
LRRK2 Kinase Activity Does Not Alter Cell-Autonomous Tau Pathology Development in Primary Neurons
Abstract
Background:
Mutations in leucine-rich repeat kinase 2 (LRRK2) are the most common cause of familial Parkinson’s disease (PD) and are also associated with genetic risk in idiopathic PD. Mutations in LRRK2, including the most common p.G2019S lead to elevated kinase activity, making LRRK2 kinase inhibitors prime targets for therapeutic development. However, the role of LRRK2 kinase activity in PD pathogenesis has remained unclear. While essentially all LRRK2-PD patients exhibit dopaminergic neuron loss, many of these patients do not have α-synuclein Lewy bodies in their brains. So, what is the neuropathological substrate of LRRK2-PD? Tau has emerged as a possible candidate due to the presence of tau pathology in the majority of LRRK2 mutation carriers and reports of hyperphosphorylated tau in LRRK2 animal models.
Objective:
In the current study, we aim to address whether a mutation in LRRK2 changes the cell-autonomous seeding of tau pathology in primary neurons. We also aim to assess whether LRRK2 kinase inhibitors are able to modulate tau pathology.
Methods/Results:
Treatment of primary neurons with LRRK2 kinase inhibitors leads to prolonged kinase inhibition but does not alter tau pathology induction. The lack of an effect of LRRK2 kinase activity was further confirmed in primary neurons expressing LRRK2G2019S and with two different forms of pathogenic tau. In no case was there more than a minor change in tau pathology induction.
Conclusion:
Together, our results indicate that LRRK2 kinase activity is not playing a major role in the induction of tau pathology in individual neurons. Understanding the impact of LRRK2 kinase inhibitors on pathology generation is important as kinase inhibitors move forward in clinical trials.
INTRODUCTION
Parkinson’s disease (PD) afflicts over 6 million people worldwide [1]. Multiple factors, including lifestyle, exposure to environmental pathogens, and genetic variants can influence risk of developing PD. Up to 27%of PD has been estimated to be heritable [2, 3] through both rare mutations and more common polymorphisms in the genome. The most common cause of familial PD and a common risk factor for idiopathic PD is a mutation in the gene encoding leucine-rich repeat kinase 2 (LRRK2) [4]. The most prevalent of these mutations, p.G2019S, confers a 25–42.5%risk of developing PD [5]. Although these patients show similar symptoms to idiopathic PD, neuropathologically, 21–54%of patients lack the hallmark α-synuclein Lewy bodies exhibited by idiopathic PD patients [6–8]. Notably, the majority of LRRK2 mutation carriers exhibit tau pathology [8, 9]. The tau pathology in idiopathic PD and LRRK2-PD is similar in conformation and distribution to AD tau and may be partially responsible for the cognitive decline seen in these patients during the disease course [8, 10]. The appearance of tau pathology in LRRK2 mutation carriers suggests that the genetic risk conferred by LRRK2 mutations may alter the development or progression of tau pathology. If LRRK2 mutations do in fact modulate tau pathology, then LRRK2 kinase inhibitors being developed for the treatment of genetic and idiopathic PD may influence tau pathology in these patients, and possibly in AD and other tauopathy patients as well.
In the current study, we seeded tau pathology in primary neuron cultures from wildtype mice and mice expressing LRRK2G2019S to determine whether tau pathology was altered in a cell-autonomous manner. We show, using two different forms of pathogenic tau, that LRRK2G2019S expression does not substantially alter the development of tau pathology. We also show that three LRRK2 kinase inhibitors show strong and sustained inhibition of LRRK2 kinase activity but have no impact on cell-autonomous tau pathology development. This study suggests that LRRK2 kinase activity does not have a substantial role in the initial development of tau pathology, but it does not preclude a role for LRRK2 in the cell-to-cell transmission of tau or long-term effects which are not recapitulated in cell culture.
MATERIALS AND METHODS
Mice
All housing, breeding, and procedures were performed according to the NIH Guide for the Care and Use of Experimental Animals and approved by the University of Pennsylvania Institutional Animal Care and Use Committee. C57BL/6J (NTG, JAX 000664, RRID: IMSR_JAX:000664) and B6.Cg-Tg(Lrrk2*G2019S)2Yue/J (G2019S, JAX 012467, RRID: IMSR_JAX:012467) mice have been previously described [11]. The current G2019S BAC line was backcrossed to C57BL/6J mice for > 10 generations and bred to homozygosity at loci as determined by quantitative PCR and outbreeding. The expression level of G2019S LRRK2 was thereby stabilized in this line of mice. All experiments shown use homozygous G2019S mice.
Primary hippocampal or neuron cultures
Primary hippocampal neuron cultures were prepared as previously described [12] from postnatal day (P) 1 non-transgenic or LRRK2G2019S transgenic mice. Dissociated hippocampal or cortical neurons were plated at 17,000 cells/well (96-well plate) or 1,000,000 cells/well (6-well plate) in neuron media (Neurobasal medium (ThermoFisher 21103049) supplemented with B27 (ThermoFisher 17504044), 2 mM GlutaMax (ThermoFisher 35050061), and 100 U/mL penicillin/ streptomycin (ThermoFisher 15140122).
Recombinant tau PFFs
Purification of recombinant human tau and gene-ration of tau X-T40 PFFs was conducted as des-cribed elsewhere [13]. The plasmid containing the gene of interest was transformed into BL21 (DE3) RIL-competent E. coli (Agilent Technologies Cat#230245). A single colony from this transformation was expanded and tau protein was purified by cationic exchange using fast protein liquid chromatography as previously described [14].
Tau fibrillization was induced in the absence of co-factors by incubation of 40μM recombinant T40 (4R2N tau) monomer with 2 mM DTT in Dulbecco’s phosphate-buffered saline (DPBS, Corning Cat#21-031-CV) and incubated shaking at 1,000 rpm and at 37°C for 7 days to create passage 1 of fibrils. A sec-ond passage was set up by incubating 10%of the first passage with 90%fresh tau monomer at a final concentration of 40μM tau. This passaging was repeated until tau was recovered in the pellet fraction in a stochastic subset of the reactions. Conversion to PFFs was validated by sedimentation at 100,000 x g for 30 minutes at 22°C. Equal volumes of supernatant and pellet fraction were loaded and analyzed by Coomassie blue staining of SDS-PAGE gels.
