
NEAT1 on the Field of Parkinson’s Disease: Offense, Defense, or a Player on the Bench?
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disease worldwide. Considering the devastating symptoms, high prevalence, and lack of definitive diagnostic test, there is an urgent need to identify possible biomarkers and new therapeutic targets. Genes identified and/or proposed to be linked to PD encode proteins that fulfill diverse roles in cellular functions. There is a growing interest in identifying common traits which lead to the disease. Long non-coding RNAs have recently emerged as possible regulatory hubs of complex molecular changes affecting PD development. Among them, NEAT1 has attracted particular interest. It is a major component and the initiator of nuclear paraspeckles, thus regulating transcription and modifying protein functions. This review summarizes data available on the role of NEAT1 in PD. NEAT1 upregulation in PD has repeatedly been reported, however, whether this is part of a protective or a damaging mechanism is still a topic of debate. It has been proposed that NEAT1 propagates PD via its interaction with PINK1 and several micro RNAs and by modulating SNCA expression. On the other hand, findings of NEAT1 acting as a bona fide LRRK2 inhibitor argue for its protective role. These contradictory results could be due to the different disease models implemented. This calls attention to the difficulties posed by the complex patho-mechanisms of neurodegenerative disorders and the limitations of disease models. However, the potential of NEAT1 as a biomarker and as a therapeutic target for PD highly warrants further research to elucidate its exact role in this neurodegenerative disorder.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disease, affecting approximately 1-2% of the population over the age of 65 [1]. The prevalence of the disease increases exponentially with age, causing millions of deaths each year [2]. The characteristic motor symptoms of PD are often accompanied by various non-motor symptoms, exacerbating disease severity. In the absence of an early diagnostic test, PD diagnosis is based on the cardinal motor symptoms. However, by the time these manifest, the majority of the dopaminergic neurons in the substantia nigra have been irreversibly lost [3–5]. Despite the intensive research focusing on development of disease-modifying therapies [6], so far no effective treatment is available. Given the devastating symptoms, high prevalence, and lack of a specific diagnostic test, there is an urgent need to identify possible biomarkers and new therapeutic targets for PD.
PD is a complex multifactorial disease, the exact patho-mechanism of which has yet to be fully elucidated. Besides various environmental and lifestyle factors identified as triggers and/or facilitators of the disease [7], several genetic alterations have been found to be related to the disorder. In addition to 21 PARK genes described in the human genome as potential direct culprits of the disease [8], genetic variants of 26 loci have been proposed to be disease risk modifiers [9, 10]. These genes encode proteins that fulfill roles in diverse cellular functions, such as synaptic transmission, vesicle transport, protein transport and degradation, autophagy, mitochondrion maintenance and energy homeostasis [11]. There is a growing interest in identifying common traits behind the diverse mechanisms causing malfunctions which lead to PD.
Due to their versatile roles in cellular functions, long non-coding RNAs (lncRNAs) have recently emerged as possible regulatory hubs of complex molecular changes affecting PD development. lncRNAs are RNA polymerase II transcripts over 200 nucleotides in length, without long open reading frames. They are frequently polyadenylated, alternatively spliced and capped, thus having an mRNA-like structure [12]. lncRNAs have gained attention in relation to neurodegenerative diseases due to the diverse mechanisms by which they can affect cellular homeostasis [13]. lncRNAs are known to exert regulatory roles on gene expression by modulating histone post-translational modifications and transcription factor activities, participating directly in post-transcriptional mRNA modifications, acting as ceRNAs (competing endogenous RNAs) that can sponge micro RNAs (miRNAs) and possibly by several other mechanisms acting at translational and post-translation levels (for a review, see [12, 14]).
NEAT1 lncRNA has attracted particular interest in the past few years since its levels have been shown to be altered in neurodegenerative diseases (reviewed in [15]). The possibility of a direct relation between NEAT1 and PD has been strengthened by recent findings on NEAT1 effects on mitochondrial function [16], detection of elevated NEAT1 levels in postmortem PD brain samples [17, 18] and recently our research group detected elevated NEAT1 levels also in the peripheral blood of PD patients [19]. However, the questions whether a change in NEAT1 level is in causal relationship with alleviation or aggravation of PD, or alternatively, NEAT1 lncRNA is a bystander in PD pathogenesis, without being actively involved in the disease course, are still unanswered. In this review we summarize recently published data related to the possible role of NEAT1 in PD. Similarly to the seemingly contradictory views which attribute both oncogenic and tumor-suppressor roles to NEAT1 lncRNA in cancer [20, 21], recently published data suggest both protective and enhancing roles for NEAT1 in neurodegeneration. We critically review these reports with particular attention to PD in order to facilitate a clearer view on the possible involvement of this lncRNA in the disease. We hope that calling attention to the topic will help clarify contrasting data and raise questions for further research.
NEAT1: DISCOVERY, GENE STRUCTURE, EXPRESSION
NEAT1 (Nuclear Enriched Abundant Transcript 1, later changed to Nuclear Paraspeckle Assembly Transcript) lncRNA was first described in 2007 as a highly abundant nuclear RNA [22]. In human, NEAT1 is transcribed from the multiple endocrine neoplasia (MEN) type I locus on the long arm of chromosome 11 [23]. Transcription results in two NEAT1 isoforms: the shorter NEAT1_1 (alias MENepsilon) is 3 684 nucleotides, while the longer NEAT1_2 (alias MENbeta) is 22 743 nucleotides. For simplicity we will refer to the former as NEAT1S and to the latter as NEAT1L. NEAT1 related genes are specific to mammals [24] and the gene sequence is well conserved across mammalian species [25], which is an uncommon feature of lncRNAs is general [22]. Mouse NEAT1 isoforms are smaller than the human ones (3.7 and 20 kb), but are in similar relation to each other as the human ones (see more on this below).
The two NEAT1 isoforms are transcribed by RNA polymerase II from the same promoter under the same transcriptional control. NEAT1S is produced by early 3’end processing of the transcript at a canonical polyadenylation site. NEAT1L results from suppression of polyadenylation at this site. Its 3’ end is formed without poly(A) tail by RNase P cleavage at a tRNA-like structure [26, 27]. Consequently, the two isoforms overlap over the full length of NEAT1S that corresponds to the 5’ end sequence of NEAT1L. The proportion of the two NEAT1 isoforms produced is determined through the regulation of poly(A) addition; however, it remains to be elucidated how this process is linked to cell homeostasis.
The shorter NEAT1 isoform is generally observed in higher quantities and in a wider range of tissues. Nonetheless, the function of NEAT1S is less clear compared to that of NEAT1L which is indisputably the major structural component of paraspeckles. Paraspeckles are subnuclear ribonucleoprotein complexes within the interchromatin space in mammalian cells [28, 29]. These complexes are assembled from RNAs and various proteins many of which have RNA binding affinity. Paraspeckles play roles in regulating transcription and RNA processing by several mechanisms which include retaining RNA and proteins, modulating RNA editing and splicing and acting as sponges for miRNAs (reviewed in [30]). Knockdown of NEAT1L production results in paraspeckle elimination even in the presence of intact NEAT1S [31]. NEAT1L folds end-to-end within paraspeckles with 5’ and 3’ ends of the lncRNA localizing on the periphery while the core is positioned in the center of the structure. As the 5’ ends of the two NEAT1 isoforms are identical, this may suggest that the short isoform is also localized in the periphery of paraspeckles [32]. However, recent findings argue against NEAT1S as a major paraspeckle component, instead revealing the short isoform to be localized in foci termed ‘micro-speckles’ [32–34]. Mice lacking the long isoform of NEAT1 show defects in female reproductive tissue development while absence of the short isoform does not cause any obvious external or histological abnormalities [35, 36]. These findings raised the possibility of NEAT1S being a by-product without any specific role [36]. However, the observations that NEAT1L and NEAT1S accumulate differently in and have different effects on some cancer types [21, 37–39] and that overproduction of NEAT1S increases resistance of cells to oxidative stress [40] refute this notion. The observation that NEAT1S is more conserved in evolution and is generally more abundant, together with it being detected outside of paraspeckles [33] may also serve as an indirect argument for an as yet unidentified paraspeckle-independent function of this isoform.
While there is a general consensus on the production of the two NEAT1 variants, the existence of further isoform(s) is less clear. The Human Genome Ensemble (GRCH38.p13) depicts nine NEAT1 splice variants. Some of these are “annotated manually” while others are products of the “manually supervised computational pipeline”. These transcripts bear small differences in their 5’regions, due to five short putative introns. As there are no reported RNA mapping results to verify the removal of these, it remains open if any of the depicted NEAT1 splice variants deserve particular attention. Among the few reports on NEAT1 isoforms Chowdhury et al. mention, 3 out of 8 NEAT1 variants to be upregulated in human endothelial cells after LPS (lipopolysaccharide) treatment [41] and Kessler et al. found differences in the expression levels of 3 variants (NEAT1-201, NEAT1-202/v2, and NEAT1-205) by comparing NEAT1 RNAs in hepato-cellular carcinoma and normal tissue samples [39].
Data on NEAT1 lncRNA expression, tissue distribution and function have been obtained primarily from mouse models which permit genome editing of the gene and from cancer related studies using tumor samples and various human cell lines. Due to space constraints these will not be reviewed here; instead we call attention only to data which exemplify the diverse, frequently contrasting effects attributed to NEAT1 lncRNAs. In the following sections we review very recent data related to possible NEAT1 functions in neurodegenerative disorders and models of these focusing primarily on PD. Excellent recent reviews on the regulation of NEAT1 lncRNA expression and the contribution of NEAT1 to tumor development can be found in [21, 42, 43].
CELLULAR FUNCTIONS AFFECTED BY NEAT1
Shortly after the description of NEAT1, it was demonstrated that the lncRNA localizes to specific nuclear ribonucleoprotein structures. Subsequent studies proved that NEAT1L knockdown leads to paraspeckle disintegration while overexpression increases paraspeckle abundance; furthermore details on the folding of the RNA within paraspeckles as well as on the protein components of the complex were revealed [32, 44]. However, the involvement of NEAT1S in paraspeckles remains disputed. NEAT1’s role in paraspeckle scaffolding imply an effect on cellular functions: paraspeckles regulate transcription and RNA maturation via accumulation of protein factors. The amount of paraspeckles affects the retention of A-I edited RNAs, mitoRNAs (mitochondrial protein coding RNAs) and miRNAs. Changes in the level of NEAT1 modulate functions via these. A further mechanism of NEAT1 action which may or may not be associated with paraspeckles is acting as ceRNA by sponging miRNAs. This seems to be a major means by which NEAT1 affects carcinogenesis (reviewed in [21]).
