
The α-Synuclein Origin and Connectome Model (SOC Model) of Parkinson’s Disease: Explaining Motor Asymmetry, Non-Motor Phenotypes, and Cognitive Decline
Abstract
A new model of Parkinson’s disease (PD) pathogenesis is proposed, the α-Synuclein Origin site and Connectome (SOC) model, incorporating two aspects of α-synuclein pathobiology that impact the disease course for each patient: the anatomical location of the initial α-synuclein inclusion, and α-synuclein propagation dependent on the ipsilateral connections that dominate connectivity of the human brain. In some patients, initial α-synuclein pathology occurs within the CNS, leading to a brain-first subtype of PD. In others, pathology begins in the peripheral autonomic nervous system, leading to a body-first subtype. In brain-first cases, it is proposed that the first pathology appears unilaterally, often in the amygdala. If α-synuclein propagation depends on connection strength, a unilateral focus of pathology will disseminate more to the ipsilateral hemisphere. Thus, α-synuclein spreads mainly to ipsilateral structures including the substantia nigra. The asymmetric distribution of pathology leads to asymmetric dopaminergic degeneration and motor asymmetry. In body-first cases, the α-synuclein pathology ascends via the vagus to both the left and right dorsal motor nuclei of the vagus owing to the overlapping parasympathetic innervation of the gut. Consequently, the initial α-synuclein pathology inside the CNS is more symmetric, which promotes more symmetric propagation in the brainstem, leading to more symmetric dopaminergic degeneration and less motor asymmetry. At diagnosis, body-first patients already have a larger, more symmetric burden of α-synuclein pathology, which in turn promotes faster disease progression and accelerated cognitive decline. The SOC model is supported by a considerable body of existing evidence and may have improved explanatory power.
INTRODUCTION
Lewy body disorders including Parkinson’s disease (PD) and dementia with Lewy bodies (DLB) are heterogeneous disorders with multi-factorial etiology. In recent years, there has been increased focus on the importance of the α-synuclein protein [1]. The misfolding, aggregation, and subsequent cell-to-cell propagation of pathological α-synuclein inclusions share some similarity with bona fide prions [2–4]. In addition to the prion-like behavior of α-synuclein, many other risk factors and molecular events play important roles in the pathogenesis of PD. The interplay between pathological α-synuclein and these other factors remains poorly understood; it is also unclear why certain types of neurons appear to be much more vulnerable than others [5, 6].
A novel model of PD etiopathogenesis is proposed herein: the α-Synuclein Origin site and Connectome (SOC) model. The SOC model shares some similarities with other recently proposed models [7–9]. However, this model posits that α-synuclein pathobiology is the core feature in PD pathogenesis with two characteristics of defining importance: (1) the anatomical location of the very first pathogenic α-synuclein, which varies among patients; and (2) the neural connectome plays a crucial role in determining how pathological α-synuclein propagates through the nervous system. These two features may explain a significant degree of phenotypic variance among patients, including different non-motor subtypes of PD, variations in motor and non-motor asymmetry, and differing rates of cognitive decline.
Furthermore, it is proposed that the SOC model applies to all Lewy pathology-positive cases, which include, by definition, all cases with DLB, and that great majority of PD patients (>99%), in which classical Lewy pathology is seen. The model also applies to Lewy pathology-positive pure autonomic failure patients (PAF), and it is assumed that patients with incidental Lewy body disease (ILBD) represent early disease stages of these disorders [10].
The model fully acknowledges the significant roles played by many other factors, including inflamma-tion, infection, microbiota, genetics, proteostatic de-ficiency, oxidative stress, mitochondrial dysfunction, calcium dysregulation, and other features [11–15]. Some of these factors may increase the probability that the first α-synuclein aggregation occurs at a specific anatomical location such as the gut. Selective transmissibility factors may promote differences in intra-neuronal and neuron-to-neuron propagation of α-synuclein pathology [16]. In addition, many factors probably influence how a neuron responds to the presence of α-synuclein inclusions. Such factors are most likely responsible for the characteristic selective neuronal vulnerability, i.e., certain neuronal populations undergo severe degeneration and cell death, whereas other neurons are more resistant to the presence of α-synuclein pathology during the course of the disease. Importantly, the SOC model should be viewed as an attempt to reconcile the α-synuclein spreading hypothesis with the selective vulnerability hypothesis [17]. The model yields a number of predictions, which can be experimentally tested in existing datasets and in future studies. As prion-like propagation is integral to the SOC model, if predictions turn out to be accurate, the prion-like behavior of pathological α-synuclein will have gained further support.
Box 1:
Topics covered
Overview of the SOC model
The human connectome and the SOC model
Utility of the SOC model
The SOC model predicts non-motor subtypes
The SOC model predicts motor asymmetry
Other asymmetries
The SOC model predicts differences in cognitive decline
The olfactory bulb
Controversies and criticisms of the SOC model
OVERVIEW OF THE SOC MODEL
The SOC model is an extended version of our previously proposed body-first versus brain-first hypothesis [18]. In body-first PD, the peripheral autonomic nervous system is severely damaged before the dopamine system is affected, while the opposite is true for brain-first PD, resulting in different clinical phenotypes (Fig.1) [19]. The SOC model (Figs.2 and 3) proposes that the following features are of particular importance in the pathogenesis of Lewy body disorders, including PD:
(1) The first α-synuclein inclusions appear stochastically in the synapses of a single or in very few closely situated neurons in varied an-atomical locations. In brain-first PD, pathology originates inside the CNS; in body-first PD, pathology begins in the peripheral autonomic nervous system.
(2) If α-synuclein propagation depends in part on connection strength, unilateral pathology will disseminate more to the ipsilateral hemisphere as the connectivity of the human brain is dominated by ipsilateral connections.
(3) In brain-first PD, pathology arises in a single location of one hemisphere and disseminates predominantly according to the ipsilateral connections, leading to asymmetric intra-cerebral propagation. Whereas in body-first PD, enteric pathology propagates retrogradely in the vagus nerves to involve the right and left dorsal monitor nucleus of the vagus (DMV) bilaterally (due to the overlapping gastrointestinal innervation of the right and left DMV); therefore, initial α-synuclein pathology and subsequent propagation within the CNS is symmetric.
(4) These features influence the degree of asymmetry in motor and non-motor symptoms and rate of progression to dementia.
(5) The prodromal phase is longer in body-first PD as multiple synapses separate the enteric nervous system from the substantia nigra (SN). In brain-first PD, the SN is involved very early due to its strong monosynaptic connections with typical brain-first origin sites (amygdala, locus coeruleus [LC], SN), leading to a shorter prodromal phase.
Fig. 1
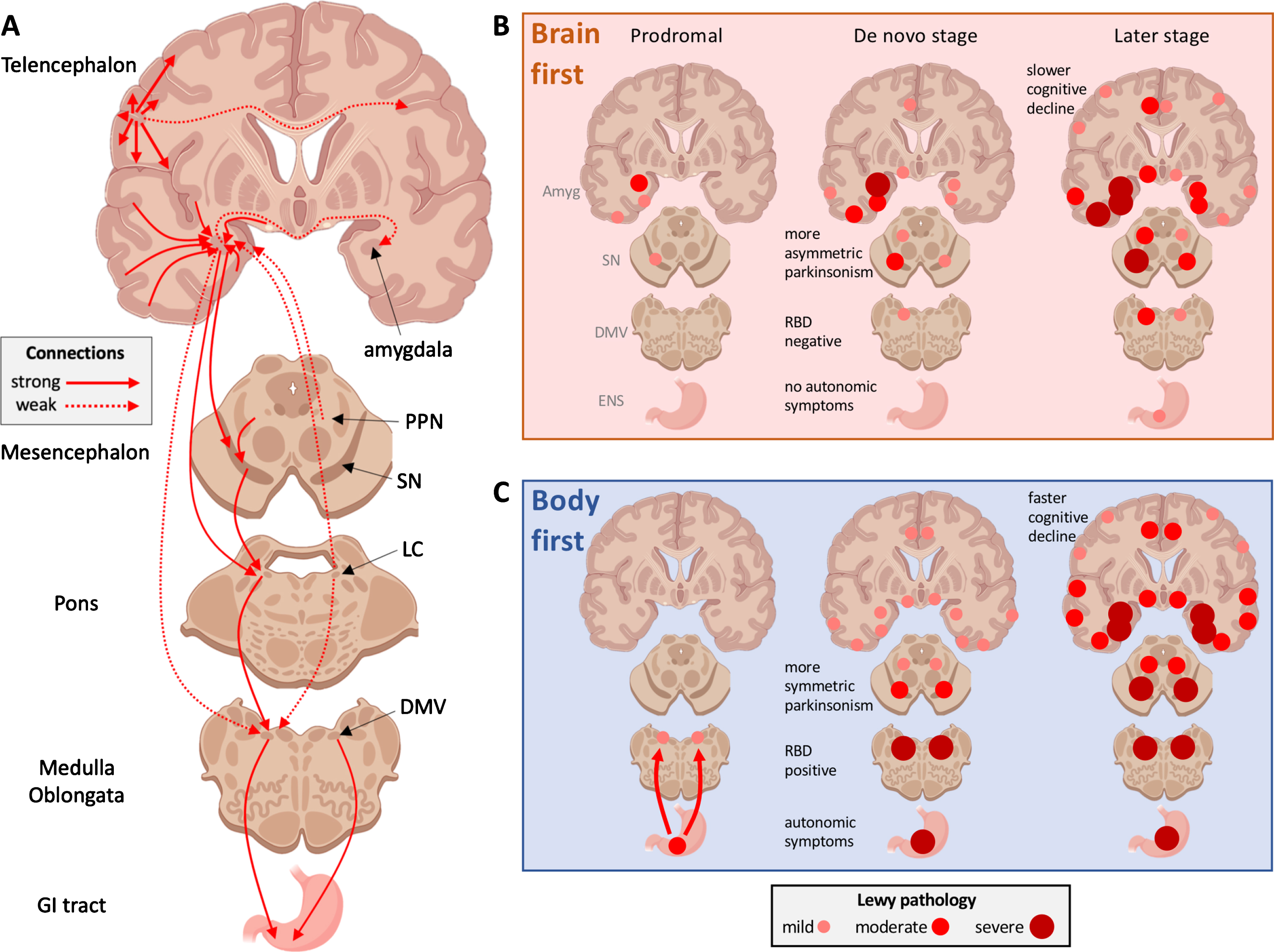
In the body-first subtype of PD, the initial α-synuclein pathology presumably originates in the enteric or autonomic nervous system and spreads to the CNS via the vagus and sympathetic connectome. A brainstem-predominant profile of Lewy pathology follows. These patients develop RBD in the prodromal phase, have more autonomic symptoms, significant hyposmia, faster motor and non-motor progression, and more rapid cognitive decline. When parkinsonism emerges, it is (on average) more symmetric. In the brain-first subtype of PD, the initial α-synuclein pathology presumably originates in the amygdala or in closely connected structures such as the olfactory bulb. An amygdala-predominant profile of Lewy pathology then develops. These patients are RBD-negative in the prodromal phase, have fewer autonomic symptoms, less frequent hyposmia, slower motor and non-motor progression, and less rapid cognitive decline. When parkinsonism emerges, it is most often asymmetric. Different genetic mutations seem to be associated with one or the other of these phenotypes [18]. Both figures illustrate the site of initial α-synuclein pathology (1) and the subsequent spatial-temporal sequence of progressive Lewy pathology (2–5).
![In the body-first subtype of PD, the initial α-synuclein pathology presumably originates in the enteric or autonomic nervous system and spreads to the CNS via the vagus and sympathetic connectome. A brainstem-predominant profile of Lewy pathology follows. These patients develop RBD in the prodromal phase, have more autonomic symptoms, significant hyposmia, faster motor and non-motor progression, and more rapid cognitive decline. When parkinsonism emerges, it is (on average) more symmetric. In the brain-first subtype of PD, the initial α-synuclein pathology presumably originates in the amygdala or in closely connected structures such as the olfactory bulb. An amygdala-predominant profile of Lewy pathology then develops. These patients are RBD-negative in the prodromal phase, have fewer autonomic symptoms, less frequent hyposmia, slower motor and non-motor progression, and less rapid cognitive decline. When parkinsonism emerges, it is most often asymmetric. Different genetic mutations seem to be associated with one or the other of these phenotypes [18]. Both figures illustrate the site of initial α-synuclein pathology (1) and the subsequent spatial-temporal sequence of progressive Lewy pathology (2–5).](https://content.iospress.com:443/media/jpd/2021/11-2/jpd-11-2-jpd202481/jpd-11-jpd202481-g001.jpg)
Fig. 2
Overview of the SOC model illustrating how the onset site of pathogenic α-synuclein leads to symmetric vs. asymmetric propagation of α-synuclein pathology. The origin site and symmetry/asymmetry aspects influence the clinical phenotype of patients including the degree of motor and non-motor asymmetry, and the progression rate to dementia. α-syn, α-synuclein; DMV, dorsal motor nucleus of vagus; GI, gastrointestinal; LC, locus coeruleus; SN, substantia nigra; NBM, nucleus basalis of Meynert; NMS, non-motor symptoms.
