
Evading the Immune System: Immune Modulation and Immune Matching in Cell Replacement Therapies for Parkinson’s Disease
Abstract
Stem cell-based therapies for Parkinson’s disease are now being applied clinically. Notably, studies have shown that controlling the graft-induced immune response improves the results. In this mini-review, we concisely summarize current approaches used for this control. We focus on four modes of stem cell-based therapies: autologous transplantation, allogeneic transplantation with human leukocyte antigen-matching and allogeneic transplantation without, and finally the application of “universal” pluripotent stem cells. We also discuss immuno-suppressive treatments and the monitoring of immune reactions in the brain.
INTRODUCTION
Clinical cell therapies for Parkinson’s disease (PD) using dopamine progenitors derived from pluripotent stem cells (PSCs), such as embryonic stem cells (ESCs) [1] and induced pluripotent stem cells (iPSCs) [2–4], have begun. Cell therapies like these in the central nervous system (CNS) are less likely to cause an immune rejection than in other organs. Indeed, one double-blind clinical trial showed that allografts of human embryonic dopamine neurons are not rejected following transplant even without immune suppression [5]. However, although the brain has been considered a site of immune privilege, this concept is being revisited [6]. Consistently, past clinical experience of cell therapies using aborted fetal tissues has shown that the use of immunosuppressants improves the results in Huntington’s disease [7]. Based on studies of organ and bone marrow transplantations, graft and host human leukocyte antigens (HLA), especially HLA-A, -B, and -DR, determine the activity of host cytotoxic T cells and B cells, which can influence graft rejection and survival.
Because the availability of fetal tissue is limited, cell therapies usually use a mixture of several fetuses as donor cells, making matching or even typing HLA haplotypes difficult. Stem cell-based transplantations may solve this problem.
PSC-BASED THERAPIES
Autologous transplantation