Paraspeckles are dispensable under normal laboratory conditions but play essential roles when cells are placed under stress. In accord with this several cellular stressors enhance NEAT1 expression and paraspeckle formation. This is well reflected by the multitude of transcription factors known to affect NEAT1 expression. A comprehensive review on this topic was recently published by [43].
NEAT1 IN PARKINSON’S DISEASE
Altered expression of NEAT1 has been reported in various neurodegenerative diseases (reviewed in [15]), among them in PD. Elevated NEAT1 levels were reported in human postmortem brain samples of various brain areas, such as in the substantia nigra and anterior cingulate gyrus [17, 18]. Upregulation of the lncRNA was found to increase with progression of the disease [17]. Besides the central nervous system (CNS), elevated NEAT1 levels were also reported in the peripheral blood of PD patients [19].
In this review we summarize data available on the role of NEAT1 in PD pathogenesis obtained from in vitro and in vivo models of the disease (Fig. 1). As demonstrated by results shown below, various stressors lead to the upregulation of NEAT1 RNA; however, the role that NEAT1 plays in PD is still a topic of debate. Some of the data indicate that NEAT1 upregulation has a detrimental effect and accelerates disease progression. Other observations suggest a compensatory mechanism by which the RNA might promote cell survival and arrest disease pathology (Figs. 1–4). Finally, it may be that NEAT1 has no significant effect on PD pathogenesis and the observed changes in RNA merely reflect a bystander effect on NEAT1 in the disease process. In the following sections we summarize available data supporting either the protective or the harmful role of NEAT1 upregulation in the course of PD. Table 1 and Fig. 1 show brief summaries of reported results obtained by alterations of NEAT1 lncRNA levels using different PD models and the mechanisms assumed, respectively. Figs. 2–4 show observed effects of NEAT1 highlighting reported data in respects of PD models (animal and cellular models: Fig. 2 vs. Fig. 3) and toxins used (Fig. 3 vs. Fig. 4).
Fig. 1
Proposed mechanisms by which NEAT1 affects the course of PD. For a detailed description please see the corresponding sections of the text. NEAT1, Nuclear Paraspeckle Assembly Transcript 1; PINK1, protein phosphatase and tensin homolog (PTEN)-induced kinase 1; SNCA, Alpha-synuclein (gene); GJB1, Gap junction beta-1 (gene); α-syn, Alpha-synuclein (protein); NLRP3, NOD-, LRR- and pyrin domain-containing protein; RAB3IP, RAB3A interacting protein (gene); LRRK2, Leucine-rich repeat kinase 2.
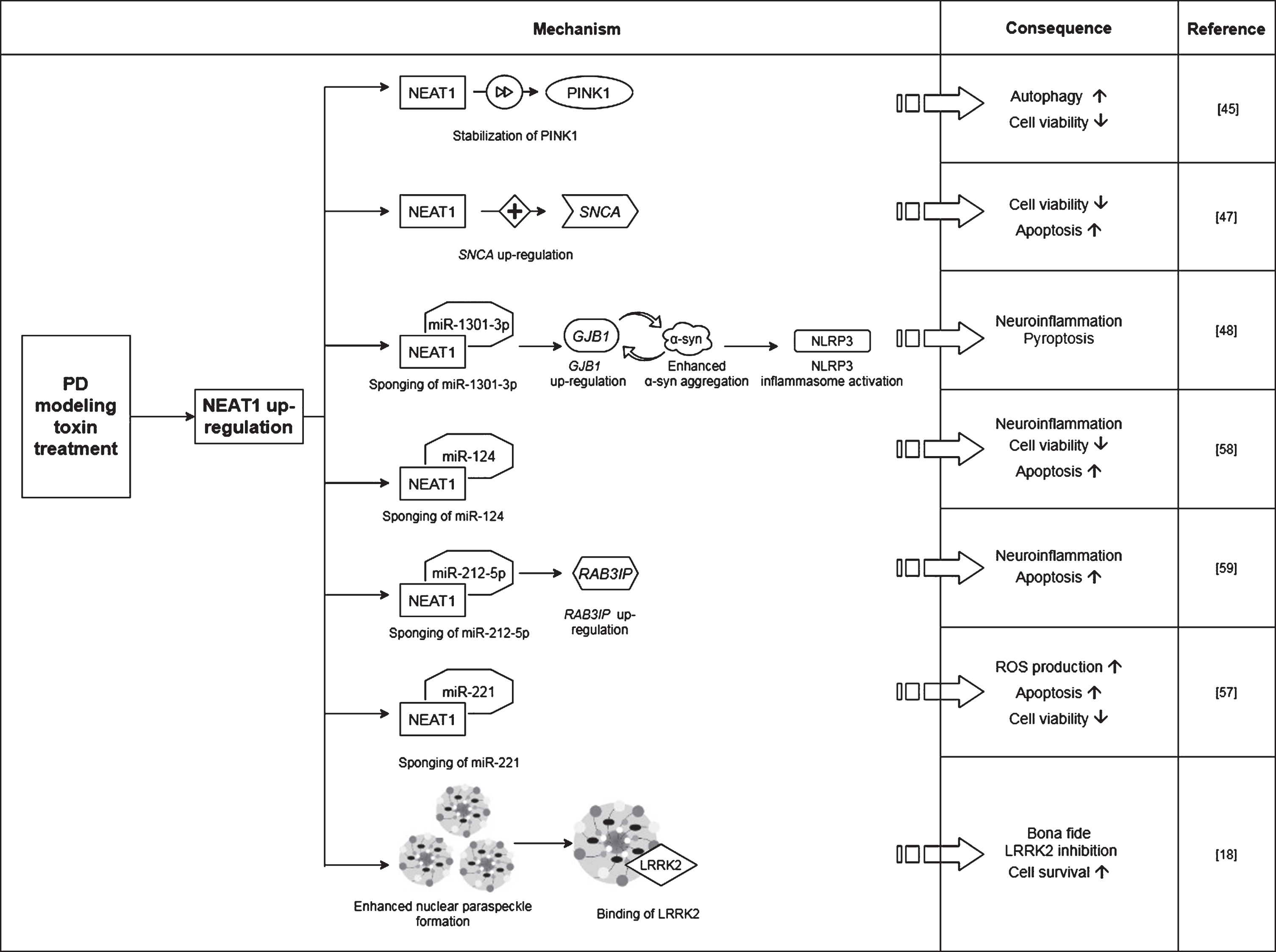
Fig. 2
Observed effects of NEAT1 in animal models of PD. For a detailed description please see the corresponding sections of the text. MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NEAT1, Nuclear Paraspeckle Assembly Transcript 1; TH, Tyrosine hydroxylase.
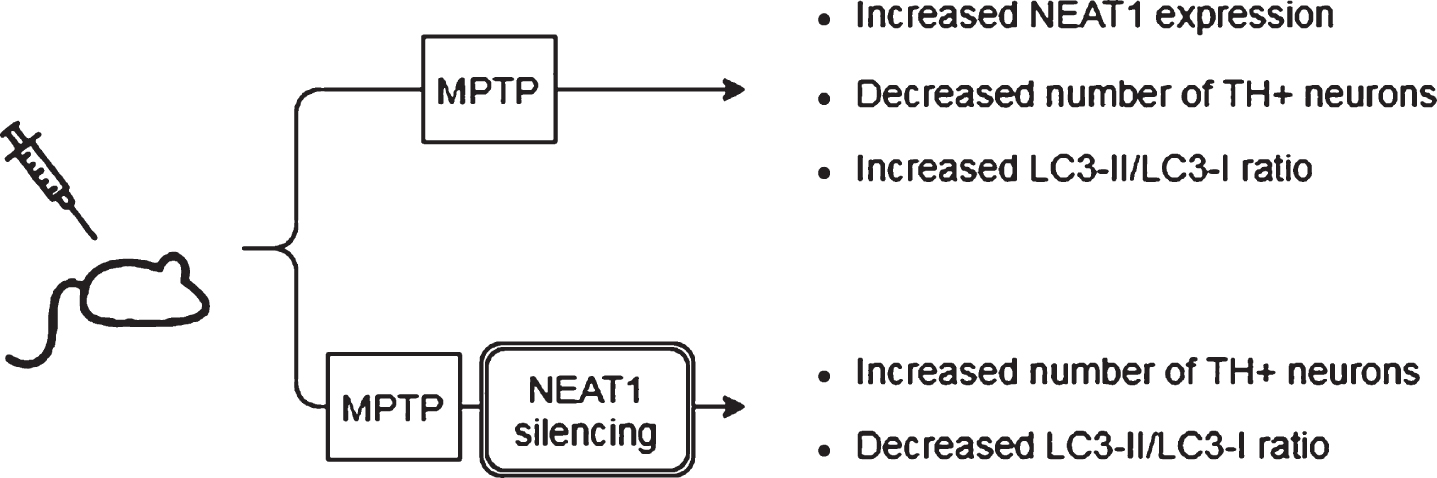
Fig. 3
Observed effects of NEAT1 in the MPP+cell model of PD. For a detailed description please see the corresponding sections of the text. MPP+, 1-methyl-4-phenylpyridinium; NEAT1, Nuclear Paraspeckle Assembly Transcript 1; PINK1, protein phosphatase and tensin homolog (PTEN)-induced kinase 1; SNCA, Alpha-synuclein (gene); NLRP3, NOD-, LRR- and pyrin domain-containing protein; GJB1, Gap junction beta-1; RAB3IP, RAB3A interacting protein (gene); ROS, Reactive oxygen species; SOD, Superoxide dismutase; LDH, Lactate dehydrogenase; IL-1β, interleukin-1β; IL-6, interleukin-6; TNF-α, Tumor necrosis factor α.
