
Infection and Risk of Parkinson’s Disease
Abstract
Parkinson’s disease (PD) is thought to be caused by a combination of genetic and environmental factors. Bacterial or viral infection has been proposed as a potential risk factor, and there is supporting although not entirely consistent epidemiologic and basic science evidence to support its role. Encephalitis caused by influenza has included parkinsonian features. Epidemiological evidence is most compelling for an association between PD and hepatitis C virus. Infection with Helicobacter pylori may be associated not only with PD risk but also response to levodopa. Rapidly evolving knowledge regarding the role of the microbiome also suggests a role of resident bacteria in PD risk. Biological plausibility for the role for infectious agents is supported by the known neurotropic effects of specific viruses, particular vulnerability of the substantia nigra and even the promotion of aggregation of alpha-synuclein. A common feature of implicated viruses appears to be production of high levels of cytokines and chemokines that can cross the blood-brain barrier leading to microglial activation and inflammation and ultimately neuronal cell death. Based on multiple avenues of evidence it appears likely that specific bacterial and particularly viral infections may increase vulnerability to PD. The implications of this for PD prevention requires attention and may be most relevant once preventive treatments for at-risk populations are developed.
ETIOLOGY OF PARKINSON’S DISEASE: CURRENT CONCEPTS
Numerous genetic and environmental factors have been associated with Parkinson’s disease (PD), which is thought to be caused by a complex interplay of multiple factors unique to an individual. In the past decade, the number of known genetic risk factors has greatly increased with 90 risk alleles now identified [1]. However, these known loci account for only approximately 20%of PD risk [1], leaving a substantial proportion of PD unexplained on the bases of currently known genetic associations. There is an urgent need to identify the missing etiologic fraction, to develop preventive and therapeutic strategies.
There is consistent or mostly consistent evidence for several environmental associations with risk of PD (e.g., inverse associations with cigarette smoking, caffeine intake, physical activity, plasma urate and positive associations with pesticide exposure) [2] and a large number of associations with less consistent evidence spanning multiple categories, including dietary factors, chemical exposures, physical and emotional trauma [3]. One of the more controversial categories of risk factors is infection, bacterial or viral.
INFECTION AND PD: AN EPIDEMIOLOGIC PERSPECTIVE
The possibility of an infectious trigger or contributor to PD has gained support from several different avenues. Early observations of clusters of individuals affected by parkinsonism following infectious outbreaks provide an example of presumed viral in-fections followed by and thus assumed to be leading to chronic neurological disease causing parkinsonism. On example of this, recognizing that the disease is pathologically distinct from PD, is encephalitis lethargica and subsequent post-encephalitic parkinsonism that has been associated with the influenza pandemic of 1918 [4]. In addition there have been other case reports of postencephalitic parkinsonism following non-influenza infections, although these have been rare [5, 6]. More recently, the role for infection gained plausibility through the Braak hypothesis of pathological spread starting in the olfactory bulb and peripheral nerves of the gastrointestinal tract [7]. Both of these locations could be portals of entry for an environmental trigger whether toxic or infectious. Constipation and olfactory impairment are two of the most common and earliest features of the prodromal phase of PD, further supporting the importance of the olfactory pathway and gastrointestinal tract in its pathogenesis [8]. Pathologically, inflammation appears to be an early feature [9] which may be consistent with a role for an infectious agent. However, there are alternative explanations for inflammation as a component of PD pathology, and whether or not this is a cause or a consequence of the neurodegenerative process is debated.
Additional evidence for a role of an infectious trigger or risk factor for PD is found in studies showing a relationship between specific occupational exposures and PD. In some studies, occupations where there is higher interpersonal exposures (e.g., teaching, clergy) show an increased risk of developing PD [10]. There is also a significant body of literature on specific infections and subsequent risk of PD, which will be discussed later in this paper. However, there are significant challenges interpreting epidemiological evidence for etiology in PD. The very long prodromal period, spanning decades, makes measurement of initiating factors difficult due to inadequate availability of records or poor recall. In addition, there is undoubtedly a complex interplay of genetic and environmental factors influenced by the neurodegenerative process during the prodromal period that make interpretation difficult. These general challenges are amplified by the complexity of considering the timing and relative impact of multiple short infectious exposures over a lifetime, the widely varying types of infectious agents, variable severity of infections and the inevitable presence of unrecognized infections. For example, a recent epidemiological study tested the “multiple microbe” hypothesis and reported that PD risk was increased compared to healthy controls in individuals who were seropositive for five or six of the pathogens studied (CMV, EBV, HSV-1, B. burgdorferi, C. pneumoniae, and H. pylori) but not less [11].
The role of infections in PD may extend beyond being a triggering event. PD is ultimately a disease of aging and the aging nervous system is vulnerable to the direct and indirect effects of infections which can influence the manifestations of PD. Age-related increases in oxidative stress and impaired energy production can render neurons vulnerable to the toxicity of infectious agents [12]. It is well-described that the symptoms of PD and other neurodegenerative diseases worsen in the context of infection and indeed any metabolic stress [13]. As often observed in clinical practice this worsening can last months and may never return to baseline.
Viral infections and PD risk
The notion of a viral etiology to PD has been mooted for many years. One early example, and poi-gnant today in light of the recent SARS-CoV-2 pandemic, was the emergence of a parkinsonian disorder, encephalitis lethargica [14], that has been linked (although not definitively causally linked) to the 1918 influenza pandemic. Viruses, particularly those that are neurotropic, are plausible causal agents of PD but have been relatively understudied compared to genetic risk factors and other environmental risk factors for PD. In this section we will consider the viruses that have been linked to PD in observational studies and evaluate the strength of evidence to support a causal link.
The most common virus associated with parkinsonism is influenza. Each strain of the influenza virus, varies in its ability to directly infect the CNS. Those that can directly infect cells in the nervous system are considered neurotropic whereas those than cannot are thought to be non-neurotropic. Most of the influenza viruses that have circulated among humans are non-neurotropic; including the 1918 H1N1 virus (Spanish flu) [15], the 1957-1958 H2N2 (Asian flu) [16], the 1968 H3N2 (Hong Kong flu) [17] and the 2009 H1N1 (Mexican or Swine flu) [18]. Despite the lack of direct infection of the CNS, each of these pandemic outbreaks has been associated with encephalitis with parkinsonian features. The common feature of each of these influenzas was their ability to induce a significant systemic infection characterized by production of significantly high levels of cytokines and chemokines [4]. The sheer volume of this cytokine/chemokine production overwhelms the body’s ability to regulate them leading to the induction of what is known as a “cytokine storm” [19]. A body of literature has demonstrated that these peripheral cytokines can pass through the blood brain barrier and communicate with the brain. In fact, inflammatory cytokines, such as TGFalpha, IFNg, and IL6, which are upregulated by influenza infection can induce activation of microglia setting off an inflammatory cascade in the brain that can lead to neuronal dysfunction and even cell death [20].
One hypothesis that has been proffered based on these findings is that the viral-based inflammation primes the CNS; thus making it more susceptible for a later insult that otherwise would have been innocuous [21]. This is often the explanation used to link the 1918 Spanish flu to development of the post-encephalitic parkinsonism [14]. In addition to signs of parkinsonism, Von Economo’s encephalopathy (or encephalitis lethargica) also included other neurological symptoms such as hypersomnolence and cranial neuropathies [22]. The appearance of this influenza-associated syndrome seems to be fairly specific to the 1918 H1N1 strain of influenza, based on the lack of a significant association between influenza infection and PD in a meta-analysis combining data from 4 small, case control studies (combined OR 1.95, 95%CI 0.77–4.94 for the risk of PD following influenza infection) [23]. This suggests that not all viruses have the same potential for CNS damage and for those non-neurotropic viruses it will be critical to understand the profile of inflammatory response induced by each individual virus. This is of particular concern due to the recent outbreak of COVID-19, whose causative agent is the SARS-CoV2 coronavirus. Preliminary studies are equivocal as to the neurotropic potential of this virus. However, what is clear is that it induces a significant “cytokine storm,” with the potential to induce an inflammatory reaction in the brain [24] and sensitize it to later insult; including in regions known to be affected in PD.
