
Effects of Alpha-Synuclein Targeted Antisense Oligonucleotides on Lewy Body-Like Pathology and Behavioral Disturbances Induced by Injections of Pre-Formed Fibrils in the Mouse Motor Cortex
Abstract
Background:
Alpha-synuclein (αsyn) characterizes neurodegenerative diseases known as synucleinopathies. The phosphorylated form (psyn) is the primary component of protein aggregates known as Lewy bodies (LBs), which are the hallmark of diseases such as Parkinson’s disease (PD). Synucleinopathies might spread in a prion-like fashion, leading to a progressive emergence of symptoms over time. αsyn pre-formed fibrils (PFFs) induce LB-like pathology in wild-type (WT) mice, but questions remain about their progressive spread and their associated effects on behavioral performance.
Objective:
To characterize the behavioral, cognitive, and pathological long-term effects of LB-like pathology induced after bilateral motor cortex PFF injection in WT mice and to assess the ability of mouse αsyn-targeted antisense oligonucleotides (ASOs) to ameliorate those effects.
Methods:
We induced LB-like pathology in the motor cortex and connected brain regions of male WT mice using PFFs. Three months post-PFF injection (mpi), we assessed behavioral and cognitive performance. We then delivered a targeted ASO via the ventricle and assessed behavioral and cognitive performance 5 weeks later, followed by pathological analysis.
Results:
At 3 and 6 mpi, PFF-injected mice showed mild, progressive behavioral deficits. The ASO reduced total αsyn and psyn protein levels, and LB-like pathology, but was also associated with some deleterious off-target effects not involving lowering of αsyn, such as a decline in body weight and impairments in motor function.
Conclusions:
These results increase understanding of the progressive nature of the PFF model and support the therapeutic potential of ASOs, though more investigation into effects of ASO-mediated reduction in αsyn on brain function is needed.
INTRODUCTION
Alpha-synuclein (αsyn) is a protein expressed throughout the brain, where it is primarily localized to the presynaptic terminal and neuronal nucleus [1, 2]. αsyn can be involved in a variety of cellular functions, including suppressing apoptosis, regulating glucose levels, maintaining SNARE complex structure, and contributing to neuronal differentiation [3–8]. At the synapse, αsyn binds to neurotransmitter-containing vesicles and can promote membrane curvature [9]. αsyn is also important for endo- and exocytosis, and vesicle clustering, which are required for neurotransmission [10–13]. The nuclear function of αsyn is less clear. αsyn associates with poly ADP-ribose, increasing pathological toxicity; inhibits histone acetylation, increasing toxicity; and protects DNA from hydroxyurea-induced stress, decreasing toxicity [14–16]. We have also shown that αsyn and psyn bind to DNA and can regulate forms of DNA repair, and that this function may be interrupted in pathological conditions [17].
αsyn was first purified from cholinergic presynaptic terminals [2] and later connected to neurodegeneration when it was identified as a component of senile plaques in Alzheimer’s disease [18]. Later, tracing of familial Parkinson’s disease (PD) led to the discovery of mutations in the gene encoding αsyn, SNCA [19, 20]. Insoluble, phosphorylated αsyn (psyn) is the primary component of Lewy bodies (LBs) and Lewy neurites, the hallmark inclusions of a class of neurodegenerative diseases known as synucleinopathies [21, 22]. Beyond several specific, but rare SNCA point mutations—such as A53T, A53E, A30P, E46K, H50Q, and G51D—or multiplication mutations in SNCA [19, 20, 23–29], the direct cause of most cases of synucleinopathy is unknown. Recent evidence supports a prion-like mechanism of αsyn aggregation and spread, whereby introduction of exogenous αsyn pre-formed fibrils (PFFs) causes endogenous αsyn to progressively take on an insoluble, aggregated conformation [30–33]. The prion-like mechanism of neurodegeneration was originally put forth by Prusiner and colleagues and has since been deeply explored [34–38]. Use of the PFF model has grown substantially to study sporadic synucleinopathies, and more thorough characterization of this relatively new model is needed.
Synucleinopathies are diagnosed, in part, based on the development of specific kinds of nervous system dysfunction, which likely do not appear until after significant pathological spread has already occurred. For example, PD is diagnosed based on the presence of motor symptoms and signs [39], yet patients retroactively report years of other symptomatic disturbances, such as decreased olfaction [40] and sleep disturbances [41, 42], and often continue to develop other additional, non-motor symptoms, such as impaired cognition. Postmortem assessments also suggest a progressive spread of LBs that could map to the timing of symptom presentation [43]. This likely translates to a delayed recognition of disease until well after pathological aggregation has started and spread.
The mouse motor cortex represents a potentially interesting model for studying PFF-induced LB-like formation and spread, which is why we chose this region for our PFF injections. It is able to generate high levels of LB-like pathology in mice compared to other brain regions after PFF injection into the dorsal striatum [44], likely due a combination of this region’s rich connectivity with the striatum and the high levels of αsyn expression found in motor cortex neurons [45]. High levels of intrinsic αsyn expression are a key determinant of a neuron’s potential to form LBs after PFF injection [46], and make the motor cortex ideal for generating this kind of pathology robustly. In addition to local uptake and seeding of LB formation after PFF injection, brain regions which are monosynaptically connected to the injection site are thought to be the next ones to develop LB pathology [44, 47, 48]. Importantly, the connectivity of the motor cortex has been elucidated in detail, and high resolution maps of the specific inputs into [49] and projections from [50] this region are now known. Interestingly, aged C57Bl/6Nc mice showed an increase in psyn accumulation in the motor cortex, but not the striatum or substantia nigra, compared to young mice [51], and PD patients with cortical myoclonus were found to have higher αsyn levels in the motor cortex than PD patients without myoclonus [52]. In our previous work, the pathological progression of LB and Lewy neurite formation after motor cortex PFF injections was studied in vivo using multiphoton imaging [53], but a corresponding detailed behavioral phenotypic characterization following these injections was not done. For these reasons, we chose to study the progression of motor and cognitive behavioral abnormalities and pathology development following bilateral motor cortex PFF injection in this work, and its response to a potential treatment.
There is currently no treatment to stop the progression of synucleinopathies. Treatments are typically focused on symptom management, such as dopamine replacement therapy and deep-brain stimulation to ameliorate the motor symptoms in PD [54]. These treatments become less effective with time and can have serious side-effects, such as levodopa-induced dyskinesia and impulsive behavior [55, 56]. In addition, these treatments do little to improve non-motor symptoms, highlighting the need to develop alternate therapeutic strategies that have true disease-modifying effects. Antisense oligonucleotides (ASOs), which impair translation of specific mRNAs to decrease target protein levels, might modify PD-related pathology. For example, PFF-injected mice treated with ASOs against leucine-rich repeat kinase 2 (LRRK2) showed reduced inclusion formation [57]. Currently, there are ongoing ASO clinical trials for a range of neurodegenerative diseases [58, 59]. However, more characterization and validation are needed to refine treatment development.
Here, we aimed to: 1) characterize the development and progression of behavioral and cognitive changes following bilateral injections of PFFs in the motor cortex of C57Bl/6J wild-type (WT) mice, and 2) assess if reducing αsyn expression throughout the CNS with a targeted ASO treatment could ameliorate the cognitive, behavioral, and pathological effects of PFF injections.
MATERIALS AND METHODS
Animals
For the full behavioral and cognitive test battery described below, 35 C57BL/6J (WT) males obtained from Jackson Laboratories at 4-5 weeks of age (Bar Harbor, ME, USA) were used. Only males were included due to the higher prevalence of PD and other Lewy-body disorders in men compared to women [60, 61]. The mice were maintained on a 12-h light/dark cycle, with lights turning on at 6:00 and off at 18:00. Food and water were provided ad libitum.
Following three days of habituation to the animal facility, mice underwent intracranial injection to induce LB-like pathology (“PFF”, PFF injections) or as a control (“mono”, monomer injections). Three months post-injection (mpi), animals underwent the first round of behavioral and cognitive testing (Fig. 1). This time was selected because WT mice with induced LB-like pathology typically begin to show early-stage αsyn aggregation in neuritic processes at 2.5–3 mpi, as well as mild behavioral alterations, such as decreased latency to remain on the wire hang [30, 33]. At 4 mpi, mice received a single, 700μg intracerebroventricular (ICV) infusion of either a mouse sequence αsyn-targeted antisense oligonucleotide (“ASO”) or a scrambled control oligonucleotide (“scramble”) into the right lateral ventricle. A second round of behavioral and cognitive testing was performed 5 weeks later to assess the effects of the targeted ASO. During periods of behavioral and cognitive testing, mice were singly housed 72 h before the start of testing; during all other periods, mice were group housed 3–5 to a cage and monitored daily for signs of fighting or distress. Throughout the duration of the experiment the researcher performing the experiment was blinded to groups.
Fig. 1
Timeline of experiments. A) Four-week-old C57Bl/6J male mice were delivered from Jackson Labs and acclimated to our facilities for 3 days. Mice were then injected with either monomeric (Mono) or fibrillized (PFF) α-synuclein bilaterally into the motor cortex (n = 17/group). At 3 months post-injection (mpi), all mice went through a battery of behavioral tests in the following order: rotarod, open field, novel object recognition, wire hang, water maze, and fear conditioning. B) After the first round of behavioral testing, mice received a single injection in the right ventricle with either an α-synuclein-targeting antisense oligonucleotide (ASO) or a scramble oligonucleotide (Scramble) (n = 8-9/group). Mice went through a second round of behavioral testing at 6 mpi (5 weeks following ASO delivery) in the following order: home cage activity monitoring, food intake, rotarod, open field, novel object recognition, wire hang, and water maze. Body weight was recorded weekly from the start of behavioral testing until animals were euthanized. Following completion of the second round of behavior, animals were euthanized and tissue collected for subsequent analysis. Italicized behavioral tests were only performed at one time point.
Following the second round of behavioral testing, mice were euthanized by cervical dislocation and their brains were quickly removed. The right hemisphere was dissected into the hippocampus, cortex, and cerebellum. These tissues were flash-frozen and stored at –80°C. The left hemisphere was post-fixed overnight in 4%paraformaldehyde, and subsequently placed in 30%sucrose.
To confirm the results and assess potential off-target effects associated with the mouse ASO used in this study, an additional 15 male mice were acquired from the Jackson Laboratory at 4-5 weeks of age. This consisted of 5 male C57BL/6N-Sncatm1Mjff/J (“αsyn-KO”), and 10 male C57Bl/6NJ (“NJ”). Half of the NJ mice received the αsyn-targeted ASO, and the other half received the control construct. All αsyn-KO mice received the ASO. None of these mice received PFF- or Mono-injections. Their body weight was tracked for 7 weeks.
All animal procedures were reviewed and approved by the Institutional Animal Care and Use Committee at the Oregon Health and Science University.
Administered agents and surgeries
Administered agents
Monomeric alpha-synuclein and pre-formed fibrils (PFFs). Mono and PFF synuclein were generously supplied by Dr. Kelvin Luk. Solutions for injection were prepared according to previously described protocols [30]. Briefly, monomeric mouse sequence αsyn (“mono”) or PFFs (endotoxin level 0.15 EU/mg protein) were diluted in PBS to 2μg/μl and sonicated prior to injection as previous described [33]. One animal was euthanized following this surgery due to recovery complications.
Antisense oligonucleotides. Mouse sequence αsyn-targeted and scrambled control antisense oligonucleotides (ASOs) were generously supplied by Ionis Pharmaceuticals. αsyn-targeted ASO was matched to the non A4 component of amyloid precursor component (#678363). Mouse αsyn ASO sequence: TTTAATTACTTCCACCA; control ASO sequence: CCTATAGGACTATCCAGGAA. ASOs were diluted in 1x PBS (without Ca2 + or Mg2 +) to 100 mg/ml stock solution. For injections, a working solution of 70 mg/ml was made and stored at 4°C. One animal was euthanized following this surgery due to recovery complications.
Surgeries
Motor cortex injections of mono/PFF. Injections of Mono and PFF αsyn were performed according to our standard protocol [33]. All mice received an injection of 0.05μg/g buprenorphine prior to being given anesthesia. Mice were induced with 5% isoflurane; following induction, mice were maintained at 1.5–2%isoflurane for the duration of surgeries. Body temperature was maintained with a water heat pad, and depth of anesthesia and breathing were monitored throughout the surgeries. Following induction, mice were placed into a custom-made stereotaxic frame and heads secured with fixation bars. Ophthalmic ointment was applied to the eyes to ensure they remained lubricated throughout the procedure. The heads of the mice were shaved, and all mice received a local subcutaneous injection of lidocaine. The heads of the mice were sterilized by alternating 3 swabs of betadine and 2 swabs of 70%isopropanol. An incision was made on the midline of the scalp to expose the skull and holes drilled bilaterally above the motor cortex (Bregma coordinates: AP = –1.0 mm, ML =±1.5 mm). A Hamilton syringe (701-RN, 26s-gauge, ref# 80330) was lowered to DV –0.6 mm, then raised back up to DV –0.3 mm for injection of 2.5μl (2μg/μl) of either monomeric mouse αsyn or PFFs at a rate of 0.2μl/minute. The syringe remained in place for 3 min following infusion and remained out for 3 min before lowering into the opposite hemisphere. The order of injecting into the left and right hemisphere was counterbalanced. Following injections, the scalp was stitched with two to three sutures. Animals were monitored until awake and received 0.05μg/g buprenorphine for the following two days.
Intracerebroventricular infusions of ASO/Control. All ASO/Control surgeries followed the same standard procedures as above. Following placement in the stereotaxic frame, a Hamilton syringe was loaded with 10μL of either ASO or Scramble control (70 mg/ml) and lowered into the right ventricle [AP: –0.3 mm, ML: +1.0 mm, DV: –2.5 mm]. Infusions occurred at a rate of 2μl/min, and the syringe was left in place for 1 min following completion of infusion. Again, animals received 2-3 sutures and buprenorphine for post-operative care.
Behavioral and cognitive assessments
Health measures: body weight, food intake, and circadian activity
Body weights were recorded weekly starting during the first round of behavioral testing until animals were euthanized.
During the second round of behavioral and cognitive testing (6 mpi), food intake was recorded. The food in each cage was weighed twice each day—once in the morning (∼8:00) and once in the evening (∼17:00)—to assess approximate amount of food eaten during daytime hours and nighttime hours.
Additionally, home-cage activity was assessed as previously described [62] over a week-long period following ASO delivery. Mice were singly housed in cages containing infrared sensors, and data were continuously collected from 12:00 PM on a Monday until 12:30 PM on a Friday (MLog, Biobserve, Bonn, Germany). Following collection in 1-second increments, data were compiled into 5 min, 30 min and 12 h bins for analyses of light and dark activity.
