
The Association Between Type 2 Diabetes Mellitus and Parkinson’s Disease
Abstract
In recent years, an emerging body of evidence has forged links between Parkinson’s disease (PD) and type 2 diabetes mellitus (T2DM). In observational studies, those with T2DM appear to be at increased risk of developing PD, as well as experiencing faster progression and a more severe phenotype of PD, with the effects being potentially mediated by several common cellular pathways. The insulin signalling pathway, for example, may be responsible for neurodegeneration via insulin dysregulation, aggregation of amyloids, neuroinflammation, mitochondrial dysfunction and altered synaptic plasticity. In light of these potential shared disease mechanisms, clinical trials are now investigating the use of established diabetes drugs targeting insulin resistance in the management of PD. This review will discuss the epidemiological links between T2DM and PD, the potential shared cellular mechanisms, and assess the relevant treatment options for disease modification of PD.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disease in the world [1]. With increasing life expectancy and an ageing global population, its prevalence is set to more than double between 2015 and 2040 [2, 3]. PD is a progressive disease of the nervous system that is characterised by the degeneration of nigrostriatal dopaminergic neurons and pathological hallmarks of the disease can be identified widely in both central and peripheral tissues. Several overlapping disease mechanisms have been identified including aberrant protein accumulation, lysosomal and mitochondrial dysfunction, and chronic systemic inflammation [4].
Type 2 diabetes mellitus (T2DM) is a chronic condition characterised by the failure of pancreatic β-cells to produce enough insulin to overcome systemic insulin resistance, which results in the dysregulation of glucose metabolism and chronic systemic inflammation. Studies suggest that similar metabolic dysregulation can occur in the brain in early PD [5]. The global prevalence of T2DM quadrupled between 1980 and 2014 and already grossly exceeds the predictions made in 2000 for the year 2030 by both the International Diabetes Federation and the World Health Organisation [6]. In contrast to T2DM, type 1 diabetes (T1DM) is an autoimmune disease involving the destruction of pancreatic β cells [7]. Limited studies have been conducted on the association between PD and autoimmune diseases including T1DM [8–10], but T1DM is not discussed further in this review.
There are previous review articles that discuss the association between T2DM and PD [4, 11–15] or the common therapeutic approaches to T2DM and PD [4, 13, 16, 17]. Our review provides an updated discussion on the epidemiological association between T2DM and PD, the shared pathways involved in T2DM and PD pathophysiology, and the common therapeutic approaches to T2DM and PD.
THE ASSOCIATION BETWEEN TYPE 2 DIABETES MELLITUS AND PARKINSON’S DISEASE
T2DM is associated with an increase in the risk of PD
An association between T2DM and PD was first reported by Sandyk in 1993, where it was noted that PD patients with co-existent T2DM had worse motor symptoms and reduced response to treatment [18]. In the same study, a high prevalence of impaired glucose tolerance tests was reported among PD patients (50–80%), however a more recent estimate suggests that overtly impaired glucose metabolism occurs in only around 20% [19].
In subsequent years, a large number of studies have explored the association between T2DM and the risk of PD. These include several prospective cohort studies which generally indicate that T2DM is associated with an increased risk of PD [20–22]. For example, a large prospective study in Finland found that patients with T2DM had an 85% increased risk of developing PD [20] and another prospective study in the US showed that patients with T2DM were 40% more likely to develop PD [21]. A meta-analysis published in 2016 combined the effect estimates from seven population-based cohort studies and concluded that patients with T2DM had an average 28% higher risk of developing PD [23].
Observational studies using different designs have also reported an association between T2DM and an increased risk of PD. A large study using routinely gathered health record data in the UK showed that T2DM was associated with an increased risk of PD by 32% [24]. Similarly, a retrospective study in Taiwan reported a 23% increased risk of PD among patients with T2DM [25], and a case-control study from Denmark found that T2DM was associated with a 36% increased risk of PD [26]. Despite these associations, it is important to acknowledge that although various studies have reported that T2DM increases the risk of developing PD, the absolute risk of developing PD among patients with T2DM appears to be below 1%. In a study of 2,017,115 T2DM participants, 14,252 also had PD observed (0.7%) [24].
Whether the association between T2DM and PD represents a truly causal link between the two conditions remains uncertain, and associations between two traits can arise through confounding and bias in observational studies. The use of oral anti-hyperglycaemic agents may influence the likelihood of developing PD in patients with T2DM, potentially masking associations between the two. Various studies in different settings have suggested that the use of metformin, thiazolidinediones and GLP-1 agonists may reduce the risk of developing PD in patients who have T2DM [27–31]. Other confounders may bias effect estimates, due to associations with both T2DM and PD, and these include other vascular risk factors or raised BMI [20, 32, 33].
Bias in observational studies may occur for several other reasons. Patients with T2DM are more likely to have increased contact with healthcare and this could result in bias from increased medical surveillance [22]. Bias may also occur through reverse causation, that is, something about PD increasing the risk of being diagnosed with T2DM. Dopaminergic neurons are involved in promoting feeding behaviour in the hypoglycaemic state, mediated by insulin receptors in the substantia nigra and therefore dopaminergic neuronal loss may alter glycaemic control [22]. It is recognised that a high proportion of dopaminergic neurons have already been lost by the time clinical signs of PD are identified and a diagnosis is made. This makes it difficult to mitigate the effects of reverse causation even in prospective studies with long follow-up periods [22].
Prospective cohort studies follow participants over time and longer durations of follow-up can lead to drop out and bias occurring through these losses. If the probability of loss to follow-up does not relate to exposure (in this case diabetes) then the effect will be a loss of precision without biasing the effect estimate. However, if diabetes is associated with being more or less likely to remain under follow-up (which is plausible), then the effect estimate may be biased in either direction.
Whilst the studies outlined above have generally reported a positive association between T2DM and PD, this has not always been the case. A recent cross-sectional study based on the Neurological Disorders in Central Spain (NEDICES) database showed no clear association between T2DM and PD [34]. However, a sub-analysis suggested that there may be a positive association between PD and T2DM among patients who had T2DM for over 10 years [34]. Separately, another prospective cohort study in the US did not find a significant association between T2DM and PD risk [35]. To make matters more confusing, a meta-analysis of fourteen case-control studies concluded that T2DM was associated with a reduced risk of PD (summary odds ratio 0.75) [36]. A very large, recent, cross-sectional study using self-reported information also found T2DM to be negatively associated with PD [37]. However, whilst cohort studies may be prone to biases as outlined above, cross-sectional and retrospective case-control studies may have additional design issues that bias effect estimates.
Several groups have reported on the differences in pooled effect estimates generated from case-control and cohort studies in the context of T2DM and PD. A meta-analysis of 4 cohort studies and 5 case-control studies reported a 1.37 pooled risk ratio for PD in diabetic patients in the prospective cohort studies and an inverse association (pooled odds ratio of 0.56) between diabetes and PD in case-control studies [32]. This was followed by a meta-analysis of 9 case-control studies and 4 cohort studies, which showed pooled effect estimates of 0.72 and 1.31 respectively [38]. Later still, the aforementioned and separate meta-analyses of 14 case-control studies (OR 0.75) and 7 cohort studies (RR 1.37) reported the same phenomenon [23, 36]. Whilst it is not uncommon to observe differential effect sizes by study design, such a clear divergence in the direction of effect warrants further consideration.
A possible explanation for the divergence observed between case-control and cohort studies is survival bias, which can be a problematic in studies with a retrospective case-control design. Higher mid-life mortality among diabetic patients could contribute towards the inverse relationship seen between T2DM and PD in these settings, and thus far has been given little consideration [36].
To explore this possibility, the case-control studies included in meta-analyses by Cereda et al., 2011, Noyce et al., 2012 and Lu et al., 2014 were selected and the mean ages of participants were plotted against the odds ratio of the risk of PD for each study (Fig. 1). The odds ratio for PD was lowest (negative) in the studies with the youngest mean age of participants and highest (positive) in studies with the highest mean age. This perhaps indicates that the risk of PD increases depending on the duration of exposure to T2DM, with most people acquiring diabetes in mid-life and PD later in older age. It is also plausible that inverse association in the studies with youngest mean age may be driven in part by low incidence of PD in mid-life and partly by premature mortality in patients with T2DM before they develop PD. There is evidence to suggest that patients diagnosed with T2DM before the age of 45 years have a higher risk of premature death than those diagnosed from the age of 45 years onwards [39]. Furthermore, patients diagnosed with T2DM at a younger age have an increased risk of acquiring complications such as nephropathy and cardiovascular disease that increase the risk of premature mortality [40, 41]. The role of survival bias in observational studies linking T2DM to PD requires further consideration.
