
GDNF and Parkinson’s Disease: Where Next? A Summary from a Recent Workshop
Abstract
The concept of repairing the brain with growth factors has been pursued for many years in a variety of neurodegenerative diseases including primarily Parkinson’s disease (PD) using glial cell line-derived neurotrophic factor (GDNF). This neurotrophic factor was discovered in 1993 and shown to have selective effects on promoting survival and regeneration of certain populations of neurons including the dopaminergic nigrostriatal pathway. These observations led to a series of clinical trials in PD patients including using infusions or gene delivery of GDNF or the related growth factor, neurturin (NRTN). Initial studies, some of which were open label, suggested that this approach could be of value in PD when the agent was injected into the putamen rather than the cerebral ventricles. In subsequent double-blind, placebo-controlled trials, the most recent reporting in 2019, treatment with GDNF did not achieve its primary end point. As a result, there has been uncertainty as to whether GDNF (and by extrapolation, related GDNF family neurotrophic factors) has merit in the future treatment of PD. To critically appraise the existing work and its future, a special workshop was held to discuss and debate this issue. This paper is a summary of that meeting with recommendations on whether there is a future for this therapeutic approach and also what any future PD trial involving GDNF and other GDNF family neurotrophic factors should consider in its design.
INTRODUCTION
The discovery and characterisation of specific neurotrophic factors in the context of neuronal development and synapse formation in the last half of the 20th century, led to the hypothesis that certain adult neuronal populations lost to chronic disease processes might be rescued and potentially regenerated by the administration of these agents [1]. This has been extensively explored in Parkinson’s disease (PD) with the use of GDNF and related factor NRTN and the dopaminergic (DA) nigrostriatal pathway- the loss of which is known to be central and critical to the development and clinical expression of this condition [2].
In this short paper, we critically appraise the pre-clinical and clinical trial work with GDNF and NRTN in patients with PD. This appraisal is based on a meeting held over 2 days in August 2019 that brought together experts who had direct and practical experience in this field. The timing of this meeting was linked to the recent publication of a UK-based clinical trial and parallel airing on the BBC of the two-part documentary “The Parkinson’s Drug Trial: A Miracle Cure? [https://www.bbc.co.uk/mediacentre/proginfo/2019/09/parkinsons-drug-trial-a-miracle-cure? The meeting was organised and funded by The Cure Parkinson’s Trust and supported by The Michael J Fox Foundation and Van Andel Institute.
THE PRECLINICAL EVIDENCE THAT GDNF CAN RESCUE THE NIGROSTRIATAL PATHWAY
The discovery of GDNF in 1993 was made at a time of great interest in the therapeutic development of neurotrophic factors which offered potential for treating a number of disease states. The search for a survival factor with high selectivity for midbrain DA neurons had already been going on for some time. As such, when Lin et al. reported the cloning and bioactivity of this new trophic factor in 1993 there was great excitement [3]. Indeed, this in part helps explain why there was such a short time span between the first pre-clinical in vivo studies (performed and published in 1994-95) and the first clinical trial with this agent [4] which started recruiting patients in July 1996.
The second member of the GDNF family of ligands, NRTN, was discovered in 1996 [5] along with the receptor signaling pathways for these 2 factors [6]. This work revealed that while GDNF and NRTN are members of the transforming growth factor beta (TGF-β) family, they signal through a completely different receptor system compared to other TGF-β family members. GDNF first binds to the glycosylphosphatidylinositol (GPI)-anchored co-receptor GDNF family receptor alpha-1 (GFRα1) and then the GDNF-GFRα1 complex binds to, and activates the transmembrane receptor tyrosine kinase RET. NRTN likewise signals to the cells via the RET receptor, but its binding to RET is mediated through the GFRα2 co-receptor although when delivered at high levels it also can bind to GFRα1 [6]. RET then activates the intracellular mitogen-activated protein kinase (MAPK), Akt (protein kinase B) and Src signaling cascades that are responsible for the survival and regeneration of DA neurons. It is important to stress that GDNF and NRTN trigger rapid responses in DA neurons through protein phosphorylation, but in addition to that they activate a number of transcription factors that have longer-lasting effects on DA neurons
The initial work with GDNF was made possible through having access to a recombinant human form of protein from Synergen and Genentech. This enabled the generation of preliminary in vivo data on DA neuroprotection in the three rodent PD models available at the time: the rat 6 hydroxydopamine (6-OHDA) model [7, 8], the mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model [9] and the knife-transection model [10]. In addition, a study performed in intact rats showed that GDNF, administered into the substantia nigra, could stimulate DA neuronal function [11]. The fact that the findings from all of these studies provided positive evidence in the same direction was re-assuring – namely a growth factor that seemed to work on DA neuronal rescue and regeneration.
These initial preclinical studies used intracerebral administration of GDNF, (single or repeated injections of microgram amounts over the substantia nigra). Nevertheless, Amgen, the company that had acquired the rights to GDNF, opted for an intraventricular delivery approach in its first clinical trial [4] (see below). A critical factor in this decision were the results of a study, sponsored by Amgen, reporting significant, dose-dependent beneficial effects obtained by monthly intraventricular injections of GDNF in MPTP-treated rhesus monkeys [12]. The results again looked promising: all the major motor features (bradykinesia, rigidity, posture and balance) were improved when assessed 4 weeks after the last (fourth) monthly injection. Doubts on this mode of administration, however, soon arose: studies on the distribution of GDNF after intraventricular delivery indicated very limited diffusion into the brain parenchyma given its strong binding to extracellular matrix and cell surface heparin sulphate proteoglycans [13, 14]. Furthermore, a follow-up study by another team, performed in the same monkey model, failed to show any protective effect on the MPTP-lesioned DA neurons [15].
While this trial was ongoing, experimental work performed in rodent and primate PD models provided further and more compelling evidence that GDNF must be administered directly into the brain parenchyma to exert its actions, and that the site, dose and timing of GDNF delivery are important. Experiments in 6-OHDA lesioned rats showed that delivery into the substantia nigra could rescue DA neuronal cell bodies against toxic damage but failed to protect their axonal projections in the striatum [16]. Rescue of both DA neuronal cell bodies and their axons projecting to the striatum required that the factor be delivered into the striatum, or into both the striatum and substantia nigra, provided that it is given before or soon after the toxin treatment [17]. Furthermore, the timing of the delivery of GDNF was also found to be important. In both rodents and primates there was evidence that delayed intrastriatal GDNF delivery, starting weeks or months after 6-OHDA or MPTP treatment, could still protect surviving DA neurons and stimulate regenerative sprouting from spared axons in the partially denervated striatum [18–21].
Based on all this experimental data it was concluded that the therapeutic potential of GDNF is due to a combination of three interacting mechanisms: 1) Protection of midbrain DA neurons against toxin-induced cell death; 2) stimulation of axonal regeneration in the area reached by GDNF; and 3) recovery of function through up regulation of DA turnover and release.
These experimental studies, performed over the first decade after GDNF’s discovery, were very encouraging and stimulated a series of clinical trials, including some where the agent was given intra-putaminally (see below). At that time, the preclinical data seemed to support such a move to the clinic based on all the studies performed in mice, rats and non-human primates. There were, however, obvious weaknesses in the pre-clinical data: Firstly, GDNF was relatively ineffective in the face of severe lesions giving >80% loss of DA neurons which more closely mimics the human condition. Secondly, the data showing efficacy was obtained in toxin-based models where 6-OHDA or MPTP are administered acutely which results in a degenerative process that is primarily driven by oxidative damage or mitochondrial dysfunction not protein aggregation. Further, these models do not replicate the late stages of PD nor the progressive, alpha-synuclein related pathophysiology that is characteristic of the human disease. Given this difference, there was a concern that the results obtained in toxin-based models may not be predictive for human PD patients receiving this experimental therapy. In addition, there were also concerns about whether the GDNF made at that time using E. coli (as opposed to human recombinant protein made in mammalian cells) would work less well in human patients.
Some of these anxieties have been borne out in more recent experiments. Over the last few years the access to alpha-synuclein-based PD models has allowed this first question to be further explored [22]. These models not only offer better opportunities to more faithfully replicate the alpha-synuclein related pathology seen in people with PD, but the lesions so induced evolve slowly over time in contrast to the far more rapid time course of acute toxin models of PD. Using such alpha-synucleinopathy models, studies designed to reproduce the type of neuroprotective and restorative effects of GDNF, consistently seen in MPTP and 6-OHDA models, have so far failed [23, 24].
In a follow-up study performed in the AAV-alpha-synuclein model [25], Decressac et al. (2012) suggested that this could be due to a failure of alpha-synuclein overexpressing nigral neurons to respond to GDNF. This, the authors postulated, could be due to down regulation of the GDNF receptor RET, mediated by a reduced expression of the DA related transcription factor, nuclear receptor related 1 protein (Nurr1). Nurr1 is known to regulate RET expression in DA neurons [26] and the two are thus closely related.
In human PD postmortem material, Nurr1 has been shown to be down-regulated in DA neurons that over express alpha-synuclein [27] and a similar down regulation of Nurr1 is also seen in alpha-synuclein overexpressing neurons in the AAV model of PD. This, in turn, is accompanied by a marked reduction in the expression of RET [25]. The dependence on Nurr1/RET expression is further supported by an experiment performed in Nurr1 deleted mice showing that the ability to respond to GDNF is abolished in DA neurons lacking Nurr1 [25].
