
Endoplasmic Reticulum Stress Regulators: New Drug Targets for Parkinson’s Disease
Abstract
Parkinson’s disease (PD) pathology involves progressive degeneration and death of vulnerable dopamine neurons in the substantia nigra. Extensive axonal arborization and distinct functions make this type of neurons particularly sensitive to homeostatic perturbations, such as protein misfolding and Ca2+ dysregulation. Endoplasmic reticulum (ER) is a cell compartment orchestrating protein synthesis and folding, as well as synthesis of lipids and maintenance of Ca2+ homeostasis in eukaryotic cells. When misfolded proteins start to accumulate in ER lumen the unfolded protein response (UPR) is activated. UPR is an adaptive signaling machinery aimed at relieving of protein folding load in the ER. When UPR is chronic, it can either boost neurodegeneration and apoptosis or cause neuronal dysfunctions. We have recently discovered that mesencephalic astrocyte-derived neurotrophic factor (MANF) exerts its prosurvival action in dopamine neurons and in an animal model of PD through the direct binding to UPR sensor inositol-requiring protein 1 alpha (IRE1α) and attenuation of UPR. In line with this, UPR targeting resulted in neuroprotection and neurorestoration in various preclinical animal models of PD. Therefore, growth factors (GFs), possessing both neurorestorative activity and restoration of protein folding capacity are attractive as drug candidates for PD treatment especially their blood-brain barrier penetrating analogs and small molecule mimetics. In this review, we discuss ER stress as a therapeutic target to treat PD; we summarize the existing preclinical data on the regulation of ER stress for PD treatment. In addition, we point out the crucial aspects for successful clinical translation of UPR-regulating GFs and new prospective in GFs-based treatments of PD, focusing on ER stress regulation.
INTRODUCTION
Is ER stress inducing PD pathology?
Parkinson’s disease (PD) is characterized by progressive loss of midbrain dopamine (DA) neurons and accumulation of proteinaceous intracellular inclusions called Lewy bodies. These inclusions are mostly composed of misfolded phosphorylated α-synuclein (αSyn), as well as other proteins, membrane fragments and dysmorphic organelles [1, 2]. To date, misfolded αSyn is considered to be the main inducer of PD pathology and neurodegeneration through multiple mechanisms, such as disruption of axonal transport and loss of synapses, as well as impairment of mitochondrial, lysosomal, proteasomal and endoplasmic reticulum (ER) functions [3].
About 7 million people worldwide have PD; the majority of the cases are sporadic and only 5–10% are familial. To date, more than 20 genes mutated in familial cases of PD and genetic risk factors have been identified; many of these genes are involved in the maintenance of ER and mitochondrial protein homeostasis (Table 1). Mutations in the GBA gene, encoding lysosomal enzyme glucocerebrosidase 1, are responsible for 5–10% of all PD cases and considered to be the most common risk factor; this highlights the importance of maintaining proteostasis through lysosomal activity. Other common risk factors for PD are mutations in the leucine-rich repeat serine/threonine-protein kinase 2 (LRRK2) gene and the SNCA gene encoding for αSyn, present in 2% and 1–2% of all PD cases, respectively. Mutations in other genes, indicated in Table 1, are risk factors for PD as well, although they are less common and autosomal recessive.
Table 1
Genes mutated in PD are involved in the maintenance of protein homeostasis
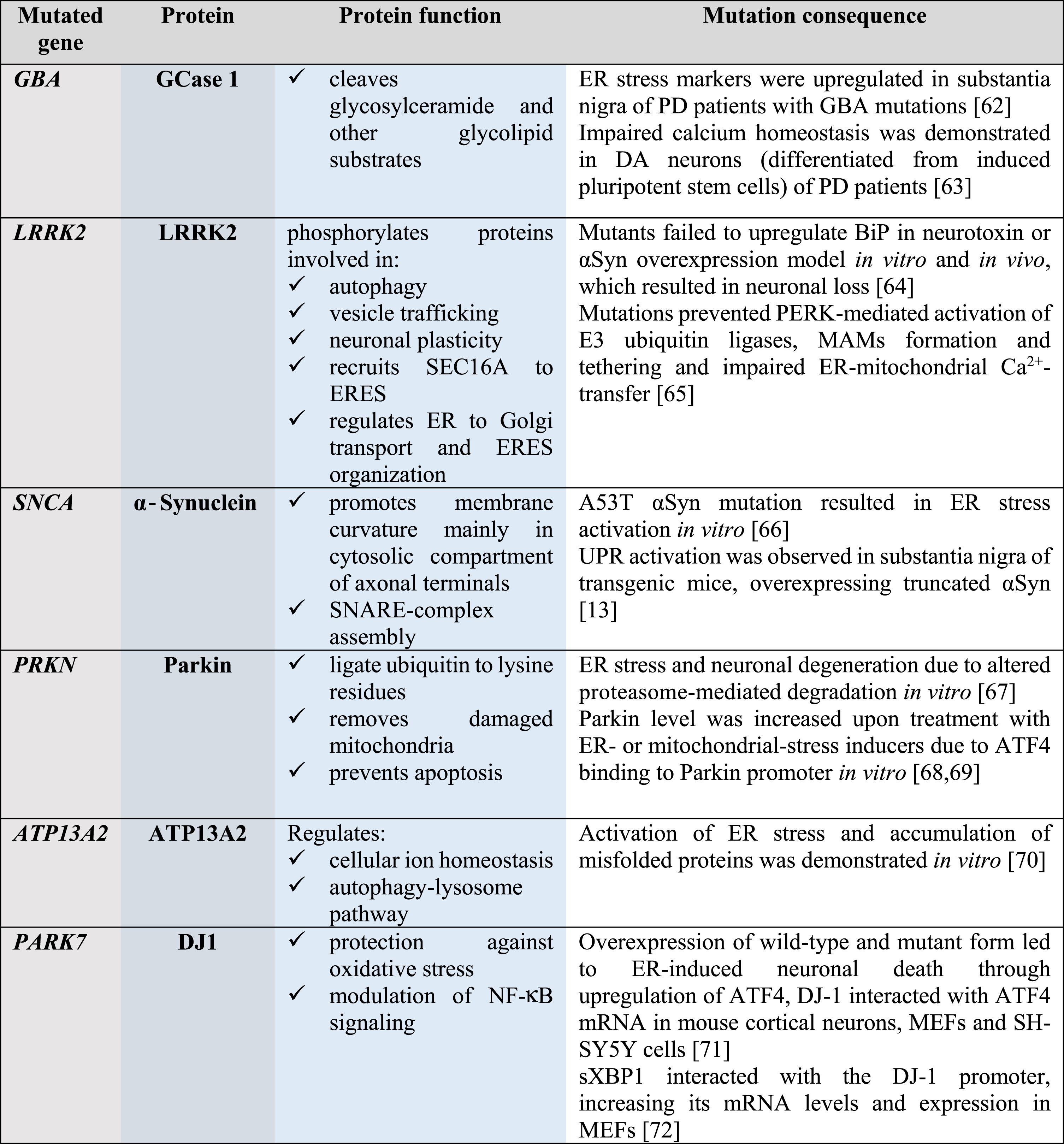 |
GCase 1, glucocerebrosidase 1; LRRK2, leucine-rich repeat kinase 2; αSyn, α-Synuclein; Parkin, E3 ubiquitin ligase; ATP13A2, polyamine-transporting ATPase 13A2; DJ1, protein/nucleic acid deglycase 1; SEC16A, protein transport protein; SNARE, soluble N-ethylmaleimide-sensitive factor attachment protein receptor; ER, endoplasmic reticulum; ERES, ER-exit sites; NF-κB, nuclear factor-κB; PD, Parkinson’s disease; BiP, immunoglobulin heavy-chain-binding protein, major ER chaperone; PERK, protein kinase RNA-like ER kinase; UPR, unfolded protein response; MAMs, mitochondria-associated membranes; ATF4, activating transcription factor 4; SH-SY5Y, neuroblastoma cell line; sXBP1, spliced X-box binding protein-1; MEFs, mouse embryonic fibroblasts.