Human tissue
All procedures were done in accordance with local institutional review board guidelines of the University of Pennsylvania. Written informed consent for autopsy and analysis of tissue sample data was obtained either from patients themselves or their next of kin. Cases used for extraction (Table 1) of PHF tau were selected based upon a high burden of tau pathology by immunohistochemical staining.
Table 1
Characterization of AD tau preparations. Descriptive information related to the cases of AD PHFs tau prepared for these studies. PMI, postmortem interval
| Case | Age | Sex | PMI (hours) | Total protein (μg/μL) | Tau (μg/μL) | Tau (%protein) | α-Syn (μg/mL) | Aβ 1–40 (ng/mL) | Aβ 1–42 (ng/mL) |
| AD Case 1 | 73 | F | 4 | 8.3 | 2.6 | 31.3 | 0.7 | 31.2 | 111.4 |
| AD Case 2 | 62 | M | 12 | 8.7 | 1.4 | 16.1 | 0.5 | 53.7 | 60.1 |
Human brain sequential detergent fractionation
Frozen postmortem human frontal or temporal cor-tex brain tissue containing abundant tau-positive in-clusions was selected for sequential extraction based on IHC examination of these samples as described [15] using previously established methods. These brains were sequentially extracted with increasing detergent strength as previously described [13]. After thawing, meninges were removed and gray matter was carefully separated from white matter. Gray matter was weighed and suspended in nine volumes (w/v) high salt (HS) buffer (10 mM Tris-HCL (pH 7.4), 800 mM NaCl, 1 mM EDTA, 2 mM dithioth-reitol [DTT], protease and phosphatase inhibitors and PMSF) with 0.1%sarkosyl and 10%sucrose, followed by homogenization with a dounce homogenizer and centrifugation at 10,000 x g for 10 min at 4°C. The resulting pellet was re-extracted with the same buffer conditions and the supernatants from all extractions were filtered and pooled.
Additional sarkosyl was added to the pooled supernatant to reach a final concentration of 1%and the supernatant was nutated for 1 h at room temperature. The samples were then centrifuged at 300,000 x g for 60 min at 4°C. The pellet, which contains pathological tau, was washed once with PBS and resuspended in 100μL of PBS per gram of gray matter by passing through a 27G/0.5-in. needle. The pellets were further suspended by brief sonication (QSonica Microson™ XL-2000; 20 pulses; setting 2; 0.5 s/pulse). The suspension was centrifuged at 100,000 x g for 30 min at 4°C. The pellet was suspended in one-fifth to one-half the pre-centrifugation volume, sonicated briefly (60–120 pulses; setting 2; 0.5 s/pulse) and centrifuged at 10,000 x g for 30 min at 4°C. The final supernatant was utilized for all studies and is referred to as AD PHF tau. All extractions were characterized by western blotting, sandwich ELISA for tau, α-synuclein and Aβ 1–40, Aβ 1–42, and validated by immunocytochemistry in primary neurons from non-transgenic mice. For the extractions used in this study, tau constituted 16.1–35.7%of the total protein, while α-synuclein and Aβ constituted 0.011%or less of total protein.
LRRK2 inhibitor treatments
LRRK2 inhibitors PF-475 and PF-360 were synthesized at Pfizer, Inc. MLi-2 was obtained from Tocris Bioscience (Cat#5756). All LRRK2 inhibitors were reconstituted at 10 mM in DMSO and stored at –20°C. They were further diluted to the final concentration indicated in neuron media with DMSO as a vehicle control.
Tau XT-40 PFF/AD PHF treatments
Primary neurons
For treatment of neurons, X-T40 PFF tau and AD PHF tau were vortexed and diluted with Dulbecco’s phosphate-buffered saline (DPBS, Corning Cat#21-031-CV). They were then sonicated on high for 10 cycles of 30 s on, followed by 30 s off (Diagenode Biorupter UCD-300 bath sonicator). Tau was then diluted in neuron media to the noted concentrations and added to neuron cultures at 7 days in-vitro (DIV). Neuron cultures were harvested 14–21 days post-treatment (DPT), as noted.
Immunoblotting
Total protein concentration in each sample was determined by a bicinchoninic acid colorimetric assay (Fisher Cat#23223 and 23224), using bovine serum albumin as a standard (Thermo Fisher Cat#23210). Protein was resolved on 5–20%gradient pol-yacrylamide gels using equal protein loading. Pro-teins were transferred to 0.2μm nitrocellulose or PVDF membranes and detected with primary antibo-dies targeting LRRK2 (ab133474, Abcam, RRID: AB_2713963, 1:500), pS935 LRRK2 (ab133450, Abcam, RRID:AB_2732035, 1:400) or GAPDH (2-RGM2, Advanced Immunological, RRID:AB_2721282, 1:5000). Primary antibodies were detected using IRDye 800 (Li-cor 925-32210) or IRDye 680 (Li-cor 925-68071) secondary antibodies, scanned on Li-cor Odyssey Imaging System and analyzed using Image Studio software. LRRK2 and pS935 LRRK2 values were normalized to GAPDH as an internal loading control, then further normalized to the mean of all control samples.
Immunocytochemistry
Primary neurons treated with X-T40 tau PFFs were fixed with 4%paraformaldehyde, 4%sucrose in phosphate-buffered saline and washed five times in PBS. Cells were permeabilized in 3%BSA + 0.3%TX-100 in PBS for 15 min at room temperature. Af-ter a PBS wash, cells were blocked for 50 min with 3%BSA in PBS prior to incubation with pri-mary antibodies for 2 h at room temperature. Primary antibodies used were targeting pS202/T205 tau (AT8, ThermoFisher Cat#MN1020, 1:1000), MAP2 (17028, CNDR, 1:2000). Cells were washed 5x with PBS and incubated with secondary antibodies for 1 h at room temperature. After 5x wash with PBS, cells were incubated in DAPI (ThermoFisher Cat#D21490, 1:10,000) in PBS. Primary neurons tre-ated with AD PHF tau were stained differently due to the possible presence of p-tau signal in the human-derived material added to cultures. Soluble protein was extract with 2%HDTA for 10 min at room temperature. Neurons were then fixed and stained as above, except that a primary antibody that selectively binds mouse tau (T49, CNDR, 1:2500) was utilized to detect neuronal tau inclusions.
96-well plates were imaged on an In Cell Analyzer 2200 (GE Healthcare) and analyzed in the accompanying software. A standard intensity-based threshold was applied to MAP2 and p-tau or mouse tau channels equally across plates and the positive area was quantified. All quantification was optimized and applied equally across all conditions.
