enrollment (following the issuance of the protocol clarification letter). 788 participants had a definitive or clinically established PD diagnosis as per MDS criteria, and 35 participants whose EMR did not include a clinically established diagnosis of PD, but included high probability or suspected of PD. Among those 788 participants with the clinically established or definitive PD diagnoses, those identified as mutation carriers for LRRK2 and GBA were 48 and 73, respectively. 667 participants were identified as idiopathic PD. At the end of trial more than 96% of the participants (807) consented to receive the genetic results, and 93% (780) of the participants wanted to be re-contacted for future research. Dx, diagnosis; PD, Parkinson’s disease.
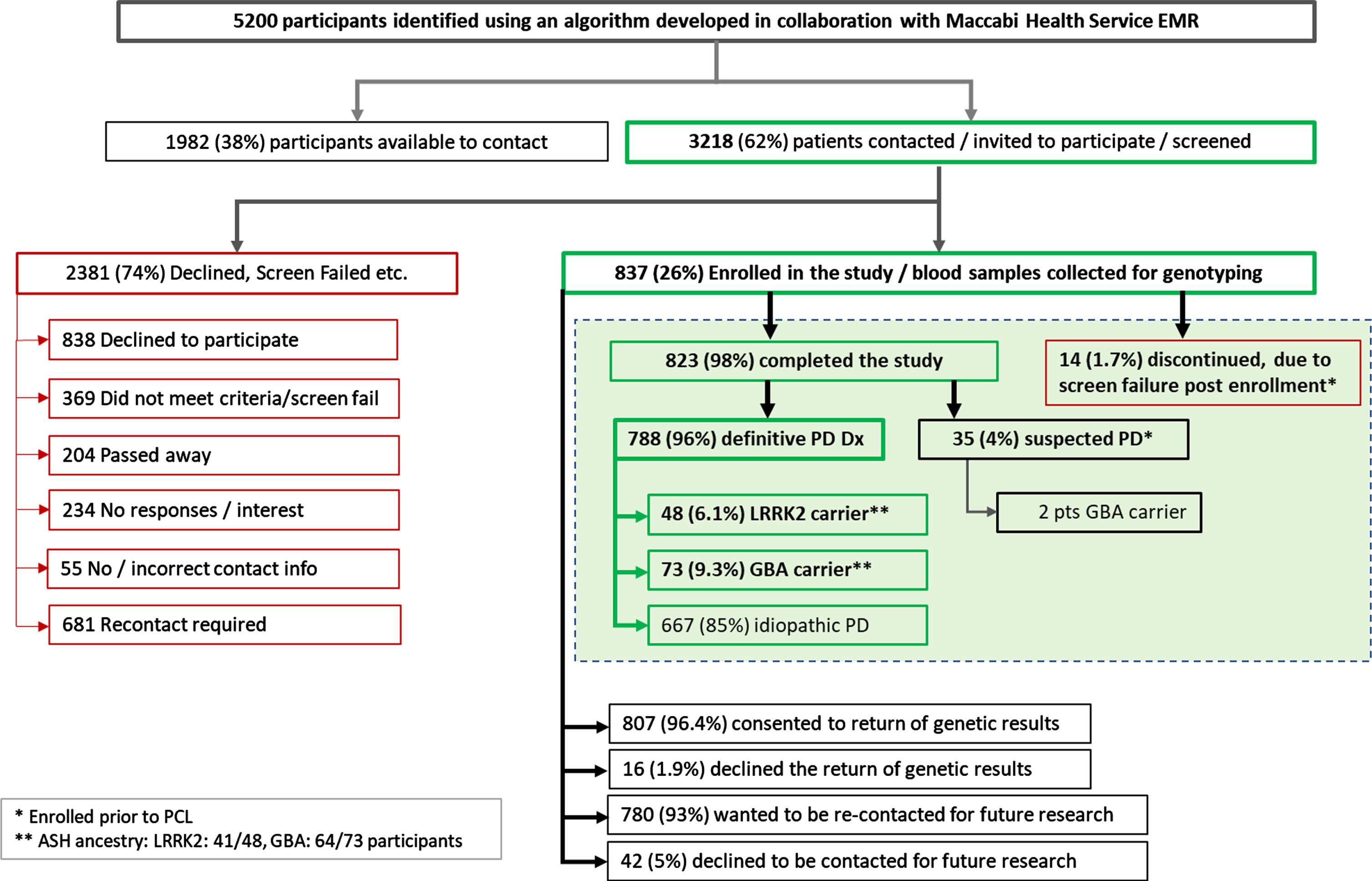
Among the 837 participants enrolled, 823 (98%) participants completed the study, and 14 (1.7%) participants were reported as discontinued, primarily for not meeting study inclusion eligibility noted in the protocol clarification letter (PCL was finalized post enrollment). The PCL required that a patient meets the inclusion criterion of having a Maccabi diagnostic code for clinical symptoms that are strongly associated with PD, therefore are consistent with a highly probable diagnosis of PD, and that they must also primarily have PD diagnosis, documented in the medical records. Thus, results from 14 discontinued subjects were not included in the final analysis. Of the 823, 96% (n = 788) participants had a definitive or clinically established or clinically probable PD as per MDS criteria, while 35 (4%) participants, whose EMR did not include a clinically established PD diagnoses (ICD-9 CM code) had a high probability or suspected of PD diagnosis (as evidenced by Maccabi diagnostic codes for PD).