Exceptionally vulnerable DA neurons of the subtantia nigra (SN) drastically differ in terms of morphology and functions from other neurons. In long and highly branched neurites of aging DA neurons, maintenance of ER protein homeostasis and axonal transport of mRNA, proteins and organelles is particularly challenging. High demand in mRNA transport and protein translation, especially translation of synaptic proteins, including αSyn, overloads the ER proteostasis machinery and results in protein misfolding. Impaired ER proteostasis, i.e., accumulation of misfolded proteins, impairment of Ca2+ homeostasis and dysregulation of axonal transport led to prolonged ER stress, playing an essential role in PD pathology [4].
ER stress was shown to accompany PD pathology; however, to date, the cause-effect relationship between ER stress and PD is uncertain. ER stress was induced in neurotoxin models of PD in vitro. DA neurons generated from human induced pluripotent stem cells derived from PD patients carrying SNCA or GBA mutations showed increased ER stress, elevated ER stress markers and impaired ER-associated degradation (ERAD) [6, 7]. Increased levels of activated ER stress sensor PERK and its downstream target phosphorylated eIF2α (p-eIF2α) were found in postmortem midbrain samples of early stage PD patients [8].
Since impaired ER proteostasis is associated with aging and PD pathology [4], re-establishment of ER proteostasis may be considered as a disease-modifying strategy. This can be achieved through the targeting of components of ERAD, the ER Ca2+ homeostasis, as well as the functioning of ER chaperones or isomerases, and ER stress-activated signaling receptors or their regulators.
Endoplasmic reticulum stress triggers unfolded protein response activation
ER is a cellular compartment responsible for synthesis of both secretory and membrane proteins, their modification, folding and secretion as well as for lipid synthesis and calcium homeostasis. Protein misfolding, aggregation and homeostasis perturbations in the ER lumen lead to ER stress triggering coordinated adaptive signaling machinery called the unfolded protein response (UPR). Adaptive UPR is aimed at restoring cellular homeostasis by increasing protein folding capacity through attenuation of protein synthesis, selective mRNA degradation, degradation of misfolded proteins and upregulation of transcription of genes involved in ERAD, autophagy, lipid biosynthesis, and Ca2+ homeostasis [9]. Prolonged terminal UPR, studied mainly in secretory cells and not well documented in neurons, can lead to activation of pro-apoptotic and pro-inflammatory signaling contributing to cell death [10]. In mammalian cells, UPR occurs through three ER transmembrane proteins termed inositol-requiring protein 1 alpha (IRE1α), protein kinase RNA-like ER kinase (PERK) and activating transcription factor 6 (ATF6) (Fig. 1).
Fig. 1
ER-stress signaling and ER stress-modulating proteins and small molecule compounds affecting neuronal survival. A) Healthy neuron, main ER chaperone BiP (alias GRP78) is bound to luminal domains (LDs) of monomeric transmembrane UPR sensors protein kinase RNA-like ER kinase (PERK), inositol-requiring protein 1 alpha (IRE1α) and activating transcription factor 6 (ATF6) and keeps these inactive. Signaling is aimed at maintaining protein homeostasis and classical UPR signaling is not triggered. B) Diseased neuron, ER is overloaded with misfolded proteins, BiP dissociates from LDs of UPR sensors to bind misfolded proteins and re-establish homeostasis; dissociation of BiP from UPR sensors leads to their dimerization and phosphorylation of IRE1α and PERK, and activates cytoplasmic UPR signaling [73]. Alternatively, upon BiP dissociation from UPR sensors their full activation occurs through the direct binding of misfolded proteins to their luminal domains, as was demonstrated for IRE1α and PERK LDs [74, 75]. BiP dissociation from PERK LD leads to dimerization of PERK LDs, allowing dimerization and autophosphorylation of the cytoplasmic kinase domain of PERK. Active PERK phosphorylates two substrates: α-subunit of eukaryotic initiation factor 2 (eIF2α) and nuclear factor erythroid 2-related factor 2 (NRF2). This step can be modulated using the PERK inhibitor GSK2606414 or the PERK activator CCT020312; both of these compounds have been shown to be neuroprotective in different in vitro and in vivo models of neurodegeneration. Through phosphorylation of eIF2α PERK attenuates global protein translation, decreasing the loading of ER with misfolded proteins. However, chronic attenuation of protein translation, continuously repressing the expression of synaptic proteins, can be detrimental for neurons. Few compounds blocking translation attenuation, such as integrated stress response inhibitor (ISRIB), its improved derivative 2BAct, trazodone hydrochloride and dibenzoylmethane (DBM) have been shown to be cytoprotective. Blockade of protein phosphatase 1 (PPI)-mediated dephosphorylation of eIF2α through growth arrest and DNA damage-inducible protein (GADD34) using salubrinal, sephin-1 or guanabenz was neuroprotective. In addition to attenuation of protein translation, phosphorylation of eIF2α selectively enhances translation of activating transcription factor 4 (ATF4), promoting transcription of genes responsible for chaperone expression, autophagy and antioxidant response. ATF4 also induces the expression of a transcriptional factor C/EBP Homologous Protein (CHOP), upregulating the expression of genes involved in apoptosis. PERK phosphorylates NRF2, which is then translocated to the nucleus and activates transcription of genes involved in antioxidant responses. BiP dissociation from IRE1α LD, similarly to dissociation from PERK LD, results in dimerization and autophosphorylation of IRE1α cytoplasmic kinase domain. Activation of the kinase domain leads to activation of IRE1α ribonuclease (RNase) domain. Activation of IRE1α kinase can be inhibited by the family of compounds called kinase-inhibiting RNase attenuators (KIRAs) and IRE1α RNase can be inhibited by 4μ8C. As an endoribonuclease, IRE1α degrades specific mRNAs via regulated IRE1α-dependent decay (RIDD), decreasing the load of misfolded proteins to ER. RIDD can also decrease apoptosis through degradation of death receptor 5 (DR5) mRNA [76]. Endoribonuclease of IRE1α also performs unconventional splicing of X-box binding protein-1 (XBP1), transcription factor spliced XBP1 (sXBP1), in turn, upregulates expression of the genes regulating chaperone, lipid biosynthesis, and ER-associated degradation (ERAD). Activation and oligomerization of IRE1α lead to the recruiting of TNF receptor-associated factor 2 (TRAF2), activating c-Jun N-terminal kinase (JNK) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathways; these regulate apoptosis and transcription-related inflammatory genes [77, 78]. Upon BiP dissociation from ATF6 LD, ATF6 is transported to the Golgi apparatus, where it is cleaved by site-specific proteases. Cleaved ATF6 translocates to the nucleus and activates transcription of the genes involved in chaperone and lipid biosynthesis, ERAD, as well as inducing XBP1 mRNA. ATF6 translocation from ER to the Golgi apparatus can be specifically inhibited by Ceapins or activated by small molecule ATF6 activators, compounds 147 and 263. Mesencephalic astrocyte-derived neurotrophic factor (MANF) directly interacts with UPR sensors and attenuates UPR signaling in DA neurons and in an animal model of PD [34]. MANF, its homologous protein CDNF and their small molecule mimetics represent a strategy to treat PD, attenuating ER stress and inflammation. ER stress-modulating interventions are shown with red color.