Quantification and statistical analysis
The number of samples analyzed in each experiment, the statistical analysis performed, as well as the p-values for all results < 0.05 are reported in the figure legends. For all cell culture experiments, “n” represents the number of separate cultures (e.g., one scraped or imaged well is reported as one “n”). All cell culture data were analyzed in GraphPad Prism 7 using the noted statistical tests.
RESULTS
LRRK2 kinase inhibitors show prolonged inhibition of phosphorylation in primary neurons
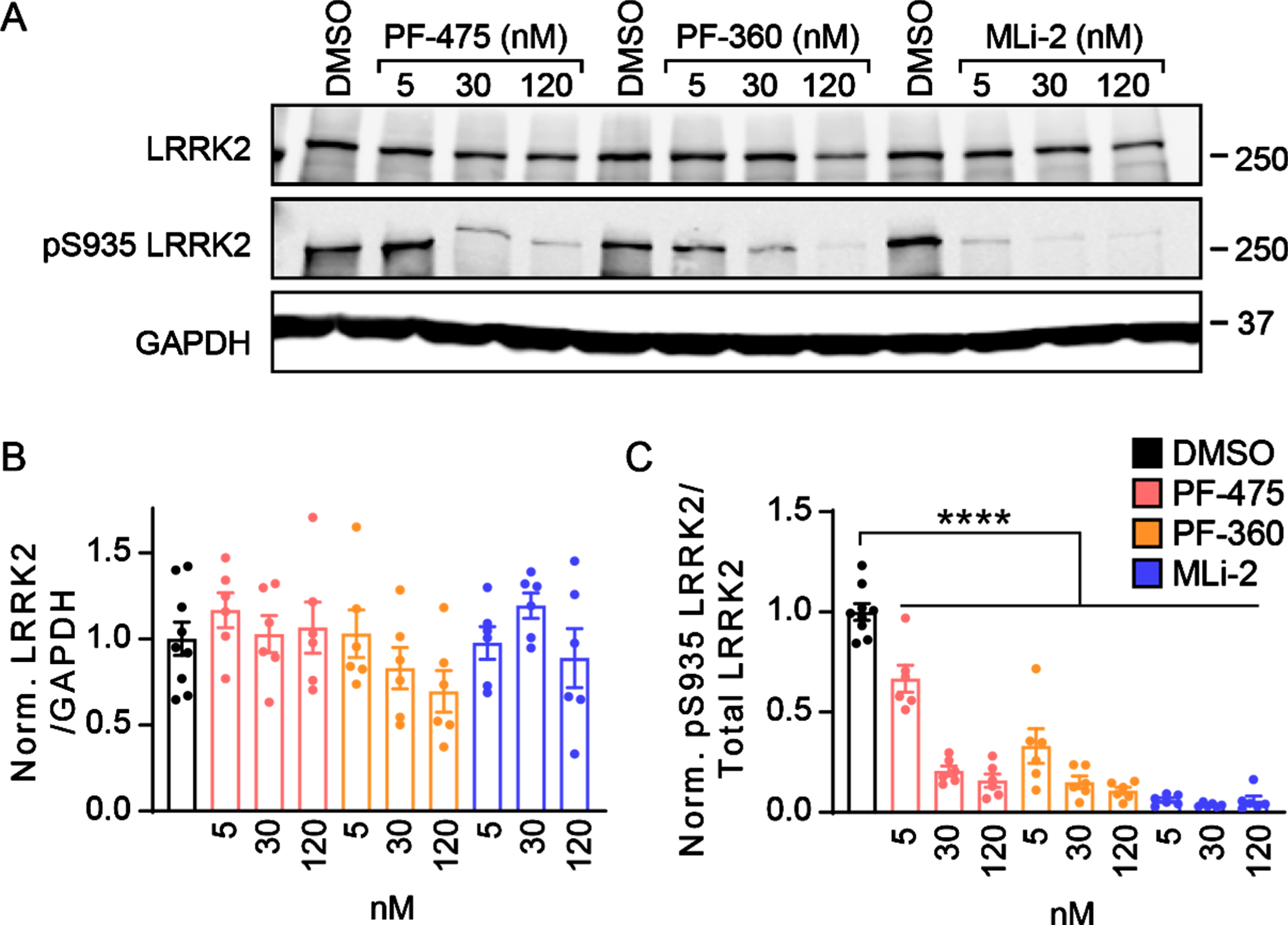
Primary neuron cultures provide a rapid and acc-essible method for the evaluation of cellular pheno-types and the efficacy of therapeutics. Several LRRK2 kinase inhibitors have been developed which show potent inhibition of LRRK2 kinase activity, but their effect on neuropathology, particularly tau, is still unknown. We first tested whether LRRK2 kinase activity could be inhibited in primary neurons over prolonged incubation periods. To ensure kinase inhibition even when LRRK2 is overexpressed, we cul-tured primary neurons from LRRK2G2019S transgenic mice (B6.Cg-Tg(Lrrk2*G2019S)2Yue/J) which ove-rexpress LRRK2G2019S [11, 12]. Primary neurons were treated at 5 DIV with one of three LRRK2 inhibitors—PF-06447475 (PF-475), PF-06685360 (PF-360), and MLi-2 at 5, 30, or 120 nM concentration. After 16 days, neuronal proteins were extracted and run by western blot to determine LRRK2 kinase activity (Fig. 1). Total LRRK2 and pS935 LRRK2 were monitored as a common and reliable readout for LRRK2 kinase activity [16–19]. All three compounds were able to reduce pS935 LRRK2 levels strongly at low nanomolar concentrations with minimal disruption of total LRRK2 levels (Fig. 1A-C).
Fig. 1
LRRK2 kinase inhibitors show prolonged inhibition of phosphorylation in primary neurons. A) Primary cortical neurons from LRRK2G2019S mice were treated at 5 days in vitro (DIV) with LRRK2 inhibitors at the noted concentrations and lysate was harvested at 21 DIV. Western blot is shown for total LRRK2 and pS935 LRRK2, the latter being a proxy for LRRK2 activity. B) Quantification of total LRRK2 normalized to GAPDH and vehicle treatment (One-way ANOVA; Dunnett’s multiple comparison test: all p > 0.05., n = 6–9 samples/group). (C) Quantification of pS935 normalized to total LRRK2 and vehicle treatment (One-way ANOVA; Dunnett’s multiple comparison test: all ****p < 0.0001 as compared to vehicle treatment, n (separate cultures) = 6–9 independent samples/group).