iPSC technology enables autologous transplanta-tion, which theoretically prevents an immune resp-onse or rejection (see Fig. 1, Autologous). In the brain of non-human primates, autologous transplantations using iPSCs cause a minimal immune response [8]. To date, one group has reported an autologous iPSC-based therapy for PD with good results, but only on one patient [2], and only one other group has performed an autologous iPSC-based therapy, which was for age-related macular degeneration [9]. The reason is that although iPSC-based autologous transplantation is ideal from an immunological point of view, it currently suffers from high cost. Furthermore, the large scalability of iPSC lines and their subsequent derivatives under good manufacturing practice (GMP) is still not practical.
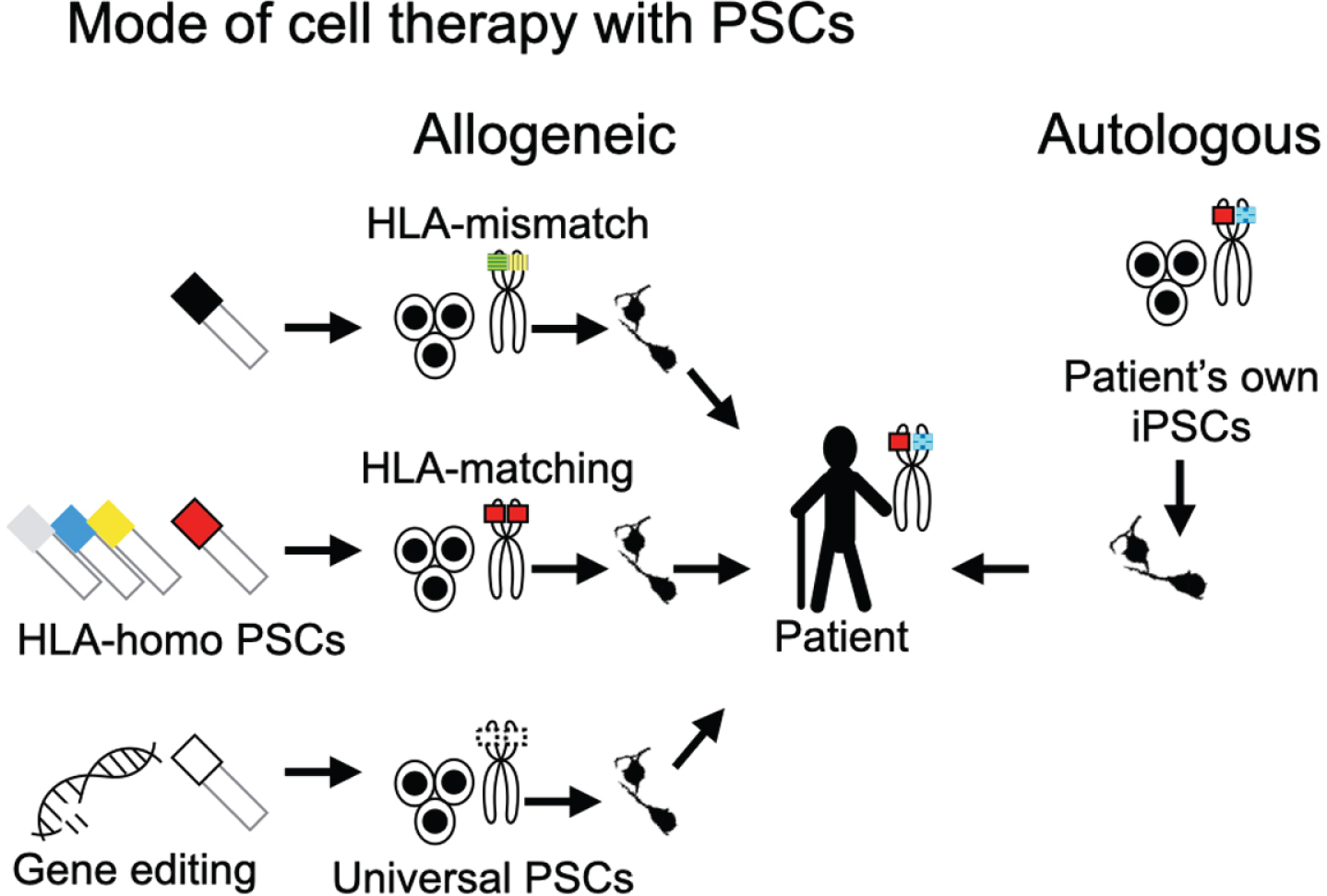
Fig. 1
Modes of PSC-based cell therapies. Cell therapies using PSCs can be divided into two types from the perspective of the donor origin: allogeneic and autologous transplantations. Allogeneic transplantations are subdivided into HLA-mismatch transplantation, HLA-matching transplantation, and universal PSC-based transplantation. The colors and the patterns in the schema of the chromosomes indicate the haplotypes of HLA. For HLA-matching transplantation, cell lines with HLA-homologous haplotypes (HLA-homo PSCs) are normally used to increase the probability of matching. For clinical application, different HLA-homo PSC lines must be banked. Universal PSCs promise transplantations without inducing an immune response to any recipient. They can be derived by editing genes for T-cell responses (e.g., HLA-A, -B, -DR, PD-L1, CTLA4) and NK cells (e.g., HLA-C, -E, -G).
Allogeneic transplantation (HLA-mismatch)
Current stem cell banks and stocks permit allogeneic transplantations but not autologous ones. In cases using healthy donor ESCs or iPSCs of which the HLAs do not match the host, immunosuppressants are recommended [1] (see Fig. 1, HLA-mismatch). This strategy, HLA-mismatch allograft with immune suppression, is practical for the CNS, because the donor cells from healthy volunteers are readily available and well characterized. Accordingly, clinical trials for PD using this strategy are ongoing [1, 4]. As for other organs, such as the heart, which are more susceptible to immune rejection [10], HLA-matching or other modes should be considered.