![ER-stress signaling and ER stress-modulating proteins and small molecule compounds affecting neuronal survival. A) Healthy neuron, main ER chaperone BiP (alias GRP78) is bound to luminal domains (LDs) of monomeric transmembrane UPR sensors protein kinase RNA-like ER kinase (PERK), inositol-requiring protein 1 alpha (IRE1α) and activating transcription factor 6 (ATF6) and keeps these inactive. Signaling is aimed at maintaining protein homeostasis and classical UPR signaling is not triggered. B) Diseased neuron, ER is overloaded with misfolded proteins, BiP dissociates from LDs of UPR sensors to bind misfolded proteins and re-establish homeostasis; dissociation of BiP from UPR sensors leads to their dimerization and phosphorylation of IRE1α and PERK, and activates cytoplasmic UPR signaling [73]. Alternatively, upon BiP dissociation from UPR sensors their full activation occurs through the direct binding of misfolded proteins to their luminal domains, as was demonstrated for IRE1α and PERK LDs [74, 75]. BiP dissociation from PERK LD leads to dimerization of PERK LDs, allowing dimerization and autophosphorylation of the cytoplasmic kinase domain of PERK. Active PERK phosphorylates two substrates: α-subunit of eukaryotic initiation factor 2 (eIF2α) and nuclear factor erythroid 2-related factor 2 (NRF2). This step can be modulated using the PERK inhibitor GSK2606414 or the PERK activator CCT020312; both of these compounds have been shown to be neuroprotective in different in vitro and in vivo models of neurodegeneration. Through phosphorylation of eIF2α PERK attenuates global protein translation, decreasing the loading of ER with misfolded proteins. However, chronic attenuation of protein translation, continuously repressing the expression of synaptic proteins, can be detrimental for neurons. Few compounds blocking translation attenuation, such as integrated stress response inhibitor (ISRIB), its improved derivative 2BAct, trazodone hydrochloride and dibenzoylmethane (DBM) have been shown to be cytoprotective. Blockade of protein phosphatase 1 (PPI)-mediated dephosphorylation of eIF2α through growth arrest and DNA damage-inducible protein (GADD34) using salubrinal, sephin-1 or guanabenz was neuroprotective. In addition to attenuation of protein translation, phosphorylation of eIF2α selectively enhances translation of activating transcription factor 4 (ATF4), promoting transcription of genes responsible for chaperone expression, autophagy and antioxidant response. ATF4 also induces the expression of a transcriptional factor C/EBP Homologous Protein (CHOP), upregulating the expression of genes involved in apoptosis. PERK phosphorylates NRF2, which is then translocated to the nucleus and activates transcription of genes involved in antioxidant responses. BiP dissociation from IRE1α LD, similarly to dissociation from PERK LD, results in dimerization and autophosphorylation of IRE1α cytoplasmic kinase domain. Activation of the kinase domain leads to activation of IRE1α ribonuclease (RNase) domain. Activation of IRE1α kinase can be inhibited by the family of compounds called kinase-inhibiting RNase attenuators (KIRAs) and IRE1α RNase can be inhibited by 4μ8C. As an endoribonuclease, IRE1α degrades specific mRNAs via regulated IRE1α-dependent decay (RIDD), decreasing the load of misfolded proteins to ER. RIDD can also decrease apoptosis through degradation of death receptor 5 (DR5) mRNA [76]. Endoribonuclease of IRE1α also performs unconventional splicing of X-box binding protein-1 (XBP1), transcription factor spliced XBP1 (sXBP1), in turn, upregulates expression of the genes regulating chaperone, lipid biosynthesis, and ER-associated degradation (ERAD). Activation and oligomerization of IRE1α lead to the recruiting of TNF receptor-associated factor 2 (TRAF2), activating c-Jun N-terminal kinase (JNK) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathways; these regulate apoptosis and transcription-related inflammatory genes [77, 78]. Upon BiP dissociation from ATF6 LD, ATF6 is transported to the Golgi apparatus, where it is cleaved by site-specific proteases. Cleaved ATF6 translocates to the nucleus and activates transcription of the genes involved in chaperone and lipid biosynthesis, ERAD, as well as inducing XBP1 mRNA. ATF6 translocation from ER to the Golgi apparatus can be specifically inhibited by Ceapins or activated by small molecule ATF6 activators, compounds 147 and 263. Mesencephalic astrocyte-derived neurotrophic factor (MANF) directly interacts with UPR sensors and attenuates UPR signaling in DA neurons and in an animal model of PD [34]. MANF, its homologous protein CDNF and their small molecule mimetics represent a strategy to treat PD, attenuating ER stress and inflammation. ER stress-modulating interventions are shown with red color.](https://content.iospress.com:443/media/jpd/2021/11-s2/jpd-11-s2-jpd212673/jpd-11-jpd212673-g001.jpg)
Upon ER stress IRE1α, possessing both serine-threonine kinase and endoribonuclease activity performs splicing of the mRNA encoding for X-box binding protein-1 (XBP1) [11]. Spliced form of XBP1 (XBP1s) activates prosurvival genes regulating ERAD, autophagy, and lipid biosynthesis. IRE1α endoribonuclease also decreases the ER load of misfolded proteins by degrading specific mRNAs in a process called regulated IRE1α-dependent decay (RIDD). PERK with cytoplasmic Ser/Thr kinase domain has two main targets: eukaryotic translational initiation factor 2α (eIF2α) and nuclear factor erythroid 2-related factor 2 (NRF2). Through the phosphorylation of eIF2α PERK along with other integrated-stress response kinases inhibits protein synthesis initiation by attenuating global protein synthesis, decreasing ER protein load. A chronic, unresolved UPR can trigger apoptosis through upregulation of C/EBP homologous protein (CHOP). During the global attenuation of protein translation, however, some proteins, such as activating transcription factor 4 (ATF4) are able to be synthesized and further regulate expression of specific UPR genes. Through the phosphorylation of NRF2, PERK triggers an antioxidant response [12]. Upon ER stress, the third UPR sensor ATF6 is transported to the Golgi apparatus where it is cleaved, and its cytoplasmic fragment enters the cell nucleus and triggers transcription of ER chaperones and ERAD genes.
