
Neuroprotective Potential of a Small Molecule RET Agonist in Cultured Dopamine Neurons and Hemiparkinsonian Rats
Abstract
Background:
Parkinson’s disease (PD) is a progressive neurological disorder where loss of dopamine neurons in the substantia nigra and dopamine depletion in the striatum cause characteristic motor symptoms. Currently, no treatment is able to halt the progression of PD. Glial cell line-derived neurotrophic factor (GDNF) rescues degenerating dopamine neurons both in vitro and in animal models of PD. When tested in PD patients, however, the outcomes from intracranial GDNF infusion paradigms have been inconclusive, mainly due to poor pharmacokinetic properties.
Objective:
We have developed drug-like small molecules, named BT compounds that activate signaling through GDNF’s receptor, the transmembrane receptor tyrosine kinase RET, both in vitro and in vivo and are able to penetrate through the blood-brain barrier. Here we evaluated the properties of BT44, a second generation RET agonist, in immortalized cells, dopamine neurons and rat 6-hydroxydopamine model of PD.
Methods:
We used biochemical, immunohistochemical and behavioral methods to evaluate the effects of BT44 on dopamine system in vitro and in vivo.
Results:
BT44 selectively activated RET and intracellular pro-survival AKT and MAPK signaling pathways in immortalized cells. In primary midbrain dopamine neurons cultured in serum-deprived conditions, BT44 promoted the survival of the neurons derived from wild-type, but not from RET knockout mice. BT44 also protected cultured wild-type dopamine neurons from MPP+-induced toxicity. In a rat 6-hydroxydopamine model of PD, BT44 reduced motor imbalance and seemed to protect dopaminergic fibers in the striatum.
Conclusion:
BT44 holds potential for further development into a novel, possibly disease-modifying, therapy for PD.
INTRODUCTION
More than 10 million people worldwide suffer from Parkinson’s disease (PD), and the number is increasing with the overall aging of the population (Parkinson’s Foundation 2019). The crucial pathological feature of PD is progressive loss of dopamine neurons within the substantia nigra pars compacta (SNpc) in the midbrain which leads to depletion of dopamine in the caudate nucleus and putamen (striatum) [1, 2]. The resultant impaired function of basal ganglia-thalamocortical neural circuitry eventually manifests the characteristic parkinsonian mo-tor symptoms such as bradykinesia, rigidity, resting tremor, and postural instability [3, 4]. Although prodromal non-motor symptoms such as hyposmia, sleep disorders, depression and constipation typically precede the motor symptoms, only the onset of the latter ones leads to the diagnosis [5, 6]. It is estimated that approximately 30% of the nigral dopamine neurons and 50–70% of the striatal dopaminergic fibers and dopamine content is already lost at the time of the diagnosis [7]. Currently, drug treatment of PD is based on dopamine receptor agonists, monoamine oxidase B (MAO-B) inhibitors and replacement of brain dopamine with levodopa [8]. Since none of these treatments can halt or slow down the progression of the disease, there is a great unmet medical need for a disease-modifying therapy for PD. The lack of efficient therapy for non-motor symptoms also represents a significant challenge in PD management.
Neurotrophic factors (NTFs) are secretory proteins promoting development, survival and regeneration of the nervous system [9, 10]. Among various NTFs, glial cell line-derived neurotrophic factor (GDNF) and neurturin (NRTN), both belonging to GDNF family ligands (GFLs), are considered promising therapeutic agents for neurodegenerative disorders such as PD. GFLs mainly signal via receptor tyrosine kinase RET (rearranged during transfection). They first bind to glycosyl phosphatidylinositol-anchored co-receptor GDNF family receptor α (GFRα1 for GDNF and GFRα2 for NRTN) and afterwards form a complex with RET. The formation of GFL-GFRα-RET complex leads to homodimerization and autophosphorylation of RET intracellular tyrosine kinase domains which subsequently activate multiple downstream signaling cascades required for cell survival and regeneration such as AKT, MAPK-ERK, PLCγ, Src, and JNK [9]. GDNF and NRTN promote survival of cultured dopamine neurons and show neu-roprotective and restorative effects in various animal models of PD [11–32]. Although some effects of GFLs can be mediated by alternative receptors such as NCAM or syndecan-3 [33, 34], RET is essential for the neuroprotective and neurorestorative effects of GDNF in animal models of PD [35].
In two open-label phase I trials, intraputamenal infusion of GDNF significantly improved motor fun-ction and was associated with increased 18F-dopa uptake in the posterior putamen of PD patients [36–38], whereas two randomized and placebo-controlled phase I–II trials did not meet their primary endpoints [39, 40]. In a recent placebo-controlled phase II study with intermittent bilateral convection-enhanced protein infusion, 43% of the patients in the GDNF group showed a clinically significant motor improvement in post hoc analysis, although the trial failed to reach its primary endpoints [41, 42]. Furthermore, both GDNF protein and adeno associated virus vector-encoded GDNF (AAV2-GDNF) increased 18F-dopa uptake in the putamen in recent clinical trials [41, 43] indicative of improved presynaptic dopaminergic integrity possibly as a results of nerve fiber restoration. Similarly, clinical trials with AAV2-NRTN failed to reach the primary efficacy endpoints [44]. Nevertheless, postmortem analysis of brain samples from some of PD patients receiving AAV2-NRTN revealed an increase in the density of dopaminergic fibers in the areas of putamen where a high level of NRTN expression was also detected [45]. However, these areas covered rather small portion of putamen and were perhaps insufficient to result in motor function improvement in patients. Importantly, all clinical trials with GDNF and NRTN, except the study conducted by Nutt and co-authors [39], reported no serious adverse effects.
Varying efficacy in the clinical trials has raised questions about optimal initiation time of the treatments, dosing and delivery methods as well as other protein and gene therapy related challenges such as immunogenicity, low bioavailability and tissue penetration of the GFLs. Furthermore, protein and gene therapy require intracranial delivery using brain surgery that increases the costs and risks of the treatment and imposes ethical restrictions for the selection of patients in clinical trials making the treatment available mainly to people with moderate or advanced PD. Therefore, developing a small molecule which mimics the function of GFLs in dopamine system by activating the same receptors and signaling pathways would be an appealing approach for an effective and safe disease-modifying therapy for PD.
Previously, we discovered the first generation RET agonist, a compound called BT13, which supports cultured sensory and dopamine neurons, alleviates pain in animal models of experimental neuropathy and enhances dopamine release in the mouse striatum [46, 47]. The objective of the present study was to investigate therapeutic potential and effects of the second generation RET agonist BT44 on dopamine system both in vitro and in vivo. We describe the activation of receptor tyrosine kinase RET and in-tracellular signaling pathways by BT44, as well as its effect on the survival of cultured midbrain dop-amine neurons from wild-type and RET knockout mice. We also show that BT44 protects cultured midbrain dopamine neurons from 1-methyl-4-phenylpyridinium (MPP+)–induced toxicity. In addition, we report the ability of BT44 to induce functional recovery and protection of striatal dopaminergic fibers in a unilateral 6-hydroxydopamine (6-OHDA) induced hemiparkinsonian model of PD in rats.
MATERIALS AND METHODS
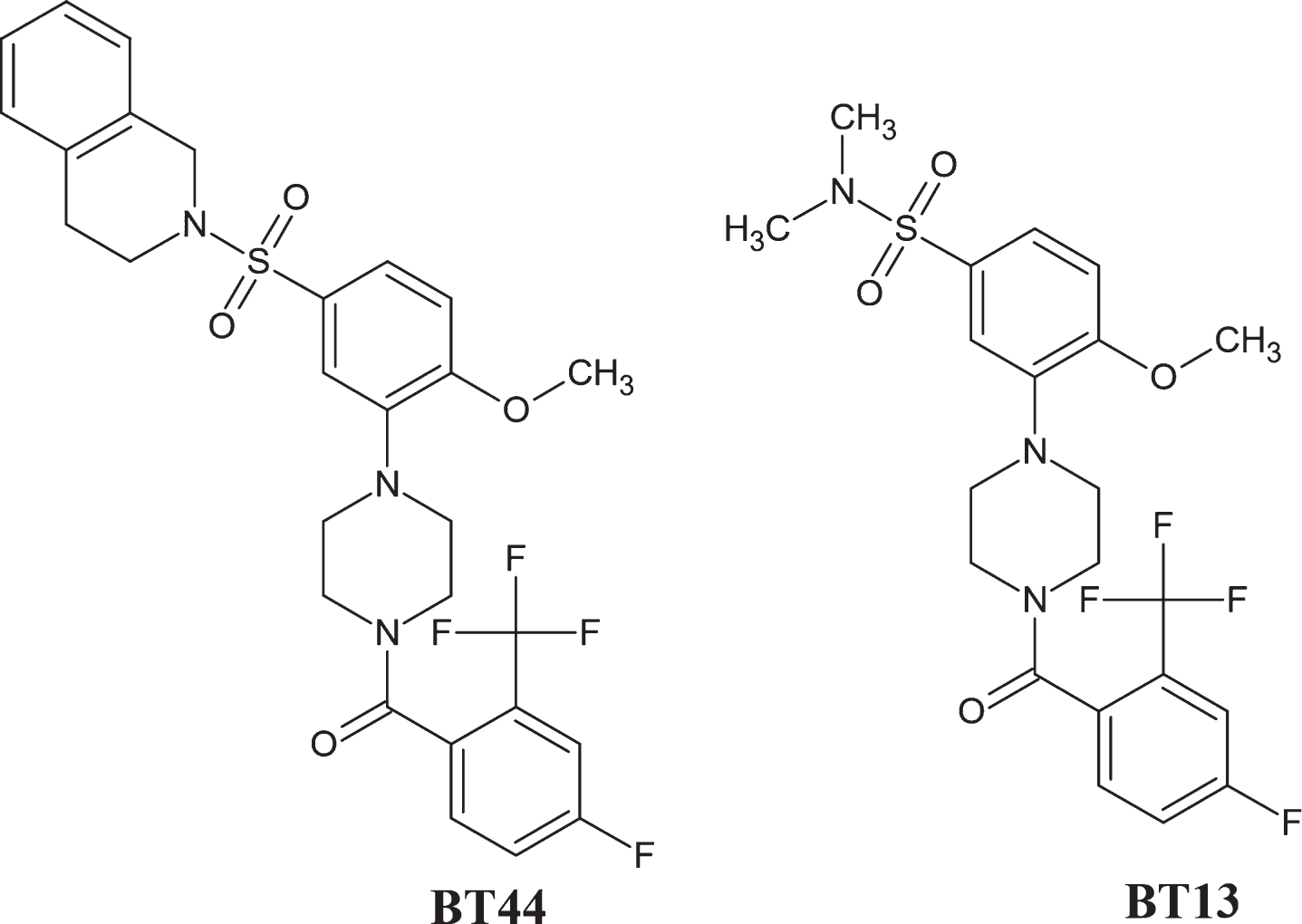
BT44 ((4-5-((3,4-dihydroisoquinolin-2(1H)-yl)sulfonyl)-2-methoxyphenyl)piperazin-1-yl (4-fluoro-2-(trifluoromethyl)phenyl)methanone, MW: 577.59; LogD pH 7.4:6.3, Fig. 1, BT stands for Baltic Technology) was ordered from EvoBlocks (Hungary, Cat# EBR-10719615). The purity of the synthesized compound determined by high-performance liquid chromatography (HPLC) was 97.3%. The structure, integrity, and purity of BT44 was verified by NMR experiments recorded on a Bruker Avance III HD NMR spectrometer operated at 1H frequency of 850.4 MHz equipped with a cryogenic probe head by NMR facility at the Institute of Biotechnology, University of Helsinki. BT44 was dissolved in cell culture medium containing 1% of dimethyl sulfoxide (DMSO) for the in vitro experiments, and in 100% propylene glycol (PG) with final concentrations of 0.0083μg/μl and 0.025μg/μl for the in vivo experiment due to limited solubility in aqueous solutions.
Fig. 1
Chemical structure of BT44 ((4-5-((3,4-dihydroisoquinolin-2(1H)-yl)sulfonyl)-2-methoxyphenyl)piperazin-1-yl(4-fluoro-2-(trifluoromethyl)phenyl)methanone) and BT13.
Proteins
Human recombinant GDNF (hrGDNF; Icosagen, Estonia, Cat# P-103-100) and human recombinant neurturin (hrNRTN; PeproTech, USA, Cat# 450-11) were used for in vitro experiments. For in vivo studies, hrGDNF was purchased from PeproTech (Cat# 450-10) and dissolved in phosphate-buffered saline, pH 7.4 (PBS) with final concentration of 0.25μg/μl.
Experimental animals
Animals used in the experiments were housed under a 12 h light-dark cycle and provided with standard rodent chow (Harlan, the Netherlands) and fresh tap water ad libitum. E13.5 embryos of NMRI mice and RET knockout mice (C57BL/6JOlaHsd) (Laboratory Animal Centre, University of Helsinki) were used for primary cultures of midbrain dopamine neurons. Genotyping PCR was used to identify embryos lacking RET [48]. Mice were housed individually or in groups of 2–10 animals per cage. Adult male Wistar rats (RccHan:WIST; Harlan), weighing 230–310 grams at the start of the experiment, were used for testing the efficacy of BT44 in the unilateral 6-OHDA lesion model of PD. Rats were initially housed in groups of 4 per cage, but after implanting brain cannulas attached to minipumps for treatment infusions they were moved into individual cages until removal of the cannulas and the minipumps. Thereafter, the rats were regrouped (4 per cage) into original cages until the end of the experiment. The wellbeing of the animals was verified on a regular basis. All experiments were carried out in accordance with the 3R principle of the EU directive 2010/63/EU on the care and use of experimental animals, local laws and regulations (Finnish Act on the Protection of Animals Used for Scientific or Educational Purposes [497/2013] and the Government Decree on the Protection of Animals Used for Scientific or Educational Purposes [564/2013]). The design of the animal experiment was approved by the National Animal Experiment Board of Finland (protocol approval number: ESAVI/198/04.10.07/2014) for experiments with living animals and the Laboratory Animal Center of the University of Helsinki (license number: KEK15-022) for collection of E13.5 embryos of NMRI and C57BL/6JolaHsd RET knockout mice.
RET phosphorylation assay by immunoprecipitation and western blotting
The level of RET phosphorylation in vitro in response to BT44, GDNF and NRTN was studied in MG87RET murine fibroblasts stably transfected with the long isoform of RET [49] as described previously [46, 47]. MG87RET cells were cultured overnight and transiently transfected with full-length human GFRα1 cDNA sub-cloned in pCDNA6 vector (Invitrogen, USA) [50], hGFRα2 cDNA in pCR3.1 or enhanced green fluorescent protein (GFP) cDNA in pEGFP-N1, using Lipofectamine 2000 (Invitrogen), as described by the manufacturer. The cells were starved for 4 h before the treatments in serum-free DMEM containing 1% DMSO and 15 mM HEPES, pH 7.2. The cells were treated with different concentrations of BT44 (7.5, 18, 36 and 75μM) and GDNF (200 ng/ml) or NRTN (200 ng/ml) dissolved in starvation medium for 15 min. Then, after washing with ice-cold PBS containing 1 mM Na3VO4 the cells were lysed with RIPA-modified buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1% TX-100, 10% glycerol, EDTA-free protease inhibitor cocktail (Roche, Switzerland), 1 mM Na3VO4, 6 mM sodium deoxycholate, 1 mM PMSF), 500μl per well. The lysates were centrifuged for 5 min (5000 rpm, 4°C) and supernatants were collected for immunoprecipitation. To immunoprecipitate RET, the supernatants (approx. 400μg of total protein) were incubated with anti-RET C-20 (2μg/ml, Santa Cruz Biotechnology, USA, Cat# sc-1290) antibody and magnetic beads coated with protein G (Dynabeads Protein G, Life Technologies, USA) on a rotator overnight at 4°C. The next day, after washing the beads with Tris-buffered saline, pH 7.4 (TBS) containing 1% Triton X-100, immunoprecipitated proteins were eluted by adding 50μl of 2×Laemmli buffer, resolved in 7.5% sodium dodecyl sulfate–polyacrylamide gel (SDS-PAAG) and then transferred on nitrocellulose membrane. The membrane was blocked with 10% skimmed milk in TBS-T (TBS with 0.15% of Tween 20) for 10 min and probed overnight with anti-pan-phosphotyrosine (1:1500, clone 4G10, Merck Millipore, Germany, Cat# 05-321) antibody in TBS-T with 3% skimmed milk. The membrane was washed and incubated with goat anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibody (Agilent Dako, USA, Cat# P044701) diluted 1:3000 in TBS-T with 3% skimmed milk for 1 h at room temperature. After washing, the bands were visualized with ECL reagent (Pierce Biotechnology, USA) using Luminescent Image Analyzer LAS-3000 (Fujifilm, Japan). Equal loading of proteins in different wells was confirmed by re-probing the membrane with primary anti-RET C-20 antibody (1:500, Santa Cruz Biotechnology, Cat# sc-1290) and secondary anti-goat antibody (1:500, Agilent Dako, Cat# P0449).