Hepatitis B and C viruses have also been investigated for their associations with PD in recent epidemiological studies. Understanding such associations are important given the prevalence of these infections. Hepatitis C virus (HCV) is an RNA virus of the Flavivirus family and is estimated to infect 143 million people worldwide. It primarily involves the liver with chronic infection resulting in cirrhosis and hepatocellular carcinoma [25]. Extrahepatic manifestations include a myriad of inflammatory and immune-mediated disorders [26–28].
An observational study from Taiwan, in a community setting, showed that prior diagnosis of HCV was associated with an increased risk of subsequent PD (adjusted odds ratio (OR) 1.39, 95%CI 1.07–1.80), but no similar association was observed with HBV [28]. A larger, prospective study followed in ∼0.25 million people from the Taiwan national health insurance research database which appeared to confirm these observations. It showed that prior diagnosis with HCV was associated with an increased risk of PD (adjusted hazard HR 1.29, 95%CI 1.06–1.56). Again, prior hepatitis B (HBV) infection was not associated with a similar increased risk [29]. Despite the consistency of these results, one limitation is that they had overlapping study periods, may have included some of the same participants, and that the clinical definition of PD was based only on diagnostic codes without any clinical confirmation. In a separate UK-based study, Pakpoor and colleagues used the Hospital Episode Statistics (HES) database to further assess this association. They reported associations for both HCV (RR 1.51 (95%CI, 1.18–1.9)) and HBV (RR 1.76 (95%CI 1.28–2.37) using standardised rate ratios [30], but there was no clear association with other causes of hepatitis. A systematic review and meta-analysis calculated a combined OR of 1.35 (95%CI 1.18–1.93) for HCV infection [31], and a later meta-analysis which included a further two studies gave an OR of 1.19 (95%CI 1.01–1.41) [23]. Finally, the most recent observational study (from Israel) further explored the relationship, reported an OR of 1.18 (95%CI 1.04–1.35) for HCV and OR 1.08 (95%CI 1.00–1.16) for HBV [32]. Thus, the currently available epidemiologic evidence would suggest a positive, although small, association between HCV and future development of PD. Direct evidence in support of an association between hepatitis and PD arises from studies showing that HCV is neurotropic; and once in the brain, the predominant cell type harboring HCV infection is macrophages/microglia [27]. Additionally, in vitro studies examining the effects of HCV infection in cultured rat brain have shown that this agent can induce loss of dopaminergic neurons [26, 28].
If there is a causal relationship between HCV and PD, it would be expected that successful treatment of HCV infection may mitigate the risk. Two recent studies, again using the Taiwanese national health insurance database, explored the role of interferon therapy for chronic HCV infection to see whether this was associated with a lower risk of PD [33, 34]. Given the probability of considerable overlap in these studies due to near-identical study periods and potentially the same patients being included, the results were perhaps unsurprisingly similar. In the first study, the investigators selected ∼0.25 million patients with recorded HCV infection and divided them into those that were treated with interferon (plus ribavirin) and those that were not treated. In the treated group, the risk of PD was lower than the untreated group (adjusted HR 0.75, 95%CI 0.59–0.96 after 5 years of follow-up), suggesting that the increased risk ass-ociated with HCV infection may be mitigated by antiviral treatment. In the second study, HCV inf-ection was again associated with parkinsonism; treatment with antivirals was associated with a 38%reduced risk (adjusted HR 0.62; 95%CI 0.50–0.77) [34]. The authors proposed that antiviral treatment reduces neuroinflammation, thereby reducing risk.
The human immunodeficiency virus (HIV) is acq-uired in similar ways to HCV and infections with the two frequently co-occur. HIV infection affects ∼37 million people worldwide and is associated with a wide spectrum of neurological disorders, either from immunodeficiency leading to opportunistic infecti-ons, malignancy, or inflammatory conditions, or di-rect neurological consequences of HIV, including HIV-associated neurocognitive disorders (HAND). Despite the prevalence of both HIV and PD, there are limited examples of high-quality observational studies exploring a link between them. This is despite parkinsonism (and other movement disorders) being well recognised in patients with chronic HIV infection and HAND [35, 36]. Manifestation of parkinsonism alongside cognitive impairment can be seen in the context of HIV encephalopathy or HIV-associated dementia, but there are reports of HIV patients with isolated parkinsonism and up to 5–10%of HIV patients may have PD-like motor deficits [37, 38]. Some of the early cases/series suggested that parkinsonism could be unmasked in HIV patients treated with antipsychotics or even with highly active antiretroviral therapy (HAART) [36, 37]. However, in general the prevalence of HAND has decreased in the HAART era [39]. A recent large-scale, observational study suggested relative protection against incident PD in HIV patients treated with antiretroviral drugs compared to those who were not (HR for neurocognitive impairment 0.41, 0.37–0.45) [40]. In the aforementioned study by Pakpoor and colleagues [30] using UK HES data, HIV was not associated with incident PD (RR 0.98; 95%CI 0.50–1.70). An uncontrolled, small, retrospective review of healthcare records of HIV patients in Brazil suggested that incident PD was no more frequent than in the general population [41]. HIV did not appear in a recent systematic review and meta-analysis of infections and risk of PD [23], and we are not aware of any other case-control or cohort studies evaluating the link. Thus, the evidence to date is mixed and further research is needed to understand the relationship.
Beyond the associations already described, there exist other examples of parkinsonism manifesting during acute viral infections including Coxsakie virus infection (a picornavirus), and more classically in patients suffering with Flaviviruses such as Japanese encephalitis, St Louis encephalitis, Western Equine Encephalitic virus (WEEV) and West Nile virus [22]. For these later examples, the predilection for basal ganglia involvement is evident, but parkinsonism often manifests alongside a wider spectrum of neurological signs and symptoms, rather than in isolation and it is unclear how the parkinsonism described in these reports may relate to PD with Lewy pathology.
One of the biggest impediments to drawing conclusions about the link between viral infections and PD is a severe lack of high-quality epidemiological studies. Most data come from small case-control studies, with the biases inherent in design and in likelihood of being published. High quality cohort studies are lacking and there are difficulties around exposure ascertainment, latency, duration, as well as confounding factors such as vaccination, treatment, and co-infection for several of the chronic viral infections.
Bacterial infections and PD risk
The ample spectrum of bacteria that may acutely or permanently infect the tissues of humans has been associated with the development and, to a lesser extent, with the progression of PD. Bacterial production of pro-inflammatory and neurotoxic factors might play a major role in the development and/or in the cascade of neurotoxic events leading to degeneration. One key player in such events is the bacterial endotoxin lipopolysaccharide (LPS). LPS stimulates production of several inflammatory factors that may contribute to neurodegeneration. Tumor necrosis factor-α (TNF-α) is released from microglia; nitric oxide (NO) is released by microglia and astrocytes in and there is hyperproduction of prostagladins. All of these phenomena may lead to neurodegeneration and gliosis [42]. Given that LPS is the endotoxin of Gram-negative bacteria, theoretically, every Gram-negative infection can induce a cascade that could trigger PD; however, the evidence on bacteria is limited to a number of specific organisms.
Borrelia Budgdoferi is a Gram-negative spirochete that is the only known cause of Lyme disease in North America. Borrelia Budgdoferi infection can affect the central nervous system and, when active, can cause a syndrome that may resemble PD [43]. Indeed, there is some initial evidence that Lyme disease can affect dopaminergic activity, especially the dopaminergic component of the frontal reward mechanism [44]. Anecdotally, it has been reported that B. burgdorferi infection can damage of the substantia nigra [45]. However, epidemiological evidence is not supportive of a link between Lyme disease and PD; when geographic locations of Lyme disease and death due to PD were compared, no correlation was found. Given the focal distribution of Lyme disease in the United States (Midwest, Northeast and mid-Atlantic) an increase of PD was expected in those regions if there was a positive correlation, but no association was suggested [46]. In addition, a study using the Danish National registry from 1986-2016 identifying 2,607 cases of Lyme neuroborreliosis did not find a significantly increased long term risk of PD and other neurodegenerative diseases [47].