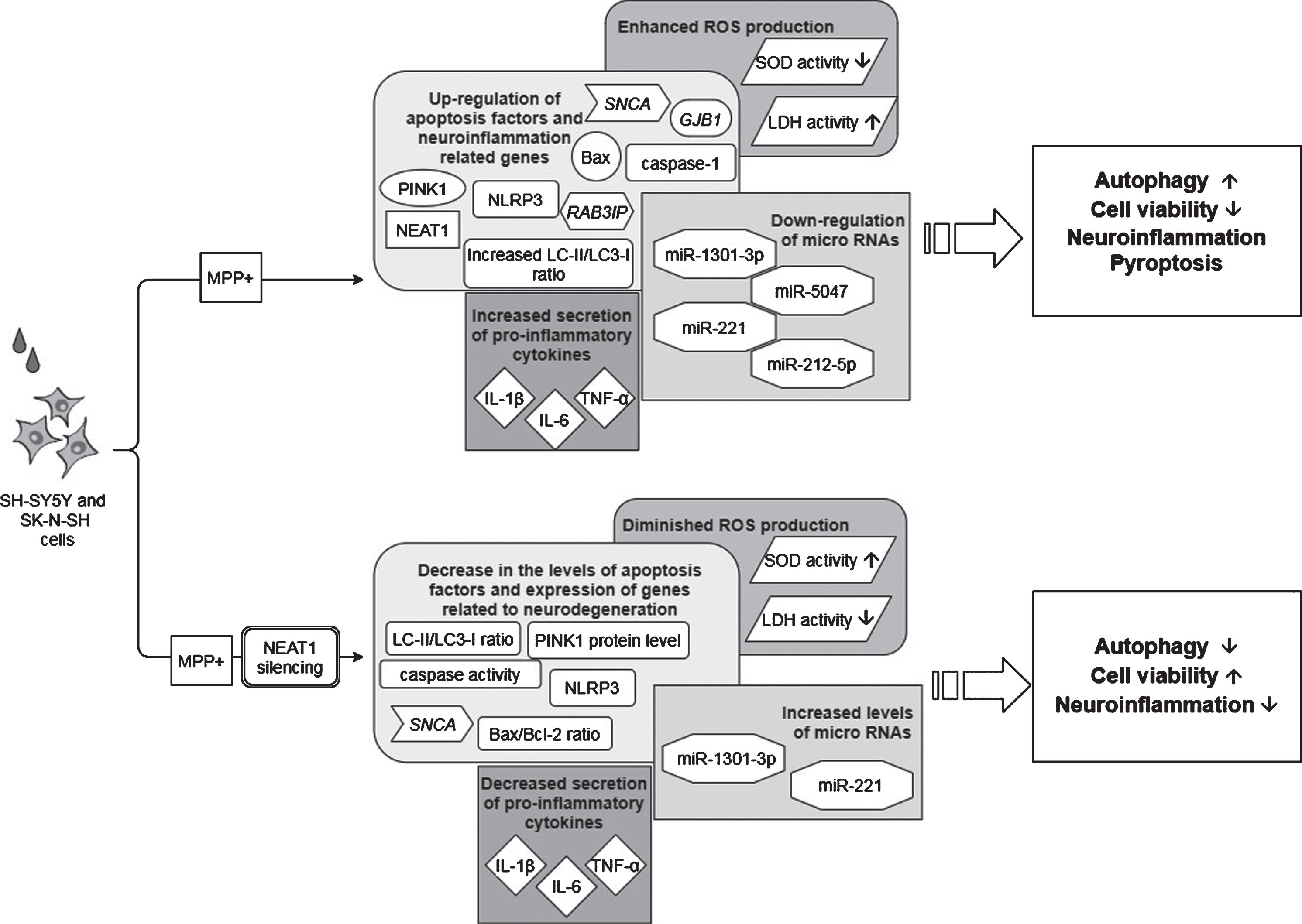
Fig. 4
Observed effects of NEAT1 in the PQ and tBHP cell models of PD. For a detailed description please see the corresponding sections of the text. PQ, Paraquat; tBHP, tert-Butyl hydroperoxide; NEAT1, Nuclear Paraspeckle Assembly Transcript 1; ROS, Reactive oxygen species.
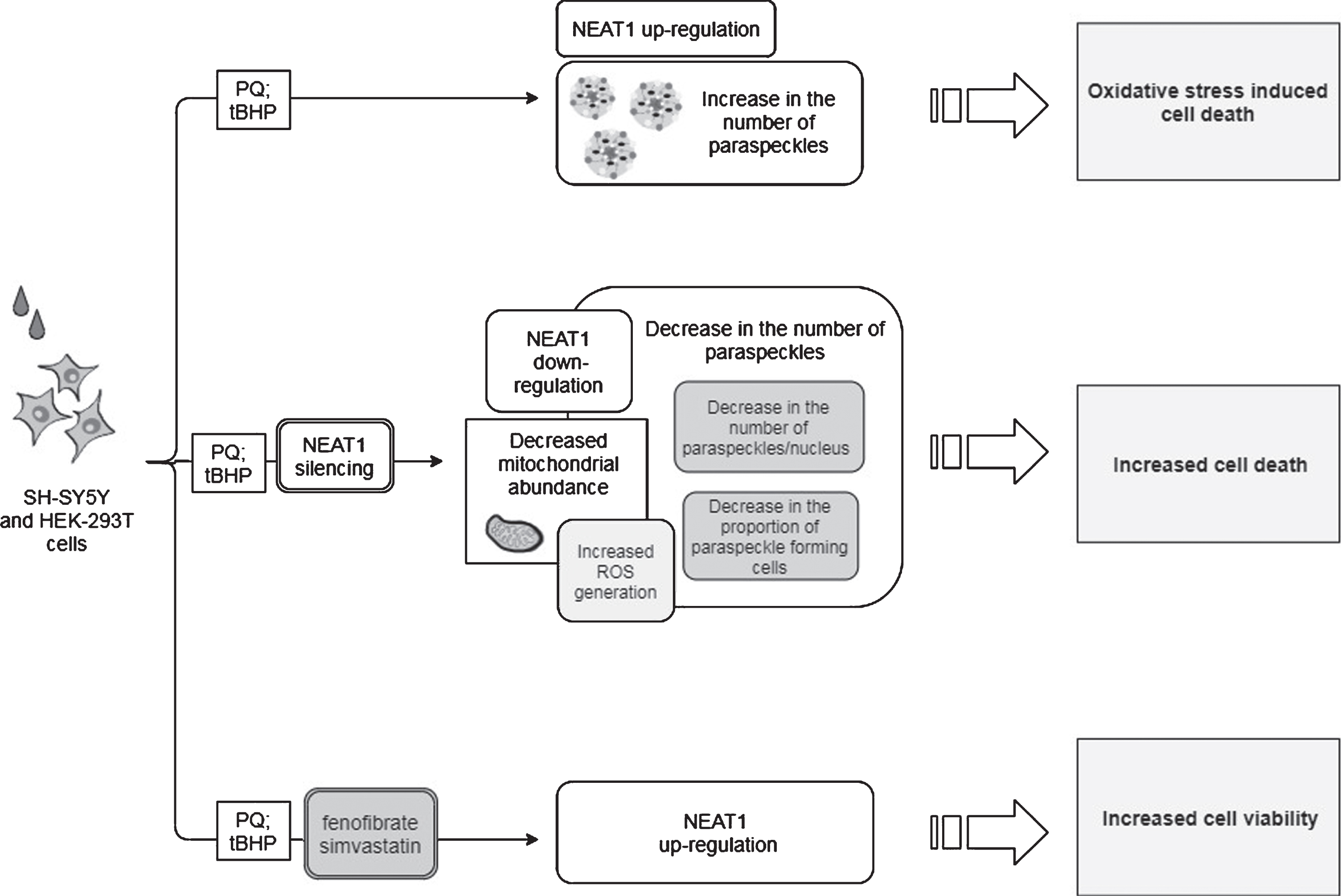
Table 1
Reported results obtained by alterations of NEAT1 lncRNA levels using different PD models and the mechanisms assumed
| Model organism | Toxin | Effect of toxin | NEAT1 intervention | Effect of NEAT1 intervention | Proposed NEAT1 mode of action | Reference |
| mouse | MPTP | increase in: | NEAT1 silencing | decrease in: | Stabilizes, thus increases the level of PINK1 protein | [45] |
| - NEAT1 expression | - PINK1 protein level | |||||
| - PINK1 protein level | - LC3-II/LC3-I ratio | |||||
| - LC3-II/LC3-I ratio | ||||||
| decrease in the number of TH+neurons | increase in the number of TH+neurons | |||||
| SH-SY5Y cells | MPP+ | increase in: | NEAT1 silencing | decrease in: | ||
| - NEAT1 expression | - PINK1 protein level | |||||
| - PINK1 protein level | - LC3-II/LC3-I ratio | |||||
| - LC3-II/LC3-I ratio | ||||||
| increase in cell viability | ||||||
| mouse | MPTP | increase in NEAT1 expression decrease in the number of TH+neurons | n.a. | n.a. | Upregulation of SNCA | [47] |
| SH-SY5Y cells | MPP+ | increase in NEAT1 expression | NEAT1 silencing | decrease in: | ||
| - Bax/Bcl-2 ratio | ||||||
| - caspase activity | ||||||
| downregulation of SNCA expression | ||||||
| improved cell viability and diminished cell apoptosis | ||||||
| SH-SY5Y cells | MPP+ | enhanced expression of: | NEAT1 silencing | decreased expression of: | ||
| - SNCA | - SNCA | Sponges miR-1301-3p thus leads to enhanced GJB1 expression and consequent α-syn induced NLRP3 inflammasome activation | [48] | |||
| - GJB | - NLRP3 | |||||
| - NLR3P | - caspase-1 | |||||
| - IL-1β | - IL-1β | |||||
| - caspase-1 | ||||||
| - Bax downregulation of: | increased miR-1301-3p expression | |||||
| - miR-1301-3p | decrease in the number of apoptotic cells | |||||
| - miR-5047 | ||||||
| SH-SY5Y cells | MPP+ | upregulation of NEAT1 and downregulation of miR-221 expression | NEAT1 silencing | increased miR-221 expression | Sponges miR-221, by this enhances ROS production, LDH release and upregulation of pro-inflammatory cytokines IL-1β, IL-6 and TNFα | [57] |
| diminished ROS generation improved cell viability and decreased apoptosis | ||||||
| SH-SY5Y cells | MPP+ | NEAT1 upregulation; increased secretion of IL-1β, IL-6 and TNF-α | NEAT1 silencing | decreased levels of: | Sponges miR-124 | [58] |
| - IL-1β | ||||||
| - IL-6 | ||||||
| - TNFα | ||||||
| improved cell viability and decreased apoptosis rate | ||||||
| SK-N-SH cells | MPP+ | downregulation of miR-212-5p and upregulation of both NEAT1 and RAB3IP; decreased SOD- and increased LDH activity | NEAT1 silencing | reversed decreased SOD- and increased LDH activity | Sponges miR-212-5p thus indirectly upregulates RAB3IP expression which promotes inflammatory processes and apoptosis | [59] |
| diminished ROS production promotion of cell viability and reduction of apoptosis | ||||||
| SH-SY5Y and HEK-293T cells | PQ and tBHP | NEAT1 upregulation; increased number of paraspeckles | NEAT1 silencing | decrease in the: | NEAT1 acts as a bona fide LRRK2 inhibitor | [18] |
| - proportion of paraspeckle forming cells | ||||||
| - number of paraspeckles/nucleus | ||||||
| - number of mitochondria exacerbated oxidative stress provoked cell death | ||||||
| NEAT1 upregulation by fenofibrate and simvastatin | increased cell viability |
NEAT1, Nuclear Paraspeckle Assembly Transcript 1; PINK, phosphatase and tensin homolog (PTEN)-induced kinase 1; TH, Tyrosine hydroxylase; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; MPP+, 1-methyl-4-phenylpyridinium; GJB, gap junction beta 1; NLR3P, nucleotide oligomerization domain-like receptor protein with pyrin domain containing 3; IL-1β, interleukin-1β; IL-6, interleukin-6; TNF-α, Tumor necrosis factor α; RAB3IP, RAB3A-interacting protein; SOD, Superoxide dismutase; LDH, Lactate dehydrogenase; SNCA, Alpha-synuclein gene; ROS, Reactive oxygen species.