Intracellular signaling assay
To investigate whether RET phosphorylation leads to the activation of downstream intracellular signaling cascades AKT and MAPK-ERK, MG87RET fibroblasts were transfected with hGFRα1, hGFRα2- or GFP-expressing plasmids, treated with BT44 (7.5, 18, 36 and 75μM) and GDNF (200 ng/ml) or NRTN (200 ng/ml) and lysed as described above. Thereafter, western blotting was used to semi-quantitatively evaluate the phosphorylation level of AKT and ERK. For that, 100μl of cell lysate was collected and mixed with the same amount of 2×Laemmli buffer. The samples were boiled for 10 min, approx. 25μg of total protein was resolved using 12% SDS-PAAG, and finally proteins were transferred to nitrocellulose membrane. The membrane containing ERK was blocked with 3% skimmed milk in TBS-T for 1 h and probed with rabbit anti-ERK antibody (1:500, Santa Cruz Biotechnology, Cat# sc-94) or mouse anti-pERK antibody (1:1000, Santa Cruz Biotechnology, Cat# sc-7383) overnight at 4°C. Similarly, the membrane containing AKT was blocked with 3% bovine serum albumin (BSA) in TBS-T for 1 h and probed with mouse anti-AKT antibody (1:2000, Cell Signaling Technology, USA, Cat# 2920) or rabbit anti-pAKT antibody (1:500, Cell Signaling Technology, USA, Cat# 9271) overnight at 4°C. The membranes were washed with TBS-T, after which ERK-containing membrane was incubated with HRP-conjugated goat anti-mouse secondary antibody (Agilent Dako, Cat# P0447), diluted 1:3000 in 3% skimmed milk in TBS-T, and AKT-containing membrane was incubated with HRP-conjugated donkey anti-rabbit antibody (GE Healthcare, Cat# NA934), diluted 1:3000 in 3% BSA in TBS-T for 1 h at room temperature. The membrane was visualized with ECL Plus Western Blotting Substrate (Pierce Biotechnology) using Luminescent Image Analyzer LAS-3000 (Fujifilm). The membrane was stripped and re-probed with mouse antibody against glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (1:4000, Merck Millipore, Cat# MAB374) in order to confirm equal loading of the samples.
Quantification of western blots
All western blot images were quantified using Image Studio 5.2 software (LI-COR Biosciences, USA) as described earlier [51]. Intensities of the bands of the phosphorylated form of RET (MW = 170 kDa) were normalized to the band intensities of RET. Similarly, band intensities of pERK and pAKT were normalized to band intensities of GAPDH. The area was kept constant in all analyses. The images from 3 to 6 independent experiments were quantified.
Survival assay for primary dopamine neurons
Midbrain neurons were isolated from E13.5 embryos of NMRI mice or C57BL/6JolaHsd RET knockout mice in Dulbecco’s medium containing 2% of BSA as described previously [47, 52]. Dissected tissue samples were washed 3 times with calcium and magnesium-free Hank’s Balanced Salt Solution (HBSS, Gibco, Life Technologies) and incubated in 5 mg/ml trypsin solution in HBSS for 20 min at 37°C. Enzymatic activity of trypsin was blocked by adding FBS containing 0.1 mg/ml of DNase I (Roche, Cat# 11284932001). Cells were triturated with a siliconized glass Pasteur pipette to get single cell suspension and centrifuged at 200 rcf for 5 min. The pellets were washed with primary neuron culture medium [(Dulbecco’s MEM/Nut mix F12 (Invitrogen/Gibco, Cat# 21331-020), 1xN2 serum supplement (Invitrogen/Gibco, Cat# 17502-048), 33 mM D-glucose (Sigma-Aldrich, Germany, Cat# G-8769), 0.5 mM L-glutamine (Invitrogen/Gibco, Cat# 25030-032), and 100μg/ml Primocin (InvivoGen, USA, Cat# ant-pm-2)] to remove any traces of serum, and thus there was practically no neurotrophic support in the beginning of the survival assay. The washed pellets were resuspended in 150–200μl of the primary neuron culture medium, and cells were counted using a TC20 automated cell counter (Bio-Rad Laboratories, USA). Finally, 30 000 cells per well were plated on a 96-well plate pre-coated with poly-DL-ornithine (0.5 mg/ml in 0.15 M borate buffer, pH 8.7, Sigma-Aldrich, Cat# P8638) and cultured at 37°C. At 1 h post plating, different concentrations of BT44 (0.2, 3.5, 7.5, 75, and 3500 nM) and GDNF (10 ng/ml, Icosagen) were dissolved in primary neuron culture medium containing 1% of DMSO and applied on cultured primary neurons for 5 days. The half of the compound-containing solution was replaced with fresh portion at 2.5 days post plating.
The survival of MPP + -challenged dopamine neurons was performed exactly as described by us before [47]. The cells were cultured for 5 days in primary neuron culture medium. On the 6th day, MPP + (2μM) together with BT44 (10 nM or 100 nM) or GDNF (10 ng/ml ≃0.33 nM) was added simultaneously for 48 hours.
Immunocytochemistry
Immunocytochemical staining with tyrosine hyd-roxylase (TH) antibody was performed in cultures of embryonic midbrain neurons either after 5 days (for survival assay) or 7 days (for neurotoxin assay) of culturing. Cells were fixed with 4% paraformaldehyde (PFA) in PBS for 20 min, washed with PBS and permeabilized with 0.2% Triton X-100 in PBS for 15 min. Blocking solution (5% horse serum in 0.2% Triton X-100 in PBS) was used to block unspecific binding sites for 1 h, followed by incubation with mouse anti-TH antibody (1:500, Merck Millipore, Cat# MAB318) overnight at 4°C. Cells were then washed and incubated with Alexa Fluor™ 647 conjugated donkey anti-mouse secondary antibody (1:500, Thermo Fisher Scientific, USA, Cat# A-31571) for 1 h at room temperature. Finally, cells were counterstained with 0.2μg/ml DAPI (4’, 6-diamidino-2-phenylindole) in PBS for 10 min at room temperature and kept in PBS until imaging with CellInsight (CX51110, Thermo Fisher Scientific) CX5 High Content Screening (HCS) using 20×magnification. The number of TH-positive cells and the total number of cells was quantified using CellProfiler image analysis software [53].
Stereotaxic surgeries
In the first stereotaxic surgery, the rats received 3 unilateral 6-OHDA micro injections (each 3μg) into the right dorsal striatum as described previously [54]. In the second surgery two weeks later, an infusion catheter was implanted into the right dorsal striatum in order to make site specific infusions of the compounds used in the study (Fig. 4B; for detailed information see below).
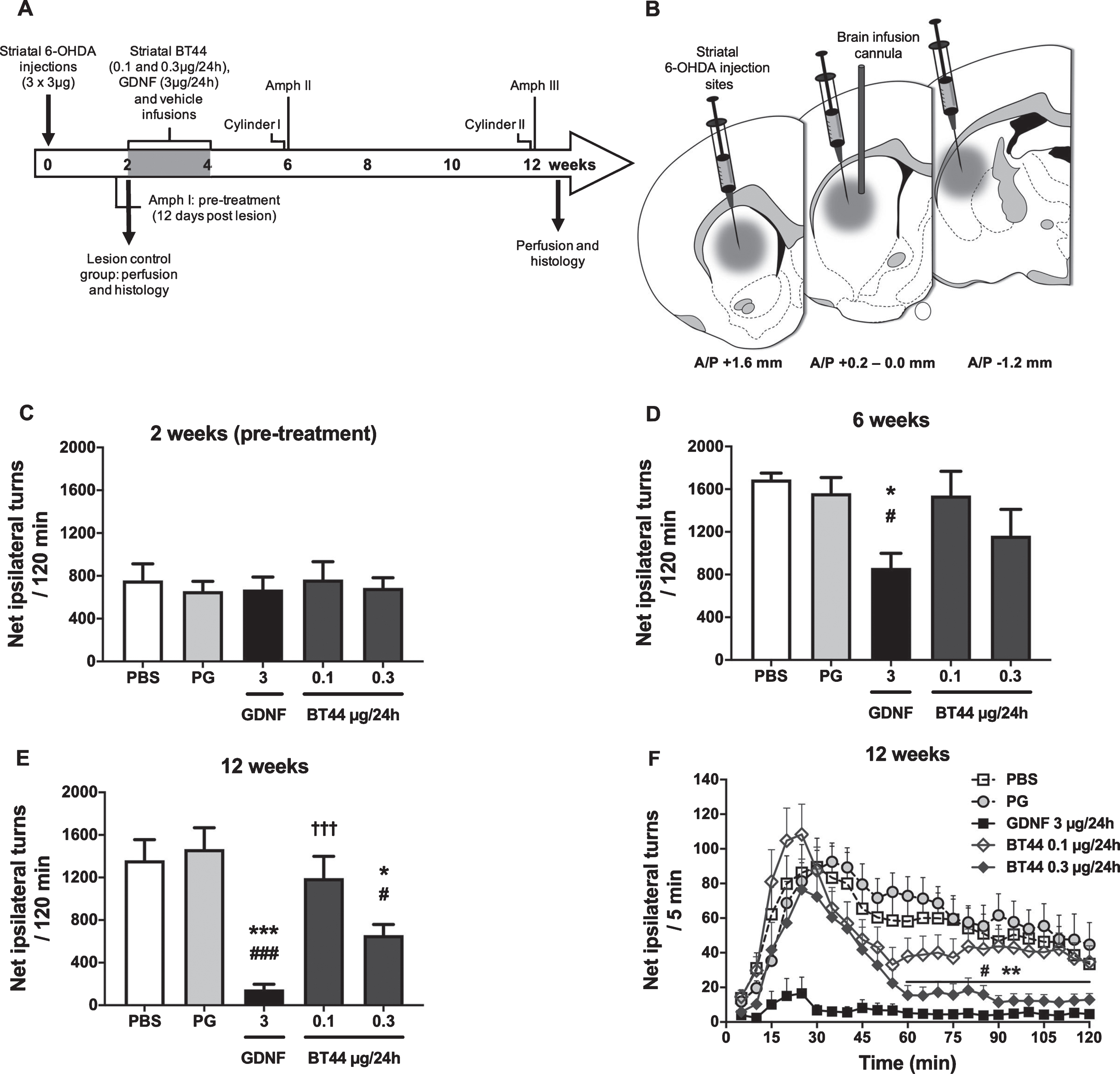
Fig. 4
BT44 (0.3μg/24h) and GDNF (3μg/24h) alleviate amphetamine-induced rotational asymmetry in 6-OHDA lesioned rats. Design of the in vivo experiment with 6-OHDA lesioned hemiparkinsonian rats (A). Unilateral striatal 6-OHDA injections, striatal drug delivery with osmotic pumps, time points for amphetamine-induced turning behavior (Amph I-III) and cylinder (Cylinder I and II) tests, and perfusion time points are depicted on the timeline. Schematic illustration of 6-OHDA injection sites and treatment infusion site in the dorsal striatum (B). All rats received 3 deposits of 6-OHDA (3μg/deposit) along the rostrocaudal axis of the right striatum. The syringes indicate the 6-OHDA injection sites and the vertical cannula the treatment infusion site. Amphetamine-induced rotational asymmetry (cumulative data for 120 min) at 2 weeks (C), 6 weeks (D), and 12 weeks (E) after 6-OHDA lesion. Rotation rate per 5 min at 12 weeks post lesion (F). PBS, phosphate buffered saline; PG, propylene glycol. *p < 0.05, ***p < 0.001 vs. PG; #p < 0.05, # # #p < 0.001 vs. PBS; †††p < 0.001 vs. GDNF, Tukey HSD after one-way ANOVA (D, E). **p < 0.01 BT44 0.3μg/24 h vs. PG; #p < 0.05 BT44 0.3μg/24 h vs. PBS, Tukey HSD after RM ANOVA (F). Mean±SEM, n = 8–11 per group.
6-Hydroxydopamine lesion
All rats received a unilateral injection of 6-OHDA (6-hydroxydopamine hydrochloride, Sigma-Aldrich) into 3 different sites in the right dorsal striatum (A/P + 1.6, L/M –2.8, D/V –6.0; A/P 0.0, L/M –4.1, D/V –5.5; and A/P –1.2, L/M –4.5, D/V –5.5, mm relative to the bregma, according to the rat brain atlas [55]). The dose of 6-OHDA injected to each site was 3μg (calculated as free base) in 1.5μl of ice-cold, de-oxygenated, saline with 0.02% ascorbic acid (the concentration of solution was 2μg/μl). All surgical procedures were performed under general isoflurane (Vetflurane® 1000 mg/g, Virbac, France) anesthesia (4.5% during induction and 2-3% during maintenance). Rats were placed in a stereotaxic frame (Stoelting, USA) and a small amount of lidocaine-adrenalin-solution (10 mg/ml; Orion Pharma, Finland) was injected under the scalp for local anesthesia and to prevent bleeding. The skull was exposed and burr holes were made using a high-speed drill. The 6-OHDA solution was injected at the flow rate of 0.5μl/min using an electronic injector (Quintessential stereotactic injector, Stoelting) and a 10μl -needle (NanoFil 33G, World Precision Instruments, USA). The needle was lowered into the brain at a 10° angle to avoid lateral ventricles. At the completion of each injection, the needle was kept in place for 5 min to minimize backflow of the solution. Desipramine (15 mg/kg i.p.; calculated as free base; Sigma-Aldrich) was administrated 30 min before the 6-OHDA injections to prevent the uptake of 6-OHDA into noradrenergic and serotonergic nerve terminals. One group of animals (lesion control group, n = 4) was sacrificed 2 weeks after the lesion. This group served as a reference showing the extent of dopamine neuron degeneration at the time of the treatment infusion initiation.