Open field
To assess locomotion, spatial learning, and anxiety-like behaviors, mice were placed into an open field (41×41 cm) and allowed to explore for 5 min over three (3 mpi) or two (6 mpi) subsequent days, following a standard protocol [63]. The enclosures were thoroughly cleaned with 0.5%acetic acid and dried between each trial. Light intensity in the enclosures ranged from 300 to 500 lux. Animals were video recorded at a rate of 15 samples per second, and total distance moved, average velocity, and time spent in the center (defined as a center square sized 20×20 cm) was analyzed using Ethovision XT 7.1 software (Noldus Information Technology, Wageningen, Netherlands). Data were analyzed using repeated measures ANOVAs, with PFF status and ASO treatment used as between group variables.
Novel object recognition
Following testing in the open field, two similar objects were secured in place in the center of the same fields [63]. Animals were allowed to explore for 15 min (novel object day 1). The next day, one object was replaced with a distinct, novel object. Again, animals were allowed to explore for 15 min (novel object day 2). Total time exploring the objects was manually recorded for both sessions, as well as time spent with each individual object. The researcher recording exploration time was blinded to the groups. Percent time exploring the novel object was calculated on day 2 as an indicator that animals could distinguish which object they had previously seen. This test was repeated at both 3 mpi and 6 mpi; all the objects used at 6 mpi were distinct from the objects used during the first round of testing. Data were analyzed using repeated measures ANOVAs and two-way ANOVAs followed by Sidak’s post-hoc comparisons when appropriate.
Rotarod
Motor function and endurance were tested using a rotarod (Rotamex, Columbia Instruments, Columbus, OH) as previously reported [63]. Mice were placed onto a rotating rod starting at 5 rotations per minute (rpm). Every 3 s, the speed increased by 1 rpm. The maximum run time was capped at 300 s, or 50 rpm. During the first round of behavioral testing, mice were tested over three consecutive days; on each day, mice received three trials, separated by 3 min each. During the second round of behavioral testing, the mice received a single day of rotarod with three trials separated by 3 min each. The rotarod was cleaned with 0.5%acetic acid between every trial. Latency to fall (in seconds) and final rpm were recorded and analyzed using a repeated measures ANOVA.
Wire hang
Motor function was also assessed using the wire hang task, adopting the “falls and reaches” method described by van Putten 2016 [64]. Mice were placed on a suspended metal wire so that they were hanging only by their front paws. In this method, mice start with a fall score of 10 and a reach score of 0. Over the duration of 180 s, the performance score was reduced by 1 point every time the mouse fell and was increased by 1 point every time they reached one of the poles holding up the wire. The time of each fall or reach event was also recorded. Each time a mouse fell or reached, the timer was paused to replace the mouse in the center of the wire again. The benefit of using this test is that it allows assessments of not only endurance and strength, but also of more complex motor coordination.
Water maze
Spatial learning and memory were assessed using the Morris water maze similar to previously described protocols [65]. A circular pool (140 cm in diameter) was filled with room temperature water (23°C±1) and made opaque with white chalk. The pool was divided into four quadrants (NE, NW, SE, and SW). A circular platform (12 cm in diameter) was placed 2 cm below the surface of the water; in order to complete the maze, mice were required to locate the platform and remain on it for a minimum of three seconds. Task learning was assessed during “visible” platform trials; a 50 mL conical tube weighed down and wrapped in colorful tape was placed as a flag on top of the platform to serve as a visual cue. Spatial learning (acquisition) was assessed during “hidden” platform trials; the conical tube was removed and spatial cues of different shapes and colors were hung on the walls surrounding the pool. Training consisted of two trials per session (separated by 10 min) and two sessions per day (separated by 3 h). Mice were taken out of the pool once they located and remained on the platform for 3 s, or after 60 s had passed without finding the platform. For training purposes, when mice did not find the platform on their own, the researcher guided the animals to the platform and stepped out of view for the animal to associate the platform with escape. Average velocity, cumulative distance to the target platform, and latency to find the platform were assessed as indications of learning.
To assess spatial memory retention, mice were tested in a series of probe trials in which the platform was removed from the pool. Probe trials lasted for a total of 60 s and ended with the researcher picking up the animals from the location where the platform would have been located. The same extra-maze cues used during hidden trials remained on the walls for probes. Cumulative distance from the target, and time spent in the target quadrant as compared to the three non-target quadrants were analyzed as measures of memory retention.
Water maze performance was assessed at both 3 and 6 mpi. At both time points, mice were first trained to locate a “hidden” platform. During the first round of testing, the platform remained in the SW quadrant for sessions 1–10. To assess reversal learning, a prefrontal cortex-dependent function, for sessions 11–14, we moved the platform to a new (the opposite) quadrant (NE). Lastly, we performed a single visible platform session to assess task learning. During the second round of water maze testing, the platform was placed in the SE quadrant for sessions 1–8. On the last day (sessions 9-10), we administered two visible platform sessions with the platform in the NW and SW quadrants, respectively.
Fear conditioning
Mice were tested for contextual and cued fear learning and memory at 3 mpi following a standard protocol [66]. In the fear conditioning test, hippocampus-dependent and -independent learning and memory can be distinguished. Contextual fear conditioning is known to be hippocampus-dependent, whereas cued fear conditioning is known to be hippocampus-independent and amygdala-dependent [67, 68]. Briefly, mice were placed into a sound-attenuating chamber. After a 120 s baseline period without any stimuli, a tone (80 dB, 2800 Hz) was played for 30 s, which co-terminated with a 2 s shock (0.5 mA). This was followed by a 90 s period with no stimuli before the tone-shock pairing was repeated again. In total, mice received 2 tone-shock pairings. The chambers were thoroughly cleaned with 0.5%acetic acid between animals. Twenty-four hours later, mice were tested for contextual and cued fear memory recall. For contextual memory recall, mice were placed into the same chamber for a period of 5 min, with no cues presented. For cued memory recall, the chambers were changed to remove any contextual cues (floors were covered with a solid, white panel, the roof and walls were changed to a black triangle, and 10%isopropanol was used for cleaning). Animals had a baseline period of 90 s with no stimuli; at 90 s, the same tone played for a total of 3 min.
All trials were recorded and analyzed with Video Freeze software (PMED-VFC-NIR-M, Med Associates, Inc., Fairfax, VT) and automatic outputs were generated to analyze average motion and time freezing (defined as a minimum of 30 frames, or 1 s, of total movement cessation). Percent time freezing was analyzed as measure of fear memory and assessed with a 1-way ANOVA between PFF and Mono groups, or with a 1-way repeated measures ANOVA when looking over time within a trial. Because we did not observe any significant differences between PFF and Mono groups at 3 mpi, we did not repeat fear conditioning testing at 6 mpi.
Postmortem analysis
Immunofluorescence and microscopy
LB-like aggregates were analyzed by immunofluorescence. Following post-fixation, the left hemisphere was sectioned using a cryostat at 40μm thickness. The sections were washed, blocked in 4%normal goat serum with 0.3%Triton-X (NGS), and incubated overnight in 4%NGS containing an antibody targeting αsyn phosphorylated at serine 129 (anti-pSer129, rabbit monoclonal, 1:600, Abcam #51253). Subsequently, the sections were incubated with secondary antibody in 4%NGS overnight (goat anti-rabbit IgG, 1:1000, AlexaFluor 594, Lifetechnologies, #A11012). The nuclear counterstain DAPI (Sigma Aldrich, St. Louis, MO, USA) was applied at a 1:200 concentration for 20 min prior to slide mounting. Coverslips were sealed with VectaShield Hard Set Antifade Mounting Media without DAPI (Vector Labs, Burlingame, CA, USA, #H-1400). Whole-hemisphere images were taken using a Zeiss AxioScan.Z1 at 20x magnification (Zeiss, Thornwood, NY, USA). The percent area occupied by immunoreactivity in the motor cortex, medial and lateral somatosensory cortices, and the hippocampus were analyzed using ImageJ Software (NIH).
Western blots
Western blot analysis was used to assess phosphorylated and total protein levels. Dissected hippocampi were dissolved in lysis buffer (1M Tris-Cl, pH 7.5; 6M NaCl; 10%SDS; 0.5M EDTA; Triton-X 100; Phosphatase Inhibitor #3, Roche, #05-892-970-001; Protease Inhibitor, Sigma-Aldrich, #P0044) by homogenizing 3×20 s and sonicating on ice for 15 s at 40 Hz. A BCA kit (Pierce, Thermo Scientific, #23227) was used to determine protein concentrations. Samples were incubated in Novex Tris-glycine SDS Sample Buffer (Invitrogen) at 95°C for 10 min and separated on 10–20%Tris-glycine gels (Invitrogen) for 70 min at 125 V. Proteins were transferred onto Immobilon-FL PVDF membranes (Millipore, #IPF00010) for 2 h at 30 V on ice. When assessing αsyn and psyn protein levels, membranes were immediately fixed in 4%PFA + 0.1%GA for 20–30 min [69, 70]. Total protein measures were assessed using REVERT™ Total Protein Stain (Li-Cor Biosciences, Lincoln, NE). Following image acquisition of total protein, blots were blocked in Odyssey Blocking Buffer (Li-Cor Biosciences) for 1 h at room temperature and incubated overnight at 4°C with primary antibodies against Syn1 (mouse, 1:1000, BD Biosciences, #610786), pSer129 (rabbit, 1:1000, Abcam, #51253), GAPDH (mouse, 1:10000, Millipore, #MAB3740), or phospho-histone H2Ax (rabbit, 1:1000, Cell Signal, #9718). Blots were then incubated in appropriate secondary antibodies (goat anti-rabbit IR800 CW, Li-Cor, #926-32211; goat anti-mouse IR680 LT, Li-Cor, #926-68050) for 2 h at room temperature. Images of hybridized blots were taken using a Li-Cor Odyssey CLx and densomitry analyses were performed using ImageJ software. The levels of the proteins of interest were normalized to the total protein loaded for that particular lane for statistical analyses.
Statistics
All data were first assessed for normality of variance. In all cases, we were able to proceed with standard parametric tests. Statistical analyses were performed using SPSS v.25 (IBM, Armonk, NY) and Prism v.7 (GraphPad, San Diego, CA). Statistical results for all measures are indicated in Table 1.
Table 1
Breakdown of statistics and significant effects and/or interactions for all measures
| Data | Type of Test | Factors | Significant Finding(s) | t or F statistic | p | Corrections Applied |
| Fig. 2D aSyn and pSyn Protein | 2-way ANOVA | PFF Status (PFF/Mono) | Main effect of ASO (asyn) | F(1,29) = 74.33 | p < 0.0001 | Sidak’s |
| ASO Staus (ASO/Scramble) | Main effect of ASO (psyn) | F(1,29) = 35.88 | p < 0.0001 | Sidak’s | ||
| Fig. 2E pSyn Inclusions: Composite Score | Independent Samples t-test | ASO Status (ASO/Scramble) | Main effect of ASO | t(65) = 2.280 | p = 0.0259 | |
| Fig. 2E pSyn Inclusions: Motor Cortex | Independent Samples t-test | ASO Status (ASO/Scramble) | No effects | t(15) = 1.117 | p = 0.281 | |
| Fig. 2E pSyn Inclusions: Hippocampus | Independent Samples t-test | ASO Status (ASO/Scramble) | No effects | t(14) = 1.067 | p = 0.304 | |
| Fig. 2E pSyn Inclusions: Medial Somatosensory Cortex | Independent Samples t-test | ASO Status (ASO/Scramble) | No effects | t(15) = 1.145 | p = 0.270 | |
| Fig. 2E pSyn Inclusions: Lateral Somatosensory Cortex | Independent Samples t-test | ASO Status (ASO/Scramble) | No effects | t(15) = 1.022 | p = 0.23 | |
| Fig. 3B-C Water Maze Cumulative Distance during Hidden Platform Location | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Time x PFF | F(8.472,245.702) = 2.025 | p = 0.041 | Greenhouse-Geisser |
| ASO Status (ASO/Scramble) Time | Main effect of PFF | F(1,29) = 6.638 | p = 0.015 | |||
| Time x ASO | F(8.472,245.702) = 2.626 | p = 0.008 | Greenhouse-Geisser | |||
| Fig. 3B-C Water Maze Cumulative Distance during Visible Platform Location | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Time x ASO | F(7.527,218.278) = 3.705 | p = 0.001 | Greenhouse-Geisser |
| ASO Status (ASO/Scramble) Time | ||||||
| Fig. 3E Water Maze Change in Cumulative Distance to Hidden Platform Locations | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Main effect of PFF | F(1,29) = 4.159 | p = 0.05 | |
| ASO Status (ASO/Scramble) Time | ||||||
| Fig. 3E Water Maze Change in Cumulative Distance to Visible Platform Locations | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Main effect of ASO | F(1,29) = 7.049 | p = 0.012 | |
| ASO Status (ASO/Scramble) Time | ||||||
| Fig. 3F Water MazeCumulative Distance during Probe Trials | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Time x PFF | F(4.283,124.217) = 3.257 | p = 0.005 | Greenhouse-Geisser |
| ASO Status (ASO/Scramble) Time | Main effect of PFF | F(1,29) = 9.372 | p = 0.012 | Greenhouse-Geisser | ||
| Fig. 3G Water Maze Percent Time in Target Quadrant during Probe Trials | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Time x PFF | F(7,203) = 3.353 | p = 0.001 | |
| ASO Status (ASO/Scramble) Time | Main effect of PFF | F(1,29) = 8.043 | p = 0.008 | |||
| Time x ASO | F(7,203) = 2.083 | p = 0.047 | ||||
| Fig. 4B Total Distance Moved in the Open Field | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Time x PFF x ASO | F(1,29) = 5.637 | p = 0.024 | |
| ASO Status (ASO/Scramble) Time | ||||||
| Average Velocity in the Open Field | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Time x PFF x ASO | F(1,29) = 5.533 | p = 0.026 | |
| ASO Status (ASO/Scramble) Time | ||||||
| Fig. 4C Novel Object 3 mpi | Paired Samples t-test | Object (Familiar/Novel) | Mono-Scramble | t(50) = 5.632 | p < 0.0001 | Sidak’s |
| Mono-ASO | t(50) = 4.706 | p < 0.0001 | Sidak’s | |||
| PFF-Scramble | t(50) = 6.796 | p < 0.0001 | Sidak’s | |||
| PFF-ASO | t(50) = 6.713 | p < 0.0001 | Sidak’s | |||
| Fig. 4D Novel Object 6 mpi | Paired Samples t-test | Object (Familiar/Novel) | Mono-Scramble | t(52) = 4.206 | p = 0.0004 | Sidak’s |
| Mono-ASO | t(52) = 1.845 | p = 0.2541 | Sidak’s | |||
| PFF-Scramble | t(52) = 1.945 | p = 0.2099 | Sidak’s | |||
| PFF-ASO | t(52) = 3.462 | p = 0.0043 | Sidak’s | |||
| Fig. 5B Latency to Fall off the Rotarod | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Time x ASO | F(3,87) = 16.145 | p < 0.0001 | |
| ASO Status (ASO/Scramble) Time | ||||||
| Fig. 5C Wire Hang Latency to Fall | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Main effect of ASO | F(1,26) = 21.388 | p < 0.001 | |
| ASO Status (ASO/Scramble) Time | Time x ASO | F(1,26) = 11.936 | p = 0.002 | |||
| PFF x ASO | F(1,26) = 6.030 | p = 0.021 | ||||
| Fig. 5C Wire Hang Fall Score | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Main effect of ASO | F(1,26) = 13.826 | p = 0.001 | |
| ASO Status (ASO/Scramble) Time | Time x ASO | F(1,26) = 12.923 | p = 0.002 | |||
| Trend towards main effect of PFF | F(1,26) = 3.739 | p = 0.064 | ||||
| Fig. 5E Wire Hang Fall Score over Time 3 mpi | Repeated Measures ANOVA | PFF Status (PFF/Mono) | No effects | |||
| ASO Status (ASO/Scramble) Time | ||||||
| Fig. 5F Wire Hang Fall Score over Time 6 mpi | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Main effect of ASO | F(1,26) = 20.089 | p < 0.001 | |
| ASO Status (ASO/Scramble) Time | Time x ASO | F(35,910) = 6.087 | p < 0.001 | |||
| Time x PFF | F(35,910) = 3.409 | p < 0.001 | ||||
| Fig. 5G Wire Hang Latency to Reach | Repeated Measures ANOVA | PFF Status (PFF/Mono) | No effects | |||
| ASO Status (ASO/Scramble) Time | ||||||
| Fig. 5H Wire Hang Reach Score | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Trend towards main effect of PFF | F(1,26) = 3.250 | p = 0.084 | |
| ASO Status (ASO/Scramble) Time | ||||||
| Fig. 5I Wire Hang Reach Score over Time 3 mpi | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Time x PFF | F(35,980) = 3.195 | p < 0.001 | |
| ASO Status (ASO/Scramble) Time | ||||||
| Fig. 5J Wire Hang Reach Score over Time 6 mpi | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Time x ASO | F(35,910) = 5.116 | p < 0.001 | |
| ASO Status (ASO/Scramble) Time | Time x PFF x ASO | F(35,910) = 4.036 | p < 0.001 | |||
| PFF x ASO | F(1,26) = 6.770 | p = 0.015 | ||||
| Trend towards main effect of ASO | F(1,26) = 3.995 | p = 0.056 | ||||
| Fig. 6A Average Daytime Activity | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Time x ASO | F(1.703,34.060) = 3.550 | p = 0.046 | Huynh-Feldt |
| ASO Status (ASO/Scramble) Time | ||||||
| Fig. 6B Average Nighttime Activity | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Trend towards a Time x ASO interaction | F(3,60) = 2.316 | p = 0.085 | |
| ASO Status (ASO/Scramble) Time | ||||||
| Fig. 6C Body Weight | Repeated Measures ANOVA | PFF Status (PFF/Mono) | Time x ASO | F(3.613,104.777) = 16.093 | p < 0.001 | Huynh-Feldt |
| ASO Status (ASO/Scramble) Time | Main effect of ASO | F(1,29) = 7.952 | p < 0.001 | |||
| Fig. 6D Food Intake | 2-way ANOVA | PFF Status (PFF/Mono) | No effects (Daytime) | |||
| ASO Staus (ASO/Scramble) | Main effect of ASO (Nighttime) | F(1,26) = 5.297 | p = 0.0296 | |||
| Fig. 6F Body Weight in aSyn KO mice | Repeated Measures ANOVA | ASO Status (ASO/Scramble) Time | Time x ASO | F(16,88) = 6.079 | p = 0.0001 | |
| Main effect of ASO | F(2,11) = 19.03 | p = 0.0003 |
Upon completion of behavioral and cognitive testing, all behavioral and cognitive measures were analyzed with PFF-status (Mono/PFF) and ASO-treatment (Control/ASO) as between group variables using ANOVAs. Most measures were analyzed with a repeated measures ANOVA (body weight; home cage activity monitoring; food intake; open field activity, velocity, and center duration; time exploring during object recognition; rotarod latency to fall; wire hang latency to fall and reach, and fall and reach scores; water maze average velocity, latency to find the platform, and cumulative distance from the platform; and time freezing during each fear conditioning trial) within and over the two time periods of testing. For analysis of effects of time and time interactions, sphericity was assessed; when violated, Greenhouse-Geisser corrections were applied.
