
Mutations in GBA and LRRK2 Are Not Associated with Increased Inflammatory Markers
Abstract
Background:
Inflammation is an integral part of neurodegeneration including in Parkinson’s disease (PD). Ashkenazi Jews have high rates of genetic PD with divergent phenotypes among GBA-PD and LRRK2-PD. The role of inflammation in the prodromal phase of PD and the association with disease phenotype has yet to be elucidated.
Objective:
To assess central and peripheral cytokines among PD patients with mutations in the LRRK2 and GBA genes and among non-manifesting carriers (NMC) of these mutations in order to determine the role of inflammation in genetic PD.
Methods:
The following cytokines were assessed from peripheral blood and cerebrospinal fluid (CSF): TNF-α, IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10 and INF- γ. A comprehensive intake including general medical conditions, use of anti-inflammatory treatments, motor and cognitive assessments and additional laboratory measures were recorded, enabling the construction of the MDS probable prodromal score.
Results:
Data from 362 participants was collected: 31 idiopathic PD (iPD), 30 LRRK2-PD, 77 GBA-PD, 3 homozygote GBA-PD, 3 GBA-LRRK2-PD, 67 LRRK2-NMC, 105 GBA-NMC, 14 LRRK2-GBA-NMC, and 32 healthy controls. No between-group differences in peripheral or CSF cytokines were detected. No correlation between disease characteristics or risk for prodromal PD could be associated with any inflammatory measure.
Conclusion:
In this study, we could not detect any evidence on dysregulated immune response among GBA and LRRK2 PD patients and non-manifesting mutation carriers.
INTRODUCTION
α-synuclein accumulation is the pathological hallmark of Parkinson’s disease (PD). It occurs within the central nervous system (CNS) and in the autonomic nervous system [1]. The neurodegenerative process of PD is associated with an inflammatory response which can be detected in peripheral cytokines, stool samples, and colonic biopsies [2]. Age-dependent spread of α-synuclein from gut to brain has been previously reported [3] raising the possibility that the inflammation and α-synuclein spread are intertwined and represent an early phenomenon in the pathogenesis of PD [4]. However, whether this process is initiated in the CNS or peripheral nervous system (PNS) is still debated [5]. Interestingly, these inflammatory processes occur in patients with a short disease duration [6] and abate with disease progression.
Neuroinflammation in PD is associated with a chronic immune response in the CNS, in which innate immune cells (astrocytes and microglia) are responsible for an inflammatory response and potentially neural death [7]. Whether the microglial response is a consequence of PD related cell death or an independent process is still debated [8]. PD has been associated with the accumulation of pro-inflammatory cytokines including tumor necrosis factor alpha (TNF-α), interferon gamma (INF-γ), and interleukins 6 (IL-6), IL-1β, IL-2, and IL-10 in both cerebrospinal fluid (CSF) and periphery [9, 10]. However, previous studies attempting to correlate motor severity and cognitive profiles of patients with PD and inflammatory markers have yielded conflicting results [9, 11–14].
Among Ashkenazi Jews (AJ), mutations in the LRRK2 and GBA genes together account for about one third of the incidence of the disease [15]. These mutations have been associated with different phenotypes in which GBA confers a more aggressive form of disease compared to a milder form of LRRK2-PD [16]. Decreased activity of the lysosomal enzyme glucocerebrosidase (GCase) results in Gaucher disease (GD) and has been detected in brain tissue and CSF of PD patients [17, 18] with a direct effect on α-synuclein accumulation [19]. Macrophages from GD patients show increased secretion of IL-1β and IL-6 [20] and GBA-PD has been associated with increased levels of IL-8 among other cytokines [21].
LRRK2 is expressed in neurons and in the innate and adaptive immune system [22, 23] with higher levels of immune cells among LRRK2-PD compared to age matched controls [24]. The concentration of peripheral pro-inflammatory cytokines was higher among LRRK2 non-manifesting carriers (NMC) compared to healthy controls [25]. In addition, mutations in the LRRK2 gene have been associated with different inflammatory and infectious diseases such as Crohn’s disease and leprosy [26, 27].
As the penetrance of the mutations in the GBA and LRRK2 genes is incomplete [28], genetic and environmental modifiers are suspected to play a role in disease risk and severity. Identifying modifiers for disease is crucial for better understanding of the mechanism linking both GBA and LRRK2 to PD potentially enabling disease modifying trials.
In this study, we aimed to characterize the inflammatory profile of patients with PD carriers of GBA or LRRK2 mutations and their first-degree unaffected NMC. We hypothesized that the level of inflammatory cytokines among GBA-PD would be greater than that of LRRK2-PD and idiopathic PD (iPD), thus partially explaining the more severe phenotype of GBA-PD. We further hypothesized that increased markers of inflammation will be evident among NMC and that this inflammatory reaction will be correlated with GCase activity and an increase burden of premotor symptoms (higher likelihood ratio for developing PD) among unaffected “at risk” individuals.
METHODS
This study evaluated demographic, laboratory and clinical data from subjects who participated in the BEAT-PD study (TLV-0204–16), a natural history study which set out to characterize LRRK2 and GBA PD as well as NMC of these mutations. For this sub-study, all PD patients and NMC who carried mutations in the GBA gene were recruited together with all LRRK2-NMC, a group of age and disease duration matched iPD and LRRK2-PD, and age matched controls. Patients were recruited if they were AJ, diagnosed with PD by a movement disorders specialist based on the MDS task force criteria [29] and were at Hoehn and Yahr stages 1-2 [30]. NMC were recruited if they were older than 40 years of age, first degree relatives of GBA-PD or LRRK2-PD patients, and not diagnosed with PD themselves. All subjects were excluded if they had additional neurological or psychiatric disorders, a malignancy or were HIV, Hepatitis B or C positive. The study was approved by the local ethics committee of the Tel-Aviv Medical Center according to the guidelines of the Helsinki declaration. All participants provided an informed written consent prior to participation.
Procedure
Participants were genotyped for the G2019S-LRRK2 mutation and 9 common mutations in the GBA gene; N370S, R496H, considered mild mutations (mGBA-PD), L444P, 84GG, IVS2 + 1G->A, V394L considered severe GBA mutations (sGBA-PD) E326K and T369M, considered risk variants and 370Rec, these mutations account for up to 96%of those found in AJ GD patients [31]. Patients without these mutations were labeled as iPD. Each subject participated in a cross-sectional assessment while 135 subjects agreed to participate in a 5-year longitudinal study, which included CSF collection, MRI and DaT scan three years apart.
Disease severity was assessed using the MDS-Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) during ON medication [32]. The Montreal Cognitive Assessment (MoCA) was used to assess global cognitive functions [33]; mood was assessed using the Beck Depression Inventory (BDI) [34]. The Non-Motor Symptoms Questionnaire (NMSQ) [35], the Scale of Autonomic Function in PD (SCOPA-AUT) [36] and the REM sleep Behavior Disorder Questionnaire (RBDQ) were collected [37]. Olfaction was tested using the University of Pennsylvania Smell Identification Test (UPSIT) [38]. These measures were used to calculate the probability for prodromal PD (Likelihood Ratio Score) for all participants without a diagnosis of PD, based on the updated MDS task force prodromal criteria guidelines [39]. Each non-manifesting subject was allocated a ratio between 0–100%for risk for future development of PD based on risk and prodromal symptoms. Levodopa equivalent daily dose (LEDD) was calculated for all patients [40]. In addition, all participants were assessed for inflammatory disease and use of anti-inflammatory medications including aspirin and antibiotics in the days prior to participation.