Wildtype and LRRK2G2019S neurons exhibit robust, inhibitor-insensitive tau pathology
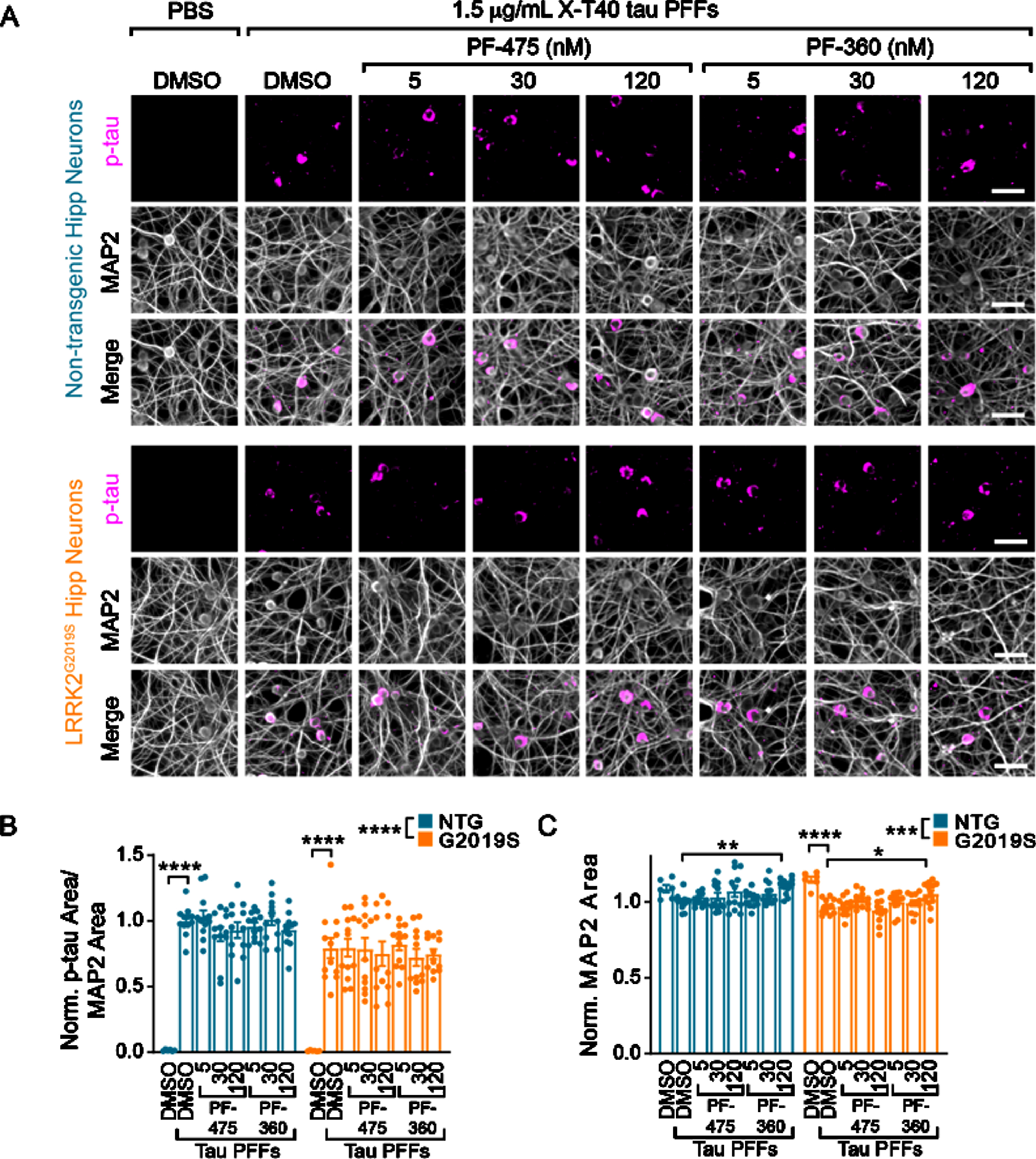
Having established the ability to inhibit LRRK2 kinase activity levels over weeks, we next sought to investigate the impact of LRRK2 kinase activity on tau pathology development (Fig. 2). Tau pathology can be induced in non-transgenic neurons by seeding pathology with recombinant tau pre-formed fibrils (PFFs). PFFs were generated by shaking recombinant tau, sonicating and passaging into monomer sequentially in a manner that stochastically generates fibrillar tau without the need for a co-factor such as heparin [13]. These “X-T40” PFFs can be sonicated and added to NTG primary neurons and will seed the misfolding of endogenous tau. Neither NTG, nor LRRK2G2019S neurons have detectable levels of pS202/T205 tau without the addition of X-T40 PFFs, but PFFs induce robust tau inclusions in neurons (Fig. 2A). Interestingly, we observed a slight reduction of tau pathology in LRRK2G2019S neurons, with no apparent effect of LRRK2 kinase inhibition (Fig. 2B). There was a small reduction in overall dendritic area (MAP2) with PFF treatment that was reversed with the highest dose of PF-360 (Fig. 2C).
Fig. 2
Wildtype and LRRK2G2019S neurons exhibit robust, inhibitor-insensitive tau pathology. A) Primary hippocampal neurons from NTG or LRRK2G2019S mice were treated with vehicle or LRRK2 inhibitors at the noted concentrations at 5 DIV. They were further treated with X-T40 tau PFFs at 1.5μg/mL at 7 DIV and fixed and stained for pS202/T205 tau (AT8, magenta) and MAP2 (gray) at 21 DIV. Scale bar = 50μm. B) Quantification of the pS202/T205 tau area normalized to MAP2 area and further normalized to NTG-DMSO-PFF. LRRK2G2019S neurons showed a small, genotype-level significant reduction in tau pathology, and each genotype showed no tau pathology without addition of X-T40 tau PFFs (Two-way ANOVA; genotype effect ****p < 0.0001, Dunnett’s multiple comparison test within genotype: NTG: DMSO-PFF vs. DMSO****p < 0.0001; G2019S: DMSO-PFF vs. DMSO ****p < 0.0001; All other values were not statistically significant). C) Quantification of the MAP2 area also showed a small genotype-level significant change as well as a reduction related to PFF treatment. Interestingly, the highest dose of PF-360 also elevated MAP2 area slightly (Two-way ANOVA; genotype effect ***p = 0.001, Dunnett’s multiple comparison test within genotype: NTG: DMSO-PFF vs. 120 nM PF-360-PFF **p = 0.0027; G2019S: DMSO-PFF vs. DMSO ****p < 0.0001; DMSO-PFF vs. 120 nM PF-360 *p = 0.0216; All other values were not statistically significant). n (separate cultures) = 7-8 independent samples/group. Data are represented as mean±SEM with individual data points plotted.