Infusions of BT44, or GDNF, or corresponding vehicles with osmotic pumps
Rats were assigned into equal treatment groups according to the first amphetamine-induced rotation rate at 12 days after the 6-OHDA lesion (see below in the Materials and Methods and Fig. 4A and C). Subsequently, osmotic infusion pumps were implanted in the second stereotaxic surgery and treatment infusions were started 2 weeks after the 6-OHDA injections. The rats were anesthetized and the skull was exposed. A brain infusion cannula (Alzet Brain infusion kit no. 2, Durect, USA) was implanted to coordinates relative to the bregma A/P + 0.2; L/M –3.0; D/V –5.0 mm and secured to the skull with 3 stainless steel screws and polycarboxylate cement (Aqualox, VOCO, Germany). The tip of the cannula was placed in the right dorsal striatum, in the middle of the 6-OHDA injection sites (Fig. 4B). The cannula was connected via a 3 cm-long catheter tubing to an osmotic infusion pump (Alzet osmotic pump model 2002, Durect) which was placed into a subcutaneous pocket between the scapulae. The pump constantly delivered BT44 0.1μg/24 h (n = 10), BT44 0.3μg/24 h (n = 10), GDNF 3μg/24 h (n = 11, positive control), PG (n = 11, vehicle control) or PBS (n = 9, vehicle control) at a flow rate of 0.5μl/h for 14 days. At the end of the treatment infusions, 14 days after the implantation of the pumps, the rats were anesthetized and fixed in the stereotaxic frame for the third time. The osmotic pumps, infusion cannulas, and screws were removed, and the incision was cleaned, disinfected, and sutured.
Pain management
Before every stereotaxic surgery the rats rec-eived buprenorphine 0.05 mg/kg s.c. (Temgesic® 0.3 mg/ml, Indivior UK Limited, United Kingdom) for analgesia. Carprofen 5 mg/kg (Rimadyl Vet® 50 mg/ml, Zoetis, USA) was injected s.c. immediately after the surgeries to relieve postoperative pain. Additional doses of buprenorphine and carprofen were given 1 day after the surgeries.
Behavioral tests
Rotational assay
Rotational asymmetry in response to amphetamine injection was used to measure motor deficits arising from the unilateral 6-OHDA lesion of the nigrostriatal dopamine system. Amphetamine-induced rotational behavior was monitored in a blinded manner at 2, 6, and 12 weeks post lesion in automated rotom-eter bowls (Med Associates, USA) as described previously in [21, 56]. After a 30 min habituation period, rats were injected with a single dose of D-amphetamine (2.5 mg/kg i.p.; calculated as free base; Division of Pharmaceutical Chemistry and Technology, University of Helsinki, Finland). The number of full (360°) uninterrupted clockwise and counterclockwise turns was recorded for 120 min. Net ipsilateral turns to the lesion side were calculated by subtracting contralateral turns from ipsilateral turns.
Cylinder test
At 6- and 12-weeks post lesion, before the amphetamine-induced rotational assay, rats underwent a cylinder test to evaluate spontaneous limb-use asymmetry. The cylinder test protocol is described in detail elsewhere [57, 58]. Briefly, rats were placed in a transparent plexiglass cylinder (diameter ∼20 cm, height ∼30 cm) and allowed to freely explore the novel environment in dim light. Exploratory activity was recorded for 10 min with a video camera placed below the cylinder. The number of individual weight-shifting movements of ipsilateral (non-impaired) and contralateral (impaired) forelimbs against the wall were scored by an observer blind to the treatments. During a rear, the first forepaw contact on the wall was scored as an independent contact for that limb. Simultaneous placement of both forepaws on the wall was scored as “both limb contact”. Lateral exploration along the wall by alternating right and left forepaw placements (wall stepping) was scored as a series of “both limb contacts” (one combination of the right and left forepaws equaled to one “both limb contact”). Hopping along the wall using only one limb was scored as individual forepaw contacts for that limb. After a full rear, the first weight-receiving contact to the ground was scored as a landing contact for the forepaw used for landing. If both paws contacted the ground simultaneously, “both limb contact” for landing was scored. Ambiguous contacts were not included in the analysis. Percentual limb-use asymmetry score was calculated separately for wall exploration and landing using the following formula:
Tissue collection
After the last behavioral tests at 12 weeks post lesion, rats were deeply anesthetized with sodium pentobarbital (90 mg/kg, i.p.; Mebunat Vet® 60 mg/ml, Orion Pharma) and transcardially perfused first with PBS for 5 min followed by 4% PFA in PBS for 10 min. The brains were removed and immersed in 4% PFA overnight at 4°C for post-fixation, and then stored in 20% sucrose in PBS at 4°C until snap freezing in dry ice-cooled isopentane and sectioning with a cryostat (Leica CM3050, Leica Biosystems, Germany). The lesion control rats were perfused at 2 weeks post lesion, and the brains were collected for histology.
Immunohistochemistry
A series of free-floating coronal sections (40μm) were collected from the striatum and midbrain, and stained for TH and dopamine transporter (DAT) to assess the number of dopamine neurons in the SNpc and the density of dopaminergic fibers in the striatum essentially as described elsewhere [59, 60]. Endogenous peroxidase activity was quenched with 3% H2O2 (in 10% methanol in PBS). For DAT staining, antigen retrieval was performed by incubating the sections in 10 mM citrate buffer (pH 6) for 30 min at 80°C followed by blocking in 5% normal goat serum, 2% BSA and 0.3% Triton X-100 in PBS for 1 h. For TH staining, the sections were blocked in 3% BSA and 0.3% Triton X-100 in PBS for 1 h. The sections were then probed with rabbit anti-TH antibody (1:2000, Merck Millipore, Cat# AB152) or rabbit anti-DAT antibody (1:500, Abcam, Cat# ab184451) overnight at 4°C. After rinsing in PBS, TH-probed sections were incubated for 1 h at room temperature in biotinylated protein A solution [1:100, prepared using protein A (MP Biomedicals, USA) and N-hydroxysuccinimido-biotin (Sigma-Aldrich)] in place of the secondary antibody. DAT-probed sections were similarly incubated in biotinylated goat anti-rabbit secondary antibody (1:500, Vector Laboratories, Cat# BA-1000). Finally, the staining was reinforced with avidin-biotin-HRP complex (Vectastain Elite ABC HRP Kit, Cat# PK-6100, Vector Laboratories, USA) and visualized using 3,3′-diaminobenzidine-tetrahydrochloride-dihydrate (DAB; 0.5 mg/ml in 0.03% H2O2 in PBS; Cat# 32750, Sigma-Aldrich) as a chromogen. Stained sections were placed on gelatin/chrome coated glass slides, dehydrated with increasing concentrations of ethanol solutions (70, 96 and 99%, respectively), cleared in xylene and mounted using DePeX® mounting medium (VWR International).
Assessment of dopamine neurons in the SNpc
TH-immunoreactive (TH-ir) cell bodies in the SNpc were counted using an automated convolutional neural networks (CNN) algorithm and cloud-embedded AiforiaTM platform (Aiforia Technologies Oy, Finland). This computer-assisted cell counting method based on supervised machine learning and automated image recognition is described in detail and validated by Penttinen and co-authors [61]. Briefly, TH-immunostained coronal midbrain sections were digitized using Pannoramic P250 Flash II whole slide scanner (3DHistech, Hun-gary) with extended focus at a resolution of 0.22μm/pixel. A total of five focal layers were acquired with 2μm intervals. The digitized images were uploaded to Aiforia™ image processing platform. In order to encompass the full rostro-caudal extent of the SNpc, the dorsal tier of the SNpc was demarcated bilaterally in every sixth section approximately between levels A/P –4.8 and –6.0 mm relative to the bregma [55] by an observer blind to the treatment groups. Subsequently, the number of TH-ir neurons within the demarcated areas was analyzed using the CNN algorithm that was trained to recognize dopaminergic cell bodies from the digital images. The algorithm consisted of two layers: the first layer segmented the TH-ir cell bodies and the second layer counted the individual cell bodies in the first layer. The number of detected TH-ir neurons was summed up from the six sections separately for both hemispheres. The data are presented as percentage of the lesioned side as compared to the intact side.
Additionally, the performance of the CNN algorithm was confirmed against stereological analysis of TH-ir cell body counts in 20 randomly selected brains from the present study. The number of TH-ir cells was counted with the CNN algorithm from 6 sections per brain as described above, and from the same brains, using unbiased optical fractionator counting method with Olympus BX51 microscope (Olympus Corporation, Japan) and Stereo Investigator platform (v11.06.2; MBF Bioscience, USA) from 3 sections per brain as described earlier [62, 63] (Supplementary Table 1). When comparing the data obtained with the CNN algorithm and the optical fractionator method, we observed a strong positive correlation of the results across the 20 brains (Pearson’s r = 0.83, p < 0.001) (Supplementary Figure 8), an observation which is in line with the earlier results by Penttinen and co-authors [61]. The number of DAT-immunoreactive (DAT-ir) cell bodies in the SNpc were measured similarly as described above for TH-ir cell bodies.
Assessment of fiber density in the striatum
Optical density of the TH-ir and DAT-ir fibers in the dorsal striatum was measured bilaterally from 3 different rostro-caudal levels through the striatum (approximately A/P + 1.2, + 0.48, and –0.26, mm relative to the bregma [55]). The total magnitude of striatal denervation was analyzed as an average reduction in the optical density at the three levels measured. Digital images of the TH or DAT-immunostained sections were acquired with Pannoramic P250 Flash II whole slide scanner (3DHistech). The images were converted to 8-bit gray scale, and colors were inverted. The dorsal part of caudate putamen limiting to the ventral edge of external capsule of corpus callosum was outlined. From this outlined area, integrated optical density divided by the size of the area was measured with Fiji ImageJ software (Media Cybernetics, USA) by a blinded observer. Optical density of nonspecific background staining was measured from corpus callosum and subtracted from the striatal optical densities. The data are presented as percentage of the lesioned striatum as compared to the intact striatum.
Pharmacokinetics and blood-brain barrier penetration of BT44
The main pharmacokinetic parameters and blood-brain barrier penetration after a single intravenous injection of BT44 at a dose of 10 mg/kg were evaluated in rats. Blood samples and brains were collected 0.5, 1, and 3 h after dosing of BT44 (n = 3 rats per group). Before the collection of brain samples animals were transcardially perfused with PBS. The concentration of BT44 in plasma and brain homogenates was measured using ultra HPLC coupled with time-of-flight mass spectrometry.
Experimental design and statistical analysis
Experiments in cultured cells were repeated 3–6 times. Experimental design of the in vivo study with 6-OHDA lesioned hemiparkinsonian rats is illustrated in Fig. 4A. The group sizes and the dose used for GDNF treated rats were selected on the basis of our previous experiments [21, 63, 64]. The doses for the efficacy assessment of BT44 were chosen based on the potency of the compound in neuronal survival in vitro assay and available biological activity data for the parent compound BT13 in a 6-OHDA model of PD [47, 65]. Pre-treatment rotational behavior at 2 weeks post lesion was used to verify proper development of the nigrostriatal lesion before the initiation of the treatment infusions. Only rats rotating more than 120 net ipsilateral turns in 120 min were included in the experiment. In the cylinder test, rats with less than 10 forepaw contacts upon wall explorations and landings during the 10 min test period were excluded from the analysis. In addition, Grubbs’ test (α= 0.05) was used to detect outliers in the behavioral tests and histological analyses. One outlier was excluded from PBS group at 6 weeks post-lesion and one outlier from GDNF 3μg/24 h and BT44 0.3μg/24 h groups at 12 weeks post-lesion in amphetamine-induced rotational asymmetry analyses. In TH-ir fiber density analysis, one outlier was identified in PBS, GDNF 3μg/24 h and BT44 0.1μg/24 h groups, and in TH-ir cell count analysis one outlier in PG group. In DAT-ir fiber density analysis, one outlier was identified in PG, PBS, GDNF 3μg/24 h and BT44 0.1μg/24 h groups, and in DAT-ir cell count analysis one outlier in PG, GDNF 3μg/24 h and BT44 0.1μg/24 h groups. In total, 11 rats died during the surgeries or behavioral follow-up, and one rat had to be sacrificed due to reaching a humane endpoint. The data from those rats were excluded from the analyses.
Results were statistically analyzed using paired t-test or one-way ANOVA followed by a Dunnett’s or Tukey HSD post hoc test. Pearson correlation coefficient was used to assess the strength of the linear relationship between amphetamine-induced turning behavior at 12 weeks post lesion and histological measurements. GraphPad Prism 6 (GraphPad, USA) or SPSS® Statistics 22 (IBM SPSS, USA) softwares were used for all statistical analyses. Results are expressed as mean±SEM and considered statistically significant at p < 0.05.
RESULTS
BT44 stimulates RET phosphorylation and activates intracellular signaling pathways
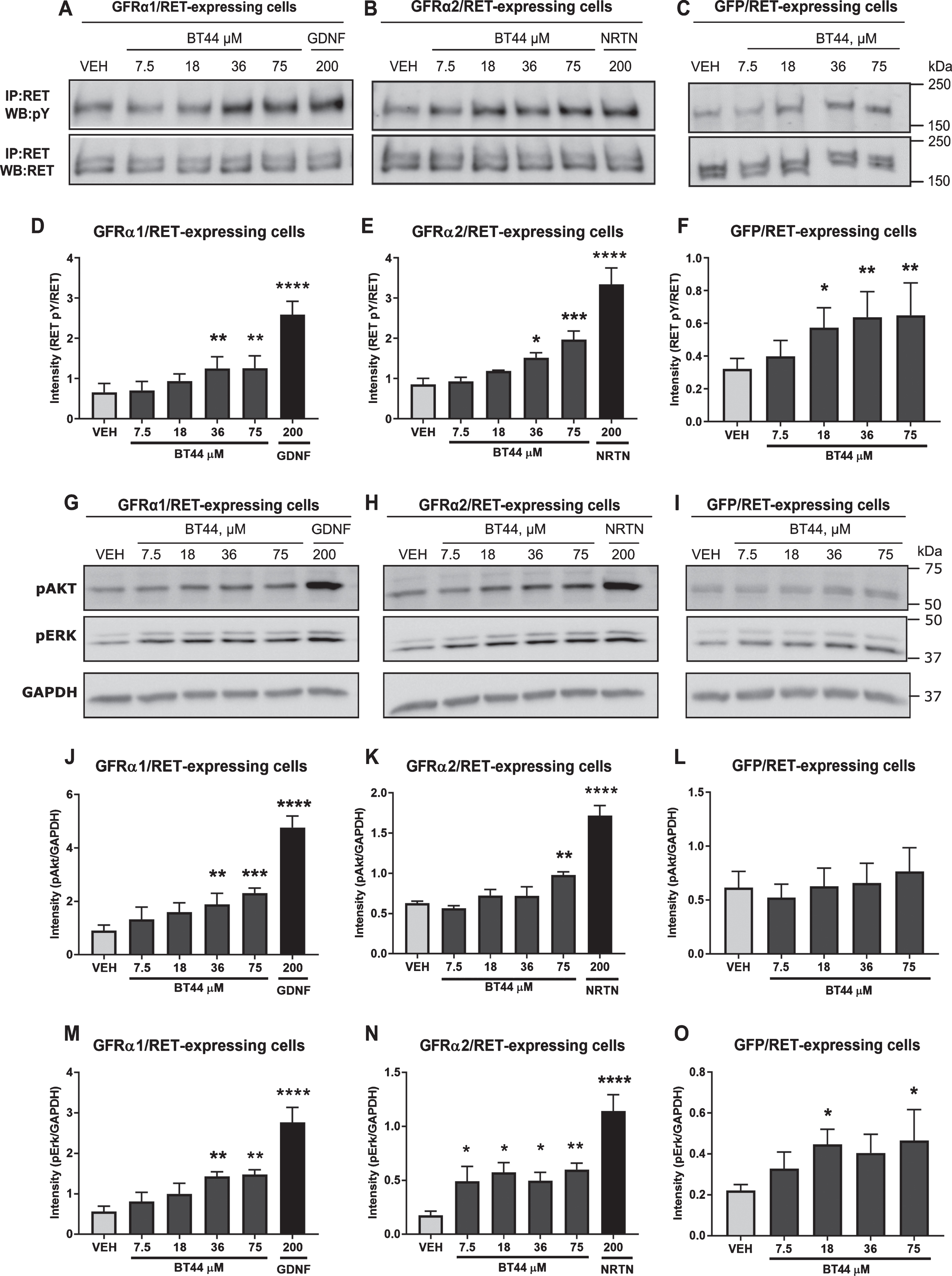
To evaluate the ability of BT44 to activate RET and its downstream signaling pathways, we used MG87RET fibroblasts transfected with GDNF co-receptor GFRα1 and NRTN co-receptor GFRα2. Both GFRα1 and GFRα2 proteins were expressed in MG87RET cells (Supplementary Figure 1). We also used GFP-transfected cells in order to assess the direct activation of RET and its intracellular targets. BT44 was able to induce RET phosphorylation in both GFRα1 and GFRα2 transfected MG87RET cells (Fig. 2A, B). BT44 also induced the activation of RET receptor in GFP transfected MG87RET cells (Fig. 2C). Repeated measures (RM) ANOVA revealed statistically significant differences between the treatments in GFRα1-RET (F5,20 = 35.67; p < 0.0001), GFRα2-RET (F5,15 = 24.04; p < 0.0001) and GFP-RET (F4,20 = 6.175; p = 0.0021) expressing cells. The level of RET phosphorylation became higher in response to 36μM (1.25±0.29) and 75μM (1.26±0.31) of BT44 (p = 0.0092 and p = 0.0085, respectively; Dunnett’s post hoc) as compared with the vehicle (0.66±0.22) in the cells expressing GFRα1-RET (Fig. 2D). Similarly, in GFRα2-RET expressing cells both 36μM (1.52±0.12) and 75μM (1.97±0.22) of BT44 increased the level of RET phosphorylation compared to vehicle (0.86±0.15; p = 0.037 and p = 0.0005, respectively; Dunnett’s post hoc) (Fig. 2E). The level of RET phosphorylation also increased in response to 18μM (0.58±0.12), 36μM (0.64±0.16), and 75μM (0.65±0.20) of BT44 (p = 0.024, p = 0.0044, and p = 0.0032, respectively; Dunnett’s post hoc) compared to vehicle (0.32±0.06) in GFP-RET expressing cells (Fig. 2F). Both GDNF (2.59±0.33) and NRTN (3.34±0.41) at 200 ng/ml elevated the RET phosphorylation level when they were applied to GFRα1 and GFRα2 transfected MG87RET cells, respectively (p < 0.0001; Dunnett’s post hoc), but as expected, GDNF did not activate RET in the absence of GFRα1 (Supplementary Figure 2). To further characterize which intracellular tyrosine residues of RET become phosphorylated in response to BT44, we used specific antibodies raised against pY905Ret, pY1015Ret, pY1062Ret, and pY1096Ret, which are shown to be involved in intracellular signaling activation by GFLs [66]. RET phosphorylation by BT44 seemed to occur mainly at Tyr1062 which has been reported to be one of the main docking sites for the intracellular adaptor proteins FRS2 and SHC leading to the activation of AKT and ERK pathways [67] (Supplementary Figure 3).