Helicobacter pylori is a Gram-negative bacterium that infects a large number of the world’s population causing mostly gastrointestinal symptoms. The association with PD has been heavily explored [48]. Increased gastric and duodenal ulcers in patients with PD have been described as early as the 1960s [49]. Before the advent of antibiotic treatment of H. pylori infection the classical treatment of H. pylori was vagotomy that interestingly has been associated with a reduction of the risk of PD [50]. A large meta-analysis reported a 1.5-2-fold increased risk of developing PD after H. pylori infection; [51] and large case-control studies reported 2-3 times increase of PD in patients with H. pylori [52]. Therefore, the association between PD and H. pylori is confirmed although a causal relationship has not been proven. Being a gastrointestinal pathogen, a causal role is biologically plausible through a gastrointestinal “portal of entry” according to the “dual hit hypothesis” [7]. On the other hand, eradicating H. pylori does not seem to alter the risk of PD. [52] In addition, H. pylori has a critical role in the absorption of L-Dopa and therefore, in symptomatic treatments. Eradication of the infection can be important to improve the response to L-Dopa [53] and, conversely, a reduction of the response to L-Dopa requires consideration of H. pylori presence [54].
The potential association between PD and bacterial infection is not restricted to Gram negative organisms. Nocardia asteroides, a weak Gram-positive ba-cterium, has been reported to potentially induce PD-like pathology in mice [55]. Another study reported the possible induction of apoptosis in the substantia nigra [56] suggesting a vulnerability of dopaminergic neurons to the effects of Nocardia infection. On the other hand, a case-control study on serum of patients with PD did not report a significant association between Nocardia asteroides and PD [57].
It is important to consider the possible role of multiple pathogens together; in fact, when multiple bacteria are colonizing and/or are acting synergistically there could be an increased, and cumulative risk of diseases, as demonstrated in stroke [58] and Alzheimer’s disease [59]. One study explored the role of the infectious burden in PD, exploring the presence of antibodies against cytomegalovirus, Epstein Barr virus, herpes simplex virus type-1, Borrelia burgdorferi, Chlamydophila pneumoniae, and Helicobacter pylori in serum of patients with PD and controls [11]. The study observed that the combination of the previous bacteria and viruses was associated with PD compared to controls; however, a causative role was not clarified and further studies are needed.
Notably, there is minimal evidence for associations between more severe infection such as sepsis and the future risk of PD. A recently published case-control study reported that there was no association between severe infections that required hospitalization and sepsis and the risk of PD later in life [60]. However, it is not yet clear whether an infectious condition proximate to the onset of PD or before the onset of PD can trigger or lower the threshold for the upcoming neurodegenerative process.
Understanding the gut microbiome and its role in PD in particular is an area of active study. The interest in the role of gut bacteria has been greatly promoted by pathological evidence for the involvement of the gut early in PD. Indeed, an increasing body of evidence suggests that PD may start in the gut or, at least that the gut may constitute a portal of entry into the nervous system that subsequently spreads to the brain [7]. In fact, the gastrointestinal tract may have a role in the development of synucleinopathies mediated by the bacterial activity of the gut microbiome. Endotoxins (LPS) produced by some gut bacteria (e.g., E. coli) have been reported to have a role in aggregating synuclein and generate toxic synuclein products that can participate in the cascade of events of PD [61–63]. The gastrointestinal tract contains about 1,000 different bacterial species, and a number of studies have shown differences in the gut microbiome between individuals with and without PD [64]. Potential mechanisms are beginning to be elucidated; an early study in mice overexpressing synuclein highlighted that microbiota extracted from PD patients caused motor symptoms and a neuro-inflammatory cascade [65]. Although the studies performed in humans have provided cont-roversial results [66], there is increasing evidence that microbiota in patients with PD may have a significant role in the development of the disease. A recent study reported that there was a dysbiotic alteration of the microbiotic bacteria in PD in the families of Bifidobacteriaceae, Christensenellaceae, Lachno-spiraceae, Lactobacillaceae, Pasteurellaceae and Verrucomicrobiaceae [67]. The study provides a possible mechanism for an indirect effect of changes in microbiota, given that this modification would cause an increase of accumulation of pesticides and other xenobiotics that are not metabolized by the modified PD microbiome; therefore, the accumulation of such molecules may lead to an increase of the risk of PD. Further studies understanding the relationship of the gut microbiome with PD are needed because the microbiome represents a potentially modifiable risk factor not only for incident disease but also disease progression.
Importantly, several studies have reported a possible reduction of risk of PD in patients with appendicectomy [68–70]. In addition, regulating and maintaining the gut flora seems to be mediated by the vermiform appendix, that is no longer considered a vestigial remnant [60]. It is indeed possible to speculate that the appendix can be not only a reservoir of synuclein but also that the lack of regulation of the appendix-mediated gut flora could be another component to increase the risk of PD.
Overall, despite the mounting evidence for a bacteria role in the development of PD (especially H. pylori and the gut microbiome) more studies are needed to understand their causative role and potential treatment opportunities.
MOLECULAR MECHANISMS AND BASIC SCIENCE EVIDENCE FOR THE ROLE OF INFECTION IN PD
Much of the evidence associating PD with infections, whether viral or bacterial, is based on obser-vational studies demonstrating increased risk to develop the disease, rather than direct evidence of infection as a singular cause. In fact, due to the complexity and multifactorial etiology of PD, identifying a single point of initiation in human PD (even in cases of known genetic mutations) is often impossible. For this reason, and the impossibility of interventional studies in humans involving infectious agents, the use of preclinical animal models of PD may provide the clearest evidence for or against a role of infectious agents in the etiology of PD.
As discussed earlier in this review, a number of viral agents have been linked to PD, including in-fluenza, Coxsackie, Japanese encephalitis B, WEEV, Herpesviruses, HCV, and HIV [22, 71]. Preclinical work examining effects of these viruses have shown significant support for their role in the etiology of PD, although in many cases, it is not known if the effects of these viral infections are direct or indirect. When viruses invade the nervous system, they are said to be neurotropic. What is interesting about these neurotropic viruses is that rather than cause a generalized infection, they often target specific regions of the nervous system. Due to this speci-ficity, specific syndromes can often be ascribed to specific viral infections. In the case of PD, WEEV and certain strains of neurotropic influenza (e.g., H5N1) have been shown to directly infect regions affected in PD including the enteric nervous system as well as CNS regions including the substantia nigra and the olfactory bulb [72–74]. In addition to direct infection of neurons, these viruses are also gliotropic, leading to an induction of both astrogliosis and microgliosis [72, 74, 75]. What makes both influenza viruses and WEEV particularly interesting is they also induce a number of changes in the brain that are prominent in PD including induction of an increase in expression of phosphorylated alpha-synuclein, down-regulation of dopamine production and dopaminergic neuron death in the substantia nigra pars compacta [72–74]. The induction of these specific parkinsonian pathologies appears to be specific to the viral infection. For example, using Lund human mesencephalic dopaminergic cells, H1N1 infection was found to lead to a build-up of alpha-synucle-in secondary to blockade of autophagosome function and impaired cellular proteostasis [76]. What was even more interesting is that this process did not affect other proteins (tau, TDP-43) known to aggregate in neurodegenerative disease showing that each virus has the potential to produce specific proteinopathies, as well as acting as a general inflammagen.
As described above, many non-neurotropic infections (both viral and bacterial) induce a significant inflammatory response throughout the body, often referred to as the innate response. During this innate response, circulating immune cells secrete a number of different proteins including interferons, interleukins, chemokines, colony stimulating factors and TNFs [77]. What is important to recognize is that within these classes of inflammatory molecules, some are considered to be pro-inflammatory and some are anti-inflammatory. It is the balance of each, as well as the way they interact with their cognate soluble receptors that determines the ultimate outcomes of the process [78]. If the pro- inflammatory response overwhelms the anti-inflammatory response one can set off a cascade that has been commonly called a “cytokine storm” [19]. The result of this overwhelming inflammatory response is often cellular toxicity. In addition to inducing this toxicity in the periphery, many of the circulating cytokines, although large in size, appear to be able to cross the blood-brain barrier using one of several mechanisms. These mechanisms include 1) a saturable transport system [79], 2) entering through regions of decreased blood-brain barrier called circumventricular organs, and 3) increasing capillary permeability [80–82]. The latter opens up the tight junctions of the blood-brain barrier, and allowing these large proteins to bypass the protections traditionally offered by this barrier.