HLA-matching
Stem cell banks and stocks also make HLA-mat-ched transplantation feasible. To maximize the number of patients who will benefit from HLA-matched stem cell products, donors with homozygous HLA (HLA-homo), i.e., the paternal and maternal alleles have the same HLA haplotypes (see Fig. 1, HLA-matching), increase the probability of HLA-matching for a cell therapy. Regarding CNS cell therapies, because the expression of HLA-class II on neural cells is minimal, it may not be necessary to consider HLA-DR. HLA-homo donors can be found by cooperating with existing cell banks. For example, in Japan, the Japan Red Cross, Japan Marrow Donor Program, and several Japanese cord blood banks are assisting in donor recruitment for an iPSC stock [11]. It is estimated that 1, 10, 75, and 140 HLA-homo iPSC lines would match approximately 17%, 50%, 80%, and 90%of the Japanese population, respectively [12]. In countries with more diverse HLA haplotypes, however, these percentages are smaller.
Nevertheless, the benefits are controversial. Major histocompatibility complex (MHC)-matched allogeneic grafts in a non-human primate PD model led to better survival but were rejected in a non-human primate Huntington’s disease model [13, 14]. One possible reason for the rejection was an immune response via minor antigens or NK cells through killer cell immunoglobulin-like receptor (KIR) signaling [15]. Thus, even HLA-matched grafts should be combined with immunosuppressants. For the heart, all MHC-matched allografts in a non-human primate myocardial infarction model survived when combined with methylprednisolone and tacrolimus [10].
Universal PSCs with genome editing
Genome editing enables the generation of PSC lines that do not express HLA molecules, allowing the PSC derivatives to evade the host’s immune response (see Fig. 1, Gene editing). One example is knocking out the beta 2 microglobulin (B2M) gene, which prevents the expression of HLA-class I molecules and allows the cells to evade T cell imm-unity. The same cells, however, may still be attacked by NK cells, because HLA molecules function as ligands for NK cell inhibitory receptors, such as KIR and NKG2A. Many subsets of NK cells express different combinations of these receptors, complicating the immune suppression. For example, although HLA-E is a ligand of NKG2A receptor, its overexpression did not inhibit KIR2D-positive NK cells [16], which are inhibited by HLA-C or HLA-G. Accordingly, to protect transplanted cells from T cell and NK cell responses, researchers have knocked out HLA-A, -B, -C genes while overexpressing HLA-G gene [17]. In another approach, HLA-A, -B and HLA-class II genes were knocked out by CRISPR-Cas9 technology, but HLA-E, -F, and -G genes were maintained [18]. More study of these cells is needed before clinical application but hampered by limitations of humanized mice models and the cost of non-human primate models. Moreover, because MHC-class I contributes to brain development and plasticity, such as neurite outgrowth, the establishment and function of cortical connections, activity-dependent refinement in the visual system, and long-term and homeostatic plasticity [19], any off-target effects by universal cells need to also be examined. In addition to HLA editing, “immune cloaking” may evade rejection. This strategy mimics how cancer cells escape immune rejection and introduces genes that encode key immunomodulatory molecules, such as the checkpoint molecules programmed cell death 1 ligand 1 (PD-L1) and cytotoxic T lymphocyte antigen 4 (CTLA4) [20].
IMMUNE SUPPRESSION
In current protocols for immune suppression in organ and bone marrow transplantations, a triple medication strategy is generally used; namely, tacrolimus or cyclosporine, mycophenolate mofetil (MMF), and a glucocorticoid. Tacrolimus and cyclosporine are calcineurin inhibitors that can suppress cytotoxic T-cell activation and the secretion of various cytokines including IL-2 and IL-3. Many of their side effects are dose-dependent and include nephrotoxicity, neurotoxicity (leukoencephalopathy), disturbances in glucose metabolism, gastrointestinal disturbance and hypertension. MMF is a prodrug of mycophenolic acid (MPA) and inhibitor of inosine monophosphate dehydrogenase (IMPDH). It preferentially inhibits the de novo pathway of guanosine nucleotide synthesis in T and B lymphocytes, which prevents their proliferation and suppresses both cell-mediated and humoral immune responses. The precise mechanism of action of glucocorticoids is not fully understood. However, the mechanism seems multifaceted, involving both direct and indirect mechanisms and affecting proximal and distal events of T cell activation.