GDNF- and NRTN-dependent RET phosphorylation activates the intracellular targets AKT and ERK which are imperative for the neuronal survi-val and neurite outgrowth. Therefore, we tested whether BT44 could also induce the phosphorylation of these downstream signaling targets in GFRα1-RET, GFRα2-RET and GFP-RET expressing cells (Fig. 2G-O). Indeed, BT44 and the GFLs increa-sed the phosphorylation of AKT in GFRα1 (F5,10 = 63.1; p < 0.0001; RM ANOVA), and GFRα2 (F5,15 = 40.09; p < 0.0001; RM ANOVA) transfected MG87RET cells. The level of AKT phosphorylation incr-eased in response to both 36μM (1.89±0.41) and 75μM (2.31±0.19) of BT44 compared to vehicle (0.91±0.20; p = 0.0099 and p = 0.0008, respectively; Dunnett’s post hoc) in the cells expressing GFRα1-RET (Fig. 2J). Similarly, in cells expressing GFRα2-RET, the phosphorylation of AKT was increased in response to 75μM (0.98±0.039) of BT44 (p = 0.0098; Dunnett’s post hoc) compared to vehicle (0.63±0.02) (Fig. 2K). BT44 did not have significant effects on AKT phosphorylation in GFP-RET expressing cells (Fig. 2L). As expected, GDNF (4.77±0.43) and NRTN (1.72±0.13) increased the level of AKT phosphorylation in GFRα1 and GFRα2 transfected cells, respectively (p < 0.0001; Dunnett’s post hoc). Importantly, in GFRα1-RET expressing cells, the protein levels of AKT and ERK did not change in response to BT44 or GDNF (Supplementary Figure 4).
BT44 also induced the phosphorylation of ERK in GFRα1-RET (F5,20 = 20.55; p < 0.0001; RM ANOVA), GFRα2-RET (F5,20 = 15.02; p < 0.0001; RM ANOVA), and GFP-RET (F4,12 = 3.439; p = 0.043; RM ANOVA) expressing cells. Both 36μM (1.43±0.11) and 75μM (1.48±0.12) of BT44 inc-reased the level of ERK phosphorylation in the cells expressing GFRα1-RET (p = 0.0083 and p = 0.0054, respectively; Dunnett’s post hoc) compared to vehicle (0.56±0.13) (Fig. 2M). In GFRα2 transfected cells, the level of ERK phosphorylation was increased in response to 7.5μM (0.49±0.14, p = 0.048), 18μM (0.57±0.089, p = 0.010), 36μM (0.50±0.077, p = 0.044), and 75μM (0.60±0.058, p = 0.0061) of BT44 compared to vehicle (0.18±0.04) (Fig. 2N). In addition, BT44 18μM (0.45±0.074) and BT44 75μM (0.47±0.15) increased the level of ERK phosphorylation in GFP-RET expressing cells compared to vehicle (0.22±0.03; p = 0.039 and p = 0.025, respectively; Dunnett’s post hoc) (Fig. 2O). The level of ERK phosphorylation was elevated in GFRα1 and in GFRα2 transfected cells treated with GDNF (2.76±0.37) and NRTN (1.14±0.15) at a concentration of 200 ng/ml (p < 0.0001; Dunnett’s post hoc). The data presented above for both the phosphorylated AKT and ERK were normalized to GAPDH. We also saw statistically significant increase in the level of phosphorylated AKT and ERK when normalized to pan-AKT and pan-ERK, respectively, with the amplitude of response (fold increase in the level of pAKT and pERK) comparable to the data normalized to GAPDH (Supplementary Figure 4).
We also investigated if the effects of BT44 were RET specific by stimulating reporter cells expressing the signaling receptor for brain-derived neurotrophic factor (BDNF), tropomyosin-related kinase B (TrkB), with BT44 and measuring luminescence. BT44 did not activate TrkB receptor. As expected, BDNF increased the luciferase activity in reporter cells expressing TrkB receptor (Supplementary Figure 5).
Fig. 2
BT44 induces RET phosphorylation and activation of AKT and ERK signaling pathways. Phosphorylation of RET (A-F) and its downstream signaling targets (G-O) analyzed by western blotting. Quantification of RET phosphorylation level (D-F) and AKT/ERK phosphorylation levels (J-O) based on corresponding western blots. GDNF (200 ng/ml, ≃ 6.6 nM) and NTRN (200 ng/ml, ≃4.2 nM) were used as positive controls in MG87RET fibroblasts transfected with GFRα1 and GFRα2 and their concentrations are provided in ng/ml. VEH, vehicle; IP, immunoprecipitation; WB, western blotting; GAPDH, glyceraldehyde 3-phosphate dehydrogenase as a loading control. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, RM ANOVA with Dunnett’s post hoc test. Mean±SEM, n = 3–6.
BT44 promotes the survival of cultured midbrain dopamine neurons
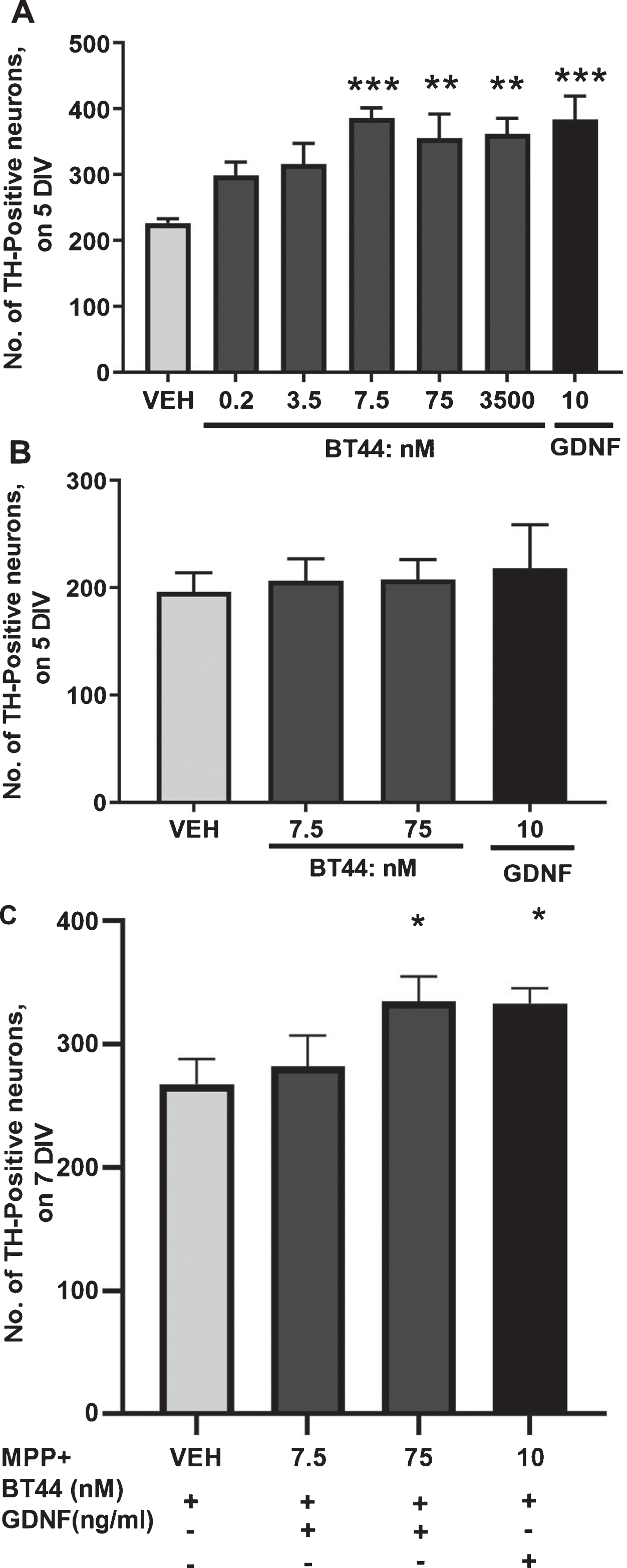
GDNF and NRTN are well-known survival-promoting NTFs for dopamine neurons [11, 68]. Therefore, we assessed whether BT44 is also able to promote the survival of cultured mouse midbrain embryonic dopamine neurons by quantifying the number of cells expressing TH, the key enzyme of dopamine synthesis. BT44 and GDNF significantly increased the survival of cultured wild-type dopamine neurons (F6,18 = 5.734; p = 0.0018, RM ANOVA). After 5 days in vitro, the number of TH-ir cells was increased in wells treated with 7.5 nM (386.6±15.0), 75 nM (355.6±36.7), and 3.5μM (361.8±23.9) of BT44 (226.3±6.6; p = 0.0008, p = 0.0061, and p = 0.0040, respectively; Dunnett’s post hoc) as compared with the vehicle-treated wells (Fig. 3A). As expected, GDNF (10 ng/ml, 0.33 nM) significantly increased the number of wild-type TH-ir cells (384.0±35.2; p = 0.0009; Dunnett’s post hoc). Representative images of mouse embryonic E13.5 wild-type dopamine neuron cultures treated with vehicle, BT44, and GDNF are provided in Supplementary Figure 6.
In order to confirm that the effect of BT44 on the survival of dopamine neurons was RET dependent, we assessed the survival of RET knockout TH-ir midbrain neurons treated with BT44 and GDNF. We did not observe survival promoting effect of BT44 (7.5 or 75 nM) or GDNF (10 ng/ml, ≃0.33 nM) in cultured RET knockout dopamine neurons on the 5th day in vitro (Fig. 3B).
We further tested the neuroprotective ability of BT44 in cultured midbrain dopamine neurons when app-lied together with MPP + neurotoxin. BT44 protec-ted dopamine neurons from MPP + -induced cell death (F3,15 = 5.410; p = 0.01, RM ANOVA) (Fig. 3C). The number of TH-ir neurons was higher in wells treated with 75 nM of BT44 (334.8±20.3) compared to vehicle (267.7±20.4; p = 0.0160; Dunnett’s post hoc). As expected, GDNF (10 ng/ml ≃0.33 nM) also significantly protected TH-ir cells from MPP + toxicity as compared to vehicle (333.0±12.6 vs. 267.7±20.4; p = 0.0191; Dunnett’s post hoc). These data demonstrate significant increase in the potency of BT44 compared to the parent compound BT13: BT13 promoted the survival of naïve dopamine neurons at concentrations 0.1-1μM and showed neuroprotective effect on MPP + challenged dopamine neurons at a concentration of 1μM [47]. Therefore, we chose BT44 for further studies.
Fig. 3
BT44 promotes the survival of cultured wild-type primary dopamine neurons, but not RET knockout dopamine neurons and protects cultured dopamine neurons against MPP + induced cell death. A) Effect of BT44 and GDNF on the number of TH-ir cells in wild-type midbrain cultures after 5 days in vitro (5 DIV). B) The number of TH-ir cells in RET knockout midbrain cultures on the 5th day in vitro (5 DIV). C) The number of TH-ir cells in wild-type midbrain cultures exposed to MPP + . The number of TH-ir cells is normalized to the total number of cells in the culture. Concentration of GDNF used as a positive control is provided in ng/ml (10 ng/ml, ≃0.33 nM). VEH, Vehicle. *p < 0.05, **p < 0.01, ***p < 0.001, RM ANOVA with Dunnett’s post hoc test. Mean±SEM, Number of independent repeats (n) = 4 for wild-type and (n) = 2 for RET knockout dopamine neuron cultures. Neuroprotection experiment was repeated 5 times with reproducible results and N is the number of wells from one experiment, N = 6.
BT44 shows no off-target activities, penetrates the blood-brain barrier, and is rapidly eliminated from the blood circulation
To further address the selectivity of BT44 towards RET and its potential safety-related off-target effects, we tested BT44 in a panel of in vitro assays by Eurofins CEREP company (Eurofins Scientific 2019). We evaluated direct effects of 1μM BT44 on a set of ion channels, G-protein coupled receptors, transporters, kinases and enzymes metabolizing dopamine (Supplementary Table 2). The concentration of BT44 for profiling assays was chosen based on its biological activity in dopamine neurons and in such a way to exceed expected maximal concentration in in vivo tests after systemic or intracranial delivery. BT44 did not affect the activity of the selected proteins: in all performed assays, we observed less than 25% of target inhibition or stimulation which, according to Eurofins CEREP’s results interpretation guidelines, reflects assay variability and indicates the lack of significant effects of a test compound. In automated patch-clamp assay 0.1–10μM BT44 inhibited human Ether-à-go-go-Related Gene (hERG) by less than 20% indicative of relative cardiac safety of the compound.