NEUROTOXIC NEAT1 EFFECTS
To date, seemingly more data support the notion of NEAT1 downregulation being protective against PD progression.
In a study Yan and colleagues found that treatment of mice with MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) led to a rise in the expression of NEAT1, alongside an increase in the protein levels of PINK1 (phosphatase and tensin homolog (PTEN)-induced kinase 1) and LC3-II/LC3-I ratio (LC3: Microtubule-associated protein light chain 3) in the midbrain of the animals [45]. The detrimental effect of MPTP on neuronal cell survival was demonstrated by the significant decrease in the number of TH+cells (Fig. 2). The tyrosine hydroxylase enzyme catalyzes the transformation of the amino acid L-tyrosine to L-3,4-dihydroxyphenylalanine (L-DOPA) and is a marker of dopaminergic neurons in the CNS. NEAT1 silencing significantly increased the number of TH+neurons and led to a significant decrease in PINK1 protein levels. These changes were accompanied by the elevation of LC3I and decrease of LC3-II protein levels. LC3-II is an autophagosome marker, converted from the cytoplasmic LC3-I. The membrane bound LC3-II protein plays a role in the formation and elongation of the autophagosome [46]. The reduced LC3-II/LC3-I ratio is an indicator of decreased autophagy. In vitro studies involving the SH-SY5Y cell model of the disease yielded similar results: elevated expression of NEAT1 and PINK1 protein and increased LC3-II/LC3-I ratio were detected upon MPP+(1-methyl-4-phenylpyridinium; the active metabolite of MPTP) exposure. Conversely, knockdown of the lncRNA decreased the MPP+induced high expression of PINK1 protein, reversed the change in LC3-II/LC3-I ratio and improved cell viability (Fig. 3). Intriguingly, overexpression of PINK1 reversed the beneficial effects of NEAT1 silencing on cell survival. This observation raised the possibility that NEAT1 exerts its effects in a PINK1-dependent manner. Yan and colleagues proposed that the lncRNA might bind directly to the protein and stabilize it by influencing its ubiquitination and preventing its degradation. Elevated NEAT1 level thus leads to an increase in PINK1 level [45] (Fig. 1).
Based on these in vivo and in vitro observations, Yan et al. concluded that NEAT1 upregulation is detrimental since by stabilizing PINK1 protein the lncRNA promotes autophagy [45]. In accord with this, knocking down the lncRNA proved to be protective against MPP+/MPTP induced cell loss.
The finding on the protective effect of NEAT1 silencing was strengthened by Liu and Lu [47]. In their experiments MPTP treatment of mice led to a reduction in the number of TH+cells in the brain and NEAT1 upregulation was observed in both in vivo and in vitro models of the disease (Figs. 2, 3). In MPP+treated SH-SY5Y cells knockdown of NEAT1 improved cell viability and diminished cell apoptosis as indicated by decreased Bax/Bcl-2 ratio and caspase activity. Upon NEAT1 silencing a downregulation in SNCA (Alpha-synuclein) expression was observed. Intriguingly, the beneficial effects of the knockdown of the lncRNA on cell survival and apoptosis could be reversed by overexpressing the SNCA gene. These findings suggest that upregulation of NEAT1 is harmful in the course of PD via an α-syn related mechanism (Fig. 1).
According to a more recent study by Sun et al. [48], MPP+treatment not only caused upregulation of NEAT1 but also enhanced expression of α-syn, GJB1 (Connexin32, Cx32; gap junction beta 1), NLRP3 (nucleotide oligomerization domain-like receptor protein with pyrin domain containing 3), IL-1β and apoptosis factors caspase-1 and Bax, while Bcl-2 and the miRNAs miR-1301-3p and miR-5047 were downregulated (Fig. 3).
NLRP3 containing inflammasome is a protein complex of NLRP3, ASC (Apoptosis-associated speck-like protein containing a CARD) and caspase-1, which has been identified to play a pathologic role in neuroinflammation related to various neurodegenerative diseases. Upon activation, inflammasomes provoke innate immune responses by secreting pro-inflammatory cytokines such as IL-1β and IL-18 and by promoting pyroptosis, a caspase 1-dependent cell death which contributes to the propagation of inflammation via the release of further inflammatory markers [49]. In murine models of PD NLRP3 inflammasome was found to be activated by fibrillar α-syn and by the degeneration of dopaminergic neurons themselves [50]. The cardinal role of inflammasome activation in PD pathology is supported by findings obtained both from studies involving animal models and human samples. Treatment with small molecule NLRP3 inhibitors inhibited inflammasome activation and effectively mitigated motor deficits, nigrostriatal dopaminergic degeneration, and accumulation of α-syn aggregates in various rodent models of the disease [50]. Further studies showed that absence of either NLRP3 or caspase 1 was protective against the development of PD symptoms and loss of neurons in the substantia nigra after treatment with rotenone and MPTP, respectively (reviewed in [51]).
GJB1 (alias connexin-32 (Cx32)) is a member of the gap junction connexin family. The protein has recently been reported to play a central role in the uptake of α-syn oligomeric assemblies in neurons and oligodendrocytes [52]. In vitro and in vivo models of PD demonstrated a correlation between the upregulation of GJB1 and accumulation of α-syn aggregates. The correlation is established by a positive feedback loop: in vitro studies demonstrated that GJB1 overexpressing cells are more prone to α-syn oligomer uptake, and both exposure to α-syn aggregates and overexpression of the SNCA gene leads to upregulation of GJB1. These findings underpin the role of GJB1 in the pathophysiology of PD and raise the possibility of GJB1 expression modulation as a feasible way of therapeutic intervention [52].
In the study of Sun and colleagues, NEAT1 knockdown in MPP+treated SH-SY5Y cells reversed the neurotoxic effects, as indicated by a significant decrease in the number of apoptotic cells and by the suppression of α-syn, NLRP3, caspase-1 and IL-1β expression (Fig. 3). Overexpression of α-syn reversed the anti-apoptotic effects of NEAT1 silencing. These findings are in line with the results of Liu and Lu as discussed earlier [47], namely that NEAT1 downregulation improves cell survival via decreasing α-syn expression by an as yet unidentified mechanism. Sun and colleagues proposed that the α-syn modulating ability of NEAT1 is linked to the miR-1301-3p/GJB1 pathway [48] (Fig. 1). This was based on their findings that NEAT1 downregulation led to increased miR-1301-3p expression, while inhibition of the micro RNA diminished the protective effects of NEAT1 silencing. The latter effects were demonstrated by the increased number of apoptotic cells and by the promotion of both transcription and translation of GJB1. Reporter gene assays revealed direct interactions between both NEAT1/ miR-1301-3p and miR-1301-3p/GJB1, leading to the conclusion that the lncRNA serves as an endogenous sponge for miR-1301-3p [48]. NEAT1 silencing prevents sponging of the miRNA thus miR-1301-3p can exert its inhibitory effect on GJB1 expression and through this prevent α-syn induced activation of the NLRP3 inflammasome.
Besides these observations, it has been proposed that NEAT1 affects the course of PD by another micro RNA related mechanism. miR-221 is one of the most abundant miRNAs in the human CNS, and plays an important role in promoting neurite outgrowth and neuronal differentiation [53]. A direct target of miR-221 micro RNA is PTEN (Phosphatase and tensin homolog), a tumor suppressor which has also been found to be involved in the course of various neurodegenerative diseases, such as Alzheimer’s disease (AD), amyotrophic lateral sclerosis and PD [54]. Several papers have reported miR-221 downregulation in serum samples of PD patients and proposed the possibility of this RNA serving as a biomarker of the disease [55, 56]. In a study Geng et al. found that MPP+exposure of SH-SY5Y cells resulted in upregulation of NEAT1 and downregulation of miR-221 expression in a dose- and time dependent manner [57] (Fig. 3). However, NEAT1 specific siRNA treatment increased miR-221 expression and diminished reactive oxygen species (ROS) generation, which resulted in improved cell viability and decreased apoptosis. Overexpression of miR-221 prior to MPP+treatment also diminished ROS production and was accompanied by decreased lactate dehydrogenase (LDH) release and downregulation of pro-inflammatory cytokines IL-1β, IL-6 and TNFα. Based on these observations NEAT1 was proposed to act as a molecular sponge for miR-221 (Fig. 1), and the conclusion was drawn that the beneficial effects of NEAT1 silencing could be related to decreased miR-221 sponging and a consequent higher availability of the micro RNA [57].