Can α-synuclein induce endoplasmic reticulum stress in dopamine neurons?
To date, the cause-effect relationships between αSyn misfolding/aggregation and ER stress/UPR activation are still unclear. It has been shown that aggregated misfolded αSyn induces BiP (alias GRP78) and ATF4 expression in vitro and in a mouse model overexpressing truncated αSyn [13]. αSyn-overexpression induced UPR, inhibited protein trafficking from ER to Golgi apparatus and resulted in impaired protein maturation in yeast cells; re-establishing of ER-Golgi trafficking was shown to be protective from αSyn-mediated toxicity in yeast, DA neurons and αSyn-overexpressing Drosophila and C. elegans models of PD [14].
Interestingly, αSyn interacted with UPR sensor ATF6 and inhibited its activation by ER stress in in vitro study [15]. Overexpressed major ER chaperone, BiP, and αSyn were shown to form a complex, likely contributing to neuroprotective effect of BiP overexpression in αSyn-overexpressing rat model of PD [16]. Interaction of αSyn with BiP, was demonstrated also under physiological conditions, i.e., in non-transgenic mice, indicating that ER stress may contribute to aggregation of αSyn and progression of PD pathology [17]. Whether αSyn is directly interacting with ER chaperons and directly activating UPR in a physiologically relevant manner remains unknown. It is important to emphasize that αSyn is a cytoplasmic protein, whereas UPR activation occurs in the lumen of the ER. Thus, is it possible that αSyn can move from the cytoplasm to the ER lumen and interact there with BiP or with the luminal domain of ATF6? Many of the previous studies used overexpression of αSyn protein, and in those cases, the protein can be localized into cell compartments that are not occurring for physiological concentrations. Although no experimental data exist on αSyn movement from the cytoplasm to the ER, it can be possible, since αSyn fibrils very efficiently penetrate through cellular membranes of various cell types [18].
Although αSyn being considered a primary suspect in PD pathology, its overexpression is not always sufficient to induce robust degeneration of DA neurons in vitro and in vivo [19]. Noteworthy, in familial cases of PD, SNCA gene duplication or triplication induces PD pathology and neurodegeneration, with the early pathology onset in the case of triplication, suggesting that the level of αSyn correlates with the disease progression [20]. In line with this, a high increase (about 5-fold) in levels of αSyn resulted in pronounced neurodegeneration in AAV-αSyn transgenic rats [21]. However, in some studies, neurodegeneration was also observed with control proteins, such as GFP, after high overexpression [22]. Significant DA neuron loss was observed in animal models combining αSyn overexpression and inoculation with preformed αSyn fibrils [23], or when aggregation-prone truncated αSyn fragments were overexpressed [24]. Although quite a few experimental data demonstrate the induction of ER stress by αSyn and even direct interactions of αSyn with the UPR machinery components, it remains unclear whether αSyn can directly trigger ER stress activation. It needs to be clarified how cytosolic αSyn can modulate the processes occurring in ER lumen.
Can we treat Parkinson’s disease targeting ER proteostasis with UPR-regulating growth factors and small molecules?
To date, classically used for PD treatment GFs, glial cell line-derived neurotrophic factor (GDNF) and neurturin (NTRN), have not been shown to be involved in the regulation of ER stress and UPR, and only a few GFs were demonstrated to have a potential for alleviation of pathology-induced UPR.
Mesencephalic astrocyte-derived neurotrophic factor (MANF) and its paralog cerebral dopamine neurotrophic factor (CDNF) have been shown to protect DA neurons both in vitro and in vivo [25–28]. These neurotrophic factors reside in the ER lumen and alleviate ER stress in DA neurons and other cell types [28–33]. Recently we have discovered that MANF directly interacts with UPR sensor IRE1α and with lower affinities also with PERK and ATF6 and competes with BiP for the interaction with IRE1α [34]. Using computational modeling, mutagenesis, biochemical and cellular assays we have demonstrated that MANF–IRE1α interaction is crucial for pro-survival activity of MANF in DA neurons. We have shown that MANF is improving motor behaviour and increasing the number of DA neurons in SN in a neurotoxin rat model of PD, whereas MANF mutant unable to bind to IRE1α lacks neuroprotective activity in vivo [34]. The ability to be secreted upon Ca2+ depletion-induced ER stress [35] and exert protective properties when added extracellularly [28] hints at the existence of extracellular receptors or other mechanisms of internalization, such as lipid-mediated internalization, which is suggested by their structures [30, 36] and data on lipid binding [37]. Recent data show that binding of MANF, but not of CDNF, to cell surface neuroplastin receptors, reduced inflammatory responses and apoptosis via suppression of NF-κB signaling [38]. Unlike many GFs possessing mainly neuroprotective properties, MANF and CDNF, similar to GDNF and NRTN, also show neurorestorative action in DA neurons [26, 27, 39–41]. Importantly, in addition to their neurorestorative action, MANF and CDNF alleviated UPR and reduced inflammation [38, 42, 43]. Moreover, CDNF reduced αSyn aggregation in neurons, although in this study rather high CDNF concentration was used [44]. CDNF interacted directly with αSyn, reduced the levels of phosphorylated at Ser129 αSyn and alleviated motor deficits in rodents challenged with α-synuclein fibrils [45].
Modulation of the UPR components was already shown to regulate the survival of DA neurons in animal PD models. Interestingly, some UPR components, such as CHOP contributed to neurodegeneration, while others, such as sXBP1 and ATF6 supported neuroprotection [46–48]. Contribution of UPR-related genes in neurodegeneration and PD pathology has been reviewed earlier [4, 49].
Targeting ER proteostasis with small molecules was mainly performed through targeting the process of translational attenuation through PERK/p-eIF2α pathway. In a mouse neurotoxin PD model, the PERK inhibitor GSK2606414 protected DA neurons and restored motor function by increasing the level of DA and synaptic proteins [50]. In prion-diseased mice, GSK2606414 and de-repression of translational attenuation with integrated stress response inhibitor (ISRIB), were neuroprotective as well [51, 52]. Translational attenuator ISRIB had good general tolerability and no toxic effects in prion-diseased mice [52], suggesting that ISRIB and its improved derivative 2BAct [53] along with other non-toxic PERK/p-eIF2α modulators can be considered for future therapeutic use. Increasing p-eIF2α levels with salubrinal, a small molecule that inhibits p-eIF2α dephosphorylation through growth arrest and DNA damage-inducible protein (GADD34), led to improvement of motor function and rescued the phenotype in a mutant αSyn-overexpressing mouse model of PD [17]. However, in prion-diseased mice, having profound neurological deficits, salubrinal treatment led to neurodegeneration likely due to a significant reduction of protein synthesis compromising production of synaptic proteins [51].
Targeting ER proteostasis through IRE1α branch is aimed at regulation of sXBP1 activation or at decreasing the ER load of misfolded proteins through RIDD. Overexpression of sXBP1 was shown to be neuroprotective in an animal neurotoxin model of PD [47]. sXBP1 was also shown to improve synaptic plasticity and cognition through increasing the expression of brain-derived neurotrophic factor (BDNF) in vivo [54]. Effective small molecule modulators of the IRE1α branch, such as KIRA6 and KIRA8, have not yet been studied in animal models of PD. However, considering their cytoprotective action in pancreatic beta cells and their potential for diabetes treatment [10, 55], further pre-clinical studies in animal PD models are warranted.