Finally, BT44 (10 mg/kg) was intravenously inj-ected to rats to assess its pharmacokinetics and the ability to cross the blood-brain barrier. BT44 was rapidly eliminated from the circulation (half-life (t1/2) = 0.72 h) and brain (t1/2 = 0.47 h) and penetr-ated the blood-brain barrier (13–26 % of serum concentration was detected in the brain) (Table 1). The main pharmacokinetic parameters of BT44 were as follows: maximal concentration (Cmax) = 963 ng/ml; area under the plasma concentration time curve from the beginning to infinity (AUC0 - ∞) = 1717 ng*h/ml; mean residence time (MRT) = 0.72 h; clearance (Cl) = 97.1 ml/min/kg; volume of distribution (Vd) = 4.6 l/kg.
Table 1
Intravenously injected BT44 (10 mg/kg) penetrates the blood-brain barrier and is rapidly eliminated from the circulation. Concentration of BT44 in the brain and plasma 0.5–3 hours after an intravenous injection. Data are presented as Mean±SEM, n = 3 rats per group
| Time, h | BT44 concentration | |
| Plasma, ng/ml | Brain, ng/g | |
| 0.5 | 963±313 | 250±31.4 |
| 1 | 449±79.1 | 80.6±10.2 |
| 3 | 77.9±29.4 | 5.6±2.1 |
BT44 reverses amphetamine-induced motor imbalance in 6-OHDA rat model of PD
Due to promising survival-promoting effects on cultured dopamine neurons, we studied the ability of BT44 to produce functional recovery in a rat model of PD with progressive unilateral 6-OHDA lesion [54]. The effects of BT44 (0.1μg/24 h and 0.3μg/24 h) and GDNF (3μg/24 h) were tested on amphetamine-induced rotational behavior and limb-use asymmetry assays. BT44, GDNF, or vehicles (PBS for GDNF and PG for BT44) were infused for 14 days into the right dorsal striatum starting 2 weeks after the ipsilateral 6-OHDA lesion (Fig. 4A, B).
The 6-OHDA administration produced a substantial lesion on dopaminergic neurons as indicated by strong amphetamine-induced turning behavior in vehicle treated rats at 2–12 weeks post-lesion (Fig. 4C-E). Statistically significant differences in motor function between the treatment groups were detected at 6 weeks (F4,45 = 3.638; p = 0.012; one-way ANOVA) (Fig. 4D) and 12 weeks following the toxin administration (F4,44 = 11.365; p < 0.001; one-way ANOVA) (Fig. 4E). BT44 0.3μg/24h (658.6±101.3) was able to significantly reduce the number of net ipsilateral turns as compared to PBS (1363.2±193.2) and PG (1467.0±199.8) treated rats (p = 0.046 and p = 0.010, respectively; Tukey HSD post hoc) only at 12 weeks after the 6-OHDA lesion (Fig. 4E), although a trend to reduction was observed already at 6 weeks. GDNF reduced rotational asymmetry both at 6 weeks and 12 weeks post lesion. The number of ipsilateral turns in GDNF treated group was 862.6±136.4 at 6 weeks, which was significantly less than in PBS (1691.4±58.9) and PG (1562.1±147.0) treated groups (p = 0.025 and p = 0.046, respectively; Tukey HSD post hoc). At 12 weeks post lesion, the turning behavior was further reduced in GDNF treated animals (149.1±47.9) and they rotated significantly less (p < 0.001; Tukey HSD post hoc) than PBS (1363.2±193.2) and PG (1467.0±199.8) treated rats. Also, BT44 0.1μg/24h (1193.9±205.6) infused rats rotated significantly more than GDNF infused rats (p < 0.001; Tukey HSD post hoc). When we analyzed the rotation rate per 5 min at 12 weeks post lesion, we detected significant difference in the time-dependence of turning behavior of BT44 0.3μg/24 h treated rats as compared to the other treatment groups (Fig. 4F). In the beginning of the experiment, the rats seemed to respond to the amphetamine challenge in a similar manner as vehicle treated rats or those treated with BT44 0.1μg/24 h. BT44 0.3μg/24 h infused rats, however, recovered faster and more completely from the strong turning response. From 60 min time point onwards, BT44 0.3μg/24 h showed its significant effect in alleviating the rotational asymmetry as compared to PBS and PG (p = 0.029 and p = 0.003, respectively; Tukey HSD after RM ANOVA 60–120 min: F4,44 = 8.491; p < 0.001). Although the effect of GDNF was more pronounced, similar profile as compared with BT44 0.3μg/24h was seen for GDNF: at first, we observed a higher number of ipsilateral rotations followed by a long-lasting decrease in the number of ipsilateral turns. Neither of the treatments had a significant effect on spontaneous limb-use asymmetry in the cylinder test (data not shown) which is in line with previously published data for GDNF [22, 69, 70].
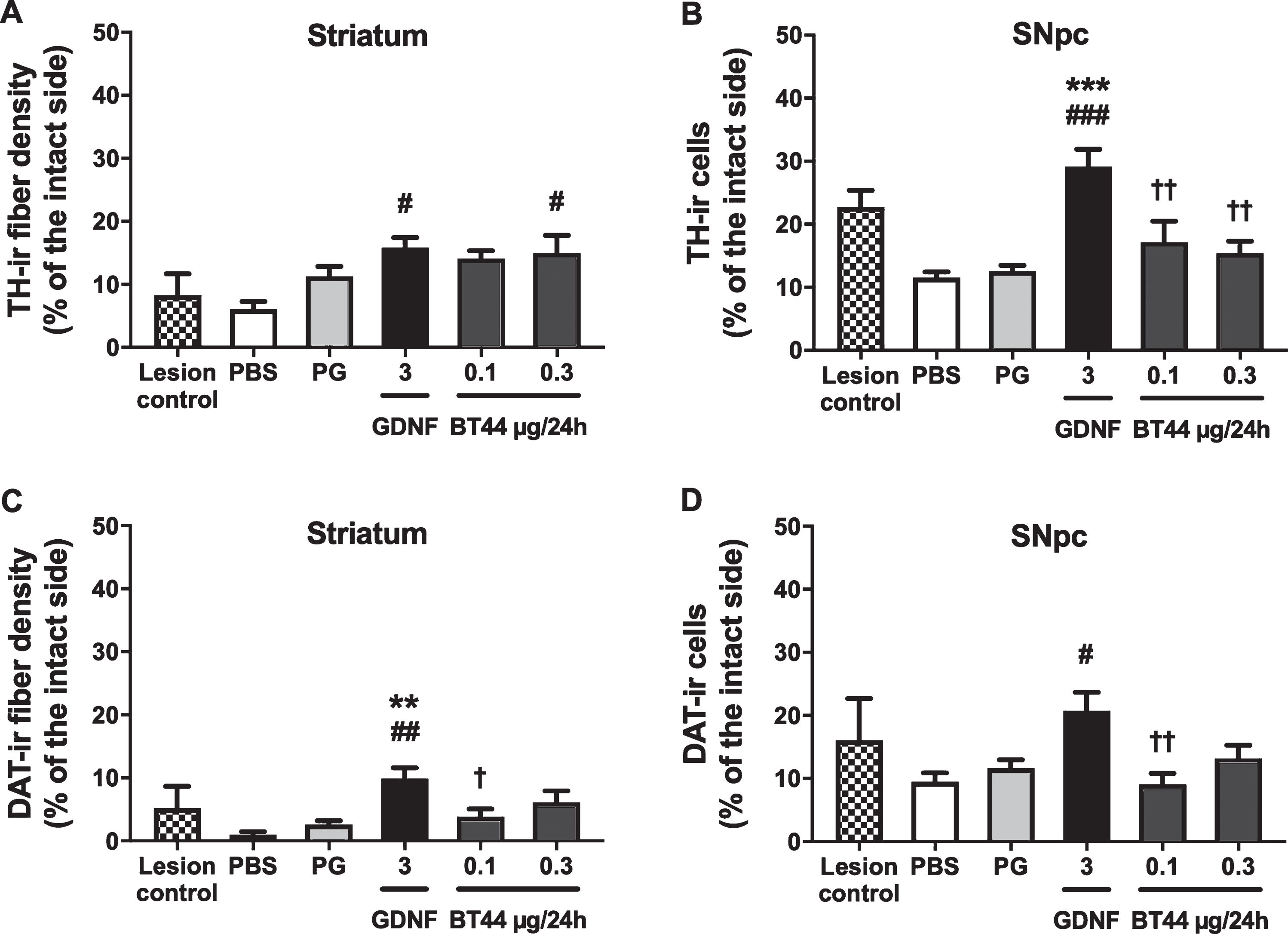
BT44 seems to protect dopaminergic fibers in the striatum but is not able to protect dopamine neurons in the SNpc of 6-OHDA lesioned rats
The ability of BT44 to protect and restore nig-rostriatal dopamine neurons was assessed by immunohistochemical analysis of the nigral and striatal sections probed with anti-TH and anti-DAT antibodies at 12 weeks post lesion. In line with previously published results [54], 6-OHDA delivery paradigm employed in this study resulted in a severe and progressive retrograde degeneration of the nigrostriatal dopamine neurons (Fig. 5). At 2 weeks post lesion, the density of TH-ir fibers in the striatum was reduced by 91.7±3.4% and the number of TH-ir cell bodies in the SNpc by 77.2±2.6% in comparison to the intact side. The density of DAT-ir fibers in the striatum was reduced by 94.8±3.5% and the number of DAT-ir cell bodies in the SNpc by 83.9±6.6% in comparison to the intact side (lesion control group).The decrease in the number of TH-ir cells in the SNpc of PBS and PG treated animals at 12 weeks post lesion constituted 88.5±0.9% and 87.4±0.9% in comparison to the intact side, respectively. The number of DAT-ir cells in the SNpc of PBS and PG treated animals at 12 weeks post lesion was reduced by 90.5±1.4% and 88.4±1.3% in comparison to the intact side, respectively. Representative images of TH-immunostained coronal sections from the striatum and the ventral midbrain are presented in Supplementary Figure 7.
At 12 weeks post lesion, the density of remaining TH-ir fibers in the striatum of rats treated with BT44 0.3μg/24 h was 2.5 times higher (15.0±2.8% of the intact side) as compared with PBS treated animals (6.1±1.2% of the intact side, p = 0.025; Tukey HSD post hoc test after one-way ANOVA F5,46 = 3.671; p = 0.007) (Fig. 5A). Also, BT44 0.1μg/24 h showed a tendency (p = 0.065; Tukey HSD post hoc) to similar neuroprotective effect on TH-ir fibers (14.1±1.3% of the intact side). In GDNF 3μg/24 h infused rats, the TH-ir fiber density was 2.6 times higher (15.8±1.6% of the intact side) as compared to PBS treated rats (p = 0.010; Tukey HSD post hoc). No statistically significant differences in the density of striatal TH-ir fibers were detected between PBS and PG treated rats. We also assessed the effect of BT44 and GDNF on TH-ir neurite number in the striatum of 6-OHDA lesioned rats using an automated CNN algorithm and cloud-embedded Aiforia™ platform. No statistically significant differences in the number of TH-ir fibers were detected in BT44 and GDNF treated rats, although this parameter was slightly higher in the striata of rats treated with BT44 0.1μg/24 h. These results were obtained using a newly developed algorithm and should be interpreted cautiously as the algorithm needs to be validated with a proper positive control (Supplementary Figure 9).
In accordance with the TH-ir fiber density results, the density of DAT-ir fibers in the striatum of BT44 0.3μg/24h treated rats was 6.1 and 2.3 times higher (6.1±1.8% of the intact side) as compared with PBS (1.0±0.5% of the intact side) and PG (2.6±0.6% of the intact side) treated animals, respectively (Fig. 5C), but these differences did not reach statistical significance. In GDNF 3μg/24 h infused rats, the DAT-ir fiber density was 9.9 times higher (9.9±1.7% of the intact side, p = 0.001; Tukey HSD post hoc test after one-way ANOVA F5,45 = 4.746; p = 0.001) as compared with PBS infused rats. DAT-ir fiber density in GDNF group was also significantly higher than in PG (p = 0.006) and BT44 0.1μg/24 h (3.9±1.2% of the intact side, p = 0.046) groups.
In the SNpc, GDNF infusion was clearly able to prevent the loss of dopamine neurons. The number of TH-ir cell bodies on the 6-OHDA lesioned side in GDNF treated rats (29.1±2.8% of the intact side) was 2.5 times higher than in PBS (11.5±0.9% of the intact side, p < 0.001), and 2.3 times higher than in PG (12.6±0.9% of the intact side, p < 0.001) treated animals (Tukey HSD post hoc test after one-way ANOVA F5,48 = 8.898; p < 0.001) (Fig. 5B, Supplementary Table 3). The number of TH-ir cells in GDNF group differed significantly also from BT44 0.1μg/24 h (17.2±3.3% of the intact side) and BT44 0.3μg/24 h (15.4±1.9% of the intact side) (p = 0.004 and p = 0.001, respectively; Tukey HSD post hoc) groups. The number of DAT-ir cell bodies in GDNF treated rats (20.7±2.9% of the intact side) was 2.2 times higher than in PBS treated animals (9.5±1.4% of the intact side, p = 0.012; Tukey HSD post hoc test after one-way ANOVA F5,46 = 3.752; p = 0.006) (Fig. 5D). The number of DAT-ir cells was also significantly higher in GDNF group than in BT44 0.1μg/24h group (9.1±1.7% of the intact side, p = 0.008). Statistically significant differences in the number of DAT-ir cell bodies on the lesioned side between GDNF and PG (11.6±1.3% of the intact side) or BT44 0.3μg/24h (13.2±2.1% of the intact side) treated animals were not detected. Intrastriatal infusion of BT44 was unable to prevent the loss of dopaminergic cell bodies in the SNpc as compared to vehicle infusion.
Fig. 5
Effect of BT44 and GDNF on the density of TH and DAT-immunoreactive fibers in the striatum and the number of TH and DAT-immunoreactive cells in the SNpc in 6-OHDA lesioned rats. A) Densitometric quantification of TH-ir fibers in the dorsal striatum. B) Number of TH-ir cells in the SNpc. C) Densitometric quantification of DAT-ir fibers in the dorsal striatum. D) Number of DAT-ir cells in the SNpc. All values are presented as percentage of the intact side. PBS, phosphate buffered saline; PG, propylene glycol. **p < 0.01, ***p < 0.001 vs. PG; #p < 0.05, # #p < 0.01, # # #p < 0.001 vs. PBS; †p < 0.05, ††p < 0.01 vs. GDNF, Tukey HSD after one-way ANOVA. Mean±SEM, lesion control group n = 4, other groups n = 8–11.