Once in the brain, these cytokines/chemokines/ Interferons/TNFs can bind to microglial cells, which induces their “activation” [83]. In addition to this indirect effect of circulating cytokines, a number of studies have provided support for PD neurodegeneration that occurs as a direct invasion of circulating lymphocytes (including T- and B-cells) that subsequently interact with the innate inflammatory cells of the brain. A number of observational studies have found increased numbers of circulating lymphocytes and monocytes in the brains of PD patients [84, 85]. Additionally, preclinical studies have directly demonstrated the critical nature of circulating immune cells in PD pathogenesis. Early studies showed that dopaminergic toxins, such as 6-OHDA or MPTP, induced T-cell infiltration into the brain; and the importance of these cells in the induction of pathology was demonstrated by the lack of pathology in Rag-1-KO mice [84, 86, 87]. Another study examined athymic mice that were deficient in mature T-cells and were injected with AAV-alpha-synuclein. These animals showed less behavioral and anatomical pathologies compared to T-cell competent mice [88]. In aggregate, these results demonstrated that mature T-cells were necessary to induce both behavioral as well as the anatomic pathologies.
Mechanistically, what might be the link between the cells of the peripheral immune system and the innate immune system in the brain? One critical component functions through recognition of MHCII; a key antigen presenting protein [89]. MHCII is critical for the presentation of antigen to both T-cells (both inside and outside the brain) as well as microglial cells situated in the CNS [90, 91]. In regard to microglia, it is interesting to note that the SNpc contains the highest microglia:neuron ratio in the brain [92]; perhaps leading to its particular sensitivity to inflammation [93].
Antigen presentation has been shown to elicit secretion of cytokines and chemokines, both in per-ipheral immune cells as well as in microglia [90]. What is important in PD pathogenesis is that once chronically elevated, these proteins can both initiate and maintain glial activation [94–96]. Once microglia and astrocytes are actively expressing their inflammatory programs they themselves secrete similar cytokines and chemokines as are produced by the peripheral immune system. Again, like in the periphery, when the proinflammatory proteins are in greater quantity than anti-inflammatory cytokines the environmental milieu of neurons becomes toxic [97]. Additionally, microglia [98, 99] as well as astrocytes [100] when activated also express MHC antigens and become phagocytic.
The susceptibility of the basal ganglia to such re-actions may relate to the density of microglia and astrocytes relative to neurons, which is highest in the substantia nigra of all other brain regions [92, 93, 101]. This is of particular concern as the mechanism of microglial activation can cause a feed-forward loop of clustering of microglia around dopaminergic neurons and a subsequent increase in activation [102]. Additionally, the overproduction of alpha-synuclein induced by viral infection can, in and of itself, induce activation of microglia [72]. which then sets up a feed forward cascade that perpetuates the effects of the infection. Related to alpha-synuclein, the finding of virally-increased levels of misfolded alpha-synuclein, to the detriment of native synuclein expression, may also provide a mechanism for the sensitivity of the dopaminergic neurons. Beatman et al. [103] showed that cells expressing native alpha-synuclein were resistant to West Nile Virus or Ven-ezuelan equine encephalitis viral infection, but when this protein was removed by gene deletion the brain was much more susceptible to infection. Another study correlated expression of alpha-synuclein to viral infection and found that patients who had undergone intestinal allografts and subsequently were infected with norovirus exhibited significantly higher expression of misfolded alpha-synuclein [104] in the gut; which has been implicated as a starting site for PD pathogenesis [105, 106] Based on these finding, one could imagine a general mechanism by which the viral-induced (or PD) increased load of misfolded oligomeric alpha-synuclein could skew the overall protein pool of native synuclein downward. The lowered levels of native synuclein would make these neurons more prone to oxidative stress and subsequent death.
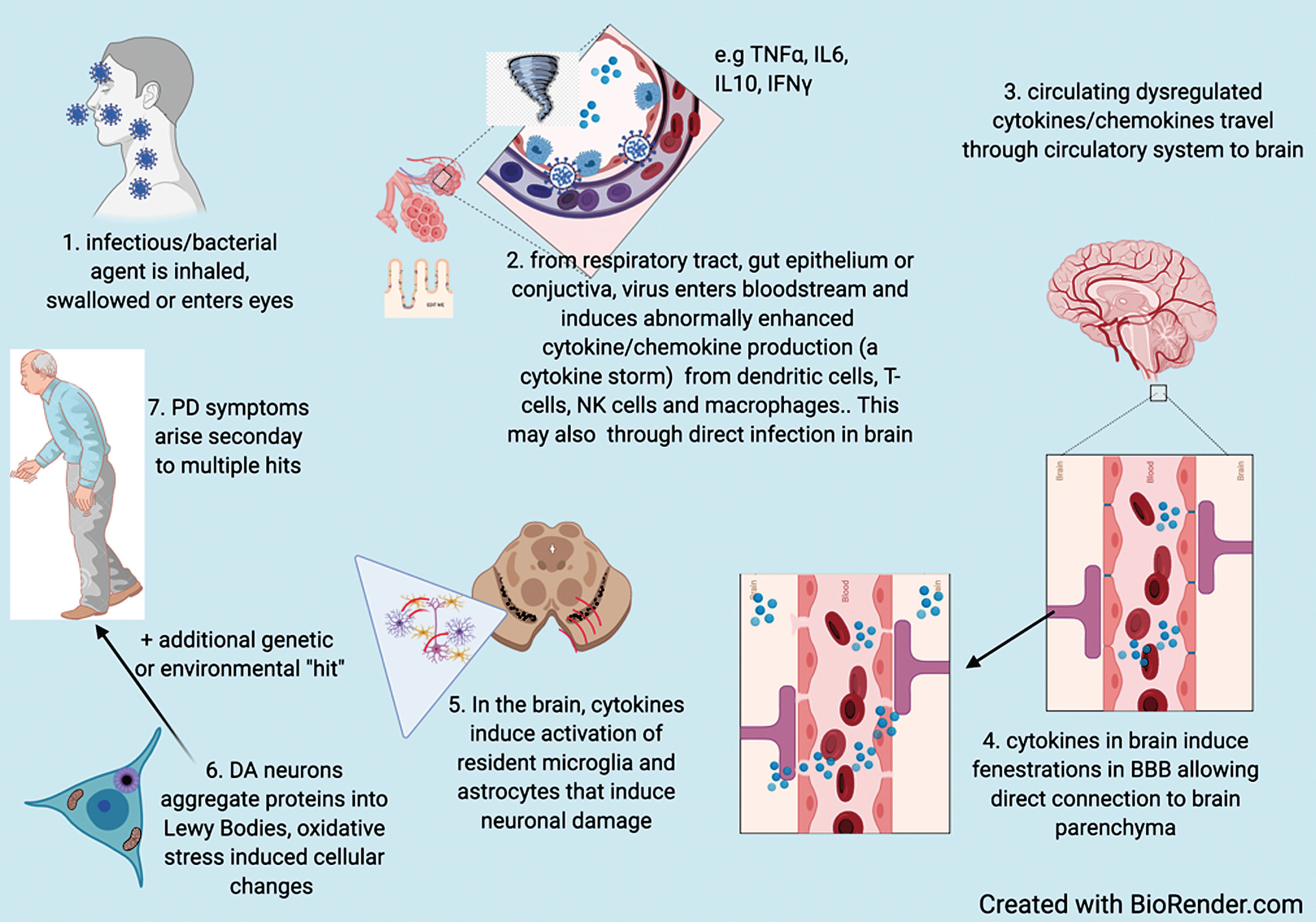
These observations provide plausible mechanisms to explain infection as either a susceptibility or cau-sative factor for PD. The hypothesized process by which infectious agents increase susceptibility to PD is summarized in Fig. 1.