Tau from AD brains induces inhibitor-insensitive tau pathology in wildtype and LRRK2G2019S neurons
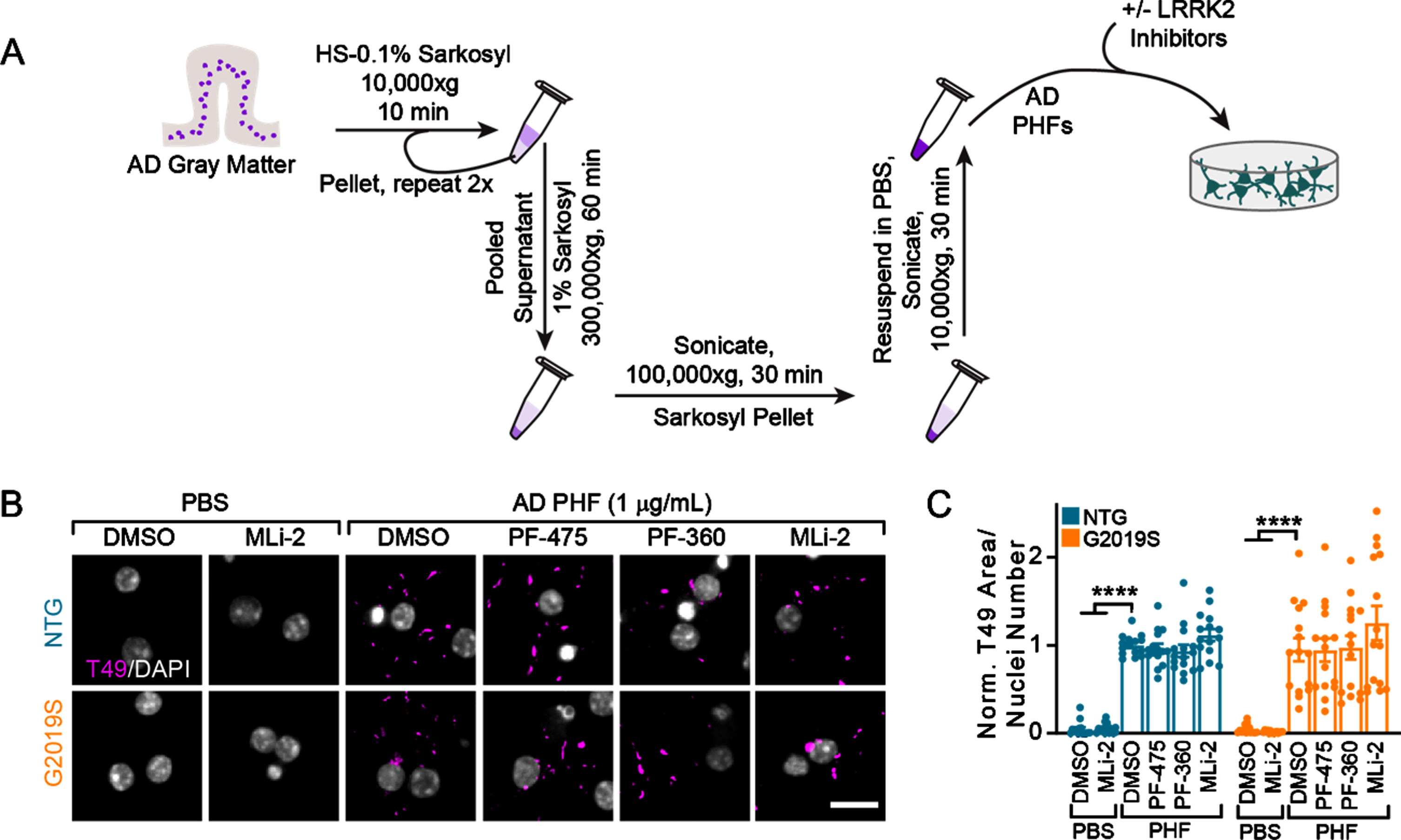
Recent structural biology studies have established that recombinant tau fibrils are not the same as human brain-derived tau fibrils [20]. Further, the structure of tau fibrils differs between diseases [20]. We therefore sought to induce pathology using human-derived tau. The majority of tau pathology in LRRK2 mutation carriers is Alzheimer’s disease (AD) type tau, as recognized by a selective AD tau antibody [8]. We utilized a biochemical sequential detergent extraction of gray matter from AD patient brains to obtain an en-riched fraction of paired helical filament (PHF) tau (Table 1, Fig. 3A) [13]. This extraction method yie-lded a final purity of 16.1–31.3%tau, with 0.01%or less α-synuclein and amyloid β (Table 1). The purified tau fraction retains the pathogenic conformation present in human disease and induces the misfolding of tau in primary neurons and mice without the overexpression of tau [13]. AD tau therefore serves as a valuable tool to assess tau pathology in different genetic backgrounds. Primary hippocampal neurons from NTG or LRRK2G2019S mice were treated with vehicle or LRRK2 inhibitors at 5 DIV followed by treatment with DPBS or AD tau at 7 DIV (Fig. 3B). Notably, neither NTG nor LRRK2G2019S neurons form tau inclusions without the addition of PHF tau. However, 14 days post-treatment with AD tau, neurons form detergent-insoluble tau inclusions from endogenous mouse tau, which do not differ by neuron genotype. Further, LRRK2 kinase inhibition had no effect on tau pathology in these neurons (Fig. 3B,3C).
Fig. 3
Tau from AD brains induces inhibitor-insensitive tau pathology in wildtype and LRRK2G2019S neurons. A) Schematic diagram of AD PHF tau extraction from human brain tissue and subsequent treatment of primary neurons. B) Primary hippocampal neurons from NTG or LRRK2G2019S mice were treated with vehicle or LRRK2 inhibitors at 300 nM at 5 DIV. They were further treated with AD PHF tau at 1μg/mL at 7 DIV, and both fixed and stained for insoluble mouse tau (T49, magenta) and DAPI (gray) at 21 DIV. Scale bar = 15μm. C) Quantification of the insoluble mouse tau area/nuclei number in each condition. No insoluble tau was present in the absence of PHF treatment and there was no overall effect of genotype (Two-way ANOVA; genotype effect p = 0.837, Dunnett’s multiple comparison test within genotype: NTG: DMSO-PHF vs. DMSO ****p < 0.0001; DMSO-PHF vs. MLi-2 ****p < 0.0001; G2019S:: DMSO-PHF vs. DMSO ****p < 0.0001; DMSO-PHF vs. MLi-2 ****p < 0.0001; all other values were not statistically significant, n (separate cultures) = 15 independent samples/group). Data are represented as mean±SEM with individual data points plotted.