Since ATF6α was shown to be involved in neuroprotection in a neurotoxin mouse model of PD [48], recently discovered specific activators of ATF6α, compounds 147 and 263 [56] and selective inhibitors of ATF6α, ceapins [57] hold great promise for PD treatment.
Modulation of UPR sensor activities is an attractive target for PD therapy not only because it is a way to prevent chronic terminal UPR, apoptosis and inflammation, but also to stimulate synaptic function and neuronal plasticity. This is why small molecules targeting different UPR components, already shown to be neuroprotective in various studies, is of particular importance (Fig. 1).
It has not escaped our notice that ER-mitochondria contact sites may play an important role in PD pathology. A few of PD-associated proteins, shown in Table 1, namely αSyn, Parkin, DJ1 and also PTEN-induced kinase 1 (PINK1) were demonstrated to localize to mitochondria-associated membranes (MAMs) and mutations in the genes encoding these proteins altered or disrupted ER-mitochondria signaling at MAMs [58]. That is why, MAMs should be considered as alternative targets for mechanistic studies and therapeutic approaches for PD.
CONCLUSION
Search for the effective UPR-regulating growth factor
Several gene therapies and protein clinical trials showed that GFs have a potential for PD treatment [41]. At the same time, the approaches and strategies for PD treatment need to be modified. The complications in the use of GFs and use of GF’s small molecule mimetics have been discussed earlier [41, 59]. We need to find, or develop, on the basis of existing proteins an effective UPR-regulating GF or even “GFs cocktail”. This potent GF should have good diffusion in brain tissue and not only protect, but also restore damaged midbrain DA neurons. In addition, it should alleviate ER-stress induced inflammation, block apoptosis and decrease the aggregation and neurotoxicity of αSyn.
UPR-regulating growth factors CDNF and MANF can protect and restore DA neurons, diffuse in the brain tissue much better, compared to classical GFs, alleviate inflammation and apoptosis and CDNF also decreases αSyn aggregation. MANF and CDNF, shown to act only on injured cells [28, 60, 61], represent a promising strategy for PD treatment. Since MANF, CDNF and likely their mimetics bind only to hyperactivated UPR sensors, when in ER stressed cells BiP has dissociated from UPR sensors, their peripheral delivery is unlikely to cause serious adverse effects. Lack of the effects in healthy cells means the absence of side effects upon peripheral delivery. Systemically delivered GFs likely may be more potent due to treatment of not only motor, but also non-motor symptoms of PD. Taking into account the above-mentioned points and lower costs compared to proteins for production and delivery, MANF/CDNF-like peptides and small molecule mimetics seem to be good alternatives to native proteins in the context of clinical translation.
Since ER proteostasis is crucial for maintenance of functioning of DA neurons, new therapeutic approaches to target and regulate ER proteostasis and UPR components, such as UPR-regulating GFs and their mimetics, should be a prime target.
ACKNOWLEDGMENTS
The authors thank Professor Barry Hoffer, Professor Mikko Airavaara, Dr. Brandon K. Harvey, Dr. Maria Lindahl, Dr. Olesja Bondarenko, and Dr. Yulia A. Sidorova for critical comments on the manuscript. V.K. and M.S. are supported by a grant from the Academy of Finland (No. 1310891) and by Jane and Aatos Erkko Foundation grant.
CONFLICT OF INTEREST
The authors have no conflict of interest to report. MS is the inventor in the MANF- and CDNF-related patents owned by Herantis Pharma Plc. and a shareholder in this company.
REFERENCES
[1] | Spillantini MG , Crowther RA , Jakes R , Hasegawa M , Goedert M ((1998) ) α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A 95: , 6469–6473. |
[2] | Shahmoradian SH , Lewis AJ , Genoud C , Hench J , Moors TE , Navarro PP , Castaño-Díez D , Schweighauser G , Graff-Meyer A , Goldie KN , Sütterlin R , Huisman E , Ingrassia A , Gier Y de , Rozemuller AJM , Wang J , Paepe A De , Erny J , Staempfli A , Hoernschemeyer J , Großerüschkamp F , Niedieker D , El-Mashtoly SF , Quadri M , Van IJcken WFJ , Bonifati V , Gerwert K , Bohrmann B , Frank S , Britschgi M , Stahlberg H , Van de Berg WDJ , Lauer ME ((2019) ) Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci 22: , 1099–1109. |
[3] | Bendor JT , Logan TP , Edwards RH ((2013) ) The function of α-synuclein. Neuron 79: , 1044–1066. |
[4] | Hetz C , Saxena S ((2017) ) ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol 13: , 477–491. |
[5] | Holtz WA , O’Malley KL ((2003) ) Parkinsonian mimetics induce aspects of unfolded protein response in death of dopaminergic neurons. J Biol Chem 278: , 19367–19377. |
[6] | Fernandes HJR , Hartfield EM , Christian HC , Emmanoulidou E , Zheng Y , Booth H , Bogetofte H , Lang C , Ryan BJ , Sardi SP , Badger J , Vowles J , Evetts S , Tofaris GK , Vekrellis K , Talbot K , Hu MT , James W , Cowley SA , Wade-Martins R ((2016) ) ER stress and autophagic perturbations lead to elevated extracellular α-synuclein in GBA-N370S Parkinson’s iPSC-derived dopamine neurons. Stem Cell Rep 6: , 342–356. |
[7] | Chung CY , Khurana V , Auluck PK , Tardiff DF , Mazzulli JR , Soldner F , Baru V , Lou Y , Freyzon Y , Cho S , Mungenast AE , Muffat J , Mitalipova M , Pluth MD , Jui NT , Schul̈e B , Lippard SJ , Tsai LH , Krainc D , Buchwald SL , Jaenisch R , Lindquist S ((2013) ) Identification and rescue of α-synuclein toxicity in Parkinson patient-derived neurons. Science 342: , 983–987. |
[8] | Hoozemans JJM , Van Haastert ES , Nijholt DAT , Rozemuller AJM , Scheper W ((2012) ) Activation of the unfolded protein response is an early event in Alzheimer’s and Parkinson’s disease. Neurodegener Dis 10: , 212–215. |
[9] | Hetz C , Zhang K , Kaufman RJ ((2020) ) Mechanisms, regulation and functions of the unfolded protein response. Nat Rev Mol Cell Biol 21: , 421–438. |