Fig. 1
Graph of the mean age of participants in case-control studies against odds ratio for risk of PD between cases and controls. This suggests that age may modify the association between T2DM and PD, potentially driven by duration of exposure, probability of PD at given ages, and/or by survival bias.
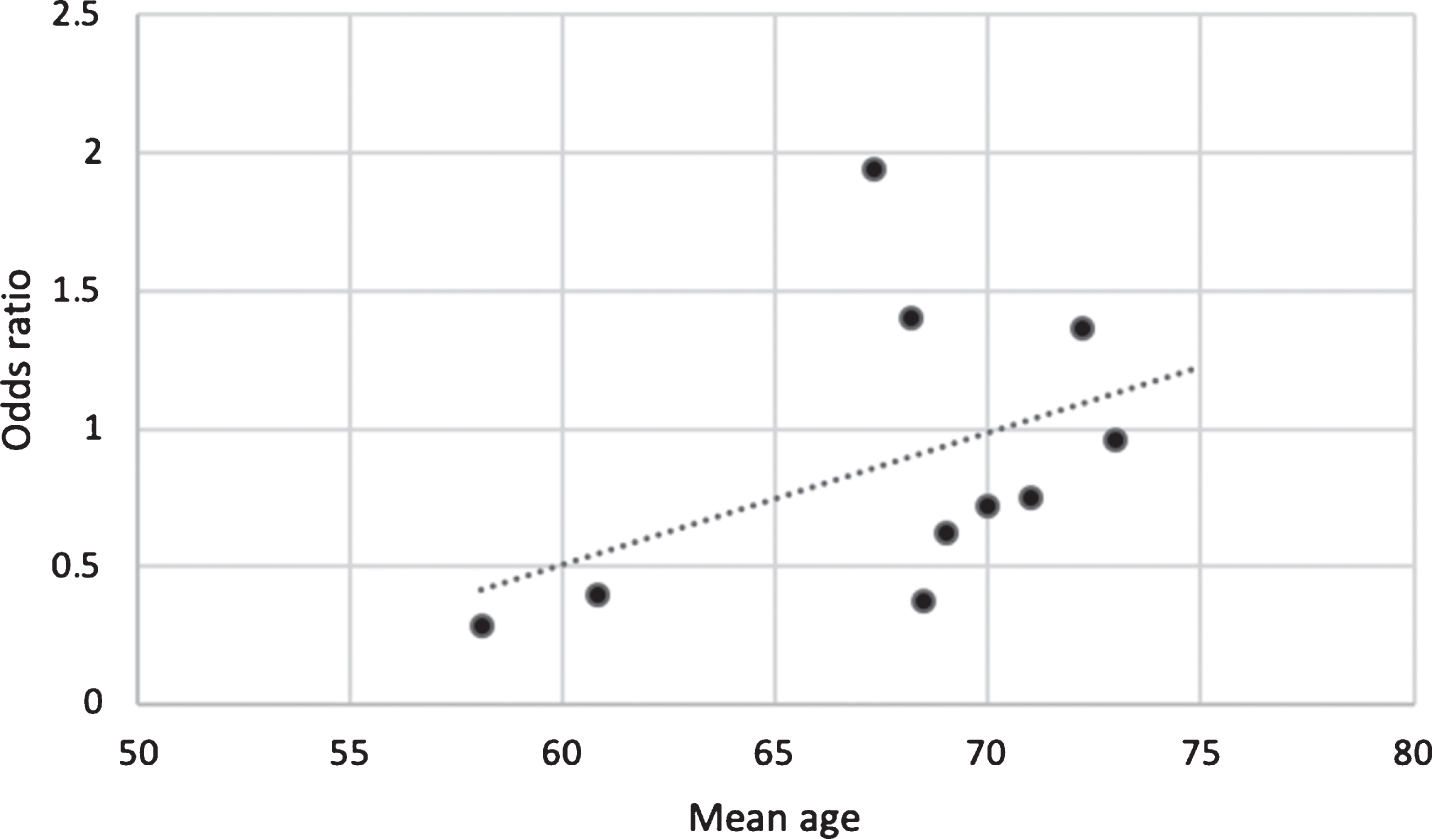
Other limitations of case-control studies include recall bias, where risk factors and the duration of exposure may not be represented accurately, especially in PD patients experiencing cognitive decline, and separately selection bias, where the controls are not drawn from the same population as the cases [32]. Overall, further study is warranted to explore the divergence in estimates arising from cohort and case-control studies and risk of PD.
T2DM worsens PD disease progression
Following the initial report by Sandyk [18], several studies have found that T2DM is also associated with a worsening of the PD phenotype, including more severe axial motor symptoms (gait disturbances and postural instability) and cognitive impairment [42–45].
A case-control study showed that patients with T2DM who subsequently developed PD scored higher on PD severity scales including the motor component of the Unified Parkinson Disease Rating Scale (UPDRS) and Hoehn and Yahr stage [46]. Another case-control study of 72 PD patients reported that those with concomitant T2DM developed motor complications on average 12 months earlier, independent of medication or other disease factors [47]. A retrospective cohort study in PD patients assessed the effect of T2DM on striatal dopamine transporter availability using 18F-FP-CIT PET imaging, cognitive performance on bedside tests, cortical thickness using MRI scans and overall disease severity. The investigators found that the presence of T2DM had a significant adverse effect on all four outcomes [33]. Similarly, a prospective cohort study found that patients with T2DM had lower striatal dopamine transporter binding and accelerated motor and cognitive decline, supporting the notion of an acceleration in the disease process [48]. However, these features are not necessarily driven by dopamine depletion, and some studies suggest that the more severe phenotype is independent of striatal dopamine, and even cholinergic deficits, and instead may be mediated by microvascular disease [43, 44].
MRI imaging has been used to study structural changes in PD associated with comorbid T2DM. A cross-sectional study showed that PD patients with T2DM have greater cortical atrophy than patients without T2DM [49]. These findings were most evident in the frontal brain region, perhaps reflecting an accelerated decline in executive function [49]. A separate study found that PD patients with T2DM had significantly more cortical thinning in the right inferior temporal cortex than those without T2DM [33]. Further replication of these findings and their clinical correlates is warranted. As a fluid biomarker, CSF tau is elevated in several neurodegenerative diseases and is indicative of neuronal loss. As such, it is a non-specific marker of the severity of a neurodegenerative process. A recent cross-sectional study, using data from the Parkinson’s Progression Markers Initiative found that PD patients with T2DM had higher tau CSF levels than patients without T2DM [48]. Similar findings have been reported in other neurodegenerative diseases, in which T2DM was associated with a higher level of tau protein in CSF in patients with mild cognitive impairment [50].
SHARED MECHANISMS IN PATHOPHYSIOLOGY OF TYPE 2 DIABETES MELLITUS AND PARKINSON’S DISEASE
Common pathogenic mechanisms of systemic and brain insulin resistance
As epidemiological evidence for a link between PD and T2DM accumulates, parallel experimental evidence indicates potential overlap in disease mechanisms and pathways. Systemic insulin resistance has long been an established key feature of T2DM. Recently, studies have found that insulin resistance is present in the brain in neurodegenerative diseases such as Alzheimer’s disease (AD) and other dementias [51], and PD [52]. Both systemic and local insulin resistance may drive pathology in the brain. Systemic insulin resistance may do so through hyperglycaemia and its consequences [10], microvascular disease, chronic inflammation, and dysfunction of the blood brain barrier, which may be compounded by associated comorbidities such as hypertension, dyslipidaemia and renal impairment [51]. Local brain insulin resistance may act via protein deposition and aggregation, and failure of clearance mechanisms, independent of systemic insulin resistance [51, 53].