Analysis of LB-like pathology was performed with independent samples t-tests comparing ASO and control in only PFF-injected animals for each brain region. To assess overall pathology load, we normalized psyn signal in each brain region to the PFF-Scramble average of that region. We then combined regions for a composite burden measure to analyze between groups. Comparisons were made with independent samples t-tests between PFF-Scramble and PFF-ASO. Mono-injected animals were not included in statistical analysis, as there was no area occupied in any brain region measured for either ASO group. For western blot analysis, bands of interest were normalized to total protein, and 2-way ANOVAs were used to compare groups.
In all cases, when significance was found with ANOVAs, post-hoc comparisons were made and Sidak’s corrections applied. In analysis of time exploring the novel vs. the familiar object, Sidak’s post-hoc comparison was applied within each group.
RESULTS
Alpha-synuclein-targeted ASO treatment reduces alpha- and phosphorylated-synuclein protein levels and LB-like pathology induced by PFF injections in the motor cortex in mice
To determine the extent to which ASO treatment reduced αsyn and psyn protein levels, we used Western blot analysis of hippocampal tissues. Neural tissue was collected after the second round of behavioral testing, 6 months post-PFF or -Mono injections and 7 weeks after ICV delivery of targeted ASO or Scramble (control) ASO (Fig. 1). Levels of both αsyn and psyn forms of the protein were decreased by greater than 50%in hippocampal tissue by the ASO treatment (Fig. 2A). A two-way ANOVA for αsyn integrated density normalized to total protein revealed a main effect of ASO (p < 0.0001; Fig. 2B) and Sidak’s post hoc comparison showed that ASO treatment decreased αsyn compared to the Scramble controls in both Mono- and PFF-injected animals (Mono: p < 0.0001; PFF: p < 0.0001). Similarly, there was a main effect of ASO on psyn integrated density (p < 0.0001; Fig. 2B), with Sidak’s post hoc testing revealing that ASO-treated animals had lower levels than Scramble-treated animals in both Mono- and PFF-injected groups (Mono: p < 0.01; PFF: p < 0.001). There was no effect of PFF on αsyn (p = 0.239) or psyn (p = 0.864) protein levels, nor any PFF by ASO interactions, suggesting the PFF treatment did not have a detectable effect on synuclein protein levels in the hippocampus (all statistics are presented in Table 1).
Fig. 2
Analysis of α-synuclein (αsyn) and phosphorylated synuclein (psyn) protein levels in the hippocampus and LB-like pathology. A) Representative blot of total protein stain (top), αsyn band at 14.4 kDa (middle) and psy band at 14.4 kDa (bottom). Lanes are indicated with “M” for Mono, “P” for PFF, “A” for ASO, and “S” for scramble. The far right lane (lane 14) is a psyn positive control. B) Left: Quantification of αsyn, normalized to total protein. A two-way ANOVA indicated a main effect of ASO treatment (F(1,29) = 74.33, p < 0.0001) and Sidak’s post-hoc tests indicated αsyn protein was decreased in Mono-ASO mice compared to Mono-Scramble (p < 0.0001) and PFF-ASO compared to PFF-Scramble (p < 0.0001). Right: Quantification of psyn, normalized to total protein levels. A two-way ANOVA indicated a main effect of ASO treatment (F(1,29) = 35.88, p < 0.0001) and Sidak’s post-hoc tests indicated psyn protein was decreased in Mono-ASO mice compared to Mono-Scramble (p < 0.01) and PFF-ASO compared to PFF-Scramble (p < 0.001). Data presented as group averages±SEM. C) Representative images from mice injected with PFF-Scramble (left) and PFF-ASO (right). D) Higher magnification representative images of the motor cortex, hippocampus, medial somatosensory cortex, and lateral somatosensory cortex from the hemispheres in (C). PFF-Scramble is on the top, PFF-ASO is on the bottom. E) Normalized percent area occupied of LB-like aggregates in the motor cortex, hippocampus, lateral somatosensory cortex, and medial somatosensory cortex. Comparison of Scramble vs. ASO revealed an overall decrease in LB-like burden (t(65) = 2.280, p < 0.05), but pathology in individual brain regions was not significantly reduced. *p < 0.05, **p < 0.01, ***p < 0.001.
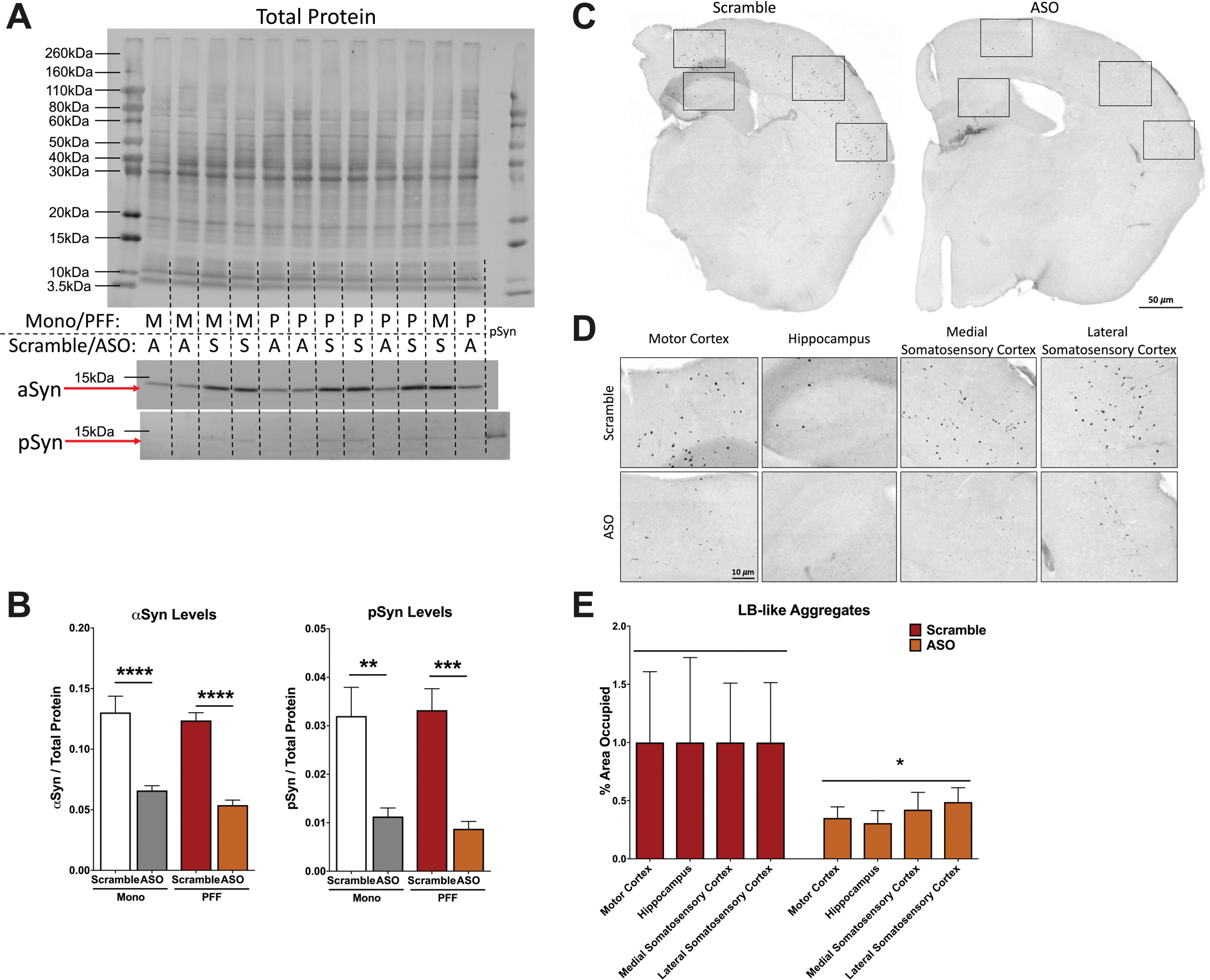
Next, we assessed if ASO treatment reduced LB-like pathology in PFF-injected animals using immunohistochemistry to examine psyn-positive aggregates. We analyzed the percent area occupied by LB-like pathology in four different brain regions (motor cortex, medial somatosensory cortex, lateral somatosensory cortex, and hippocampus) to create a composite pathology burden measure. Using this composite score, we detected a significant reduction in LB-like aggregate load in ASO-treated mice compared to control (p = 0.0259; Fig. 2C-E). However, when we analyzed each individual region separately, we did not observe significant differences between PFF-injected animals treated with ASO or control (Table 1), suggesting that LB-like pathology variability was relatively high.
PFF injections in the motor cortex do not alter fear learning and memory
Prior to delivery of αsyn-targeted ASO or Scramble, we assessed all animals in a battery of behavioral and cognitive tests to determine the effects of PFF injections in the motor cortex on motor abilities, anxiety-like behavior, and learning and memory. This first round of behavioral testing took place 3 mpi, when mature LB-like inclusions have appeared in the PFF group (Fig. 1A) [33]. Following the first round of behavioral tests, the Mono- and PFF-injected groups were split, with half in each group receiving either Scramble or αsyn-targeted ASO injections (n = 8-9/group). Seven weeks after ASO delivery, animals were tested in a second battery of behavioral tests (Fig. 1B).
In the first round of testing (pre-ASO), we assessed fear learning and memory to examine possible hippocampus-dependent and -independent effects. At 3 mpi, we trained mice in a cued fear conditioning paradigm, and assessed their contextual and cued recall 24 h later. We did not detect differences between groups in acquisition of fear, or in contextual or cued recall, indicating no fear memory impairments (Supplementary Figure 1). Because of this, we did not repeat fear conditioning in the second round of behavioral testing.
PFF injections in the motor cortex are associated with hippocampus-dependent spatial learning and memory impairments
During the first round of behavioral testing, we assessed hippocampus-dependent spatial learning and memory using the Morris water maze (WM). For the first 5 days, the platform was located in the SW quadrant with extra-maze cues hung on the wall (“hidden” platform trials); for the subsequent 2 days, the platform was moved to the NE quadrant; and for the final day, we assessed visible platform learning (Fig. 3A).