[10] | Ghosh R , Wang L , Wang ES , Perera BGK , Igbaria A , Morita S , Prado K , Thamsen M , Caswell D , Macias H , Weiberth KF , Gliedt MJ , Alavi M V. , Hari SB , Mitra AK , Bhhatarai B , Schürer SC , Snapp EL , Gould DB , German MS , Backes BJ , Maly DJ , Oakes SA , Papa FR ((2014) ) Allosteric inhibition of the IRE1α RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 158: , 534–548. |
[11] | Walter P , Ron D ((2011) ) The unfolded protein response: From stress pathway to homeostatic regulation. Science 334: , 1081–1086. |
[12] | Cullinan SB , Zhang D , Hannink M , Arvisais E , Kaufman RJ , Diehl JA ((2003) ) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 23: , 7198–7209. |
[13] | Bellucci A , Navarria L , Zaltieri M , Falarti E , Bodei S , Sigala S , Battistin L , Spillantini M , Missale C , Spano P ((2011) ) Induction of the unfolded protein response by α-synuclein in experimental models of Parkinson’s disease. J Neurochem 116: , 588–605. |
[14] | Cooper AA , Gitler AD , Cashikar A , Haynes CM , Hill KJ , Bhullar B , Liu K , Xu K , Strathearn KE , Liu F , Cao S , Caldwell KA , Caldwell GA , Marsischky G , Kolodner RD , LaBaer J , Rochet JC , Bonini NM , Lindquist S ((2006) ) α-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313: , 324–328. |
[15] | Credle JJ , Forcelli PA , Delannoy M , Oaks AW , Permaul E , Berry DL , Duka V , Wills J , Sidhu A ((2015) ) α-Synuclein-mediated inhibition of ATF6 processing into COPII vesicles disrupts UPR signaling in Parkinson’s disease. Neurobiol Dis 76: , 112–125. |
[16] | Gorbatyuk MS , Shabashvili A , Chen W , Meyers C , Sullivan LF , Salganik M , Lin JH , Lewin AS , Muzyczka N , Gorbatyuk OS ((2012) ) Glucose regulated protein 78 diminishes α-synuclein neurotoxicity in a rat model of parkinson disease. Mol Ther 20: , 1327–1337. |
[17] | Colla E , Coune P , Liu Y , Pletnikova O , Troncoso JC , Iwatsubo T , Schneider BL , Lee MK ((2012) ) Endoplasmic reticulum stress is important for the manifestations of α-synucleinopathy in vivo. J Neurosci 32: , 3306–3320. |
[18] | Ihse E , Yamakado H , Van Wijk XM , Lawrence R , Esko JD , Masliah E ((2017) ) Cellular internalization of alpha-synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci Rep 7: , 9008. |
[19] | Albert K , Voutilainen MH , Domanskyi A , Airavaara M ((2017) ) AAV vector-mediated gene delivery to substantia nigra dopamine neurons: Implications for gene therapy and disease models. Genes (Basel) 8: , 63. |
[20] | Chartier-Harlin MC , Kachergus J , Roumier C , Mouroux V , Douay X , Lincoln S , Levecque C , Larvor L , Andrieux J , Hulihan M , Waucquier N , Defebvre L , Amouyel P , Farrer M , Destée A ((2004) ) α-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364: , 1167–1169. |
[21] | Decressac M , Mattsson B , Lundblad M , Weikop P , Björklund A ((2012) ) Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of α-synuclein in midbrain dopamine neurons. Neurobiol Dis 45: , 939–953. |
[22] | Albert K , Voutilainen MH , Domanskyi A , Piepponen TP , Ahola S , Tuominen RK , Richie C , Harvey BK , Airavaara M ((2019) ) Downregulation of tyrosine hydroxylase phenotype after AAV injection above substantia nigra: Caution in experimental models of Parkinson’s disease. J Neurosci Res 97: , 346–361. |
[23] | Thakur P , Breger LS , Lundblad M , Wan OW , Mattsson B , Luk KC , Lee VMY , Trojanowski JQ , Björklund A ((2017) ) Modeling Parkinson’s disease pathology by combination of fibril seeds and α-synuclein overexpression in the rat brain. Proc Natl Acad Sci U S A 114: , E8284–E8293. |
[24] | Wegrzynowicz M , Bar-On D , Calo’ L , Anichtchik O , Iovino M , Xia J , Ryazanov S , Leonov A , Giese A , Dalley JW , Griesinger C , Ashery U , Spillantini MG ((2019) ) Depopulation of dense α-synuclein aggregates is associated with rescue of dopamine neuron dysfunction and death in a new Parkinson’s disease model. Acta Neuropathol 138: , 575–595. |
[25] | Petrova PS , Raibekas A , Pevsner J , Vigo N , Anafi M , Moore MK , Peaire AE , Shridhar V , Smith DI , Kelly J , Durocher Y , Commissiong JW ((2003) ) MANF: A new mesencephalic, astrocyte-derived neurotrophic factor with selectivity for dopaminergic neurons. J Mol Neurosci 20: , 173–187. |
[26] | Lindholm P , Voutilainen MH , Laurén J , Peränen J , Leppänen VM , Andressoo JO , Lindahl M , Janhunen S , Kalkkinen N , Timmusk T , Tuominen RK , Saarma M ((2007) ) Novel neurotrophic factor CDNF protects and rescues midbrain dopamine neurons in vivo. Nature 448: , 73–77. |
[27] | Voutilainen MH , Bäck S , Pörsti E , Toppinen L , Lindgren L , Lindholm P , Peränen J , Saarma M , Tuominen RK ((2009) ) Mesencephalic astrocyte-derived neurotrophic factor is neurorestorative in rat model of Parkinson’s disease. J Neurosci 29: , 9651–9659. |
[28] | Eesmaa A , Yu L-Y , Göös H , Nõges K , Kovaleva V , Hellman M , Zimmermann R , Jung M , Permi P , Varjosalo M , Lindholm P , Saarma M ((2021) ) The cytoprotective protein MANF promotes neuronal survival independently from its role as a GRP78 cofactor. J Biol Chem 100295. |
[29] | Apostolou A , Shen Y , Liang Y , Luo J , Fang S ((2008) ) Armet, a UPR-upregulated protein, inhibits cell proliferation and ER stress-induced cell death. Exp Cell Res 314: , 2454–2467. |
[30] | Parkash V , Lindholm P , Peränen J , Kalkkinen N , Oksanen E , Saarma M , Leppänen VM , Goldman A ((2009) ) The structure of the conserved neurotrophic factors MANF and CDNF explains why they are bifunctional. Protein Eng Des Sel 22: , 233–241. |
[31] | Hellman M , Arumäe U , Yu LY , Lindholm P , Peränen J , Saarma M , Permi P ((2011) ) Mesencephalic astrocyte-derived neurotrophic factor (MANF) has a unique mechanism to rescue apoptotic neurons. J Biol Chem 286: , 2675–2680. |
[32] | Lindahl M , Danilova T , Palm E , Lindholm P , Võikar V , Hakonen E , Ustinov J , Andressoo JO , Harvey BK , Otonkoski T , Rossi J , Saarma M ((2014) ) MANF is indispensable for the proliferation and survival of pancreatic β cells. Cell Rep 7: , 366–375. |