The findings obtained in the AAV-alpha-synuclein model should however be interpreted with caution. The cellular levels of alpha-synuclein obtained in this model are artificially high (4-5-fold above normal) and thus may not reflect the milieu in the affected DA neurons in the PD brain. Furthermore, showing that RET expression is reduced in the human PD brain has proven inconclusive (see Su et al. 2017 for a dissenting view [28]).
In summary, the preclinical data suggests that GDNF can rescue DA neurons and their projections in the nigrostriatal pathway in a range of toxin animal models but the ability to rescue may be different in the context of the alpha synuclein pathology that is seen in the brain of people with PD.
THE EARLY CLINICAL TRIALS
The move from the lab to the clinic is always challenging and in order to assess progress and success, new agents are often evaluated against four key elements. These include whether the drug (in this case GDNF for PD):
• Reaches its proposed site of action at sufficient concentrations (namely the DA nigrostriatal pathway);
• Shows target engagement at that site in a measurable way (GFRa1/RET signalling leading to positive changes in this DA system);
• Displays functional downstream pharmacological effects (shows sprouting, growth and /or survival of DA fibers/synapses in the presence of an ongoing degenerative disease process);
• Exhibits improvement in the relevant phenotype of the treated individuals (better motor performance around measures known to be sensitive to this DA network).
The first of these criteria falls under the umbrella of delivery, the latter three provide a basis for potential efficacy, if delivery sufficient to cover the putamen can be achieved. In the sections below, we consider the open-label and double-blind, placebo-controlled clinical trials to-date in which recombinant human GDNF has been directly administered to people with PD. This will be followed by a description and discussion of the clinical studies where a related trophic factor NRTN was administered as viral vector injections to the basal ganglia as well as an ongoing GDNF gene therapy trial.
The initial double-blind randomised control trial of intracerebroventricular injections of GDNF protein
The first clinical trial of GDNF was conducted by Amgen and was a multicentre, randomized, double-blind, placebo-controlled study of intracerebroventricular (ICV) administration of GDNF [4]. Monthly ICV injections were given via an intraventricular cannula inserted in the right frontal horn using standard stereotactic techniques. This was a dose escalation study with five dosage arms (25 μg, 75 μg, 150 μg, 300 μg, and 4000 μg) with 7-8 patients receiving active drug and 2-3 patients receiving placebo for a period of 8 months followed by an open-label extension period of up to an additional 20 months giving maximum single doses of up to 4000 μg in 16 subjects. The primary outcome variables, the change in “practically defined OFF” and ON motor Unified Parkinson’s Disease Rating Scale (UPDRS) scores, were not significantly different from placebo in any of the active treatment groups apart from a mild but significant worsening in OFF scores in the 75 μg group and ON scores in the 300 μg group. Adverse effects were more common in the active treatment groups and included anorexia, weight loss (>5% body weight), hyponatremia and the unexpected finding (at the time) of sensory symptoms such as paraesthesia and Lhermitte’s phenomenon.
Although the adverse effect profile indicated that GDNF administered by ICV injection was having biological effects (the anorexia and weight loss were thought to be due to its action in the hypothalamus), this approach did not improve the clinical state of the patient.
It was postulated that this lack of benefit could relate to a failure of GDNF to reach and mediate effects in the target tissue (putamen and indirectly the substantia nigra) [4]. Supporting this hypothesis was a report of the postmortem assessment of a single patient from this trial [29]. In contrast to experiments in monkeys, where GDNF immunoreactivity was observed within the caudate nucleus ipsilateral to the infused ventricular frontal-horn and in the septum bilaterally (although whether this was sufficient to activate RET signalling was not assessed), the human postmortem evaluation demonstrated no intra-parenchymal diffusion of GDNF across the cerebrospinal fluid:brain barrier from the ventricular cavity to the relevant basal ganglia structures. As such, it was to be expected that the autopsied tissue failed to demonstrate evidence of significant regeneration of nigral neurons and their fibres [29].
While this first in-human trial failed to hit its primary end point, the above results showed that monthly infusions of a biologic-agent unable to penetrate the blood-brain-barrier was well tolerated and “relatively” safe even when high doses of GDNF were given (4000 μg) [4]. However, the lack of parenchymal penetration coupled with an absence of motor benefit led on to further trials with infusions directly to the putamen.
The initial open label trials of intraputaminal Injections of GDNF protein
To ensure that GDNF reached the DA terminal plexus within the posterior-dorsal striatum, two small open label studies evaluated direct-to-putamen continuous (rather than bolus) catheter infusions of GDNF [30, 31]. To effect continuous infusions, GDNF was administered from subcutaneous pumps placed in the abdomen connected to a single catheter to each putamen in a 5-patient cohort in Bristol, UK and to a single-sided unilateral catheter only in a 10-patient cohort studied in Kentucky, USA. The Bristol group initially reported after 6 and 12 months as did the Kentucky group [30, 31]. Doses in the Kentucky study were escalated to 30 μg per day and in the Bristol trial, patient’s doses were on average 30 μg per day. Some patients did receive even higher doses (>30 μg GDNF per day) but this produced high signal changes on MRI in the putamen—changes which resolved with dose reduction.
Both of these small open label studies reported marked benefits in UPDRS motor (part III) scores in the practically defined OFF state with a mean reported improvement of approximately 30%–40%. Changes in diary fluctuations were equally encouraging at this open-label stage [30, 31] although it should be noted that the changes were bilateral even in patients who had been in receipt of unilateral infusions for reasons that are not clear.
In the Bristol study,18-fluorine-dihydroxyphenylalanine ([18F]DOPA) positron emission tomography (PET) scans showed an increase in tracer uptake mainly around the catheter tip, which potentially represented sprouting of remaining terminals, supported in part by a subsequent single case postmortem study [32].
In the Kentucky study, there was one serious adverse event (SAE) when the catheter became exposed which was associated with oedema around the catheter track in the putamen of this same patient. Three patients reported mild tingling sensations in the forehead, neck and lower back and two patients experienced transient Lhermitte’s phenomenon. Seven patients developed antibodies to GDNF without clinical sequelae. High resolution MRI scans revealed that there was no evidence of GDNF-induced cerebellar toxicity, which became more of a concern in some of the later preclinical non-human primate studies with GDNF (see below). Finally, all improvements in UPDRS scores were lost within 9-months of stopping the GDNF infusions.
Based on these encouraging open label observations, a prospective, double-blind, placebo-controlled trial of continuously infusing GDNF to the putamen was initiated.
DOUBLE-BLIND, PLACEBO-CONTROLLED TRIALS
The Amgen trial
Amgen sponsored the first double-blind trial involving direct intra-putaminal delivery of GDNF. In this multicentre trial, patients were randomized 1 : 1 to receive bilateral continuous intra-putaminal infusions of either GDNF at a dose of 15 μg/putamen/day or placebo [33]. One catheter was stereotactically placed on each side with its tip targeted to the posterior-dorsal putamen and attached to a separate SynchroMed pump (Medtronic), implanted subcutaneously over the patient’s abdomen. The primary end point was the change in UPDRS motor score in the practically defined OFF condition at 6 months. Secondary end points included other UPDRS scores, motor tests, dyskinesia ratings, patient diaries, and [18F]-DOPA uptake on PET imaging. Patients were stratified by baseline UPDRS OFF motor score (<44, >45) and 30 subjects (15 in each group) were calculated to be needed to give a 90% power to detect a between group difference of 25% in the percent change in UPDRS OFF motor score.
34 patients were implanted and randomized; 17 received GDNF (all completed the trial), and 17 received placebo (with 16 completing the trial, and one discontinuing due to pump site infection). At 6 months, the mean percentage change in OFF UPDRS motor score was –10% in the GDNF group compared to –4.3% in the placebo group which was not statistically significant. Secondary end point results were also similar between the groups. There was no significant relationship between the change in motor scores and the catheter tip location. In the two thirds of paired evaluable PET scans (1/3 of paired scans were excluded due to head movement artefact) there was a 32.5% treatment difference favouring GDNF in mean [18F]DOPA influx constant (p = 0.019) but this did not correlate with changes in the OFF UPDRS motor scores. Procedure- and device-related complications were not uncommon while treatment related complications were infrequent. The marked anorexia and weight loss observed in the higher dose ICV study were not seen. Serious, device-related adverse events required surgical repositioning of catheters in two patients and removal of devices in another [33].
Three patients, one in the double-blind phase and two in the open label extension, developed neutralizing anti-GDNF antibodies—again without any obvious clinical sequelae- which may relate to the way the GDNF was delivered with leakage to the periphery, and activation of the immune system. Furthermore, contemporaneously, new toxicological studies in non-human primates (NHPs) found focal limited loss of Purkinje cells and near complete loss of molecular and granule cell layers in 3/5 monkeys rapidly withdrawn from 3 months of unilateral infusions of much higher doses of GDNF (100 μg/putamen/day) while one monkey continuing on treatment was found to have milder cerebellar cortical pathology [34].
As a result of this combination of a negative clinical double-blind placebo-controlled trial result, the finding of neutralizing antibodies in a small number of patients and concerns about the NHP toxicological findings, Amgen chose to terminate their GDNF program for PD. This led to a vigorous debate between various researchers and patient groups as to why the double-blind trial and the open-label studies had come to different conclusions. These included:
1. The potential for a major placebo effect in open-label trials given it involved an invasive surgical approach and problems in maintaining true clinical equipoise. However, it should be noted that in the double-blind study there was no major placebo effect, but rather an absence of a positive clinical effect in either group.