Overview of insulin cellular signalling pathways
Insulin may play a key role in neuroprotection [4] via its receptor (IR), which activates insulin receptor substrates (IRS) 1 and 2. IRS1 is particularly expressed in skeletal muscle, adipose tissue and cerebral cortex, whereas IRS2 has a particular role in the liver and the hypothalamus [51]. Insulin binds to IR/IRS to stimulate various downstream pathways (summarised in Fig. 2). These in turn activate downstream secondary messengers via three main pathways:
1. Activation of the IR-Shc-MAP kinase (MAPK) pathway, which is involved in promoting genetic expression of proteins for cell growth and maintenance, as well as synapse plasticity. Insulin also acts via the MAPK pathway to regulate transcription, translation and post-translational modification of proteins [51]. Insulin is able to influence learning and memory via this pathway [54].
2. Activation of the phosphatidylinositol 3-kinase (PI3K) pathway directly influences neurotransmission via cyclic nucleotide phosphodiesterase 3B (cPD3B), which in turn regulates information processing, cognitive function and memory [55]. The PI3K-Akt pathway is involved in the inhibition of apoptosis [56]. Additional downstream effectors of PI3K-Akt include glycogen synthase kinase 3β (GSK3β), forkhead box O1 (FOXO1), nuclear factor κB (NFκB) and mammalian target of rapamycin (mTOR).
3. Mediation of neurotransmission via direct activation of NMDA glutamate receptors to increase the opening of calcium channels at synapses and promote NMDA-mediated neurotransmission [57]. This increases the recruitment of functional GABA receptors to postsynaptic sites to enhance GABA transmission [58] and therefore regulate synaptic inhibition for neuronal functions involved in learning and memory [57].
Fig. 2
Diagrammatic summary of main pathways involved in insulin signalling in the brain. IR, insulin receptor; IRS, insulin receptor substrate; PI3K, phosphoinositide-3-kinase; PDK, 3-phosphoinositide-dependent protein kinase; Akt, Protein kinase B (PKB), plays a key role in activating downstream regulators of cell metabolism, proliferation and survival; PTEN, phosphatase and tensin homolog, regulates PI3K/Akt pathway by inhibiting Akt; mTOR, mammalian target of rapamycin, regulates cell metabolism and proliferation and synapse regeneration in neurons; GSK3β, glycogen synthase kinase 3, downstream mediator involved in IDE inactivation, leading to an increase in α synuclein expression, which aggregate into amyloid fibres; FOXO1, Forkhead box O1, involved in maintaining the mitochondrial electron transport chain for ATP generation and fatty acid oxidation, preventing oxidative stress; NFκB, nuclear factor κB regulates microglial activation and the expression of inflammatory mediators such as IL1β and TNFα; cPD3β, cyclic nucleotide phosphodiesterase 3β; Shc, an adaptor protein involved in the MAPK pathway; Grb2/SOS, downstream adaptor proteins in MAPK pathway; Ras, downstream protein in MAPK pathway that recruits Raf; Raf, Ras effector that stimulates a downstream signalling cascade through phosphorylation of MAPK; MAPK, mitogen-activated protein kinase, modulates downstream protein kinases involved in regulating cell proliferation, differentiation and apoptosis, maintaining neuronal growth and survival.
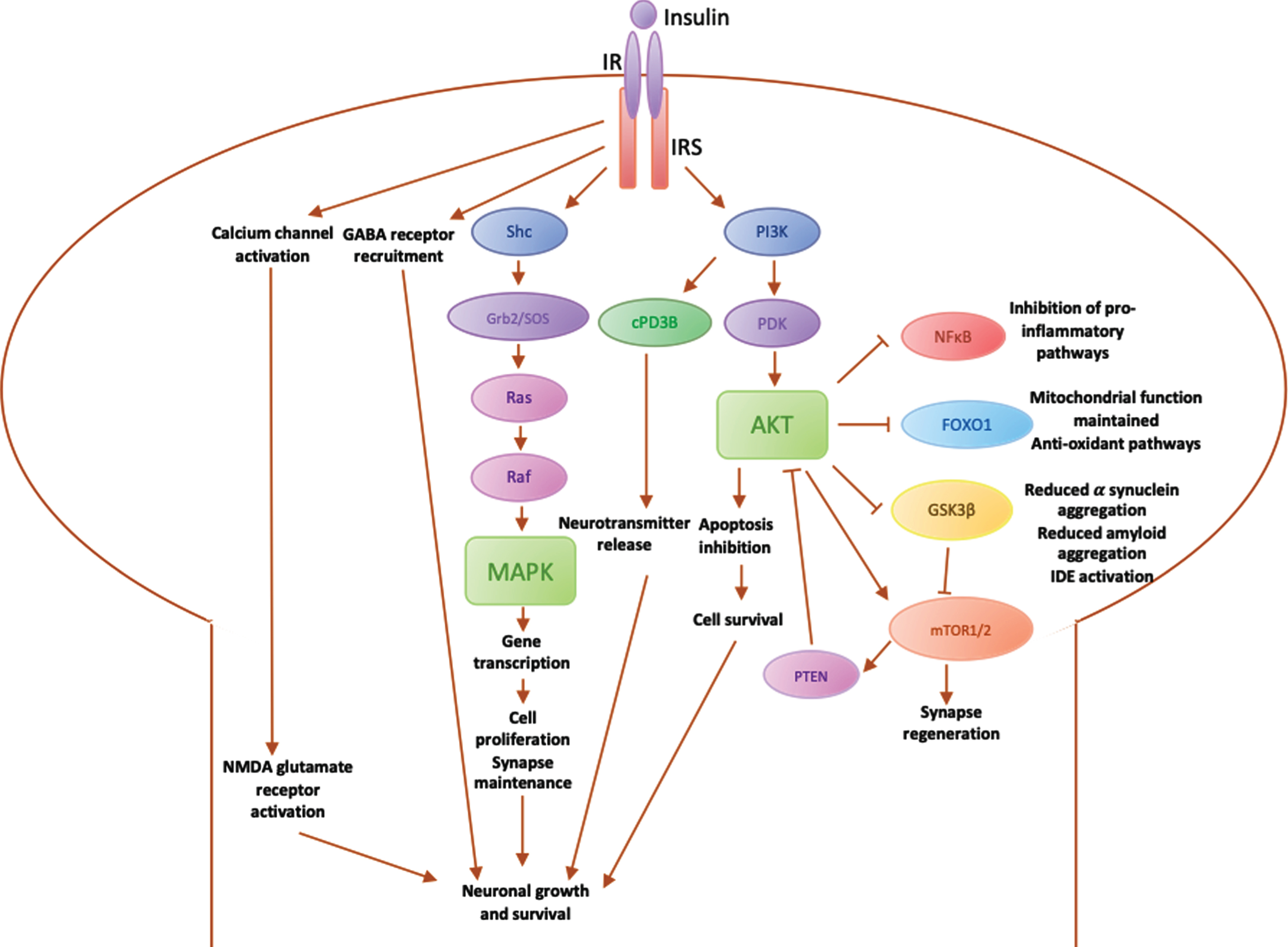
Insulin dysregulation may be involved in pathophysiology of PD and T2DM
Insulin receptors are expressed in the basal ganglia [4] and in the substantia nigra [59], which are the areas of the brain most affected in patients with PD. Studies using rodent models have shown that insulin resistance may cause reduced expression of surface dopamine transporters in the striatum [60], reduced dopamine turnover [61], and reduced insulin-dependent dopamine release in the striatum [62]. 1-methyl 4-phenyl 1,2,3,6-tetrahydropyridine (MPTP) is a toxin that induces parkinsonism by producing oxidative stress in dopaminergic neurons, resulting in mitochondrial dysfunction and cell death, and MPTP treated rodents are one of the most commonly used animal models for PD [63]. MPTP-treated mice have been observed to have simultaneous increases in pancreatic and midbrain expression of pro-inflammatory cytokines and α-synuclein, hinting at potential organ-specific links between PD and T2DM [64].
Very few mechanistic studies have been conducted on human subjects to date. A study in 1996 that analysed the mRNA levels of insulin receptors in the substantia nigra of PD human brains post-mortem, found a reduction in insulin receptor mRNA compared to control brains, which were likely associated with neuronal loss in the substantia nigra [65]. However, due to a small sample size and flaws in methodology, these results are difficult to interpret reliably. Separately, a functional brain imaging study on 63 elderly subjects found that insulin resistance was increased in the brains of PD patients [52]. Future studies investigating the common pathophysiology between insulin dysregulation and PD in human subjects would be beneficial in furthering our understanding.