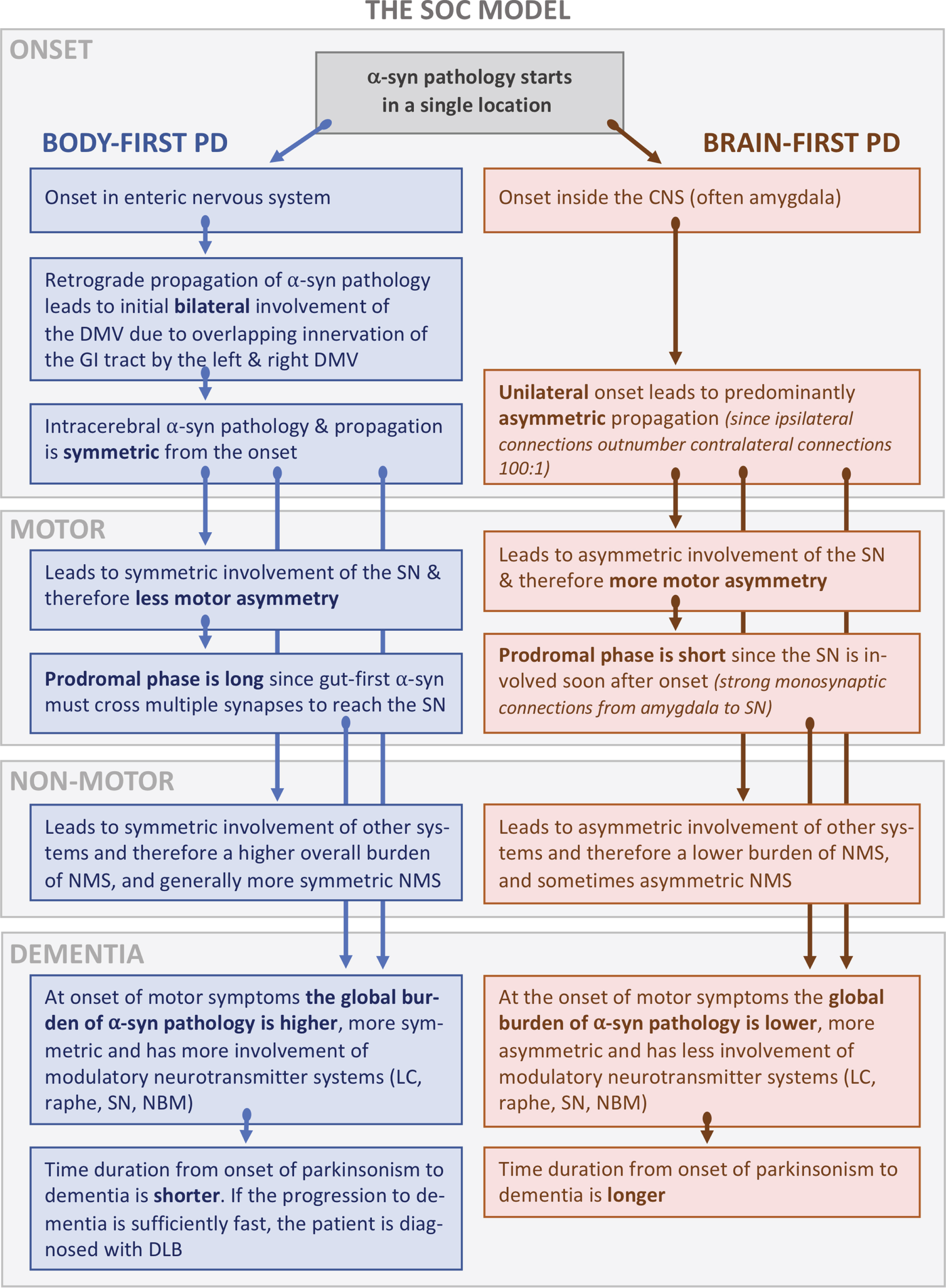
Fig. 3
A) Schematic representation of important connectome details in PD. Ipsilateral connections outnumber contralateral projections approximately 100-to-1. The commissural projections often show most intense innervation to their contralateral homologs, termed homotypic innervation. The diagram also shows key mono-synaptic connections of the amygdala and brainstem nuclei, which are also predominantly ipsilateral. Importantly, the autonomic innervation of the GI tract from the right and left DMV shows a great deal of overlap. B) Brain-first PD. The originating α-synuclein pathology appears unilaterally, often in the amygdala or closely connected structures such as the olfactory bulb. Due to the predominantly ipsilateral connections in one hemisphere, the pathology spreads initially to ipsilateral structures. At the time of diagnosis (middle column), the ipsilateral degeneration of the substantia nigra gives rise to asymmetric motor symptoms. The asymmetric distribution of α-synuclein persists into later disease stages. C) Body-first PD. The originating α-synuclein pathology, which can arise anywhere in the enteric nervous system, propagates to the DMV bilaterally due to the overlapping vagal innervation. The symmetric α-synuclein pathology then propagates rostrally, and leads to a more symmetric loss of nigrostriatal innervation and more symmetric motor symptoms. When parkinsonism emerges, the global burden of α-synuclein pathology is higher in body-first PD due to the more symmetric involvement of both hemispheres, possibly further promoted by the more marked involvement of ascending, neuromodulatory brainstem nuclei. Measured from the time of parkinsonism onset, the body-first patient is therefore at elevated risk of faster progression and accelerated cognitive decline. Amyg, amygdala; SN, substantia nigra; DMV, dorsal motor nucleus of vagus; ENS, enteric nervous system; PPN, pedunculopontine nuclei.
The SOC model assumes that the quantity of α-synuclein inclusions in a given nucleus or cortical region increases over time [20–22]. This gives rise to the phenomenon of gradients: the initially affected neurons tend to show the most severe pathology, and the most recently affected neurons have the lowest density of α-synuclein pathology. Once a neuron has accumulated a sufficient number of α-synuclein in-clusions, it becomes dysfunctional and is at risk of degenerating and dying. Different types of neurons display different degrees of vulnerability towards α-synuclein pathology [17]. Thus, a strong relationship should not always be expected between either the quantity of α-synuclein inclusions in a neuron or the length of time those inclusions have been present and the degree of dysfunction or degeneration of those neurons. Importantly, the density of α-synuclein pat-hology in a given nucleus over time may follow an inverted U-shape: once most of the synapses and cell bodies have degenerated, there is nowhere to harbor intra-cellular α-synuclein pathology. This is most likely to be observed in very vulnerable neuron populations such as the postganglionic sympathetic neurons innervating the heart [23], and the dopamine neurons of the substantia nigra pars compacta [24].
THE HUMAN CONNECTOME AND THE SOC MODEL
The human brain contains an estimated 86 billion neurons. Perhaps surprisingly, only 17–25 billion are located in the cerebrum, whereas the cerebellum contains around 69 billion neurons [25, 26]. The corpus callosum contains close to 95% of commissural fibers and comprises approximately 190 million axons [27–29]. It follows that only approximately 1% of cerebral neurons project to the contralateral hemisphere. This assertion is supported by modern data from the human connectome project showing that cerebral connectivity is completely dominated by ipsilateral connections [30], and that less than 1–2% of hippocampal projections, for example, are contralateral [31]. The fraction of crossing connections from brainstem nuclei in humans has not been reported in detail. However, studies in primates, cats, rodents, and other mammals universally show that reciprocal connections (afferent and efferent) of im-portant subcortical structures, including the amygdala, SN, basal forebrain nuclei, and LC are almost exclusively ipsilateral (Fig.3A) [32–44].
Therefore, if α-synuclein pathology appears sto-chastically in a single unilateral location in the CNS, and the subsequent spreading to neighboring neurons is influenced by connection strength, initial disse-mination will occur mainly to the ipsilateral hemisphere. Thus, one hemisphere in a brain-first PD case will initially develop a considerably larger burden of α-synuclein pathology compared with the other hemisphere. A small fraction of the propagating α-synuclein will eventually spread to the contralateral hemisphere and begin the spreading process there, but with a significant time delay. Moreover, the ori-ginator hemisphere will accumulate pathological α-synuclein at a higher rate, and the threshold for neuronal dysfunction and degeneration will be crossed earlier in this hemisphere.
The scarce contralateral projections underlie an-other element of the SOC model. Most cortical re-gions and some subcortical structures send most of their commissural projections to their contralateral homologs, so called homotypic connections (illustrated in Fig.3A) [30]. If brain-first PD is most com-monly initiated in a single unilateral structure, such as the entorhinal cortex or amygdala, the first structures to be affected in the contralateral hemisphere will also be the amygdala or entorhinal cortex [33, 45]. As the α-synuclein pathology propagates to additional structures in the ipsilateral hemisphere, many of those structures will seed their contralateral homologs. Therefore, the contralateral hemisphere will be a ti-me-shifted mirror image of the ipsilateral, originator hemisphere (Fig.3B). In support of this, a recent longitudinal PET study in Alzheimer’s disease revealed that progressive build-up of pathological tau protein occurs disproportionally more via such homotypic connections to the contralateral hemisphere [46].
For body-first PD, it is important to consider the autonomic connectome with regards to the gastroin-testinal (GI) tract. The preganglionic parasympathe-tic motor neurons located in the DMV are particularly prone to α-synuclein pathology formation, much more so than the sensory neurons of the nucleus of the solitary tract [20]. The DMV innervation of the GI tract is most intense in the distal esophagus and stomach, moderate in the small intestine and proximal colon, and weak in the transverse colon of rats [47]. The recto-sigmoid colon is innervated from parasympathetic nuclei situated in the sacral spinal cord. Sympathetic innervation of the GI tract is provided by postganglionic neurons in the midline coeliac and mesenteric ganglia.
When retrograde neuronal tracers such as horser-adish peroxidase, fast blue, and fluorogold are inje-cted into the rat duodenum, jejunum, ileum, cecum, and the ascending, transverse and descending colon, the two DMV nuclei show symmetric labelling [48–51]. It has been shown that the ventral stomach of rats is innervated more by the left DMV than the right [52], but almost the entire GI tract is doubly innervated by both the left and right DMV. Human data on DMV innervation of the GI tract is lacking, but the innervation pattern of this phylogenetically ancient system is presumably similar in humans. Thus, if an initial patch of α-synuclein pathology appears almost anywhere in the GI tract, α-synuclein pathology will propagate via both the left and right DMV. Consequently, the first instance of subsequent α-synuclein pathology within the CNS will be bilateral and further intra-cerebral propagation will lead to a relatively symmetric pathology compared with that seen in brain-first PD (Fig.3C).
Animal studies provide some support for the validity of these concepts. When peripheral α-synuclein pathology is triggered in the gut by injecting recombinant preformed α-synuclein fibrils locally into the upper GI tract of rodents, a very symmetric initial involvement of the DMV is seen, followed by symmetric involvement of the LC, SN, limbic, and cortical regions [53, 54]. Similar symmetric involvement is seen when intra-peritoneal injections of seeds are used [55]. In contrast, unilateral injections of α-synuclein seeds into the olfactory bulb leads to a much more asymmetric involvement of the hemispheres, and the same asymmetry is seen following unilateral injections into the striatum [56, 57]. Similarly, injections of adeno-associated virus into one SN of rats also lead to widespread distribution in the brain, which is almost entirely ipsilateral [58]. Figure4A shows an example of symmetric α-synuclein pathology in the DMV of rats following gut-injection of α-synuclein seeds, while Fig.4B shows asymmetric involvement of the limbic system and SN following unilateral seeding in the amygdala. Of note, in the published rodent studies, the pattern of α-synuclein pathology in the non-injected hemisphere generally resembles the pattern in the injected hemisphere at the previous timepoint, supporting the SOC model of spreading to the contralateral hemisphere happens in part via commissural, homotypic projections from the involved regions to their homologs in the contralateral hemisphere (see Fig.4C, D).
Fig. 4
A) The DMV nuclei in rats show symmetric deposits of pathological, hyperphosphorylated α-synuclein inclusions following injection of α-synuclein seeds in the duodenum. B) Following unilateral injection of α-synuclein fibrils into the amygdala, α-synuclein inclusions are seen in ipsilateral limbic and cortical structures, with early involvement of the ipsilateral substantia nigra (red insert). No contralateral pathology is seen at this early timepoint. These wild-type rats were both sacrificed three months post-injection. C) Schematic representation of published data from injection of α-synuclein seeds unilaterally into a single CNS location [56, 57, 59]. Markedly asymmetric α-synuclein pathology is evident at the early time point 1. At the later time point 2, progressive accumulation of α-synuclein is seen in nearly all affected regions, but the asymmetry persists. D) The distribution of α-synuclein pathology in the ipsilateral hemisphere at time point 1 is very similar to the α-synuclein distribution in the contralateral hemisphere at time point 2. This suggests that the α-synuclein pathology in the contralateral hemisphere is, in part, promoted by commissural homolog-to-homolog connections from the ipsilateral hemisphere.