Fig. 1
Based on available evidence, hypothesized process by which infectious agents increase susceptibility to PD.
CONCLUSIONS
Infections are a plausible risk factor for parkinsonism and PD from both epidemiologic and basic science evidence. The magnitude of the risk is unknown for most agents but from the available epidemiologic studies appears to be overall small, with a less than doubling of risk observed in most studies. In addition, the latency from infection to PD appears to be highly variable and often long. From these data it is difficult to justify recommendations for specific monitoring for PD in individuals with previous or ongoing infection.
On the other hand, the pathophysiological insights provided by infection and its apparent relationship to dopaminergic neuron loss and thus PD susceptibility provide important leads for treatment strategies that could be effective in the prodromal period. As suggested by epidemiologic evidence related to hepatitis C, treatment does appear to mitigate the risk. Furthermore, the mechanisms discussed above by which infection may contribute to PD are not unique to infection but share features with other environmental insults such as chemical exposures or head trauma, as each appear to induce pathology by a mechanism that has neuroinflammation as a key part of the process [42, 107–110]. Heretofore, treatments have not been routinely applied to exposed individuals with a view to managing risk of future neurodegenerative disease, but this could be a useful and feasible strategy if used in a targeted way.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
Dr. Smeyne receives research support from the National Institute of Neurological Disorders and Str-oke and the National Institute of Environmental Health Science.
Dr. Marras Receives research support from the Parkinson’s Foundation (US), Michael J Fox Foundation, Canadian Institutes of Health Research and is a site investigator for a clinical trial supported by Theravance.
Dr. Savica receives research support from the National Institute on Aging, the National Institute of Neurological Disorders and Stroke, and the Mayo Clinic Small Grants Program National Center for Advancing Translational Sciences (NCATS) and Acadia Pharmaceuticals Inc.
Dr. Noyce is funded by the Barts Charity. He reports additional grants from Parkinson’s UK, Ali-gning Science Across Parkinson’s (ASAP) and Michael J Fox Foundation (MJFF), the Virginia Kieley Benefaction, grants and non-financial support from GE Healthcare, and personal fees from Bial, Britannia, AbbVie, Profile, F. Hoffmann-La Roche and Biogen.
REFERENCES
[1] | Nalls MA , Blauwendraat C , Vallerga CL , Heilbron K , Bandres-Ciga S , Chang D , Tan M , Kia DA , Noyce AJ , Xue A , Bras J , Young E , von Coelln R , Simón-Sánchez J , Schulte C , Sharma M , Krohn L , Pihlstrøm L , Siitonen A , Iwaki H , Leonard H , Faghri F , Gibbs JR , Hernandez DG , Scholz SW , Botia JA , Martinez M , Corvol JC , Lesage S , Jankovic J , Shulman LM , Sutherland M , Tienari P , Majamaa K , Toft M , Andreassen OA , Bangale T , Brice A , Yang J , Gan-Or Z , Gasser T , Heutink P , Shulman JM , Wood NW , Hinds DA , Hardy JA , Morris HR , Gratten J , Visscher PM , Graham RR , Singleton AB ((2019) ) Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol 18: , 1091–1102. |
[2] | Chen H , Ritz B ((2018) ) The search for environmental causes of Parkinson’s disease: Moving forward. J Parkinsons Dis 8: , S9–s17. |
[3] | Marras C , Canning CG , Goldman SM ((2019) ) Environment, lifestyle, and Parkinson’s disease: Implications for prevention in the next decade. Mov Disord 34: , 801–811. |
[4] | Henry J , Smeyne RJ , Jang H , Miller B , Okun MS ((2010) ) Parkinsonism and neurological manifestations of influenza throughout the 20th and 21st centuries. Parkinsonism Relat Disord 16: , 566–571. |
[5] | Casals J , Elizan TS , Yahr MD ((1998) ) Postencephalitic parkinsonism–a review. J Neural Transm (Vienna) 105: , 645–676. |
[6] | Rebai I , Ben Rhouma H , Kraoua I , Klaa H , Rouissi A , Ben Youssef-Turki I , Gouider-Khouja N ((2015) ) Postencephalitic parkinsonism and selective involvement of substantia nigra in childhood. Brain Dev 37: , 153–157. |
[7] | Hawkes CH , Del Tredici K , Braak H ((2007) ) Parkinson’s disease: A dual-hit hypothesis. Neuropathol Appl Neurobiol 33: , 599–614. |
[8] | Pont-Sunyer C , Hotter A , Gaig C , Seppi K , Compta Y , Katzenschlager R , Mas N , Hofeneder D , Brücke T , Bayés A , Wenzel K , Infante J , Zach H , Pirker W , Posada IJ , Álvarez R , Ispierto L , De Fábregues O , Callén A , Palasí A , Aguilar M , Martí MJ , Valldeoriola F , Salamero M , Poewe W , Tolosa E ((2015) ) The onset of nonmotor symptoms in Parkinson’s disease (the ONSET PD study). Mov Disord 30: , 229–237. |
[9] | Pradhan S , Andreasson K ((2013) ) Commentary: Progressive inflammation as a contributing factor to early development of Parkinson’s disease. Exp Neurol 241: , 148–155. |
[10] | Tsui JK , Calne DB , Wang Y , Schulzer M , Marion SA ((1999) ) Occupational risk factors in Parkinson’s disease. Can J Public Health 90: , 334–337. |
[11] | Patrick KL , Bell SL , Weindel CG , Watson RO ((2019) ) Exploring the “Multiple-Hit Hypothesis” of neurodegenerative disease: Bacterial infection comes up to bat. Front Cell Infect Microbiol 9: , 138. |
[12] | Mattson MP ((2004) ) Infectious agents and age-related neurodegenerative disorders. Ageing Res Rev 3: , 105–120. |
[13] | Umemura A , Oeda T , Tomita S , Hayashi R , Kohsaka M , Park K , Sugiyama H , Sawada H ((2014) ) Delirium and high fever are associated with subacute motor deterioration in Parkinson disease: A nested case-control study. PLoS One 9: , e94944. |
[14] | von Economo K ((1917) ) Encepahlitis lethargica. Wien Klin Wochenschr 30: , 581–585. |
[15] | McCall S , Henry JM , Reid AH , Taubenberger JK ((2001) ) Influenza RNA not detected in archival brain tissues from acute encephalitis lethargica cases or in postencephalitic Parkinson cases. J Neuropathol Exp Neurol 60: , 696–704. |
[16] | Kapila CC , Kaul S , Kapur SC , Kalayanam TS , Banerjee D ((1958) ) Neurological and hepatic disorders associated with influenza. Br Med J 2: , 1311–1314. |
[17] | Hosseini S , Wilk E , Michaelsen-Preusse K , Gerhauser I , Baumgärtner W , Geffers R , Schughart K , Korte M ((2018) ) Long-term neuroinflammation induced by influenza A virus infection and the impact on hippocampal neuron morphology and function. J Neurosci 38: , 3060–3080. |
[18] | Sadasivan S , Zanin M , O’Brien K , Schultz-Cherry S , Smeyne RJ ((2015) ) Induction of microglia activation after infection with the non-neurotropic A/CA/04/2009 H1N1 influenza virus. PLoS One 10: , e0124047. |