Amyloid aggregation occurs in PD and T2DM
Both T2DM and PD are associated with the accumulation of misfolded proteins which form amyloid aggregates. In T2DM, islet amyloid polypeptide (IAPP) aggregation in pancreatic β cells leads to cellular dysfunction and death [66, 67]. In PD, α-synuclein aggregates initially into oligomeric and latterly fibrillar structures, which in turn aggregate into Lewy bodies, the pathological hallmark of PD [68].
A recent study of IAPP and α-synuclein found cross-reactivity between the two proteins and demonstrated that IAPP in T2DM can promote α-synuclein aggregation [69]. Further evidence of the interaction between these proteins was shown using proximity ligation assays in pancreatic tissue [70]. Phosphorylated α-synuclein aggregates were found in pancreatic β-cells in the majority of patients with PD or T2DM, and α-synuclein deposits showed co-localization and interaction with IAPP. Another study suggested that insulin degrading enzyme (IDE) can prevent α-synuclein aggregation by binding to α-synuclein oligomers [53]. In patients with T2DM, insulin resistance can competitively inhibit IDE and therefore promote the formation of α-synuclein fibrils, potentially predisposing patients to PD or potentiating the disease process [53].
Separately, a study involving co-immunoprecipitation experiments on α-synuclein and the Kir6.2 subunit of the ATP-sensitive potassium channel in pancreatic beta cells found that α-synuclein interacts with Kir6.2 to reduce insulin secretion [71]. Neuronal Kir6.2 is involved in the downregulation of dopamine secretion in the brain [72], possibly indicating a role in PD. Another study in an MPTP mouse model demonstrated that α-synuclein can activate GSK3β via the PI3K/Akt pathway, which is involved in increasing α-synuclein expression and aggregation and inactivation of IDE [73]. This observation was further supported by a study on post-mortem PD brains, which reported an increase in α-synuclein and GSK3β as well as an increase in tau hyperphosphorylation in PD patients [74].
Microglial activation and systemic chronic inflammation increase the risk of T2DM and PD
Microglia are mononuclear, phagocytic immune cells in the central nervous system. They are normally involved in removal of damaged neurons, and they release neuroprotective factors to promote synaptic regeneration [75]. Microglia can be activated towards either an anti-inflammatory or inflammatory phenotype. For example, microglia can be stimulated by lipopolysaccharide to enter an activated inflammatory state and express pro-inflammatory cytokines such as TNFα, interleukin (IL) 1β and IL6, triggering neuroinflammation [76]. A study of 14 patients with PD found evidence of increased microglial activation on PET imaging [77]. Microglial activation is a key contributor to neuroinflammation through the release of inflammatory cytokines [78], and patients with PD have been shown to have high concentrations of inflammatory mediators such as IL1β, IL6 and TNFα in the brain [78].
Initial microglial activation is generally associated with neuroprotection [79], however, prolonged microglial activation may have deleterious effects on PD progression [80]. Insulin resistance has an effect on microglial activation and neuroinflammation via NFκB and the PI3K/Akt pathway, that regulates microglial activation and the expression of inflammatory mediators [76] (Fig. 2). In addition, inflammatory cytokines such as TNFα have been found to induce the inactivation of IRS1, which inhibits subsequent activation of downstream mediators in a ‘vicious cycle’ [57]. In patients with T2DM, insulin resistance may result in the formation of advanced glycation end-products (AGEs) [4], including in regions of the brain such as the substantia nigra [81]. AGEs interact with their receptor (RAGE) to activate downstream pathways, leading to oxidative stress, inflammation and neuronal cell death [81]. Interestingly, AGEs have been found alongside α-synuclein in Lewy bodies [16]. Glycated α-synuclein may worsen PD progression in various ways. Glycation promotes the aggregation of α-synuclein by inducing cross-links and the formation of α-synuclein oligomers, which appear more toxic than larger aggregations of α-synuclein [81]. Furthermore, glycation also inhibits α-synuclein degradation normally regulated by ubiquitin, proteasomes and lysosomes, resulting in the accumulation of α-synuclein [16]. Methylglyoxal (MGO), a glycation agent, inhibits the ubiquitin-proteasome system involved in the degradation of α-synuclein, further increasing the accumulation of α-synuclein [81], and potentially worsening PD progression.
Involvement of oxidative stress/mitochondrial dysfunction in T2DM and PD pathogenesis
Mitochondrial proteins, when dysfunctional, produce an increase in oxidative stress [82] and cell death [83]. MPTP exerts its Parkinson’s-like effects in rodent models by selectively inhibiting complex I, the first enzyme in the mitochondrial respiratory chain pathway, leading to neuronal death and neurodegeneration [13]. The features of mitochondrial dysfunction may be shared in T2DM and PD [82]. In PD, dysfunctional insulin signalling has been found to increase oxidative stress [84, 85], while a recent study showed that chronic insulin resistance in diabetic db/ db mice can cause mitochondrial disruption and dopaminergic neuronal degeneration [86]. Studies using rodent models show that IRS1 and IRS2 inhibit FOXO1 via the PI3K/Akt pathway [85, 87], resulting in dysfunctional ATP generation and fatty acid oxidation, and the generation of ROS and oxidative stress. While the exact mechanism by which mitochondrial dysfunction and oxidative stress contribute towards PD remains uncertain, its role is likely to be important in PD pathogenesis and potentially relevant to the link with T2DM.
Insulin resistance impairs synaptic plasticity in PD
Dopamine depletion in PD causes changes in synaptic plasticity that result in an upregulation of factors that suppress movement and contribute to a downregulation of factors that initiate movement [88]. The two main components of synaptic plasticity, long-term depression (LTD) and long-term potentiation (LTP), are also involved in memory formation and storage through synaptic restructuring [89]. The combined activation of mTORC1 and mTORC2 is required for dendritic regrowth, neuronal shape restructuring and synaptic plasticity via actin aggregation, for consolidating long-term memory [90].
Insulin promotes NMDA-mediated neurotransmission by directly activating glutamate NMDA receptors [57] and increasing the extra-synaptic transport of GluA1 AMPA receptors in neurons [91] involved in increasing synaptic strength and regulating LTP [92]. A study on streptozotocin-induced diabetic rats found that NMDA and AMPA receptor expression was reduced [93], impairing synaptic transmission.
The common pathophysiological processes linking T2DM and PD offer new avenues for research into the use of T2DM therapeutic approaches repurposed for use in PD.
COMMON THERAPEUTIC APPROACHES TO T2DM AND PD
The current focus of pharmacological management of PD is to relieve symptoms by increasing circulating levodopa, inhibiting levodopa breakdown or stimulating dopamine receptors [94]. Non-oral approaches to management are medical (such as apomorphine or levodopa carbidopa intestinal gel), surgical (such as deep brain stimulation), or therapies-led (physiotherapy, occupational therapy and speech and language therapy), and they play an increasing role as the disease progresses. The current weight of evidence for therapeutic options for PD supports an effect on symptoms only and no modification of disease progression. However, recent studies on the use of T2DM drugs to treat PD have shown promising results.
Insulin
Given the neuroprotective effects of insulin outlined above, the administration of exogenous insulin may offer potential benefits in PD patients. Insulin can be administered nasally to avoid effects on peripheral glucose levels [95], enabling use in non-diabetic PD patients. In rodent models, intranasal insulin is reported to improve neuronal stem cell activation and repair, dendritic sprouting and neuroprotection from inflammation and oxidative stress [95].
Clinical trials of insulin have been conducted in other neurodegenerative diseases. A double-blind, placebo-controlled, pilot clinical trial in AD patients found that intranasal administration of insulin improved memory and preserved cognition. These benefits were retained at 2 months of follow-up after stopping treatment, but the authors acknowledged that as insulin was administered for only 4 months, long-term effects and safety of intranasal insulin could not be determined [96]. Moreover, the small size of the trial may have affected the accuracy of the results, and a longer and larger trial may be useful in studying this further [96]. It should however be noted that the chronic use of insulin may promote insulin desensitisation in the brain [95].