Fig. 3
Performance in the water maze. A) Schematic of the water maze paradigm used at 3 mpi (left) and 6 mpi (right). Animals were trained to locate a hidden platform over the course of 5 days (2 sessions per day). At 3 mpi, the escape platform was located in the SW Quadrant for days 1-5 and in the NE Quadrant for days 6-7; at 6 mpi, the escape platform was located in the SE Quadrant. On the last day at each time point, a visible cue was placed on the platform. Probe trials were performed in the morning to assess spatial memory. B, C) Cumulative distance to the target platform at 3 mpi (B, pre-ASO) and 6 mpi (C, post-ASO). Mono-injected animals had a lower cumulative distance to the target compared to PFF-injected animals during training for the hidden platforms across both time points (F(1,29) = 6.638, p < 0.05). Additionally, a time-by-PFF interaction suggests that PFF animals had impaired learning over time compared to Mono animals (F(8.472,245.702) = 2.025, p < 0.05), and a time-by-ASO interaction suggests altered learning before and after ASO delivery compared to Scramble-treated animals (F(8.472,245.702) = 2.626, p < 0.01). A time by ASO interaction was found when visible platform location was analyzed, indicating that ASO treatment significantly impaired task performance compared to the prior time point (F(1.521,43.853) = 5.078, p = 0.017). D) Average cumulative distance to the target for the distinct platform locations at 3 and 6 mpi, and average visible cumulative distance to the target. PFF animals had a higher overall cumulative distance to the target during the hidden sessions (F(1,29) = 6.638, p < 0.05) compared to Mono animals. No differences were detected during the visible sessions, and no effects of ASO were detected. E) Change in water maze performance, calculated by subtracting the last session from the first session for a given platform location. PFF mice had a smaller change in water maze performance during the hidden platform locations compared to Mono mice (F(1,29) = 4.159, p < 0.05), indicating impaired learning from the first to last sessions. During the visible sessions, ASO animals showed a negative change that was significantly different than Scramble-treated animals (F(1,29) = 7.049, p < 0.05), indicating worse performance at 6 mpi (post-treatment) compared to 3 mpi (pre-treatment). F) Cumulative distance to the target location during probe trials at 3 mpi and 6 mpi. PFF mice did not show improvement over time, in contrast to the Mono mice (F(4.283,124.217) = 3.257, p < 0.05). Mono-Scramble and Mono-ASO mice swam closer to the target location than PFF-Scramble and PFF-ASO across all probe trials (F(1,29) = 9.372, p < 0.05). G) Percent time spent in the target quadrant during probe trials. PFF mice spent less time in the target quadrants across all probe trials (F(1,29) = 8.043, p = 0.008). Additionally, PFF mice showed a slower improvement in memory, indicated by a time by PFF interaction (F(7,203) = 3.535, p = 0.001). We also found a time by ASO interaction, where Scramble-injected animals continued to improve after ICV delivery, but ASO animals did not (F(7,203) = 2.083, p = 0.047). *p < 0.05, PFF vs. Mono; **p < 0.01, PFF vs. Mono; &p < 0.05, ASO vs. Scramble.
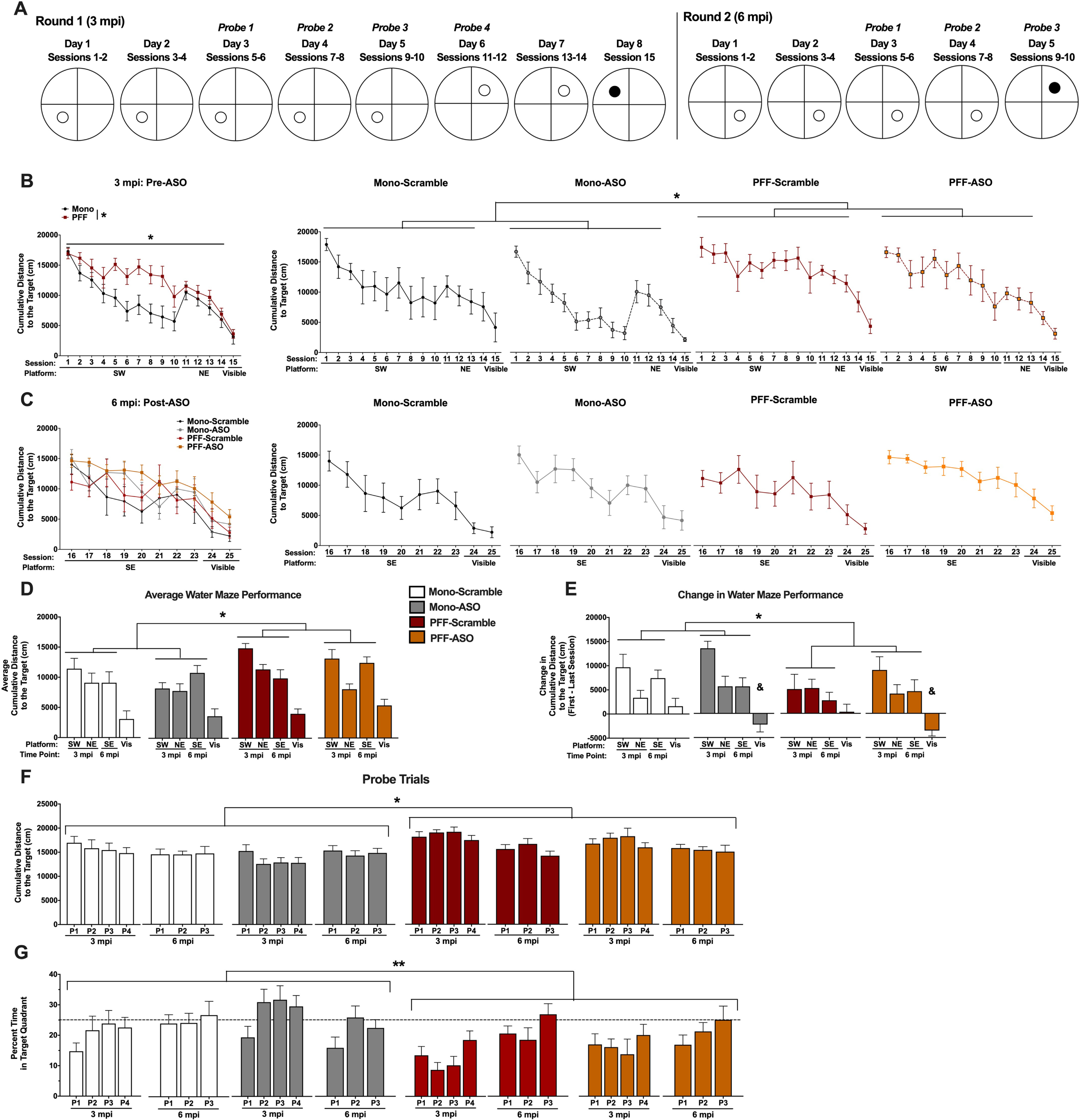
After ASO delivery, we performed the WM test again, with new platform locations not used in the first round of testing (Fig. 3A). When spatial learning was assessed in this way, we discovered that PFF-injected mice displayed impaired hippocampus-dependent learning indicated by a greater cumulative distance to the target platform compared to Mono-injected mice (p = 0.015; Fig. 3B, D). PFF-injected animals also did not improve as much as Mono animals over the course of training sessions, shown by a significant time by PFF interaction (p = 0.041). ASO treatment also affected learning as measured by the cumulative distance to the target, since ASO-injected animals displayed altered learning before versus after ASO injection (p = 0.008; Fig. 3C, D). There was no overall main effect of ASO (p = 0.465) and no PFF by ASO interaction (p = 0.790) on cumulative distance to the target, however, indicating that ASO treatment did not change learning compared to Scramble-injected animals at the 6 mpi time point. Averaged performance for each platform location and indications of significance are shown in Fig. 3D.
To ensure that the differences in cumulative distance to the target were not due to changes in swim speeds, average velocity was assessed over the hidden platform sessions (Supplementary Figure 2). ASO-injected mice swam slower than Scramble-injected mice following treatment, indicated by a significant time by ASO interaction (p = 0.001). There was no time by PFF interaction (p = 0.434), main effect of PFF (p = 0.179), main effect of ASO (p = 0.517), or PFF by ASO interaction (p = 0.435), suggesting that the PFF-dependent differences found in cumulative distance to the target were not due to motor impairments.
We also assessed performance during the visible platform sessions with the cumulative distance to the target platform performance measure. Similar to the hidden platform sessions, ASO-injected mice swam farther from the platform after receiving treatment, shown by a significant time by ASO interaction (p = 0.017). There were no significant differences based on PFF (p = 0.233) or ASO (p = 0.395) status in ability to locate the visible platform, nor any time by PFF interaction (p = 0.272) or PFF by ASO interaction (p = 0.731; Fig. 3B-D), again indicating that these differences in task performance were not due to the PFF injection. We also analyzed the average velocity during the visible platform sessions. ASO again animals swam slower after ICV delivery compared to before (p = 0.039). There were no significant differences detected between PFF (p = 0.492) or ASO (p = 0.626) groups, nor time by PFF interaction (p = 0.507) or PFF by ASO interaction (p = 0.845), overall suggesting that ASO delivery, but not PFF injections, led to motor impairments during the water maze test.
In order to further clarify the time interactions found during analysis of the hidden platform sessions, we calculated the difference (delta) in the cumulative distance to the target platform between the first and last sessions for each platform location. A repeated measures ANOVA revealed that PFF-injected mice had a smaller change in cumulative distance to the target location than Mono-injected mice (p = 0.05, Fig. 3E), indicating less improvement over the course of training. There was no main effect of ASO (p = 0.292) or PFF by ASO interaction detected (p = 0.599), suggesting that ASO treatment did not affect hippocampus-dependent memory over time. When the change in cumulative distance was calculated for the visible sessions between 3 and 6 mpi, we found that ASO-injected mice performed worse than Scramble-injected mice (p = 0.012). Together with the average velocity data, this suggests that ASO treatment led to deficits in motor abilities, while PFF injections did not, as no effect of PFF (p = 0.394) or PFF by ASO interaction (p = 0.990) were detected.
Throughout training, we periodically assessed spatial memory retention using probe trials, during which the platform was removed. Similar to training, PFF-injected mice had a higher cumulative distance to the target location than Mono-injected mice (p = 0.005; Fig. 3F) and did not improve over probe trials like the Mono-injected mice did (p = 0.012). There were no differences seen based on ASO treatment (p = 0.186), nor interactions of PFF and ASO status (p = 0.836), or time and ASO (p = 0.259), indicating that while PFF injection worsened spatial memory, ASO treatment had no effect. This was supported when we analyzed percent time spent in the target quadrant during probe trials, where we also found that PFF-injected mice spent less time in the target quadrants than Mono mice (p = 0.008, Fig. 3G). Here, we did find a time by ASO interaction (p = 0.047), as Scramble-injected mice continued to increase in time spent in the target quadrant after delivery, but ASO-injected mice did not, and a time by PFF interaction (p = 0.001), indicating a slower improvement in memory than Mono mice. Post-hoc tests did not indicate differences when each group was compared to all others.
We also assessed average velocity during the probe trials. Animals swam slower after ASO delivery than before (p < 0.0001; Supplementary Figure 2), but there was no main effect of ASO (p = 0.926), PFF by ASO interaction (p = 0.689), time by PFF (p = 0.073) or main effect of PFF (p = 0.055), again indicating that ASO, but not PFF, affected motor performance.
Novel object recognition and spatial habituation are impaired in mice at 6 months following PFF injections in the motor cortex, but rescued by targeted ASO treatment
We tested animals in the open field test both pre- and post-ASO delivery to assess anxiety-like behavior as well as spatial habituation learning (Fig. 4A). At 3 mpi, there were no differences detected in total distance moved, average velocity, or time spent in the center, with both PFF- and Mono-injected animals showing typical habituation to the enclosures. At 6 mpi, though, there was an intriguing time by PFF by ASO interaction in the total distance moved (p = 0.024; Fig. 4B) and average velocity (p = 0.026), where all groups, except for PFF-Scramble mice, showed the expected decrease in overall activity and velocity. The absence of a decrease in PFF-Scramble animals indicates an impairment in this habituation behavior, and that this targeted ASO treatment restored normal levels of habituation in PFF-injected mice.
Fig. 4
Performance in the open field and novel object recognition. A) Schematic of the behavioral protocol used at 3 mpi (left) and 6 mpi (right). Distinct objects were used between the two time points. B) Total distance moved during the open field on day 1 and day 2 at 3 mpi and 6 mpi. No differences were detected in total distance moved at 3 mpi. However, at 6 mpi there was a significant time-by-PFF-by-ASO interaction (F(1,29) = 5.637, p = 0.024). PFF-Scramble animals did not decrease their total distance moved from day 1 to day 2, while all other groups did. C) Percent time spent with the objects at 3 mpi; while Scramble and ASO groups are indicated here, this time point is prior to ASO delivery. All groups showed a preference for the novel object: Mono-Scramble familiar vs. novel, t(50) = 5.632, p < 0.0001; Mono-ASO familiar vs. novel, t(50) = 4.706, p < 0.0001; PFF-Scramble familiar vs. novel, t(50) = 6.796, p < 0.0001; PFF-ASO familiar vs. novel, t(50) = 6.713, p < 0.0001. D) Percent time spent with the objects at 6 mpi; this time point is post-ASO delivery. Mono-Scramble and PFF-ASO animals showed a preference for the novel object (t(52) = 4.206, p = 0.0004; t(52) = 3.462, p = 0.0043, respectively), whereas Mono-ASO and PFF-Scramble animals did not (t(52) = 1.845, p = 0.2541; t(52) = 1.945, p = 0.2099, respectively). Data are presented as group averages±SEM. Animals that explored < 2s were excluded from analysis. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
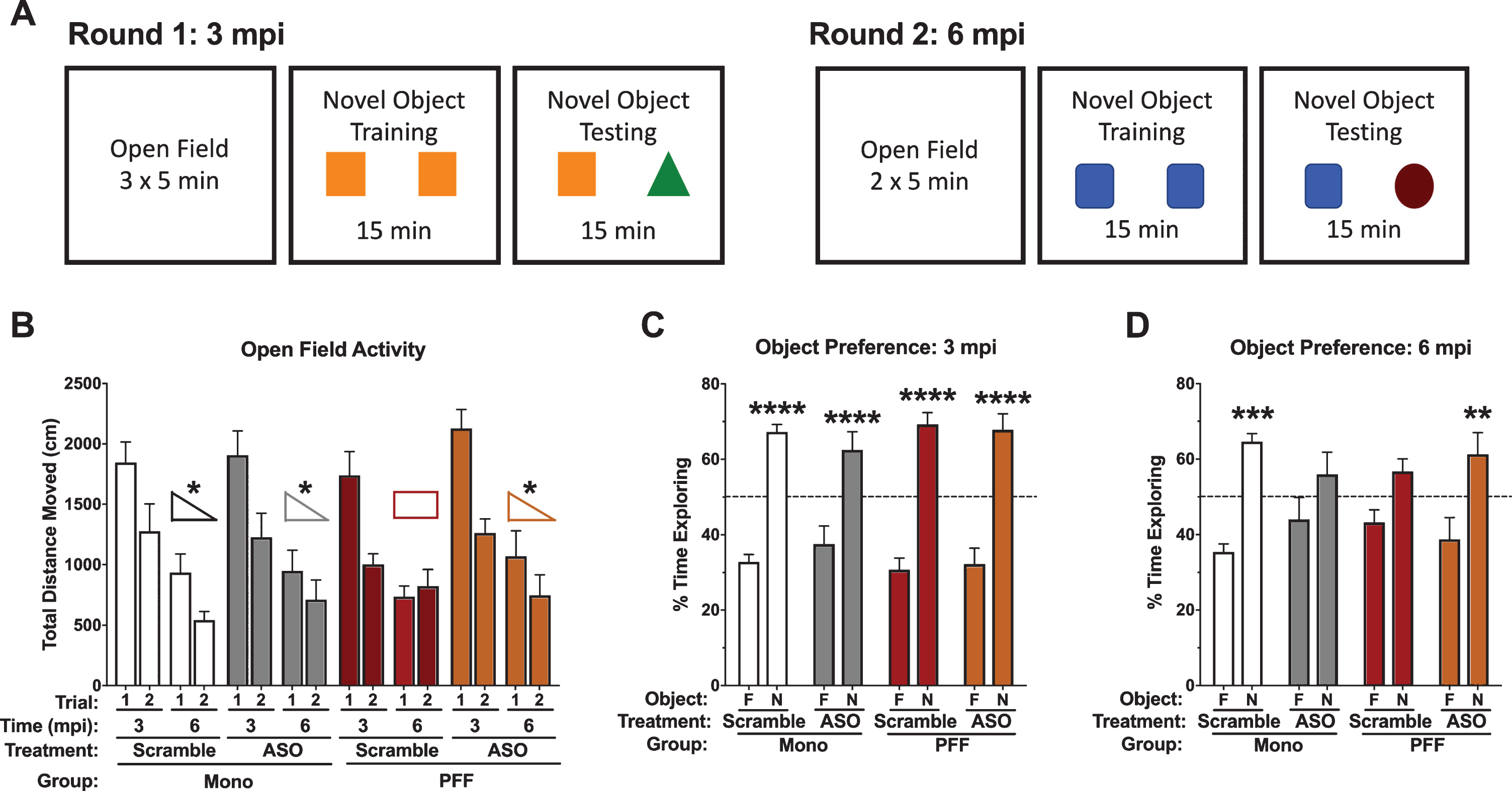
Following open field testing, we assessed novel object recognition in the same enclosures at both 3 and 6 mpi. Time spent with the novel object compared to the familiar object was analyzed as a measure of memory. At 3 mpi, there were no differences seen in object recognition memory, with all groups showing robust preference for the novel object (Fig. 4C). At 6 mpi, we again saw differential effects of ASO based on PFF status. Mono-Scramble and PFF-ASO animals both showed a preference for exploring the novel object (Fig. 4D). However, neither Mono-ASO nor PFF-Scramble showed a preference for exploring the novel object (Fig. 4D). This interaction suggests that the ASO successfully rescued hippocampus-dependent memory impairments in mice injected with PFFs, but impaired hippocampus-dependent memory in mice with no psyn pathology.