2. Differences in dosages given, in particular higher doses were generally used in the open label studies, although benefit had also been claimed with lower doses in these early studies;
3. Differences in delivery including catheter dimension and design. In general, catheter dimensions including its external diameter, design (no step, stepped or recessed stepped) and number of catheters inserted along with implantation technique could all have an effect on the extent to which the agent was delivered and remained at the target site. In addition, differences in the diffusion of the agent across the target structure could also have impacted on the total volume of putaminal tissue exposed to study drug (see below) [35] and thus its potential therapeutic effectiveness. All of this has led to the development of new convection enhanced delivery systems (see below).
4. Differences in the patients selected for trials, in particular whether more advanced patients with more severe DA losses were recruited to the double-blind study.
Given this uncertainty, there was a feeling in some quarters that this therapy should not be abandoned at this stage, a position reinforced by further observations from the original open-label Bristol cohort [32, 36, 37]. This included the fact that the original five subjects who continued to receive continuous infusions from 12 to 24 months and beyond, all maintained their improved UPDRS part II and part III OFF scores compared to baseline, consistent with their improved [18F]DOPA PET data [36]. Finally, one subject who had been infused continuously for 39 months and then reviewed at 36 months after GDNF cessation, continued to experience a major clinical benefit. This benefit was accompanied by [18F]DOPA PET putaminal uptake that continued to show an improvement compared to pre-treatment scans [37]. Although a single case, this did support the concept that GDNF might still work if methodological aspects of its administration were improved. Thus, a new GDNF trial was proposed.
The recent Bristol study
This new double-blind investigation of directly administered GDNF took the form of a randomised, placebo-controlled, single-centre trial sponsored by the UK National Health Service (and funded by Parkinson’s UK and The Cure Parkinson’s Trust) which started in 2012 [38] (NCT03652363). Patients selected were 35–75 years old, had motor symptoms for 5 or more years, with moderate disease severity in the OFF state (Hoehn and Yahr stage 2–3 and a UPDRS motor score between 25–45) and motor fluctuations (average of at least 2.5 hours of OFF time per day on 3-day fluctuation diaries). They all had marked levodopa responsiveness as defined by a>40% improvement in UPDRS motor score following a levodopa challenge after a practically defined OFF period. Importantly, the major difference with this trial with what had gone before was the use of a new delivery device designed to establish greater coverage of the putamen.
Once implanted with this new intermittent enhanced drug administration system that enabled convection enhanced delivery (CED), patients were randomised. Post-randomisation, patients received a total of 10 study treatments at 4-weekly intervals (Weeks 0 to 36). At each treatment, 400 μL of infusate (300 μL GDNF or placebo, followed by 100 μL aCSF) was delivered per catheter into the post-commissural putamen at a GDNF concentration of 0.2 μg/μL. Thus, the total GDNF dose given every 4 weeks was 240 μg (120 μg/putamen given as 60 μg/catheter).
The results of this trial were published in February 2019 [38] and they revealed that the trial did not reach its prespecified primary endpoint; the mean OFF state UPDRS motor score decreased by 17.3±17.6% in the active group and 11.8±15.8% in the placebo group. A range of secondary and supplementary efficacy endpoints also failed to show significant differences between the groups as well. In contrast to the non-significant clinical results, the [18F]DOPA PET findings were positive. Between baseline and week 40 there was no change in the placebo group, whereas in the GDNF group there were significant changes across the putamen (in a graded fashion ranging from 25% anteriorly to 100% in the posterior putamen) but not in the caudate (which acted as an internal control). These marked relative percentage increases, while statistically significant, still meant that the absolute improvement was only to a level that was 50–60% of that seen in the normal intact posterior putamen which may explain why the treatment did not result in significant clinical changes.
At the conclusion of this double-blind study, all patients had the chance to enrol into an open label extension trial that used the same GDNF dose regimen and intermittent infusion parameters as for the initial double-blind study. This open label extension trial, also lasted 40 weeks, and was initiated before the results from the double-blind parent investigation were known. It was undertaken primarily to gain longer term safety data and to gather further exploratory information on GDNF clinical effects over a more prolonged period of repeated tissue exposures [39].
The primary endpoint of this extension study was the percentage change from baseline (Week 0) to Week 80 in the practically defined OFF state UPDRS motor score, comparing the group that had received GDNF in the initial trial followed by open-label GDNF (GDNF/GDNF) versus the group that received placebo in the parent investigation followed by open-label GDNF (placebo/GDNF) (in effect a delayed-start design). Secondary endpoints included absolute change from baseline in OFF and ON state UPDRS part II and part III scores and change from baseline in diary ratings. A further pre-specified secondary endpoint included comparing Week 80 UPDRS scores in the GDNF/GDNF group against Week 40 scores in the placebo/GDNF group (i.e., at the end of the placebo treatment).
All 41 parent study participants were enrolled into the extension study, and all were included in the analyses. Again, there were no significant differences. Comparing baseline (Week 0) to the end of treatment (Week 80), the OFF state UPDRS motor score improved by 26.7±20.7% (mean±standard deviation) in the GDNF/GDNF group and by 27.6±23.6% in the placebo/GDNF group. Likewise, none of the secondary or supplementary outcomes spanning the entire 80-week period were significantly different outside changes in L-DOPA equivalent dose (the increase in the daily L-DOPA equivalent dose from baseline to Week 80 was smaller in the GDNF/GDNF group (59±194 mg) than in the placebo/GDNF group (289±365 mg) [35].
Treatment emergent adverse events (TEAEs, events commencing post initiation of GDNF or placebo infusions) were reported for all 41 patients. No patient had a TEAE that led to discontinuation of study medication. Of the eight serious TEAEs, three were considered to be device related and included two occurrences of a hypertrophic skin reaction around the port site that required surgical skin thinning and a possible port site infection that occurred approximately 15 weeks into the treatment phase and required inpatient treatment with oral antibiotics.
Two patients enrolled into the double-blind study did not proceed to randomisation and were withdrawn prior to the start of treatment because they failed the post-surgery eligibility criteria. One patient experienced a mildly symptomatic putaminal ischemic stroke coincident with the initial test infusion. The patient recovered completely but was withdrawn to avoid unnecessary risks. The second patient suffered a small asymptomatic haemorrhage in both putamina during the initial test infusion.
Blood sample analyses showed no measurable GDNF plasma concentrations and no GDNF-binding serum antibodies in GDNF-treated patients at any point. This contrasts with the double-blind Amgen study and the earlier open label studies and may relate to the different delivery devices and delivery regimens that were used in each trial.
In summary, these two studies have shown that direct infusions of GDNF administered in a manner to achieve CED can be given every 4-Weeks over 18 months in a fashion that patients found tolerable. Employing this approach, as evidenced by a combination of direct Gadolinium infusion through the delivery system and improvement in [18F]DOPA PET uptake, appeared to achieve accurate and whole putamen-wide target tissue delivery with some evidence of target receptor engagement using PET imaging. Despite this apparent optimisation of delivery, however, the clinical primary and secondary endpoints in both trials were negative. Whilst the partial restoration in PET signal may alleviate some of the concerns around insensitivity to GDNF in the face of an alpha-synucleinopathy or that patients more than 5 years from the point of diagnosis have no terminals left to restore, the fact remains that improvement in [18F]DOPA PET signal cannot be used as evidence for improvement in functional pharmacology, especially as this tracer has also been said to label inflammation [40]. Questions therefore remain over whether the lack of significant benefit in placebo-controlled trials to date reflects therapeutic ineffectiveness or whether this would be resolved with an increased dose and exposure of mammalian cell made GDNF coupled to treating patients with earlier stage disease.
GENE THERAPY TRIALS WITH NRTN AND GDNF
In contrast to the immense logistical challenges and potential safety concerns associated with continuous or repeated long-term delivery of recombinant GDNF protein, gene therapy promises sustained, durable and localized production of properly folded biologically active GDNF following a one-time dosing procedure. Several clinical studies have now been conducted in PD, including a multi-phase program of NRTN gene transfer, a homolog of GDNF, and more recently a Phase 1 clinical safety trial of GDNF gene transfer. Both the NRTN and GDNF gene therapy programs utilized gene transfer vectors derived from the non-pathogenic adeno-associated virus serotype 2 (AAV2) with a constitutive CMV promoter. These vectors appear to have a favourable safety profile for neurotrophic factor gene delivery in PD, in addition to which AAV2 has an exclusive neuronal tropism and restricted distribution when directly delivered to the brain [41], thus minimising off target side effects.
The initial preclinical studies exploring this approach demonstrated that GDNF and NRTN gene delivery conveyed efficient protection against MPTP/6-OHDA lesions when the gene transfer was performed prior to, or shortly after, neurotoxin exposure. However, in an attempt to more closely mimic both early and later stages of PD a more refined MPTP model was developed in non-human primates (NHPs [21, 42]). Using this model, animals with established parkinsonian signs were randomized to receive either 1) AAV2-GDNF (9.9×1011 vector genomes, vg; n = 8), or 2) sham PBS (n = 7) intraputaminal infusions via CED [21], and were followed for 1, 6, 14, or 24 months.