Within the MHS EMR database, ethnic origin/ancestry is not recorded. Hence, all participants were asked to complete a questionnaire on their ancestry as well as if they could recall if any relatives or family members, had been previously diagnosed with PD. The summary of results of ancestry, genotype frequencies, GBA mutation variants, and family history are presented in Tables 1–3 and Supplementary Table 3, respectively. Most of the participants in the study (72%, 571 out of 788 participants), self-reported as having either all 4 grandparents or at least 1 grandparent of Ashkenazi Jewish (AJ) ancestry (68.5%, and 3.9% respectively). Approximately 27% (213 out of 788) of the participants reported having no relatives of AJ ancestry (Supplementary Table 3). Among those of AJ ancestry, less than 16% (89 out of 571) reported having relatives with PD (Table 1).
Table 1
Report of relatives with or without Parkinson’s disease among genotyped participants with definitive Parkinson’s disease
| Ancestry | Relativesa with PD n/N (%) | Relatives without PD n/N (%) | Unknown familial disease history n/NT (%) |
| Ashkenazi | 89/571 (15.6%) | 384/571 (67.3%) | 97/788 (12.3%) |
| Non-Ashkenazib | 38/213 (17.8%) | 154/213 (72.3%) | 20/788 (2.5%) |
| Unknown Ancestry | 1/788 (0.13%) | 2/788 (0.25%) | 1/788 (0.13%) |
| Totalc | 127/784 (16.2%) | 538/784 (68.6%) | 118/788 (15.0%) |
N, Total number of participants with definitive Parkinson’s disease of reported ancestry (784); NT, Total number of participants with definitive Parkinson’s disease (788); n, number of participants. aRelatives include paternal/maternal grandparents, biological mother, father, siblings (half/full), children. bSelf-report of having no relatives of Ashkenazi ancestry. cUnknown includes those participants who declined to answer or did not know status to response. 4 participants (out of 788) who reported unknown ancestry and PD status were not included in the total N = 784.
Table 2
Mutation frequencies among participants with Parkinson’s disease
| Mutation Frequency | ||||||
| Mutation Type | Number of Mutations Observed | Frequency of Mutation Overall | With 4 ASH Grandparents | With 1–3 ASH Grandparents | With 0 ASH Grandparents | Unknown Grandparents |
| n | Fraction n/N (%) | Fraction n/N (%) | Fraction n/N (%) | Fraction n/N (%) | Fraction n/N (%) | |
| LRRK2a | 48 | 48/788 (6.1%) | 38/540 (7.0%) | 3/31 (9.7%) | 6/213 (2.8%) | 1/4 (25%) |
| GBAb | 75 | 73/788 (9.3%) | 60/540 (11.1%) | 4/31 (12.9%) | 9/213 (4.2%) | 0/4 (0%) |
| Total | 121 | 121 (15.4%) | 98 (18.1%) | 7 (22.6%) | 15 (7%) | 1 (25%) |
ASH, Ashkenazi Jewish ancestry; GBA, β-glucocerebrosidase gene (7 variants for GBA: p.(Leu29Alafs*18), c.(115 + 1G>A), p.(Glu326Lys), p.(Leu444Pro), p.(Asn370Ser), p.(Val389Leu), and p.(Arg496His)); LRRK2, leucine-rich repeat kinase 2 gene (p.(Gly2019Ser)); N, Total number of participants with definitive PD of known ancestry within each column category; n, number of participants with mutation type within each column category. aOne participant had 1 GBA (p.(Asn370Ser)) mutation and LRRK2 (p.(Gly2019Ser)) mutation. bTwo participants have 2 GBA mutations (p.(Asn370Ser) and p.(Arg496His)) each but were counted once.