Both PFF injections in the motor cortex and intracerebroventricular ASO injections are associated with impairments in motor function
To assess the potential effects of PFF and ASO administration on motor function, we tested all animals in the rotarod and wire hang tests at two time points (3 mpi and 6 mpi; Fig. 5A). Latency to fall off the rotarod at the two time points was assessed with a repeated measures ANOVA, which revealed that mice fell off the rotarod much sooner after ASO delivery compared to before, shown by a time by ASO interaction (p < 0.0001). No differences were seen between PFF- and Mono-injected mice (p = 0.702), nor were there differences in how well PFF mice did over time (p = 0.797). There was no overall effect of ASO detected (p = 0.451, Fig. 5B).
Fig. 5
Motor performance in the rotarod and wire hang. A) Schematic of the behavioral protocols for rotarod (top) and the falls-and-reaches wire hang method (bottom). B) Latency to fall off of the rotarod by day. The 3 mpi time point is before ASO delivery; the ASO groups indicate the animals that will have ASO in the future. We found a significant time-by-ASO interaction (F(3,87) = 16.145, p < 0.001), indicated by the arrows. C) Latency to fall off the wire hang for the first time. We found a significant time-by-ASO interaction (F(1,26) = 11.936, p = 0.002), PFF-by-ASO interaction (F(1,26) = 6.030, p = 0.021), and main effect of ASO (F(1,26) = 21.388, p < 0.001). The arrows indicate the time-by-ASO interaction. D) The average final fall score. We found a significant time-by-ASO interaction (F(1,26) = 12.923, p = 0.002), and main effect of ASO (F(1,26) = 13.826, p = 0.001). The arrows indicate the time-by-ASO interaction. Additionally, we found a trend towards a main effect of PFF (F(1,26) = 3.739, p = 0.064). E) Fall score plotted over time at 3 mpi (pre-ASO). No significant differences were detected. F) Fall score plotted over time at 6 mpi (post-ASO). A repeated measures ANOVA revealed a time by ASO interaction (F(35,910) = 6.087, p < 0.001) and a time by PFF interaction (F(35,910) = 3.409, p < 0.001), with Mono-Scramble falling the least over time, PFF-Scramble animals falling more over time, and both ASO groups falling the most. A significant main effect of ASO was also detected (F(1,26) = 20.089, p < 0.001). G) Latency to escape (reach) the wire hang for the first time. There was no main effect of PFF (p = 0.925), ASO (p = 0.513), or PFF by ASO interaction (p = 0.174) detected. No time interactions were found, either. H) The average final reach score. A trend towards a main effect of PFF was found (F(1,26) = 3.250, p = 0.084). No time interactions, main effect of ASO (p = 0.328), or PFF by ASO interaction (p = 0.328) were detected. I) Reach score plotted over time at 3 mpi (pre-ASO). A time-by-PFF interaction indicated that Mono animals reached the edge of the wire hang more over time than PFF animals (F(35,980) = 3.195, p < 0.001). No main effect of PFF (p = 0.094), ASO (p = 0.625), or PFF by ASO interaction (p = 0.895) were found. J) Reach score plotted over time at 6 mpi (post-ASO). A repeated measures ANOVA showed a time-by-ASO-by-PFF interaction (F(35,910) = 4.036, p < 0.001), a time-by-ASO interaction (F(35,910) = 5.116, p < 0.001) and a PFF-by-ASO interaction (F(1,26) = 6.770, p < 0.05). There was also a trend toward a main effect of ASO (F(1,26) = 3.995, p = 0.056), but no main effect of PFF (p = 0.475). Mono-Scramble animals reached more over time than PFF animals, and Mono-ASO animals almost never reached. Data are presented as group averages±SEM; individual points are individual animals. ∧p < 0.08; *p < 0.05; ***p < 0.001; ****p < 0.0001; &p < 0.05, Time*ASO.
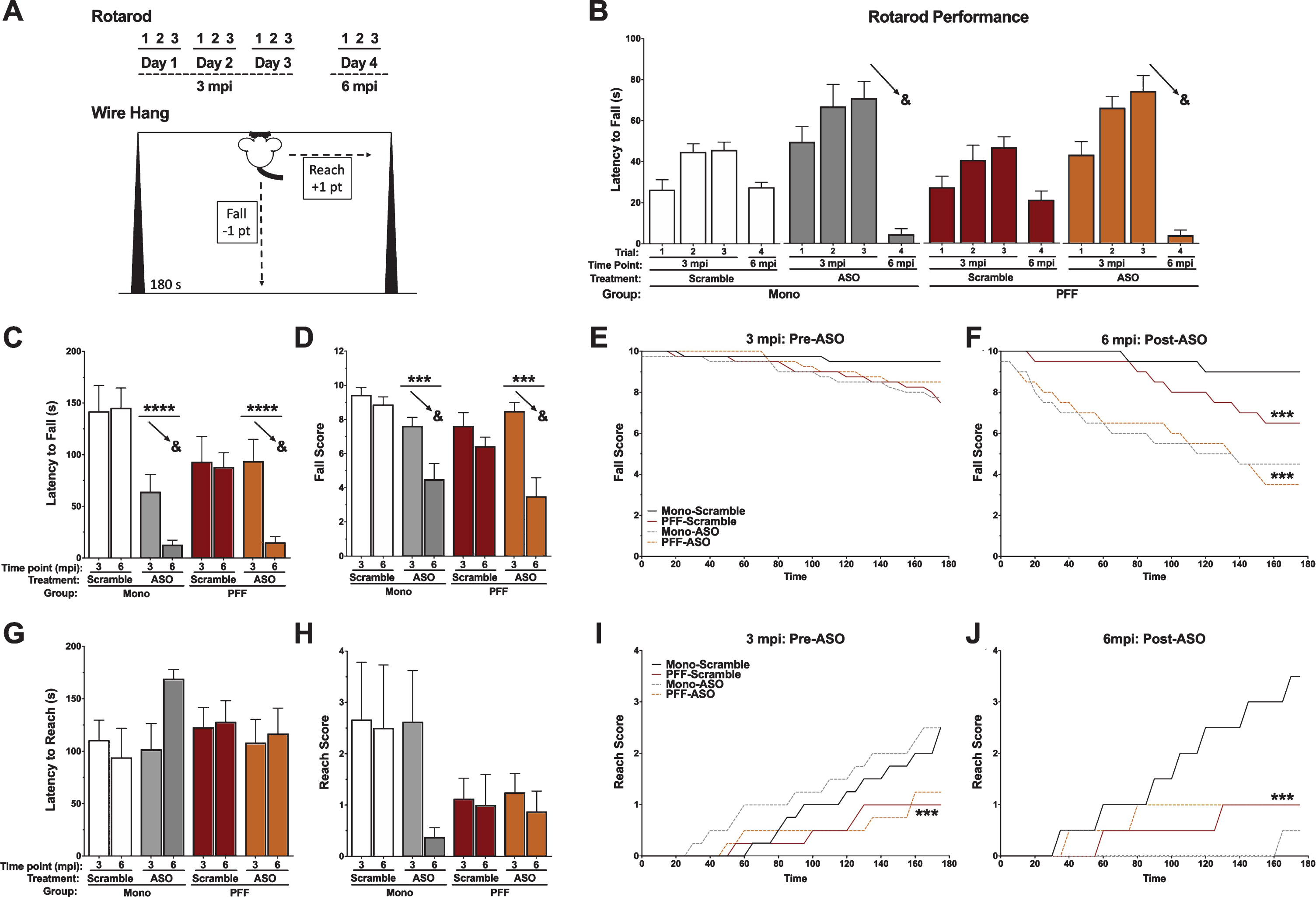
Similarly, latency to the first fall from the wire hang was analyzed with a repeated measures ANOVA (Fig. 5C). ASO treatment resulted in mice falling off the wire hang sooner than prior to treatment (p = 0.002), in addition to ASO mice falling sooner than Scramble mice (p < 0.0001). There was also a significant PFF by ASO interaction (p = 0.021), likely due to differences in wire hang performance seen between Mono mice that would receive ASO or Scramble at 3 mpi. PFF mice did not fall off the wire hang sooner (p = 0.323), nor did PFF injections lead to worse performance between 3 and 6 mpi (p = 0.212). The final fall score in the wire hang was also analyzed, again showing that ASO treatment led to worse performance over time (p = 0.002) and that ASO mice performed worse than Scramble mice (p = 0.001). There was a trend towards PFF-injected mice having a lower score than Mono-injected mice (p = 0.064; Fig. 5D). For this measure, there was no significant PFF by ASO interaction (p = 0.080). To further investigate performance at each time point, we assessed the fall score over the 180 s of each trial. At 3 mpi, all groups performed similarly, indicated by no significant effects or interactions (Fig. 5E). Conversely, at 6 mpi both PFF-injected animals (p < 0.001) and ASO treated animals fell more over time (p < 0.001). ASO animals also had overall lower fall scores than Scramble animals (p < 0.001; Fig. 5F), indicating worse performance.
The latency to first escape (“reach”) during the wire hang was also assessed, though no differences between groups or decline over time were detected (Fig. 5G). Reach score was similarly assessed, and revealed a trend towards PFF-injected animals having a lower reach score than Mono-injected animals (p = 0.08; Fig. 5H). ASO treatment did not affect reach score, nor was there a decline in reach score over time. However, when we analyzed the reach score over the 180 s of each trial, PFF mice escaped the wire hang at a slower rate than Mono mice at 3 mpi (p < 0.001; Fig. 5I). There were no differences based on future ASO treatment (p = 0.625) or in the final reach score based on PFF (p = 0.094). At 6 mpi, we saw more pronounced effects, with ASO treatment differentially affecting mice based on PFF status (p = 0.015; Fig. 5J). Mono-Scramble mice escaped the wire hang at a faster rate than Mono-ASO mice and both groups of PFF-injected mice (p < 0.001), with a trend towards an overall lower score in the ASO-treated groups (p = 0.056). Similar to the novel object recognition data, the PFF-dependent effects of the ASO treatment indicate that amount of available, soluble αsyn may be important.
Together, these tests show a slight progression of coordinated motor impairments 6 months following PFF seeding and that ASO treatment was associated with impaired motor function, which may be due to unknown off-target effects not involving ASO-mediated lowering of αsyn (see below).
WT and αsyn knock out mice treated with ASO show alterations in sleep patterns, feeding behavior, and weight loss, indicating possible off-target effects
Sleep disturbances are often reported in patients with LB pathology, with evidence that sleep disorders can appear nearly a decade before motor symptoms emerge [41]. To determine whether there were alterations in circadian activity levels, animals were placed into home-cage monitoring devices to assess undisturbed activity over a week. Analysis of average activity during the light cycle over the week revealed a time by ASO interaction (p = 0.046), indicating that ASO animals failed to habituate during daylight over the course of the week, where mice treated with ASO showed increased activity over the week compared to Scramble animals that decreased over the week (Fig. 6A). There were no differences based on PFF status (p = 0.867), and both PFF- and Mono-injected mice showed similar responses to the ASO (p = 0.189). The difference in habituation was unique to the daytime, though, as this same time by ASO interaction was not observed during nighttime activity over the week (p = 0.085; Fig. 6B). Again, there were no effects of PFF (p = 0.665) or ASO (p = 0.848), nor a PFF by ASO interaction (p = 0.324).
Fig. 6
Health-related data. A) Average daytime activity over the course of 5 days. A repeated measures ANOVA for daytime activity indicated an interaction of day and ASO (F(1.703,34.060) = 3.550, p < 0.05), where Scramble animals decreased their daytime activity over the week, but ASO animals increased. No main effect of PFF (p = 0.867), ASO (p = 0.705), or PFF by ASO interaction (p = 0.189) were detected. B) Average nighttime activity over the course of 6 nights. A repeated measures ANOVA for nighttime activity suggested a similar pattern. Scramble animals showed a significant decrease in activity over nights (Effect of night, p = 0.005), whereas ASO animals did not (no effect of night, p = 0.639). No effect of PFF (p = 0.665), ASO (p = 0.848), or PFF by ASO interaction (p = 0.324) were detected. C) Body weights were recorded weekly beginning at the first round of behavioral testing. Prior to ASO delivery, a repeated-measures ANOVA showed that all animals increased in body weight over time (F(3.229,96.862) = 122.880, p < 0.001) with no group differences in weight. Following ASO delivery, ASO animals showed a steady decline in body weight over time (F(3.613,104.777) = 16.093, p < 0.001), and an overall reduced body weight compared to Scramble animals (F(1,29) = 7.952, p < 0.001). D) Food intake was recorded over a 5-day period. Food was weighed in the morning (∼8:00 AM) and the evening (∼17:00 PM) to approximate nighttime and daytime food intake. A 2-way ANOVA revealed no group differences in daytime food intake, but did show a main effect of ASO for nighttime food intake (F(1,26) = 5.297, p < 0.05). E) Schematic of the control experiment to assess specificity of the ASO. C57Bl/6NJ (WT-NJ) and αsyn knock out mice (KO) were injected with the ASO construct, and compared against WT-NJ mice injected with scramble. F) A repeated measures ANOVA indicated a significant time by ASO interaction (F(16,88) = 6.079, p = 0.0001) and main effect of ASO (F(2,11) = 19.03, p = 0.0003), suggesting off-target effects. Data presented as group averages±SEM; individual points are individual animals. *p < 0.05; ***p < 0.001.; &p < 0.05, Time*ASO.