![A) The DMV nuclei in rats show symmetric deposits of pathological, hyperphosphorylated α-synuclein inclusions following injection of α-synuclein seeds in the duodenum. B) Following unilateral injection of α-synuclein fibrils into the amygdala, α-synuclein inclusions are seen in ipsilateral limbic and cortical structures, with early involvement of the ipsilateral substantia nigra (red insert). No contralateral pathology is seen at this early timepoint. These wild-type rats were both sacrificed three months post-injection. C) Schematic representation of published data from injection of α-synuclein seeds unilaterally into a single CNS location [56, 57, 59]. Markedly asymmetric α-synuclein pathology is evident at the early time point 1. At the later time point 2, progressive accumulation of α-synuclein is seen in nearly all affected regions, but the asymmetry persists. D) The distribution of α-synuclein pathology in the ipsilateral hemisphere at time point 1 is very similar to the α-synuclein distribution in the contralateral hemisphere at time point 2. This suggests that the α-synuclein pathology in the contralateral hemisphere is, in part, promoted by commissural homolog-to-homolog connections from the ipsilateral hemisphere.](https://content.iospress.com:443/media/jpd/2021/11-2/jpd-11-2-jpd202481/jpd-11-jpd202481-g004.jpg)
The present claims about potential asymmetric distribution of α-synuclein pathology may seem surprising to some, but it is important to remember that the entire modern literature on postmortem staging of α-synuclein in human brains is based on single-hemisphere studies. For decades, it has been common practice to preserve one hemisphere freshly frozen to allow alternative types of analysis precluded by formaldehyde fixation. We can find no published study that systematically performed staging of Lewy pathology in both hemispheres from the same individuals, at least not with modern immunohistochemical techniques targeting α-synuclein. The SOC model predicts that cases with an asymmetric burden of α-synuclein are common, especially in brain-first PD cases at earlier disease stages (Fig.3B).
We previously hypothesized that brain-first PD is equivalent to those postmortem cases with a limbic (amygdala) predominant profile of α-synuclein path-ology, and that body-first PD is equivalent to a brainstem-predominant distribution of α-synuclein pathology [18, 19, 21, 60, 61]. The SOC model pre-dicts that those postmortem cases with a brainstempr-edominant profile will tend to have more symmetric α-synuclein pathology compared with the more asymmetric pathology seen in limbic-predominant cases. This asymmetry may be most evident during the prodromal and early disease stages, but disappear at later stages as the brain becomes bilaterally saturated with Lewy pathology.
Currently, these predictions can only be empirically tested in rare brain bank material, where both hemispheres have been processed in the same way. However, it will be relatively straightforward to test once a suitable α-synuclein positron emission tomography (PET) tracer becomes available. Such studies would also determine if asymmetric α-syn-uclein pathology in the hemispheres correlates with asymmetric loss of nigrostriatal dopaminergic inn-ervation. Finally, it is noted that 18F-fluoro-deoxy-glucose PET scans of later stage PD patients and DLB patients often show asymmetric patterns of hypometabolism in the hemispheres of individual cases [62]. This asymmetric metabolism may reflect underlying asymmetric Lewy pathology in those subjects, although co-existing Alzheimer pathology will also affect the metabolic patterns seen in these scans.
UTILITY OF THE SOC MODEL
The SOC model predicts non-motor subtypes
Body-first PD. If the originating α-synuclein pat-hology arises in the enteric nervous system, and uses the parasympathetic and sympathetic connectome to invade the brain bilaterally, this would most likely impose a certain degree of uniformity on the pattern of α-synuclein spreading within the CNS. If the initial pathological locus is situated anywhere in the distal esophagus, stomach, small intestine, or proximal two-thirds of the colon, the propagating pathology will be siphoned towards identical CNS nuclei, nam-ely the DMV, and via the sympathetic ganglia to the intermediolateral cell column (IML) of the spinal cord. The pathology then spreads bilaterally according to connection strength and other factors in a ca-udo-rostral direction and affects pontine nuclei before the SN is involved [20]. Importantly, it is believed that the prodromal phase of PD can last 20 years or more, which suggests that neuron-to-neuron propagation of α-synuclein pathology may be very slow [63]. Thus, there is ample time for the α-synuclein pathology to induce some neuronal dysfunction and degeneration at each station along the spreading route before the next station is sufficiently involved. The SOC model therefore predicts a specific sequence of developing non-motor symptoms prior to motor dysfunction in body-first PD (Figs.1 and 3C).
Given that autonomic structures (including the DMV and sympathetic ganglia) are involved first, the initial symptoms would often be autonomic, such as constipation and orthostatic hypotension. Once pontine structures (including the magnocellularis and subcoeruleus nuclei) are sufficiently dysfunctional, isolated REM sleep behavior disorder (iRBD) appears [64]. Involvement of other brainstem nuclei may give rise to prodromal neuropsychiatric symptoms including anxiety and mood disorders [65, 66]. Supporting this, a recent study reported that depression and anxiety were more prevalent in iRBD pat-ients compared with a group of unselected de novo PD patients [67]. This timeline of events has also been confirmed in epidemiological studies [63]. The mesencephalon is next in line and once approximately 50% of the nigrostriatal dopaminergic terminals have degenerated, motor symptoms appear [68]. Importan-tly, the median time to motor phenotype conversion in iRBD patients is eight years, which underscores that the propagation of pathological α-synuclein, and perhaps especially the deleterious effects on neuron function, is a slow process [69].
Not every single body-first PD patient will develop all these symptoms, and not necessarily in the predicted order. Conceivably, variations could arise due to differing genetic make-up, variable co-morbidities, and other factors, which determine the relative vulnerability of each of the affected neuronal systems to the presence of pathological α-synuclein. However, it is beyond dispute that the overall burden of autonomic symptoms, including constipation and orthostatic hypotension, is markedly greater in RBD-positive PD patients than in RBD-negative patients [69–72]. Furthermore, imaging studies have shown that ∼95% of iRBD patients exhibit a near complete loss of sympathetic cardiac innervation [73–77] and significantly decreased uptake of 11C-donepezil in the colon, as a marker of parasympathetic denervation [73]. Importantly, when de novo PD patients are studied with multi-modal imaging, those patients who developed RBD during the pre-motor phase (i.e., body-first) show ubiquitous loss of the cardiac sympathetic signal and low 11C-donepezil in the colon [19]. In contrast, the majority of RBD-negative de novo PD patients (i.e., brain-first) display normal or near-normal cardiac and colonic imaging parameters.
In addition, Derkinderen’s group found that 64% of RBD-positive PD patients show α-synuclein pathology in colon biopsies compared with only 13% of RBD-negative patients [78]. This indicates that wid-espread enteric α-synuclein pathology occurs earlier in RBD-positive PD patients, supporting a body-first etiology. Another study reported that 90% of skin samples from PD patients with orthostatic hypotension contained α-synuclein pathology, compared with only 38% of samples from patients without orth-ostatic hypotension [71]. In the former group, 86% had RBD, compared with 36% in the latter group. Taken together, these clinical, imaging, and biopsy studies strongly support the notion that the body-first PD prodromal phase is characterized by early autonomic and sleep symptoms, widespread α-synuclein pathology in peripheral nerves, and measurable degeneration of the autonomic nervous system.
Brain-first PD. According to the SOC model, brain-first PD patients will show more heterogene-ous symptoms than body-first patients. Whereas the DMV and sympathetic nuclei serve as natural, ana-tomical bottlenecks in body-first patients leading to uniform progression patterns inside the CNS, this is not the case in brain-first patients. Wherever path-ology starts, it can propagate in multiple directions. Thus, the sequence of symptom development in brain-first PD patients might be more variable, dep-ending on where the first α-synuclein is formed. However, due to the predicted predominantly unilateral distribution of α-synuclein in brain-first PD, prodromal non-motor symptoms are generally expected to be subclinical or mild. In addition, the SOC model predicts that the prodromal phase in brain-first PD is short, providing little time for non-motor symptoms to appear before overt motor symptoms.
Studies of ILBD patients show that ‘single-site’ Lewy pathology is, when excluding the olfactory bulb, most commonly seen in the amygdala, the LC, and the SN, suggesting that these structures are the most common origin sites of brain-first α-synuclein pathology [21, 22, 79]. The olfactory bulb may be a special case, as discussed below.
Importantly, neuropathology studies have documented that the distribution of Lewy pathology shows two general profiles: (1) a brainstem-predominant pattern with the most intense pathology in the lower brainstem, even when the limbic system and cortex are involved; and (2) a limbic-predominant pattern with the most intense pathology in the amygdala and entorhinal cortex, even when the brainstem and cortex are involved (Fig.5) [21, 22, 61]. Furthermore, nearly all postmortem human data is compatible with the interpretation that α-synuclein pathology in an affected structure accumulates over time. Therefore, it can be hypothesized that when a gradient of α-synuclein pathology is seen, the peak of that gradient often points to where the pathology originated. This concept would identify the DMV and sympathetic trunk as the first affected structures in brainstem-predominant cases, since these structures generally show the most pathology. In limbic-predominant cases, the amygdala and entorhinal cortex would be the most likely starting points. One caveat here is the common occurrence of Alzheimer co-pathology in limbic structures, as discussed below.
Fig. 5
Stylized sketch of the two typical graded profiles of α-synuclein pathology seen in postmortem studies [20–22]. Pathology ranges from none to very severe in 11 anatomical regions sorted according to Braak stages. Cases showing most pathology in the autonomic nuclei and brainstem are hypothesized to be body-first (blue). Cases showing most pathology in the amygdala and entorhinal cortex, but relatively less pathology in the lower brainstem are hypothesized to be brain-first (red). Amyg, amygdala; Cing, cingulate; DMV, dorsal motor nucleus of vagus; Ento, trans-entorhinal cortex; Front, frontal cortex; IML, sympathetic intermediolateral cell column; LC, locus coeruleus; Pariet, parietal cortex; SN, substantia nigra; S-Para, sacral parasympathetic nuclei; Temp, temporal cortex.
![Stylized sketch of the two typical graded profiles of α-synuclein pathology seen in postmortem studies [20–22]. Pathology ranges from none to very severe in 11 anatomical regions sorted according to Braak stages. Cases showing most pathology in the autonomic nuclei and brainstem are hypothesized to be body-first (blue). Cases showing most pathology in the amygdala and entorhinal cortex, but relatively less pathology in the lower brainstem are hypothesized to be brain-first (red). Amyg, amygdala; Cing, cingulate; DMV, dorsal motor nucleus of vagus; Ento, trans-entorhinal cortex; Front, frontal cortex; IML, sympathetic intermediolateral cell column; LC, locus coeruleus; Pariet, parietal cortex; SN, substantia nigra; S-Para, sacral parasympathetic nuclei; Temp, temporal cortex.](https://content.iospress.com:443/media/jpd/2021/11-2/jpd-11-2-jpd202481/jpd-11-jpd202481-g005.jpg)
The SOC model predicts motor asymmetry
Ever since the original description by James Parkinson, PD has been defined by asymmetric motor symptoms. However, a significant fraction (30–40%) of newly diagnosed PD patients display only minor or no obvious motor asymmetry [80–85]. The SOC model proposes a simple explanation for both the presence of and the variation in this motor asymmetry.
If the initial α-synuclein pathology in brain-first PD arises unilaterally in the amygdala or the LC, the ipsilateral SN would be involved soon thereafter, since the SN has strong ipsilateral connections to both the LC and amygdala [33, 34, 38, 39, 86, 87]. The dopaminergic neurons in the SN ipsilateral to the origin site would therefore get a head start and degenerate earlier than the contralateral SN. Such patients would show clinical motor asymmetry (Fig.3B). In support of this, postmortem studies of patients with ILBD report that some cases only have pathology in the amygdala and very few other locations [21, 88]. Those other locations are most commonly the SN, nucleus basalis of Meynert, and entorhinal cortex, all of which are mono-synaptically connected to the amygdala.
On the other hand, in body-first patients, the initial α-synuclein pathology originates in the enteric nervous system and ascends to involve the DMV bilaterally. The subsequent symmetric propagation through the brainstem would lead to a more symmetric involvement of the SN. Such patients would, on average, present with less or sometimes no motor asymmetry compared with brain-first patients (Fig.3C).
Importantly, this explanation should not be under-stood as an absolute symmetry/asymmetry dichot-omy. The model predicts that body-first patients are less asymmetric than brain-first PD, not that they are always fully symmetric. First, it should be appreciated that motor symptoms occupy a special place in the phenomenology of neurological disorders. Motor symptoms are very visible and striking, so even a min-or degree of asymmetry is easy to recognize and qua-ntify. In contrast, it is more difficult to appreciate or quantify asymmetries in most non-motor symptoms, including autonomic disturbance, depression, and cognitive function. Second, the involvement of the DMV in body-first PD may not necessarily be completely symmetric, due to an uneven amount of α-synuclein seeds being transported through the left and right vagus. As mentioned above, the ventral stomach of rats is innervated more by the left than the right DMV, so a hypothetical insult in the ventral stomach may lead to more pathology in the left DMV [52]. There may also be intrinsic factors in important neuronal stations in the brainstem, which could facilitate non-uniform propagation in the left and right side of the brainstem. Third, some of the many previous hypothetical explanations for motor asymmetry, including handedness and hemisphere dominance may contribute to a more rapid breakthrough of motor symptoms on one side of the body [80, 89]. However, the explanation proposed by the SOC model is simple and potentially powerful in that it is derived from a general model, which seems capable of explaining many other facets of PD symptomatology. In any case, the predictions about motor asymmetry can be tested experimentally in animal models of PD featuring propagating α-synuclein, and can be fully tested in human patients, once an α-synuclein PET tracer becomes available.