Blood tests were collected from all participants without an overnight fast. A complete blood count was drawn with white blood count (WBC), hemoglobin, platelet count, absolute lymphocyte and neutrophil levels recorded, and a ratio of lymphocyte/neutrophil (NLR) calculated. Uric acid, urine microalbumin, and CRP were assessed as well.
Blood samples were centrifuged at 1500×g at 4°C for 15 min, 30 min after blood draw. The serum was separated and aliquoted into 2 ml tubes and placed in a –80°C freezer. For the cytokines panel assay, serum samples were thawed on wet ice and sub aliquoted to 105μl in cryotubes.
Lumbar punctures were performed in a sub-group of participants as detailed previously [41]. CSF was collected in 15 ml conical tubes and centrifuged at 2000×g at 25°C for 10 min. The supernatant was collected to a conical tube, aliquoted into 0.5 ml and frozen at –80°C. For the cytokine measurement, the CSF aliquots were thawed on wet ice and sub-aliquoted to 100μl in cryotubes. The CSF samples were stored in –80°C and shipped in dry ice. CSF and serum were taken at the same day for those who underwent a lumbar puncture.
Human high sensitivity cytokine Luminex custom 8-plex kits were used to assess cytokine levels. The assays were performed in 96-well plates according to the manufacturer’s instructions (R&D Systems, Inc. Minneapolis, MN USA). Standards were run in duplicates, while samples were run in singles. Briefly, the Luminex Assays utilize color-coded superparamagnetic beads coated with analyte-specific antibodies. Beads recognizing different target analytes were mixed together and incubated with the bio-fluid sample. Captured analytes are subsequently detected using a cocktail of biotinylated detection antibodies and a streptavidin-Phycoerythrin conjugate. Magnetic beads were isolated and measured by using the Luminex MAGPIX® Analyzer. The following cytokines were detected in serum and CSF: TNF-α IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, and INF-γ. CSF was assessed for the same cytokines as well.
For the GCase analysis blood samples were collected in a 10 ml EDTA tube. Fifty microliters of whole blood were dropped on each of ten circles on a filter paper and dried at room temperature for at least 4 hours. The filter paper was then stored in a sealed plastic bag at room temperature until the samples were shipped for processing at Centogene (Rostock, Germany). A specific synthetic substrate was added, and the plates were incubated further at 37°C for 16 h. The reaction was stopped by changing the pH to 10.5. The enzymatic product was quantified by fluorimetry using an external standard curve. The results of the enzymatic activity determination were calculated in μmol/L/h. As quality parameters for the assays, standard blood samples were added to each batch to ensure the accuracy of the determination.
Statistical analysis
Descriptive statistics (means and standard deviations (SD) for continuous variables) were computed for all measures. Missing cytokine variables were treated with two-imputation methods: 1) Taking the panels’ threshold as the value; 2) Imputed them as 0. The two methods allowed us to examine the sensitivity of the analysis in two extreme ends of possible values.
Log transformation was performed to reduce the natural skewness of the following variables: peripheral cytokines: TNF-α, IL-2, IL-1-β, IL-6 and IL-10; CSF cytokines: TNF-α, IL-1β, IL-6 INF-γ. The following laboratory measures were transformed as well: WBC, urine microalbumin, N/L ratio and CRP.
We first created a linear model based on age, disease duration, body-mass index (BMI), use of anti-inflammatory medication and presence of additional inflammatory diseases for each of the CSF/peripheral cytokines. Next, the residuals of each group were compared using Wilcoxon Test in order to avoid any assumption about the residuals’ distribution. Due to the limited number of observations and natural skewness of the measures, we used the non-parametric Wilcoxon’s test which is appropriate when there is no evidence for normal distribution. This test is known to be more powerful than the T-test when the number of observations is small and when the normality assumption does not necessarily hold.
p-values were adjusted using the Benjamini-Hochberg procedure to control the false discovery rate at level 0.05.
Non-parametric Spearman-rho correlations (used to identify monotonic relationships) between cytokines and additional laboratory and behavioral measures for each study group were assessed. The resulting p-values per group were adjusted for multiplicity using the Benjamini-Hochberg method. The threshold of significance was set at 0.05.
Finally, random forest classification was used on the cytokines to classify the participant into genetic groups. This was performed separately for the PD subgroups and the non-manifesting subjects. In this model we used the original cytokine values as explanatory variables in conjunction with the covariates of the linear model detailed above. Since random forest is based on decision trees, it can be used to identify inflammatory profile for each group of participants. Statistical analysis was performed using R version 4.0.3.
RESULTS
Data was collected from 356 participants: 31 idiopathic PD (iPD), 30 LRRK2-PD, 77 GBA-PD, 67 LRRK2-NMC, 105 GBA-NMC, 14 LRRK2-GBA-NMC and 32 healthy controls. Cohort characteristics are presented in Tables 1 and 2.
Table 1
Characteristics of the PD cohort
| iPD | LRRK2-PD | GBA-PD | p | |
| Mutation type | N370S –51 | |||
| R496H –5 | ||||
| 84GG –5 | ||||
| IVS2 –3 | ||||
| V394L –3 | ||||
| Rec370 –2 | ||||
| L444P –1 | ||||
| E326K –5 | ||||
| T369M –2 | ||||
| N | 31 | 30 | 77 | |
| Age (y) | 65.77 (10.44) | 65.21 (9.99) | 64.83 (10.15) | 0.906 |
| Gender M/F (%male) | 20/11 (64.5%) | 18/12 (60.0%) | 54/27 (66.3%) | 0.828 |
| BMI (kg/m2) | 25.50 (3.22) | 25.04 (3.97) | 26.25 (4.57) | 0.354 |
| Disease duration (y) | 3.69 (1.91) | 3.82 (2.66) | 3.78 (2.58) | 0.980 |
| Age at diagnosis (y) | 62.13 (10.74) | 61.70 (9.98) | 61.49 (10.03) | 0.957 |
| LEDD | 360.98 (296.65) | 443.69 (389.38) | 456.41 (419.68) | 0.506 |
| MDS-UPDRS | 41.87 (15.73) | 35.87 (16.94) | 40.54 (21.42) | 0.430 |
| MoCA | 23.71 (3.73) | 25.03 (3.95) | 23.32 (4.40) | 0.160 |
| UPSIT | 15.42 (9.85) | 20.07 (9.86) | 15.43 (9.20) | 0.078 |
| RBDQ | 3.13 (3.13) | 3.06 (2.29) | 3.84 (3.05) | 0.377 |
| SCOPA-AUT | 18.34 (9.75) | 15.58 (10.87) | 15.95 (10.39) | 0.613 |
| GCase (μmol/L/h) | 4.77 (1.25) | 5.06 (1.25) | 3.02 (0.86) | < 0.001* |
| WBC (103/ μL) | 6.91 (1.52) | 7.35 (1.83) | 7.13 (1.65) | 0.605 |
| Lymphocytes (103/ μL) | 1.75 (0.55) | 1.50 (0.45) | 1.67 (0.59) | 0.219 |
| Neutrophils 103/ μL) | 4.46 (1.21) | 5.09 (1.72) | 4.71 (1.35) | 0.239 |
| Platelets (103/ μl) | 213.00 (53.64) | 212.21 (44.42) | 209.26 (51.88) | 0.926 |
| C-Reactive Protein (mg/l) | 1.44 (1.23) | 3.39 (6.50) | 1.98 (4.30) | 0.204 |
| Uric Acid (mg/dL) | 5.22 (1.24) | 5.45 (1.39) | 5.41 (1.41) | 0.634 |
| Urine Microalbumin (mg) | 6.32 (7.55) | 17.26 (58.44) | 15.61 (41.42) | 0.420 |
| Presence of any inflammatory disease | 7 (22.6%) | 4 (13.3%) | 10 (12.0%) | 0.357 |
| Use of any anti-inflammatory medication | 9 (29.0%) | 10 (33.3%) | 29 (34.9%) | 0.838 |
iPD, idiopathic Parkinson’s disease; M, male; F, female; BMI, body mass index; LEDD, LevoDopa Equivalent Daily Dose; MDS-UPDRS, Movement Disorder Society - Unified Parkinson’s Disease Rating Scale; MoCA, Montreal Cognitive Assessment; UPSIT, University of Pennsylvania Smell Identification Test; RBDQ, REM Sleep Behavior Questionnaire; SCOPA-AUT, Scales of Outcomes in PD –autonomic; GCase, beta glucocerebrosidase; WBC, white blood cells. Data is shown as mean±standard deviation or percentage. *p≤0.05. *significant difference between GBA-PD and LRRK2-PD, iPD.