Regulation of neuroinflammation by NEAT1 was proposed to occur via a further mechanism. Results of experiments by Xie et al. involving the MPP+treated SH-SY5Y cell model of the disease show that silencing of NEAT1 attenuated neuroinflammation as indicated by the decreased levels of IL-1β, IL-6 and TNFα [58] (Fig. 3). In line with findings of others, NEAT1 knockdown improved cell viability and decreased apoptosis rate. RNA pull down and immunoprecipitation assays revealed a direct interaction between NEAT1 and the micro RNA miR-124. Silencing both NEAT1 and miR-124 in MPP+exposed cells led to decreased cell viability and an increase in the levels of pro-inflammatory cytokines compared to that seen in the case on NEAT1 silencing only. These observations led to the conclusion that NEAT1 regulates MPP+induced neuronal injury in a miR-124-dependent manner [58] (Fig. 1).
According to recent findings of Liu et al., NEAT1 also interacts with miR-212-5p, thus modulating the course of MPP+induced neurodegeneration via the miR-212-5p/ RAB3IP miR-1301-3p and miR-221 pathway [59] (Figs. 1, 3). Treatment of SK-N-SH cells with MPP+caused the downregulation of miR-212-5p and upregulation of both NEAT1 and RAB3IP (RAB3A-interacting protein). RAB3IP is known to be involved in various cell functions such as autophagy, cell growth and apoptosis [59]. Similarly to the observations made in the in vitro PD models mentioned previously, NEAT1 knockdown in MPP+exposed cells reversed the decreased superoxide dismutase and increased LDH activity and diminished ROS production, thus promoting cell viability and reducing the rate of apoptosis. Interestingly, overexpression of miR-212-5p also improved cell survival and alleviated MPP+linked inflammation and cytotoxicity. Based on their findings, Liu and colleagues suggested that similarly to the situation discussed above in relation to miRNAs miR-1301-3p and miR-221, NEAT1 acts as a molecular sponge for miR-212-5p as well, leading to the downregulation of this miRNA. Dual-luciferase reporter gene assays showed that miR-212-5p directly binds to RAB3IP mRNA and by this negatively regulates the expression of RAB3IP. In their study Liu and colleagues also showed that overexpression of RAB3IP promoted inflammatory processes and apoptosis of MPP+treated SK-N-SH cells. These findings led to the conclusion that a possible mechanism of the neuroprotective effect that NEAT1 knockdown shows against MPP+toxicity is the higher level of available miR-212-5p miRNA. The diminishment of miR-212-5p miRNA sponging with NEAT1 exerts beneficial effects on cell survival and apoptosis by indirectly causing the downregulation of RAB3IP.
NEAT1 IN NEUROPROTECTIVE ROLE
Opposite to the studies discussed above, the findings of Simchovitz and colleagues argue for a protective role of NEAT1 upregulation in the course of PD [18]. They reported that in postmortem substantia nigra PD samples NEAT1 was significantly upregulated compared to healthy controls. The significant difference was found to be due to the upregulation of the long NEAT1 variant, as upregulation of NEAT1L was more prominent than the expression change of both isoforms together (fold change: 2.3 and 1.7, NEAT1L and NEAT1L+S, respectively). In vitro experiments yielded similar results: upon paraquat (PQ) and tBHP (t-butyl hydroperoxide) induced oxidative stress significant NEAT1 upregulation was observed in HEK-293T and SH-SY5Y cell lines, primarily due to the increased expression of the long variant (fold change: 7 and 2.5, NEAT1L and NEAT1L+S, respectively) (Fig. 4). In murine neuronal primary cultures (GSE70368), α-syn overexpressing cells also manifested upregulated NEAT1 expression as compared to their non-overexpressing counterparts.
Investigation of PQ effect on paraspeckle formation revealed that the mean number of paraspeckles in a nucleus was increased by 60% in HEK-293T cells following PQ exposure, while no change was observed either in the number of paraspeckle forming cells or in the nuclear localization of NEAT1L. Thus, upregulation of the lncRNA upon PQ exposure seemed to be in correlation with the elevation in the number of paraspeckles. In light of this, it was proposed that in PD substantia nigra the elevated NEAT1L expression could be a cellular response to neuronal stress in order to promote enhanced formation of paraspeckles [18]. Silencing of NEAT1 decreased both the proportion of cells forming paraspeckles and the number of paraspeckles/nucleus. In addition, this also led to a decrease in the number of mitochondria, indicating that depletion of the lncRNA also affects mitochondrial abundance (Fig. 4). Treatments with NEAT1 siRNA exacerbated oxidative stress provoked cell death; however, this could be reversed by the LRRK2 (Leucine-rich repeat kinase 2) inhibitor PF-06447475. This observation gave ground to the suggestion that NEAT1 improves cell viability by an LRRK2-dependent manner. The finding that LRRK2 protein interacts with the paraspeckle proteins NONO and SFPQ supports this assumption [18, 60]. Simchovitz and colleagues proposed that NEAT1 acts as a bona fide LRRK2 inhibitor via binding the LRRK2 protein in paraspeckles. Mutations of the LRRK2 gene are one of the most common genetic causes of both sporadic and familial PD [61]. Several pathogenic LRRK2 mutations have been identified to cause increased kinase activity, and overactivation of LRRK2 has been found to cause disturbances in lysosomal homeostasis, microglial overactivation, phosphorylated tau accumulation and mitochondrial function (reviewed in [61, 62]). Since LRRK2 dysfunction plays crucial role in PD pathology [63], restoration of the impaired function of the kinase is an appealing approach for the treatment of the disease. There has been intensive research focusing on the development of kinase inhibitors for PD therapy (reviewed in [64]), and the finding of NEAT1 acting as a natural LRRK2 inhibitor could make upregulation of NEAT1 a target of such drug research. The promoter region of NEAT1 lncRNA contains a PPARα (Peroxisome proliferator-activated receptor alpha) binding site thus NEAT1 expression induction could be achieved by the use of PPARα activators. Indeed, treatment with both PPARα agonist fenofibrate and 3-hydroxy-3-methylglutaryl-coenzyme A inhibitor simvastatin led to the upregulation of NEAT1 expression, leading to a more prominent rise in the amount of the long lncRNA variant. In vitro experiments demonstrated that administration of fenofibrate and simvastatin increased viability of PQ and tBHP treated cells (Fig. 4). In HEK-293T cells, the beneficial effect of NEAT1 upregulation on cell survival was abolished after co-treatment with PQ and LRRK2 inhibitor, strengthening the notion that NEAT1 exerts its neuroprotective effects via mediating LRRK2 function (Fig. 1).
Combining the results obtained from human samples and in vitro models of the diseases it was proposed that NEAT1 upregulation in the substantia nigra reflects the accumulation of the lncRNA and the enhanced formation of paraspeckles in the dying neurons, and is therefore a hallmark of neurodegeneration. Simchovitz et al. proposed that the reason behind the upregulation of NEAT1 in dopaminergic neurons could be to enhance the formation of nuclear paraspeckles as a mechanism of protecting neurons from the damage mediated by LRRK2 [18]. The fact that HOTAIR (Hox transcript antisense intergenic RNA), another lncRNA has been previously identified as an LRRK2-dependent modifier of PD pathology also support this notion [65]. Opposite to NEAT1, however, HOTAIR was reported to enhance LRRK2 gene expression thus propagating the disease.
DISCUSSION
The diverse interaction of NEAT1 with a broad range of molecules demonstrates well the complex ways in which this lncRNA can regulate cell functions. Despite intensive research and a rapidly growing body of evidence of the involvement of NEAT1 in PD, it is still not elucidated whether this lncRNA has an ameliorating or an exacerbating effect on disease progression. The controversial results of different research groups may originate from the different disease models implemented. The observation that the effect of NEAT1 upregulation varies depending on the agent used for disease modeling raises the possibility that the contrasting results may at least partly reflect differences of causative or consequential nature of PD insults. Studies with genetic models (either knockout or transgene) of the disease which are more likely to represent pathological changes that are causative in the development of the disorder might be useful to clarify questions in this respect. This calls attention to difficulties stemming from the complex patho-mechanism behind neurodegenerative disorders: even the acknowledged and well established in vitro and in vivo models are hardly, if at all, able to mimic precisely the complexity of pathological processes. Thus, results obtained from disease models should always be interpreted with great caution.
It is worth pointing out that although in the context of PD NEAT1 downregulation improved cell viability and decreased apoptosis in MPTP/MPP+models of the disease, NEAT1 upregulation was found to have a protective effect in in vitro models induced by oxidative stressors such as PQ and tBPH. This implies that the effect of NEAT1 is likely context dependent. MPTP/MPP+is a mitochondrial toxin which inhibits complex I of the mitochondrial respiratory chain, resulting in the disruption of ATP synthesis and ROS generation. MPTP also damages dopamine storage of cells, a feature considered to play a key role in the selective loss of dopaminergic neurons (reviewed in [66]). PQ is a herbicid, which, by interfering with photosynthetic electron transport in plants, leads to the production of superoxide. Though PQ has been linked to the production of ROS and accumulation of α-syn aggregates in dopaminergic neurons in experimental models of PD, the exact way by which it damages dopaminergic cells is not fully elucidated [67, 68]. Such ambiguous results regarding the role of NEAT1 in different PD models could be partly due to the different pathological effects the implemented toxins exert.
The role of NEAT1 is controversial not only in PD, but in cancer and other neurodegenerative diseases as well, such as Huntington’s disease (HD) and AD.
Sunwoo et al. found NEAT1 to be upregulated in brain samples of both HD patients and the R6/2 HD mouse model of the disease. However, various in vitro models, such as mutant huntingtin (mHtt)-transfected neuro2A cells and mouse striatal neuron-derived cell lines (STHdh) did not show upregulation of the lncRNA. Despite the fact that no change was observed in NEAT1 expression in the above in vitro HD models, transfection with the NEAT1 short isoform vector in the mouse neuroblastoma cell line Neuro2A improved cell viability under H2O2-induced oxidative stress [69]. These ambiguous findings were proposed to reflect the lack of in vitro models’ ability to portray the complex underlying pathophysiological mechanisms of HD [69]. This again calls attention to the complexity of neurodegenerative diseases and might offer explanation for the seemingly controversial results acquired from studies implementing different models.