The present hypothesis already has some support from published studies. When evaluating the literature, it is important to remember that isolated RBD (i.e., RBD developed before onset of parkinsonism) is considered a robust sign of the body-first subtype, since ascending pathology will affect the pons before the SN [19]. In a seminal study by Postuma’s group, PD patients were divided into three groups using cluster analysis. In cluster I, only 19% of patients had RBD, whereas 93% in cluster III had RBD [70]. Thus, according to the SOC model, cluster I contained mainly RBD-negative brain-first patients, whereas cluster III comprised mainly RBD-positive, body-first patients. The average motor asymmetry score in cluster I was twice as large as in cluster III. Also, only 12% of patients in cluster I had bilateral onset of motor symptoms compared to nearly three times as many patients (35%) in cluster III. These results support the notion that body-first (RBD-positive) PD patients generally have more symmetric parkinsonism at onset. Furthermore, dopamine transporter imaging data from the Parkinson’s Progression Markers Initiative (PPMI) confirms that RBD-positive PD patients show a more symmetric decline in putaminal binding compared to RBD-negative PD patients [90]. Taken together, these clinical and imaging data support the predictions of the SOC model.
Other asymmetries
Whereas motor asymmetry in PD has been studied intensely for decades, asymmetries in non-motor symptoms have received only limited attention. The SOC model predicts that asymmetric non-motor symptoms and signs might be more common than first expected. Supporting this, studies have found asymmetric responses in the sympathetic skin wrinkling test and in olfactory function of early-stage PD patients [91–93]. In accordance with the SOC model, the skin responses and olfaction were deficient ipsilateral to the most diseased hemisphere. A number of studies have compared cognitive deficit in the visual-spatial domain of left-sided vs. right-sided hemiparkinsonian PD patients, including optic flow, mental rotation, facial emotion recognition, hierarchical pattern recognition, sustained attention, and complex figure copying [94–99]. Patients with left-sided hemiparkinsonism were more impaired in tasks involving their left visual hemifield, whereas patients with right-sided hemiparkinsonism showed impairment in their right visual hemifield. Another study reported correlation between brainstem auditory-evoked potential and cervical vestibular-evoked myogenic potential and motor symptom asymmetry [100]. In summary, these studies suggest that asymmetric symptoms are present throughout the neuraxis ranging all the way from the olfactory bulb to sympathetic autonomic responses in the extremities.
The SOC model predicts differences in cognitive decline
It might be reasoned that brain-first PD, where the initial α-synuclein pathology arises inside the CNS, would predispose to rapid cognitive decline. However, a large body of evidence disagrees with this. On the contrary, it is well established that increased risk of cognitive decline and dementia clusters with RBD, constipation, and orthostatic hypotension—all markers of the body-first subtype [70, 101–103]. Longitudinally, constipation and RBD predicts faster cognitive decline [104]. A recent study found that benign, slowly progressing PD had earlier onset, more motor asymmetry, and low prevalence of depression, pointing to a brain-first etiology. In contrast, a malignant, fast progressing group had less motor asymmetry, and more depression, hallucinations, dysautonomia and RBD, compatible with body-first PD [105]. Interestingly, agricultural occupation was associated with the malignant group, supporting the reasonable assumption that pesticides trigger a body-first subtype via a toxic influence on the enteric nervous system and possibly also via the olfactory epithelium. Other studies reported that the frequencies of constipation, orthostatic hypotension, pathological MIBG heart scans, and RBD are higher in de novo DLB compared with de novo PD [102, 106]. All these observations suggest that rapid cognitive decline is more closely associated with a body-first type of neuronal α-synuclein pathology. Indeed, the risk of rapid conversion to dementia in the body-first type could be so high that many cases will be diagnosed with DLB rather than PD, because of the ‘one-year rule’.
The SOC model suggests that this association is explained, at least in part, by the proposal that body-first patients, at the time of overt motor symptom development, have a larger, overall burden of α-synuclein pathology, primarily because it is more symmetric. Here it must be remembered that most clinical follow-up studies and epidemiological studies use the appearance of motor symptoms as the reference time point for diagnosis of PD. According to the SOC model, at the time of diagnosis, body-first PD has severe, bilateral α-synuclein pathology and damage in pontine structures including the LC and raphe nuclei, and in the medulla oblongata. Presumably, the amygdala already contains significant bilateral pathology, because of the mono-synaptic connections between the amygdala and the SN, LC, and DMV [33]. By contrast, if the pathology starts unilaterally in brain-first PD in the amygdala or the LC, the ipsilateral SN will be invaded as one of the first structures due to the strong, ipsilateral connections between these nuclei. Once ipsilateral dopaminergic neurons are sufficiently degenerated to trigger parkinsonism, the α-synuclein pathology have not spread as far as in body-first PD, especially not into the contralateral hemisphere. The SOC model also predicts that the prodromal phase of body-first PD is longer than for brain-first PD, since more synapses have to be crossed for pathology originating in the gut in comparison to the amygdala. Thus, when motor symptoms appear, the total burden of α-synuclein pathology is overall greater in de novo body-first PD, facilitated by the symmetric involvement.
Importantly, this discussion can also be framed in a different way with potentially far-reaching consequences. The studies cited above demonstrated that RBD and dysautonomia predict faster cognitive decline, thereby suggesting that such patients have a malignant phenotype. However, these studies all used onset of parkinsonism as ‘time zero’. This may lead to significant bias if the prodromal phase is considerably longer in body-first PD, as predicted by the SOC model. If it were possible to use the ‘true time zero’ as the starting point (i.e., the timepoint when the first pathological α-synuclein appeared), it is possible that brain-first PD actually does progress to dementia faster than body-first PD.
As mentioned above, there is no available postmortem evidence from human brains to explore the symmetry vs. asymmetry predictions of the SOC model, since nearly all (if not all) available data were derived from single hemispheres. However, one postmortem study may be supportive of the SOC model, albeit indirectly. Here, the authors compared the magnitude of α-synuclein pathology at autopsy in ten different brain regions of 41 RBD-positive PD patients versus 40 RBD-negative PD patients [107]. The patients were quite late-stage with an average disease duration of 14.5 years. Thus, any clear differences in initial asymmetry of α-synuclein pathology would most likely be significantly diminished due to progressive involvement of the contralateral hemisphere in the RBD-negative (brain-first) cases. Nevertheless, the study reported that RBD-positive patients had greater pathology in 9 of the 10 examined regions.
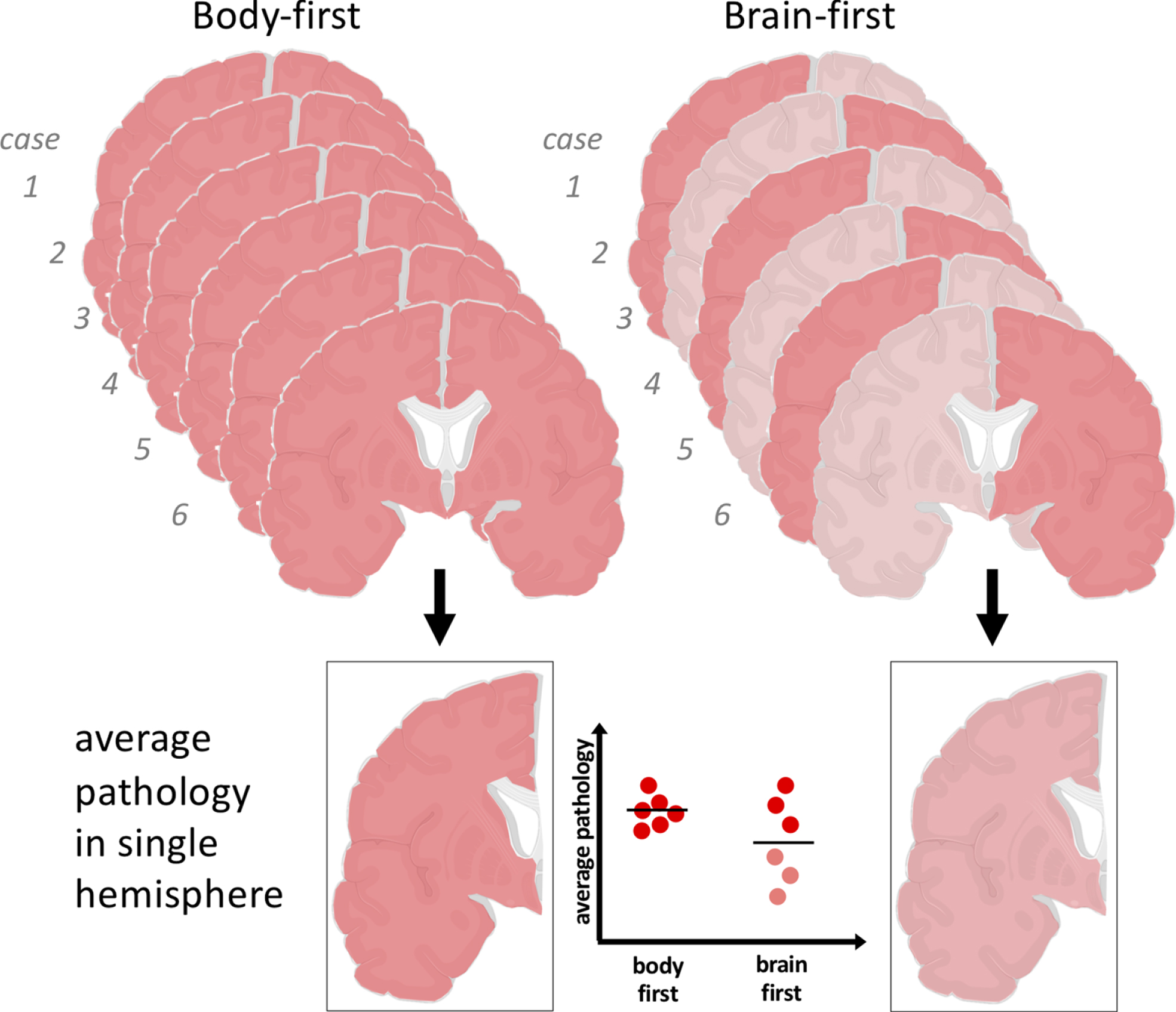
This finding would be predicted by the SOC model because of a random sampling effect (Fig.6). In short, the patients were carefully matched on disease duration (=motor symptom duration), and the SOC model proposes that the RBD-positive cases originally had more, bilateral pathology at motor symptom onset. Therefore, body-first patients will also have more symmetric pathology at this later time point. However, since only single hemispheres were investigated [108], approximately half of the hemispheres from the RBD-negative, brain-first group would simply by chance be the less affected ones. This interpretation is also supported by the fact that the α-synuclein severity data in the two groups showed more overlap than separation across all anatomical regions. The other half of the sampled hemispheres from the RBD-negative patients were the more affected ones, and they presumably contained a similar degree of pathology to that present in both hemispheres of the RBD-positive group. This interpretation is speculative, but illustrates how to explore the SOC model in current postmortem data. It also highlights the urgent need for bihemispheric postmortem studies, where both hemispheres have been processed and fixated with identical methodologies.
Fig. 6
The SOC model predicts that body-first PD shows symmetric α-synuclein pathology, whereas brain-first PD shows asymmetric pathology in the hemispheres. Since only single hemispheres are studied in postmortem studies, this can give rise to the impression that brain-first PD have less Lewy pathology on average. In reality, the most severely affected hemisphere in each brain-first case may show a similar amount of pathology as that seen in both hemispheres of body-first cases.
It is important to note the impact of Alzheimer type co-pathology, which is more common in demented than non-demented PD cases [109–111]. Also, limbic Alzheimer pathology is more common in Lewy body-positive cases with a limbic predominant Lewy pathology profile compared with patients with a brainstem predominant profile [21]. It is likely that Alzheimer co-pathology contributes towards the dev-elopment of dementia in Lewy-body positive cases, since two concomitant neurodegenerative diseases are most likely worse than one. Moreover, in vitro and in vivo studies have shown that amyloid-β and tau fibrils can induce α-synuclein monomers to aggregate [112, 113]. Approximately 10% of cognitively normal people at 50 years are amyloid-β positive, rising to 33% at 80 years [114]. Thus, it is possible that Alzheimer pathology could be responsible for inducing a number of brain-first Lewy pathology cases through cross-seeding. Of note, 10–15% of DLB cases have normal dopamine transporter scans at diagnosis [115]. This observation underscores that it is not uncommon to develop severe, symptomatic Lewy body disease with dementia before noticeable dysfunction of the SN.
In addition, because Alzheimer co-pathology is so common in the elderly, the disease course of body-first PD is also likely to be modified. Once the wave of propagating, symmetric α-synuclein pathology in body-first patients ascends above the brainstem and encounters a pre-existing Alzheimer pathology, this very likely puts the patient in a poor prognostic category. It is possible that the fraction of body-first cases, who just happens to have pre-existing Alzheimer pathology, might develop rapid cognitive decline and therefore be much more likely to receive a DLB diagnosis than body-first cases without Alzheimer pathology. The exponentially increasing prevalence of AD-pathology in the elderly fits with the observation that DLB patients are older than PD patients at time of diagnosis. Moreover, it has also been suggested that Alzheimer co-pathology exacerbates Lewy pathology, thereby setting up a vicious circle leading to rapid cognitive decline and dementia [116].