Table 2
Characteristics of the Non manifesting participants
| Controls | LRRK2NMC | GBA NMC | LRRK2- GBANMC | Significance | |
| N370S –59 | G2019S + N370S –5 | ||||
| R496H –10 | G2019S + R496H –2 | ||||
| 84GG –13 | G2019S + E326K –5 | ||||
| IVS2 –4 | G2019S + T369M –1 | ||||
| V394L –5 | G2019S + V394L - 1 | ||||
| Rec370 –4 | |||||
| L444P –2 | |||||
| E326K –4 | |||||
| T369M –4 | |||||
| N | 32 | 67 | 105 | 14 | |
| Age (y) | 54.82 (10.03) | 54.45 (12.46) | 54.88 (10.03) | 52.00 (11.48) | 0.803 |
| Gender M/F (%male) | 13/20 (39.4%) | 33/34 (49.3%) | 38/68 (35.8%) | 5/9 (35.7%) | 0.368 |
| BMI (kg/m2) | 26.16 (4.05) | 26.14 (4.23) | 26.70 (4.73) | 25.77 (3.94) | 0.771 |
| Probability of prodromal (%) | 9.81 (24.60) | 20.84 (31.52) | 29.10 (33.45) | 14.40 (23.72) | 0.014* |
| Probability above 80%n (%) | 0 | 8 (11.94) | 18 (17.15) | 1 (7.14) | 0.058 |
| MDS-UPDRS | 4.85 (4.76) | 5.33 (4.29) | 5.94 (6.85) | 7.69 (9.25) | 0.437 |
| MoCA | 27.39 (2.96) | 26.05 (3.34) | 25.87 (3.13) | 26.31 (3.48) | 0.142 |
| UPSIT | 31.30 (6.97) | 31.23 (5.07) | 29.88 (6.52) | 32.36 (3.91) | 0.308 |
| RBDQ | 1.79 (1.45) | 1.40 (1.46) | 1.95 (1.91) | 1.76 (1.09) | 0.122 |
| SCOPA-AUT | 7.66 (5.74) | 8.58 (7.05) | 8.29 (7.43) | 9.80 (7.41) | 0.344 |
| GCase (μmol/L/h) | 4.86 (1.41) | 4.89 (1.31) | 3.08 (0.89) | 3.05 (0.79) | < 0.001∧ |
| WBC (103/ μL) | 7.52 (2.18) | 6.60 (1.50) | 6.94 (2.01) | 6.18 (1.16) | 0.056 |
| Lymphocytes (103/ μL) | 2.09 (1.25) | 1.81 (0.48) | 1.91 (0.56) | 1.90 (0.55) | 0.299 |
| Neutrophils 103/ μL) | 4.72 (1.76) | 4.11 (1.19) | 4.35 (1.8) | 3.59 (0.77) | 0.097 |
| Platelets (103/ μl) | 228.09 (56.14) | 226.09 (54.16) | 229.60 (58.28) | 222.80 (37.25) | 0.961 |
| C-Reactive Protein (mg/l) | 2.56 (5.67) | 1.78 (2.03) | 2.71 (4.21) | 1.87 (2.07) | 0.449 |
| Uric Acid (mg/dL) | 4.79 (1.16) | 5.24 (1.42) | 5.07 (1.41) | 5.26 (1.66) | 0.446 |
| Urine Microalbumin (mg) | 8.93 (20.45) | 18.45 (55.95) | 5.62 (15.22) | 5.50 (9.82) | 0.384 |
| Presence of any Inflammatory disease | 7 (21.2%) | 7 (10.4%) | 19 (17.9%) | 4 (25.0%) | 0.351 |
| Use of any Anti-inflammatory medication | 8 (24.2%) | 14 (20.9%) | 24 (22.6%) | 2 (12.5%) | 0.800 |
NMC, non-manifesting carrier; M, male; F, female; BMI, body mass index; MDS-UPDRS, Movement Disorder Society - Unified Parkinson’s Disease Rating Scale; MoCA, Montreal Cognitive Assessment; UPSIT, University of Pennsylvania Smell Identification Tes;, GCase, beta glucocerebrosidase; WBC, white blood cells. Data is shown as mean±standard deviation or percentage. *Significant difference between GBA-NMC, LRRK2-NMC and controls, LRRK2-GBA-NMC. ∧Significant difference between GBA-NMC, LRRK2-GBA-NMC and LRRK2-NMC, controls.
CSF samples were collected from a subgroup of participants that underwent lumber puncture (n = 80); 9 iPD, 12 LRRK2-PD, 15 GBA-PD, 10 LRRK2-NMC, 19 GBA-NMC, 5 LRRK-GBA-NMC and 10 controls.
We compared behavioral and laboratory measures within the three groups of PD participants (iPD, LRRK2-PD and GBA-PD) and within the four groups of non-manifesting participants (controls, LRRK2-NMC, GBA-NMC and LRRK2-GBA-NMC).
GCase activity was significantly lower among GBA-PD compared with LRRK2-PD and iPD (3.02±0.86, 5.06±1.25 and 4.77±1.25, p < 0.001) and among GBA-NMC and LRRK2-GBA-NMC compared to LRRK2-NMC and controls (3.08±0.89, 3.05±0.79, 4.89±1.31, 4.86±1.41, p < 0.001), no other behavioral characteristics reached between group significance when correcting for multiplicity (Tables 1 and 2).
Peripheral INF-γ and IL-2 were not included in the analysis as less than 30%of their readings were above threshold. Data regarding missing variables is presented in Supplementary Tables 1 and 2. Data regarding coefficients of variation for each cytokine in serum and CSF is presented as Supplementary Table 3, the inter-plate variability for each cytokine is presented in Supplementary Table 4.