Table 3
GBA mutation variants in Ashkenazi and Non-Ashkenazi populations
| Variant | SNP | Ashkenazi n, (%) | Non-Ashkenazi n, (%) | Unknown (n) | Totala (n) |
| p.(Leu29Alafs*18) | rs387906315 | 1, (1.5) | 0 | 0 | 1 |
| c.(115 + 1G > A) | rs104886460 | 1, (1.5) | 0 | 0 | 1 |
| p.(Glu326Lys) | rs2230288 | 6, (9.3) | 2, (22.2) | 0 | 8 |
| p.(Asn370Ser) | rs76763715 | 48, (75) | 3, (33.3) | 0 | 51 |
| p.(val394Leu) | rs80356769 | 1, (1.5) | 0 | 0 | 1 |
| p.(Leu444Pro) | rs421016 | 0 | 2, (22.2) | 0 | 2 |
| p.(Arg496His) | rs80356773 | 7, (10.9) | 2, (22.2) | 0 | 9 |
| Total | 64 | 9 | 0 | 73b |
n, number of participants; GBA, β-glucocerebrosidase gene; SNP, single nucleotide polymorphism. aTotal of number of GBA mutations represent all genotyped participants. bTwo subjects have two GBA mutations each, thus total observed participants with GBA mutations are 73.
The overall combined mutation rate for both LRRK2 and GBA genes among those with a definitive PD diagnosis, was approximately 15.4% (121 participants). LRRK2 p.(Gly2019Ser) mutation was 6.1% overall and 7.1% in patients with four AJ grandparents. Participants identified as GBA mutation carriers were 9.3% overall and 11.1% in patients with four AJ grandparents. Two from this study population had 2 GBA mutation variants, p.(Asn370Ser) and p.(Arg496His) each. One participant had both a LRRK2 p.(Gly2019Ser) and a GBA p.(Asn370Ser) mutation (Table 2). Moreover, mutation carriers were enriched in participants with a family history of AJ ancestry (with at least 1 grandparent), in that both LRRK2 and GBA mutation frequencies increased 2-3-fold (from 3% to 7% for LRRK2; 4% to 11% for GBA) among patients with AJ ancestry compared to those of non-AJ ancestry. Among the GBA variants interrogated, p.(Asn370Ser) was the most prominent (48 out of 64 observations), followed by p.(Arg496His) (7/64) and p.(Glu326Lys) (6/64) among those with AJ ancestry (Table 3). Among the 35 participants with a suspected PD diagnosis, 2 were identified as GBA mutation carriers (Fig. 1).
Most of the participants (96.4%, N = 807) in the study, requested the return of their genetic results, which was communicated via a letter (approved previously by the Israel MoH and ethical committee review), along with an offer of genetic counseling. Thirty-eight patients who received the letter contacted the PI for additional information. Most of the participants (93%, N = 780) consented to be re-contacted for future clinical trials (Fig. 1).
DISCUSSION
This study examined the feasibility and success rate of using a large EMR system to screen and to enrich for a genetically defined cohort of PD. The Maccabi Healthcare System (MHS) in Israel was selected due to its robust EMR system and for previously observed high rate of LRRK2 and GBA genetic mutations in the AJ population with PD than in other populations. The results of this study indicate that there is an opportunity to utilize EMRs to rapidly identify and recruit patients with PD, particularly identifying those with genetic mutations with AJ ancestry. Approximately 26% of participants with PD identified from the EMR were enrolled in the study, and more than 98% of these participants completed the study and importantly 93% gave their consent to be re-contacted for future clinical trials.