Together, these results suggest that LRRK2G2019S and LRRK2 kinase inhibition have a minimal effect on the cell-autonomous production of tau pathology in neurons. However, primary neuron cultures do not allow for the assessment of the impact of LRRK2 on tau pathology over long time periods, effects on cell-to-cell spread, or non-cell autonomous effects.
DISCUSSION
PD is a heterogenous clinical syndrome. In contrast, patients with a similar genetic risk factor of-ten have more homogenous clinical symptoms, and therefore are a good group in which to investigate underlying disease mechanisms [21]. It is therefore surprising that disease pathology is so heterogenous in LRRK2-PD patients, with 21–54%of patients lacking the pathognomonic Lewy body inclusions [6–8]. These neuropathological findings make it un-likely that LRRK2 leads to neuron loss only through α-synuclein aggregation. This supposition is bolstered by the lack of α-synuclein pathology in LRRK2 mutant mice [22, 23].
In contrast, most LRRK2 PD patients harbor some degree of tau pathology [6, 8, 9]. While LRRK2 mutations are rare in primary tauopathies, such as pro-gressive supranuclear palsy (PSP) [24, 25], a recent genome-wide association study found that genetic variance in the LRRK2 locus is associated with survival in PSP patients [26]. Cell [27] and animal [28–30] models expressing mutant LRRK2 have consistently been reported to accumulate hyperphosphorylated tau. Several reports have also shown that under certain conditions LRRK2 can directly phosphorylate tau [31–34]. These studies suggest that LRRK2 mutations may be predisposing neurons to the development of tau pathology. However, in the current study, we found no evidence in primary neurons that LRRK2 kinase activity can cause direct development of tau pathology, as evidenced by the lack of phosphorylated tau in the absence of tau seeding. Further, elevated LRRK2 kinase activity or LRRK2 kinase inhibition did not substantially alter the development of tau pathology following patho-genic tau seeding. Therefore, on the cell-autonomous level, LRRK2 kinase activity does not seem to play a major role in tau pathology development.
A limitation of this work is the nature of neuron cell culture. While neuron culture allows the rapid assessment of kinase inhibition and cellular phenotypes, cultures largely consist of homogenous neuron types with minimal glial growth. This precludes assessment of multiple neuron types, network spread of pathology, and non-cell autonomous effects of glia or peripheral immune cells. LRRK2 is highly expressed in the innate immune system and LRRK2 mutations have been associated with dysregulation of immune inflammatory pathways [35]. It is therefore possible that LRRK2G2019S impacts intact organisms through modulation of inflammation. Another possible mechanism by which LRRK2 could facilitate tau pathology progression is through enhanced neuronal release of tau, a mechanism which would make little difference in a cell culture dish. Enhanced synaptic vesicle release is one phenotype which has been associated with LRRK2G2019S expression in mice. Because tau is released from neurons [36–38] in an activity-dependent manner, changes in physiology by LRRK2 could also change pathology spread in a non-cell autonomous manner. One study in mice [39] showed that viral overexpression of tau in LRRKG2019S mice led to elevated tau in sites distal from the injection site, supporting enhanced spread of tau pathology with a LRRK2 mutation. Finally, multiple studies crossing tau transgenic mice to LRRK2 transgenic mice have found little effect of LRRK2 on tau pathology [39, 40], suggesting that when tau pathology develops synchronously across the brain, there is little effect of LRRK2 on pathology.
In conclusion, evidence suggests that tau pathology is not regulated in a cell-autonomous manner by LRRK2 kinase activity. However, there is substantial evidence from human case studies, cell and animal models, that LRRK2 is somehow promoting tau pathology spread. Future studies should investigate the mechanisms of tau pathology spread and how they relate to LRRK2 kinase activity.
ACKNOWLEDGMENTS
We would like to thank members of the laboratory for their feedback in developing this manuscript and Dr. Jing Guo for the initial description and development of X-T40 tau fibrils. This study was supported by The Michael J. Fox Foundation for Parkinson’s Research grant 16879 (M.X.H.); NIH grants: T32-AG000255 (V.M.Y.L), P30-AG10124 (J.Q.T.) U19-AG062418 (J.Q.T.), P50-NS053488 (J.Q.T.).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | GBD 2016 Parkinson’s Disease Collaborators ((2018) ) Global, regional, and national burden of Parkinson’s disease, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 17: , 939–953. |
[2] | Nalls MA , Blauwendraat C , Vallerga CL , Heilbron K , Bandres-Ciga S , Chang D , Tan M , Kia DA , Noyce AJ , Xue A , Bras J , Young E , von Coelln R , Simon-Sanchez J , Schulte C , Sharma M , Krohn L , Pihlstrom L , Siitonen A , Iwaki H , Leonard H , Faghri F , Gibbs JR , Hernandez DG , Scholz SW , Botia JA , Martinez M , Corvol JC , Lesage S , Jankovic J , Shulman LM , Sutherland M , Tienari P , Majamaa K , Toft M , Andreassen OA , Bangale T , Brice A , Yang J , Gan-Or Z , Gasser T , Heutink P , Shulman JM , Wood NW , Hinds DA , Hardy JA , Morris HR , Gratten J , Visscher PM , Graham RR , Singleton AB , 23andMe Research Team; System Genomics of Parkinson’s Disease Consortium; International Parkinson’s Disease Genomics Consortium ((2019) ) Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol 18: , 1091–1102. |