No significant difference in CSF or peripheral cytokine levels were detected between any of the PD subgroups or the non-manifesting subgroups when missing levels were computed as “0” or as the specific batch threshold (Figs. 1 and 2). Furthermore, assessing cytokine differences between mGBA-PD and sGBA-PD and between mGBA-NMC and sGBA-NMC did not yield any differences.
Fig. 1
Levels of peripheral cytokines among study participants iPD, idiopathic Parkinson’s disease; NMC, non-manifesting carriers; IL, interleukin; TNF, tumor necrosis factor.
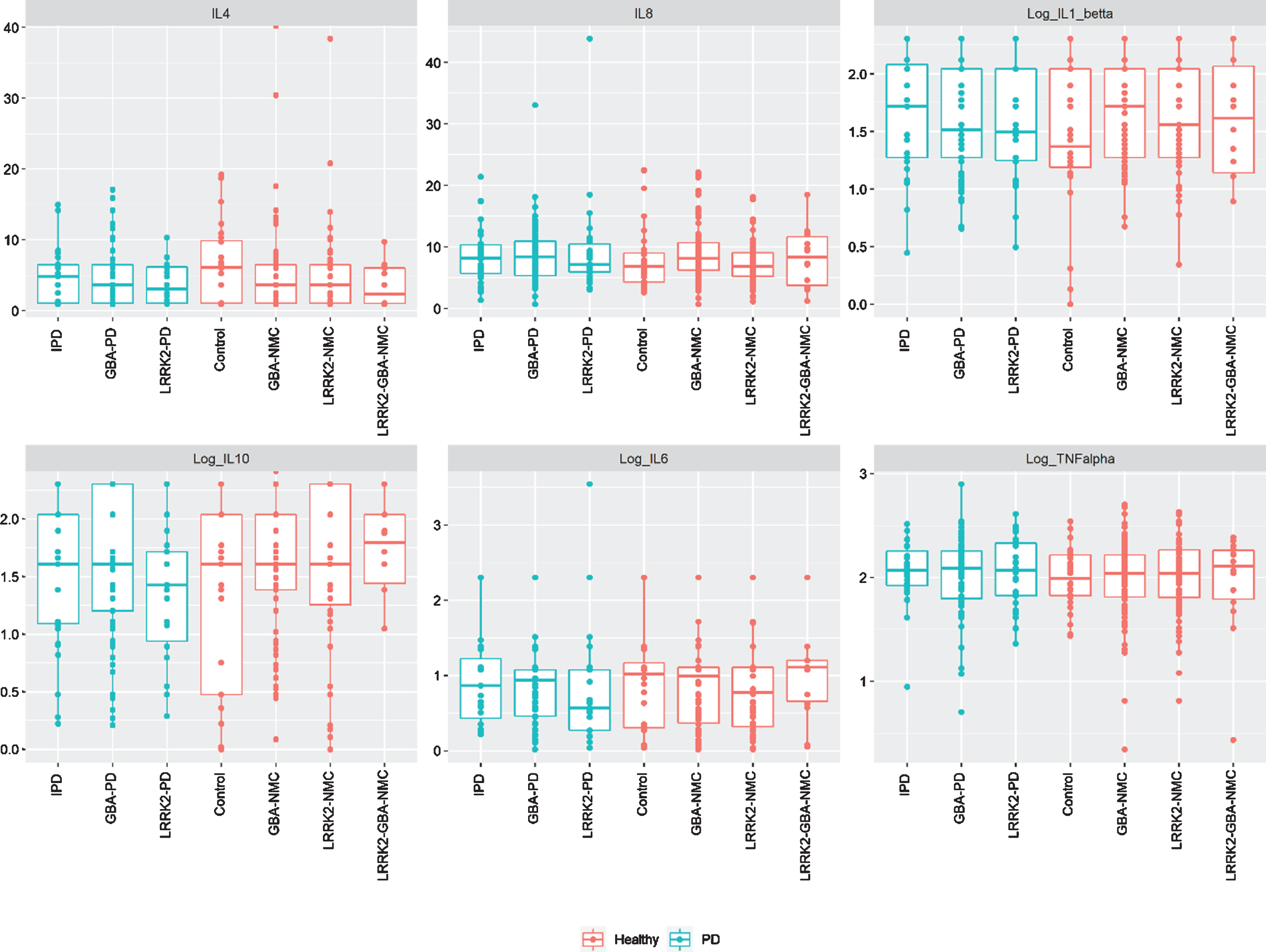
Fig. 2
Levels of CSF cytokines among study participants iPD, idiopathic Parkinson’s disease; NMC, non-manifesting carriers; IL, interleukin; INF, interferon; TNF, tumor necrosis factor.
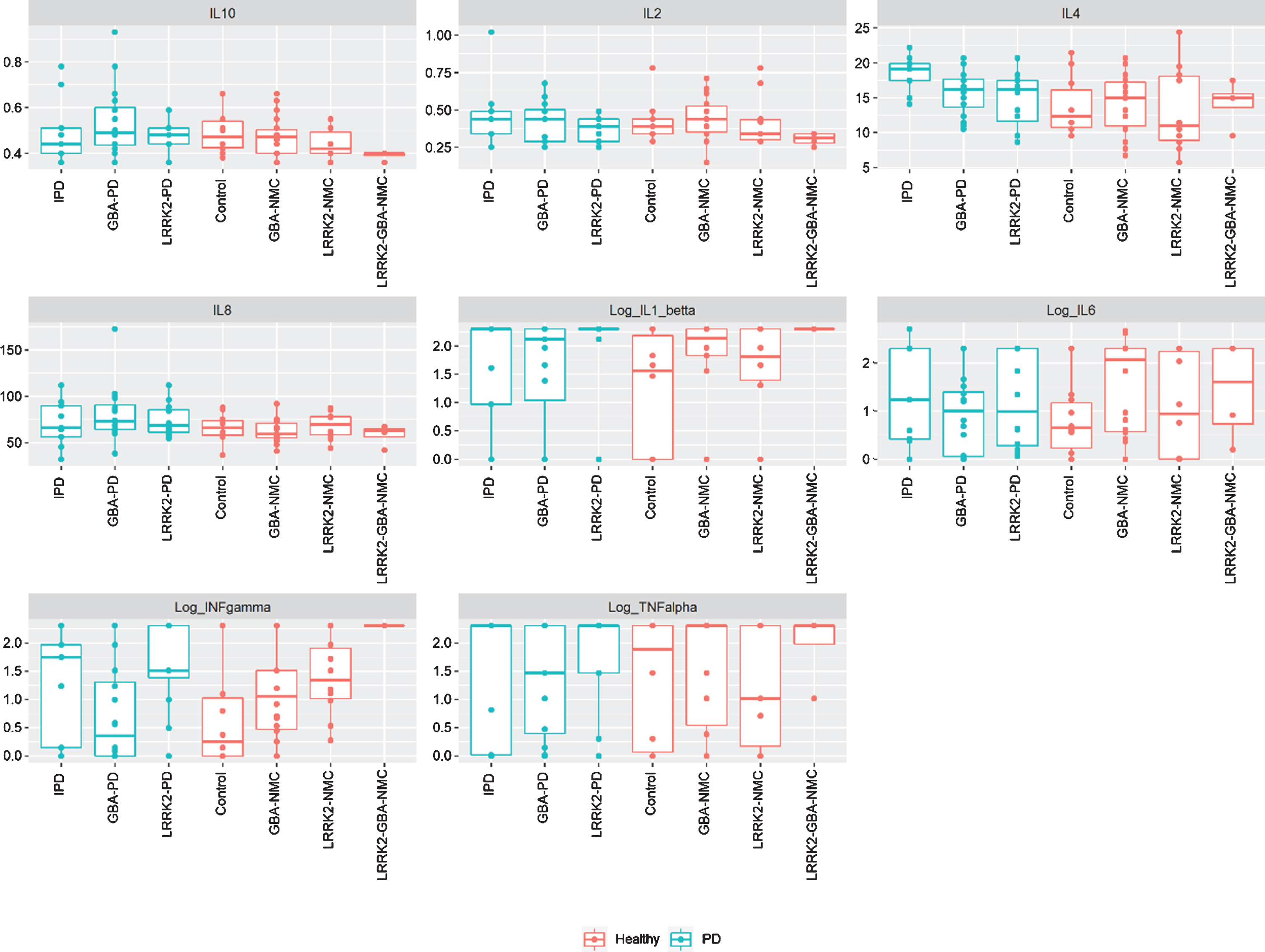
CSF cytokine levels were not correlated with any behavioral or laboratory measures or with risk for prodromal PD among any of the sub-groups.