THE OLFACTORY BULB
The dual-hit hypothesis, initially formulated by Braak, proposed that external insults lead to initial accumulation of α-synuclein in the enteric nervous system and olfactory bulb, presumably via the olf-actory epithelium [117]. It is perhaps less well recognized that Braak, in his seminal 2003 staging study, stated that the olfactory spreading route seems less important, since the propagating inclusions do not appear to spread beyond the olfactory cortex [20]. He therefore based his staging system entirely on the DMV as the starting point. In any case, he proposed that the olfactory bulb pathology is essentially part of the body-first type, even if it doesn’t propagate very far. Other researchers have suggested that the olfactory spreading route is important, and may be responsible for limbic-predominant Lewy pathology and associated with DLB, whereas the DMV-route and brainstem-predominant pathology would be causative of PD [61, 118].
This topic is still a matter of debate, but some evidence supports that olfactory bulb dysfunction is indeed more closely related to the body-first type than the brain-first type. First, the majority of iRBD patients are hyposmic, nearly all have lost the cardiac sympathetic innervation, but many patients have normal or only slightly pathological dopamine imaging scans [69, 73]. This constellation of findings support that both the olfactory bulb and lower brainstem is more involved than is the SN during prodromal body-first PD. Moreover, a recent study reported that only 48% of de novo PD patients with normal MIBG were hyposmic, compared to 71% of those patients with abnormal MIBG scintigraphy [119]. This study also corroborates that hyposmia is significantly more common in body-first cases with pathological MIBG than in brain-first cases with normal MIBG scans.
If the olfactory bulb route were to be responsible for the majority or even all brain-first cases, it is necessary to consider how the pathology would propagate from the bulb. If an inhaled virus or other toxic agent affected the olfactory epithelium bilaterally, this would likely lead to bilateral α-synuclein pathology in the olfactory bulbs. The SOC model would predict symmetric propagation from this initial symmetric origin site, eventually leading to relatively little or no motor asymmetry. However, as discussed above, it seems clear that PD patients with the brain-first phenotype are characterized by more asymmetric parkinsonism than body-first cases; they also have more normal olfaction. Therefore, it does not seem probable that a bilateral olfactory bulb origin would be closely associated with brain-first PD. However, if the initial olfactory bulb pathology were to arise unilaterally, this would set the stage for more asymmetric propagation of α-synuclein pathology leading to asymmetric motor symptoms. Such patients would presumably also have more normal olfaction, since one olfactory bulb would still be relatively intact. Of note, Brundin’s group has elegantly shown that unilateral injections of α-synuclein seeds into a single olfactory bulb of mice leads to markedly asymmetric spreading of α-synuclein pathology [57]. Thus, the olfactory bulb could theoretically be a common origin site for brain-first PD, but it would then have to be a relatively unilateral pathology and propagation.
Moreover, a large brain bank study has found that the most common single-site location of Lewy pat-hology in ILBD cases is the olfactory bulb [79]. However, a recent large autopsy study of 178 Lewy body-positive cases reported only three cases with isolated olfactory bulb pathology, whereas 29 cases had pathology elsewhere but not in the bulb [22]. Thus, the true frequency of isolated α-synuclein pathology in the bulb is unclear and its importance is not resolved. Isolated olfactory bulb pathology may, for unknown reasons, be a very common and rel-atively non-specific finding, in similarity to the freq-uently occurring α-synuclein pathology seen in the majority of appendix-specimens from otherwise hea-lthy adults [120–122]. For instance, air pollution seems to induce pathological α-synuclein inclusions in the olfactory bulb—even in children [123]. In add-ition, many of those patients with isolated olfactory pathology could in fact be body-first cases, as pro-posed by Braak. The fact that the DMV in such cases is still negative does not rule out that there are one or more small patches of α-synuclein in the enteric nervous system, which did not yet spread to the DMV. As described below, it is very difficult to find a small patch of α-synuclein pathology in the GI tract at postmortem. Finally, it is noted that 12 of the 20 stage 1 cases in Braak’s original staging study had pathology only in the DMV but not in the olfactory bulb [20]. Those data further suggest that at the earliest stages of body-first PD, it is possible that only one of the two ‘dual-hit structures’ have developed sufficient Lewy pathology to be detectable with standard histological measures.
CONTROVERSIES AND CRITICISMS OF THE SOC MODEL
The brain-first versus body-first hypothesis and the SOC model are intricately connected, both requiring that the initial α-synuclein pathology can form in the peripheral nervous system in the first place. We know that pathological α-synuclein inclusions have been detected in the gut of PD patients 20 years before diagnosis, and truncal vagotomy decreases the risk of PD by 40–50% [120, 124–127]. Also, the DMV and sympathetic ganglia generally show the highest amount of Lewy pathology in brainstem-predominant cases [21], suggesting that these are the first structures to be affected. Animal studies show that injecting α-synuclein seeds into the gut leads to propagating α-synuclein pathology through the autonomic connectome and results in a brainstem pattern of pathology highly similar to that seen in brainstem-predominant cases [53, 54].
Nevertheless, some skepticism about gut-first PD remains. More than 600 whole-body autopsies have been performed as part of the Arizona PD consortium [79, 108]. In this cohort, not a single case of ‘gut-only’ α-synuclein pathology has been found. This observation has so far only been reported in review papers, and the full dataset and methodology have yet to be published in detail. Of note, other studies have reported cases with isolated α-synuclein pathology in the sympathetic ganglia and heart [22], a high frequency of aggregated α-synuclein in the colon and appendix of normal controls [120, 121] and in the gut of children with norovirus infections [128]. Thus, differences in detection sensitivity needs clarification [129]. Moreover, 46% of cases in the Arizona study already had cerebral Lewy pathology, and such cases are not eligible, when searching for gut-only pathology, thereby cutting their overall sample size nearly in half [130].
It is reasonable to assume that the initial α-syn-uclein pathology could form in a very small patch anywhere in the GI tract. Such pathology could arise stochastically or be induced by the microbiome in the colon, bacterial overgrowth in the small intestine, helicobacter pylori in the stomach, inflammatory processes, or toxic insults anywhere in the GI tract and so on [128, 131–134]. Of note, specific microbiome signatures have been linked to RBD and constipation [135].
The human GI tract measures approximately 8–10 meters and has a geometric surface area of at least 7000cm2. Thus, if the initial patch of α-synuclein pathology is small, hundreds or even thousands of tissue sections throughout the GI tract are required to detect it with any degree of certainty. Of note, the terminal end field of a parasympathetic motor neuron probably covers only a few cm2 [136]. The exact methodology applied to those 300+eligible cases in the Arizona study has not been published, but in an early seminal study of 92 whole body autopsies, the same authors used one single slide from 9 locations in the GI tract, followed by up to five 80μm-thick slides in the lower esophagus [137]. The probability of finding a small patch (a few cm2) of randomly located α-synuclein pathology with this number of slides would be considerably less than 1%. To make matters worse, the time window from initial formation of enteric α-synuclein pathology to the appearance of the first DMV inclusion could be very short (weeks) [138]. As soon as the first DMV pathology is seen, the patient would no longer be defined as a ‘gut-only case’ and thus discarded. Finally, we do not yet know how to define α-synuclein pathology in the peripheral nervous system. It is probable that the early enteric pathology might be immature and consist of oligomers or proto-fibrils. These seeds could nevertheless travel to the DMV and start the forma-tion of inclusions here, which might then appear simultaneous with inclusions in the GI tract. Importantly, oligomeric α-synuclein is known to travel more readily than mature fibrils in the vagus nerve [139–141]. In support of this, a recent study used protein misfolding cyclic amplification on ENS and CNS tissue from PD and ILBD patients and showed that this fixed postmortem tissue seeds the aggregation of monomeric α-synuclein into fibrillar assemblies, even when no visible Lewy pathology were present on immunohistochemistry [142]. Another recent st-udy of transgenic Alzheimer mice showed that pat-hogenic amyloid-β seeds exist well in advance of the formation of mature amyloid-β plaques [143]. Thus, if similar pathogenic α-synuclein seeds are transported to the DMV, it may be impossible to find a gut-only case when applying immunohistochemical techniques validated to detect mature brainstem-type inclusions. For these reasons, the lack of gut-only cases in the Arizona cohort does not constitute strong evidence against the existence of body-first PD. It only precludes initial enteric α-synuclein pathology consisting of widespread, mature α-synuclein inclusions, which persist for a significant time before spreading to the DMV, and which are amenable to detection with standard immunohistochemistry protocols validated for brain-type Lewy pathology [137].
Other models of PD pathogenesis have been proposed such as the threshold theory model [144]. This model argues that prion-like propagation does not occur, but that the sequence of neurodegeneration and symptoms are explained by variable symptomatic thresholds in different neuronal populations. However, this model has a number of problems. First, it does not explain why some PD patients show drastic denervation of the autonomic nervous system many years before the dopamine system is damaged, whereas other PD patients show the opposite sequence [19]. Second, it does not attempt to explain why gradients of α-synuclein pathology are seen in nearly all PD and DLB cases postmortem. In short, in ILBD cases with minimal Lewy pathology, the inclusions are nearly always seen in neighboring neurons, for example, in the DMV and the LC, or in the amygdala and the entorhinal cortex or SN. In ILBD or PD/DLB cases with more pathology, most cases conform to either a brainstem-predominant or limbic-predominant profile, where the magnitude of pathology decreases in proportion to distance (i.e., number of synapses) from either the DMV or the limbic structures. These observations are easily explained by synuclein-spreading models such as the SOC model, but are very difficult to explain using the threshold theory model and no explanation was presented by the authors. Within the threshold theory, one might expect more random patterns of α-synuclein pathology. This same criticism applies to all theories of PD pathogenesis, where it is suggested that PD is primarily initiated by blood-borne factors crossing the blood-brain barrier, for instance by components of the immune system. Such models also cannot readily explain the graded and progressive α-synuclein pathology profiles, which seems to occur in most patients. Needless to say, such factors may still be very important, perhaps as secondary aggravating factors [7].
SUMMARY
Here, a new model of PD pathogenesis is proposed that takes into account the origin site of the initiating α-synuclein pathology and the important role played by the highly lateralized connectivity of the human brain. The SOC model may have greater explanatory power than previously proposed models as it is readily compatible with the two main profiles of α-synuclein pathology seen in postmortem studies: brainstem-predominant and limbic-predominant profiles. It is in full agreement with Braak’s gut-first hypothesis, but suggests that his hypothesis only applies to the body-first fraction of patients. The SOC model also readily explains why truncal vagotomy would decrease the risk of PD by 40–50%, and the observation that the peripheral autonomic nervous system is almost completely degenerated in RBD-positive prodromal PD, while the substantia nigra remains mostly intact.
In addition, the SOC model proposes a framework to explain known variations in the patterns of non-motor symptoms, variable degrees of motor asymmetry, and the variable rates of cognitive decline seen in different subtypes of PD. The predictions of the SOC model already have some support from existing clinical data, but prospective studies are needed to test the model further. Most importantly, careful bihemispheric studies of biobanked brains from Lewy-body positive cases with rich antemortem clinical data will be a crucial step in critically assessing the model, as will the development of an α-synuclein-specific PET tracer. Finally, it is emphasized that the SOC model only pertains to aggregation and propagation of pathological α-synuclein. The model fully acknowledges and is compatible with the crucial importance of a long list of other pathogenic factors in PD, including selective neuronal vulnerability. Elucidating how these factors contribute to initiating PD and to aggravating the disease course is as critical as ever, since those factors represent important disease-modifying treatment targets.
CONFLICT OF INTEREST
The author has no conflict of interest to report.
ACKNOWLEDGMENTS
The author holds grants from the Lundbeck foundation.