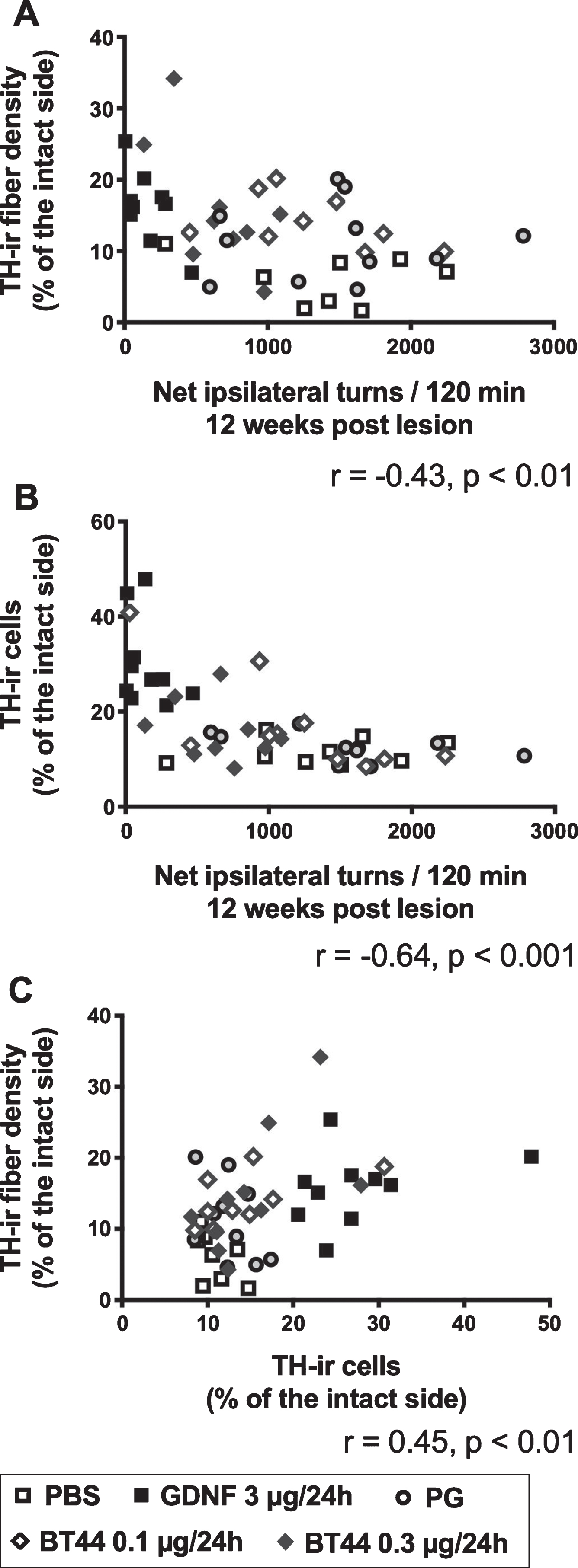
Amphetamine-induced rotational behavior poorly correlates with TH immunohistological measures
Due to discrepancies between amphetamine-induced turning behavior and immunohistochemical measures at 12 weeks post lesion, we analyzed how well rotational asymmetry correlates with TH-ir fiber density in the striatum and cells counts in the SNpc. Amphetamine-induced rotations showed only low, but significant, negative correlation with TH-ir fiber density in the striatum (Pearson’s r = –0.43, p < 0.01), and moderate negative correlation with TH-ir cell number in the SNpc (Pearson’s r = –0.64, p < 0.001) when data from all treatment groups were pooled together (Fig. 6A, B). We also evaluated the correlation between the striatal fiber density and nigral cell numbers and found a low correlation between these two histological read-outs when data from all treatment groups were analyzed together (Pearson’s r = 0.45, p < 0.01) (Fig. 6C). Notably, GDNF-treated rats rotated significantly less as compared to the other rats, but this behavioral improvement was not accompanied by axonal regeneration in the striatum to the same extent (Figs. 4E, 5A). This is visualized in Fig. 6A as a grouping of GDNF-treated rats (black squares) close to the y-axis.
Fig. 6
Correlations between TH immunohistochemical measures and amphetamine-induced turning rate. A) Amphetamine-induced turning behavior at 12 weeks post lesion plotted against TH-ir fiber density in the lesioned striatum for each experimental animal. B) Amphetamine-induced turning behavior at 12 weeks post lesion plotted against TH-ir cell number in the SNpc on the lesion side for each experimental animal. (C) TH-ir cell numbers in the SNpc plotted against TH-ir fiber densities in the striatum. r, Pearson correlation coefficient, all treatment groups analyzed together.
DISCUSSION
PD patients await disease-modifying therapies that would slow down the progression of the disorder. Neurotrophic factors GDNF and NRTN have been tested in clinical trials in PD patients, but conclusive results are yet to be obtained. These NTFs have been delivered into the brain tissue either as a protein or gene therapy using a technically challenging stereotaxic surgery, which may constrain their clinical availability to the late-stage patients. At the same time, early-stage patients seem to be the proper target group for GFL-related treatments [71]. Both GDNF and NRTN activate intracellular signaling through receptor tyrosine kinase RET which therefore is an apparent target for a small molecule compound. Our novel small molecule compound BT44 activates RET and its intracellular signaling pathways. It also alleviates amphetamine-induced motor dysfunction in the 6-OHDA induced neurotoxin model of PD in rats at 12 weeks post lesion. In addition, BT44 may have protective effects on dopaminergic fibers in the striatum of 6-OHDA lesioned rats. Safety profiling raised no concerns regarding off-target effects of BT44.
Our results indicate that BT44 activates RET and RET-dependent intracellular signaling both in the presence and absence of co-receptors (GFRα1 and GFRα2) which is in line with previously published data for the parent compound BT13 [46, 47]. Interestingly, however, the intracellular signaling induced by BT44 can be modified by the co-receptors. In cells expressing the co-receptors GFRα1 and GFRα2 together with RET, BT44 activates both ERK and AKT pathways, but only ERK pathway is activated in the absence of the co-receptors. It has been shown that phosphotyrosine 1062 (predominantly phosphorylated in response to BT44) in RET provides a docking site for adaptor proteins FRS2 and SHC necessary for both ERK and AKT activation, respectively [72]. These adaptor proteins compete for binding to phosphorylated tyrosine 1062 and this can affect the pattern of activation of intracellular signaling cascades. Recent molecular docking and molecular dynamic simulations suggest that the possible binding site for compounds belonging to BT scaffold is located on the interface of GFRα1 and RET [51]. It can be speculated that in the absence of GFRα1, activated RET has a conformation favoring the binding of FRS2 and subsequent activation of ERK signaling cascades. In the presence of GFRα1, the GFRα1/RET complex is promiscuous to both FRS2 and SHC, thus providing mechanistic basis for biased effect of BT44 observed in the present study in MG87RET cells. Further extensive experiments are needed to confirm this speculation.
The pattern of phosphorylated tyrosine residues in RET differed in the BT44 treated cells compared with GFL treated cells. In contrast to GFLs, BT44 stimulated the phosphorylation of Y905 only weakly and failed to phosphorylate Y1096. This can provide further evidence for biased agonism observed in the cell signaling in response to BT44. As Y905 is possibly important for the activation of JNK [73–75], a signaling pathway leading to cell death, the reduced ability of BT44 to increase pY905 level can be beneficial for supporting neuronal survival and axonal regeneration.
Although BT44 is indiscriminate to GFRα co-re-ceptors and can elicit signaling even in their absence, it is selective to RET. In addition to RET, dopamine neurons express several other receptor tyrosine kinases including BDNF receptor TrkB [76, 77] and vascular endothelial growth factor (VEGF) receptors 1-2 (VEGFR 1–2) [78], the activation of which promotes the survival of cultured dopamine neurons [78–80]. Our data show that BT44 only supports the survival of RET-expressing wild-type dopamine neurons similarly to GDNF, whereas RET knockout neurons do not respond to BT44. In accordance with these data, BT44 also failed to activate ERK-related signaling in TrkB-expressing cells. Additionally, no interaction with other proteins was observed in CEREP screening, further supporting the notice of BT44 selectivity towards RET.
In cultured dopamine neurons, BT44 not only supported the survival of naïve serum-deprived cells, but also showed significant neuroprotection against MPP + induced toxicity similarly to GDNF. Both BT44 and GDNF promoted the survival of cultured dopamine neurons at much lower concentrations as compared to those required to elicit RET phosphorylation in cultured cells. Although we did not study concentration-dependence of GDNF effects in RET phosphorylation assay and dopamine neuron survival assay in this study, we usually see reproducible and detectible increase in the level of phosphorylated RET upon the treatment with≥50 ng/ml of GDNF [81]. At the same time, the effects of GDNF on neuronal survival are detected at much lower concentration of 5 ng/ml [81] or even at 1 ng/ml with somewhat increase detectable already at 0.1 ng/ml of GDNF [52]. RET phosphorylation assay reflects the number of phosphorylated RET residues after a short-term (15 min) treatment with the growth factor compared to the control, while dopamine neuron survival assay represents an integral result of prolonged (5 days) RET activation. Therefore, a low concentration of the ligand is sufficient to elicit biological response in the second case. GDNF concentrations for in vitro tests were chosen on the basis of previous results to reproducibly produce robust effects [81].
In PD, dopamine neuron cell death occurs in a ‘dying back’ manner where striatal nerve terminals degenerate before the death of nigral cell bodies [7]. The disease is diagnosed mainly based on the onset of characteristic motor symptoms, which appear when the neuronal damage of the nigrostriatal pathway is already pronounced. Therefore, in the present study we used the neurorestoration paradigm with progressive retrograde degeneration of the nigrostriatal dopamine neurons [54]. This paradigm may reflect the status of patients with moderate to advanced PD, i.e., a few years after the diagnosis. Both BT44 and GDNF were able to induce functional recovery assessed with amphetamine-induced rotational asymmetry test. GDNF also clearly restored dopaminergic processes in the striatum. BT44 stimulated some increase in the density of dopamine nerve terminals in the striatum although these changes were smaller as compared to the ones elicited by GDNF and not always statistically significant. Our observations of robust reduction in amphetamine-induced turning behavior in GDNF treated rats, together with significant sparing of nigrostriatal dopamine neurons, are consistent with earlier reports of GDNF induced potentiation of dopaminergic activity and restoration of motor behavior in unilateral lesion models. Therapeutic intervention with GDNF or BT44 at an earlier stage of the neurodegenerative process could possibly have produced more pronounced effects on striatal innervation and motor performance.
Pharmacokinetic profiling of BT44 suggests its ability to cross the blood-brain barrier when injected intravenously to rats. Here, however, we wanted to investigate local effects of BT44 after infusing into the dorsal striatum, which allowed us to make direct comparisons with the effects of GDNF. After lead optimization, RET agonists could be administered systemically to avoid risky intracranial administration and overcome the problematic question about the most effective delivery site in the brain. Indeed, our study shows proof of principle for a RET agonist having similar, but less potent, effect as GDNF in an experimental animal model of PD.
Interestingly, at 12 weeks post lesion BT44 0.3μg/24 h treatment resulted in a divergent time-dependence of rotational behavior (analyzed as turns per 5 min) as compared to the other treatment groups. The rats treated with BT44 (0.3μg/24 h) initially showed strong turning behavior but recovered faster from the amphetamine challenge. A similar profile with considerably smaller initial increase in the turning rate was observed also in GDNF treated animals. It can be speculated, that BT44 at 0.3μg/24 h has an effect on dopamine dynamics. In this respect changes in DAT activity or dopamine metabolizing enzymes, for example, are possible. RET signaling is suggested to negatively regulate the cell surface trafficking and activity of DAT [82–84]. In addition, the parent compound BT13 was shown to acutely enhance dopamine release in vivo which may reflect the ability of BT compounds to modify striatal dopamine dynamics. We cannot exclude the possibility that as a lipophilic compound BT44 is able to diffuse to the non-lesioned hemisphere during the 14-day infusion period which may compromise the results of amphetamine challenge. Further experiments are needed to clarify these possible effects of BT44.
As discussed above, in vitro experiments revealed that BT44 agonism is somewhat biased towards ERK signaling cascade which is predominantly responsible for the promotion of neurite outgrowth and regeneration in neurons [9]. Neuronal survival is controlled mainly by PI3 kinase-AKT signaling cascade, which is activated by BT44 only when both a GFRα co-receptor and RET are expressed by the cells. In addition, in GFRα2/RET expressing cells higher concentration of BT44 was necessary to activate AKT as compared to ERK. These data can exp-lain why BT44 may have some protective effect on dopamine fibers in the striatum rather than increase the number of dopamine cell bodies in the SNpc. In addition, BT44 is rapidly degraded in the tissues. Since it was infused to the striatum, its concentration was higher there than in the SNpc. Therefore, BT44 may have elicited more pronounced effects on dopaminergic fibers in the striatum as compared to the cell bodies in the SNpc.
In agreement with earlier reports, amphetamine-induced rotational behavior poorly correlated with the histological measures of TH-ir cells in the SNpc and TH-ir fibers in the striatum [63, 85–88]. BT44 may regulate the cell surface expression or activity of DAT as it was shown for GDNF [82, 89]. Also, other neural circuits than nigrostriatal dopaminergic pathway may be responsible for the functional recovery of the rats and provide an explanation for the discrepancy in the read-outs. For example, amphetamine induces dopamine release also from dendritic vesicles in the substantia nigra pars reticulata (SNr) [90, 91]. In the SNr, dopamine acts primarily at presynaptic D1 receptors of the GABAergic striatonigral medium spiny neurons (i.e., direct pathway) facilitating GABA transmission in the SNr. This reduces the overactive firing of GABAergic SNr neurons projecting to the ventromedial nucleus of thalamus that is characteristic to PD. In consequence, amphetamine-induced dendritic dopamine release in the SNr may have been elevated in GDNF treated rats whose dopaminergic cell bodies in the SNpc were more preserved as compared to the other treatments. This might explain why rotational asymmetry was balanced more than could be expected from the striatal fiber restoration in GDNF treated rats and why the efficacy of BT44 in the behavioral assay was lower than that of GDNF.
Important to note, that in addition to the motor symptoms, PD patients suffer from a number of non-motor symptoms such as olfaction deficit, constipation, depression, sleep disturbances and cognitive impairments. Many of these symptoms are caused by neuronal dysfunction and degeneration in different organs (e.g., bowel) and brain regions (e.g., olfactory bulb). GDNF signaling is important for olfactory [92] and enteric neurons [93, 94] and its role in memory and learning has also been described [95]. The effects of GFL proteins on non-motor symptoms have not been evaluated, because they have limited bioavailability and biodistribution failing to target multiple organs. Systemically acting small molecules stimulating GFL receptors may, however, overcome these limitations and relieve both motor and non-motor symptoms of PD.Some adverse effects, e.g., weight loss, were reported in one of phase II clinical trials with GDNF protein [39]. Indeed, RET signaling was recently shown to be important for appetite control by the protein called GDF15. GDF15 activates RET in complex with its co-receptor GFRAL expressed in chemoreceptor trigger zone in the brain in stress and disease conditions [96]. It is unclear if compounds belonging to BT scaffold can activate GFRAL/RET, but we saw no effects on weight gain in rats treated subcutaneously with relatively high doses of either BT13 [46] or BT44 [97]. It is possible that GFRAL blocks BT compound binding site on RET interface and, therefore, they do not influence weight, or 6-OHDA treatment fails to produce stress in the brainstem involved in weight control. Also, concerns regarding oncogenic potential of RET activation might have been raised by some researchers based on the gain-of-function mutations in RET gene constitutively activating RET as seen in multiple endocrine neoplasia type 2 (MEN2) and familial medullary thyroid cancer (FMTC) syndromes. Here, we would like to stress, that mice overexpressing GDNF did not to develop tumors during their lifespan [98]. Therefore, it is unlikely that RET agonists of relatively low potency and efficacy such as BT44 would be oncogenic. Indeed, we observed no tumors in rats treated with BT13 or BT44 for 2–6 weeks [46, 97]. An attractive avenue for further development in this field is the design of selective positive allosteric modulators of GDNF signaling. The first compound possessing such biological activity has been described [99, 100], but it has not been tested in PD models thus far. Such compounds can reduce the risk of known or potential side-effects caused by GFL proteins and prevent the disruption of endogenous GFL signaling.
Taken together, the functional and histological effects of BT44 observed in the animal model of PD are in alignment with those of GDNF but the efficacy of BT44 is considerably lower. With further optimization this promising lead compound could be developed into a novel and efficacious disease-modifying therapy for PD.
CONCLUSIONS
BT44 is a small molecule RET agonist with drug-like properties. It activates RET and its downstream signaling pathways AKT and ERK, promotes the survival of RET-expressing cultured midbrain dopamine neurons and protects them from the neurotoxin induced cell death. In rats modelling an advanced stage of PD, BT44 is the first-in-class small molecule capable of promoting functional recovery of the nigrostriatal dopamine system and alleviating motor deficits. The functional improvements seem to be accompanied by protection of dopaminergic processes innervating the striatum. Consequently, BT44 serves as a valuable lead compound that paves the way to develop disease-modifying treatments for PD.