[33] | Pakarinen E , Danilova T , Võikar V , Chmielarz P , Piepponen P , Airavaara M , Saarma M , Lindahl M ((2020) ) MANF ablation causes prolonged activation of the UPR without neurodegeneration in the mouse midbrain dopamine system. eNeuro 7: , ENEURO.0477-19.2019. |
[34] | Kovaleva V , Yu L-Y , Ivanova L , Nam J , Eesmaa A , Kumpula E-P , Huiskonen J , Lindholm P , Voutilainen M , Karelson M , Saarma M ((2020) ) MANF regulates unfolded protein response and neuronal survival through its ER-located receptor IRE1α. bioRxiv, doi: https://doi.org/10.1101/2020.09.22.307744. |
[35] | Glembotski CC , Thuerauf DJ , Huang C , Vekich JA , Gottlieb RA , Doroudgar S ((2012) ) Mesencephalic astrocyte-derived neurotrophic factor protects the heart from ischemic damage and is selectively secreted upon sarco/endoplasmic reticulum calcium depletion. J Biol Chem 287: , 25893–25904. |
[36] | Lindholm P , Saarma M ((2010) ) Novel CDNF/MANF family of neurotrophic factors. Dev Neurobiol 70: , 360–371. |
[37] | Bai M , Vozdek R , Hnízda A , Jiang C , Wang B , Kuchar L , Li T , Zhang Y , Wood C , Feng L , Dang Y , Ma DK ((2018) ) Conserved roles of C. elegans and human MANFs in sulfatide binding and cytoprotection. Nat Commun 9: , 897. |
[38] | Yagi T , Asada R , Kanekura K , Eesmaa A , Lindahl M , Saarma M , Urano F ((2020) ) Neuroplastin modulates anti-inflammatory effects of MANF. iScience 23: , 101810. |
[39] | Ren X , Zhang T , Gong X , Hu G , Ding W , Wang X ((2013) ) AAV2-mediated striatum delivery of human CDNF prevents the deterioration of midbrain dopamine neurons in a 6-hydroxydopamine induced parkinsonian rat model. Exp Neurol 248: , 148–156. |
[40] | Voutilainen MH , De Lorenzo F , Stepanova P , Bäck S , Yu LY , Lindholm P , Pörsti E , Saarma M , Männistö PT , Tuominen RK ((2017) ) Evidence for an additive neurorestorative effect of simultaneously administered CDNF and GDNF in hemiparkinsonian rats: Implications for different mechanism of action. eNeuro 4: , ENEURO.0117-16.2017. |
[41] | Sidorova YA , Saarma M ((2020) ) Can growth factors cure Parkinson’s disease? Trends Pharmacol Sci 41: , 909–922. |
[42] | Nadella R , Voutilainen MH , Saarma M , Gonzalez-Barrios JA , Leon-Chavez BA , Jimnez JDM , Jimnez SDH , Escobedo L , Martinez-Fong D ((2014) ) Transient transfection of human CDNF gene reduces the 6-hydroxydopamine-induced neuroinflammation in the rat substantia nigra. J Neuroinflammation 11: , 209. |
[43] | Neves J , Zhu J , Sousa-Victor P , Konjikusic M , Riley R , Chew S , Qi Y , Jasper H , Lamba DA ((2016) ) Immune modulation by MANF promotes tissue repair and regenerative success in the retina. Science 353: , aaf3646. |
[44] | Latge C , Cabral KMS , De Oliveira GAP , Raymundo DP , Freitas JA , Johanson L , Romão LF , Palhano FL , Herrmann T , Almeida MS , Foguel D ((2015) ) The solution structure and dynamics of full-length human cerebral dopamine neurotrophic factor and its neuroprotective role against α-synuclein oligomers. J Biol Chem 290: , 20527–20540. |
[45] | Albert K , Raymundo DP , Panhelainen A , Eesmaa A , Shvachiy L , Araújo GR , Chmielarz P , Yan X , Singh A , Cordeiro Y , Palhano FL , Foguel D , Luk KC , Domanskyi A , Voutilainen MH , Huttunen HJ , Outeiro TF , Saarma M , Almeida MS , Airavaara M ((2021) ) Cerebral dopamine neurotrophic factor reduces α-synuclein aggregation and propagation and alleviates behavioural alterations in vivo. Mol Ther, doi: 10.1016/j.ymthe.2021.04.035. |
[46] | Silva RM , Ries V , Oo TF , Yarygina O , Jackson-Lewis V , Ryu EJ , Lu PD , Marciniak SJ , Ron D , Przedborski S , Kholodilov N , Greene LA , Burke RE ((2005) ) CHOP/GADD153 is a mediator of apoptotic death in substantia nigra dopamine neurons in an in vivo neurotoxin model of parkinsonism. J Neurochem 95: , 974–986. |
[47] | Valdés P , Mercado G , Vidal RL , Molina C , Parsons G , Court FA , Martinez A , Galleguillos D , Armentano D , Schneider BL , Hetz C ((2014) ) Control of dopaminergic neuron survival by the unfolded protein response transcription factor XBP1. Proc Natl Acad Sci U S A 111: , 6804–6809. |
[48] | Egawa N , Yamamoto K , Inoue H , Hikawa R , Nishi K , Mori K , Takahashi R ((2011) ) The endoplasmic reticulum stress sensor, ATF6α, protects against neurotoxin-induced dopaminergic neuronal death. J Biol Chem 286: , 7947–7957. |
[49] | Martinez A , Lopez N , Gonzalez C , Hetz C ((2019) ) Targeting of the unfolded protein response (UPR) as therapy for Parkinson’s disease. Biol Cell 111: , 161–168. |
[50] | Mercado G , Castillo V , Soto P , López N , Axten JM , Sardi SP , Hoozemans JJM , Hetz C ((2018) ) Targeting PERK signaling with the small molecule GSK2606414 prevents neurodegeneration in a model of Parkinson’s disease. Neurobiol Dis 112: , 136–148. |
[51] | Moreno JA , Radford H , Peretti D , Steinert JR , Verity N , Martin MG , Halliday M , Morgan J , Dinsdale D , Ortori CA , Barrett DA , Tsaytler P , Bertolotti A , Willis AE , Bushell M , Mallucci GR ((2012) ) Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 485: , 507–511. |
[52] | Halliday M , Radford H , Sekine Y , Moreno J , Verity N , Le Quesne J , Ortori CA , Barrett DA , Fromont C , Fischer PM , Harding HP , Ron D , Mallucci GR ((2015) ) Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis 6: , e1672. |
[53] | Wong YL , LeBon L , Basso AM , Kohlhaas KL , Nikkel AL , Robb HM , Donnelly-Roberts DL , Prakash J , Swensen AM , Rubinstein ND , Krishnan S , McAllister FE , Haste N V. , O’Brien JJ , Roy M , Ireland A , Frost JM , Shi L , Riedmaier S , Martin K , Dart MJ , Sidrauski C ((2019) ) eIF2B activator prevents neurological defects caused by a chronic integrated stress response. Elife 8: , e42940. |
[54] | Martínez G , Vidal RL , Mardones P , Serrano FG , Ardiles AO , Wirth C , Valdés P , Thielen P , Schneider BL , Kerr B , Valdés JL , Palacios AG , Inestrosa NC , Glimcher LH , Hetz C ((2016) ) Regulation of memory formation by the transcription factor XBP1. Cell Rep 14: , 1382–1394. |