The finding that NEAT1 transfection improved cell viability in H2O2-induced oxidative stress is in line with the findings of Simchovitz et al., who also found that NEAT1 upregulation increased cell viability after treatment with ROS generators PQ or tBHP [18].
Chanda and colleagues detected consistent and significant upregulation of NEAT1 not only in animal models, but also in mHtt expressing in vitro models of the disease. Knockdown of NEAT1 led to a significant decrease in mHtt aggregates and decreased expression of TP53 (Tumor protein 53) [70].
In addition to HD, NEAT1L (but not NEAT1S) upregulation was reported by Chang et al. in other polyglutamine (polyQ) repeat diseases, such as spinocerebellar ataxia types 1, 2 and 7 [71]. Upregulation of NEAT1 in mHtt expressing SH-SY5Y cells was protective against mHtt induced toxicity, while inhibition of the lncRNA decreased cell viability. Interestingly, NEAT1 silencing not only increased mHtt sensitivity of the cells but also augmented viability upon treatment with the mitochondrial toxin 3-nitropropionic acid (3-NP) [71].
Some of the observations made using AD models seem to be more directly linked to and supporting the beneficial role of NEAT1 silencing in MPTP/MPP+PD models. In in vitro models of AD Aβ (amyloid beta)-exposure enhanced NEAT1 expression, and knockdown of the lncRNA promoted cell viability and diminished apoptosis [72]. NEAT1 was identified as a decoy for miR-107, and the lncRNA was proposed to aggravate Aβ-induced cell damage by sponging the micro RNA [72].
Recently Huang and colleagues proposed a further mechanism by which NEAT1 regulates Aβ metabolism and modifies AD pathology [73]. In the APP/PS1 transgenic mouse model, NEAT1 overexpression was found to exacerbate Aβ production, whereas knockdown of the lncRNA inhibited the generation of amyloid deposits [73]. In the same animal model knockdown of the lncRNA led to an increase in the levels of PINK1 as well as those of other autophagy markers such as P62, OPTN and LC3. NEAT1 overexpression promoted the ubiquitination and consequent degradation of PINK1—just the opposite of what was seen in PD models, where NEAT1 was identified as a stabilizer of the protein [45]. Based on their findings Huang et al. proposed that via facilitating PINK1 degradation, NEAT1 causes the inhibition of autophagy signaling thus impairing Aβ clearance. This results in the accumulation of amyloid aggregates and propagates disease pathology [73].
NEAT1 was also proposed to modulate AD pathology by epigenetic regulation of various genes due to its interaction with the PC300/CBP lysine acetyltransferases [74]. Knocking down the lncRNA affected both the acetylation and crotonylation of H3K27, thus impacting the transcription of several genes involved in endocytosis. In vitro studies involving the human astrocytic U251 cell line showed that inhibition of NEAT1 impeded Aβ uptake and degradation, suggesting a negative role of the lncRNA in AD pathology [74].
Changes in NEAT1 level and the responses presumably evoked by this have been reported to affect several further neurological conditions: NEAT1 upregulation was observed in hypoxic-ischemic brain damage (HIBD). The change in NEAT1 expression was proposed to be part of a protective response reaction [75]. In neonatal HIBD mice, NEAT1 was identified to competitively bind to the micro RNA miR-339-5p. Sponging of miR-339-5p led to the upregulation of homeobox A1 (HOXA1), promotion of cell viability and decreased apoptosis.
NEAT1 silencing was also reported to have a beneficial effect on age-related memory impairment [76]. Knockdown of NEAT1 caused disruption of histone 3 lysine 9 demethylation (H3K9me2), a repressive histone modification mark which increases with age in rodent hippocampus [76]. NEAT1 overexpression led to memory impairment of young mice, similar to that observed in their older counterparts. NEAT1 knockdown, on the other hand, improved behavior test–associated memory of mice of both age groups.
NEAT1 depletion was reported to ameliorate memory impairment related to AD as well: knockdown of NEAT1 led to improvement of learning and cognitive functions of APP/PS1 transgenic mice [73]. The question of whether these effects could be causally linked to the changes in NEAT1 expression and whether they relate to the effects observed in PD models remains to be answered.
In addition to NEAT1 various other lncRNAs play role in pathological processes of PD as it has been indicated and/or proved by findings of numerous in vivo and in vitro studies (recent reviews on these: [14, 77, 78]). Several lncRNAs are implied to have protective effects against disease development (including UCHL1-AS, MAPT-AS1, Mirt2), while others are likely to play a detrimental role (such as HOTAIR, MALAT1, lincRNA-p21, BACE1-AS, HAGLROS and SNHG1) ([14, 78] and references in there). The mode of action of these transcripts resemble those proposed for NEAT1: among them are regulation of SNCA expression and α-syn aggregation by MALAT1 (alias NEAT2) [79] and SNHG1 [80], respectively, regulation of MAPT promoter activity by MAPT-AS1 [81], enhancement of UCHL1 gene (alias PARK5) via its anti-sense pair UCHL1-AS [82] and modulation of LRRK2 mRNA stability through HOTAIR [65]. Besides transcriptional and post-transcriptional regulation of PARK genes, lncRNAs can influence processes related to neuroinflammation partly via their interaction with miRNAs (such as Mirt2 lncRNA and miR-101 [83]; lincRNA-p21 and miR-1277-5p [84]). Further modes of action of PD related lncRNAs are autophagosomy system balance maintenance, oxidative stress and dopaminergic cell loss [85, 86] (reviewed in [14] and [78]).
CONCLUSION
Despite the fact that PD is one of the most common neurodegenerative diseases worldwide, causing tremendous burden not only on the individual but on society as well, the exact underlying patho-mechanism of the disease is still unknown. In the past few years lncRNAs have emerged as intriguing subjects of PD research due to the diverse functions they fulfill. Among lncRNAs, NEAT1 attracted particular interest, since its expression was found to be elevated both in different brain regions and also in peripheral blood of PD patients. Upregulation of NEAT1 has been detected in various in vitro and in vivo models of the disease however data on whether its role in disease progression is protective or detrimental is conflicting. Upregulated NEAT1 level was proposed to have a damaging effect via the interaction of the RNA with PINK1 protein and various micro RNAs such as miR-1303-3p, miR-124, miR-212-5p and miR-221 and by the upregulation of SNCA expression. On the other hand, results of Simchovitz et al. argue for the protective role of NEAT1, based on the finding that the lncRNA acts as a natural LRRK2 inhibitor.
The effects of NEAT1 on disease progression are contradictory in other neurodegenerative diseases such as HD and AD as well. The cause of this could be in the different models implemented by different research groups. Due to the complexity of these disorders, to date no in vitro or in vivo model exists that is capable of precisely mimicking the pathological mechanisms of neurodegeneration. Inconsistent data regarding NEAT1 effects also imply that the RNA acts in context dependent modes: based on the toxin used for modeling PD, both NEAT1 upregulation or knockdown can prove to be protective. Research aiming to clarify the role and mode of action of this lncRNA in PD is highly warranted, since NEAT1 shows promise to emerge as both a promising biomarker and a potential therapeutic target for this neurodegenerative disease.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We would like to thank Katalin Boros (Manchester, United Kingdom) for the help in English language editing.
The current work was supported by Hungarian Brain Research Program (Grant No. 2017-1.2.1-NKP-2017-00002), Economic Development and Innovation Operational Programme (Grant No. GINOP-2.3.2-15-2016-00034) and TUDFO/47138-1/2019-ITM. F.A.B was supported by The ÚNKP-20-4 - New National Excellence Program of The Ministry for Innovation and Technology from the Source of the National Research, Development and Innovation Fund.