In previous open label trials of fetal transplantation for PD, several embryos with different HLA types were transplanted at the same time. In many cases, triple medication protocols were used and withdrawn 2-3 years post-operatively without any graft rejection [21]. A milder immune suppression might be enough for stem cell-based transplantations in the brain, because 1) an immune response is less likely to occur in the brain than in other organs, 2) the population of donor cells is not mixed with the population causing graft versus host-disease (GVHD) such as lymphocytes, and 3) normally one stem cell line is sufficient to prepare the donor cells [1, 4].
Other side effects when performing immune suppression include infection, cancer, renal dysfunction, and leukoencephalopathy. Impaired renal function may limit the surgical indications of a cell therapy due to the risk of renal failure during the immune suppression. During a pandemic, such as COVID-19, it is necessary to consider the increased risk of infection, the reactivation of latent virus, and the failure of vaccination due to immune suppression, which could postpone the surgery until after the pandemic has subsided. We also have to take it into consideration that immune suppression might affect the disease status.
MONITORING IMMUNE REACTIONS
In liver transplantation, liver function markers obtained from blood examinations are used to mon-itor the immune response and as indices of graft rejection. In kidney transplantation, histological mo-nitoring is performed by needle biopsy. In PD, however, it is difficult to monitor the immune response in the brain after the cell therapy. At present, a combination of neurological evaluation of motor functions and diagnostic imaging (functional imaging of dopamine metabolism and inflammation) is used. Animal studies have shown the possibility of monitoring graft-derived immune responses by positron emission tomography (PET) [8, 13]. PET scans with a translocator protein (TSPO) tracer are used clinically to detect inflammation in the brain [22]. TSPO is a protein that accumulates on the outer membrane of mitochondria in activated microglia and is capable of capturing the immunoinflammatory response of the brain. Other PET biomarkers under development include P2X ligand-gated ion channel type 7 (P2X7R), cannabinoid receptor 2 (CB2R), cyclooxygenase (COX) enzymes, colony-stimulating factor 1 receptor (CSF1R), purinergic metabotropic 12 receptor (P2Y12R), triggering receptor expressed on myeloid cell 1 (TREM1), cluster differentiation factor (CD)19, and CD20 [23]. Clinical responses to graft-induced immune responses in the brain include increasing the dose of the immunosuppressants or adding other drugs. However, PET studies are expensive and not practical to perform on all patients at multiple time points. Therefore, cheaper and easier monitoring of the immune response, such as blood tests, is desired. One group showed the benefits of blood tests to detect donor specific antibodies and also of lymphocytes-grafts immune reaction (LGIR) tests, in which the proliferation of the patient’s blood cells is estimated in a co-culture with donor cells in vitro after the cell therapy [24–26]. Donor-derived cell-free DNA (dd-cfDNA) in the plasma is another potential marker of allogeneic graft injury [27, 28]. Currently, however, the amount of cfDNA in a cell therapy for PD that marks the rejection state has not been established.
COMPARISONS OF CELL THERAPY STRATEGIES
In principle, an autologous transplantation only requires one cell line be characterized extensively and confirmed for efficacy and safety. However, establishing a cell line that is optimized for the cell therapy is not trivial and takes some time, during which the patient’s condition may worsen. As for allogeneic HLA-mismatch transplantations, one cell line is also enough, because the protocol has been optimized beforehand for that line. The cell lines and protocols used in the HLA-matched strategy have also been optimized beforehand, but multiple lines are required. This adds to cost, but is still cheaper than the autologous strategy, since one line can serve many patients (as much as 17%of a population, as stated above). Ultimately, off-the-shelf products made from universal PSCs are expected to provide all the benefits of the above three strategies (see Table 1).
Table 1
Modes of PSC-based cell therapies
| allogeneic HLA-mismatch | allogeneic HLA-match | autologous | universal PSCs | |
| risk of immune rejection | ++ | + | - | - (?) |
| NK cell activation | + | ++ | - | - (?) |
| immune suppression | necessary | recommended | unnecessary | unnecessary (?) |
| off-the-shelf | yes | yes | no | yes |
| relative cell production cost per patient | low (but high immune suppression costs) | middle | high | low |
| widely usable | yes, but immune suppression limits patient eligibility | depends on population | no | yes |
| clinical application for Parkinson’s disease | ref. [4] | ref. [4] | ref. [2] | |
| ref. [1] |
(?) indicates more research is needed.