[55] | Morita S , Villalta SA , Feldman HC , Register AC , Rosenthal W , Hoffmann-Petersen IT , Mehdizadeh M , Ghosh R , Wang L , Colon-Negron K , Meza-Acevedo R , Backes BJ , Maly DJ , Bluestone JA , Papa FR ((2017) ) Targeting ABL-IRE1α signaling spares ER-stressed pancreatic β cells to reverse autoimmune diabetes. Cell Metab 25: , 883–897.e8. |
[56] | Plate L , Cooley CB , Chen JJ , Paxman RJ , Gallagher CM , Madoux F , Genereux JC , Dobbs W , Garza D , Spicer TP , Scampavia L , Brown SJ , Rosen H , Powers ET , Walter P , Hodder P , Luke Wiseman R , Kelly JW ((2016) ) Small molecule proteostasis regulators that reprogram the ER to reduce extracellular protein aggregation. Elife 5: , e15550. |
[57] | Gallagher CM , Garri C , Cain EL , Ang KKH , Wilson CG , Chen S , Hearn BR , Jaishankar P , Aranda-Diaz A , Arkin MR , Renslo AR , Walter P ((2016) ) Ceapins are a new class of unfolded protein response inhibitors, selectively targeting the ATF6α branch. Elife 5: , e11878. |
[58] | Gómez-Suaga P , Bravo-San Pedro JM , González-Polo RA , Fuentes JM , Niso-Santano M ((2018) ) ER-mitochondria signaling in Parkinson’s disease review-article. Cell Death Dis 9: , 337. |
[59] | Sidorova YA , Saarma M ((2020) ) Small molecules and peptides targeting glial cell line-derived neurotrophic factor receptors for the treatment of neurodegeneration. Int J Mol Sci 21: , 1–20. |
[60] | Airavaara M , Harvey BK , Voutilainen MH , Shen H , Chou J , Lindholm P , Lindahl M , Tuominen RK , Saarma M , Hoffer B , Wang Y ((2012) ) CDNF protects the nigrostriatal dopamine system and promotes recovery after MPTP treatment in mice. Cell Transplant 21: , 1213–1223. |
[61] | Voutilainen MH , Bäck S , Peränen J , Lindholm P , Raasmaja A , Männistö PT , Saarma M , Tuominen RK ((2011) ) Chronic infusion of CDNF prevents 6-OHDA-induced deficits in a rat model of Parkinson’s disease. Exp Neurol 228: , 99–108. |
[62] | Gegg ME , Burke D , Heales SJR , Cooper JM , Hardy J , Wood NW , Schapira AHV ((2012) ) Glucocerebrosidase deficiency in substantia nigra of Parkinson disease brains. Ann Neurol 72: , 455–463. |
[63] | Schöndorf DC , Aureli M , McAllister FE , Hindley CJ , Mayer F , Schmid B , Sardi SP , Valsecchi M , Hoffmann S , Schwarz LK , Hedrich U , Berg D , Shihabuddin LS , Hu J , Pruszak J , Gygi SP , Sonnino S , Gasser T , Deleidi M ((2014) ) IPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun 5: , 4028. |
[64] | Yuan Y , Cao P , Smith MA , Kramp K , Huang Y , Hisamoto N , Matsumoto K , Hatzoglou M , Jin H , Feng Z ((2011) ) Dysregulated LRRK2 signaling in response to endoplasmic reticulum stress leads to dopaminergic neuron degeneration in C. elegans. PLoS One 6: , e322354. |
[65] | Toyofuku T , Okamoto Y , Ishikawa T , Sasawatari S , Kumanogoh A ((2020) ) LRRK 2 regulates endoplasmic reticulum–mitochondrial tethering through the PERK-mediated ubiquitination pathway. EMBO J 39: , e100875. |
[66] | Smith WW , Jiang H , Pei Z , Tanaka Y , Morita H , Sawa A , Dawson VL , Dawson TM , Ross CA ((2005) ) Endoplasmic reticulum stress and mitochondrial cell death pathways mediate A53T mutant alpha-synuclein-induced toxicity. Hum Mol Genet 14: , 3801–3811. |
[67] | Takahashi R , Imai Y ((2003) ) Pael receptor, endoplasmic reticulum stress, and Parkinson’s disease. J Neurol 250 Suppl 3: , III25–29. |
[68] | Bouman L , Schlierf A , Lutz AK , Shan J , Deinlein A , Kast J , Galehdar Z , Palmisano V , Patenge N , Berg D , Gasser T , Augustin R , Trümbach D , Irrcher I , Park DS , Wurst W , Kilberg MS , Tatzelt J , Winklhofer KF ((2011) ) Parkin is transcriptionally regulated by ATF4: Evidence for an interconnection between mitochondrial stress and ER stress. Cell Death Differ 18: , 769–782. |
[69] | Sun X , Liu J , Crary JF , Malagelada C , Sulzer D , Greene LA , Levy OA ((2013) ) ATF4 protects against neuronal death in cellular Parkinson’s disease models by maintaining levels of parkin. J Neurosci 33: , 2398–2407. |
[70] | Ugolino J , Fang S , Kubisch C , Monteiro MJ ((2011) ) Mutant Atp13a2 proteins involved in parkinsonism are degraded by ER-associated degradation and sensitize cells to ER-stress induced cell death. Hum Mol Genet 20: , 3565–3577. |
[71] | Yang J , Kim KS , Iyirhiaro GO , Marcogliese PC , Callaghan SM , Qu D , Kim WJ , Slack RS , Park DS ((2019) ) DJ-1 modulates the unfolded protein response and cell death via upregulation of ATF4 following ER stress. Cell Death Dis 10: , 135. |
[72] | Duplan E , Giaime E , Viotti J , Sévalle J , Corti O , Brice A , Ariga H , Qi L , Checler F , Da Costa CA ((2013) ) ER-stress-associated functional link between Parkin and DJ-1 via a transcriptional cascade involving the tumor suppressor p53 and the spliced X-box binding protein XBP-1. J Cell Sci 126: , 2124–2133. |
[73] | Bertolotti A , Zhang Y , Hendershot LM , Harding HP , Ron D ((2000) ) Dynamic interaction of BiP and ER stress transducers in the unfolded-protein response. Nat Cell Biol 2: , 326–332. |
[74] | Karagöz GE , Acosta-Alvear D , Nguyen HT , Lee CP , Chu F , Walter P ((2017) ) An unfolded protein-induced conformational switch activates mammalian IRE1. Elife 6: , e30700. |
[75] | Wang P , Li J , Tao J , Sha B ((2018) ) The luminal domain of the ER stress sensor protein PERK binds misfolded proteins and thereby triggers PERK oligomerization. J Biol Chem 293: , 4110–4121. |
[76] | Lu M , Lawrence DA , Marsters S , Acosta-Alvear D , Kimmig P , Mendez AS , Paton AW , Paton JC , Walter P , Ashkenazi A ((2014) ) Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 345: , 98–101. |
[77] | Urano F , Wang XZ , Bertolotti A , Zhang Y , Chung P , Harding HP , Ron D ((2000) ) Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287: , 664–666. |
[78] | Hu P , Han Z , Couvillon AD , Kaufman RJ , Exton JH ((2006) ) Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1α-mediated NF-κB activation and down-regulation of TRAF2 expression. Mol Cell Biol 26: , 3071–3084. |