GLP-1 receptor agonists
Among the most promising treatments in this area are the GLP1 receptor (GLP1R) agonists, which do not act on insulin receptors, and thus avoid insulin desensitisation over time [12]. GLP1R agonists, including exenatide, liraglutide and lixisenatide, are currently licensed for the treatment of T2DM [29]. Compared to endogenous GLP1, GLP1R agonists have a longer half-life as they are not metabolised by the protease dipeptidyl peptidase 4 (DPP-4) [97]. GLP1R agonists bind to GLP1R and activate the PI3K/Akt pathway, which regulates various downstream mediators involved in the insulin signalling pathway (Fig. 3).
Fig. 3
Diagrammatic summary of main pathways involved in GLP1 signalling in the brain. PI3K, phosphoinositide-3-kinase; PDK, 3-phosphoinositide-dependent protein kinase; Akt, protein kinase B (PKB), plays a key role in activating downstream regulators of cell metabolism, proliferation and survival; GSK3β, glycogen synthase kinase 3, downstream mediator involved in IDE inactivation, leading to an increase in α synuclein expression, which aggregate into amyloid fibres; FOXO1, Forkhead box O1, involved in maintaining the mitochondrial electron transport chain for ATP generation and fatty acid oxidation, preventing oxidative stress; NFκB, nuclear factor κB regulates microglial activation and the expression of inflammatory mediators such as IL1β and TNFα; cAMP, cyclic AMP, activated by binding of GLP1 to GLP1 receptor; PKA, protein kinase A, activates downstream processes via MAPK pathway; MAPK, mitogen-activated protein kinase, modulates downstream protein kinases involved in regulating cell proliferation, differentiation and apoptosis, maintaining neuronal growth and survival.
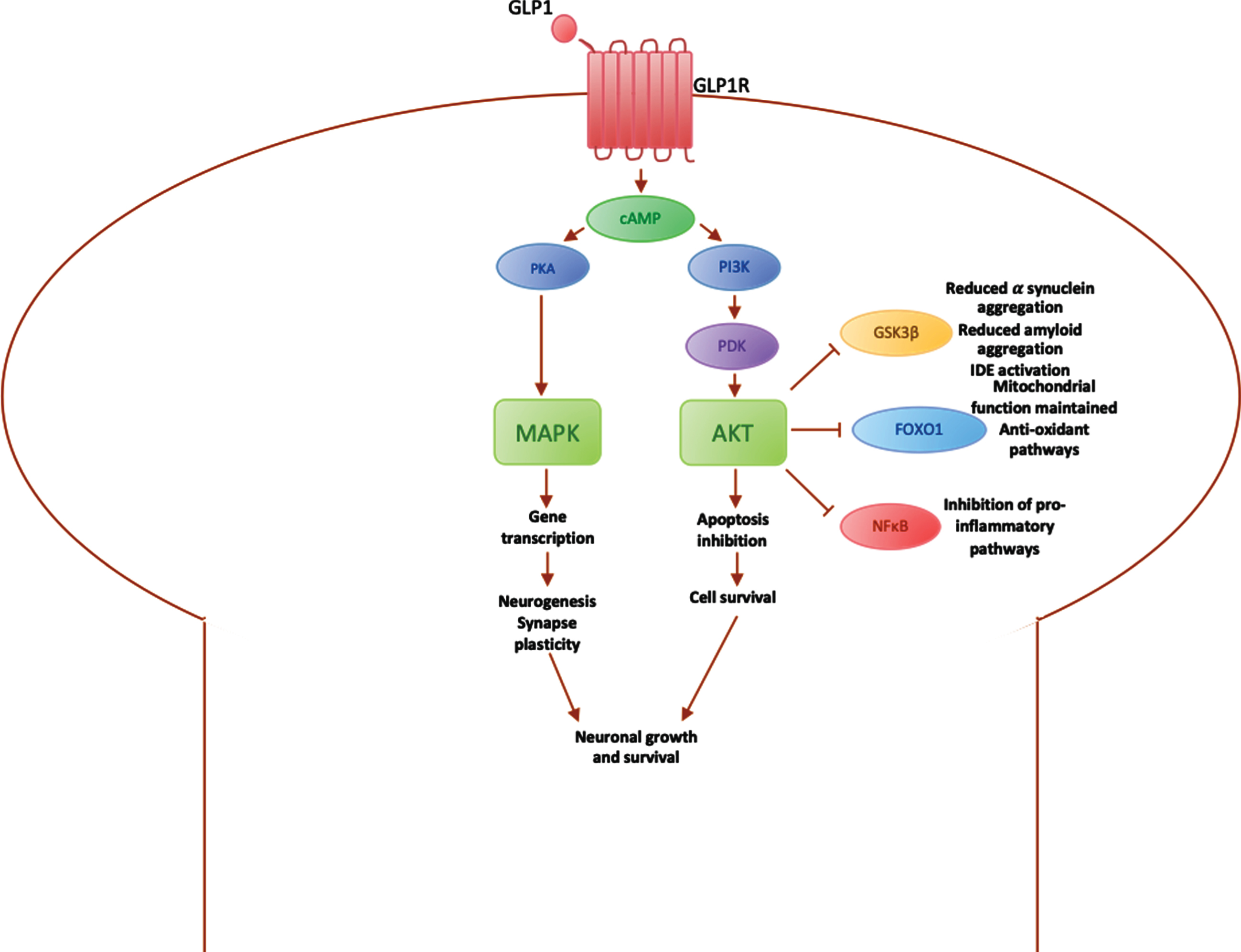
In the brain, GLP1R are mainly expressed on pyramidal neurons in the cortex or hippocampus and Purkinje neurons in the cerebellum and substantia nigra [98]. Activation of the PI3K/Akt pathway results in various downstream effects, including the inactivation of GSK3β, which reduces α-synuclein aggregation [29], the inactivation of FOXO1 [99], which prevents apoptosis and promotes cell survival, and inactivation of NFκB, which leads to microglial cell inactivation and a reduction in inflammatory mediators. GLP1R agonists also activate the expression of genes involved in cell growth and repair, improving neuroprotection against stress factors [95], such as α-synuclein, inflammatory mediators and cell death via the MAPK pathway [29] (Fig. 3).
Studies carried out using GLP1R agonists have shown promising results in slowing disease progression in PD rodent models [100]. An in vivo study found that Exendin-4 promotes neural progenitor cell numbers in the subventricular area that may compensate for the loss of dopaminergic neurons in the substantia nigra [101]. Another study using MPTP rodents showed that exenatide increased tyrosine hydroxylase levels in primary dopaminergic neurons, producing more dopamine [63]. A recent study on the effect of GLP1 analogues on MPTP rodents found improvements in striatal dopamine levels and reduced neuronal damage via inhibition of inflammatory cytokines and stimulation of anti-oxidant enzymes [102].
In-human clinical trials have also reported the therapeutic effects of GLP1R agonists on PD, even extending beyond motor benefits. An open label clinical trial found clinically significant amelioration of PD manifestations, including motor and cognitive decline [103], with improvements on the Movement Disorders Society Unified PD Rating Scale (MDS-UPDRS) and the Mattis DRS-2 cognitive score. A follow-up study also showed improvements were still present after one year [104]. Recently, a landmark double blind placebo-controlled trial also reported similar findings with patients treated with exenatide scoring on average 3.5 points less on the MDS-UPDRS than those not treated with exenatide, following a washout period to exclude a symptomatic effect [105]. In a novel approach to assessing target engagement using brain-derived exosomes purified from serum samples, the same group showed that patients treated with exenatide had an increase in levels of Akt and mTOR protein activation during the period of drug exposure in comparison with placebo-treated patients [106].
Thiazolidinediones
Thiazolidinediones (TZDs) are another class of oral anti-hyperglycaemic agents used in the treatment of T2DM. They act primarily on Peroxisome proliferator-activater receptor γ (PPARγ) to regulate genes involved in insulin sensitivity [13] and reduce insulin resistance. These receptors are known to be expressed in insulin sensitive organs such as the pancreas, as well as in regions of the brain including the substantia nigra and putamen [107]. Through their activation of PGC1α, a mitochondrial regulator, TZDs are thought to moderate mitoNEET, a mitochondrial membrane protein, to regulate neuronal complex I activity [108], reducing oxidative stress and cell death. They are also thought to inhibit microglial activation and reduce oxidative stress in neurons, enhancing mitochondrial function [28] and preventing neurodegeneration. Some studies have shown a significant decrease in the risk of PD with TZD use in T2DM patients [30, 31]. However, a retrospective cohort study in the USA showed that there was no significant decrease in PD risk with TZD use [109]. Moreover, a recent phase 2, double-blinded, clinical trial found 1 year exposure to pioglitazone had no significant benefit on PD progression at two doses (15 mg and 45 mg) compared to placebo [110]. The effectiveness of TZDs as a potential PD treatment option may also be limited by adverse effects, including cardiovascular effects, fracture risk and an association with bladder cancer [4].