RISKS
All the above stem cell therapies for PD have surgical risks, such as bleeding, donor cell-related risks, such as graft induced dyskinesia and tumorigenicity, and risks related to host immune responses, such as graft dysfunction and rejection. Immunosuppressants risk susceptibility to infection and cancer generation and also drug toxicity. It should be remembered that the immune rejection of a PD cell therapy is not always lethal, which is unlike bone marrow and organ transplantation rejections. Therefore, sometimes the discontinuation of the immune suppressive therapy is advised even if it results in graft rejection.
CONCLUSION
Compared with fetal tissues, stem cell technologies are providing cells that have more predictable effects and survival in a cell therapy, leading to more uniform clinical results. Currently, HLA-matching and immunosuppressants to control the immune response are recommended to improve neural graft survival [1, 4, 8, 13]. At the same time, sometimes mismatched allogeneic grafts survive without immunosuppressants [5, 8, 13], demonstrating that protocols to control the immune response are to be decided per patient. Ultimately, because they are expected to com-bine the benefits of autologous and allogeneic strategies, universal PSCs are being pursued.
ACKNOWLEDGMENTS
We thank all lab members and collaborators, esp-ecially Dr. Ryosuke Takahashi, Dr. Nobukatsu Saw-amoto (Neurology, Kyoto University), Dr. Takero Shindo (Hematology/Oncology, Kyoto University), and Dr. Takayuki Anazawa (Surgery, Kyoto University). We also thank Dr. Peter Karagiannis (CiRA, Kyoto University) for critical reading of the manuscript. The authors have received funding from the Highway Project for Realization of Regenerative Medicine from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), the Research Center Network for Realization of Regenerative Medicine from the Japan Agency for Medical Research and Development (AMED), and the Research Project for Practical Applications of Regenerative Medicine from AMED.
CONFLICT OF INTEREST
Jun Takahashi receives a grant for collaborative research by Sumitomo Dainippon Pharma Co., Ltd. Asuka Morizane has no conflict of interest to report.
REFERENCES
[1] | Piao J , Zabierowski S , Dubose BN , Hill EJ , Navare M , Claros N , Rosen S , Ramnarine K , Horn C , Fredrickson C , Wong K , Safford B , Kriks S , El Maarouf A , Rutishauser U , Henchcliffe C , Wang Y , Riviere I , Mann S , Bermudez V , Irion S , Studer L , Tomishima M , Tabar V ((2021) ) Preclinical efficacy and safety of a human embryonic stem cell-derived midbrain dopamine progenitor product, MSK-DA01. Cell Stem Cell 28: , 217–229.e7. |
[2] | Schweitzer JS , Song B , Herrington TM , Park TY , Lee N , Ko S , Jeon J , Cha Y , Kim K , Li Q , Henchcliffe C , Kaplitt M , Neff C , Rapalino O , Seo H , Lee IH , Kim J , Kim T , Petsko GA , Ritz J , Cohen BM , Kong SW , Leblanc P , Carter BS , Kim KS ((2020) ) Personalized iPSC-derived dopamine progenitor cells for Parkinson’s disease. N Engl J Med 382: , 1926–1932. |
[3] | Doi D , Magotani H , Kikuchi T , Ikeda M , Hiramatsu S , Yoshida K , Amano N , Nomura M , Umekage M , Morizane A , Takahashi J ((2020) ) Pre-clinical study of induced pluripotent stem cell-derived dopaminergic progenitor cells for Parkinson’s disease. Nat Commun 11: , 3369. |