REFERENCES
[1] | Spillantini MG , Schmidt ML , Lee VM , Trojanowski JQ , Jakes R , Goedert M ((1997) ) Alpha-synuclein in Lewy bodies. Nature 388: , 839–840. |
[2] | Uchihara T , Giasson BI ((2016) ) Propagation of alpha-synuclein pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol 131: , 49–73. |
[3] | Brundin P , Melki R ((2017) ) Prying into the prion hypothesis for Parkinson’s disease. J Neurosci 37: , 9808–9818. |
[4] | Goedert M , Masuda-Suzukake M , Falcon B ((2017) ) Like prions: The propagation of aggregated tau and alpha-synuclein in neurodegeneration. Brain 140: , 266–278. |
[5] | Giguere N , Burke Nanni S , Trudeau LE ((2018) ) On cell loss and selective vulnerability of neuronal populations in Parkinson’s disease. Front Neurol 9: , 455. |
[6] | Gonzalez-Rodriguez P , Zampese E , Surmeier DJ ((2020) ) Selective neuronal vulnerability in Parkinson’s disease. Prog Brain Res 252: , 61–89. |
[7] | Johnson ME , Stecher B , Labrie V , Brundin L , Brundin P ((2019) ) Triggers, facilitators, and aggravators: Redefining Parkinson’s disease pathogenesis. Trends Neurosci 42: , 4–13. |
[8] | Tarutani A , Hasegawa M ((2019) ) Prion-like propagation of alpha-synuclein in neurodegenerative diseases. Prog Mol Biol Transl Sci 168: , 323–348. |
[9] | Pang SY , Ho PW , Liu HF , Leung CT , Li L , Chang EES , Ramsden DB , Ho SL ((2019) ) The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl Neurodegener 8: , 23. |
[10] | Coughlin DG , Petrovitch H , White LR , Noorigian J , Masaki KH , Ross GW , Duda JE ((2019) ) Most cases with Lewy pathology in a population-based cohort adhere to the Braak progression pattern but ‘failure to fit’ is highly dependent on staging system applied. Parkinsonism Relat Disord 64: , 124–131. |
[11] | Dias V , Junn E , Mouradian MM ((2013) ) The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis 3: , 461–491. |
[12] | Allen Reish HE , Standaert DG ((2015) ) Role of alpha-synuclein in inducing innate and adaptive immunity in Parkinson disease. J Parkinsons Dis 5: , 1–19. |
[13] | Bose A , Beal MF ((2016) ) Mitochondrial dysfunction in Parkinson’s disease. J Neurochem 139: (Suppl 1), 216–231. |
[14] | Hirsch EC , Standaert DG ((2021) ) Ten unsolved questions about neuroinflammation in Parkinson’s disease. Mov Disord 36: , 16–24. |
[15] | Surmeier DJ , Schumacker PT , Guzman JD , Ilijic E , Yang B , Zampese E ((2017) ) Calcium and Parkinson’s disease. Biochem Biophys Res Commun 483: , 1013–1019. |
[16] | Henrich MT , Geibl FF , Lakshminarasimhan H , Stegmann A , Giasson BI , Mao X , Dawson VL , Dawson TM , Oertel WH , Surmeier DJ ((2020) ) Determinants of seeding and spreading of alpha-synuclein pathology in the brain. Sci Adv 6: , eabc2487. |
[17] | Surmeier DJ , Obeso JA , Halliday GM ((2017) ) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18: , 101–113. |
[18] | Borghammer P , Van Den Berge N ((2019) ) Brain-first versus gut-first Parkinson’s disease: A hypothesis. J Parkinsons Dis 9: , S281–S295. |
[19] | Horsager J , Andersen KB , Knudsen K , Skjaerbaek C , Fedorova TD , Okkels N , Schaeffer E , Bonkat SK , Geday J , Otto M , Sommerauer M , Danielsen EH , Bech E , Kraft J , Munk OL , Hansen SD , Pavese N , Goder R , Brooks DJ , Berg D , Borghammer P ((2020) ) Brain-first versus body-first Parkinson’s disease: A multimodal imaging case-control study. Brain 143: , 3077–3088. |
[20] | Braak H , Del Tredici K , Rub U , de Vos RA , Jansen Steur EN , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: , 197–211. |
[21] | Raunio A , Kaivola K , Tuimala J , Kero M , Oinas M , Polvikoski T , Paetau A , Tienari PJ , Myllykangas L ((2019) ) Lewy-related pathology exhibits two anatomically and genetically distinct progression patterns: A population-based study of Finns aged 85. Acta Neuropathol 138: , 771–782. |
[22] | Tanei ZI , Saito Y , Ito S , Matsubara T , Motoda A , Yamazaki M , Sakashita Y , Kawakami I , Ikemura M , Tanaka S , Sengoku R , Arai T , Murayama S ((2021) ) Lewy pathology of the esophagus correlates with the progression of Lewy body disease: A Japanese cohort study of autopsy cases. Acta Neuropathol 141: , 25–37. |
[23] | Orimo S , Uchihara T , Nakamura A , Mori F , Kakita A , Wakabayashi K , Takahashi H ((2008) ) Axonal alpha-synuclein aggregates herald centripetal degeneration of cardiac sympathetic nerve in Parkinson’s disease. Brain 131: , 642–650. |
[24] | Braak H , Del Tredici K ((2008) ) Invited Article: Nervous system pathology in sporadic Parkinson disease. Neurology 70: , 1916–1925. |
[25] | Azevedo FA , Carvalho LR , Grinberg LT , Farfel JM , Ferretti RE , Leite RE , Jacob Filho W , Lent R , Herculano-Houzel S ((2009) ) Equal numbers of neuronal and nonneuronal cells make the human brain an isometrically scaled-up primate brain. J Comp Neurol 513: , 532–541. |
[26] | Songthawornpong N , Teasdale TW , Olesen MV , Pakkenberg B ((2021) ) Is there a correlation between the number of brain cells and IQ? Cereb Cortex 31: , 650–657. |
[27] | Zhou J , Wen Y , She L , Sui YN , Liu L , Richards LJ , Poo MM ((2013) ) Axon position within the corpus callosum determines contralateral cortical projection. Proc Natl Acad Sci U S A 110: , E2714–2723. |
[28] | Lamantia AS , Rakic P ((1990) ) Cytological and quantitative characteristics of four cerebral commissures in the rhesus monkey. J Comp Neurol 291: , 520–537. |
[29] | Tomasch J ((1954) ) Size, distribution, and number of fibres in the human corpus callosum. Anat Rec 119: , 119–135. |
[30] | Rosen BQ , Halgren E ((2020) ) A whole-cortex probabilistic diffusion tractography connectome. BioRxiv, pp. 1-25. https://doi.org/10.1101/2020.06.22.166041 |
[31] | Maller JJ , Welton T , Middione M , Callaghan FM , Rosenfeld JV , Grieve SM ((2019) ) Revealing the hippocampal connectome through super-resolution 1150-direction diffusion MRI. Sci Rep 9: , 2418. |
[32] | Bunney BS , Aghajanian GK ((1976) ) The precise localization of nigral afferents in the rat as determined by a retrograde tracing technique. Brain Res 117: , 423–435. |
[33] | Volz HP , Rehbein G , Triepel J , Knuepfer MM , Stumpf H , Stock G ((1990) ) Afferent connections of the nucleus centralis amygdalae. A horseradish peroxidase study and literature survey. Anat Embryol (Berl) 181: , 177–194. |
[34] | Usunoff KG , Itzev DE , Rolfs A , Schmitt O , Wree A ((2006) ) Brain stem afferent connections of the amygdala in the rat with special references to a projection from the parabigeminal nucleus: A fluorescent retrograde tracing study. Anat Embryol (Berl) 211: , 475–496. |
[35] | Wallace DM , Magnuson DJ , Gray TS ((1989) ) The amygdalo-brainstem pathway: Selective innervation of dopaminergic, noradrenergic and adrenergic cells in the rat. Neurosci Lett 97: , 252–258. |
[36] | Schwaber JS , Kapp BS , Higgins GA , Rapp PR ((1982) ) Amygdaloid and basal forebrain direct connections with the nucleus of the solitary tract and the dorsal motor nucleus. J Neurosci 2: , 1424–1438. |
[37] | Ottersen OP ((1980) ) Afferent connections to the amygdaloid complex of the rat and cat: II. Afferents from the hypothalamus and the basal telencephalon. J Comp Neurol 194: , 267–289. |
[38] | Fritschy JM , Grzanna R ((1990) ) Distribution of locus coeruleus axons within the rat brainstem demonstrated by Phaseolus vulgaris leucoagglutinin anterograde tracing in combination with dopamine-beta-hydroxylase immunofluorescence. J Comp Neurol 293: , 616–631. |
[39] | Cedarbaum JM , Aghajanian GK ((1978) ) Afferent projections to the rat locus coeruleus as determined by a retrograde tracing technique. J Comp Neurol 178: , 1–16. |
[40] | Westlund KN , Coulter JD ((1980) ) Descending projections of the locus coeruleus and subcoeruleus/medial parabrachial nuclei in monkey: Axonal transport studies and dopamine-beta-hydroxylase immunocytochemistry. Brain Res 2: , 235–264. |
[41] | Ter Horst GJ , Toes GJ , Van Willigen JD ((1991) ) Locus coeruleus projections to the dorsal motor vagus nucleus in the rat. Neuroscience 45: , 153–160. |
[42] | Semba K , Reiner PB , McGeer EG , Fibiger HC ((1988) ) Brainstem afferents to the magnocellular basal forebrain studied by axonal transport, immunohistochemistry, and electrophysiology in the rat. J Comp Neurol 267: , 433–453. |
[43] | Luiten PG , Spencer DG Jr. , Traber J , Gaykema RP ((1985) ) The pattern of cortical projections from the intermediate parts of the magnocellular nucleus basalis in the rat demonstrated by tracing with Phaseolus vulgaris-leucoagglutinin. Neurosci Lett 57: , 137–142. |
[44] | Dinopoulos A , Papadopoulos GC , Parnavelas JG , Antonopoulos J , Karamanlidis AN ((1989) ) Basal forebrain projections to the lower brain stem in the rat. Exp Neurol 105: , 316–319. |
[45] | Nitecka L , Amerski L , Narkiewicz O ((1981) ) Interamygdaloid connections in the rat studied by the horseradish peroxidase method. Neurosci Lett 26: , 1–4. |
[46] | Franzmeier N , Neitzel J , Rubinski A , Smith R , Strandberg O , Ossenkoppele R , Hansson O , Ewers M , Alzheimer’s Disease Neuroimaging Initiative ((2020) ) Functional brain architecture is associated with the rate of tau accumulation in Alzheimer’s disease. Nat Commun 11: , 347. |
[47] | Berthoud HR , Carlson NR , Powley TL ((1991) ) Topography of efferent vagal innervation of the rat gastrointestinal tract.R. Am J Physiol 260: , 200–207. |
[48] | Zhang X , Renehan WE , Fogel R ((2000) ) Vagal innervation of the rat duodenum. J Auton Nerv Syst 79: , 8–18. |
[49] | Zhang XG , Fogel R , Simpson P , Renehan W ((1991) ) The target specificity of the extrinsic innervation of the rat small intestine. J Auton Nerv Syst 32: , 53–62. |
[50] | Shapiro RE , Miselis RR ((1985) ) The central organization of the vagus nerve innervating the stomach of the rat. J Comp Neurol 238: , 473–488. |
[51] | Altschuler SM , Escardo J , Lynn RB , Miselis RR ((1993) ) The central organization of the vagus nerve innervating the colon of the rat. Gastroenterology 104: , 502–509. |
[52] | Rinaman L , Roesch MR , Card JP ((1999) ) Retrograde transynaptic pseudorabies virus infection of central autonomic circuits in neonatal rats. Brain Res Dev Brain Res 114: , 207–216. |
[53] | Van Den Berge N , Ferreira N , Gram H , Mikkelsen TW , Alstrup AKO , Casadei N , Tsung-Pin P , Riess O , Nyengaard JR , Tamguney G , Jensen PH , Borghammer P ((2019) ) Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol 138: , 535–550. |
[54] | Kim S , Kwon SH , Kam TI , Panicker N , Karuppagounder SS , Lee S , Lee JH , Kim WR , Kook M , Foss CA , Shen C , Lee H , Kulkarni S , Pasricha PJ , Lee G , Pomper MG , Dawson VL , Dawson TM , Ko HS ((2019) ) Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103: , 627–641 e627. |
[55] | Breid S , Bernis ME , Babila JT , Garza MC , Wille H , Tamguney G ((2016) ) Neuroinvasion of alpha-synuclein prionoids after intraperitoneal and intraglossal inoculation. J Virol 90: , 9182–9193. |
[56] | Henderson MX , Cornblath EJ , Darwich A , Zhang B , Brown H , Gathagan RJ , Sandler RM , Bassett DS , Trojanowski JQ , Lee VMY ((2019) ) Spread of alpha-synuclein pathology through the brain connectome is modulated by selective vulnerability and predicted by network analysis. Nat Neurosci 22: , 1248–1257. |
[57] | Rey NL , Steiner JA , Maroof N , Luk KC , Madaj Z , Trojanowski JQ , Lee VM , Brundin P ((2016) ) Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med 213: , 1759–1778. |