In one of these NHP studies, it was demonstrated that there were marked functional motor improvements following AAV2-GDNF (mean 56% reduction of motor rating scores) in both the moderately and severely lesioned MPTP monkeys. This motor recovery directly correlated with increased 6-[18F]Fluoro-L-M-tyrosine (FMT) PET uptake that remained stable throughout the 24-month time point and which also correlated with enhanced dopamine and dopamine metabolites when assayed from tissue homogenates from these same animals. Increased tyrosine hydroxylase-immunoreactive (TH-IR) fibre density was also seen in the partially lesioned hemisphere (equivalent to “early” PD) receiving the AAV2-GDNF but was much less prominent in the severely lesioned side (comparable to advanced PD). Together these findings suggested that intraputaminal infusions of AAV2-GDNF were safe and that greater parenchymal GDNF levels (∼24 ng/mg protein) were well-tolerated, without the adverse effects seen with protein infusions of GDNF (e.g., weight loss) [4, 33]. This also indicates that GDNF is capable of restoring dopaminergic terminals with an associated significant recovery of motor function, particularly in the partially lesioned conditions. In addition, there was strong evidence that GDNF delivery provided greater potential for intrinsic TH-IR positive sprouting in earlier rather than later stages of nigrostriatal degeneration.
These and related studies [43] also found that there was anterograde transport of AAV2-GDNF, via direct and indirect connections, which was independent of the degenerating nigrostriatal dopaminergic (DA) neurons [43, 44] and their capacity to retrogradely transport GDNF protein. This mechanism resulted in the broad expression of GDNF from the putamen to the substantia nigra (SN) pars reticulata, despite varying degrees of nigrostriatal DA neurodegeneration and raised the potential that this therapeutic may provide distinct advantages through rebuilding DA nigrostriatal networks within the PD brain.
This NRTN preclinical work led to clinical trials that were performed by Ceregene Inc. using the AAV-2 serotype and the NRTN transgene, the first of which was an open label clinical trial [45] (NCT00252850). Twelve patients aged 35–75 years with a diagnosis of PD for at least 5 years, in accordance with the Parkinson’s UK Brain Bank Criteria, received bilateral, stereotactic, intraputaminal injections of AAV2-neurturin (CERE-120). The first six patients received doses of 1.3×1011 vector genomes (vg)/patient, and the next six patients received 5.4×1011 vg/patient. The treatment was well tolerated with no side effects and a number of clinical endpoints suggested improvement. However, disappointingly there was no increase in 18F-DOPA uptake on PET imaging.
This initial trial was followed by a multi-centre randomized (2 : 1) double-blind trial comparing intraputaminal injections of AAV2-neurturin to sham surgery in 58 PD patients [46]. An infusion volume of 40 μl of vector was injected into each putamen with subjects in the active treatment arm receiving a dose of 5.4×1011 vg/patient. Disappointingly, there was no significant difference between the two groups based on UPDRS Part III motor scores in the OFF state at 12 months post-transduction, the primary endpoint. However, a significant placebo effect was noted, with a 6-point reduction in UPDRS seen at 3 months in the sham group, which persisted for the duration of the study. However, a pre-specified post-hoc analysis suggested that those patients blindly assessed at the 15–18 months post-treatment time point may have had some benefit, although there was no controlling for multiple comparisons. In addition, it should be noted that the whole cohort could not be followed blindly out to these time points due to the ending of the trial and the blind being broken at 12 months. This may have created a bias in the effect seen.
Histological data from patients who died from events unrelated to the procedure, indicated that NRTN was being expressed within the vicinity of the injection sites, and that this resulted in focal upregulation of TH, but to an extent that was probably insufficient to provide any clinical benefit. Additionally, there was very limited NRTN seen in nigral neurons, suggesting that the severity of the nigrostriatal axonopathy in these advanced PD patients did not allow sufficient retrograde transport of NRTN to the nigral perikarya to provide neurorestorative effects [47].
Based on the small area of transduction, the lack of NRTN expression in nigral neurons, the perceived defect in retrograde transport and the potential for changes to occur at a longer time-point, a second randomized double-blind trial comparing higher putaminal volumes plus a direct injection into the nigra was undertaken [48]. Fifty-one patients were enrolled in this multi-centre trial and randomly assigned (1 : 1) to receive either bilateral AAV2-NRTN (180 μl injection volume per hemisphere) into the substantia nigra (2.0×1011 vg/patient, 15 μL×2 infusions) and putamen (1.0×1012 vg/patient, 50 μL×3 infusions), or sham surgery. Again, no statistically significant clinical differences were seen in UPDRS Part III motor OFF scores at 15-months (primary endpoint) between the active treatment and sham operated arms.
Following the NRTN studies, an open-label, dose-escalation Phase 1 study of AAV2-GDNF was initiated in 2013 (NCT01621581) [49]. In this GDNF gene therapy study, 13 (of an intended 24) participants with advanced PD received bilateral magnetic resonance imaging (MRI)-guided, CED intraputaminal infusions of AAV2-GDNF (9×1010 (n = 6), 3×1011 (n = 6), 9×1011 vg (n = 1); delivered in a 450 μL volume per putamen. Safety and tolerability of AAV2-GDNF intraputaminal delivery by CED was confirmed by real-time MRI and postoperative monitoring, with no serious adverse events (SAEs) attributed either to the procedure, or to the investigational product. Increased PET uptake values of 18F-DOPA were noted at the documented infusion sites at the 6-month time point as compared to baseline values, with further enhanced uptake observed at 18-months post-treatment time point. A trend was noted for earlier and more marked increases in patients with shorter disease duration. No significant differences were seen at 18-months between the three treated cohorts in terms of their UPDRS Part III motor scores or total levodopa equivalent doses. The unchanged PD motor scores and stabilisation of their anti-parkinsonian medications following putaminal AAV2-GDNF delivery might support possible biological effects of this therapy in participants with advanced PD but this remains unproven.
Several key changes were made as part of the AAV2-GDNF Phase 1 study design compared to prior direct infusion studies in PD conducted in the early 2000s, including:
a) the use of intraoperative MRI-guidance, and gadolinium co-infusion with AAV-GDNF
b) using a reflux-resistant delivery cannula with a stepped design to increase distribution within the target putamen while reducing off-target leakage;
c) allowing the visualisation and monitoring of CED infusions in real-time with an ability to surgically modify the cannula position and infusion parameters to maximise the putaminal coverage, and
d) increasing the infusion volumes up to 450 μL per putamen, 3 times greater than the volume delivered in the Phase II AAV2-NRTN study [46, 48].
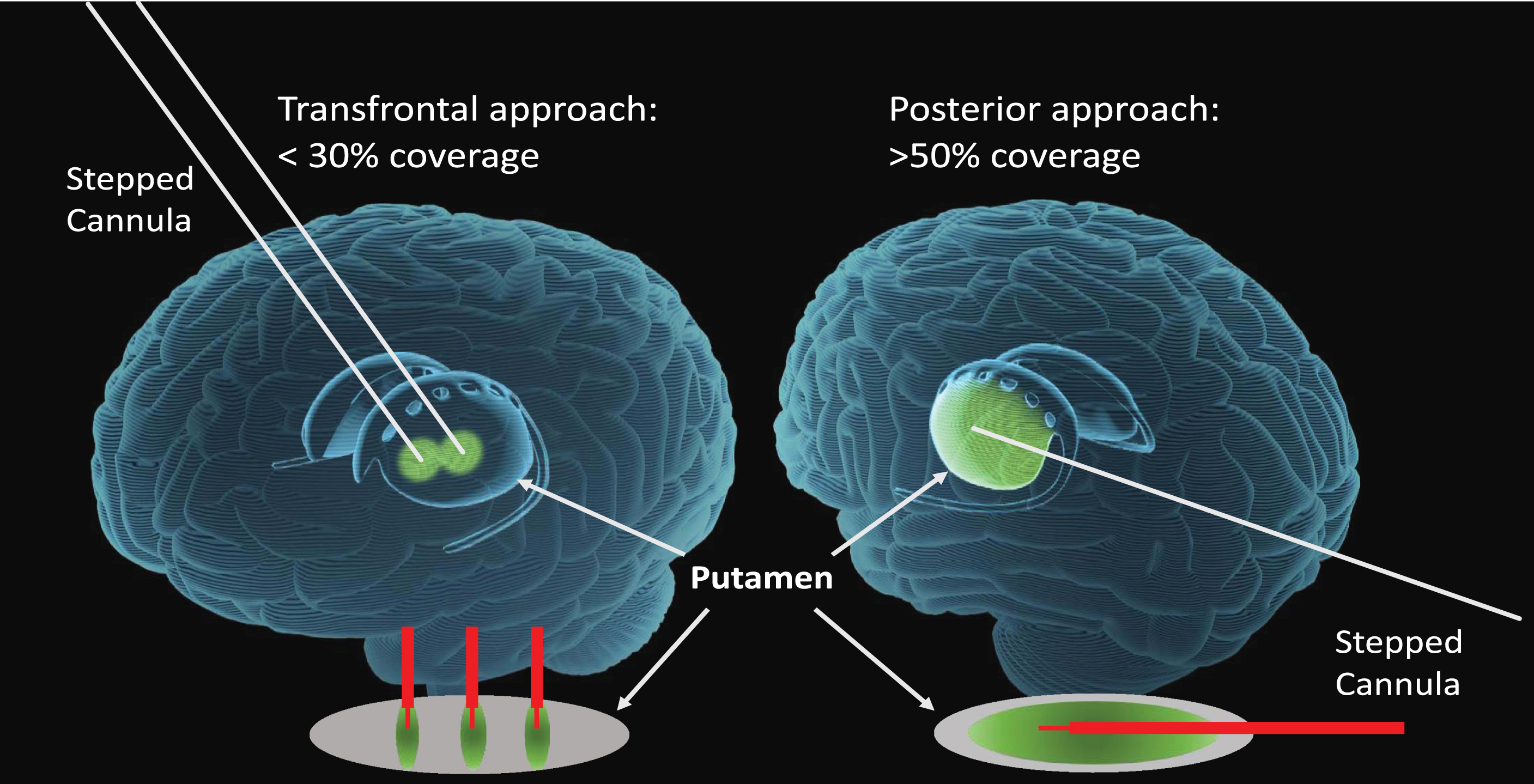
Despite these key modifications in methods, the average putaminal volumetric coverage documented by retrospective interim analysis of MRIs was only 26%, much lower than that anticipated to be required for a meaningful clinical benefit. In part, this limitation in putaminal coverage may have been due to the transfrontal surgical approach to the putamen, where the trajectories are perpendicular to the long axis of the target volume. This inability to broadly cover the putamen with this standard surgical approach may have also been a relevant disadvantage in the AAV2-NRTN and earlier recombinant GDNF protein infusion studies, where there was minimal putaminal transgene expression or effects with small localised changes relative to the radiographic improvement displayed via18F-DOPA PET imaging.