REFERENCES
[1] | Goedert M ((2001) ) Alpha-synuclein and neurodegenerative diseases. Neuroscience 2: , 492–501. |
[2] | Fahn S ((2003) ) Description of Parkinson’s disease as a clinical syndrome. N Y Acad Sci 991: , 1–14. |
[3] | Berg D , Postuma RB , Bloem B , Chan P , Dubois B , Gasser T , Goetz CG , Halliday GM , Hardy J , Lang AE , Litvan I , Marek K , Obeso J , Oertel W , Olanow CW , Poewe W , Stern M , Deuschl G ((2014) ) Time to redefine PD? Introductory statement of the MDS Task Force on the definition of Parkinson’s disease. Mov Disord 29: , 454–462. |
[4] | Sulzer D ((2007) ) Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci 30: , 244–250. |
[5] | Surmeier JD , Obeso JA , Halliday GM ((2017) ) Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci 18: , 101–113. |
[6] | Savitt D , Jankovic J ((2019) ) Targeting α-synuclein in Parkinson’s disease: Progress towards the development of disease-modifying therapeutics. Drugs 79: , 797–810. |
[7] | Johnson ME , Stecher B , Labrie V , Brundin L , Brundin P ((2019) ) Triggers, facilitators, and aggravators: Redefining Parkinson’s disease pathogenesis. Trends Neurosci 42: , 4–13. |
[8] | Marras C , Lang A , van de Warrenburg BP , Sue CM , Tabrizi SJ , Bertram L , Mercimek-Mahmutoglu S , Ebrahimi-Fakhari D , Warner TT , Durr A , Assmann B , Lohmann K , Kostic V , Klein C ((2016) ) Nomenclature of genetic movement disorders: Recommendations of the international Parkinson and movement disorder society task force. Mov Disord 31: , 436–457. |
[9] | Nalls MA , Pankratz N , Lill CM , Do CB , Hernandez DG , Saad M , DeStefano AL , Kara E , Bras J , Sharma M , Schulte C , Keller MF , Arepalli S , Letson C , Edsall C , Stefansson H , Liu X , Pliner H , Lee JH , Cheng R ; International Parkinson’s Disease Genomics Consortium (IPDGC); Parkinson’s Study Group (PSG) Parkinson’s Research: The Organized GENetics Initiative (PROGENI); 23andMe; GenePD; NeuroGenetics Research Consortium (NGRC); Hussman Institute of Human Genomics (HIHG); Ashkenazi Jewish Dataset Investigator; Cohorts for Health and Aging Research in Genetic Epidemiology (CHARGE); North American Brain Expression Consortium (NABEC); United Kingdom Brain Expression Consortium (UKBEC); Greek Parkinson’s Disease Consortium; Alzheimer Genetic Analysis Group, Ikram MA , Ioannidis JP , Hadjigeorgiou GM , Bis JC , Martinez M , Perlmutter JS , Goate A , Marder K , Fiske B , Sutherland M , Xiromerisiou G , Myers RH , Clark LN , Stefansson K , Hardy JA , Heutink P , Chen H , Wood NW , Houlden H , Payami H , Brice A , Scott WK , Gasser T , Bertram L , Eriksson N , Foroud T , Singleton AB ((2014) ) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46: , 989–993. |
[10] | Boros FA , Török R , Vágvölgyi-Sümegi E , Pesei ZG , Klivényi P , Vécsei L ((2019) ) Assessment of risk factor variants of LRRK2, MAPT, SNCA and TCEANC2 genes in Hungarian sporadic Parkinson’s disease patients. Neurosci Lett 706: , 140–145. |
[11] | Benson DL , Huntley GW ((2019) ) Are we listening to everything the PARK genes are telling us? J Comp Neurol 527: , 1527–1540. |
[12] | Zhang X , Wang W , Zhu W , Dong J , Cheng Y , Yin Z , Shen F ((2019) ) Mechanisms and functions of long non-coding RNAs at multiple regulatory levels. Int J Mol Sci 20: , 5573. |
[13] | Oe S , Kimura T , Yamada H ((2019) ) Regulatory non-coding RNAs in nervous system development and disease. Front Biosci Landmark 24: , 1203–1240. |
[14] | Lv Q , Wang Z , Zhong Z , Huang W ((2020) ) Role of long noncoding RNAs in Parkinson’s disease: Putative biomarkers and therapeutic targets. Parkinsons Dis 2020: , 5374307. |
[15] | Prinz F , Kapeller A , Pichler M , Klec C ((2019) ) The implications of the long non-coding RNA NEAT1 in non-cancerous diseases. Int J Mol Sci 20: , 627. |
[16] | Wang Y , Hu S-B , Wang M-R , Yao R-W , Wu D , Yang L , Chen L-L ((2018) ) Genome-wide screening of NEAT1 regulators reveals cross-regulation between paraspeckles and mitochondria. Nat Cell Biol 20: , 1145–1158. |
[17] | Kraus TFJ , Haider M , Spanner J , Steinmaurer M , Dietinger V , Kretzschmar HA ((2017) ) Altered long noncoding RNA expression precedes the course of Parkinson’s disease-a preliminary report. Mol Neurobiol 54: , 2869–2877. |
[18] | Simchovitz A , Hanan M , Niederhoffer N , Madrer N , Yayon N , Bennett ER , Greenberg DS , Kadener S , Soreq H ((2019) ) NEAT1 is overexpressed in Parkinson’s disease substantia nigra and confers drug-inducible neuroprotection from oxidative stress. FASEB J 33: , 11223–11234. |
[19] | Boros FA , Maszlag-Török R , Vécsei L , Klivényi P ((2020) ) Increased level of NEAT1 long non-coding RNA is detectable in peripheral blood cells of patients with Parkinson’s disease. Brain Res 1730: , 146672. |
[20] | Mello SS , Attardi LD ((2018) ) Neat-en-ing up our understanding of p53 pathways in tumor suppression. Cell Cycle 17: , 1527–1535. |
[21] | Dong P , Xiong Y , Yue J , Hanley SJB , Kobayashi N , Todo Y , Watari H ((2018) ) Long non-coding RNA NEAT1: A novel target for diagnosis and therapy in human tumors. Front Genet 9: , 471. |
[22] | Hutchinson JN , Ensminger AW , Clemson CM , Lynch CR , Lawrence JB , Chess A ((2007) ) A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8: , 39. |
[23] | Guru SC , Agarwal SK , Manickam P , Olufemi SE , Crabtree JS , Weisemann JM , Kester MB , Kim YS , Wang Y , Emmert-Buck MR , Liotta LA , Spiegel AM , Boguski MS , Roe BA , Collins FS , Marx SJ , Burns L , Chandrasekharappa SC ((1997) ) A transcript map for the 2.8-Mb region containing the multiple endocrine neoplasia type 1 locus. Genome Res 7: , 725–735. |
[24] | Fox AH , Lamond AI ((2010) ) Paraspeckles. Cold Spring Harb Perspect Biol 2: , a000687. |
[25] | An H , Williams NG , Shelkovnikova TA ((2018) ) NEAT1 and paraspeckles in neurodegenerative diseases: A missing lnc found? Noncoding RNA Res 3: , 243–252. |
[26] | Sunwoo H , Dinger ME , Wilusz JE , Amaral PP , Mattick JS , Spector DL ((2009) ) Men ɛ/β nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res 19: , 347–359. |
[27] | Naganuma T , Nakagawa S , Tanigawa A , Sasaki YF , Goshima N , Hirose T ((2012) ) Alternative 3′-end processing of long noncoding RNA initiates construction of nuclear paraspeckles. EMBO J 31: , 4020–4034. |
[28] | Fox AH , Fox AH , Lam YW , Lam YW , Leung AKL , Leung AKL , Lyon CE , Lyon CE , Andersen J , Andersen J , Mann M , Mann M , Lamond AI , Lamond AI ((2002) ) Paraspeckles: A novel nuclear domain. Curr Biol 12: , 13–25. |
[29] | Andersen JS , Lyon CE , Fox AH , Leung AKL , Lam YW , Steen H , Mann M , Lamond AI ((2002) ) Directed proteomic analysis of the human nucleolus. Curr Biol 12: , 1–11. |
[30] | Bond CS , Fox AH ((2009) ) Paraspeckles: Nuclear bodies built on long noncoding RNA. J Cell Biol 186: , 637–644. |
[31] | Sasaki YTF , Ideue T , Sano M , Mituyama T , Hirose T ((2009) ) MENɛ/β noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc Natl Acad Sci U S A 106: , 2525–2530. |
[32] | Lin Y , Schmidt BF , Bruchez MP , McManus CJ ((2018) ) Structural analyses of NEAT1 lncRNAs suggest long-range RNA interactions that may contribute to paraspeckle architecture. Nucleic Acids Res 46: , 3742–3752. |
[33] | Li R , Harvey AR , Hodgetts SI , Fox AH ((2017) ) Functional dissection of NEAT1 using genome editing reveals substantial localization of the NEAT1-1 isoform outside paraspeckles. RNA 23: , 872–881. |
[34] | Nakagawa S , Naganuma T , Shioi G , Hirose T ((2011) ) Paraspeckles are subpopulation-specific nuclear bodies that are not essential in mice. J Cell Biol 193: , 31–39. |
[35] | Nakagawa S , Yamazaki T , Hirose T ((2018) ) Molecular dissection of nuclear paraspeckles: Towards understanding the emerging world of the RNP milieu. Open Biol 8: , 180150. |
[36] | Isobe M , Toya H , Mito M , Chiba T , Asahara H , Hirose T , Nakagawa S ((2020) ) Forced isoform switching of Neat1_1 to Neat1_2 leads to the loss of Neat1_1 and the hyperformation of paraspeckles but does not affect the development and growth of mice. RNA 26: , 251–264. |
[37] | Wu Y , Yang L , Zhao J , Li C , Nie J , Liu F , Zhuo C , Zheng Y , Li B , Wang Z , Xu Y ((2015) ) Nuclear-enriched abundant transcript 1 as a diagnostic and prognostic biomarker in colorectal cancer. Mol Cancer 14: , 191. |
[38] | Knutsen E , Lellahi SM , Aure MR , Nord S , Fismen S , Larsen KB , Gabriel MT , Hedberg A , Bjørklund SS ; Oslo Breast Cancer Research Consortium (OSBREAC), Bofin AM , Mælandsmo GM , Sørlie T , Mortensen ES , Perander M ((2020) ) The expression of the long NEAT1_2 isoform is associated with human epidermal growth factor receptor 2-positive breast cancers. Sci Rep 10: , 1277. |
[39] | Kessler SM , Hosseini K , Hussein UK , Kim KM , List M , Schultheiß CS , Schulz MH , Laggai S , Jang KY , Kiemer AK ((2019) ) Hepatocellular carcinoma and nuclear paraspeckles: Induction in chemoresistance and prediction for poor survival. Cell Physiol Biochem 52: , 787–801. |
[40] | Sunwoo J-S , Lee S-T , Im W , Lee M , Byun J-I , Jung K-H , Park K-I , Jung K-Y , Lee SK , Chu K , Kim M ((2016) ) Altered expression of the long noncoding RNA NEAT1 in Huntington’s disease. Mol Neurobiol 54: , 1577–1586. |
[41] | Chowdhury IH , Narra HP , Sahni A , Khanipov K , Schroeder CLC , Patel J , Fofanov Y , Sahni SK ((2017) ) Expression profiling of long noncoding RNA splice variants in human microvascular endothelial cells: Lipopolysaccharide effects in vitro. Mediators Inflamm 2017: , 3427461. |