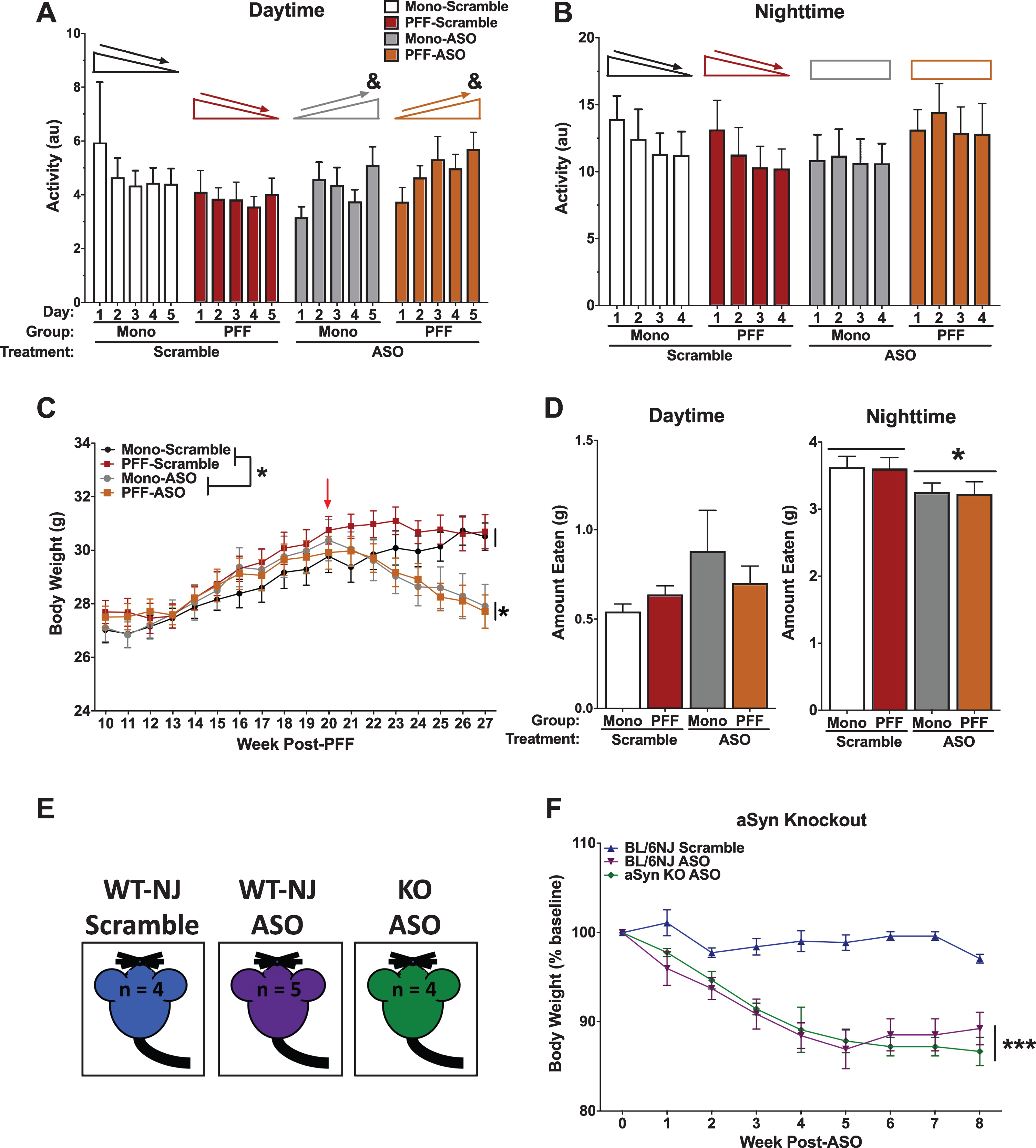
Body weights were recorded once a week over the duration of the experiment. Prior to ASO injections, there were no differences observed in body weight based on PFF status (p = 0.281), with all groups showing an expected increase over time (p < 0.001; Fig. 6C). However, following ASO or Scramble treatment, ASO-treated animals significantly decreased in body weight over time until the end of the experiment (p < 0.001). Overall, ASO mice weighed significantly less than Scramble mice (p < 0.001). PFF injections did not affect body weight (p = 0.5440), nor was there an interaction of PFF and ASO (p = 0.517).
Based on the observed body weight changes following ASO injections, we next measured food intake over a period of 5 days. ASO animals ate significantly less between 17:00 and 8:00 (“overnight”) than Scramble animals (p = 0.0296; Fig. 6D). There were no differences in daytime food intake (from 8:00 to 17:00) based on PFF (p = 0.726) or ASO (p = 0.106), nor a PFF by ASO interaction (p = 0.26).
To test whether this ASO could have off-target effects, we injected it into αsyn-knock out (KO) mice and C57Bl/6NJ (WT-NJ) matched controls (Fig. 6E). Following ICV delivery, both the αsyn-KO mice and WT-NJ mice that received ASO progressively lost body weight compared to the Scramble mice, indicated by a time by ASO interaction (p = 0.0001) and a main effect of ASO (p = 0.0003; Fig. 6F). These data suggest αsyn-independent off-target effects with the ASO used.
DISCUSSION
We set out to assess the progressive cognitive and behavioral effects of PFF injections in the motor cortex of male WT mice, as well as explore if reducing αsyn expression throughout the CNS with a targeted ASO could ameliorate cognitive, behavioral, and pathological effects of PFF injections. The PFF model of synucleinopathies has become more common and is gaining in popularity since it was first described [30, 32]. αsyn expression is abundant throughout the central nervous system, though is especially high in regions such as the substantia nigra, striatum, hippocampus, and cortex [45]. In this study, we observed LB-like aggregates in the motor cortex, as well as in the medial and lateral somatosensory cortices and the hippocampus at 6 months post-injection. Mice that received PFF injections displayed mild behavioral deficits starting at 3 mpi. We also found that a single, intracerebroventricular injection of an αsyn-targeted ASO was sufficient to reduce αsyn and psyn expression for months, as well as reduce psyn inclusions. Yet, off-target effects of this particular ASO were detrimental to animal health.
Substantial evidence is pointing to a trans-synaptic progression of psyn aggregation [53, 71, 72]. The results of the current study provide evidence of impairments in behavioral and cognitive performance following motor cortex PFF injections, as PFF-injected mice showed mild motor and cognitive impairments at 3 mpi and a mild progression of motor impairment at 6 mpi. For example, PFF-injected mice performed similarly to Mono-injected controls in the wire hang at 3 mpi, but fell sooner and more often and were unable to “escape” at similar rates at 6 mpi. Clinically, the motor deficits observed in PD are likely reflective of a combination of problems, including bradykinesia and rigidity—which result in the difficulty to execute motor tasks, especially repetitive tasks—as escaping from the wire hang requires fine, repetitive motor control of all four paws [39, 64, 73, 74]. These results are similar to results from other groups that have assessed motor performance following striatal injections: original characterization of striatal injections in 2-3-month-old C57Bl6/C3H WT mice indicated that wire hang deficits began at 3 mpi, while rotarod deficits did not appear until 6 mpi and locomotion in the open field remained unaffected up to 6 mpi [30]. Minor deficits in the balance beam have been reported in C57Bl6/C3H WT mice 2 months post-striatal injection without differences in rotarod or open field locomotion, as well [75].
Additionally, we observed hippocampus-dependent cognitive impairments at both 3 mpi and 6 mpi, and clear spread of pathology to the hippocampus when the animals were sacrificed at 6 mpi. The hippocampus has only recently become a region of interest in PD, with evidence showing that disrupted dopaminergic signaling impairs hippocampal plasticity [76]. Dopamine signaling from the midbrain to the hippocampus has been shown in both rodents and humans to modulate long-term learning. Healthy adults who were placed in an MRI during an image-recognition task showed increased BOLD signal in the midbrain and hippocampus in response to previously-seen, reward-associated images [77]. In rodents, both a toxin-induced model and a transgenic model of PD revealed impaired long-term potentiation in the hippocampus that was associated with decreased dopamine transmission and impaired hippocampus-dependent spatial memory [78].
Here, we saw hippocampus-dependent novel object recognition in PFF animals at 3 mpi, but not at 6 mpi; Mono-injected mice showed object preference at both time points. Post-mortem analysis of LB-like pathology confirmed that psyn-positive inclusions had spread into the hippocampus. The motor cortex communicates with the hippocampus via the somatosensory cortex and the basal forebrain [79–81] and these may be the trans-synaptic routes of spread in our system. In the water maze, PFF mice showed learning and memory impairments, indicated by the higher cumulative distance to the target platform both during hidden platform training and probe trials. This was unlikely due to differences in motor abilities, as there were no PFF-dependent differences in average swim speeds during the hidden platform, visible platform, or probe trials. Thus, while the hippocampus did not have the most psyn pathology in the regions we analyzed, our results indicate that hippocampal function was disrupted. LB-like pathology in the hippocampus, pathology in other regions disrupting typical communication to the hippocampus, and other pathological changes may have contributed to the impaired hippocampal function.
Assessment of the αsyn-targeted ASO indicated that a single dose was able to persistently reduce αsyn and psyn protein levels and LB-like pathology through the cortex and hippocampus. The ASO we used reduced hippocampal αsyn and psyn by > 50%, and successfully lowered the amount of psyn-positive inclusions. This reduction in LB-like pathology occurred after LB-like inclusions had begun forming, supporting this approach’s potential in the clinic, where patients begin treatment well after LBs are thought to have formed and spread to many brain regions [43, 54]. Our timing for ASO administration was aimed at reflecting the timing that is typical of diagnosis of synucleinopathies in the clinic, where the goal is to affect a robust αsyn reduction. A recent report using similar ASO treatments has also reported comparable results, showing successful reduction of pathology and rescued dopaminergic function [82]. However, in our experiments this ASO was also associated with possible off-target effects. We observed detrimental side effects after ASO treatment on several measures in our study, including disruptions in light-dark cycle activity, reduced body weight, food intake, and motor abilities. These results highlight the importance to not only assessing pathological changes but also behavioral performance following ASO treatment. Others have reported treatment with a different αsyn-specific ASO which caused no changes in general health, although a lower dose (300μg) administered intranasally was use in this case [83]. The ASO dose was higher in our study (700μg), administered via ICV surgery, and caused a more substantial αsyn and psyn reduction (> 50%) than this previous work (20–40%), which could potentially account for the adverse effects we measured. This previous experiment also did not report testing their ASO in αsyn knock-out mice. Our observation of similar weight loss after ASO treatment in αsyn knock-out animals strongly suggests that at least this detrimental effect is due to off-target effects of the ASO, and not attributable to ASO-mediated lowering of αsyn.
The most profound motor deficits we observed were in the wire hang and rotarod tests, with ASO-treated mice falling significantly more in both tasks and showing impaired motor coordination, as measured by their ability to escape the wire hang. These motor deficits were substantial compared to performance prior to ASO delivery, which was also reflected in the decreased average swim speeds during the water maze test at the post-ASO time point. PFF-injected mice also showed a decreased number of escapes from the wire hang and an increased number of falls, most noticeably when we assessed performance over the 180 s trial at 6 mpi. Our ASO delivery did not further worsen the escape scores of the PFF-injected mice, but did significantly impair the Mono-injected mice, suggesting that potential off-target effects had similar consequences as the PFF injections. Others have reported, using similar ASO treatments, an improvement in wire hang performance when the ASO was given before PFF injection and a trend towards improvement in wire hang performance when the ASO was administered after PFF injections [82].
We did observe some differential effects of ASO delivery dependent on Mono- or PFF-status. ASO treatment was associated with improved performance in the novel object recognition test: while PFF-scramble animals did not show a preference, PFF-ASO animals demonstrated intact hippocampus-dependent object recognition. Additionally, PFF-ASO mice displayed typical spatial habituation in the open field following treatment, which was absent in PFF-Scramble animals. Others have reported evidence that ASOs could rescue disrupted dopamine signaling: administration of a distinct αsyn-specific ASO to WT mice, which resulted in 20–40%decrease of αsyn, increased dopamine and serotonin release in the forebrain, whereas αsyn over-expression decreased signaling [83]. Although we did not assess dopaminergic signaling, a similar ASO treatment has been reported to normalize striatal dopamine levels [82]. Therefore, it is possible that ASO treatment similarly improved dopaminergic signaling to the hippocampus in our mice, resulting in rescued hippocampus-dependent memory. Conversely, this ASO was associated with impairments in novel object recognition in control mice in our experiments. αsyn has been reported to be important in hippocampal neurogenesis [84, 85], with recent evidence suggesting that healthy αsyn has important roles in DNA damage repair that are disrupted in pathological states [17, 86]. It was recently reported that a particular single-nucleotide polymorphism in the SNCA-AS1 gene that lead to upregulation αsyn was inversely correlated with risk for Lewy body dementia (LDB), suggesting that higher levels of αsyn is protective against LBD [87]. Thus, disruption to normal αsyn levels either up or down could lead to particular disease states, and possibly explain our disease-state dependent results. As ASO treatment rescued novel object recognition in PFF mice, it might have selective beneficial effects in the context of disease when psyn levels are elevated.
While our findings in the αsyn-KO mice suggest that the particular ASO we used had off-target effects, the question of whether normal soluble αsyn has important functional roles that are compromised when its levels are lowered is potentially important to consider as well. There is evidence of αsyn pathological toxicity being mediated by both a potential gain- and loss-of-function [88–91]. For example, substantial data from many groups suggests that αsyn aggregates can be toxic [90], and this is supported by the fact that αsyn overexpression leads to PD (as seen in SNCA duplication and triplication mutations), and by evidence that knocking out αsyn in mice does not appear to cause deficits until end-stages of their lifespan [92, 93]. There are relatively few tolerability concerns reported for constitutive genetic Snca deletion in Snca knockout mice [94–98] or haploinsufficient mice where the one normal Snca allele is deleted in adult animals [99]. However, these constitutive and haploinsufficient genetic knock-outs do not fully address the question of possible compensatory mechanisms that may mask an essential role of αsyn. At this point in time, we do not likely understand all the pathways that are disrupted by decreasing αsyn in the healthy adult CNS. One possibility is that αsyn has an essential role, and when it is reduced beyond a specific threshold, compromise of this role leads to decline in an important function for the CNS. Recent work from our lab has indicated that αsyn may be influential for the response to DNA damage, including having the ability to bind DNA and modulate repair processes [17]. Detrimental effects of lowering αsyn below a certain threshold could also be driven by impaired functions at the synapse, where αsyn modulates endo- and exocytosis and regulates neurotransmitter release [10–12].