Further developments
Several therapies in development act on other mediators in the insulin pathway. One of these is glucose-dependent insulinotropic polypeptide (GIP). GIP is an incretin, a hormone involved in reducing blood glucose levels, that triggers various downstream insulin pathways, promoting insulin biosynthesis and secretion [111]. A promising development is that of dual GLP1/GIP receptor agonists, which have similar affinity for the activation of both GLP1 and GIP receptors. Both GLP1 and GIP are important hormones involved in promoting insulin signalling [112]. They are both able to cross the blood brain barrier and exert neuroprotective effects [113]. Compared to single GLP1R agonists, the dual GLP1/GIP receptor agonist has been found to have better efficacy in enhancing insulin pathways and producing neuroprotective effects in rodent models [114], and fewer side effects [115].
The GLP1/GIP receptor agonist is thought to work through activation of the PI3K/Akt pathway and Erk1/2 pathway [116, 117]. There is an increase in tyrosine hydroxylase and reduction in microglial activation, promoting dopamine production and offering neuroprotection to dopaminergic neurons [116]. A study reported reduction in neuroinflammation in MPTP rodent models after treatment with GLP1/GIP receptor agonist [118]. Significantly, in the same study, insulin sensitivity was also reported to be restored with treatment [118]. The dual GLP1/GIP receptor agonist is currently in clinical trials for the treatment of T2DM and has shown promising results compared to existing treatment options [119], further clinical trials in PD patients may be pursued in the near future.
Table 1
Summary of common pathways between T2DM and PD and therapeutics targeting each pathway
| Mechanism | Elaboration | Therapeutics |
| Amyloid aggregation | • Inhibition of insulin degrading enzyme: formation of α synuclein. | GLP1R agonists |
| • Activation of PI3K/Akt pathway: activation of GSK3B, which increases alpha synuclein aggregation. | Metformin | |
| Microglial activation &chronic inflammation | • Activation of PI3K/Akt pathway: activation of NFκB, which causes microglial activation and expression of inflammatory mediators. | Insulin |
| GLP1R agonists | ||
| Thiazolidinediones | ||
| Metformin | ||
| GLP1/GIP receptor agonist | ||
| Oxidative stress &mitochondrial dysfunction | • Activation of PI3K/Akt pathway: activation of FOXO1, causing mitochondrial dysfunction and generation of ROS. | Insulin |
| GLP1R agonists | ||
| Metformin | ||
| Impaired synaptic plasticity | • Activation of PI3K/Akt pathway: inhibition of mTOR, preventing synaptic regeneration and dendritic regrowth. | Insulin GLP1R agonists GLP1/GIP receptor agonist |
| • Inhibition of NMDA and AMPA receptor expression: reduction in synaptic strength and long-term potentiation. | ||
| • Inhibition of MAPK pathway: synthesis of inflammatory cytokines |
The common pathways between T2DM and PD and therapeutics targeting each pathway are summarised in the table above (Table 1).
CONCLUSION
Epidemiological links between T2DM and PD have been increasingly studied, but there remain many unanswered questions about the role of T2DM on both PD risk and progression. The underlying pathways and common mechanisms remain a focus of research, and through their modulation new opportunities may arise to alter the neurodegenerative trajectory.
There are still several questions that further research must aim to address. Firstly, whilst it is promising that the effectiveness of GLP1 agonists has been demonstrated in various rodent models [120–123], it remains true that these do not adequately recapitulate the human disease, and further research should also focus on clinical trials in human participants.
Secondly, although GLP1R agonists have longer half-lives than endogenous GLP1, the aim would be to avoid the requirement for multiple daily dosing. Assuming efficacy in larger phase 3 clinical trials, newer formulations such as Bydureon, a once-weekly preparation of exenatide, and an oral preparation of semaglutide, offer tangible practical advantages. Incretin analogues with a slower release or longer half-life such as the dual GLP1/GIP receptor agonist and long-acting GIP analogues are also promising [124]. Finally, the long-term safety and efficacy of these treatments must be fully established. The issues of trial design, participant and outcome selection, and timing and duration of intervention, remain major issues in the exploration of these approaches and should be considered in future studies.
CONFLICT OF INTEREST
Prof. Foltynie is principal investigator on clinical trials of Exenatide. Drs. Noyce and de Pablo Fernandez have published observational studies on the links between T2DM and PD.
ACKNOWLEDGMENTS
The Preventive Neurology Unit is funded by the Barts Charity.
REFERENCES
[1] | Tysnes OB , Storstein A ((2017) ) Epidemiology of Parkinson’s disease, J Neural Transm 124: , 901–905. |
[2] | Dorsey ER , Elbaz A , Nichols E , Abd-Allah F , Abdelalim A , Adsuar JC , Ansha MG , Brayne C , Choi JYJ , Collado-Mateo D , Dahodwala N , Do HP , Edessa D , Endres M , Fereshtehnejad SM , Foreman KJ , Gankpe FG , Gupta R , Hankey GJ , Hay SI , Hegazy MI , Hibstu DT , Jasaejan A , Khader Y , Khalil I , Khang YH , Kim YJ , Kokubo Y , Logroscino G , Massano J , Mohamed Ibrahim N , Mohammed MA , Mohammadi A , Moradi-Lakeh M , Naghavi M , Nguyen BT , Ninayo YL , Ogbo FA , Owolabi MO , Pereira DM , Postma MJ , Oorbani M , Rahman MA , Roba KT , Safari H , Safiri S , Satpathy M , Sawhney M , Shafieesabet A , Shiferaw MS , Smith M , Szoeke CEI , Tabarés-Seisdedos R , Truong NT , Ukwaja KN , Venketasubramanian N , VIllafaina S , Weldegwergs DG , Westerman R , Wijeratne T , Winkler AS , Xuan BT , Yonemoto N , Feigin VL , Vos T , Murray CJL ((2018) ) Global, regional, and national burden of Parkinson’s disease, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016, Lancet Neurol 17: , 939–953. |
[3] | Marras C , Beck JC , Bower JH , Roberts E , Ritz B , Ross GW , Abbott RD , Savica R , Van Den Eeden SK , Willis AW , Tanner CM ((2018) ) Prevalence of Parkinson’s disease across North America, NPJ Parkinsons Dis 4: , 21. |
[4] | Athauda D , Foltynie T ((2016) ) Insulin resistance and Parkinson’s disease: A new target for disease modification?, Prog Neurobiol 145-146: , 98–120. |
[5] | Dunn L , Allen GFG , Mamais A , Ling H , Li A , Duberley KE , Hargreaves IP , Pope S , Holton JL , Lees A , Heales SJ , Bandopadhyay R ((2014) ) Dysregulation of glucose metabolism is an early event in sporadic Parkinson’s disease, Neurobiol Aging 35: , 1111–1115. |
[6] | Zheng Y , Ley SH , Hu FB ((2018) ) Global aetiology and epidemiology of type 2 diabetes mellitus and its complications, Nat Rev Endocrinol 14: , 88–98. |
[7] | Maahs DM , West NA , Lawrence JM , Mayer-Davis EJ ((2010) ) Chapter 1: Epidemiology of type 1 diabetes, Endocrinol Metab Clin North Am 39: , 481–497. |