[42] | Li S , Li J , Chen C , Zhang R , Wang K ((2018) ) Pan-cancer analysis of long non-coding RNA NEAT1 in various cancers. Genes Dis 5: , 27–35. |
[43] | Wang Z , Li K , Huang W ((2020) ) Long non-coding RNA NEAT1-centric gene regulation. Cell Mol Life Sci 77: , 3769–3779. |
[44] | Yamazaki T , Souquere S , Chujo T , Kobelke S , Chong YS , Fox AH , Bond CS , Nakagawa S , Pierron G , Hirose T ((2018) ) Functional domains of NEAT1 architectural lncRNA induce paraspeckle assembly through phase separation. Mol Cell 70: , 1038–1053. |
[45] | Yan W , Chen ZY , Chen JQ , Chen HM ((2018) ) LncRNA NEAT1 promotes autophagy in MPTP-induced Parkinson’s disease through stabilizing PINK1 protein. Biochem Biophys Res Commun 496: , 1019–1024. |
[46] | Fahmy AM , Labonté P ((2017) ) The autophagy elongation complex (ATG5-12/16L1) positively regulates HCV replication and is required for wild-type membranous web formation. Sci Rep 7: , 40351. |
[47] | Liu Y , Lu Z ((2018) ) Long non-coding RNA NEAT1 mediates the toxic of Parkinson’s disease induced by MPTP/MPP+via regulation of gene expression. Clin Exp Pharmacol Physiol 45: , 841–848. |
[48] | Sun Q , Zhang Y , Wang S , Yang F , Cai H , Xing Y , Chen Z , Chen J ((2020) ) NEAT1 decreasing suppresses Parkinson’s disease progression via acting as miR-1301-3p sponge. J Mol Neurosci, doi: 10.1007/s12031-020-01660-2. |
[49] | Voet S , Srinivasan S , Lamkanfi M , van Loo G ((2019) ) Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med 11: , e10248. |
[50] | Gordon R , Albornoz EA , Christie DC , Langley MR , Kumar V , Manotovani S , Robertson AAB , Butler MS , Rowe DB , O’Neill LA , Kanthasamy AG , Schroder K , Cooper MA , Woodruff TM ((2018) ) Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci Transl Med 10: , eaah4066. |
[51] | von Herrmann KM , Salas LA , Martinez EM , Young AL , Howard JM , Feldman MS , Christensen BC , Wilkins OM , Lee SL , Hickey WF , Havrda MC ((2018) ) NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson’s disease. NPJ Park Dis 4: , 24. |
[52] | Reyes JF , Sackmann C , Hoffmann A , Svenningsson P , Winkler J , Ingelsson M , Hallbeck M ((2019) ) Binding of α-synuclein oligomers to Cx32 facilitates protein uptake and transfer in neurons and oligodendrocytes. Acta Neuropathol 138: , 23–47. |
[53] | Oh SE , Park H-J , He L , Skibiel C , Junn E , Mouradian MM ((2018) ) The Parkinson’s disease gene product DJ-1 modulates miR-221 to promote neuronal survival against oxidative stress. Redox Biol 19: , 62–73. |
[54] | Ismail A , Ning K , Al-Hayani A , Sharrack B , Azzouz M ((2012) ) PTEN: A molecular target for neurodegeneratIve disorders. Transl Neurosci 3: , 132–142. |
[55] | Ding H , Huang Z , Chen M , Wang C , Chen X , Chen J , Zhang J ((2016) ) Identification of a panel of five serum miRNAs as a biomarker for Parkinson’s disease. Parkinsonism Relat Disord 22: , 68–73. |
[56] | Ma W , Li Y , Wang C , Xu F , Wang M , Liu Y ((2016) ) Serum miR-221 serves as a biomarker for Parkinson’s disease. Cell Biochem Funct 34: , 511–515. |
[57] | Geng L , Zhao J , Liu W , Chen Y ((2019) ) Knockdown of NEAT1 ameliorated MPP+-induced neuronal damage by sponging miR-221 in SH-SY5Y cells. RSC Adv 9: , 25257–25265. |
[58] | Xie SP , Zhou F , Li J , Duan SJ ((2019) ) NEAT1 regulates MPP+-induced neuronal injury by targeting miR-124 in neuroblastoma cells. Neurosci Lett 708: , 134340. |
[59] | Liu R , Li F , Zhao W ((2020) ) Long noncoding RNA NEAT1 knockdown inhibits MPP+-induced apoptosis, inflammation and cytotoxicity in SK-N-SH cells by regulating miR-212-5p/RAB3IP axis. Neurosci Lett 731: , 135060. |
[60] | |
[61] | Singh A , Zhi L , Zhang H ((2019) ) LRRK2 and mitochondria: Recent advances and current views. Brain Res 1702: , 96–104. |
[62] | Araki M , Ito G , Tomita T ((2018) ) Physiological and pathological functions of LRRK2: Implications from substrate proteins. Neuronal Signal 2: , NS20180005. |
[63] | Ray S , Liu M ((2012) ) Current understanding of LRRK2 in Parkinson’s disease: Biochemical and structural features and inhibitor design. Futur Med Chem 4: , 1701–1713. |
[64] | Taymans J-M , Greggio E ((2016) ) LRRK2 kinase inhibition as a therapeutic strategy for Parkinson’s disease, where do we stand?. Curr Neuropharmacol 14: , 214–225. |
[65] | Wang S , Zhang X , Guo Y , Rong H , Liu T ((2017) ) The long noncoding RNA HOTAIR promotes parkinson’s disease by upregulating LRRK2 expression. Oncotarget 8: , 24449–24456. |
[66] | Langston JW ((2017) ) The MPTP story. J Parkinsons Dis 7: , S11–S19. |
[67] | Vaccari C , El Dib R , de Camargo JLV ((2017) ) Paraquat and Parkinson’s disease: A systematic review protocol according to the OHAT approach for hazard identification. Syst Rev 6: , 98. |
[68] | Richardson JR , Quan Y , Sherer TB , Greenamyre JT , Miller GW ((2005) ) Paraquat neurotoxicity is distinct from that of MPTP and rotenone. Toxicol Sci 88: , 193–201. |
[69] | Sunwoo JS , Lee S-T , Im W , Lee M , Byun J-I , Jung K-H , Park K-I , Jung K-Y , Lee SK , Chu K , Kim M ((2017) ) Altered Expression of the long noncoding RNA NEAT1 in Huntington’s disease. Mol Neurobiol 54: , 1577–1586. |
[70] | Chanda K , Das S , Chakraborty J , Bucha S , Maitra A , Chatterjee R , Mukhopadhyay D , Bhattacharyya NP ((2018) ) Altered levels of long ncRNAs Meg3 and Neat1 in cell and animal models of Huntington’s disease. RNA Biol 15: , 1348–1363. |
[71] | Cheng C , Spengler RM , Keiser MS , Monteys AM , Rieders JM , Ramachandran S , Davidson BL ((2018) ) The long non-coding RNA NEAT1 is elevated in polyglutamine repeat expansion diseases and protects from disease gene-dependent toxicities. Hum Mol Genet 27: , 4303–4314. |
[72] | Ke S , Yang Z , Yang F , Wang X , Tan J , Liao B ((2019) ) Long noncoding RNA NEAT1 aggravates Aβ-induced neuronal damage by targeting miR-107 in Alzheimer’s disease. Yonsei Med J 60: , 640–650. |
[73] | Huang Z , Zhao J , Wang W , Zhou J , Zhang J ((2020) ) Depletion of LncRNA NEAT1 rescues mitochondrial dysfunction through NEDD4L-dependent PINK1 degradation in animal models of Alzheimer’s disease. Front Cell Neurosci 14: , 28. |
[74] | Wang Z , Zhao Y , Xu N , Zhang S , Wang S , Mao Y , Zhu Y , Li B , Jiang Y , Tan Y , Xie W , Yang BB , Zhang Y ((2019) ) NEAT1 regulates neuroglial cell mediating Aβ clearance via the epigenetic regulation of endocytosis-related genes expression. Cell Mol Life Sci 76: , 3005–3018. |
[75] | Zhao J , He L , Yin L ((2020) ) lncRNA NEAT1 binds to miR-339-5p to increase HOXA1 and alleviate ischemic brain damage in neonatal mice. Mol Ther Nucleic Acids 20: , 117–127. |
[76] | Butler AA , Johnston DR , Kaur S , Lubin FD ((2019) ) Long noncoding RNA NEAT1 mediates neuronal histone methylation and age-related memory impairment. Sci Signal 12: , eaaw9277. |
[77] | Oe S , Kimura T , Yamada H ((2019) ) Regulatory non-coding RNAs in nervous system development and disease. Front Biosci 24: , 1203–1240. |
[78] | Acharya S , Salgado-somoza A , Stefanizzi FM , Lumley AI , Zhang L , Glaab E , May P , Devaux Y ((2020) ) Non-coding RNAs in the brain-heart axis: The case of Parkinson’s disease. Int J Mol Sci 21: , 6513. |
[79] | Zhang QS , Wang ZH , Zhang JL , Duan YL , Li GF , Zheng DL ((2016) ) Beta-asarone protects against MPTP-induced Parkinson’s disease via regulating long non-coding RNA MALAT1 and inhibiting α-synuclein protein expression. Biomed Pharmacother 83: , 153–159. |
[80] | Chen Y , Lian Y , Ma Y , Wu C , Zheng Y , Xie N ((2018) ) LncRNA SNHG1 promotes α-synuclein aggregation and toxicity by targeting miR-15b-5p to activate SIAH1 in human neuroblastoma SH-SY5Y cells. Neurotoxicology 68: , 212–221. |
[81] | Coupland KG , Kim WS , Halliday GM , Hallupp M , Dobson-Stone C , Kwok JBJ ((2016) ) Role of the long non-coding RNA MAPT-AS1 in regulation of microtubule associated protein tau (MAPT) expression in Parkinson’s disease. PLoS One 11: , e0157924. |
[82] | Carrieri C , Forrest ARR , Santoro C , Persichetti F , Carninci P , Zucchelli S , Gustincich S ((2015) ) Expression analysis of the long non-coding RNA antisense to Uchl1 (AS Uchl1) during dopaminergic cells’ differentiation in vitro and in neurochemical models of Parkinson’s disease. Front Cell Neurosci 9: , 114. |
[83] | Han Y , Kang C , Kang M , Quan W , Gao H , Zhong Z ((2019) ) Long non-coding RNA Mirt2 prevents TNF-α-triggered inflammation via the repression of microRNA-101. Int Immunopharmacol 76: , 105878. |
[84] | Xu X , Zhuang C , Wu Z , Qiu H , Feng H , Wu J ((2018) ) LincRNA-p21 inhibits cell viability and promotes cell apoptosis in Parkinson’s disease through activating α-synuclein expression. Biomed Res Int 2018: , 8181374. |
[85] | Li Y , Fang J , Zhou Z , Zhou Q , Sun S , Jin Z , Xi Z , Wei J ((2020) ) Downregulation of lncRNA BACE1-AS improves dopamine-dependent oxidative stress in rats with Parkinson’s disease by upregulating microRNA-34b-5p and downregulating BACE1. Cell Cycle 19: , 1158–1171. |
[86] | Peng T , Liu X , Wang J , Liu Y , Fu Z , Ma X , Li J , Sun G , Ji Y , Lu J , Wan W , Lu H ((2019) ) Long noncoding RNA HAGLROS regulates apoptosis and autophagy in Parkinson’s disease via regulating miR-100/ATG10 axis and PI3K/Akt/mTOR pathway activation. Artif Cells Nanomed Biotechnol 47: , 2764–2774. |