Although long term follow-up for the Phase 1 adeno associated virus (AAV) 2-GDNF cohorts is ongoing, enrolment was closed following the interim analysis, due to the insufficient putaminal coverage (mean of 26%).Other studies of a AAV2-L-aromatic amino acid decarboxylase (AADC) gene therapy for PD [50], however have shown that delivering volumes up to 1800 μL per putamen using a CED approach is feasible with a good safety profile and providing putaminal volumetric coverage of >50%. More importantly, this AAV2-AADC PD gene therapy investigation has provided convincing evidence that the clinical benefit improved concurrently with increases in volume of vector delivered and thereby the extent of putaminal coverage. These findings underscore the importance of optimising the transduced tissue volume and putaminal infusion coverage as factors correlating directly with clinical efficacy in PD [49].
These latter efforts have prompted the design of a new Phase 1b trial to assess a higher dose of the AAV2-GDNF therapeutic in moderately advanced PD patients, (similar to those in Phase 1), as well as in subjects with early disease (namely within 5 years of PD onset). Furthermore, this new study will be using a posterior (occipital) trajectory to each putamen, (paralleling the long axis), that allows shape-fitting CED of higher infusion volumes, thereby improving putaminal coverage and GDNF production levels more uniformly throughout the putamen [51].This posterior putaminal approach is similar to that recently reported in the GDNF protein infusion study [38] and has also been safely performed using MRI-guidance and CED, in the ongoing AAV2-AADC study (NCT03562494) (see Fig. 1).
Fig.1
Transfrontal versus Posterior (occipital) trajectories utilizing CED. MRI-guided stereotactic approaches for vector delivery to the putamen utilizing convection enhanced delivery (CED) and stepped cannulae. This approach allows for precise targeting, shape-fitting infusions, and larger delivery volumes to improve the extent of transduced putaminal tissue, thereby increasing transgene production capacity. With the transfrontal approach, the cannulae are oriented to the short axis of the putamen limiting vector coverage to <30%, often requiring 2-3 tracts. The posterior (occipital) approach maximizes the delivery by paralleling the putaminal long-axis, requiring only 1 tract, which achieves >50% putaminal vector coverage with larger infusion volumes.
WHERE NEXT?
The question as to whether GDNF has a competitive future in the treatment of PD is still unclear. A number of conclusions can be drawn from the studies undertaken to date with GDNF and related factors (see Table 1) along with a number of recommendations about what another trial with GDNF should consider and thus might look like (see Table 2). In particular, it was felt that a viral delivery system using some of the newer modified approaches would be advantageous given the one-off nature of the surgery compared to the relative complexity of the neurosurgery needed to implant the infusion delivery systems used in the recent GDNF trial and consequent requirement for on-going infusions. That said, the efficacy of GDNF may require intermittent rather than continuous RET receptor stimulation and, whilst in the development phase, understanding the exact dose administered and retaining the ability to reduce and stop dosing may have utility. In addition, it seems logical to assume that the individuals most likely to benefit from such a treatment would be those individuals with most neurons and fibres left to rescue, namely patients with early stage PD with evidence of fiber loss restricted to the dorsal striatum [52]—where the therapeutic agent would be targeted. If such an approach were recommended, then ensuring the patient actually has Dopa-responsive PD will be critical, and the use of imaging to help support such a diagnosis would be essential, including both DA imaging as well possibly fluorodeoxyglucose (FDG) PET (for both diagnostic stratification and corroboration of functional target engagement) [53]. In addition, the exclusion of certain genetic forms of parkinsonism may be wise, for example GBA heterozygote patients, given that they progress more quickly especially with pathology outside of the DA nigrostriatal pathway [54, 55].
Table 1
Summary of main findings on the effects of GDNF and related factors in models and clinical trials in PD
| •Studies have shown a statistically significant response in some patients, but these are not consistent, and the majority of studies have been negative in terms of reaching their primary outcome; |
| •Striatal dopamine has increased in most patients in receipt of GDNF as evidenced using 18F–dopa PET imaging; |
| •There is little evidence of sufficient retrograde transport of GDNF/NRTN to the substantia nigra in patients when the agent is delivered into the striatum; |
| •Postmortem studies show that where there is expression of GDNF/NRTN there is some upregulation of TH. |
Table 2
Factors to consider in future clinical trials with GDNF and related factors in PD
| What form should the GDNF be given in? |
| •A gene therapy approach was favoured over protein infusions given the complexity of the neurosurgical intervention required for the former and the burden this places on the patients with Parkinson’s disease. |
| •Consider using mammalian cell produced GDNF and NRTN proteins. |
| Patient type |
| •Younger patients with marked L-dopa response and no major ventral striatal dopamine loss on dopamine imaging. |
| •Avoid certain genetic forms/variants associated with Parkinsonism (Parkin; GBA). |
| Disease stage |
| •Avoid late stage disease. |
| Dose given |
| •Depends on the neurotrophic factor that is being delivered, but probably need higher doses than have been trialed to date (with the exception of the first ICV trial of GDNF). |
| Volume given |
| •Depends on delivery system but need to give up to ∼1 ml per striatum treated. |
| Delivery device |
| •Several now in existence, e.g., Renishaw, Clearpoint system. |
| Need for adjunct therapies |
| •Not proven to be needed, although preclinical data suggests that Nurr 1 agonists may enhance the efficacy of GDNF—so perhaps this should be included as part of further trials. |
| Need for imaging? If so with what? |
| •F-dopa PET imaging seems to have provided useful information in trials to date, but need for other PET markers looking at DA turnover/release as well as network reconstruction. |
| Trial end points: What and when? |
| •Standard measures UPDRS part 2±PDCore scores at 18–24 month as the primary end point. |
| •Sample size currently undecided given lack of major effects seen to date which would allow one to power such a study. |
| Trial design |
| •Consider a delayed start design to the trial or a randomized double-blind placebo-controlled study. |
| •Keep the trial outcomes and measures simple. |
| •In postmortem samples, it is important to show that GDNF and NRTN have activated RET-dependent signalling pathways or to show direct RET receptor activation. |
| Health economics for this agent? |
| •Depends on where it is positioned |
| BUT although it works uniquely to restore the dopaminergic nigrostriatal, it will nevertheless have to compete with other “DA” therapies/interventions- new dopamine drugs; DuoDopa®; Deep Brain Stimulation etc and the newer dopaminergic gene or cell-based therapies should they be shown to work. |
| •Currently it would not be competitive given the size of effects seen to date, but this may relate to suboptimal delivery, etc. |
Whether an adjunct therapy to up regulate Nurr1 would be required is still unclear given the clinical data to date, but the preclinical data would suggest this. However, there is no such agent that has yet been identified with a safety profile that is acceptable and even if one existed, questions still arise as to how long such a therapy should be given. Coupled to this is a need to better understand the optimal dose of GDNF and volume of its distribution, to ensure that the treatment has the best chance of showing a clinical effect, and that this effect is the maximal one that one could expect for that agent. Finally, since E. coli cannot form seven disulphide bonds correctly and glycosylate GDNF, the use of mammalian cells to make GDNF should be considered if protein infusions are being considered although this brings with it major cost implications.
As to what any trial should look like, there is still much debate as to what primary end-point should be used and at what time point, and input from the patient community on this will be vital going forward. However, this end point should reflect changes in those clinical aspects of PD that respond to dopaminergic interventions given this is the pathway being targeted by these treatments. A double-blind sham surgery trial would be the preferred design for future studies, although whether more optimisation of the delivery of GDNF should be carried out before such a trial is undertaken is debatable. Overall there was a consensus from the workshop, that longer trials may be better for fully exploring whether this agent can mediate neurorestoration and thus waiting at least 18 months from the start of any therapeutic intervention would increase the chances of seeing any such effects. In addition, using composite end points may also have some merit given that the use of any single one, such as the UPDRS part III score, has limitations. As to what that composite clinical end point should look like is unclear as regulatory agencies are currently not accepting these for licensing purposes. However, one that has recently been proposed relating to the recent Bristol GDNF trial, PDCORE, embraces good quality on-time; activities of daily living and reflects previous participant feedback [56].