In this study, the percentage of participants who reported a family history of PD and the percentage of LRRK2 and GBA mutations among the AJ PD population was lower than anticipated. Specifically, the observed frequency of LRRK2 and GBA mutations among overall PD cases of AJ ancestry was approximately 7% and 11%, respectively (Table 2), compared to previous publications showing approximately 10–20% for LRRK2 (p.(Gly2019Ser)) and 12–20% for aggregate GBA mutations [4, 5, 9–15]. It is unclear why the observed frequencies of the mutations in this study were lower than expected based on prior estimates. Despite this discrepancy in overall rate, the distribution of different disease-associated mutations observed were generally consistent with reported distributions. Specifically, the dominant GBA mutation observed in this study was the p.(Asn370Ser) mutation among both AJ (n = 48) and non-AJ ancestry subjects (n = 9), which is consistent with previous publications [10]. Moreover, other frequent GBA mutations among AJ ancestry, which include p.(Arg496His) (n = 7), p.(Glu326Lys) (n = 6), and p.(Leu29Alafs*18) (n = 1), were also observed in this study. The GBA variant, p.(Leu444Pro), was not observed in this study.
The lower frequency of LRRK2 and GBA mutations observed in this study was not due to genotyping assay performance. All LRRK2 p.(Gly2019Ser) subjects identified by the TaqMan assay were assessed additionally through the use of Sanger sequencing, confirming that they were not false positives. To determine if there were any false negatives using the TaqMan assay, we confirmed the negative TaqMan result from a sample of 92 participants who did not carry the LRRK2 mutation using Sanger sequencing (data not shown). In addition, the assay for the GBA mutations was specifically designed not to detect variants in the known pseudogene, a common reason for erroneous genotyping results in this gene.
The differences between observed frequencies in this study and previous publications may reflect underlying differences in study ascertainments as well as other study biases. First, there are 4 state-mandated HMO in Israel: Clalit, Maccabi, Meuhedet, and Leumit Health Services. Clalit serves almost 4.6 million Israelis (or 50%) and is the largest health care provider in Israel. MHS is the second largest HMO, and serves approximately 2.5 million, hence the majority of patients with PD may have been enrolled in the other HMOs, limiting the patient pool to approximately 25% of the population. Second, those participants who have been genotyped previously and/or have knowledge of their genetic status, may have declined to participate, or may be participating in an interventional study for PD therapy (n = 838 declined to participate). Third, self-reporting of their AJ ancestry may introduce potential inaccuracies, e.g., based on recollection of deceased family members (grandparents and other relatives), which cannot be corroborated; some may have immigrated from geographically “Ashkenazi” parts of Europe yet may not be of Ashkenazi Jewish ancestry.
It is likely that based on the algorithm defined initially to prescreen early/moderate stage of the disease (e.g., exclusion of PD patient with mental illness including dementia, psychiatric problems, or long-term use of dopamine blocking agents, or patients with stroke 6 month prior to PD diagnosis), may reflect lower mutation rates observe in this study, as more advanced stage or severe cases of PD were likely to be under-represented (these patients may be more likely to harbor GBA mutations). In addition, the mutation rate of those participants who either declined to participate (n = 838), or screen failed (N = 369) in the study is not known.
Despite the lower frequency of observed mutations, 93% of the participants who completed the study, were interested in obtaining their genetic results and agreed to be re-contacted for future clinical trials, indicating that recruitment using an EMR approach for future trial enrollment would not only be feasible but may be high, especially for small, focused trials. However, a limitation of this study is that the study protocol did not include follow-up visits to further engage the PD participants for an additional PD genetic clinical study, thus we were unable to ascertain the actual rate of potential recruitment to demonstrate the effectiveness of this recruitment method.
An additional advantage of using the EMR approach is that there are important invaluable clinical information/data about the disease progression, age of onset, stage of disease, imaging and lab data, medical history including concomitant medications and comorbidities etc. With the additional availability of genetic information in the EMR, future algorithms may be designed to include genetic data to enrich the patient population to target and identify more individuals based on the genetic data. Taken together, having a database of both clinical and genetic information, may be truly advantageous for future studies.