[3] | Goldman SM , Marek K , Ottman R , Meng C , Comyns K , Chan P , Ma J , Marras C , Langston JW , Ross GW , Tanner CM ((2019) ) Concordance for Parkinson’s disease in twins: A 20-year update. Ann Neurol 85: , 600–605. |
[4] | Healy DG , Falchi M , O’Sullivan SS , Bonifati V , Durr A , Bressman S , Brice A , Aasly J , Zabetian CP , Goldwurm S , Ferreira JJ , Tolosa E , Kay DM , Klein C , Williams DR , Marras C , Lang AE , Wszolek ZK , Berciano J , Schapira AH , Lynch T , Bhatia KP , Gasser T , Lees AJ , Wood NW ((2008) ) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol 7: , 583–590. |
[5] | Lee AJ , Wang Y , Alcalay RN , Mejia-Santana H , Saunders-Pullman R , Bressman S , Corvol JC , Brice A , Lesage S , Mangone G , Tolosa E , Pont-Sunyer C , Vilas D , Schule B , Kausar F , Foroud T , Berg D , Brockmann K , Goldwurm S , Siri C , Asselta R , Ruiz-Martinez J , Mondragon E , Marras C , Ghate T , Giladi N , Mirelman A , Marder K ((2017) ) Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov Disord 32: , 1432–1438. |
[6] | Poulopoulos M , Levy OA , Alcalay RN ((2012) ) The neuropathology of genetic Parkinson’s disease. Mov Disord 27: , 831–842. |
[7] | Kalia LV , Lang AE , Hazrati LN , Fujioka S , Wszolek ZK , Dickson DW , Ross OA , Van Deerlin VM , Trojanowski JQ , Hurtig HI , Alcalay RN , Marder KS , Clark LN , Gaig C , Tolosa E , Ruiz-Martinez J , Marti-Masso JF , Ferrer I , Lopez de Munain A , Goldman SM , Schule B , Langston JW , Aasly JO , Giordana MT , Bonifati V , Puschmann A , Canesi M , Pezzoli G , Maues De Paula A , Hasegawa K , Duyckaerts C , Brice A , Stoessl AJ , Marras C ((2015) ) Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol 72: , 100–105. |
[8] | Henderson MX , Sengupta M , Trojanowski JQ , Lee VMY ((2019) ) Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol Commun 7: , 183. |
[9] | Blauwendraat C , Pletnikova O , Geiger JT , Murphy NA , Abramzon Y , Rudow G , Mamais A , Sabir MS , Crain B , Ahmed S , Rosenthal LS , Bakker CC , Faghri F , Chia R , Ding J , Dawson TM , Pantelyat A , Albert MS , Nalls MA , Resnick SM , Ferrucci L , Cookson MR , Hillis AE , Troncoso JC , Scholz SW ((2019) ) Genetic analysis of neurodegenerative diseases in a pathology cohort. Neurobiol Aging 76: , 214 e211–214 e219. |
[10] | Irwin DJ , Grossman M , Weintraub D , Hurtig HI , Duda JE , Xie SX , Lee EB , Van Deerlin VM , Lopez OL , Kofler JK , Nelson PT , Jicha GA , Woltjer R , Quinn JF , Kaye J , Leverenz JB , Tsuang D , Longfellow K , Yearout D , Kukull W , Keene CD , Montine TJ , Zabetian CP , Trojanowski JQ ((2017) ) Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: A retrospective analysis. Lancet Neurol 16: , 55–65. |
[11] | Li X , Patel JC , Wang J , Avshalumov MV , Nicholson C , Buxbaum JD , Elder GA , Rice ME , Yue Z ((2010) ) Enhanced striatal dopamine transmission and motor performance with LRRK2 overexpression in mice is eliminated by familial Parkinson’s disease mutation G2019S. J Neurosci 30: , 1788–1797. |
[12] | Henderson MX , Peng C , Trojanowski JQ , Lee VMY ((2018) ) LRRK2 activity does not dramatically alter alpha-synuclein pathology in primary neurons. Acta Neuropathol Commun 6: , 45. |
[13] | Guo JL , Narasimhan S , Changolkar L , He Z , Stieber A , Zhang B , Gathagan RJ , Iba M , McBride JD , Trojanowski JQ , Lee VM ((2016) ) Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med 213: , 2635–2654. |
[14] | Li W , Lee VM ((2006) ) Characterization of two VQIXXK motifs for tau fibrillization in vitro. Biochemistry 45: , 15692–15701. |
[15] | Irwin DJ , White MT , Toledo JB , Xie SX , Robinson JL , Van Deerlin V , Lee VM , Leverenz JB , Montine TJ , Duda JE , Hurtig HI , Trojanowski JQ ((2012) ) Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 72: , 587–598. |
[16] | Fell MJ , Mirescu C , Basu K , Cheewatrakoolpong B , DeMong DE , Ellis JM , Hyde LA , Lin Y , Markgraf CG , Mei H , Miller M , Poulet FM , Scott JD , Smith MD , Yin Z , Zhou X , Parker EM , Kennedy ME , Morrow JA ((2015) ) MLi-2, a potent, selective, and centrally active compound for exploring the therapeutic potential and safety of LRRK2 kinase inhibition. J Pharmacol Exp Ther 355: , 397–409. |
[17] | Sheng Z , Zhang S , Bustos D , Kleinheinz T , Le Pichon CE , Dominguez SL , Solanoy HO , Drummond J , Zhang X , Ding X , Cai F , Song Q , Li X , Yue Z , van der Brug MP , Burdick DJ , Gunzner-Toste J , Chen H , Liu X , Estrada AA , Sweeney ZK , Scearce-Levie K , Moffat JG , Kirkpatrick DS , Zhu H ((2012) ) Ser1292 autophosphorylation is an indicator of LRRK2 kinase activity and contributes to the cellular effects of PD mutations. Sci Transl Med 4: , 164ra161. |
[18] | Dzamko N , Deak M , Hentati F , Reith AD , Prescott AR , Alessi DR , Nichols RJ ((2010) ) Inhibition of LRRK2 kinase activity leads to dephosphorylation of Ser(910)/Ser(935), disruption of 14-3-3 binding and altered cytoplasmic localization. Biochem J 430: , 405–413. |
[19] | Di Maio R , Hoffman EK , Rocha EM , Keeney MT , Sanders LH , De Miranda BR , Zharikov A , Van Laar A , Stepan AF , Lanz TA , Kofler JK , Burton EA , Alessi DR , Hastings TG , Greenamyre JT ((2018) ) LRRK2 activation in idiopathic Parkinson’s disease. Sci Transl Med 10: , eaar5429. |