[4] | Takahashi J ((2020) ) iPS cell-based therapy for Parkinson’s disease: A Kyoto trial. Regen Ther 13: , 18–22. |
[5] | Freed CR , Greene PE , Breeze RE , Tsai WY , DuMouchel W , Kao R , Dillon S , Winfield H , Culver S , Trojanowski JQ ((2001) ) Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N Engl J Med 344: , 710–719. |
[6] | Louveau A , Smirnov I , Keyes TJ , Eccles JD , Rouhani SJ , Peske JD , Derecki NC , Castle D , Mandell JW , Lee KS , Harris TH , Kipnis J ((2015) ) Structural and functional features of central nervous system lymphatic vessels. Nature 523: , 337–341. |
[7] | Krystkowiak P , Gaura V , Labalette M , Rialland A , Remy P , Peschanski M , Bachoud-Lévi A-C ((2007) ) Alloimmunisation to donor antigens and immune rejection following foetal neural grafts to the brain in patients with Huntington’s disease. PLoS One 2: , e166. |
[8] | Morizane A , Doi D , Kikuchi T , Okita K , Hotta A , Kawasaki T , Hayashi T , Onoe H , Shiina T , Yamanaka S , Takahashi J . ((2013) ) Direct comparison of autologous and allogeneic transplantation of iPSC-derived neural cells in the brain of a nonhuman primate. Stem Cell Reports 1: , 283–292. |
[9] | Mandai M , Watanabe A , Kurimoto Y , Hirami Y , Morinaga C , Daimon T , Fujihara M , Akimaru H , Sakai N , Shibata Y , Terada M , Nomiya Y , Tanishima S , Nakamura M , Kamao H , Sugita S , Onishi A , Ito T , Fujita K , Kawamata S , Go MJ , Shinohara C , Hata K-I , Sawada M , Yamamoto M , Ohta S , Ohara Y , Yoshida K , Kuwahara J , Kitano Y , Amano N , Umekage M , Kitaoka F , Tanaka A , Okada C , Takasu N , Ogawa S , Yamanaka S , Takahashi M ((2017) ) Autologous induced stem-cell-derived retinal cells for macular degeneration. N Engl J Med 376: , 1038–1046. |
[10] | Shiba Y , Gomibuchi T , Seto T , Wada Y , Ichimura H , Tanaka Y , Ogasawara T , Okada K , Shiba N , Sakamoto K , Ido D , Shiina T , Ohkura M , Nakai J , Uno N , Kazuki Y , Oshimura M , Minami I , Ikeda U ((2016) ) Allogeneic transplantation of iPS cell-derived cardiomyocytes regenerates primate hearts. Nature 538: , 388–391. |
[11] | Umekage M , Sato Y , Takasu N ((2019) ) Overview: An iPS cell stock at CiRA. Inflamm Regen 39: , 17. |
[12] | Okita K , Matsumura Y , Sato Y , Okada A , Morizane A , Okamoto S , Hong H , Nakagawa M , Tanabe K , Tezuka K-I , Shibata T , Kunisada T , Takahashi M , Takahashi J , Saji H , Yamanaka S ((2011) ) A more efficient method to generate integration-free human iPS cells. Nat Methods 8: , 409–412. |
[13] | Morizane A , Kikuchi T , Hayashi T , Mizuma H , Takara S . ((2011) ) MHC matching improves engraftment of iPSC-derived neurons in non-human primates. Nat Commun 8: , 385. |
[14] | Aron Badin R , Bugi A , Williams S , Vadori M , Michael M , Jan C , Nassi A , Lecourtois S , Blancher A , Cozzi E , Hantraye P , Perrier AL ((2019) ) MHC matching fails to prevent long-term rejection of iPSC-derived neurons in non-human primates. Nat Commun 10: , 4357. |
[15] | Ichise H , Nagano S , Maeda T , Miyazaki M , Miyazaki Y , Kojima H , Yawata N , Yawata M , Tanaka H , Saji H , Masuda K , Kawamoto H ((2017) ) NK cell alloreactivity against KIR-ligand-mismatched HLA-haploidentical tissue derived from HLA haplotype-homozygous iPSCs. Stem Cell Reports 9: , 853–867. |