[8] | Witoelar A , Jansen IE , Wang Y , Desikan RS , Gibbs JR , Blauwendraat C , Thompson WK , Hernandez DG , Djurovic S , Schork AJ , Bettella F , Ellinghaus D , Franke A , Lie BA , McEvoy LK , Karlsen TH , Lesage S , Morris HR , Brice A , Wood NW , Heutink P , Hardy J , Singleton AB , Dale AM , Gasser T , Andreassen OA , Sharma M , Nalls MA , Plagnol V , Sheerin UM , Saad M , SImon-Sanchez J , Schulte C , Sveinbjornsdottir S , Arepalli S , Barker R , Ben-Shlomo Y , Berendse HW , Berg D , Bhatia K , De Bie RMA , Biffi A , Bloem B , Bochdanovits Z , Bonin M , Bras JM , Brockmann K , Brooks J , Burn DJ , Bajounie E , Charlesworth G , Lungu C , Chen H , Chinnery PF , Chong S , Clarke CE , Cookson MR , Cooper JM , Corvol JC , Counsell C , Damier P , Dartigues JF , Deloukas P , Deuschl G , Dexter DT , Van Dijk KD , Dillman A , Durif F , Durr A , Edkins S , Evans JR , Foltynie T , Dong J , Gardner M , Goate A , Gray E , Guerreiro R , Harris C , Van Hilten JJ , Hofman A , Hollenbeck A , Holten J , Hu M , Huang X , Wurster I , Matzler W , Hudson G , Hunt SE , Huttenlocker J , Illig T , Jonsson PV , Lambert JC , Langford C , Lees A , Lichtner P , Limousin P , Lopez G , Lorenz D , McNeill A , Moorby C , Moor M , Morrison KE , Escott-Price V , Mudanohwo E , O’Sullivan SS , Pearson K , Perlmutter JS , Petursson H , Pollak P , Post B , Potter S , Ravina B , Revesz T , Riess O , Rivadeneira F , RIzzu P , Rvten M , Sawcer S , Schapira A , Scheffer H , Shaw K , Shoulson I , Shulman J , Sidransky E , Smith C , S, Pencer CCA , Stefansson H , Stockton JD , Strange A , Talbot K , Tanner CM , Tashakkori-Ghanbaria A , TIson F , Trabzuni D , Traynor BJ , Uitterlinden AG , Velseboer D , Vidailhet M , Walker R , Van De Warrenburg B , Wickremaratchi M , Williams N , Williams-Gray CH , Winder-Rhodes S , Stefansson K , Martinez M , Ferrucci L , Johnson R , Longo DL , Nalls MA , O’Brien R , Troncoso J , Van Der Brug M , Zielke HR , Zonderman A , Hardy JA , Weale M ((2017) ) Genome-wide pleiotropy between Parkinson disease and autoimmune diseases, JAMA Neurol 74: , 780–792. |
[9] | Rugbjerg K , Friis S , Ritz B , Schernhammer ES , Korbo L , Olsen JH ((2009) ) Autoimmune disease and risk for Parkinson disease, Neurology 73: , 1462–1468. |
[10] | Li X , Sundquist J , Sundquist K ((2012) ) Subsequent risks of Parkinson disease in patients with autoimmune and related disorders: A nationwide epidemiological study from Sweden, Neurodegener DIs 10: , 277–284. |
[11] | Santiago JA , Potashkin JA ((2013) ) Shared dysregulated pathways lead to Parkinson’s disease and diabetes, Trends Mol Med 9: , 176–186. |
[12] | Hölscher C ((2014) ) Drugs developed for treatment of diabetes show protective effects in Alzheimer’s and Parkinson’s diseases, Acta Physiol Sin 66: , 497–510. |
[13] | Aviles-Olmos I , Limousin P , Lees A , Foltynie T ((2013) ) Parkinson’s disease, insulin resistance and novel agents of neuroprotection, Brain 136: , 374–384. |
[14] | Craft S , Watson GS ((2004) ) Insulin and neurodegenerative disease: Shared and specific mechanisms, Lancet Neurol 3: , 169–178. |
[15] | Biosa A , Outeiro TF , Bubacco L , Bisaglia M ((2018) ) Diabetes mellitus as a risk factor for Parkinson’s disease: A molecular point of view, Mol Neurobiol 55: , 8754–8763. |
[16] | Konig A , Miranda HV , Outeiro TF ((2018) ) Alpha-synuclein glycation and the action of anti-diabetic agents in Parkinson’s disease, J Parkinsons Dis 8: , 33–43. |
[17] | Palleria C , Leporini C , Maida F , Succurro E , De Sarro G , Arturi F , Russo E ((2016) ) Potential effects of current drug therapies on cognitive impairment in patients with type 2 diabetes, Front Neuroendocrinol 42: , 76–92. |
[18] | Sandyk R ((1993) ) The relationship between diabetes mellitus and Parkinson’s disease, Int J Neurosci 69: , 125–130. |
[19] | Marques A , Dutheil F , Durand E , Rieu I , Mulliez A , Fantini ML , Boirie Y , Durif F ((2018) ) Glucose dysregulation in Parkinson’s disease: Too much glucose or not enough insulin?, Parkinsonism Relat Disord 5: , 122–127. |
[20] | Hu G , Jousilahti P , Bidel S , Antikainen R , Tuomilehto J ((2007) ) Type 2 diabetes and the risk of Parkinson’s disease, Diabetes Care 30: , 842–347. |
[21] | Xu Q , Park Y , Huang X , Hollenbeck A , Blair A , Schatzkin A , Chen H ((2011) ) Diabetes and risk of Parkinson’s disease, Diabetes Care 34: , 910–915. |
[22] | Driver JA , Smith A , Buring JE , Gaziano JM , Kurth T , Logroscino G ((2008) ) Prospective cohort study of type 2 diabetes and the risk of Parkinson’s disease, Diabetes Care 31: , 2003–2005. |
[23] | Yue X , Li H , Yan H , Zhang P , Chang L , Li T ((2016) ) Risk of Parkinson disease in diabetes mellitus, Medicine (Baltimore) 95: , e3549. |
[24] | De Pablo-Fernandez E , Goldacre R , Pakpoor J , Noyce AJ , Warner TT ((2018) ) Association between diabetes and subsequent Parkinson disease, Neurology 91: , e139–142. |
[25] | Yang YW , Hsieh TF , Li CI , Liu CS , Lin WY , Chiang JH , Li TC , Lin CC ((2017) ) Increased risk of Parkinson disease with diabetes mellitus in a population-based study, Medicine (Baltimore) 96: , e5921. |
[26] | Schernhammer E , Hansen J , Rugbjerg K , Wermuth L , Ritz B ((2011) ) Diabetes and the risk of developing Parkinson’s disease in Denmark, Diabetes Care 34: , 1102–1108. |
[27] | Lu M , Su C , Qiao C , Bian Y , Ding J , Hu G ((2016) ) Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson’s disease via autophagy and mitochondrial ROS clearance, Int J Neuropsychopharmacol 19: , pyw047. |
[28] | Hunter RL , Dragicevic N , Seifert K , Choi DY , Liu M , Kim HC , Cass WA , Sullivan PG , Bing G ((2007) ) Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system, J Neurochem 100: , 1375–1386. |
[29] | Athauda D , Foltynie T ((2017) ) Protective effects of the GLP-1 mimetic exendin-4 in Parkinson’s disease, Neuropharmacology 136: , 260–270. |
[30] | Brauer R , Bhaskaran K , Chaturvedi N , Dexter DT , Smeeth L , Douglas I ((2015) ) Glitazone treatment and incidence of Parkinson’s disease among people with diabetes: A retrospective cohort study, PLOS Med 12: , e1001854. |
[31] | Brakedal B , Flønes I , Reiter SF , Torkildsen Ø , Dölle C , Assmus J , Haugarvoll K , Tzoulis C ((2017) ) Glitazone use associated with reduced risk of Parkinson’s disease, Mov Disord 32: , 1594–1599. |
[32] | Cereda E , Barichella M , Pedrolli C , Klersy C , Cassani E , Caccialanza R , Pezzoli G ((2011) ) Diabetes and risk of Parkinson’s disease: A systematic review and meta-analysis, Diabetes Care 34: , 2614–2623. |
[33] | Chung SJ , Jeon S , Yoo HS , Kim G , Oh JS , Kim JS , Evans AC , Sohn YH , Lee PH ((2018) ) Detrimental effect of type 2 diabetes mellitus in a large case series of Parkinson’s disease, Parkinsonism Relat Disord 64: , 54–59. |
[34] | De Pablo-Fernandez E , Sierra-Hidalgo F , Benito-León J , Bermejo-Pareja F ((2017) ) Association between Parkinson’s disease and diabetes: Data from NEDICES study, Acta Neurol Scand 136: , 732–736. |