In addition to the use of wild type GDNF and NRTN given as protein deliveries or a gene therapy, other similar approaches for treating PD were also discussed. In pre-clinical studies, new GDNF and NRTN mutants with improved diffusion and stability have shown beneficial effects [57, 58]. Furthermore, to overcome the limitations of some of the pharmacokinetic properties of the GDNF and NRTN proteins, a blood-brain-barrier penetrating small molecule GDNF receptor agonist has recently been developed. This compound activates RET-dependent intracellular signaling cascades in DA neurons both in vitro and in vivo and also stimulates the release of dopamine in the mouse striatum—all of which suggests that this agent could be a novel future treatment of PD [59].
In this respect, cerebral dopamine neurotrophic factor (CDNF) is a relatively recently discovered endoplasmic reticulum (ER) located, but also secreted, protein that protects and restores the function of DA neurons in rodent and non-human primate models of PD and does so more effectively than GDNF [60]. CDNF is very different from other known trophic factors–it has a unique structure and mode of action protecting neurons by inhibiting cell death, regulating ER stress, the unfolded protein response (UPR) and reducing inflammation [61]. In addition, CDNF rescues only ER-stressed or degenerating neurons and does not influence naïve healthy neurons. This agent is now the subject of an EUH2020 funded phase I–II clinical trial in PD patients (NCT03295786) [61].
CONCLUSIONS
This special workshop comprehensively covered the studies evaluating GDNF and the related trophic factor NRTN in PD both preclinically and clinically. It critically appraised the work so that conclusions could be drawn as to what has been shown and what has not been shown with these agents. It was generally agreed that GDNF and NRTN have worked relatively well in neurotoxic animal models of PD but that their translation to the clinic has so far failed to show a major impact—perhaps highlighting the predictive limitations of toxin animal models being commonly used in the preclinical space in PD and the way we plan clinical trials.
Clinically, there is evidence that these neurotrophic factors are able to rescue the expression of TH in the human PD brain with some suggestion of a clinical correlate. Nevertheless, the current size of any such effect is not competitive compared to what is already clinically available for the DA-related features of PD (DuoDopa®; deep brain stimulation; lesion surgery such as pallidotomy; apomorphine pumps, etc.) and this may also be the case with new agents that are currently being trialled in PD around dopamine rescue (stem cell derived DA neurons; CDNF, “dopamine” gene therapies). However, it must be realised that these agents are uniquely designed to restore and regenerate the dopaminergic pathway which is very different from these other symptomatic therapies.
In conclusion, further work is needed to understand better what can be achieved with GDNF and related factors in the clinic to improve the lives of patients with PD, although ultimately whether it will ever have a competitive place for treating people with PD remains unclear.
CONFLICTS OF INTEREST
Roger Barker is a member of the advisory board for Novo Nordisk; Living Cell Technologies; FCDI; BlueRock Therapeutics, Aspen Neuroscience and UCB. He receives royalties from Springer-Nature and Wiley. He has received grant support from the EU; NIHR; MRC; Wellcome Trust; HDA; Rosetrees Trust; Cure Parkinson’s Trust; Parkinson’s UK and Evelyn Trust. He is Co-Editor in Chief of the Journal of Neurology and an Associate Editor for the Journal of Parkinson’s Disease.
Don Gash is a founding Partner in Avast Therapeutics Inc. with patents on drug candidates for treating neurological disorders.
Alan Whone was a PI on the recent UK GDNF trial that received support from Parkinson’s UK and the Cure Parkinson’s Trust
Anthony Lang reports consultancy support from Abbvie, Acorda, AFFiRis, Biogen, Denali, Janssen, Intracellular, Kallyope, Lundbeck, Paladin, Retrophin, Roche, Sun Pharma, Theravance, and Corticobasal Degeneration Solutions; advisory board support form Jazz Pharma, PhotoPharmics, Sunovion; other honoraria from Sun Pharma, AbbVie, Sunovion, American Academy of Neurology and the International Parkinson and Movement Disorder Society; grants from Brain Canada, Canadian Institutes of Health Research, Corticobasal Degeneration Solutions, Edmond J Safra Philanthropic Foundation, Michael J. Fox Foundation, the Ontario Brain Institute, Parkinson Foundation, Parkinson Canada, and W. Garfield Weston Foundation and royalties from Elsevier, Saunders, Wiley-Blackwell, Johns Hopkins Press, and Cambridge University Press.
Amber van Laar is an employee of Brain Neurotherapy Bio, Inc.
Adrian Kells is an employee and stock holder of Brain Neurotherapy Bio, Inc.
Massimo Fiandaca is co-founder and Vice President, Clinical Affairs at Brain Neurotherapy Bio, Inc. He has an equity interest in this company.
Jeff Kordower serves as a consultant and scientific advisor to Fuji-Cellular dynamics Inc., a company performing preclinical, and is in the planning stages of clinical IPSC dopamine grafts in patients with PD. Dr. Kordower is also a consultant for Seelos- Inc., NsGene Inc., Brainstorm Inc., Abbvie, Inc., Biogen Inc., Brain Neurotherapy Bio, Clintrex Inc., Encora Inc., and Inhibikase Inc. Dr. Kordower is also an Associate Editor for Journal of Comparative Neurology, Neurobiology of Aging, and Journal of Parkinson’s Disease.
Kris Bankiewicz is the Founder and CEO of Brain Neurotherapy Bio that is developing AAV2-GDNF for PD, co-founder of Voyager Therapeutics that is developing AAV2AADC for PD, co-founder of MedGenesis Therapeutics that is developing rec. GDNF protein for PD and also has financial interest in all the companies.
Karl Kieburtz is a consultant for Clintrex Research Corp, Roche/Genentech, Novartis, Blackfynn LLC. He has grant support from NIH (NINDS, NCATS), Michael J Fox Foundation and has ownership inClintrex Research Corp, Hoover Brown LLC, Blackfynn LLC, Safe Therapeutics LLC.
Sigrid Booms is an employee of Herantis Pharma Plc.
Henri Huttunen is co-founder, shareholder and an employee of Herantis Pharma Plc.
A. Jon Stoessl reports personal fees from AbbVie, personal fees from Voyager/Neurocrine Biosciences, and is Editor-in-Chief, Movement Disorders (stipend).
Howard Federoff is a co-founder of Med Genesis Therapetix and Brain Neurotherapy Bio. He also serves as the CEO of Aspen Neuroscience. He is a consultant for Juvenescence, a scientific advisory board member for Ovid Therapeutics, chair of the board for Souvien and director for Perthera. He holds equity interests in all of the above.
Mart Saarma is founder and shareholder in the Herantis Pharma Plc. This company is testing CDNF in clinical trials.
Merja H. Voutilainen is an inventor in the CDNF-patent, which is owned by Herantis Pharma Plc.
Codrin Lungu has received honoraria from Elsevier, Inc. for editorial work. The work has been conducted as part of official duty in the course of employment with the NIH, and agency of the US Federal Government.
Anders Björklund, David Eidelberg, David Dexter, Jamie Eberling, Richard Wyse, Simon Stott, Eros Bresolin, Lyndsey Issacs, Patrik Brundin, Brain Fiske, Camille Carroll, Alasdair Coles, Leah Mursaleen all have no disclosures.
Patrik Brundin has received commercial support as a consultant from Renovo Neural, Inc., Lundbeck A/S, AbbVie, Fujifilm-Cellular Dynamics International, Axial Biotherapeutics, CuraSen, Idorsia Pharmaceuticals Ltd and Living Cell Technologies. He has received commercial support for research from Lundbeck A/S and Roche. He has ownership interests in Acousort AB and Axial Biotherapeutics.
ACKNOWLEDGMENTS
The working group would like to thank The Cure Parkinson’s Trust for funding and organising the event and the Michael J. Fox Foundation for Parkinson’s Research and Van Andel institute for supporting and hosting it.