[16] | Gornalusse GG , Hirata RK , Funk SE , Riolobos L , Lopes VS , Manske G , Prunkard D , Colunga AG , Hanafi LA , Clegg DO , Turtle C , Russell DW ((2017) ) HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol 35: , 765–772. |
[17] | Han X , Wang M , Duan S , Franco PJ , Kenty JH , Hedrick P , Xia Y , Allen A , Ferreira LMR , Strominger JL , Melton DA , Meissner TB , Cowan CA ((2019) ) Generation of hypoimmunogenic human pluripotent stem cells. Proc Natl Acad Sci U S A 116: , 10441–10446. |
[18] | Xu H , Wang B , Ono M , Kagita A , Fujii K , Sasakawa N , Ueda T , Gee P , Nishikawa M , Nomura M , Kitaoka F , Takahashi T , Okita K , Yoshida Y , Kaneko S , Hotta A ((2019) ) Targeted disruption of HLA genes via CRISPR-Cas9 generates iPSCs with enhanced immune compatibility. Cell Stem Cell 24: , 566–578.e. |
[19] | Elmer BM , McAllister AK ((2012) ) Major histocompatibility complex class I proteins in brain development and plasticity. Trends Neurosci 35: , 660–670. |
[20] | Rong Z , Wang M , Hu Z , Stradner M , Zhu S , Kong H , Yi H , Goldrath A , Yang YG , Xu Y , Fu X ((2014) ) An effective approach to prevent immune rejection of human ESC-derived allografts. Cell Stem Cell 14: , 121–130. |
[21] | Piccini P , Pavese N , Hagell P , Reimer J , Björklund A , Oertel WH , Quinn NP , Brooks DJ , Lindvall O ((2005) ) Factors affecting the clinical outcome after neural transplantation in Parkinson’s disease. Brain 128: , 2977–2986. |
[22] | Wickstrøm T , Clarke A , Gausemel I , Horn E , Jørgensen K , Khan I , Mantzilas D , Rajanayagam T , in‘t Veld D-J , Trigg W ((2014) ) The development of an automated and GMP compliant FASTlab™ Synthesis of [(18) F]GE-180; a radiotracer for imaging translocator protein (TSPO). J Labelled Comp Radiopharm 57: , 42–48. |
[23] | Jain P , Chaney AM , Carlson ML , Jackson IM , Rao A , James ML ((2020) ) Neuroinflammation PET imaging: Current opinion and future directions. J Nucl Med 61: , 1107–1112. |
[24] | Sugita S , Makabe K , Fujii S , Iwasaki Y , Kamao H , Shiina T , Ogasawara K , Takahashi M ((2017) ) Detection of retinal pigment epithelium-specific antibody in iPSC-derived retinal pigment epithelium transplantation models. Stem Cell Reports 9: , 1501–1515. |
[25] | Sugita S , Iwasaki Y , Makabe K , Kamao H , Mandai M , Shiina T , Ogasawara K , Hirami Y , Kurimoto Y , Takahashi M ((2016) ) Successful transplantation of retinal pigment epithelial cells from MHC homozygote iPSCs in MHC-matched models. Stem Cell Reports 7: , 635–648. |
[26] | Sugita S , Mandai M , Hirami Y , Takagi S , Maeda T , Fujihara M , Matsuzaki M , Yamamoto M , Iseki K , Hayashi N , Hono A , Fujino S , Koide N , Sakai N , Shibata Y , Terada M , Nishida M , Dohi H , Nomura M , Amano N , Sakaguchi H , Hara C , Maruyama K , Daimon T , Igeta M , Oda T , Shirono U , Tozaki M , Totani K , Sugiyama S , Nishida K , Kurimoto Y , Takahashi M ((2020) ) HLA-matched allogeneic iPS cells-derived RPE transplantation for macular degeneration. J Clin Med 9: , 2217. |
[27] | Snyder TM , Khush KK , Valantine HA , Quake SR ((2011) ) Universal noninvasive detection of solid organ transplant rejection. Proc Natl Acad Sci U S A 108: , 6229–6234. |
[28] | Dengu F ((2020) ) Next-generation sequencing methods to detect donor-derived cell-free DNA after transplantation. Transplant Rev (Orlando) 34: , 100542. |