[19] | Ferrara JL , Abhyankar S , Gilliland DG ((1993) ) Cytokine storm of graft-versus-host disease: A critical effector role for interleukin-1. Transplant Proc 25: , 1216–1217. |
[20] | Harry GJ , Kraft AD ((2008) ) Neuroinflammation and microglia: Considerations and approaches for neurotoxicity assessment. Expert Opin Drug Metab Toxicol 4: , 1265–1277. |
[21] | Sulzer D ((2007) ) Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci 30: , 244–250. |
[22] | Jang H , Boltz DA , Webster RG , Smeyne RJ ((2009) ) Viral parkinsonism. Biochim Biophys Acta 1792: , 714–721. |
[23] | Wang H , Liu X , Tan C , Zhou W , Jiang J , Peng W , Zhou X , Mo L , Chen L ((2020) ) Bacterial, viral, and fungal infection-related risk of Parkinson’s disease: Meta-analysis of cohort and case–control studies. Brain Behav 10: , e01549. |
[24] | Polidoro RB , Hagan RS , de Santis Santiago R , Schmidt NW ((2020) ) Overview: Systemic inflammatory response derived from lung injury caused by SARS-CoV-2 infection explains severe outcomes in COVID-19. Front Immunol 11: , 1626. |
[25] | GBD 2015 Disease and Injury Incidence and Prevalence Collaborators ((2016) ) Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 388: , 1545–1602. |
[26] | Forton DM , Allsop JM , Main J , Foster GR , Thomas HC , Taylor-Robinson SD ((2001) ) Evidence for a cerebral effect of the hepatitis C virus. Lancet 358: , 38–39. |
[27] | Wilkinson J , Radkowski M , Laskus T ((2009) ) Hepatitis C virus neuroinvasion: Identification of infected cells. J Virol 83: , 1312–1319. |
[28] | Wu WY-Y , Kang K-H , Chen SL-S , Chiu SY-H , Yen AM-F , Fann JC-Y , Su C-W , Liu H-C , Lee C-Z , Fu W-M , Chen H-H , Liou H-H ((2015) ) Hepatitis C virus infection: A risk factor for Parkinson’s disease. J Viral Hepatitis 22: , 784–791. |
[29] | Tsai HH , Liou HH , Muo CH , Lee CZ , Yen RF , Kao CH ((2016) ) Hepatitis C virus infection as a risk factor for Parkinson disease: A nationwide cohort study. Neurology 86: , 840–846. |
[30] | Pakpoor J , Noyce A , Goldacre R , Selkihova M , Mullin S , Schrag A , Lees A , Goldacre M ((2017) ) Viral hepatitis and Parkinson disease: A national record-linkage study. Neurology 88: , 1630–1633. |
[31] | Wijarnpreecha K , Chesdachai S , Jaruvongvanich V , Ungprasert P ((2018) ) Hepatitis C virus infection and risk of Parkinson’s disease: A systematic review and meta-analysis. Eur J Gastroenterol Hepatol 30: , 9–13. |
[32] | Goldstein L , Fogel-Grinvald H , Steiner I ((2019) ) Hepatitis B and C virus infection as a risk factor for Parkinson’s disease in Israel-A nationwide cohort study. J Neurol Sci 398: , 138–141. |
[33] | Lin W-Y , Lin M-S , Weng Y-H , Yeh T-H , Lin Y-S , Fong P-Y , Wu Y-R , Lu C-S , Chen R-S , Huang Y-Z ((2019) ) Association of antiviral therapy with risk of Parkinson disease in patients with chronic hepatitis C virus infection. JAMA Neurol 76: , 1019–1027. |
[34] | Su T-H , Yang H-C , Tseng T-C , Chou S-W , Lin C-H , Liu C-H , Liu C-J , Chen C-L , Kao J-H ((2019) ) Antiviral therapy in patients with chronic hepatitis C is associated with a reduced risk of parkinsonism. Mov Disord 34: , 1882–1890. |
[35] | Working Group of the American Academy of Neurology AIDS Task Force ((1991) ) Nomenclature and research case definitions for neurologic manifestations of human immunodeficiency virus-type 1 (HIV-1) infection. Neurology 41: , 778–778. |
[36] | Mirsattari SM , Power C , Nath A ((1998) ) Parkinsonism with HIV infection. Mov Disord 13: , 684–689. |
[37] | Tisch S , Brew BJ ((2010) ) HIV, HAART, and Parkinson’s disease: Co-incidence or pathogenetic link? Mov Disord 25: , 2257–2258. |
[38] | Tse W , Cersosimo MG , Gracies JM , Morgello S , Olanow CW , Koller W ((2004) ) Movement disorders and AIDS: A review. Parkinsonism Relat Disord 10: , 323–334. |
[39] | Heaton RK , Franklin DR , Ellis RJ , McCutchan JA , Letendre SL , Leblanc S , Corkran SH , Duarte NA , Clifford DB , Woods SP , Collier AC , Marra CM , Morgello S , Mindt MR , Taylor MJ , Marcotte TD , Atkinson JH , Wolfson T , Gelman BB , McArthur JC , Simpson DM , Abramson I , Gamst A , Fennema-Notestine C , Jernigan TL , Wong J , Grant I ((2011) ) HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: Differences in rates, nature, and predictors. J Neurovirol 17: , 3–16. |
[40] | Siangphoe U , Archer KJ , Nguyen C , Lee KR ((2020) ) Associations of antiretroviral therapy and comorbidities with neurocognitive outcomes in HIV-1-infected patients. AIDS 34: , 893–902. |
[41] | Dehner LF , Spitz M , Pereira JS ((2016) ) Parkinsonism in HIV infected patients during antiretroviral therapy - data from a Brazilian tertiary hospital. Braz J Infect Dis 20: , 499–501. |
[42] | Liu B , Gao HM , Hong JS ((2003) ) Parkinson’s disease and exposure to infectious agents and pesticides and the occurrence of brain injuries: Role of neuroinflammation. Environ Health Perspect 111: , 1065–1073. |
[43] | Pische G , Koob M , Wirth T , Quenardelle V , Lagha-Boukbiza O , Renaud M , Anheim M , Tranchant C ((2017) ) Subacute parkinsonism as a complication of Lyme disease. J Neurol 264: , 1015–1019. |
[44] | Blum K , Modestino EJ , Febo M , Steinberg B , McLaughlin T , Fried L , Baron D , Siwicki D , Badgaiyan RD ((2017) ) Lyme and dopaminergic function: Hypothesizing reduced reward deficiency symptomatology by regulating dopamine transmission. J Syst Integr Neurosci 3: , doi: 10.15761/JSIN.1000163 |
[45] | Cassarino DS , Quezado MM , Ghatak NR , Duray PH ((2003) ) Lyme-associated parkinsonism: A neuropathologic case study and review of the literature. Arch Pathol Lab Med 127: , 1204–1206. |
[46] | Forrester JD , Kugeler KJ , Perea AE , Pastula DM , Mead PS ((2015) ) No geographic correlation between lyme disease and death due to 4 neurodegenerative disorders, United States, 2001-2010. Emerg Infect Dis 21: , 2036–2039. |
[47] | Haahr R , Tetens MM , Dessau RB , Krogfelt KA , Bodilsen J , Andersen NS , Moller JK , Roed C , Christiansen CB , Ellermann-Eriksen S , Bangsborg JM , Hansen K , Benfield TL , Ostergaard Andersen C , Obel N , Lebech AM , Omland LH ((2019) ) Risk of neurological disorders in patients with European Lyme neuroborreliosis. A nationwide population-based cohort study. Clin Infect Dis 71: , 1511–1516. |
[48] | McGee DJ , Lu XH , Disbrow EA ((2018) ) Stomaching the possibility of a pathogenic role for Helicobacter pylori in Parkinson’s disease. J Parkinsons Dis 8: , 367–374. |
[49] | Schwab RS ((1961) ) Symptomatology and medical treatment of Parkinson’s disease. Int J Neurol 2: , 61–75. |
[50] | Svensson E , Horvath-Puho E , Thomsen RW , Djurhuus JC , Pedersen L , Borghammer P , Sorensen HT ((2015) ) Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol 78: , 522–529. |
[51] | Shen X , Yang H , Wu Y , Zhang D , Jiang H ((2017) ) Meta-analysis: Association of Helicobacter pylori infection with Parkinson’s diseases. Helicobacter 22: , e12398. |