REFERENCES
[1] | Paul G , Sullivan AM ((2019) ) Trophic factors for Parkinson’s disease: Where are we and where do we go from here? Eur J Neurosci 49: , 440–452. |
[2] | Kalia LV , Lang AE ((2015) ) Parkinson’s disease. Lancet 386: , 896–912. |
[3] | Lin LF , Doherty DH , Lile JD , Bektesh S , Collins F ((1993) ) GDNF: A glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science 260: , 1130–1132. |
[4] | Nutt JG , Burchiel KJ , Comella CL , Jankovic J , Lang AE , Laws ER Jr. , Lozano AM , Penn RD , Simpson RK Jr. , Stacy M , Wooten GF , ICV GDNF Study Group. Implanted intracerebroventricular. Glial cell line-derived neurotrophic factor ((2003) ) Randomized, double-blind trial of glial cell line-derived neurotrophic factor (GDNF) in PD. Neurology 60: , 69–73. |
[5] | Kotzbauer PT , Lampe PA , Heuckeroth RO , Golden JP , Creedon DJ , Johnson EM Jr. , Milbrandt J ((1996) ) Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature 384: , 467–470. |
[6] | Airaksinen MS , Saarma M ((2002) ) The GDNF family: Signalling, biological functions and therapeutic value. Nat Rev Neurosci 3: , 383–394. |
[7] | Kearns CM , Gash DM ((1995) ) GDNF protects nigral dopamine neurons against 6-hydroxydopamine in vivo. Brain Res 672: , 104–111. |
[8] | Sauer H , Rosenblad C , Bjorklund A ((1995) ) Glial cell line-derived neurotrophic factor but not transforming growth factor beta 3 prevents delayed degeneration of nigral dopaminergic neurons following striatal 6-hydroxydopamine lesion. Proc Natl Acad Sci U S A 92: , 8935–8939. |
[9] | Tomac A , Lindqvist E , Lin LF , Ogren SO , Young D , Hoffer BJ , Olson L ((1995) ) Protection and repair of the nigrostriatal dopaminergic system by GDNF in vivo. Nature 373: , 335–339. |
[10] | Beck KD , Valverde J , Alexi T , Poulsen K , Moffat B , Vandlen RA , Rosenthal A , Hefti F ((1995) ) Mesencephalic dopaminergic neurons protected by GDNF from axotomy-induced degeneration in the adult brain. Nature 373: , 339–341. |
[11] | Hebert MA , Van Horne CG , Hoffer BJ , Gerhardt GA ((1996) ) Functional effects of GDNF in normal rat striatum: Presynaptic studies using in vivo electrochemistry and microdialysis. J Pharmacol Exp Ther 279: , 1181–1190. |
[12] | Zhang Z , Miyoshi Y , Lapchak PA , Collins F , Hilt D , Lebel C , Kryscio R , Gash DM ((1997) ) Dose response to intraventricular glial cell line-derived neurotrophic factor administration in parkinsonian monkeys. J Pharmacol Exp Ther 282: , 1396–1401. |
[13] | Hamilton JF , Morrison PF , Chen MY , Harvey-White J , Pernaute RS , Phillips H , Oldfield E , Bankiewicz KS ((2001) ) Heparin coinfusion during convection-enhanced delivery (CED) increases the distribution of the glial-derived neurotrophic factor (GDNF) ligand family in rat striatum and enhances the pharmacological activity of neurturin. Exp Neurol 168: , 155–161. |
[14] | Bespalov MM , Sidorova YA , Tumova S , Ahonen-Bishopp A , Magalhaes AC , Kulesskiy E , Paveliev M , Rivera C , Rauvala H , Saarma M ((2011) ) Heparan sulfate proteoglycan syndecan-3 is a novel receptor for GDNF, neurturin, and artemin. J Cell Biol 192: , 153–169. |
[15] | Gerhardt GA , Cass WA , Huettl P , Brock S , Zhang Z , Gash DM ((1999) ) GDNF improves dopamine function in the substantia nigra but not the putamen of unilateral MPTP-lesioned rhesus monkeys. Brain Res 817: , 163–171. |
[16] | Winkler C , Sauer H , Lee CS , Bjorklund A ((1996) ) Short-term GDNF treatment provides long-term rescue of lesioned nigral dopaminergic neurons in a rat model of Parkinson’s disease. J Neurosci 16: , 7206–7215. |
[17] | Kirik D , Rosenblad C , Bjorklund A ((2000) ) Preservation of a functional nigrostriatal dopamine pathway by GDNF in the intrastriatal 6-OHDA lesion model depends on the site of administration of the trophic factor. Eur J Neurosci 12: , 3871–3882. |
[18] | Rosenblad C , Kirik D , Devaux B , Moffat B , Phillips HS , Bjorklund A ((1999) ) Protection and regeneration of nigral dopaminergic neurons by neurturin or GDNF in a partial lesion model of Parkinson’s disease after administration into the striatum or the lateral ventricle. Eur J Neurosci 11: , 1554–1566. |
[19] | Maswood N , Grondin R , Zhang Z , Stanford JA , Surgener SP , Gash DM , Gerhardt GA ((2002) ) Effects of chronic intraputamenal infusion of glial cell line-derived neurotrophic factor (GDNF) in aged Rhesus monkeys. Neurobiol Aging 23: , 881–889. |
[20] | Eberling JL , Kells AP , Pivirotto P , Beyer J , Bringas J , Federoff HJ , Forsayeth J , Bankiewicz KS ((2009) ) Functional effects of AAV2-GDNF on the dopaminergic nigrostriatal pathway in parkinsonian rhesus monkeys. Hum Gene Ther 20: , 511–518. |
[21] | Kells AP , Eberling J , Su X , Pivirotto P , Bringas J , Hadaczek P , Narrow WC , Bowers WJ , Federoff HJ , Forsayeth J , Bankiewicz KS ((2010) ) Regeneration of the MPTP-lesioned dopaminergic system after convection-enhanced delivery of AAV2-GDNF. J Neurosci 30: , 9567–9577. |
[22] | Jiang P , Dickson DW ((2018) ) Parkinson’s disease: Experimental models and reality. Acta Neuropathol 135: , 13–32. |
[23] | Decressac M , Ulusoy A , Mattsson B , Georgievska B , Romero-Ramos M , Kirik D , Bjorklund A ((2011) ) GDNF fails to exert neuroprotection in a rat alpha-synuclein model of Parkinson’s disease. Brain 134: , 2302–2311. |
[24] | Lo Bianco C , Deglon N , Pralong W , Aebischer P ((2004) ) Lentiviral nigral delivery of GDNF does not prevent neurodegeneration in a genetic rat model of Parkinson’s disease. Neurobiol Dis 17: , 283–289. |
[25] | Decressac M , Kadkhodaei B , Mattsson B , Laguna A , Perlmann T , Bjorklund A ((2012) ) alpha-Synuclein-induced down-regulation of Nurr1 disrupts GDNF signaling in nigral dopamine neurons. Sci Transl Med 4: , 163ra156. |
[26] | Galleguillos D , Fuentealba JA , Gomez LM , Saver M , Gomez A , Nash K , Burger C , Gysling K , Andres ME ((2010) ) Nurr1 regulates RET expression in dopamine neurons of adult rat midbrain. J Neurochem 114: , 1158–1167. |
[27] | Chu Y , Le W , Kompoliti K , Jankovic J , Mufson EJ , Kordower JH ((2006) ) Nurr1 in Parkinson’s disease and related disorders. J Comp Neurol 494: , 495–514. |
[28] | Su X , Fischer DL , Li X , Bankiewicz K , Sortwell CE , Federoff HJ ((2017) ) Alpha-synuclein mRNA is not increased in sporadic PD and alpha-synuclein accumulation does not block GDNF signaling in Parkinson’s disease and disease models. Mol Ther 25: , 2231–2235. |
[29] | Kordower JH , Palfi S , Chen EY , Ma SY , Sendera T , Cochran EJ , Cochran EJ , Mufson EJ , Penn R , Goetz CG , Comella CD ((1999) ) Clinicopathological findings following intraventricular glial-derived neurotrophic factor treatment in a patient with Parkinson’s disease. Ann Neurol 46: , 419–424. |
[30] | Gill SS , Patel NK , Hotton GR , O’Sullivan K , McCarter R , Bunnage M , Brooks DJ , Svendsen CN , Heywood P ((2003) ) Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med 9: , 589–595. |
[31] | Slevin JT , Gerhardt GA , Smith CD , Gash DM , Kryscio R , Young B ((2005) ) Improvement of bilateral motor functions in patients with Parkinson disease through the unilateral intraputaminal infusion of glial cell line-derived neurotrophic factor. J Neurosurg 102: , 216–222. |
[32] | Love S , Plaha P , Patel NK , Hotton GR , Brooks DJ , Gill SS ((2005) ) Glial cell line-derived neurotrophic factor induces neuronal sprouting in human brain. Nat Med 11: , 703–704. |
[33] | Lang AE , Gill S , Patel NK , Lozano A , Nutt JG , Penn R , Brooks DJ , Hotton G , Moro E , Heywood P , Brodsky MA , Burchiel K , Kelly P , Dalvi A , Scott B , Stacy M , Turner D , Wooten VG , Elias WJ , Laws ER , Dhawan V , Stoessl AJ , Matcham J , Coffey RJ , Traub M ((2006) ) Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol 59: , 459–466. |
[34] | Hovland DN Jr. , Boyd RB , Butt MT , Engelhardt JA , Moxness MS , Ma MH , Emery MG , Ernst NB , Reed RP , Zeller JR , Gash DM , Masterman DM , Potter BM , Cosenza ME , Lightfoot RM ((2007) ) Six-month continuous intraputamenal infusion toxicity study of recombinant methionyl human glial cell line-derived neurotrophic factor (r-metHuGDNF in rhesus monkeys. Toxicol Pathol 35: , 1013–1029. |