Weak, but statistically significant correlations were detected among the total cohort of participants between peripheral cytokine levels and the following measures: CRP and IL-6 (r = 0.417, p < 0.001), CRP and TNF-α (r = 0.167, p = 0.028), neutrophil levels and IL-8 (r = 0.16, p = 0.035), neutrophil levels and IL-6 (r = 0.188, p = 0.017), uric acid and TNF-α (r = 0.263, p < 0.001), urine microalbumin and IL-6 (r = 0.186, p = 0.017), urine microalbumin and TNF-α (r = 0.178, p = 0.021). IL-6 was correlated with CRP among controls (r = 0.557, p < 0.001), LRRK2-PD (r = 0.466, p = 0.014) and iPD (r = 0.629, p = 0.018). CSF cytokines were not correlated with peripheral cytokines in any of the subgroups of participants.
The probability score for prodromal PD did not correlate with any of the cytokines among all non-manifesting participants or sub-groups assessed separately. GCase activity was not correlated with cytokine levels in any subgroup of participants either.
Performing the same analysis, excluding participants that reported concurrent inflammatory conditions or use of anti-inflammatory medications did not yield any significant findings.
The random forest analysis resulted with high error rates, meaning the explanatory variables alone could not be used to distinguish between the genetic sub-groups.
DISCUSSION
In this study assessing a large group of GBA-PD, non-manifesting LRRK2 and GBA mutation carriers, we could not detect a dysregulated immune response based on a panel of both pro and anti-inflammatory cytokines. In addition, disease severity and risk for probability of prodromal PD were not associated with specific cytokine levels or combination of cytokines.
In the central nervous system, resident immune system is comprised mostly of innate immune cells, with microglia and astrocytes as the main components. Under certain conditions, peripheral innate and adaptive immune cell can enter the CNS [42]. This reaction can be beneficial or deleterious depending on its severity and duration. Although cytokines participate in the inflammatory process, most have multiple biological targets and activities. The specific role of any cytokine in a clinical setup is dependent on the spectrum of polypeptides that are secreted alongside it.
With accumulating evidence that GI symptoms are present from the earliest stages of PD, it has been proposed that PD pathology may originate in the gut and later spread to the CNS. Immune activity may advance this progression, as intestinal inflammation can promote systemic and also CNS inflammation [43] contributing to PD-related neurodegeneration [44]. Immune activation promotes increased α-synuclein expression and aggregation [45] and α-synuclein in turn stimulates proinflammatory immune responses [46], resulting in a positive feedback loop.
However, data regarding cytokine expression in PD is conflicting: The ICICLE study assessed early PD patients and detected elevation of TNF-α, IL-1β, IL-2 and IL-10 compared with controls [13]. While elevated levels of cytokines including IL-1β, IL-2, TNF-α, IL-6 IL-10 and CRP have been reported among patients with PD compared with controls in a comprehensive meta-analysis [10]. However, the pooling of different studies, included patients in different disease stages, disease duration, use of medication and concomitant medical condition, is a limitation of this meta-analysis.
As CSF is in close contact with the CNS, paired plasma and CSF assessments may provide insight into the relationship between peripheral and central inflammation in PD. However, our study, in line with previous reports, failed to detect any correlation between cytokines analyzed in the CSF and serum of PD patients and NMC [47]. A recent post-mortem study examined mRNA levels of cytokines and mediators of the immune response from PD patients across different disease stages and found increased expression of IL-6 in the substantia nigra of patients in early disease stage while at later stages of the disease the same marker was downregulated [48]. In this work however, we could not detect evidence of central or peripheral inflammation, at least as reflected in ambient cytokine concentrations, among patients with PD or NMC of disease related mutations when correcting for anti-inflammatory medications or additional inflammatory conditions or when excluding participants with these conditions. We did detect correlations between pro-inflammatory cytokines and pro-inflammatory laboratory measures such as CRP, uric acid and microalbumin [49, 50] indicating that states that are associated with increased inflammation were associated with increased pro-inflammatory cytokines.
The pathological hallmark of GD is the accumulation of characteristic macrophages containing glycolipids (Gaucher cells) leading to chronic inflammation [51]. Glycolipid accumulation and lysosomal dysfunction in GD prime macrophages to release pro-inflammatory cytokines [52, 53] and indeed, in GD cell lines, activated microglia and astrocytes were detected prior to cell loss with an increase in pro-inflammatory cytokines [54]. Patients with type 1 GD showed elevated serum IL-6 [53]. Homozygote PD GBA carriers were found to have elevated plasma levels of ferritin, CCL18 and MIP1α but heterozygotes PD and NMC did not differ from controls in their inflammatory profile [55]. While a small study with 17 GBA-PD detected increased levels of IL-8 among other cytokines [21]. We could not detect any differences in cytokine profiles between GBA-PD and iPD or between GBA-NMC and controls even when separating the GBA cohort based on type of mutation (mild, variant, or severe), within a large cohort of participants.
Data regarding inflammation among LRRK2-PD and LRRK2-NMC is conflicting. While platelet-derived growth factor (PDGF) was higher among LRRK2-PD compared to iPD [56], other studies detected an increase in both pro-inflammatory markers (IL-12-p40) and anti-inflammatory markers (IL-10 and brain-derived neurotrophic factor (BDNF)) among iPD compared with LRRK2-PD [57]. Among LRRK2-NMC, peripheral IL-1β was significantly higher compared with controls when corrections for age, sex, current and past use of anti-inflammatory medications [25], CSF TNF-α was reported to be higher in LRRK2-NMC than in controls [58]. However, these findings were not replicated in other studies [57] or herein, possibly due to the lower age and number of controls in our study, use of concomitant inflammatory conditions as a covariate, and use of more stringent corrections for multiplicity in our study.
The penetrance of the mutations in the GBA and LRRK2 genes is incomplete [28], thus including all non-manifesting mutation carriers together as one group has the potential of combining participants that will and will not convert to PD within different timeframes. An MDS task force has suggested and updated criteria for detection of the prodromal stages of the disease [39]. Potentially, it is only those participants that are at the prodromal stage of the disease that might have an elevated inflammatory response. Comparing the 27 NMC with a probability score of above 80 to those with a lower score, did not yield any significant difference in cytokine levels. However, this might be related to the low number of NMC with high probability scores.