ACKNOWLEDGMENTS
We are grateful to Jenni Montonen, Kati Rautio and Liisa Lappalainen for the great help with cell-based experiments, brain sectioning and immunohistological stainings. We also thank Dr. Satu Kuure for her help and advice with RET-knockout dopamine neurons and Dr. Harri Jäälinoja from Light Microscopy Unit, University of Helsinki for designing an automated analysis of dopamine neurons. We further thank NMR core fac-ility supported by University of Helsinki, Biocenter Finland and Helsinki Institute of Life Science (HiLIFE) for verification of the structure of BT44.
This work was supported by FP7-HEALTH-2013-INNOVATION-1 GA N602919, FP7-PEOPLE-2013-IAPP GA N612275, Gene Code Ltd., Lundbeck Foundation, Sigrid Jusélius Foundation (MS), Par-kinson’s UK Innovation grant - K-1408 (MS and YAS), Academy of Finland Project Grant 1325555 (YAS). Academy of Finland Grants 126744, 309708 and 314233 (MV). Centre of Excellence in Molecular Cell Engineering, Estonia, 2014-2020.4.01.15-013 (MK), Finnish Parkinson’s Foundation fellowship (AKM) and The Finnish Funding Agency for Innovation (Tekes, 3iRegeneration, Project 40395/13) (RKT and MS).
CONFLICT OF INTEREST
MK and MS are inventors in composition of matter patents of BT compounds US Patent No. 8901129B2 and European Patent No. 2509953 owned by Gene Code Ltd. MK and MS are inventors in patent application WO 2014041179 A1 on the treatment of peripheral neuropathy using GFRα3 type receptor agonists.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JPD-202400.
WEB REFERENCES
Eurofins France 2019. http://www.cerep.fr/cerep/utilisateurs/index.asp, visited 15.12.2019
Eurofins Scientific 2019. https://www.eurofins.com/biopharma-services, visited 15.12.2019
Parkinson’s Foundation 2019. https://www.parkinson.org, visited 20.6.2019
REFERENCES
[1] | Dauer W , Przedborski S ((2003) ) Parkinson’s disease: Mechanisms and models. Neuron 39: , 889–909. |
[2] | Kalia LV , Lang AE ((2015) ) Parkinson’s disease. Lancet 386: , 896–912. |
[3] | Kravitz AV , Freeze BS , Parker PRL , Kay K , Thwin MT , Deisseroth K , Kreitzer AC ((2010) ) Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 466: , 622–626. |
[4] | Mallet N , Ballion B , Le Moine C , Gonon F ((2006) ) Cortical inputs and GABA interneurons imbalance projection neurons in the striatum of parkinsonian rats. J Neurosci 26: , 3875–3884. |
[5] | Duncan GW , Khoo TK , Yarnall AJ , O’Brien JT , Coleman SY , Brooks DJ , Barker RA , Burn DJ ((2014) ) Health-related quality of life in early Parkinson’s disease: The impact of nonmotor symptoms. Mov Disord 29: , 195–202. |
[6] | Postuma RB , Aarsland D , Barone P , Burn DJ , Hawkes CH , Oertel W , Ziemssen T ((2012) ) Identifying prodromal Parkinson’s disease: Pre-motor disorders in Parkinson’s disease. Mov Disord 27: , 617–626. |
[7] | Burke RE , O’Malley K ((2013) ) Axon degeneration in Parkinson’s disease. Exp Neurol 246: , 72–83. |
[8] | Schapira AHV , Olanow CW ((2008) ) Drug selection and timing of initiation of treatment in early Parkinson’s disease. Ann Neurol 64 Suppl 2: , S47–55. |
[9] | Airaksinen MS , Saarma M ((2002) ) The GDNF family: Signalling, biological functions and therapeutic value. Nat Rev Neurosci 3: , 383–394. |
[10] | Huang EJ , Reichardt LF ((2001) ) Neurotrophins: Roles in neuronal development and function. Annu Rev Neurosci 24: , 677–736. |
[11] | Lin LF , Doherty DH , Lile JD , Bektesh S , Collins F ((1993) ) GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 260: , 1130–1132. |
[12] | Hoffer BJ , Hoffman A , Bowenkamp K , Huettl P , Hudson J , Martin D , Lin L-FH , Gerhardt GA ((1994) ) Glial cell line-derived neurotrophic factor reverses toxin-induced injury to midbrain dopaminergic neurons in vivo. Neurosci Lett 182: , 107–111. |
[13] | Bowenkamp KE , Hoffman AF , Gerhardt GA , Henry MA , Biddle PT , Hoffer BJ , Granholm AC ((1995) ) Glial cell line-derived neurotrophic factor supports survival of injured midbrain dopaminergic neurons. J Comp Neurol 355: , 479–489. |
[14] | Tomac A , Lindqvist E , Lin LF , Ogren SO , Young D , Hoffer BJ , Olson L ((1995) ) Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature 373: , 335–339. |
[15] | Kearns CM , Gash DM ((1995) ) GDNF protects nigral dopamine neurons against 6-hydroxydopamine in vivo. Brain Res 672: , 104–111. |
[16] | Choi-Lundberg DL , Lin Q , Chang YN , Chiang YL , Hay CM , Mohajeri H , Davidson BL , Bohn MC ((1997) ) Dopaminergic neurons protected from degeneration by GDNF gene therapy. Science 275: , 838–841. |
[17] | Kojima H , Abiru Y , Sakajiri K , Watabe K , Ohishi N , Takamori M , Hatanaka H , Yagi K ((1997) ) Adenovirus-mediated transduction with human glial cell line-derived neurotrophic factor gene prevents 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopamine depletion in striatum of mouse brain. Biochem Biophys Res Commun 238: , 569–573. |
[18] | Rosenblad C , Martinez-Serrano A , Björklund A ((1998) ) Intrastriatal glial cell line-derived neurotrophic factor promotes sprouting of spared nigrostriatal dopaminergic afferents and induces recovery of function in a rat model of Parkinson’s disease. Neuroscience 82: , 129–137. |
[19] | Kirik D , Rosenblad C , Björklund A ((2000) ) Preservation of a functional nigrostriatal dopamine pathway by GDNF in the intrastriatal 6-OHDA lesion model depends on the site of administration of the trophic factor. Eur J Neurosci 12: , 3871–3882. |
[20] | Brizard M , Carcenac C , Bemelmans A-P , Feuerstein C , Mallet J , Savasta M ((2006) ) Functional reinnervation from remaining DA terminals induced by GDNF lentivirus in a rat model of early Parkinson’s disease. Neurobiol Dis 21: , 90–101. |
[21] | Lindholm P , Voutilainen MH , Laurén J , Peränen J , Leppänen V-M , Andressoo J-O , Lindahl M , Janhunen S , Kalkkinen N , Timmusk T , Tuominen RK , Saarma M ((2007) ) Novel neurotrophic factor CDNF protects and rescues midbrain dopamine neurons in vivo. Nature 448: , 73–77. |
[22] | Yue X , Hariri DJ , Caballero B , Zhang S , Bartlett MJ , Kaut O , Mount DW , Wüllner U , Sherman SJ , Falk T ((2014) ) Comparative study of the neurotrophic effects elicited by VEGF-B and GDNF in preclinical in vivo models of Parkinson’s disease. Neuroscience 258: , 385–400. |
[23] | Gash DM , Zhang Z , Ovadia A , Cass WA , Yi A , Simmerman L , Russell D , Martin D , Lapchak PA , Collins F , Hoffer BJ , Gerhardt GA ((1996) ) Functional recovery in parkinsonian monkeys treated with GDNF. Nature 380: , 252–255. |
[24] | Zhang Z , Miyoshi Y , Lapchak PA , Collins F , Hilt D , Lebel C , Kryscio R , Gash DM ((1997) ) Dose response to intraventricular glial cell line-derived neurotrophic factor administration in parkinsonian monkeys. J Pharmacol Exp Ther 282: , 1396–1401. |
[25] | Kordower JH , Emborg ME , Bloch J , Ma SY , Chu Y , Leventhal L , McBride J , Chen EY , Palfi S , Roitberg BZ , Brown WD , Holden JE , Pyzalski R , Taylor MD , Carvey P , Ling Z , Trono D , Hantraye P , Déglon N , Aebischer P ((2000) ) Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson’s disease. Science 290: , 767–773. |
[26] | Costa S , Iravani MM , Pearce RK , Jenner P ((2001) ) Glial cell line-derived neurotrophic factor concentration dependently improves disability and motor activity in MPTP-treated common marmosets. Eur J Pharmacol 412: , 45–50. |
[27] | Grondin R , Zhang Z , Yi A , Cass WA , Maswood N , Andersen AH , Elsberry DD , Klein MC , Gerhardt GA , Gash DM ((2002) ) Chronic, controlled GDNF infusion promotes structural and functional recovery in advanced parkinsonian monkeys. Brain 125: , 2191–2201. |
[28] | Kishima H , Poyot T , Bloch J , Dauguet J , Condé F , Dollé F , Hinnen F , Pralong W , Palfi S , Déglon N , Aebischer P , Hantraye P ((2004) ) Encapsulated GDNF-producing C2C12 cells for Parkinson’s disease: A pre-clinical study in chronic MPTP-treated baboons. Neurobiol Dis 16: , 428–439. |
[29] | Gash DM , Zhang Z , Ai Y , Grondin R , Coffey R , Gerhardt GA ((2005) ) Trophic factor distribution predicts functional recovery in parkinsonian monkeys. Ann Neurol 58: , 224–233. |
[30] | Emborg ME , Moirano J , Raschke J , Bondarenko V , Zufferey R , Peng S , Ebert AD , Joers V , Roitberg B , Holden JE , Koprich J , Lipton J , Kordower JH , Aebischer P ((2009) ) Response of aged parkinsonian monkeys to in vivo gene transfer of GDNF. Neurobiol Dis 36: , 303–311. |
[31] | Kirik D , Georgievska B , Björklund A ((2004) ) Localized striatal delivery of GDNF as a treatment for Parkinson disease. Nat Neurosci 7: , 105–110. |
[32] | Kirik D , Georgievska B , Rosenblad C , Björklund A ((2001) ) Delayed infusion of GDNF promotes recovery of motor function in the partial lesion model of Parkinson’s disease. Eur J Neurosci 13: , 1589–1599. |
[33] | Bespalov MM , Sidorova YA , Tumova S , Ahonen-Bishopp A , Magalhães AC , Kulesskiy E , Paveliev M , Rivera C , Rauvala H , Saarma M ((2011) ) Heparan sulfate proteoglycan syndecan-3 is a novel receptor for GDNF, neurturin, and artemin. J Cell Biol 192: , 153–169. |
[34] | Paratcha G , Ledda F , Ibáñez CF ((2003) ) The neural cell adhesion molecule NCAM is an alternative signaling receptor for GDNF family ligands. Cell 113: , 867–879. |
[35] | Drinkut A , Tillack K , Meka DP , Schulz JB , Kügler S , Kramer ER ((2016) ) Ret is essential to mediate GDNF’s neuroprotective and neuroregenerative effect in a Parkinson disease mouse model. Cell Death Dis 7: , e2359. |
[36] | Gill SS , Patel NK , Hotton GR , O’Sullivan K , McCarter R , Bunnage M , Brooks DJ , Svendsen CN , Heywood P ((2003) ) Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med 9: , 589–595. |
[37] | Slevin JT , Gerhardt GA , Smith CD , Gash DM , Kryscio R , Young B ((2005) ) Improvement of bilateral motor functions in patients with Parkinson disease through the unilateral intraputaminal infusion of glial cell line-derived neurotrophic factor. J Neurosurg 102: , 216–222. |
[38] | Slevin JT , Gash DM , Smith CD , Gerhardt GA , Kryscio R , Chebrolu H , Walton A , Wagner R , Young AB ((2006) ) Unilateral intraputaminal glial cell line-derived neurotrophic factor in patients with Parkinson disease: Response to 1 year each of treatment and withdrawal. Neurosurg Focus 20: , E1. |
[39] | Nutt JG , Burchiel KJ , Comella CL , Jankovic J , Lang AE , Laws ER , Lozano AM , Penn RD , Simpson RK , Stacy M , Wooten GF , ICV GDNF Study Group. Implanted intracerebroventricular. Glial cell line-derived neurotrophic factor ((2003) ) Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology 60: , 69–73. |
[40] | Lang AE , Gill S , Patel NK , Lozano A , Nutt JG , Penn R , Brooks DJ , Hotton G , Moro E , Heywood P , Brodsky MA , Burchiel K , Kelly P , Dalvi A , Scott B , Stacy M , Turner D , Wooten VGF , Elias WJ , Laws ER , Dhawan V , Stoessl AJ , Matcham J , Coffey RJ , Traub M ((2006) ) Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol 59: , 459–466. |
[41] | Whone A , Luz M , Boca M , Woolley M , Mooney L , Dharia S , Broadfoot J , Cronin D , Schroers C , Barua NU , Longpre L , Barclay CL , Boiko C , Johnson GA , Fibiger HC , Harrison R , Lewis O , Pritchard G , Howell M , Irving C , Johnson D , Kinch S , Marshall C , Lawrence AD , Blinder S , Sossi V , Stoessl AJ , Skinner P , Mohr E , Gill SS ((2019) ) Randomized trial of intermittent intraputamenal glial cell line-derived neurotrophic factor in Parkinson’s disease. Brain 142: , 512–525. |
[42] | Whone AL , Boca M , Luz M , Woolley M , Mooney L , Dharia S , Broadfoot J , Cronin D , Schroers C , Barua NU , Longpre L , Barclay CL , Boiko C , Johnson GA , Fibiger HC , Harrison R , Lewis O , Pritchard G , Howell M , Irving C , Johnson D , Kinch S , Marshall C , Lawrence AD , Blinder S , Sossi V , Stoessl AJ , Skinner P , Mohr E , Gill SS ((2019) ) Extended treatment with glial cell line-derived neurotrophic factor in Parkinson’s disease. J Parkinsons Dis 9: , 301–313. |
[43] | Heiss JD , Lungu C , Hammoud DA , Herscovitch P , Ehrlich DJ , Argersinger DP , Sinharay S , Scott G , Wu T , Federoff HJ , Zaghloul KA , Hallett M , Lonser RR , Bankiewicz KS ((2019) ) Trial of magnetic resonance-guided putaminal gene therapy for advanced Parkinson’s disease. Mov Disord 34: , 1073–1078. |
[44] | Marks WJ , Bartus RT , Siffert J , Davis CS , Lozano A , Boulis N , Vitek J , Stacy M , Turner D , Verhagen L , Bakay R , Watts R , Guthrie B , Jankovic J , Simpson R , Tagliati M , Alterman R , Stern M , Baltuch G , Starr PA , Larson PS , Ostrem JL , Nutt J , Kieburtz K , Kordower JH , Olanow CW ((2010) ) Gene delivery of AAV2-neurturin for Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol 9: , 1164–1172. |
[45] | Chu Y , Bartus RT , Manfredsson FP , Olanow CW , Kordower JH ((2020) ) Long-term post-mortem studies following neurturin gene therapy in patients with advanced Parkinson’s disease. Brain 143: , 960–975. |
[46] | Sidorova YA , Bespalov MM , Wong AW , Kambur O , Jokinen V , Lilius TO , Suleymanova I , Karelson G , Rauhala PV , Karelson M , Osborne PB , Keast JR , Kalso EA , Saarma M ((2017) ) A novel small molecule GDNF receptor RET agonist, BT13, promotes neurite growth from sensory neurons in vitro and attenuates experimental neuropathy in the rat. Front Pharmacol 8: , 365. |