On the other hand, it is important to note that it is challenging to pre-identify PD patients with genetic mutations in a PD cohort enriched for mutations, especially for larger POC clinical studies. For example, in this study more than 3,200 participants were contacted, and approximately 26% (n = 837) were enrolled (30% enrollment is considered a best-case scenario) and genotyped, resulting in a total of 151 participants with either a LRRK2 or GBA mutation. In general, depending on the study design, a far greater number of PD patients, would be needed to adequately conduct a Phase 2/3 clinical trial. For example, if success rates scale, this would indicate a need for substantially increased resourcing of initial screening efforts in a much larger patient pool. Moreover, study criteria for interventional PD trials may be more stringent (compared to this study) which may significantly limit the pre-identified patient pool using this type of approach. This study provides a good example of genetic screening using available EMR databases and some of the challenges involved. Therefore, the identification (and selection) of PD patients through EMRs with pre-genotyping is a viable approach 1) to establish a genetically defined cohort of patients with PD for clinical research; 2) to enrich for PD patients with pre-identified specific genetic mutation; 3) to supplement conventional clinical recruitment methods, especially for smaller focused trials; and 4) to allow patients with PD the opportunity to participate in subsequent clinical trials targeting LRRK2 or GBA therapeutics.
Until genetic testing for LRRK2, GBA and potentially other genetic factors associated with PD becomes routine for all PD patients, utilization of EMR is a feasible option for rapidly identifying a potential pool of patients with PD with genetic mutations for recruitment into clinical trials. However significant challenges may surface depending on expected frequencies of for variants in a given population.
ACKNOWLEDGMENTS
The authors would like to thank Sheila Erespe of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ USA, for her assistance with submission.
The study was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
CONFLICT OF INTEREST
Susan J. Lee, Peter M. Shaw, Bob Thornton, Amit Kumar, Jennifer K. Pai, Karina Bienfait, Diane Levitan, K. Chris Min, Daniel Jonathan, Tiffini Voss, Caroline S. Fox, S. Aubrey Stoch, and Arie F. Struyk are employees or former employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and may own stock/stock options in Merck & Co., Inc., Kenilworth, NJ, USA. Michal Eizik, Dan Goldstaub, Tali Braun, and Gally Teper are employees of MSD Israel. Gabriel Chodick, Daniella Beller, and Gabriel Vainstein have nothing to disclose.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JPD-212703.
REFERENCES
[1] | de Lau LM , Breteler MM ((2006) ) Epidemiology of Parkinson’s disease. Lancet Neurol 5: , 525–535. |
[2] | Nussbaum RL , Ellis CE ((2003) ) Alzheimer’s disease and Parkinson’s disease. N Engl J Med 348: , 1356–1364. |
[3] | Chang D , Nalls MA , Hallgrimsdottir IB , Hunkapiller J , van der Brug M , Cai F , International Parkinson’s Disease Genomics Consortium; 23 and Me Research Team, Kerchner GA , Ayalon G , Bingol B , Sheng M , Hinds D , Behrens TW , Singleton AB , Bhangale TR , Graham RR ((2017) ) Ameta-analysis of genome-wide association studies identifies 17 new Parkinson’sdisease risk loci. Nat Genet 49: , 1511–1516. |
[4] | Healy Healy DG , Falchi M , O’Sullivan SS , Bonifati V , Durr A , Bressman S , Brice A , Aasly J , Zabetian CP , Goldwurm S , Ferreira JJ , Tolosa E , Kay DM , Klein C , Williams DR , Marras C , Lang AE , Wszolek ZK , Berciano J , Schapira AH , Lynch T , Bhatia KP , Gasser T , Lees AJ , Wood NW , International LRRK2 Consortium ((2008) ) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol 7: , 583–590. |