While this is the largest study to date to assess inflammatory measures in GBA related PD and NMC, several limitations need to be addressed. First, the cross-sectional set-up is problematic when assessing the inflammatory profile of this cohort as daily changes in cytokine levels are well recognized. We used a targeted approach for the assessment of mutations in the LRRK2 and GBA genes and did not perform a complete sequencing of these genes as the 9 mutations in the GBA gene assessed herein account for over 96%of the known AJ mutations in GD [31] and the vast majority of LRRK2 mutations, notwithstanding, this method might have missed rare mutation carriers and thus labeled them as iPD. The sub-group of participants who underwent lumbar puncture and CSF assessment and longitudinal analysis was relatively small. While the choice of pro and anti-inflammatory cytokine was based on previous disease related studies, the use of high sensitivity kits with no related analysis of immune cells remains a limitation especially when some of the cytokines were below detection levels among the majority of participants. We thawed our blood and CSF samples for sub-aliquotation of 100μl before cytokine analysis. While all samples were aliquoted on dry ice and using the same protocol, this extra thaw-freeze cycle could affect cytokines stability differently. However, this would have a similar effect on all groups.
The role of inflammation in neurodegeneration and PD specifically seems robust with epidemiological evidence of reduced risk for PD among Inflammatory bowel disease patients treated with anti-TNF medication [59], indicating a role for monocyte activation factors and disease progression [60]. There still exists an unmet need for establishing a reliable approach for the study of neuroinflammation in PD patients and prodromal subjects. A combination of blood and CSF cytokines, cellular immunity and brain imaging might help to elucidate the relationship between inflammation, neurodegeneration and α-synuclein, but is extremely difficult to execute.
In this comprehensive analysis of central and peripheral inflammatory measures among LRRK2 and GBA PD and NMC, we could not detect any pro-inflammatory signal related to disease risk or phenotype. Future studies assessing other components of the immune system are warranted to establish the role of inflammation in the prodromal period and after disease diagnosis to determine whether the immune system might be targeted with interventions aimed to modify PD.
ACKNOWLEDGMENTS
This work was funded by the MJFF grant #16503, the Silverstein Foundation for Parkinson’s with GBA and by Biogen Inc.
CONFLICT OF INTEREST
AT –Receiving honoraria from Abbvie Israel.
NO –Nothing to disclose
NG –Serves on the Editorial Board of the Journal of Parkinson’s Disease. Serves as consultant to Biogen, Genzyme-Sanofi, Sionara, NeuroDerm, Intec Pharma, Pharma2B, Denali Neuron23 and Abbvie. Receives royalties from Lysosomal Therapeutics (LTI) and payment for lectures from Abbvie, Sanofi-Genzyme, Bial and Movement Disorder Society. Received research support from the Michael J Fox Foundation, the National Parkinson Foundation, the European Union 7th Framework Program, the Israel Science Foundation, Teva NNE program, Biogen, LTI, and Ionis.
TG –Advisory board membership with honoraria from Abbvie, Neuroderm, Medison, Allergan, Cytora and Synnerva, research support from Phonetica Ltd., Israeli Innovation Authority, Sagol School of Neuroscience and Parkinson’s Foundation. Receiving travel support from Abbvie, Allergan, Medisson and Medtronic.
ABS –Nothing to disclose
MGW –Nothing to disclose
OG –Nothing to disclose
MK –Payment for lectures from Abbvie and Teva.
JCS –Employee and shareholder in Biogen, Inc.
JMC –A former employee and shareholder in Biogen, Inc.
AOU –Research support from the Michael J Fox Foundation, Chaya Charitable Fund and Biogen and payment for lectures from Sanofi Genzyme and Pfizer.
KR –Lecture fees and travel grants from Teva, BMS, Biogen, Sanofi Genzyme, Roche, Medison, Novartis and Serono.
SST –Nothing to disclose
AM- Serving as advisor to Neuroderm.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JPD-212624.
REFERENCES
[1] | Braak H , Del Tredici K ((2008) ) Invited Article: Nervous system pathology in sporadic Parkinson disease. Neurology 70: , 1916–1925. |
[2] | Devos D , Lebouvier T , Lardeux B , Biraud M , Rouaud T , Pouclet H , Coron E , Bruley des Varannes S , Naveilhan P , Nguyen JM , Neunlist M , Derkinderen P ((2013) ) Colonic inflammation in Parkinson’s disease. Neurobiol Dis 50: , 42–48. |
[3] | Challis C , Hori A , Sampson TR , Yoo BB , Challis RC , Hamilton AM , Mazmanian SK , Volpicelli-Daley LA , Gradinaru V ((2020) ) Gut-seeded alpha-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci 23: , 327–336. |
[4] | Sulzer D , Alcalay RN , Garretti F , Cote L , Kanter E , Agin-Liebes J , Liong C , McMurtrey C , Hildebrand WH , Mao X , Dawson VL , Dawson TM , Oseroff C , Pham J , Sidney J , Dillon MB , Carpenter C , Weiskopf D , Phillips E , Mallal S , Peters B , Frazier A , Lindestam Arlehamn CS , Sette A ((2017) ) T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 546: , 656–661. |
[5] | Holmqvist S , Chutna O , Bousset L , Aldrin-Kirk P , Li W , Bjorklund T , Wang ZY , Roybon L , Melki R , Li JY ((2014) ) Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol 128: , 805–820. |
[6] | Rolli-Derkinderen M , Leclair-Visonneau L , Bourreille A , Coron E , Neunlist M , Derkinderen P ((2020) ) Is Parkinson’s disease a chronic low-grade inflammatory bowel disease? J Neurol 267: , 2207–2213. |
[7] | Hirsch EC , Vyas S , Hunot S ((2012) ) Neuroinflammation in Parkinson’s disease. Parkinsonism Relat Disord 18 Suppl 1: , S210–212. |
[8] | Su X , Maguire-Zeiss KA , Giuliano R , Prifti L , Venkatesh K , Federoff HJ ((2008) ) Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging 29: , 1690–1701. |
[9] | Reale M , Iarlori C , Thomas A , Gambi D , Perfetti B , Di Nicola M , Onofrj M ((2009) ) Peripheral cytokines profile in Parkinson’s disease. Brain Behav Immun 23: , 55–63. |