[52] | Huang HK , Wang JH , Lei WY , Chen CL , Chang CY , Liou LS ((2018) ) Helicobacter pylori infection is associated with an increased risk of Parkinson’s disease: A population-based retrospective cohort study. Parkinsonism Relat Disord 47: , 26–31. |
[53] | Tan AH , Mahadeva S , Marras C , Thalha AM , Kiew CK , Yeat CM , Ng SW , Ang SP , Chow SK , Loke MF , Vadivelu JS , Ibrahim N , Yong HS , Tan CT , Fox SH , Lang AE , Lim SY ((2015) ) Helicobacter pylori infection is associated with worse severity of Parkinson’s disease. Parkinsonism Relat Disord 21: , 221–225. |
[54] | Rahne KE , Tagesson C , Nyholm D ((2013) ) Motor fluctuations and Helicobacter pylori in Parkinson’s disease. J Neurol 260: , 2974–2980. |
[55] | Kohbata S , Beaman BL ((1991) ) L-dopa-responsive movement disorder caused by Nocardia asteroides localized in the brains of mice. Infect Immun 59: , 181–191. |
[56] | Tam S , Barry DP , Beaman L , Beaman BL ((2002) ) Neuroinvasive Nocardia asteroides GUH-2 induces apoptosis in the substantia nigra in vivo and dopaminergic cells in vitro. Exp Neurol 177: , 453–460. |
[57] | Kohbata S , Shimokawa K ((1993) ) Circulating antibody to Nocardia in the serum of patients with Parkinson’s disease. Adv Neurol 60: , 355–357. |
[58] | Elkind MS , Ramakrishnan P , Moon YP , Boden-Albala B , Liu KM , Spitalnik SL , Rundek T , Sacco RL , Paik MC ((2010) ) Infectious burden and risk of stroke: The northern Manhattan study. Arch Neurol 67: , 33–38. |
[59] | Bu XL , Yao XQ , Jiao SS , Zeng F , Liu YH , Xiang Y , Liang CR , Wang QH , Wang X , Cao HY , Yi X , Deng B , Liu CH , Xu J , Zhang LL , Gao CY , Xu ZQ , Zhang M , Wang L , Tan XL , Xu X , Zhou HD , Wang YJ ((2015) ) A study on the association between infectious burden and Alzheimer’s disease. Eur J Neurol 22: , 1519–1525. |
[60] | Hasan S , Mielke MM , Ahlskog JE , Bower J , Turcano P , Savica R ((2020) ) Infections or sepsis preceding clinically diagnosed α-synucleinopathies: A case-control study. Mov Disord 35: , 1684–1689. |
[61] | Bhattacharyya D , Mohite GM , Krishnamoorthy J , Gayen N , Mehra S , Navalkar A , Kotler SA , Ratha BN , Ghosh A , Kumar R , Garai K , Mandal AK , Maji SK , Bhunia A ((2019) ) Lipopolysaccharide from Gut Microbiota Modulates alpha-Synuclein Aggregation and Alters Its Biological Function. ACS Chem Neurosci 10: , 2229–2236. |
[62] | Kim C , Lv G , Lee JS , Jung BC , Masuda-Suzukake M , Hong CS , Valera E , Lee HJ , Paik SR , Hasegawa M , Masliah E , Eliezer D , Lee SJ ((2016) ) Exposure to bacterial endotoxin generates a distinct strain of alpha-synuclein fibril. Sci Rep 6: , 30891. |
[63] | Chen SG , Stribinskis V , Rane MJ , Demuth DR , Gozal E , Roberts AM , Jagadapillai R , Liu R , Choe K , Shivakumar B , Son F , Jin S , Kerber R , Adame A , Masliah E , Friedland RP ((2016) ) Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged Fischer 344 rats and Caenorhabditis elegans. Sci Rep 6: , 34477. |
[64] | Zhernakova A , Kurilshikov A , Bonder MJ , Tigchelaar EF , Schirmer M , Vatanen T , Mujagic Z , Vila AV , Falony G , Vieira-Silva S , Wang J , Imhann F , Brandsma E , Jankipersadsing SA , Joossens M , Cenit MC , Deelen P , Swertz MA , LifeLines cohort study, Weersma RK , Feskens EJ , Netea MG , Gevers D , Jonkers D , Franke L , Aulchenko YS , Huttenhower C , Raes J , Hofker MH , Xavier RJ , Wijmenga C , Fu J ((2016) ) Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352: , 565–569. |
[65] | Sampson TR , Debelius JW , Thron T , Janssen S , Shastri GG , Ilhan ZE , Challis C , Schretter CE , Rocha S , Gradinaru V , Chesselet MF , Keshavarzian A , Shannon KM , Krajmalnik-Brown R , Wittung-Stafshede P , Knight R , Mazmanian SK ((2016) ) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167: , 1469–1480 e1412. |
[66] | Scheperjans F , Aho V , Pereira PA , Koskinen K , Paulin L , Pekkonen E , Haapaniemi E , Kaakkola S , Eerola-Rautio J , Pohja M , Kinnunen E , Murros K , Auvinen P ((2015) ) Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord 30: , 350–358. |
[67] | Hasegawa S , Goto S , Tsuji H , Okuno T , Asahara T , Nomoto K , Shibata A , Fujisawa Y , Minato T , Okamoto A , Ohno K , Hirayama M ((2015) ) Intestinal dysbiosis and lowered serum lipopolysaccharide-binding protein in Parkinson’s disease. PLoS One 10: , e0142164. |
[68] | Killinger BA , Madaj Z , Sikora JW , Rey N , Haas AJ , Vepa Y , Lindqvist D , Chen H , Thomas PM , Brundin P , Brundin L , Labrie V ((2018) ) The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci Transl Med 10: , eaar5280. |
[69] | Marras C , Lang AE , Austin PC , Lau C , Urbach DR ((2016) ) Appendectomy in mid and later life and risk of Parkinson’s disease: A population-based study. Mov Disord 31: , 1243–1247. |
[70] | Svensson E , Horváth-Puhó E , Stokholm MG , Sørensen HT , Henderson VW , Borghammer P ((2016) ) Appendectomy and risk of Parkinson’s disease: A nationwide cohort study with more than 10 years of follow-up. Mov Disord 31: , 1918–1922. |
[71] | Schultz DR , Barthal JS , Garrett G ((1977) ) Western equine encephalitis with rapid onset of parkinsonism. Neurology 27: , 1095–1096. |
[72] | Bantle CM , Phillips AT , Smeyne RJ , Rocha SM , Olson KE , Tjalkens RB ((2019) ) Infection with mosquito-borne alphavirus induces selective loss of dopaminergic neurons, neuroinflammation and widespread protein aggregation. NPJ Parkinsons Dis 5: , 20. |
[73] | Jang H , Boltz D , McClaren J , Pani AK , Smeyne M , Korff A , Webster R , Smeyne RJ ((2012) ) Inflammatory effects of highly pathogenic H5N1 influenza virus infection in the CNS of mice. J Neurosci 32: , 1545–1559. |
[74] | Jang H , Boltz D , Sturm-Ramirez K , Shepherd KR , Jiao Y , Webster R , Smeyne RJ ((2009) ) Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A 106: , 14063–14068. |
[75] | Phillips AT , Stauft CB , Aboellail TA , Toth AM , Jarvis DL , Powers AM , Olson KE ((2013) ) Bioluminescent imaging and histopathologic characterization of WEEV neuroinvasion in outbred CD-1 mice. PLoS One 8: , e53462. |
[76] | Marreiros R , Müller-Schiffmann A , Trossbach SV , Prikulis I , Hänsch S , Weidtkamp-Peters S , Moreira AR , Sahu S , Soloviev I , Selvarajah S , Lingappa VR , Korth C ((2020) ) Disruption of cellular proteostasis by H1N1 influenza A virus causes α-synuclein aggregation. Proc Natl Acad Sci U S A 117: , 6741–6751. |
[77] | Tisoncik JR , Korth MJ , Simmons CP , Farrar J , Martin TR , Katze MG ((2012) ) Into the eye of the cytokine storm. Microbiol Mol Biol Rev 76: , 16–32. |
[78] | Park WY , Goodman RB , Steinberg KP , Ruzinski JT , Radella F , 2nd Park DR , Pugin J , Skerrett SJ , Hudson LD , Martin TR ((2001) ) Cytokine balance in the lungs of patients with acute respiratory distress syndrome. Am J Respir Crit Care Med 164: , 1896–1903. |