[47] | Mahato AK , Kopra J , Renko J-M , Visnapuu T , Korhonen I , Pulkkinen N , Bespalov MM , Domanskyi A , Ronken E , Piepponen TP , Voutilainen MH , Tuominen RK , Karelson M , Sidorova YA , Saarma M ((2020) ) Glial cell line-derived neurotrophic factor receptor Rearranged during transfection agonist supports dopamine neurons in Vitro and enhances dopamine release In Vivo. Mov Disord 35: , 245–255. |
[48] | Schuchardt A , D’Agati V , Pachnis V , Costantini F ((1996) ) Renal agenesis and hypodysplasia in ret-k- mutant mice result from defects in ureteric bud development. Development 122: , 1919–1929. |
[49] | Eketjäll S , Fainzilber M , Murray-Rust J , Ibáñez CF ((1999) ) Distinct structural elements in GDNF mediate binding to GFRalpha1 and activation of the GFRalpha1-c-Ret receptor complex. EMBO J 18: , 5901–5910. |
[50] | Sidorova YA , Mätlik K , Paveliev M , Lindahl M , Piranen E , Milbrandt J , Arumäe U , Saarma M , Bespalov MM ((2010) ) Persephin signaling through GFRalpha1: The potential for the treatment of Parkinson’s disease. Mol Cell Neurosci 44: , 223–232. |
[51] | Ivanova L , Tammiku-Taul J , Sidorova Y , Saarma M , Karelson M ((2018) ) Small-molecule ligands as potential GDNF family receptor agonists. ACS Omega 3: , 1022–1030. |
[52] | Planken A , Porokuokka LL , Hänninen A-L , Tuominen RK , Andressoo J-O ((2010) ) Medium-throughput computer aided micro-island method to assay embryonic dopaminergic neuron cultures in vitro. J Neurosci Methods 194: , 122–131. |
[53] | Jones TR , Kang IH , Wheeler DB , Lindquist RA , Papallo A , Sabatini DM , Golland P , Carpenter AE ((2008) ) CellProfiler Analyst: Data exploration and analysis software for complex image-based screens. BMC Bioinformatics 9: , 482. |
[54] | Penttinen A-M , Suleymanova I , Albert K , Anttila J , Voutilainen MH , Airavaara M ((2016) ) Characterization of a new low-dose 6-hydroxydopamine model of Parkinson’s disease in rat. J Neurosci Res 94: , 318–328. |
[55] | Paxinos G , Watson C ((1998) ) The rat brain in stereotaxic coordinates, Academic Press, Inc., San Diego. |
[56] | Ungerstedt U , Arbuthnott GW ((1970) ) Quantitative recording of rotational behavior in rats after 6-hydroxy-dopamine lesions of the nigrostriatal dopamine system. Brain Res 24: , 485–493. |
[57] | Schallert T , Fleming SM , Leasure JL , Tillerson JL , Bland ST ((2000) ) CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology 39: , 777–787. |
[58] | Tillerson JL , Cohen AD , Philhower J , Miller GW , Zigmond MJ , Schallert T ((2001) ) Forced limb-use effects on the behavioral and neurochemical effects of 6-hydroxydopamine. J Neurosci 21: , 4427–4435. |
[59] | Bäck S , Raki M , Tuominen RK , Raasmaja A , Bergström K , Männistö PT ((2013) ) High correlation between [123I]β-CIT SPECT/CT imaging and post-mortem immunohistochemical findings in the evaluation of lesions induced by 6-OHDA in rats. EJNMMI Res 3: , 46. |
[60] | Kopra J , Vilenius C , Grealish S , Härma M-A , Varendi K , Lindholm J , Castrén E , Võikar V , Björklund A , Piepponen TP , Saarma M , Andressoo J-O ((2015) ) GDNF is not required for catecholaminergic neuron survival in vivo. Nat Neurosci 18: , 319–322. |
[61] | Penttinen A-M , Parkkinen I , Blom S , Kopra J , Andressoo J-O , Pitkänen K , Voutilainen MH , Saarma M , Airavaara M ((2018) ) Implementation of deep neural networks to count dopamine neurons in substantia nigra. Eur J Neurosci 48: , 2354–2361. |
[62] | West MJ , Slomianka L , Gundersen HJ ((1991) ) Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec 231: , 482–497. |
[63] | Voutilainen MH , Bäck S , Pörsti E , Toppinen L , Lindgren L , Lindholm P , Peränen J , Saarma M , Tuominen RK ((2009) ) Mesencephalic astrocyte-derived neurotrophic factor is neurorestorative in rat model of Parkinson’s disease. J Neurosci 29: , 9651–9659. |
[64] | Voutilainen MH , Bäck S , Peränen J , Lindholm P , Raasmaja A , Männistö PT , Saarma M , Tuominen RK ((2011) ) Chronic infusion of CDNF prevents 6-OHDA-induced deficits in a rat model of Parkinson’s disease. Exp Neurol 228: , 99–108. |
[65] | Renko J-M , Voutilainen MH , Visnapuu T , Sidorova YA , Tuominen RK ((2020) ) GDNF receptor agonist alleviates motor imbalance in unilateral 6- hydroxydopamine model of Parkinson’s disease. Front Neurol and Neurosci Res 1: , 11. |
[66] | Tsui-Pierchala BA , Ahrens RC , Crowder RJ , Milbrandt J , Johnson EM ((2002) ) The long and short isoforms of Ret function as independent signaling complexes. J Biol Chem 277: , 34618–34625. |
[67] | Paratcha G , Ledda F , Baars L , Coulpier M , Besset V , Anders J , Scott R , Ibáñez CF ((2001) ) Released GFRalpha1 potentiates downstream signaling, neuronal survival, and differentiation via a novel mechanism of recruitment of c-Ret to lipid rafts. Neuron 29: , 171–184. |
[68] | Horger BA , Nishimura MC , Armanini MP , Wang L-C , Poulsen KT , Rosenblad C , Kirik D , Moffat B , Simmons L , Johnson E , Milbrandt J , Rosenthal A , Bjorklund A , Vandlen RA , Hynes MA , Phillips HS ((1998) ) Neurturin exerts potent actions on survival and function of midbrain dopaminergic neurons. J Neurosci 18: , 4929–4937. |
[69] | Gasmi M , Brandon EP , Herzog CD , Wilson A , Bishop KM , Hofer EK , Cunningham JJ , Printz MA , Kordower JH , Bartus RT ((2007) ) AAV2-mediated delivery of human neurturin to the rat nigrostriatal system: Long-term efficacy and tolerability of CERE-120 for Parkinson’s disease. Neurobiol Dis 27: , 67–76. |
[70] | Georgievska B , Kirik D , Björklund A ((2002) ) Aberrant sprouting and downregulation of tyrosine hydroxylase in lesioned nigrostriatal dopamine neurons induced by long-lasting overexpression of glial cell line derived neurotrophic factor in the striatum by lentiviral gene transfer. Exp Neurol 177: , 461–474. |
[71] | Bartus RT , Johnson EM ((2017) ) Clinical tests of neurotrophic factors for human neurodegenerative diseases, part 2: Where do we stand and where must we go next? Neurobiol Dis 97: , 169–178. |
[72] | Kurokawa K , Iwashita T , Murakami H , Hayashi H , Kawai K , Takahashi M ((2001) ) Identification of SNT/FRS2 docking site on RET receptor tyrosine kinase and its role for signal transduction. Oncogene 20: , 1929–1938. |
[73] | Chiariello M , Visconti R , Carlomagno F , Melillo RM , Bucci C , de Franciscis V , Fox GM , Jing S , Coso OA , Gutkind JS , Fusco A , Santoro M ((1998) ) Signalling of the Ret receptor tyrosine kinase through the c-Jun NH2-terminal protein kinases (JNKs): Evidence for a divergence of the ERKs and JNKs pathways induced by Ret. Oncogene 16: , 2435–2445. |
[74] | Hayashi H , Ichihara M , Iwashita T , Murakami H , Shimono Y , Kawai K , Kurokawa K , Murakumo Y , Imai T , Funahashi H , Nakao A , Takahashi M ((2000) ) Characterization of intracellular signals via tyrosine 1062 in RET activated by glial cell line-derived neurotrophic factor. Oncogene 19: , 4469–4475. |
[75] | Jhiang SM ((2000) ) The RET proto-oncogene in human cancers. Oncogene 19: , 5590–5597. |
[76] | Hyman C , Hofer M , Barde YA , Juhasz M , Yancopoulos GD , Squinto SP , Lindsay RM ((1991) ) BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 350: , 230–232. |
[77] | Knüsel B , Winslow JW , Rosenthal A , Burton LE , Seid DP , Nikolics K , Hefti F ((1991) ) Promotion of central cholinergic and dopaminergic neuron differentiation by brain-derived neurotrophic factor but not neurotrophin 3. Proc Natl Acad Sci U S A 88: , 961–965. |
[78] | Piltonen M , Planken A , Leskelä O , Myöhänen TT , Hänninen A-L , Auvinen P , Alitalo K , Andressoo J-O , Saarma M , Männistö PT ((2011) ) Vascular endothelial growth factor C acts as a neurotrophic factor for dopamine neurons in vitro and in vivo. Neuroscience 192: , 550–563. |
[79] | Falk T , Yue X , Zhang S , McCourt AD , Yee BJ , Gonzalez RT , Sherman SJ ((2011) ) Vascular endothelial growth factor-B is neuroprotective in an in vivo rat model of Parkinson’s disease. Neurosci Lett 496: , 43–47. |
[80] | Pitzer MR , Sortwell CE , Daley BF , McGuire SO , Marchionini D , Fleming M , Collier TJ ((2003) ) Angiogenic and neurotrophic effects of vascular endothelial growth factor (VEGF165): Studies of grafted and cultured embryonic ventral mesencephalic cells. Exp Neurol 182: , 435–445. |
[81] | Saarenpää T , Kogan K , Sidorova Y , Mahato AK , Tascón I , Kaljunen H , Yu L , Kallijärvi J , Jurvansuu J , Saarma M , Goldman A ((2017) ) Zebrafish GDNF and its co-receptor GFRα1 activate the human RET receptor and promote the survival of dopaminergic neurons in vitro. PLoS One 12: , e0176166. |
[82] | Kopra JJ , Panhelainen A , Af Bjerkén S , Porokuokka LL , Varendi K , Olfat S , Montonen H , Piepponen TP , Saarma M , Andressoo J-O ((2017) ) Dampened amphetamine-stimulated behavior and altered dopamine transporter function in the absence of brain GDNF. J Neurosci 37: , 1581–1590. |
[83] | Zhu S , Zhao C , Wu Y , Yang Q , Shao A , Wang T , Wu J , Yin Y , Li Y , Hou J , Zhang X , Zhou G , Gu X , Wang X , Bustelo XR , Zhou J ((2015) ) Identification of a Vav2-dependent mechanism for GDNF/Ret control of mesolimbic DAT trafficking. Nat Neurosci 18: , 1084–1093. |
[84] | Littrell OM , Pomerleau F , Huettl P , Surgener S , McGinty JF , Middaugh LD , Granholm A-C , Gerhardt GA , Boger HA ((2012) ) Enhanced dopamine transporter activity in middle-aged Gdnf heterozygous mice. Neurobiol Aging 33: , 427.e1–427.e14. |
[85] | Winkler C , Sauer H , Lee CS , Björklund A ((1996) ) Short-term GDNF treatment provides long-term rescue of lesioned nigral dopaminergic neurons in a rat model of Parkinson’s disease. J Neurosci 16: , 7206–7215. |
[86] | Kirik D , Rosenblad C , Björklund A ((1998) ) Characterization of behavioral and neurodegenerative changes following partial lesions of the nigrostriatal dopamine system induced by intrastriatal 6-hydroxydopamine in the rat. Exp Neurol 152: , 259–277. |
[87] | Kozlowski DA , Connor B , Tillerson JL , Schallert T , Bohn MC ((2000) ) Delivery of a GDNF gene into the substantia nigra after a progressive 6-OHDA lesion maintains functional nigrostriatal connections. Exp Neurol 166: , 1–15. |
[88] | Tronci E , Shin E , Björklund A , Carta M ((2012) ) Amphetamine-induced rotation and L-DOPA-induced dyskinesia in the rat 6-OHDA model: A correlation study. Neurosci Res 73: , 168–172. |
[89] | Kumar A , Kopra J , Varendi K , Porokuokka LL , Panhelainen A , Kuure S , Marshall P , Karalija N , Härma M-A , Vilenius C , Lilleväli K , Tekko T , Mijatovic J , Pulkkinen N , Jakobson M , Jakobson M , Ola R , Palm E , Lindahl M , Strömberg I , Võikar V , Piepponen TP , Saarma M , Andressoo J-O ((2015) ) GDNF overexpression from the native locus reveals its role in the nigrostriatal dopaminergic system function. PLoS Genet 11: , e1005710. |
[90] | Rommelfanger KS , Wichmann T ((2010) ) Extrastriatal dopaminergic circuits of the basal ganglia. Front Neuroanat 4: , 139. |
[91] | Timmerman W , Abercrombie ED ((1996) ) Amphetamine-induced release of dendritic dopamine in substantia nigra pars reticulata: D1-mediated behavioral and electrophysiological effects. Synapse 23: , 280–291. |
[92] | Paratcha G , Ibáñez CF , Ledda F ((2006) ) GDNF is a chemoattractant factor for neuronal precursor cells in the rostral migratory stream. Mol Cell Neurosci 31: , 505–514. |
[93] | Chalazonitis A , Rothman TP , Chen J , Gershon MD ((1998) ) Age-dependent differences in the effects of GDNF and NT-3 on the development of neurons and glia from neural crest-derived precursors immunoselected from the fetal rat gut: Expression of GFRalpha-1 in vitro and in vivo. Dev Biol 204: , 385–406. |
[94] | Gianino S , Grider JR , Cresswell J , Enomoto H , Heuckeroth RO ((2003) ) GDNF availability determines enteric neuron number by controlling precursor proliferation. Development 130: , 2187–2198. |
[95] | Revilla S , Ursulet S , Álvarez-López MJ , Castro-Freire M , Perpiñá U , García-Mesa Y , Bortolozzi A , Giménez-Llort L , Kaliman P , Cristòfol R , Sarkis C , Sanfeliu C ((2014) ) Lenti-GDNF gene therapy protects against Alzheimer’s disease-like neuropathology in 3xTg-AD mice and MC65 cells. CNS Neurosci Ther 20: , 961–972. |
[96] | Saarma M , Goldman A ((2017) ) Obesity: Receptors identified for a weight regulator. Nature 550: , 195–197. |
[97] | Viisanen H , Nuotio U , Kambur O , Mahato AK , Jokinen V , Lilius T , Li W , Santos HA , Karelson M , Rauhala P , Kalso E , Sidorova YA ((2020) ) Novel RET agonist for the treatment of experimental neuropathies. Mol Pain 16: , 1744806920950866. |
[98] | Turconi G , Kopra J , Võikar V , Kulesskaya N , Vilenius C , Piepponen TP , Andressoo J-O ((2020) ) Chronic 2-fold elevation of endogenous GDNF levels is safe and enhances motor and dopaminergic function in aged mice. Mol Ther Methods Clin Dev 17: , 831–842. |
[99] | Tokugawa K , Yamamoto K , Nishiguchi M , Sekine T , Sakai M , Ueki T , Chaki S , Okuyama S ((2003) ) XIB4035, a novel nonpeptidyl small molecule agonist for GFRalpha-1. Neurochem Int 42: , 81–86. |
[100] | Hedstrom KL , Murtie JC , Albers K , Calcutt NA , Corfas G ((2014) ) Treating small fiber neuropathy by topical application of a small molecule modulator of ligand-induced GFRα/RET receptor signaling. Proc Natl Acad Sci U S A 111: , 2325–2330. |