[10] | Qin XY , Zhang SP , Cao C , Loh YP , Cheng Y ((2016) ) Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: A systematic review and meta-analysis. JAMA Neurol 73: , 1316–1324. |
[11] | Menza M , Dobkin RD , Marin H , Mark MH , Gara M , Bienfait K , Dicke A , Kusnekov A ((2010) ) The role of inflammatory cytokines in cognition and other non-motor symptoms of Parkinson’s disease. Psychosomatics 51: , 474–479. |
[12] | Rocha NP , Teixeira AL , Scalzo PL , Barbosa IG , de Sousa MS , Morato IB , Vieira EL , Christo PP , Palotas A , Reis HJ ((2014) ) Plasma levels of soluble tumor necrosis factor receptors are associated with cognitive performance in Parkinson’s disease. Mov Disord 29: , 527–531. |
[13] | Williams-Gray CH , Wijeyekoon R , Yarnall AJ , Lawson RA , Breen DP , Evans JR , Cummins GA , Duncan GW , Khoo TK , Burn DJ , Barker RA , ICICLE-PD study group ((2016) ) Serum immune markers and disease progression in an incident Parkinson’s disease cohort (ICICLE-PD). Mov Disord 31: , 995–1003. |
[14] | Kouchaki E , Kakhaki RD , Tamtaji OR , Dadgostar E , Behnam M , Nikoueinejad H , Akbari H ((2018) ) Increased serum levels of TNF-alpha and decreased serum levels of IL-27 in patients with Parkinson disease and their correlation with disease severity. Clin Neurol Neurosurg 166: , 76–79. |
[15] | Gan-Or Z , Bar-Shira A , Mirelman A , Gurevich T , Kedmi M , Giladi N , Orr-Urtreger A ((2010) ) LRRK2 and GBA mutations differentially affect the initial presentation of Parkinson disease. Neurogenetics 11: , 121–125. |
[16] | Kozlovski T , Mitelpunkt A , Thaler A , Gurevich T , Orr-Urtreger A , Gana-Weisz M , Shachar N , Galili T , Marcus-Kalish M , Bressman S , Marder K , Giladi N , Benjamini Y , Mirelman A ((2019) ) Hierarchical data-driven analysis of clinical symptoms among patients with Parkinson’s disease. Front Neurol 10: , 531. |
[17] | Parnetti L , Chiasserini D , Persichetti E , Eusebi P , Varghese S , Qureshi MM , Dardis A , Deganuto M , De Carlo C , Castrioto A , Balducci C , Paciotti S , Tambasco N , Bembi B , Bonanni L , Onofrj M , Rossi A , Beccari T , El-Agnaf O , Calabresi P ((2014) ) Cerebrospinal fluid lysosomal enzymes and alpha-synuclein in Parkinson’s disease. Mov Disord 29: , 1019–1027. |
[18] | Gegg ME , Burke D , Heales SJ , Cooper JM , Hardy J , Wood NW , Schapira AH ((2012) ) Glucocerebrosidase deficiency in substantia nigra of Parkinson disease brains. Ann Neurol 72: , 455–463. |
[19] | Mazzulli JR , Xu YH , Sun Y , Knight AL , McLean PJ , Caldwell GA , Sidransky E , Grabowski GA , Krainc D ((2011) ) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146: , 37–52. |
[20] | Aflaki E , Moaven N , Borger DK , Lopez G , Westbroek W , Chae JJ , Marugan J , Patnaik S , Maniwang E , Gonzalez AN , Sidransky E ((2016) ) Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell 15: , 77–88. |
[21] | Chahine LM , Qiang J , Ashbridge E , Minger J , Yearout D , Horn S , Colcher A , Hurtig HI , Lee VM , Van Deerlin VM , Leverenz JB , Siderowf AD , Trojanowski JQ , Zabetian CP , Chen-Plotkin A ((2013) ) Clinical and biochemical differences in patients having Parkinson disease with vs without GBA mutations. JAMA Neurol 70: , 852–858. |
[22] | Gardet A , Benita Y , Li C , Sands BE , Ballester I , Stevens C , Korzenik JR , Rioux JD , Daly MJ , Xavier RJ , Podolsky DK ((2010) ) LRRK2 is involved in the IFN-gamma response and host response to pathogens. J Immunol 185: , 5577–5585. |
[23] | Thevenet J , Pescini Gobert R , Hooft van Huijsduijnen R , Wiessner C , Sagot YJ ((2011) ) Regulation of LRRK2 expression points to a functional role in human monocyte maturation. PLoS One 6: , e21519. |
[24] | Cook DA , Kannarkat GT , Cintron AF , Butkovich LM , Fraser KB , Chang J , Grigoryan N , Factor SA , West AB , Boss JM , Tansey MG ((2017) ) LRRK2 levels in immune cells are increased in Parkinson’s disease. NPJ Parkinsons Dis 3: , 11. |
[25] | Dzamko N , Rowe DB , Halliday GM ((2016) ) Increased peripheral inflammation in asymptomatic leucine-rich repeat kinase 2 mutation carriers. Mov Disord 31: , 889–897. |
[26] | Umeno J , Asano K , Matsushita T , Matsumoto T , Kiyohara Y , Iida M , Nakamura Y , Kamatani N , Kubo M ((2011) ) Meta-analysis of published studies identified eight additional common susceptibility loci for Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis 17: , 2407–2415. |
[27] | Marcinek P , Jha AN , Shinde V , Sundaramoorthy A , Rajkumar R , Suryadevara NC , Neela SK , van Tong H , Balachander V , Valluri VL , Thangaraj K , Velavan TP ((2013) ) LRRK2 and RIPK2 variants in the NOD 2-mediated signaling pathway are associated with susceptibility to Mycobacterium leprae in Indian populations. PLoS One 8: , e73103. |
[28] | Trinh J , Guella I , Farrer MJ ((2014) ) Disease penetrance of late-onset parkinsonism: A meta-analysis. JAMA Neurol 71: , 1535–1539. |
[29] | Postuma RB , Berg D , Stern M , Poewe W , Olanow CW , Oertel W , Obeso J , Marek K , Litvan I , Lang AE , Halliday G , Goetz CG , Gasser T , Dubois B , Chan P , Bloem BR , Adler CH , Deuschl G ((2015) ) MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 30: , 1591–1601. |
[30] | Hoehn MM , Yahr MD ((1967) ) Parkinsonism: Onset, progression and mortality. Neurology 17: , 427–442. |
[31] | Beutler E , Nguyen NJ , Henneberger MW , Smolec JM , McPherson RA , West C , Gelbart T ((1993) ) Gaucher disease: Gene frequencies in the Ashkenazi Jewish population. Am J Hum Genet 52: , 85–88. |
[32] | Goetz CG , Tilley BC , Shaftman SR , Stebbins GT , Fahn S , Martinez-Martin P , Poewe W , Sampaio C , Stern MB , Dodel R , Dubois B , Holloway R , Jankovic J , Kulisevsky J , Lang AE , Lees A , Leurgans S , LeWitt PA , Nyenhuis D , Olanow CW , Rascol O , Schrag A , Teresi JA , van Hilten JJ , LaPelle N , Movement Disorder Society UPDRS Revision Task Force ((2008) ) Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Scale presentation and clinimetric testing results. Mov Disord 23: , 2129–2170. |
[33] | Nasreddine ZS , Phillips NA , Bedirian V , Charbonneau S , Whitehead V , Collin I , Cummings JL , Chertkow H ((2005) ) The Montreal Cognitive Assessment, MoCA: A brief screening tool for mild cognitive impairment. J Am Geriatr Soc 53: , 695–699. |