[35] | Salvatore MF , Ai Y , Fischer B , Zhang AM , Grondin RC , Zhang Z , Gerhardt GA , Gash DM ((2006) ) Point source concentration of GDNF may explain failure of phase II clinical trial. Exp Neurol 202: , 497–505. |
[36] | Patel NK , Bunnage M , Plaha P , Svendsen CN , Heywood P , Gill SS ((2005) ) Intraputamenal infusion of glial cell line-derived neurotrophic factor in PD: A two-year outcome study. Ann Neurol 57: , 298–302. |
[37] | Patel NK , Pavese N , Javed S , Hotton GR , Brooks DJ , Gill SS ((2013) ) Benefits of putaminal GDNF infusion in Parkinson disease are maintained after GDNF cessation. Neurology 81: , 1176–1178. |
[38] | Whone A , Luz M , Boca M , Woolley M , Mooney L , Dharia S , Broadfoot J , Cronin D , Schroers C , Barua NU , Longpre L , Barclay CL , Boiko C , Johnson GA , Fibiger HC , Harrison R , Lewis O , Pritchard G , Howell M , Irving C , Johnson D , Kinch S , Marshall C , Lawrence AD , Blinder S , Sossi V , Stoessl AJ , Skinner P , Mohr E , Gill SS ((2019) ) Randomized trial of intermittent intraputamenal glial cell line-derived neurotrophic factor in Parkinson’s disease. Brain 142: , 512–525. |
[39] | Whone AL , Boca M , Luz M , Woolley M , Mooney L , Dharia S , Broadfoot J , Cronin D , Schroers C , Barua NU , Longpre L , Barclay CL , Boiko C , Johnson GA , Fibiger HC , Harrison R , Lewis O , Pritchard G , Howell M , Irving C , Johnson D , Kinch S , Marshall C , Lawrence AD , Blinder S , Sossi V , Stoessl AJ , Skinner P , Mohr E , Gill SS ((2019) ) Extended treatment with glial cell line-derived neurotrophic factor in Parkinson’s disease. J Parkinsons Dis 9: , 301–313. |
[40] | Sala Q , Metellus P , Taieb D , Kaphan E , Figarella-Branger D , Guedj E ((2014) ) 18F-DOPA, a clinically available PET tracer to study brain inflammation? Clin Nucl Med 39: , e283–285. |
[41] | Fiandaca MS , Varenika V , Eberling J , McKnight T , Bringas J , Pivirotto P , Beyer J , Hadaczek P , Bowers W , Park J , Federoff H , Forsayeth J , Bankiewicz KS ((2009) ) Real-time MR imaging of adeno-associated viral vector delivery to the primate brain. Neuroimage 47: (2), T27–35. |
[42] | Bankiewicz KS , Forsayeth J , Eberling JL , Sanchez-Pernaute R , Pivirotto P , Bringas J , Herscovitch P , Carson RE , Eckelman W , Reutter B , Cunningham J ((2006) ) Long-term clinical improvement in MPTP-lesioned primates after gene therapy with AAV-hAADC. Mol Ther 14: , 564–570. |
[43] | Kells AP , Forsayeth J , Bankiewicz KS ((2012) ) Glial-derived neurotrophic factor gene transfer for Parkinson’s disease: Anterograde distribution of AAV2 vectors in the primate brain. Neurobiol Dis 48: , 228–235. |
[44] | Salegio EA , Samaranch L , Kells AP , Mittermeyer G , San Sebastian W , Zhou S , Beyer J , Forsayeth J , Bankiewicz KS ((2013) ) Axonal transport of adeno-associated viral vectors is serotype-dependent. Gene Ther 20: , 348–352. |
[45] | Marks WJ Jr. , Ostrem JL , Verhagen L , Starr PA , Larson PS , Bakay RA , Taylor R , Cahn-Weiner DA , Stoessl AJ , Olanow CW , Bartus RT ((2008) ) Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: An open-label, phase I trial. Lancet Neurol 7: , 400–408. |
[46] | Marks WJ Jr. , Bartus RT , Siffert J , Davis CS , Lozano A , Boulis N , Vitek J , Stacy M , Turner D , Verhagen L , Bakay R , Watts R , Guthrie B , Jankovic J , Simpson R , Tagliati M , Alterman R , Stern M , Baltuch G , Starr PA , Larson PS , Ostrem JL , Nutt J , Kieburtz K , Kordower JH , Olanow CW ((2010) ) Gene delivery of AAV2-neurturin for Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol 9: , 1164–1172. |
[47] | Bartus RT , Kordower JH , Johnson EM Jr. , Brown L , Kruegel BR , Chu Y , Baumann TL , Lang AE , Olanow CW , Herzog CD ((2015) ) Post-mortem assessment of the short and long-term effects of the trophic factor neurturin in patients with alpha-synucleinopathies. Neurobiol Dis 78: , 162–171. |
[48] | Warren Olanow C , Bartus RT , Baumann TL , Factor S , Boulis N , Stacy M , Turner DA , Marks W , Larson P , Starr PA , Jankovic J , Simpson R , Watts R , Guthrie B , Poston K , Henderson JM , Stern M , Baltuch G , Goetz CG , Herzog C , Kordower JH , Alterman R , Lozano AM , Lang AE ((2015) ) Gene delivery of neurturin to putamen and substantia nigra in Parkinson disease: A double-blind, randomized, controlled trial. Ann Neurol 78: , 248–257. |
[49] | Heiss JD , Lungu C , Hammoud DA , Herscovitch P , Ehrlich DJ , Argersinger DP , Sinharay S , Scott G , Wu T , Federoff HJ , Zaghloul KA , Hallett M , Lonser RR , Bankiewicz KS ((2019) ) Trial of magnetic resonance-guided putaminal gene therapy for advanced Parkinson’s disease. Mov Disord 34: , 1073–1078. |
[50] | Christine CW , Bankiewicz KS , Van Laar AD , Richardson RM , Ravina B , Kells AP , Boot B , Martin AJ , Nutt J , Thompson ME , Larson PS ((2019) ) Magnetic resonance imaging-guided phase 1 trial of putaminal AADC gene therapy for Parkinson’s disease. Ann Neurol 85: , 704–714. |
[51] | Bankiewicz KS , Sudhakar V , Samaranch L , San Sebastian W , Bringas J , Forsayeth J ((2016) ) AAV viral vector delivery to the brain by shape-conforming MR-guided infusions. J Control Release 240: , 434–442. |
[52] | Kordower JH , Olanow CW , Dodiya HB , Chu Y , Beach TG , Adler CH , Halliday GM , Bartus RT ((2013) ) Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136: , 2419–2431. |
[53] | Tomse P , Peng S , Pirtosek Z , Zaletel K , Dhawan V , Eidelberg D , Ma Y , Trost M ((2018) ) The effects of image reconstruction algorithms on topographic characteristics, diagnostic performance and clinical correlation of metabolic brain networks in Parkinson’s disease. Phys Med 52: , 104–112. |
[54] | Liu G , Boot B , Locascio JJ , Jansen IE , Winder-Rhodes S , Eberly S , Elbaz A , Brice A , Ravina B , van Hilten JJ , Cormier-Dequaire F , Corvol JC , Barker RA , Heutink P , Marinus J , Williams-Gray CH , Scherzer CR , International Genetics of Parkinson Disease Progression (IGPP) Consortium ((2016) ) Specifically neuropathic Gaucher’s mutations accelerate cognitive decline in Parkinson’s. Ann Neurol 80: , 674–685. |
[55] | Winder-Rhodes SE , Evans JR , Ban M , Mason SL , Williams-Gray CH , Foltynie T , Duran R , Mencacci NE , Sawcer SJ , Barker RA ((2013) ) Glucocerebrosidase mutations influence the natural history of Parkinson’s disease in a community-based incident cohort. Brain 136: , 392–399. |
[56] | Luz M , Whone A , Bassani N , Wyse RK , Stebbins GT , Mohr E ((2020) ) The Parkinson’s Disease Comprehensive Response (PDCORE): A composite approach integrating three standard outcome measures. Brain Commun, fcaa046. |
[57] | Piltonen M , Bespalov MM , Ervasti D , Matilainen T , Sidorova YA , Rauvala H , Saarma M , Mannisto PT ((2009) ) Heparin-binding determinants of GDNF reduce its tissue distribution but are beneficial for the protection of nigral dopaminergic neurons. Exp Neurol 219: , 499–506. |
[58] | Runeberg-Roos P , Piccinini E , Penttinen AM , Matlik K , Heikkinen H , Kuure S , Bespalov MM , Peranen J , Garea-Rodriguez E , Fuchs E , Airavaara M , Kalkkinen N , Penn R , Saarma M ((2016) ) Developing therapeutically more efficient Neurturin variants for treatment of Parkinson’s disease. Neurobiol Dis 96: , 335–345. |
[59] | Mahato AK , Kopra J , Renko JM , Visnapuu T , Korhonen I , Pulkkinen N , Bespalov MM , Domanskyi A , Ronken E , Piepponen TP , Voutilainen MH , Tuominen RK , Karelson M , Sidorova YA , Saarma M ((2020) ) Glial cell line-derived neurotrophic factor receptor Rearranged during transfection agonist supports dopamine neurons in vitro and enhances dopamine release in vivo. Mov Disord 35: , 245–255. |
[60] | Lindholm P , Voutilainen MH , Lauren J , Peranen J , Leppanen VM , Andressoo JO , Lindahl M , Janhunen S , Kalkkinen N , Timmusk T , Tuominen RK , Saarma M ((2007) ) Novel neurotrophic factor CDNF protects and rescues midbrain dopamine neurons in vivo. Nature 448: , 73–77. |
[61] | Huttunen HJ , Saarma M ((2019) ) CDNF protein therapy in Parkinson’s disease. Cell Transplant 28: , 349–366. |