One potential way to test this is by using RNAi-based approaches to reduce αsyn in adult animals, but to date this approach has produced somewhat conflicting results. RNAi-mediated αsyn silencing in the substantia nigra of rats led to degeneration of dopamine cells and neuroinflammation [100–102]. However, other groups have shown that knockdown of αsyn in the substantia nigra of adult rats using a RNAi was long-lasting and safe, with significant reduction of αsyn and no neurodegeneration observed 12 months later [103]. In addition, multiple other groups using siRNAs, shRNAs, or ASOs against Snca in mice report no cell death in vivo [82, 83, 103, 104]. Two αsyn antibodies, PRX002 and BIIB054, completed first-in-human trials in which no serious adverse events were found with αsyn lowering [105, 106]. It will be important in future studies to better define the physiological and functional importance of soluble αsyn.
In summary, our data suggest that there are mild, progressive deficits associated with PFF injections in the motor cortex, and that this particular ASO can successfully reduce LB-like pathology in mice months after a single injection but that there are detrimental, off-target side effects as well. While our results support a promising future for the use of ASOs to decrease the aggregation of psyn inclusions, ongoing clinical trials and future efforts with more advanced, target-specific ASOs are warranted for determining the extent to which targeted ASO treatment can slow or halt disease progression.
ACKNOWLEDGMENTS
The authors would like to thank those who contributed by assisting with data acquisition and analysis (Eileen Ruth Torres, Joanne S. Lee, Crystal Chaw, Stefanie Kaech Petrie, Esha M. Patel), teaching techniques (Sydney Elise Dent, Allison J. Schaser, Teresa Stackhouse, Valerie Osterberg), and supplying reagents (Hien Tran Zhao, Kelvin Luk).
The following funding sources contributed to this project: P30NS061800, NSF GVPRS0015A, NSCOR NNX15AK13G, NIA RF1 AG059088, and NINDS NS102227, NS096190.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare. No funding was supplied by Ionis Pharmaceuticals.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JPD-212566.
REFERENCES
[1] | Vivacqua G , Casini A , Vaccaro R , Fornai F , Yu S , D’Este L ((2011) ) Different sub-cellular localization of alpha-synuclein in the C57BL∖6J mouse’s central nervous system by two novel monoclonal antibodies. J Chem Neuroanat 41: , 97–110. |
[2] | Maroteaux L , Campanelli JT , Scheller RH ((1988) ) Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J Neurosci 8: , 2804–2815. |
[3] | Jin H , Kanthasamy A , Ghosh A , Yang Y , Anantharam V , Kanthasamy AG ((2011) ) alpha-Synuclein negatively regulates protein kinase Cdelta expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J Neurosci 31: , 2035–2051. |
[4] | Geng X , Lou H , Wang J , Li L , Swanson AL , Sun M , Beers-Stolz D , Watkins S , Perez RG , Drain P ((2011) ) alpha-Synuclein binds the K(ATP) channel at insulin-secretory granules and inhibits insulin secretion. Am J Physiol Endocrinol Metab 300: , E276–286. |
[5] | Rodriguez-Araujo G , Nakagami H , Takami Y , Katsuya T , Akasaka H , Saitoh S , Shimamoto K , Morishita R , Rakugi H , Kaneda Y ((2015) ) Low alpha-synuclein levels in the blood are associated with insulin resistance. Sci Rep 5: , 12081. |
[6] | Burre J , Sharma M , Tsetsenis T , Buchman V , Etherton MR , Sudhof TC ((2010) ) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329: , 1663–1667. |
[7] | Burre J , Sharma M , Sudhof TC ((2014) ) alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A 111: , E4274–4283. |
[8] | Ostrerova N , Petrucelli L , Farrer M , Mehta N , Choi P , Hardy J , Wolozin B ((1999) ) alpha-Synuclein shares physical and functional homology with 14-3-3 proteins. J Neurosci 19: , 5782–5791. |
[9] | Wang C , Zhao C , Li D , Tian Z , Lai Y , Diao J , Liu C ((2016) ) Versatile structures of alpha-synuclein. Front Mol Neurosci 9: , 48. |
[10] | Xu J , Wu XS , Sheng J , Zhang Z , Yue HY , Sun L , Sgobio C , Lin X , Peng S , Jin Y , Gan L , Cai H , Wu LG ((2016) ) alpha-synuclein mutation inhibits endocytosis at mammalian central nerve terminals. J Neurosci 36: , 4408–4414. |
[11] | Wu LG , Hamid E , Shin W , Chiang HC ((2014) ) Exocytosis and endocytosis: Modes, functions, and coupling mechanisms. Annu Rev Physiol 76: , 301–331. |
[12] | Huang M , Wang B , Li X , Fu C , Wang C , Kang X ((2019) ) alpha-synuclein: A multifunctional player in exocytosis, endocytosis, and vesicle recycling. Front Neurosci 13: , 28. |
[13] | Jensen PH , Nielsen MS , Jakes R , Dotti CG , Goedert M ((1998) ) Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J Biol Chem 273: , 26292–26294. |
[14] | Kam TI , Mao X , Park H , Chou SC , Karuppagounder SS , Umanah GE , Yun SP , Brahmachari S , Panicker N , Chen R , Andrabi SA , Qi C , Poirier GG , Pletnikova O , Troncoso JC , Bekris LM , Leverenz JB , Pantelyat A , Ko HS , Rosenthal LS , Dawson TM , Dawson VL ((2018) ) Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson’s disease. Science 362: , eaat8407. |
[15] | Kontopoulos E , Parvin JD , Feany MB ((2006) ) Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet 15: , 3012–3023. |
[16] | Liu X , Lee YJ , Liou LC , Ren Q , Zhang Z , Wang S , Witt SN ((2011) ) Alpha-synuclein functions in the nucleus to protect against hydroxyurea-induced replication stress in yeast. Hum Mol Genet 20: , 3401–3414. |
[17] | Schaser AJ , Osterberg VR , Dent SE , Stackhouse TL , Wakeham CM , Boutros SW , Weston LJ , Owen N , Weissman TA , Luna E , Raber J , Luk KC , McCullough AK , Woltjer RL , Unni VK ((2019) ) Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci Rep 9: , 10919. |
[18] | Ueda K , Fukushima H , Masliah E , Xia Y , Iwai A , Yoshimoto M , Otero DA , Kondo J , Ihara Y , Saitoh T ((1993) ) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 90: , 11282–11286. |
[19] | Polymeropoulos MH , Lavedan C , Leroy E , Ide SE , Dehejia A , Dutra A , Pike B , Root H , Rubenstein J , Boyer R , Stenroos ES , Chandrasekharappa S , Athanassiadou A , Papapetropoulos T , Johnson WG , Lazzarini AM , Duvoisin RC , Di Iorio G , Golbe LI , Nussbaum RL ((1997) ) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276: , 2045–2047. |
[20] | Kruger R , Kuhn W , Muller T , Woitalla D , Graeber M , Kosel S , Przuntek H , Epplen JT , Schols L , Riess O ((1998) ) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 18: , 106–108. |
[21] | Spillantini MG , Crowther RA , Jakes R , Hasegawa M , Goedert M ((1998) ) alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A 95: , 6469–6473. |
[22] | Spillantini MG , Crowther RA , Jakes R , Cairns NJ , Lantos PL , Goedert M ((1998) ) Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 251: , 205–208. |
[23] | Zarranz JJ , Alegre J , Gomez-Esteban JC , Lezcano E , Ros R , Ampuero I , Vidal L , Hoenicka J , Rodriguez O , Atares B , Llorens V , Gomez Tortosa E , del Ser T , Munoz DG , de Yebenes JG ((2004) ) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55: , 164–173. |
[24] | Lesage S , Anheim M , Letournel F , Bousset L , Honore A , Rozas N , Pieri L , Madiona K , Durr A , Melki R , Verny C , Brice A , French Parkinson’s Disease Genetics Study Group ((2013) ) G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol 73: , 459–471. |
[25] | Fujioka S , Ogaki K , Tacik PM , Uitti RJ , Ross OA , Wszolek ZK ((2014) ) Update on novel familial forms of Parkinson’s disease and multiple system atrophy. Parkinsonism Relat Disord 20 Suppl 1: , S29–34. |
[26] | Pasanen P , Myllykangas L , Siitonen M , Raunio A , Kaakkola S , Lyytinen J , Tienari PJ , Poyhonen M , Paetau A ((2014) ) Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging 35: , 2180 e2181–2185. |
[27] | Lemkau LR , Comellas G , Kloepper KD , Woods WS , George JM , Rienstra CM ((2012) ) Mutant protein A30P alpha-synuclein adopts wild-type fibril structure, despite slower fibrillation kinetics. J Biol Chem 287: , 11526–11532. |
[28] | Appel-Cresswell S , Vilarino-Guell C , Encarnacion M , Sherman H , Yu I , Shah B , Weir D , Thompson C , Szu-Tu C , Trinh J , Aasly JO , Rajput A , Rajput AH , Jon Stoessl A , Farrer MJ ((2013) ) Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord 28: , 811–813. |
[29] | Proukakis C , Dudzik CG , Brier T , MacKay DS , Cooper JM , Millhauser GL , Houlden H , Schapira AH ((2013) ) A novel alpha-synuclein missense mutation in Parkinson disease. Neurology 80: , 1062–1064. |
[30] | Luk KC , Kehm V , Carroll J , Zhang B , O’Brien P , Trojanowski JQ , Lee VM ((2012) ) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338: , 949–953. |
[31] | Luk KC , Kehm VM , Zhang B , O’Brien P , Trojanowski JQ , Lee VM ((2012) ) Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med 209: , 975–986. |
[32] | Volpicelli-Daley LA , Luk KC , Patel TP , Tanik SA , Riddle DM , Stieber A , Meaney DF , Trojanowski JQ , Lee VM ((2011) ) Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72: , 57–71. |
[33] | Osterberg VR , Spinelli KJ , Weston LJ , Luk KC , Woltjer RL , Unni VK ((2015) ) Progressive aggregation of alpha-synuclein and selective degeneration of lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep 10: , 1252–1260. |
[34] | Prusiner SB ((2012) ) Cell biology. A unifying role for prions in neurodegenerative diseases. Science 336: , 1511–1513. |
[35] | Brettschneider J , Del Tredici K , Lee VM , Trojanowski JQ ((2015) ) Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nat Rev Neurosci 16: , 109–120. |
[36] | Tran HT , Chung CH , Iba M , Zhang B , Trojanowski JQ , Luk KC , Lee VM ((2014) ) Alpha-synuclein immunotherapy blocks uptake and templated propagation of misfolded alpha-synuclein and neurodegeneration. Cell Rep 7: , 2054–2065. |
[37] | Brundin P , Melki R ((2017) ) Prying into the prion hypothesis for Parkinson’s disease. J Neurosci 37: , 9808–9818. |
[38] | Brundin P , Ma J , Kordower JH ((2016) ) How strong is the evidence that Parkinson’s disease is a prion disorder? Curr Opin Neurol 29: , 459–466. |
[39] | Jankovic J ((2008) ) Parkinson’s disease: Clinical features and diagnosis. J Neurol Neurosurg Psychiatry 79: , 368–376. |
[40] | Ansari KA , Johnson A ((1975) ) Olfactory function in patients with Parkinson’s disease. J Chronic Dis 28: , 493–497. |
[41] | Loddo G , Calandra-Buonaura G , Sambati L , Giannini G , Cecere A , Cortelli P , Provini F ((2017) ) The treatment of sleep disorders in Parkinson’s disease: From research to clinical practice. Front Neurol 8: , 42. |
[42] | Chaudhuri KR , Healy DG , Schapira AH , National Institute for Clinical E ((2006) ) Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol 5: , 235–245. |
[43] | Braak H , Del Tredici K , Rub U , de Vos RA , Jansen Steur EN , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: , 197–211. |
[44] | Henderson MX , Cornblath EJ , Darwich A , Zhang B , Brown H , Gathagan RJ , Sandler RM , Bassett DS , Trojanowski JQ , Lee VMY ((2019) ) Spread of alpha-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat Neurosci 22: , 1248–1257. |
[45] | Caputo A , Liang Y , Raabe TD , Lo A , Horvath M , Zhang B , Brown HJ , Stieber A , Luk KC ((2020) ) Snca-GFP knock-in mice reflect patterns of endogenous expression and pathological seeding. eNeuro 7: , ENEURO.0007-20.2020. |
[46] | Luna E , Decker SC , Riddle DM , Caputo A , Zhang B , Cole T , Caswell C , Xie SX , Lee VMY , Luk KC ((2018) ) Differential alpha-synuclein expression contributes to selective vulnerability of hippocampal neuron subpopulations to fibril-induced toxicity. Acta Neuropathol 135: , 855–875. |
[47] | Rey NL , Petit GH , Bousset L , Melki R , Brundin P ((2013) ) Transfer of human alpha-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol 126: , 555–573. |
[48] | Mezias C , Rey N , Brundin P , Raj A ((2020) ) Neural connectivity predicts spreading of alpha-synuclein pathology in fibril-injected mouse models: Involvement of retrograde and anterograde axonal propagation. Neurobiol Dis 134: , 104623. |
[49] | Luo P , Li A , Zheng Y , Han Y , Tian J , Xu Z , Gong H , Li X ((2019) ) Whole brain mapping of long-range direct input to glutamatergic and GABAergic neurons in motor cortex. Front Neuroanat 13: , 44. |
[50] | Jeong M , Kim Y , Kim J , Ferrante DD , Mitra PP , Osten P , Kim D ((2016) ) Comparative three-dimensional connectome map of motor cortical projections in the mouse brain. Sci Rep 6: , 20072. |
[51] | Takahashi K , Ohsawa I , Shirasawa T , Takahashi M ((2016) ) Early-onset motor impairment and increased accumulation of phosphorylated alpha-synuclein in the motor cortex of normal aging mice are ameliorated by coenzyme Q. Exp Gerontol 81: , 65–75. |
[52] | Caviness JN , Lue LF , Beach TG , Hentz JG , Adler CH , Sue L , Sadeghi R , Driver-Dunckley E , Evidente VG , Sabbagh MN , Shill HA , Walker DG ((2011) ) Parkinson’s disease, cortical dysfunction, and alpha-synuclein. Mov Disord 26: , 1436–1442. |