[34] | Levin BE , Llabre MM , Weiner WJ ((1988) ) Parkinson’s disease and depression: Psychometric properties of the Beck Depression Inventory. J Neurol Neurosurg Psychiatry 51: , 1401–1404. |
[35] | Storch A , Schneider CB , Klingelhofer L , Odin P , Fuchs G , Jost WH , Martinez-Martin P , Koch R , Reichmann H , Chaudhuri KR , NoMoFlu-PD study group; Ebersbach G ((2015) ) Quantitative assessment of non-motor fluctuations in Parkinson’s disease using the Non-Motor Symptoms Scale (NMSS). J Neural Transm (Vienna) 122: , 1673–1684. |
[36] | Visser M , Marinus J , Stiggelbout AM , Van Hilten JJ ((2004) ) Assessment of autonomic dysfunction in Parkinson’s disease: The SCOPA-AUT. Mov Disord 19: , 1306–1312. |
[37] | Nomura T , Inoue Y , Kagimura T , Uemura Y , Nakashima K ((2011) ) Utility of the REM sleep behavior disorder screening questionnaire (RBDSQ) in Parkinson’s disease patients. Sleep Med 12: , 711–713. |
[38] | Doty RL , Frye RE , Agrawal U ((1989) ) Internal consistency reliability of the fractionated and whole University of Pennsylvania Smell Identification Test. Percept Psychophys 45: , 381–384. |
[39] | Heinzel S , Berg D , Gasser T , Chen H , Yao C , Postuma RB , MDS Task Force on the Definition of Parkinson’s Disease ((2019) ) Update of the MDS research criteria for prodromal Parkinson’s disease. Mov Disord 34: , 1464–1470. |
[40] | Tomlinson CL , Stowe R , Patel S , Rick C , Gray R , Clarke CE ((2010) ) Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov Disord 25: , 2649–2653. |
[41] | Droby A , Omer N , Gurevich T , Kestenbaum M , Mina Y , Cedarbaum JM , Aizenstein O , Giladi N , Mirelman A , Thaler A ((2020) ) Low cerebrospinal fluid volume and the risk for post-lumbar puncture headaches. J Neurol Sci 417: , 117059. |
[42] | Ransohoff RM , Brown MA ((2012) ) Innate immunity in the central nervous system. J Clin Invest 122: , 1164–1171. |
[43] | Villaran RF , Espinosa-Oliva AM , Sarmiento M , De Pablos RM , Arguelles S , Delgado-Cortes MJ , Sobrino V , Van Rooijen N , Venero JL , Herrera AJ , Cano J , Machado A ((2010) ) Ulcerative colitis exacerbates lipopolysaccharide-induced damage to the nigral dopaminergic system: Potential risk factor in Parkinson’s disease. J Neurochem 114: , 1687–1700. |
[44] | Lim S , Chun Y , Lee JS , Lee SJ ((2016) ) Neuroinflammation in synucleinopathies. Brain Pathol 26: , 404–409. |
[45] | Stolzenberg E , Berry D , Yang , Lee EY , Kroemer A , Kaufman S , Wong GCL , Oppenheim JJ , Sen S , Fishbein T , Bax A , Harris B , Barbut D , Zasloff MA ((2017) ) A role for neuronal alpha-synuclein in gastrointestinal immunity. J Innate Immun 9: , 456–463. |
[46] | Allen Reish HE , Standaert DG ((2015) ) Role of alpha-synuclein in inducing innate and adaptive immunity in Parkinson disease. J Parkinsons Dis 5: , 1–19. |
[47] | Wijeyekoon RS , Moore SF , Farrell K , Breen DP , Barker RA , Williams-Gray CH ((2020) ) Cerebrospinal fluid cytokines and neurodegeneration-associated proteins in Parkinson’s disease. Mov Disord 35: , 1062–1066. |
[48] | Garcia-Esparcia P , Llorens F , Carmona M , Ferrer I ((2014) ) Complex deregulation and expression of cytokines and mediators of the immune response in Parkinson’s disease brain is region dependent. Brain Pathol 24: , 584–598. |
[49] | Luis-Rodriguez D , Donate-Correa J , Martin-Nunez E , Ferri C , Tagua VG , Perez Castro A , Mora-Fernandez C , Navarro-Gonzalez JF ((2021) ) Serum urate is related to subclinical inflammation in asymptomatic hyperuricaemia. Rheumatology (Oxford) 60: , 371–379. |
[50] | Raj DS , Dominic EA , Pai A , Osman F , Morgan M , Pickett G , Shah VO , Ferrando A , Moseley P ((2005) ) Skeletal muscle, cytokines, and oxidative stress in end-stage renal disease. Kidney Int 68: , 2338–2344. |
[51] | Pandey MK , Jabre NA , Xu YH , Zhang W , Setchell KD , Grabowski GA ((2014) ) Gaucher disease: Chemotactic factors and immunological cell invasion in a mouse model. Mol Genet Metab 111: , 163–171. |
[52] | Michelakakis H , Spanou C , Kondyli A , Dimitriou E , Van Weely S , Hollak CE , Van Oers MH , Aerts JM ((1996) ) Plasma tumor necrosis factor-a (TNF-a) levels in Gaucher disease. Biochim Biophys Acta 1317: , 219–222. |
[53] | Allen MJ , Myer BJ , Khokher AM , Rushton N , Cox TM ((1997) ) Pro-inflammatory cytokines and the pathogenesis of Gaucher’s disease: Increased release of interleukin-6 and interleukin-10. QJM 90: , 19–25. |
[54] | Ginns EI , Mak SK , Ko N , Karlgren J , Akbarian S , Chou VP , Guo Y , Lim A , Samuelsson S , LaMarca ML , Vazquez-DeRose J , Manning-Bog AB ((2014) ) Neuroinflammation and alpha-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol Genet Metab 111: , 152–162. |
[55] | Galper J , Balwani M , Fahn S , Waters C , Krohn L , Gan-Or Z , Dzamko N , Alcalay RN (2021) Cytokines and Gaucher biomarkers in glucocerebrosidase carriers with and without Parkinson disease. Mov Disord, doi: 10.1002/mds.28525 |
[56] | Ahmadi Rastegar D , Ho N , Halliday GM , Dzamko N ((2019) ) Parkinson’s progression prediction using machine learning and serum cytokines. NPJ Parkinsons Dis 5: , 14. |
[57] | Brockmann K , Apel A , Schulte C , Schneiderhan-Marra N , Pont-Sunyer C , Vilas D , Ruiz-Martinez J , Langkamp M , Corvol JC , Cormier F , Knorpp T , Joos TO , Gasser T , Schule B , Aasly JO , Foroud T , Marti-Masso JF , Brice A , Tolosa E , Marras C , Berg D , Maetzler W ((2016) ) Inflammatory profile in LRRK2-associated prodromal and clinical PD. J Neuroinflammation 13: , 122. |
[58] | Majbour NK , Aasly JO , Hustad E , Thomas MA , Vaikath NN , Elkum N , van de Berg WDJ , Tokuda T , Mollenhauer B , Berendse HW , El-Agnaf OMA ((2020) ) CSF total and oligomeric alpha-Synuclein along with TNF-alpha as risk biomarkers for Parkinson’s disease: A study in LRRK2 mutation carriers. Transl Neurodegener 9: , 15. |
[59] | Peter I , Dubinsky M , Bressman S , Park A , Lu C , Chen N , Wang A ((2018) ) Anti-tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol 75: , 939–946. |
[60] | Nissen SK , Ferreira SA , Nielsen MC , Schulte C , Shrivastava K , Hennig D , Etzerodt A , Graversen JH , Berg D , Maetzler W , Panhelainen A , Moller HJ , Brockmann K , Romero-Ramos M ((2021) ) Soluble CD163 changes indicate monocyte association with cognitive deficits in Parkinson’s disease. Mov Disord 36: , 963–976. |