[20] | Scheres SH , Zhang W , Falcon B , Goedert M ((2020) ) Cryo-EM structures of tau filaments. Curr Opin Struct Biol 64: , 17–25. |
[21] | McFarthing K , Buff S , Rafaloff G , Dominey T , Wyse RK , Stott SRW ((2020) ) Parkinson’s disease drug therapies in the clinical trial pipeline: 2020. J Parkinsons Dis 10: , 757–774. |
[22] | Herzig MC , Bidinosti M , Schweizer T , Hafner T , Stemmelen C , Weiss A , Danner S , Vidotto N , Stauffer D , Barske C , Mayer F , Schmid P , Rovelli G , van der Putten PH , Shimshek DR ((2012) ) High LRRK2 levels fail to induce or exacerbate neuronal alpha-synucleinopathy in mouse brain. PLoS One 7: , e36581. |
[23] | Henderson MX , Cornblath EJ , Darwich A , Zhang B , Brown H , Gathagan RJ , Sandler RM , Bassett DS , Trojanowski JQ , Lee VMY ((2019) ) Spread of α-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat Neurosci 22: , 1248–1257. |
[24] | Ross OA , Whittle AJ , Cobb SA , Hulihan MM , Lincoln SJ , Toft M , Farrer MJ , Dickson DW ((2006) ) Lrrk2 R1441 substitution and progressive supranuclear palsy. Neuropathol Appl Neurobiol 32: , 23–25. |
[25] | Sanchez-Contreras M , Heckman MG , Tacik P , Diehl N , Brown PH , Soto-Ortolaza AI , Christopher EA , Walton RL , Ross OA , Golbe LI , Graff-Radford N , Wszolek ZK , Dickson DW , Rademakers R ((2017) ) Study of LRRK2 variation in tauopathy: Progressive supranuclear palsy and corticobasal degeneration. Mov Disord 32: , 115–123. |
[26] | Jabbari E , Koga S , Valentino RR , Reynolds RH , Ferrari R , Tan MMX , Rowe JB , Dalgard CL , Scholz SW , Dickson DW , Warner TT , Revesz T , Hoglinger GU , Ross OA , Ryten M , Hardy J , Shoai M , Morris HR , PSP Genetics Group ((2021) ) Genetic determinants of survival in progressive supranuclear palsy: A genome-wide association study. Lancet Neurol 20: , 107–116. |
[27] | Ohta E , Nihira T , Uchino A , Imaizumi Y , Okada Y , Akamatsu W , Takahashi K , Hayakawa H , Nagai M , Ohyama M , Ryo M , Ogino M , Murayama S , Takashima A , Nishiyama K , Mizuno Y , Mochizuki H , Obata F , Okano H ((2015) ) I2020T mutant LRRK2 iPSC-derived neurons in the Sagamihara family exhibit increased Tau phosphorylation through the AKT/GSK-3beta signaling pathway. Hum Mol Genet 24: , 4879–4900. |
[28] | Li Y , Liu W , Oo TF , Wang L , Tang Y , Jackson-Lewis V , Zhou C , Geghman K , Bogdanov M , Przedborski S , Beal MF , Burke RE , Li C ((2009) ) Mutant LRRK2(R1441G) BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat Neurosci 12: , 826–828. |
[29] | Melrose HL , Dachsel JC , Behrouz B , Lincoln SJ , Yue M , Hinkle KM , Kent CB , Korvatska E , Taylor JP , Witten L , Liang YQ , Beevers JE , Boules M , Dugger BN , Serna VA , Gaukhman A , Yu X , Castanedes-Casey M , Braithwaite AT , Ogholikhan S , Yu N , Bass D , Tyndall G , Schellenberg GD , Dickson DW , Janus C , Farrer MJ ((2010) ) Impaired dopaminergic neurotransmission and microtubule-associated protein tau alterations in human LRRK2 transgenic mice. Neurobiol Dis 40: , 503–517. |
[30] | Lin CH , Tsai PI , Wu RM , Chien CT ((2010) ) LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3ss. J Neurosci 30: , 13138–13149. |
[31] | Kawakami F , Yabata T , Ohta E , Maekawa T , Shimada N , Suzuki M , Maruyama H , Ichikawa T , Obata F ((2012) ) LRRK2 phosphorylates tubulin-associated tau but not the free molecule: LRRK2-mediated regulation of the tau-tubulin association and neurite outgrowth. PLoS One 7: , e30834. |
[32] | Bailey RM , Covy JP , Melrose HL , Rousseau L , Watkinson R , Knight J , Miles S , Farrer MJ , Dickson DW , Giasson BI , Lewis J ((2013) ) LRRK2 phosphorylates novel tau epitopes and promotes tauopathy. Acta Neuropathol 126: , 809–827. |
[33] | Shanley MR , Hawley D , Leung S , Zaidi NF , Dave R , Schlosser KA , Bandopadhyay R , Gerber SA , Liu M ((2015) ) LRRK2 Facilitates tau Phosphorylation through Strong Interaction with tau and cdk5. Biochemistry 54: , 5198–5208. |
[34] | Hamm M , Bailey R , Shaw G , Yen SH , Lewis J , Giasson BI ((2015) ) Physiologically relevant factors influence tau phosphorylation by leucine-rich repeat kinase 2. J Neurosci Res 93: , 1567–1580. |
[35] | Ahmadi Rastegar D , Dzamko N ((2020) ) Leucine Rich Repeat Kinase 2 and Innate Immunity. Front Neurosci 14: , 193. |
[36] | Saman S , Kim W , Raya M , Visnick Y , Miro S , Saman S , Jackson B , McKee AC , Alvarez VE , Lee NC , Hall GF ((2012) ) Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem 287: , 3842–3849. |
[37] | Wang Y , Balaji V , Kaniyappan S , Kruger L , Irsen S , Tepper K , Chandupatla R , Maetzler W , Schneider A , Mandelkow E , Mandelkow EM ((2017) ) The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener 12: , 5. |
[38] | Katsinelos T , Zeitler M , Dimou E , Karakatsani A , Muller HM , Nachman E , Steringer JP , Ruiz de Almodovar C , Nickel W , Jahn TR ((2018) ) Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep 23: , 2039–2055. |
[39] | Nguyen APT , Daniel G , Valdes P , Islam MS , Schneider BL , Moore DJ ((2018) ) G2019S LRRK2 enhances the neuronal transmission of tau in the mouse brain. Hum Mol Genet 27: , 120–134. |
[40] | Mikhail F , Calingasan N , Parolari L , Subramanian A , Yang L , Flint Beal M ((2015) ) Lack of exacerbation of neurodegeneration in a double transgenic mouse model of mutant LRRK2 and tau. Hum Mol Genet 24: , 3545–3556. |