[5] | Gan-Or Z , Giladi N , Rozovski U , Shifrin C , Rosner S , Gurevich T , Bar-Shira A , Orr-Urtreger A ((2008) ) Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 70: , 2277–2283. |
[6] | Hawkes CH ((2008) ) The prodromal phase of sporadic Parkinson’s disease: Does it exist and if so how long is it? Mov Disord 23: , 1799–1807. |
[7] | Schapira AH , Tolosa E ((2010) ) Molecular and clinical prodrome of Parkinson disease: Implications for treatment. Nat Rev Neurol 6: , 309–317. |
[8] | Inzelberg R , Hassin-Baer S , Jankovic J ((2014) ) Genetic movement disorders in patients of Jewish ancestry. JAMA Neurol 71: , 1567–1572. |
[9] | Sidransky E , Nalls MA , Aasly JO , Aharon-Peretz J , Annesi G , Barbosa ER , Bar-Shira A , Berg D , Bras J , Brice A , Chen CM , Clark LN , Condroyer C , De Marco EV , Durr A , Eblan MJ , Fahn S , Farrer MJ , Fung HC , Gan-Or Z , Gasser T , Gershoni-Baruch R , Giladi N , Griffith A , Gurevich T , Januario C , Kropp P , Lang AE , Lee-Chen GJ , Lesage S , Marder K , Mata IF , Mirelman A , Mitsui J , Mizuta I , Nicoletti G , Oliveira C , Ottman R , Orr-Urtreger A , Pereira LV , Quattrone A , Rogaeva E , Rolfs A , Rosenbaum H , Rozenberg R , Samii A , Samaddar T , Schulte C , Sharma M , Singleton A , Spitz M , Tan EK , Tayebi N , Toda T , Troiano AR , Tsuji S , Wittstock M , Wolfsberg TG , Wu YR , Zabetian CP , Zhao Y , Ziegler SG ((2009) ) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361: , 1651–1661. |
[10] | Ozelius LJ , Senthil G , Saunders-Pullman R , Ohmann E , Deligtisch A , Tagliati M , Hunt AL , Klein C , Henick B , Hailpern SM , Lipton RB , Soto-Valencia J , Risch N , Bressman SB ((2006) ) LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med 354: , 424–425. |
[11] | Hassin-Baer S , Laitman Y , Azizi E , Molchadski I , Galore-Haskel G , Barak F , Cohen OS , Friedman E ((2009) ) The leucine rich repeat kinase 2 (LRRK2) G2019S substitution mutation. Association with Parkinson disease, malignant melanoma and prevalence in ethnic groups in Israel. J Neurol 256: , 483–487. |
[12] | Alcalay RN , Mirelman A , Saunders-Pullman R , Tang MX , Mejia Santana H , Raymond D , Roos E , Orbe-Reilly M , Gurevich T , Bar Shira A , Gana Weisz M , Yasinovsky K , Zalis M , Thaler A , Deik A , Barrett MJ , Cabassa J , Groves M , Hunt AL , Lubarr N , San Luciano M , Miravite J , Palmese C , Sachdev R , Sarva H , Severt L , Shanker V , Swan MC , Soto-Valencia J , Johannes B , Ortega R , Fahn S , Cote L , Waters C , Mazzoni P , Ford B , Louis E , Levy O , Rosado L , Ruiz D , Dorovski T , Pauciulo M , Nichols W , Orr-Urtreger A , Ozelius L , Clark L , Giladi N , Bressman S , Marder KS ((2013) ) Parkinson disease phenotype in Ashkenazi Jews with and without LRRK2 G2019S mutations. Mov Disord 28: , 1966–1971. |
[13] | Orr-Urtreger A , Shifrin C , Rozovski U , Rosner S , Bercovich D , Gurevich T , Yagev-More H , Bar-Shira A , Giladi N ((2007) ) The LRRK2 G2019S mutation in Ashkenazi Jews with Parkinson disease: Is there a gender effect? Neurology 69: , 1595–1602. |
[14] | Saunders-Pullman R , Hagenah J , Dhawan V , Stanley K , Pastores G , Sathe S , Tagliati M , Condefer K , Palmese C , Brüggemann N , Klein C , Roe A , Kornreich R , Ozelius L , Bressman S ((2010) ) Gaucher disease ascertained through a Parkinson’s center: Imaging and clinical characterization. Mov Disord 25: , 1364–1372. |
[15] | Dagan E , Schlesinger I , Kurolap A , Ayoub M , Nassar M , Peretz-Aharon J , Gershoni-Baruch ((2016) ) LRRK2, GBA and SMPD1 founder mutations and Parkinson’s disease in Ashkenazi Jews. Dement Geriatr Cogn Disord 42: , 1–6. |