[58] | Ulusoy A , Phillips RJ , Helwig M , Klinkenberg M , Powley TL , Di Monte DA ((2017) ) Brain-to-stomach transfer of alpha-synuclein via vagal preganglionic projections. Acta Neuropathol 133: , 381–393. |
[59] | Henderson MX , Sedor S , McGeary I , Cornblath EJ , Peng C , Riddle DM , Li HL , Zhang B , Brown HJ , Olufemi MF , Bassett DS , Trojanowski JQ , Lee VMY ((2020) ) Glucocerebrosidase activity modulates neuronal susceptibility to pathological alpha-synuclein insult. Neuron 105: , 822–836 e827. |
[60] | Kosaka K , Tsuchiya K , Yoshimura M ((1988) ) Lewy body disease with and without dementia: A clinicopathological study of 35 cases. Clin Neuropathol 7: , 299–305. |
[61] | Beach TG , Adler CH , Lue L , Sue LI , Bachalakuri J , Henry-Watson J , Sasse J , Boyer S , Shirohi S , Brooks R , Eschbacher J , White CL 3rd , Akiyama H , Caviness J , Shill HA , Connor DJ , Sabbagh MN , Walker DG , Arizona Parkinson’s Disease Consortium ((2009) ) Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117: , 613–634. |
[62] | Chen D , Jiang J , Lu J , Wu P , Zhang H , Zuo C , Shi K ((2019) ) Brain network and abnormal hemispheric asymmetry analyses to explore the marginal differences in glucose metabolic distributions among Alzheimer’s disease, Parkinson’s disease dementia, and Lewy body dementia. Front Neurol 10: , 369. |
[63] | Savica R , Boeve BF , Mielke MM ((2018) ) When do alpha-synucleinopathies start? An epidemiological timeline: A review. JAMA Neurol 75: , 503–509. |
[64] | Boeve BF ((2013) ) Idiopathic REM sleep behaviour disorder in the development of Parkinson’s disease. Lancet Neurol 12: , 469–482. |
[65] | Chiu NKH , Ehgoetz Martens KA , Mok VCT , Lewis SJG , Matar E ((2020) ) Prevalence and predictors of mood disturbances in idiopathic REM sleep behaviour disorder. J Sleep Res, e13040. |
[66] | Szatmari S Jr. , Ajtay A , Oberfrank F , Dobi B , Bereczki D ((2020) ) The prevalence of psychiatric symptoms before the diagnosis of Parkinson’s disease in a nationwide cohort: A comparison to patients with cerebral infarction. PLoS One 15: , e0236728. |
[67] | Barber TR , Lawton M , Rolinski M , Evetts S , Baig F , Ruffmann C , Gornall A , Klein JC , Lo C , Dennis G , Bandmann O , Quinnell T , Zaiwalla Z , Ben-Shlomo Y , Hu MTM ((2017) ) Prodromal parkinsonism and neurodegenerative risk stratification in REM sleep behavior disorder. Sleep 40: , zsx071. |
[68] | Cheng HC , Ulane CM , Burke RE ((2010) ) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol 67: , 715–725. |
[69] | Postuma RB , Iranzo A , Hu M , Hogl B , Boeve BF , Manni R , Oertel WH , Arnulf I , Ferini-Strambi L , Puligheddu M , Antelmi E , Cochen De Cock V , Arnaldi D , Mollenhauer B , Videnovic A , Sonka K , Jung KY , Kunz D , Dauvilliers Y , Provini F , Lewis SJ , Buskova J , Pavlova M , Heidbreder A , Montplaisir JY , Santamaria J , Barber TR , Stefani A , St Louis EK , Terzaghi M , Janzen A , Leu-Semenescu S , Plazzi G , Nobili F , Sixel-Doering F , Dusek P , Bes F , Cortelli P , Ehgoetz Martens K , Gagnon JF , Gaig C , Zucconi M , Trenkwalder C , Gan-Or Z , Lo C , Rolinski M , Mahlknecht P , Holzknecht E , Boeve AR , Teigen LN , Toscano G , Mayer G , Morbelli S , Dawson B , Pelletier A ((2019) ) Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: A multicentre study. Brain 142: , 744–759. |
[70] | Fereshtehnejad SM , Romenets SR , Anang JB , Latreille V , Gagnon JF , Postuma RB ((2015) ) New clinical subtypes of Parkinson disease and their longitudinal progression: A prospective cohort comparison with other phenotypes. JAMA Neurol 72: , 863–873. |
[71] | Donadio V , Incensi A , Del Sorbo F , Rizzo G , Infante R , Scaglione C , Modugno N , Fileccia E , Elia AE , Cencini F , Liguori R ((2018) ) Skin nerve phosphorylated alpha-synuclein deposits in Parkinson disease with orthostatic hypotension. J Neuropathol Exp Neurol 77: , 942–949. |
[72] | Liu Y , Zhu XY , Zhang XJ , Kuo SH , Ondo WG , Wu YC ((2017) ) Clinical features of Parkinson’s disease with and without rapid eye movement sleep behavior disorder. Transl Neurodegener 6: , 35. |
[73] | Knudsen K , Fedorova TD , Hansen AK , Sommerauer M , Otto M , Svendsen KB , Nahimi A , Stokholm MG , Pavese N , Beier CP , Brooks DJ , Borghammer P ((2018) ) staging of pathology in REM sleep behaviour disorder: A multimodality imaging case-control study. Lancet Neurol 17: , 618–628. |
[74] | Kashihara K , Imamura T , Shinya T ((2010) ) Cardiac 123I-MIBG uptake is reduced more markedly in patients with REM sleep behavior disorder than in those with early stage Parkinson’s disease. Parkinsonism Relat Disord 16: , 252–255. |
[75] | Gabilondo I , Llorens V , Rodriguez T , Fernandez M , Concha TP , Acera M , Tijero B , Murueta-Goyena A , Del Pino R , Cortes J , Gomez-Esteban JC ((2019) ) Myocardial MIBG scintigraphy in genetic Parkinson’s disease as a model for Lewy body disorders. Eur J Nucl Med Mol Imaging 46: , 376–384. |
[76] | Sakakibara R , Tateno F , Aiba Y , Ogata T , Kishi M , Terada H , Inaoka T , Nakatsuka T , Matsuoka K ((2019) ) MIBG myocardial scintigraphy identifies premotor PD/DLB during a negative DAT scan period: Second report. Mov Disord Clin Pract 6: , 46–50. |
[77] | Miyamoto T , Miyamoto M , Inoue Y , Usui Y , Suzuki K , Hirata K ((2006) ) Reduced cardiac 123I-MIBG scintigraphy in idiopathic REM sleep behavior disorder. Neurology 67: , 2236–2238. |
[78] | Leclair-Visonneau L , Clairembault T , Coron E , Le Dily S , Vavasseur F , Dalichampt M , Pereon Y , Neunlist M , Derkinderen P ((2017) ) REM sleep behavior disorder is related to enteric neuropathology in Parkinson disease. Neurology 89: , 1612–1618. |
[79] | Adler CH , Beach TG ((2016) ) Neuropathological basis of nonmotor manifestations of Parkinson’s disease. Mov Disord 31: , 1114–1119. |
[80] | Djaldetti R , Ziv I , Melamed E ((2006) ) The mystery of motor asymmetry in Parkinson’s disease. Lancet Neurol 5: , 796–802. |
[81] | Gelb DJ , Oliver E , Gilman S ((1999) ) Diagnostic criteria for Parkinson disease. Arch Neurol 56: , 33–39. |
[82] | Toth C , Rajput M , Rajput AH ((2004) ) Anomalies of asymmetry of clinical signs in parkinsonism. Mov Disord 19: , 151–157. |
[83] | Elbaz A , Bower JH , Peterson BJ , Maraganore DM , McDonnell SK , Ahlskog JE , Schaid DJ , Rocca WA ((2003) ) Survival study of Parkinson disease in Olmsted County, Minnesota. Arch Neurol 60: , 91–96. |
[84] | Uitti RJ , Baba Y , Wszolek ZK , Putzke DJ ((2005) ) Defining the Parkinson’s disease phenotype: Initial symptoms and baseline characteristics in a clinical cohort. Parkinsonism Relat Disord 11: , 139–145. |
[85] | Tanner CM , Ross GW , Jewell SA , Hauser RA , Jankovic J , Factor SA , Bressman S , Deligtisch A , Marras C , Lyons KE , Bhudhikanok GS , Roucoux DF , Meng C , Abbott RD , Langston JW ((2009) ) Occupation and risk of parkinsonism: A multicenter case-control study. Arch Neurol 66: , 1106–1113. |
[86] | Wang ZY , Lian H , Cai QQ , Song HY , Zhang XL , Zhou L , Zhang YM , Zheng LF , Zhu JX ((2014) ) No direct projection is observed from the substantia nigra to the dorsal vagus complex in the rat. J Parkinsons Dis 4: , 375–383. |
[87] | Lee HS , Kim MA , Waterhouse BD ((2005) ) Retrograde double-labeling study of common afferent projections to the dorsal raphe and the nuclear core of the locus coeruleus in the rat. J Comp Neurol 481: , 179–193. |
[88] | Parkkinen L , Pirttila T , Alafuzoff I ((2008) ) Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol 115: , 399–407. |
[89] | Riederer P , Jellinger KA , Kolber P , Hipp G , Sian-Hulsmann J , Kruger R ((2018) ) Lateralisation in Parkinson disease. Cell Tissue Res 373: , 297–312. |
[90] | Cao R , Chen X , Xie C , Hu P , Wang K ((2020) ) Serial dopamine transporter imaging of nigrostriatal function in Parkinson’s disease with probable REM sleep behavior disorder. Front Neurosci 14: , 349. |
[91] | Djaldetti R , Melamed E , Gadoth N ((2001) ) Abnormal skin wrinkling in the less affected side in hemiparkinsonism-a possible test for sympathetic dysfunction in Parkinson’s disease. Biomed Pharmacother 55: , 475–478. |
[92] | Zucco G , Zeni MT , Perrone A , Piccolo I ((2001) ) Olfactory sensitivity in early-stage Parkinson patients affected by more marked unilateral disorder. Percept Mot Skills 92: , 894–898. |
[93] | Heckmann JG , Höcherl C , C. L , Platsch G , Hummel T ((2004) ) Hemihyposmia in a case of hemiparkinsonism. Eur J Gen Med 1: , 72–73. |
[94] | Davidsdottir S , Wagenaar R , Young D , Cronin-Golomb A ((2008) ) Impact of optic flow perception and egocentric coordinates on veering in Parkinson’s disease. Brain 131: , 2882–2893. |
[95] | Amick MM , Schendan HE , Ganis G , Cronin-Golomb A ((2006) ) Frontostriatal circuits are necessary for visuomotor transformation: Mental rotation in Parkinson’s disease. Neuropsychologia 44: , 339–349. |
[96] | Lee AC , Harris JP , Atkinson EA , Fowler MS ((2001) ) Evidence from a line bisection task for visuospatial neglect in left hemiparkinson’s disease. Vision Res 41: , 2677–2686. |
[97] | Karadi K , Lucza T , Aschermann Z , Komoly S , Deli G , Bosnyak E , Acs P , Horvath R , Janszky J , Kovacs N ((2015) ) Visuospatial impairment in Parkinson’s disease: The role of laterality. Laterality 20: , 112–127. |
[98] | Agosta S , Magnago D , Galante E , Ferraro F , Magherini A , Di Giacopo R , Miceli G , Battelli L ((2020) ) Lateralized cognitive functions in Parkinson’s patients: A behavioral approach for the early detection of sustained attention deficits. Brain Res 1726: , 146486. |
[99] | Cronin-Golomb A ((2010) ) Parkinson’s disease as a disconnection syndrome. Neuropsychol Rev 20: , 191–208. |
[100] | Shalash AS , Hassan DM , Elrassas HH , Salama MM , Mendez-Hernandez E , Salas-Pacheco JM , Arias-Carrion O ((2017) ) Auditory- and vestibular-evoked potentials correlate with motor and non-motor features of Parkinson’s disease. Front Neurol 8: , 55. |
[101] | Tanaka R , Yamashiro K , Ogawa T , Oyama G , Nishioka K , Umemura A , Shimo Y , Hattori N ((2020) ) The absence of orthostatic heart rate increase is associated with cognitive impairment in Parkinson’s disease. PLoS One 15: , e0240491. |
[102] | Oka H , Umehara T , Nakahara A , Matsuno H ((2020) ) Comparisons of cardiovascular dysautonomia and cognitive impairment between de novo Parkinson’s disease and de novo dementia with Lewy bodies. BMC Neurol 20: , 350. |
[103] | Pilotto A , Romagnolo A , Tuazon JA , Vizcarra JA , Marsili L , Zibetti M , Rosso M , Rodriguez-Porcel F , Borroni B , Rizzetti MC , Rossi C , Vizcarra-Escobar D , Molano JR , Lopiano L , Ceravolo R , Masellis M , Espay AJ , Padovani A , Merola A ((2019) ) Orthostatic hypotension and REM sleep behaviour disorder: Impact on clinical outcomes in alpha-synucleinopathies. J Neurol Neurosurg Psychiatry 90: , 1257–1263. |
[104] | Kong WL , Huang Y , Qian E , Morris MJ ((2020) ) Constipation and sleep behaviour disorder associate with processing speed and attention in males with Parkinson’s disease over five years follow-up. Sci Rep 10: , 19014. |
[105] | Merola A , Romagnolo A , Dwivedi AK , Padovani A , Berg D , Garcia-Ruiz PJ , Fabbri M , Artusi CA , Zibetti M , Lopiano L , Pilotto A , Bonacina S , Morgante F , Zeuner K , Griewing C , Schaeffer E , Rodriguez-Porcel F , Kauffman M , Turcano P , de Oliveira LM , Palermo G , Shanks E , Del Sorbo F , Bonvegna S , Savica R , Munhoz RP , Ceravolo R , Cilia R , Espay AJ ((2020) ) Benign versus malignant Parkinson disease: The unexpected silver lining of motor complications. J Neurol 267: , 2949–2960. |