[53] | Schaser AJ , Stackhouse TL , Weston LJ , Kerstein PC , Osterberg VR , Lopez CS , Dickson DW , Luk KC , Meshul CK , Woltjer RL , Unni VK ((2020) ) Trans-synaptic and retrograde axonal spread of Lewy pathology following pre-formed fibril injection in an in vivoA53T alpha-synuclein mouse model of synucleinopathy. Acta Neuropathol Commun 8: , 150. |
[54] | Del Rey NL , Quiroga-Varela A , Garbayo E , Carballo-Carbajal I , Fernandez-Santiago R , Monje MHG , Trigo-Damas I , Blanco-Prieto MJ , Blesa J ((2018) ) Advances in Parkinson’s disease: 200 years later. Front Neuroanat 12: , 113. |
[55] | Weintraub D , Siderowf AD , Potenza MN , Goveas J , Morales KH , Duda JE , Moberg PJ , Stern MB ((2006) ) Association of dopamine agonist use with impulse control disorders in Parkinson disease. Arch Neurol 63: , 969–973. |
[56] | Pandey S , Srivanitchapoom P ((2017) ) Levodopa-induced dyskinesia: Clinical features, pathophysiology, and medical management. Ann Indian Acad Neurol 20: , 190–198. |
[57] | Zhao HT , John N , Delic V , Ikeda-Lee K , Kim A , Weihofen A , Swayze EE , Kordasiewicz HB , West AB , Volpicelli-Daley LA ((2017) ) LRRK2 antisense oligonucleotides ameliorate alpha-synuclein inclusion formation in a Parkinson’s disease mouse model. Mol Ther Nucleic Acids 8: , 508–519. |
[58] | Evers MM , Toonen LJ , van Roon-Mom WM ((2015) ) Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv Drug Deliv Rev 87: , 90–103. |
[59] | Schoch KM , Miller TM ((2017) ) Antisense oligonucleotides: Translation from mouse models to human neurodegenerative diseases. Neuron 94: , 1056–1070. |
[60] | Moisan F , Kab S , Mohamed F , Canonico M , Le Guern M , Quintin C , Carcaillon L , Nicolau J , Duport N , Singh-Manoux A , Boussac-Zarebska M , Elbaz A ((2016) ) Parkinson disease male-to-female ratios increase with age: French nationwide study and meta-analysis. J Neurol Neurosurg Psychiatry 87: , 952–957. |
[61] | Miller IN , Cronin-Golomb A ((2010) ) Gender differences in Parkinson’s disease: Clinical characteristics and cognition. Mov Disord 25: , 2695–2703. |
[62] | Torres ERS , Akinyeke T , Stagaman K , Duvoisin RM , Meshul CK , Sharpton TJ , Raber J ((2018) ) Effects of sub-chronic MPTP exposure on behavioral and cognitive performance and the microbiome of wild-type and mGlu8 knockout female and male mice. Front Behav Neurosci 12: , 140. |
[63] | McGinnis GJ , Friedman D , Young KH , Torres ER , Thomas CR Jr. , Gough MJ , Raber J ((2017) ) Neuroinflammatory and cognitive consequences of combined radiation and immunotherapy in a novel preclinical model. Oncotarget 8: , 9155–9173. |
[64] | van Putten M (2016) The use of hanging wire tests to monitor muscle strength and condition over time. SOP (ID) Number: DMD M.2.1.004. |
[65] | Johnson LA , Olsen RH , Merkens LS , DeBarber A , Steiner RD , Sullivan PM , Maeda N , Raber J ((2014) ) Apolipoprotein E-low density lipoprotein receptor interaction affects spatial memory retention and brain ApoE levels in an isoform-dependent manner. Neurobiol Dis 64: , 150–162. |
[66] | Olsen RH , Johnson LA , Zuloaga DG , Limoli CL , Raber J ((2013) ) Enhanced hippocampus-dependent memory and reduced anxiety in mice over-expressing human catalase in mitochondria. J Neurochem 125: , 303–313. |
[67] | Bocchio M , Nabavi S , Capogna M ((2017) ) Synaptic plasticity, engrams, and network oscillations in amygdala circuits for storage and retrieval of emotional memories. Neuron 94: , 731–743. |
[68] | Curzon P , Rustay NR , Browman KE ((2009) ) Cued and contextual fear conditioning for rodents. In Methods of Behavior Analysis in Neuroscience, Buccafusco JJ, ed. CRC Press/Taylor & Francis, Boca Raton (FL). |
[69] | Lee BR , Kamitani T ((2011) ) Improved immunodetection of endogenous alpha-synuclein. PLoS One 6: , e23939. |
[70] | Sasaki A , Arawaka S , Sato H , Kato T ((2015) ) Sensitive western blotting for detection of endogenous Ser129-phosphorylated alpha-synuclein in intracellular and extracellular spaces. Sci Rep 5: , 14211. |
[71] | Van Den Berge N , Ferreira N , Gram H , Mikkelsen TW , Alstrup AKO , Casadei N , Tsung-Pin P , Riess O , Nyengaard JR , Tamguney G , Jensen PH , Borghammer P ((2019) ) Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol 138: , 535–550. |
[72] | Ferreira N , Goncalves NP , Jan A , Jensen NM , van der Laan A , Mohseni S , Vaegter CB , Jensen PH ((2021) ) Trans-synaptic spreading of alpha-synuclein pathology through sensory afferents leads to sensory nerve degeneration and neuropathic pain. Acta Neuropathol Commun 9: , 31. |
[73] | Giovannoni G , van Schalkwyk J , Fritz VU , Lees AJ ((1999) ) Bradykinesia akinesia inco-ordination test (BRAIN TEST): An objective computerised assessment of upper limb motor function. J Neurol Neurosurg Psychiatry 67: , 624–629. |
[74] | Berardelli A , Rothwell JC , Thompson PD , Hallett M ((2001) ) Pathophysiology of bradykinesia in Parkinson’s disease. Brain 124: , 2131–2146. |
[75] | Karampetsou M , Ardah MT , Semitekolou M , Polissidis A , Samiotaki M , Kalomoiri M , Majbour N , Xanthou G , El-Agnaf OMA , Vekrellis K ((2017) ) Phosphorylated exogenous alpha-synuclein fibrils exacerbate pathology and induce neuronal dysfunction in mice. Sci Rep 7: , 16533. |
[76] | Calabresi P , Castrioto A , Di Filippo M , Picconi B ((2013) ) New experimental and clinical links between the hippocampus and the dopaminergic system in Parkinson’s disease. Lancet Neurol 12: , 811–821. |
[77] | Wittmann BC , Schott BH , Guderian S , Frey JU , Heinze HJ , Duzel E ((2005) ) Reward-related FMRI activation of dopaminergic midbrain is associated with enhanced hippocampus-dependent long-term memory formation. Neuron 45: , 459–467. |
[78] | Costa C , Sgobio C , Siliquini S , Tozzi A , Tantucci M , Ghiglieri V , Di Filippo M , Pendolino V , de Iure A , Marti M , Morari M , Spillantini MG , Latagliata EC , Pascucci T , Puglisi-Allegra S , Gardoni F , Di Luca M , Picconi B , Calabresi P ((2012) ) Mechanisms underlying the impairment of hippocampal long-term potentiation and memory in experimental Parkinson’s disease. Brain 135: , 1884–1899. |
[79] | Burman DD ((2019) ) Hippocampal connectivity with sensorimotor cortex during volitional finger movements: Laterality and relationship to motor learning. PLoS One 14: , e0222064. |
[80] | Sahni V , Engmann A , Ozkan A , Macklis JD ((2020) ) Chapter 8 - Motor cortex connections. In Neural Circuit and Cognitive Development (Second Edition), Rubenstein J, Rakic P, Chen B, Kwan KY, eds. Academic Press, pp. 167–199. |
[81] | Ballinger EC , Ananth M , Talmage DA , Role LW ((2016) ) Basal forebrain cholinergic circuits and signaling in cognition and cognitive decline. Neuron 91: , 1199–1218. |
[82] | Cole TA , Zhao H , Collier TJ , Sandoval I , Sortwell CE , Steece-Collier K , Daley BF , Booms A , Lipton J , Welch M , Berman M , Jandreski L , Graham D , Weihofen A , Celano S , Schulz E , Cole-Strauss A , Luna E , Quach D , Mohan A , Bennett CF , Swayze EE , Kordasiewicz HB , Luk KC , Paumier KL ((2021) ) alpha-Synuclein antisense oligonucleotides as a disease-modifying therapy for Parkinson’s disease. JCI Insight 6: , e135633. |
[83] | Alarcon-Aris D , Recasens A , Galofre M , Carballo-Carbajal I , Zacchi N , Ruiz-Bronchal E , Pavia-Collado R , Chica R , Ferres-Coy A , Santos M , Revilla R , Montefeltro A , Farinas I , Artigas F , Vila M , Bortolozzi A ((2018) ) Selective alpha-synuclein knockdown in monoamine neurons by intranasal oligonucleotide delivery: Potential therapy for Parkinson’s disease. Mol Ther 26: , 550–567. |
[84] | Winner B , Regensburger M , Schreglmann S , Boyer L , Prots I , Rockenstein E , Mante M , Zhao C , Winkler J , Masliah E , Gage FH ((2012) ) Role of alpha-synuclein in adult neurogenesis and neuronal maturation in the dentate gyrus. J Neurosci 32: , 16906–16916. |
[85] | Desplats P , Spencer B , Crews L , Pathel P , Morvinski-Friedmann D , Kosberg K , Roberts S , Patrick C , Winner B , Winkler J , Masliah E ((2012) ) alpha-Synuclein induces alterations in adult neurogenesis in Parkinson disease models via p53-mediated repression of Notch1. J Biol Chem 287: , 31691–31702. |
[86] | Vasudevaraju P , Guerrero E , Hegde ML , Collen TB , Britton GB , Rao KS ((2012) ) New evidence on alpha-synuclein and Tau binding to conformation and sequence specific GC* rich DNA: Relevance to neurological disorders. J Pharm Bioallied Sci 4: , 112–117. |
[87] | Chia R , Sabir MS , Bandres-Ciga S , Saez-Atienzar S , Reynolds RH , Gustavsson E , Walton RL , Ahmed S , Viollet C , Ding J , et al. ((2021) ) Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat Genet 53: , 294–303. |
[88] | Collier TJ , Redmond DE Jr. , Steece-Collier K , Lipton JW , Manfredsson FP ((2016) ) Is alpha-synuclein loss-of-function a contributor to Parkinsonian pathology? Evidence from non-human primates. Front Neurosci 10: , 12. |
[89] | Kanaan NM , Manfredsson FP ((2012) ) Loss of functional alpha-synuclein: A toxic event in Parkinson’s disease? J Parkinsons Dis 2: , 249–267. |
[90] | Oikawa T , Nonaka T , Terada M , Tamaoka A , Hisanaga S , Hasegawa M ((2016) ) alpha-synuclein fibrils exhibit gain of toxic function, promoting tau aggregation and inhibiting microtubule assembly. J Biol Chem 291: , 15046–15056. |
[91] | Rajagopalan S , Andersen JK ((2001) ) Alpha synuclein aggregation: Is it the toxic gain of function responsible for neurodegeneration in Parkinson’s disease? Mech Ageing Dev 122: , 1499–1510. |
[92] | Al-Wandi A , Ninkina N , Millership S , Williamson SJ , Jones PA , Buchman VL ((2010) ) Absence of alpha-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol Aging 31: , 796–804. |
[93] | Tagliafierro L , Chiba-Falek O ((2016) ) Up-regulation of SNCA gene expression: Implications to synucleinopathies. Neurogenetics 17: , 145–157. |
[94] | Abeliovich A , Schmitz Y , Farinas I , Choi-Lundberg D , Ho WH , Castillo PE , Shinsky N , Verdugo JM , Armanini M , Ryan A , Hynes M , Phillips H , Sulzer D , Rosenthal A ((2000) ) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25: , 239–252. |
[95] | Chandra S , Gallardo G , Fernandez-Chacon R , Schluter OM , Sudhof TC ((2005) ) Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123: , 383–396. |
[96] | Cabin DE , Shimazu K , Murphy D , Cole NB , Gottschalk W , McIlwain KL , Orrison B , Chen A , Ellis CE , Paylor R , Lu B , Nussbaum RL ((2002) ) Synaptic vesicle depletion correlates with attenuated synaptic responses to prolonged repetitive stimulation in mice lacking alpha-synuclein. J Neurosci 22: , 8797–8807. |
[97] | Goldberg MS , Lansbury PT , Jr. ((2000) ) Is there a cause-and-effect relationship between alpha-synuclein fibrillization and Parkinson’s disease? Nat Cell Biol 2: , E115–119. |
[98] | Greten-Harrison B , Polydoro M , Morimoto-Tomita M , Diao L , Williams AM , Nie EH , Makani S , Tian N , Castillo PE , Buchman VL , Chandra SS ((2010) ) alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A 107: , 19573–19578. |
[99] | Ninkina N , Tarasova TV , Chaprov KD , Roman AY , Kukharsky MS , Kolik LG , Ovchinnikov R , Ustyugov AA , Durnev AD , Buchman VL ((2020) ) Alterations in the nigrostriatal system following conditional inactivation of alpha-synuclein in neurons of adult and aging mice. Neurobiol Aging 91: , 76–87. |
[100] | Gorbatyuk OS , Li S , Nash K , Gorbatyuk M , Lewin AS , Sullivan LF , Mandel RJ , Chen W , Meyers C , Manfredsson FP , Muzyczka N ((2010) ) In vivo RNAi-mediated alpha-synuclein silencing induces nigrostriatal degeneration. Mol Ther 18: , 1450–1457. |
[101] | Khodr CE , Sapru MK , Pedapati J , Han Y , West NC , Kells AP , Bankiewicz KS , Bohn MC ((2011) ) An alpha-synuclein AAV gene silencing vector ameliorates a behavioral deficit in a rat model of Parkinson’s disease, but displays toxicity in dopamine neurons. Brain Res 1395: , 94–107. |
[102] | Benskey MJ , Sellnow RC , Sandoval IM , Sortwell CE , Lipton JW , Manfredsson FP ((2018) ) Silencing alpha synuclein in mature nigral neurons results in rapid neuroinflammation and subsequent toxicity. Front Mol Neurosci 11: , 36. |
[103] | Zharikov A , Bai Q , De Miranda BR , Van Laar A , Greenamyre JT , Burton EA ((2019) ) Long-term RNAi knockdown of alpha-synuclein in the adult rat substantia nigra without neurodegeneration. Neurobiol Dis 125: , 146–153. |
[104] | Uehara T , Choong CJ , Nakamori M , Hayakawa H , Nishiyama K , Kasahara Y , Baba K , Nagata T , Yokota T , Tsuda H , Obika S , Mochizuki H ((2019) ) Amido-bridged nucleic acid (AmNA)-modified antisense oligonucleotides targeting alpha-synuclein as a novel therapy for Parkinson’s disease. Sci Rep 9: , 7567. |
[105] | Schenk DB , Koller M , Ness DK , Griffith SG , Grundman M , Zago W , Soto J , Atiee G , Ostrowitzki S , Kinney GG ((2017) ) First-in-human assessment of PRX002, an anti-alpha-synuclein monoclonal antibody, in healthy volunteers. Mov Disord 32: , 211–218. |
[106] | Brys M , Fanning L , Hung S , Ellenbogen A , Penner N , Yang M , Welch M , Koenig E , David E , Fox T , Makh S , Aldred J , Goodman I , Pepinsky B , Liu Y , Graham D , Weihofen A , Cedarbaum JM ((2019) ) Randomized phase I clinical trial of anti-alpha-synuclein antibody BIIB054. Mov Disord 34: , 1154–1163. |