[79] | Banks WA , Kastin AJ , Broadwell RD ((1995) ) Passage of cytokines across the blood-brain barrier. Neuroimmunomodulation 2: , 241–248. |
[80] | Basler CF ((2017) ) Molecular pathogenesis of viral hemorrhagic fever. Semin Immunopathol 39: , 551–561. |
[81] | Steinberg BE , Goldenberg NM , Lee WL ((2012) ) Do viral infections mimic bacterial sepsis? The role of microvascular permeability: A review of mechanisms and methods. Antiviral Res 93: , 2–15. |
[82] | da Fonseca AC , Matias D , Garcia C , Amaral R , Geraldo LH , Freitas C , Lima FR ((2014) ) The impact of microglial activation on blood-brain barrier in brain diseases. Front Cell Neurosci 8: , 362. |
[83] | Benveniste EN , Benos DJ ((1995) ) TNF-alpha- and IFN-gamma-mediated signal transduction pathways: Effects on glial cell gene expression and function. FASEB J 9: , 1577–1584. |
[84] | Brochard V , Combadiere B , Prigent A , Laouar Y , Perrin A , Beray-Berthat V , Bonduelle O , Alvarez-Fischer D , Callebert J , Launay JM , Duyckaerts C , Flavell RA , Hirsch EC , Hunot S ((2009) ) Infiltration of CD4+lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 119: , 182–192. |
[85] | McGeer PL , Itagaki S , Akiyama H , McGeer EG ((1988) ) Rate of cell death in parkinsonism indicates active neuropathological process. Ann Neurol 24: , 574–576. |
[86] | Ip CW , Beck SK , Volkmann J ((2015) ) Lymphocytes reduce nigrostriatal deficits in the 6-hydroxydopamine mouse model of Parkinson’s disease. J Neural Transm (Vienna) 122: , 1633–1643. |
[87] | Dutta D , Kundu M , Mondal S , Roy A , Ruehl S , Hall DA , Pahan K ((2019) ) RANTES-induced invasion of Th17 cells into substantia nigra potentiates dopaminergic cell loss in MPTP mouse model of Parkinson’s disease. Neurobiol Dis 132: , 104575. |
[88] | Subbarayan MS , Hudson C , Moss LD , Nash KR , Bickford PC ((2020) ) T cell infiltration and upregulation of MHCII in microglia leads to accelerated neuronal loss in an α-synuclein rat model of Parkinson’s disease. J Neuroinflammation 17: , 242. |
[89] | Jimenez-Ferrer I , Swanberg M ((2018) ) Immunogenetics of Parkinson’s disease. In Parkinson’s Disease: Pathogenesis and Clinical Aspects, Stoker TB, Greenland JC, eds. Codon Publications, Brisbane. |
[90] | Harms AS , Cao S , Rowse AL , Thome AD , Li X , Mangieri LR , Cron RQ , Shacka JJ , Raman C , Standaert DG ((2013) ) MHCII is required for alpha-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J Neurosci 33: , 9592–9600. |
[91] | Sulzer D , Alcalay RN , Garretti F , Cote L , Kanter E , Agin-Liebes J , Liong C , McMurtrey C , Hildebrand WH , Mao X , Dawson VL , Dawson TM , Oseroff C , Pham J , Sidney J , Dillon MB , Carpenter C , Weiskopf D , Phillips E , Mallal S , Peters B , Frazier A , Lindestam Arlehamn CS , Sette A ((2017) ) T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 546: , 656–661. |
[92] | Kim WG , Mohney RP , Wilson B , Jeohn GH , Liu B , Hong JS ((2000) ) Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J Neurosci 20: , 6309–6316. |
[93] | Smeyne M , Jiao Y , Shepherd KR , Smeyne RJ ((2005) ) Glia cell number modulates sensitivity to MPTP in mice. Glia 52: , 144–152. |
[94] | Barcia C , Ros CM , Annese V , Gómez A , Ros-Bernal F , Aguado-Yera D , Martínez-Pagán ME , de Pablos V , Fernandez-Villalba E , Herrero MT ((2011) ) IFN-γ signaling, with the synergistic contribution of TNF-α, mediates cell specific microglial and astroglial activation in experimental models of Parkinson’s disease. Cell Death Dis 2: , e142. |
[95] | Tansey MG , Goldberg MS ((2010) ) Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis 37: , 510–518. |
[96] | Tansey MG , McCoy MK , Frank-Cannon TC ((2007) ) Neuroinflammatory mechanisms in Parkinson’s disease: Potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol 208: , 1–25. |
[97] | Rothwell NJ ((1999) ) Annual review prize lecture cytokines - killers in the brain? J Physiol 514: (Pt 1), 3–17. |
[98] | Hayes GM , Woodroofe MN , Cuzner ML ((1988) ) Microglia express MHC class II in normal and demyelinating human white matter. Ann N Y Acad Sci 540: , 501–503. |
[99] | Hayes GM , Woodroofe MN , Cuzner ML ((1987) ) Microglia are the major cell type expressing MHC class II in human white matter. J Neurol Sci 80: , 25–37. |
[100] | Rostami J , Fotaki G , Sirois J , Mzezewa R , Bergström J , Essand M , Healy L , Erlandsson A ((2020) ) Astrocytes have the capacity to act as antigen-presenting cells in the Parkinson’s disease brain. J Neuroinflammation 17: , 119. |
[101] | Yang TT , Lin C , Hsu CT , Wang TF , Ke FY , Kuo YM ((2013) ) Differential distribution and activation of microglia in the brain of male C57BL/6J mice. Brain Struct Funct 218: , 1051–1060. |
[102] | Yao YY , Ling EA , Lu D ((2020) ) Microglia mediated neuroinflammation - signaling regulation and therapeutic considerations with special reference to some natural compounds. Histol Histopathol, doi: 10.14670/HH-18-239. |
[103] | Beatman EL , Massey A , Shives KD , Burrack KS , Chamanian M , Morrison TE , Beckham JD ((2015) ) Alpha-synuclein expression restricts RNA viral infections in the brain. J Virol 90: , 2767–2782. |
[104] | Stolzenberg E , Berry D , Yang D , Lee EY , Kroemer A , Kaufman S , Wong GCL , Oppenheim JJ , Sen S , Fishbein T , Bax A , Harris B , Barbut D , Zasloff MA ((2017) ) A role for neuronal alpha-synuclein in gastrointestinal immunity. J Innate Immun 9: , 456–463. |
[105] | Kim S , Kwon SH , Kam TI , Panicker N , Karuppagounder SS , Lee S , Lee JH , Kim WR , Kook M , Foss CA , Shen C , Lee H , Kulkarni S , Pasricha PJ , Lee G , Pomper MG , Dawson VL , Dawson TM , Ko HS ((2019) ) Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103: , 627–641.e627. |
[106] | Braak H , Rüb U , Gai WP , Del Tredici K ((2003) ) Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm (Vienna) 110: , 517–536. |
[107] | Acosta SA , Tajiri N , de la Pena I , Bastawrous M , Sanberg PR , Kaneko Y , Borlongan CV ((2015) ) Alpha-synuclein as a pathological link between chronic traumatic brain injury and Parkinson’s disease. J Cell Physiol 230: , 1024–1032. |
[108] | Costa LG , Cole TB , Dao K , Chang YC , Coburn J , Garrick JM ((2020) ) Effects of air pollution on the nervous system and its possible role in neurodevelopmental and neurodegenerative disorders. Pharmacol Ther 210: , 107523. |
[109] | De Miranda BR , Greenamyre JT ((2020) ) Trichloroethylene, a ubiquitous environmental contaminant in the risk for Parkinson’s disease. Environ Sci Process Impacts 22: , 543–554. |
[110] | Kanthasamy A , Jin H , Charli A , Vellareddy A , Kanthasamy A ((2019) ) Environmental neurotoxicant-induced dopaminergic neurodegeneration: A potential link to impaired neuroinflammatory mechanisms. Pharmacol Ther 197: , 61–82. |