[106] | van de Beek M , van Steenoven I , van der Zande JJ , Barkhof F , Teunissen CE , van der Flier WM , Lemstra AW ((2020) ) Prodromal dementia with Lewy bodies: Clinical characterization and predictors of progression. Mov Disord 35: , 859–867. |
[107] | Postuma RB , Adler CH , Dugger BN , Hentz JG , Shill HA , Driver-Dunckley E , Sabbagh MN , Jacobson SA , Belden CM , Sue LI , Serrano G , Beach TG ((2015) ) REM sleep behavior disorder and neuropathology in Parkinson’s disease. Mov Disord 30: , 1413–1417. |
[108] | Beach TG , Adler CH , Sue LI , Serrano G , Shill HA , Walker DG , Lue L , Roher AE , Dugger BN , Maarouf C , Birdsill AC , Intorcia A , Saxon-Labelle M , Pullen J , Scroggins A , Filon J , Scott S , Hoffman B , Garcia A , Caviness JN , Hentz JG , Driver-Dunckley E , Jacobson SA , Davis KJ , Belden CM , Long KE , Malek-Ahmadi M , Powell JJ , Gale LD , Nicholson LR , Caselli RJ , Woodruff BK , Rapscak SZ , Ahern GL , Shi J , Burke AD , Reiman EM , Sabbagh MN ((2015) ) Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology 35: , 354–389. |
[109] | Compta Y , Parkkinen L , O’Sullivan SS , Vandrovcova J , Holton JL , Collins C , Lashley T , Kallis C , Williams DR , de Silva R , Lees AJ , Revesz T ((2011) ) Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: Which is more important? Brain 134: , 1493–1505. |
[110] | Jellinger KA , Attems J ((2006) ) Does striatal pathology distinguish Parkinson disease with dementia and dementia with Lewy bodies? Acta Neuropathol 112: , 253–260. |
[111] | Donaghy P , Thomas AJ , O’Brien JT ((2015) ) Amyloid PET Imaging in Lewy body disorders. Am J Geriatr Psychiatry 23: , 23–37. |
[112] | Lim KH ((2019) ) Diverse misfolded conformational strains and cross-seeding of misfolded proteins implicated in neurodegenerative diseases. Front Mol Neurosci 12: , 158. |
[113] | Williams T , Sorrentino Z , Weinrich M , Giasson BI , Chakrabarty P ((2020) ) Differential cross-seeding properties of tau and alpha-synuclein in mouse models of tauopathy and synucleinopathy. Brain Commun 2: , fcaa-090. |
[114] | Jansen WJ , Ossenkoppele R , Knol DL , Tijms BM , Scheltens P , Verhey FR , Visser PJ , Amyloid Biomarker Study G, Aalten P , Aarsland D , Alcolea D , Alexander M , Almdahl IS , Arnold SE , Baldeiras I , Barthel H , van Berckel BN , Bibeau K , Blennow K , Brooks DJ , van Buchem MA , Camus V , Cavedo E , Chen K , Chetelat G , Cohen AD , Drzezga A , Engelborghs S , Fagan AM , Fladby T , Fleisher AS , van der Flier WM , Ford L , Forster S , Fortea J , Foskett N , Frederiksen KS , Freund-Levi Y , Frisoni GB , Froelich L , Gabryelewicz T , Gill KD , Gkatzima O , Gomez-Tortosa E , Gordon MF , Grimmer T , Hampel H , Hausner L , Hellwig S , Herukka SK , Hildebrandt H , Ishihara L , Ivanoiu A , Jagust WJ , Johannsen P , Kandimalla R , Kapaki E , Klimkowicz-Mrowiec A , Klunk WE , Kohler S , Koglin N , Kornhuber J , Kramberger MG , Van Laere K , Landau SM , Lee DY , de Leon M , Lisetti V , Lleo A , Madsen K , Maier W , Marcusson J , Mattsson N , de Mendonca A , Meulenbroek O , Meyer PT , Mintun MA , Mok V , Molinuevo JL , Mollergard HM , Morris JC , Mroczko B , Van der Mussele S , Na DL , Newberg A , Nordberg A , Nordlund A , Novak GP , Paraskevas GP , Parnetti L , Perera G , Peters O , Popp J , Prabhakar S , Rabinovici GD , Ramakers IH , Rami L , Resende de Oliveira C , Rinne JO , Rodrigue KM , Rodriguez-Rodriguez E , Roe CM , Rot U , Rowe CC , Ruther E , Sabri O , Sanchez-Juan P , Santana I , Sarazin M , Schroder J , Schutte C , Seo SW , Soetewey F , Soininen H , Spiru L , Struyfs H , Teunissen CE , Tsolaki M , Vandenberghe R , Verbeek MM , Villemagne VL , Vos SJ , van Waalwijk van Doorn LJ , Waldemar G , Wallin A , Wallin AK , Wiltfang J , Wolk DA , Zboch M , Zetterberg H ((2015) ) Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 313: , 1924–1938. |
[115] | McKeith I , O’Brien J , Walker Z , Tatsch K , Booij J , Darcourt J , Padovani A , Giubbini R , Bonuccelli U , Volterrani D , Holmes C , Kemp P , Tabet N , Meyer I , Reininger C , DLB Study Group ((2007) ) Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: A phase III, multicentre study. Lancet Neurol 6: , 305–313. |
[116] | Marui W , Iseki E , Nakai T , Miura S , Kato M , Ueda K , Kosaka K ((2002) ) Progression and staging of Lewy pathology in brains from patients with dementia with Lewy bodies. J Neurol Sci 195: , 153–159. |
[117] | Braak H , Rub U , Gai WP , Del Tredici K ((2003) ) Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm (Vienna) 110: , 517–536. |
[118] | Rey NL , Bousset L , George S , Madaj Z , Meyerdirk L , Schulz E , Steiner JA , Melki R , Brundin P ((2019) ) alpha-Synuclein conformational strains spread, seed and target neuronal cells differentially after injection into the olfactory bulb. Acta Neuropathol Commun 7: , 221. |
[119] | Kim JS , Park HE , Park IS , Oh YS , Ryu DW , Song IU , Jung YA , Yoo IR , Choi HS , Lee PH , Lee KS ((2017) ) Normal ‘heart’ in Parkinson’s disease: Is this a distinct clinical phenotype? Eur J Neurol 24: , 349–356. |
[120] | Stokholm MG , Danielsen EH , Hamilton-Dutoit SJ , Borghammer P ((2016) ) Pathological alpha-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann Neurol 79: , 940–949. |
[121] | Killinger BA , Madaj Z , Sikora JW , Rey N , Haas AJ , Vepa Y , Lindqvist D , Chen H , Thomas PM , Brundin P , Brundin L , Labrie V ((2018) ) The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci Transl Med 10: , eaar5280. |
[122] | Gray MT , Munoz DG , Gray DA , Schlossmacher MG , Woulfe JM ((2014) ) Alpha-synuclein in the appendiceal mucosa of neurologically intact subjects. Mov Disord 29: , 991–998. |
[123] | Calderon-Garciduenas L , Solt AC , Henriquez-Roldan C , Torres-Jardon R , Nuse B , Herritt L , Villarreal-Calderon R , Osnaya N , Stone I , Garcia R , Brooks DM , Gonzalez-Maciel A , Reynoso-Robles R , Delgado-Chavez R , Reed W ((2008) ) Long-term air pollution exposure is associated with neuroinflammation, an altered innate immune response, disruption of the blood-brain barrier, ultrafine particulate deposition, and accumulation of amyloid beta-42 and alpha-synuclein in children and young adults. Toxicol Pathol 36: , 289–310. |
[124] | Hilton D , Stephens M , Kirk L , Edwards P , Potter R , Zajicek J , Broughton E , Hagan H , Carroll C ((2014) ) Accumulation of alpha-synuclein in the bowel of patients in the pre-clinical phase of Parkinson’s disease. Acta Neuropathol 127: , 235–241. |
[125] | Shannon KM , Keshavarzian A , Dodiya HB , Jakate S , Kordower JH ((2012) ) Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov Disord 27: , 716–719. |
[126] | Svensson E , Horvath-Puho E , Thomsen RW , Djurhuus JC , Pedersen L , Borghammer P , Sorensen HT ((2015) ) Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol 78: , 522–529. |
[127] | Liu B , Fang F , Pedersen NL , Tillander A , Ludvigsson JF , Ekbom A , Svenningson P , Chen H , Wirdefeldt K ((2017) ) Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study. Neurology 88: , 1996–2002. |
[128] | Stolzenberg E , Berry D , Yang , Lee EY , Kroemer A , Kaufman S , Wong GCL , Oppenheim JJ , Sen S , Fishbein T , Bax A , Harris B , Barbut D , Zasloff MA ((2017) ) A role for neuronal alpha-synuclein in gastrointestinal immunity. J Innate Immun 9: , 456–463. |
[129] | Corbille AG , Letournel F , Kordower JH , Lee J , Shanes E , Neunlist M , Munoz DG , Derkinderen P , Beach TG ((2016) ) Evaluation of alpha-synuclein immunohistochemical methods for the detection of Lewy-type synucleinopathy in gastrointestinal biopsies. Acta Neuropathol Commun 4: , 35. |
[130] | Shprecher DR , Adler CH , Zhang N , Hentz JG , Serrano GE , Dugger BN , Shill HA , Savica R , Caviness JN , Sabbagh MN , Belden CM , Driver-Dunckley E , Mehta SH , Sue LI , Davis KJ , Zamrini E , Beach TG ((2018) ) Predicting alpha-synuclein pathology by REM sleep behavior disorder diagnosis. Parkinsonism Relat Disord 55: , 92–96. |
[131] | Nerius M , Doblhammer G , Tamguney G ((2020) ) GI infections are associated with an increased risk of Parkinson’s disease. Gut 69: , 1154–1156. |
[132] | Nielsen HH , Qiu J , Friis S , Wermuth L , Ritz B ((2012) ) Treatment for Helicobacter pylori infection and risk of Parkinson’s disease in Denmark. Eur J Neurol 19: , 864–869. |
[133] | Fu P , Gao M , Yung KKL ((2020) ) Association of intestinal disorders with Parkinson’s disease and Alzheimer’s disease: A systematic review and meta-analysis. ACS Chem Neurosci 11: , 395–405. |
[134] | Prigent A , Lionnet A , Durieu E , Chapelet G , Bourreille A , Neunlist M , Rolli-Derkinderen M , Derkinderen P ((2019) ) Enteric alpha-synuclein expression is increased in Crohn’s disease. Acta Neuropathol 137: , 359–361. |
[135] | Heinzel S , Aho VTE , Suenkel U , von Thaler AK , Schulte C , Deuschle C , Paulin L , Hantunen S , Brockmann K , Eschweiler GW , Maetzler W , Berg D , Auvinen P , Scheperjans F ((2020) ) Gut microbiome signatures of risk and prodromal markers of Parkinson disease. Ann Neurol 88: , 320–331. |
[136] | Holst MC , Kelly JB , Powley TL ((1997) ) Vagal preganglionic projections to the enteric nervous system characterized with Phaseolus vulgaris-leucoagglutinin. J Comp Neurol 381: , 81–100. |
[137] | Beach TG , Adler CH , Sue LI , Vedders L , Lue L , White Iii CL , Akiyama H , Caviness JN , Shill HA , Sabbagh MN , Walker DG ((2010) ) Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol 119: , 689–702. |
[138] | Holmqvist S , Chutna O , Bousset L , Aldrin-Kirk P , Li W , Bjorklund T , Wang ZY , Roybon L , Melki R , Li JY ((2014) ) Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol 128: , 805–820. |
[139] | Peelaerts W , Bousset L , Van der Perren A , Moskalyuk A , Pulizzi R , Giugliano M , Van den Haute C , Melki R , Baekelandt V ((2015) ) alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522: , 340–344. |
[140] | Reyes JF , Rey NL , Bousset L , Melki R , Brundin P , Angot E ((2014) ) Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 62: , 387–398. |
[141] | Rey NL , Petit GH , Bousset L , Melki R , Brundin P ((2013) ) Transfer of human alpha-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol 126: , 555–573. |
[142] | Fenyi A , Duyckaerts C , Bousset L , Braak H , Tredici KD , Melki R , On Behalf Of The Brainbank Neuro-Ceb Neuropathology Network ((2021) ) Seeding propensity and characteristics of pathogenic alphaSyn assemblies in formalin-fixed human tissue from the enteric nervous system, olfactory bulb, and brainstem in cases staged for Parkinson’s Disease. Cells 10: , 139. |
[143] | Uhlmann RE , Rother C , Rasmussen J , Schelle J , Bergmann C , Ullrich Gavilanes EM , Fritschi SK , Buehler A , Baumann F , Skodras A , Al-Shaana R , Beschorner N , Ye L , Kaeser SA , Obermuller U , Christensen S , Kartberg F , Stavenhagen JB , Rahfeld JU , Cynis H , Qian F , Weinreb PH , Bussiere T , Walker LC , Staufenbiel M , Jucker M ((2020) ) Acute targeting of pre-amyloid seeds in transgenic mice reduces Alzheimer-like pathology later in life. Nat Neurosci 23: , 1580–1588. |
[144] | Engelender S , Isacson O ((2017) ) The threshold theory for Parkinson’s disease. Trends Neurosci 40: , 4–14. |