[35] | Palacios N , Gao X , Mccullough ML , Jacobs EJ , Patel A V , Mayo T , Schwarzschild MA , Ascherio A ((2011) ) Obesity, diabetes, and risk of Parkinson’s disease, Mov Disord 26: , 2253–2259. |
[36] | Lu L , Fu DL , Li HQ , Liu AJ , Li JH , Zheng GQ ((2014) ) Diabetes and risk of Parkinson’s disease: An updated meta-analysis of case-control studies, PLoS One 9: , e85781. |
[37] | Heilbron K , Noyce AJ , Fontanillas P , Alipanahi B , Nalls MA , Cannon P ((2019) ) The Parkinson’s phenome-traits associated with Parkinson’s disease in a broadly phenotyped cohort, NPJ Parkinsons Dis 5: , 4. |
[38] | Noyce AJ , Bestwick JP , Silveira-Moriyama L , Hawkes CH , Giovannoni G , Lees AJ , Schrag A ((2012) ) Meta-analysis of early nonmotor features and risk factors for Parkinson disease, Ann Neurol 72: , 893–901. |
[39] | Hui Y , Wang J , An Y , Gong Q , Li H , Zhang B , Shuai Y , Chen Y , Hu Y , Li G ((2019) ) Premature death and risk of cardiovascular disease in young-onset diabetes: A 23-year follow-up of the Da Qing Diabetes Study, Endocrine 65: , 46–52. |
[40] | Saydah SH , Siegel KR , Imperatore G , Mercado C , Gregg EW ((2019) ) The cardiometabolic risk profile of young adults with diabetes in the U.S, Diabetes Care 42: , 1895–1902. |
[41] | Gregg EW , Zhuo X , Cheng YJ , Albright AL , Narayan KMV , Thompson TJ ((2014) ) Trends in lifetime risk and years of life lost due to diabetes in the USA, 1985-2011: A modelling study, Lancet Diabetes Endocrinol 2: , 867–874. |
[42] | Bosco D , Plastino M , Cristiano D , Colica C , Ermio C , De Bartolo M , Mungari P , Fonte G , Consoli D , Consoli A , Fava A ((2012) ) Dementia is associated with Insulin Resistance in patients with Parkinson’s Disease, J Neurol Sci 315: , 39–43. |
[43] | Kotagal V , Albin RL , Müller MLTM , Koeppe RA , Frey KA , Bohnen NI ((2013) ) Diabetes is associated with postural instability and gait difficulty in Parkinson disease, Parkinsonism Relat Disord 19: , 522–526. |
[44] | Bohnen MI , Kotagal V , Muller MLTM , Koeppe RA , Scott PJ , Albin RL , Frey KA , Petrou M ((2014) ) Diabetes mellitus is independently associated with more severe cognitive impairment in Parkinson disease, Parkinsonism Relat Disord 20: , 1394–1398. |
[45] | Giuntini M , Baldacci F , Del Prete E , Bonuccelli U , Ceravolo R ((2014) ) Diabetes is associated with postural and cognitive domains in Parkinson’s disease. Results from a single-center study, Parkinsonism Relat Disord 20: , 671–672. |
[46] | Cereda E , Barichella M , Cassani E , Caccialanza R , Pezzoli G ((2012) ) Clinical features of Parkinson disease when onset of diabetes came first: A case-control study, Neurology 78: , 1507–1511. |
[47] | Mohamed Ibrahim N , Ramli R , Koya Kutty S , Shah SA ((2018) ) Earlier onset of motor complications in Parkinson’s patients with comorbid diabetes mellitus, Mov Disord 33: , 1967–1968. |
[48] | Pagano G , Polychronis S , Wilson H , Giordano B , Ferrara N , Niccolini F , Politis M ((2018) ) Diabetes mellitus and Parkinson disease, Neurology 90: , e1654–e1662. |
[49] | Petrou M , Davatzikos C , Hsieh M , Albin R , Kotagal V , Müller M , Koeppe RA , Herman WH , Frey KA , Bohnen NI ((2016) ) Diabetes, gray matter loss and cognition in the setting of Parkinson Disease, Acad Radiol 23: , 577–581. |
[50] | Moran C , Beare R , Phan TG , Bruce DG , Callisaya ML , Srikanth V ((2015) ) Type 2 diabetes mellitus and biomarkers of neurodegeneration, Neurology 85: , 1123–1130. |
[51] | Arnold SE , Arvanitakis Z , Macauley-Rambach SL , Koenig AM , Wang HY , Ahima RS , Craft S , Gandy S , Buettner C , Stoeckel LE , Holtzman DM , Nathan DM ((2018) ) Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums, Nat Rev Neurol 14: , 168–181. |
[52] | Morris JK , Vidoni ED , Perea RD , Rada R , Johnson DK , Lyons K , Pahwa R , Burns JM , Honea RA ((2014) ) Insulin resistance and gray matter volume in neurodegenerative disease, Neuroscience 270: , 139–147. |
[53] | Sharma AN , Ligade SS , Sharma JN , Shukla P , Elased KM , Lucot JB ((2015) ) GLP-1 receptor agonist liraglutide reverses long-term atypical antipsychotic treatment associated behavioral depression and metabolic abnormalities in rats, Metab Brain Dis 30: , 519–527. |
[54] | Yao WD , Gainetdinov RR , Arbuckle MI , Sotnikova TD , Cyr M , Beaulieu JM , Torres GE , Grant SGN , Caron MG ((2004) ) Identification of PSD-95 as a regulator of dopamine-mediated synaptic and behavioral plasticity, Neuron 41: , 625–638. |
[55] | Zhao WQ , Chen H , Quon MJ , Alkon DL ((2004) ) Insulin and the insulin receptor in experimental models of learning and memory, Eur J Pharmacol 490: , 71–81. |
[56] | Brunet A , Bonni A , Zigmond MJ , Lin MZ , Juo P , Hu LS , Anderson MJ , Arden KC , Blenis J , Greenberg ME ((1999) ) Akt promotes cell survival by phosphorylating and inhibiting a forkhead transcription factor, Cell 96: , 857–868. |
[57] | Hariharan M ((2012) ) Effects of insulin on synapse formation and function; A possible role for insulin resistance. Dalhousie University Department of Biochemistry and Molecular Biology. Available from: http://www.dt.co.kr/contents.html?article_no=2012071302010531749001 |
[58] | Wan Q , Xiong ZG , Man HY , Ackerley CA , Braunton J , Lu WY , Becker LE , MacDonald JF , Wang YT ((1997) ) Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin, Nature 388: , 686–690. |
[59] | Unger JW , Livingston JN , Moss AM ((1991) ) Insulin receptors in the central nervous system: Localization, signalling mechanisms and functional aspects, Prog Neurobiol 36: , 343–362. |
[60] | Jones KT , Woods C , Zhen J , Antonio T , Carr K , Reith MEA ((2017) ) Effects of diet and insulin on dopamine transporter activity and expression in rat caudate-putamen, nucleus accumbens and midbrain, J Neurochem 140: , 728–740. |
[61] | Baladi MG , Horton RE , Owens WA , Daws LC , France CP ((2015) ) Eating high fat chow decreases dopamine clearance in adolescent and adult Male rats but selectively enhances the locomotor stimulating effects of cocaine in adolescents, Int J Neuropsychopharmacol 18: , 1–11. |
[62] | Stouffer MA , Woods CA , Patel JC , Lee CR , Witkovsky P , Bao L , Machold RP , Jones KT , De Vaca SC , Reith MEA , Carr KD , Rice ME ((2015) ) Insulin enhances striatal dopamine release by activating cholinergic interneurons and thereby signals reward, Nat Commun 6: , 8543. |
[63] | Li Y , Perry T , Kindy MS , Harvey BK , Tweedie D , Holloway HW , Powers K , Shen H , Egan JM , Sambamurti K , Brossi A , Lahiri DK , Mattson MP , Hoffer BJ , Wang Y , Greig NH ((2009) ) GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism, Proc Natl Acad Sci U S A 106: , 1285–1290. |
[64] | Wang L , Zhai YQ , Xu LL , Qiao C , Sun XL , Ding JH , Lu M , Hu G ((2014) ) Metabolic inflammation exacerbates dopaminergic neuronal degeneration in response to acute MPTP challenge in type 2 diabetes mice, Exp Neurol 251: , 22–29. |
[65] | Takahashi M , Yamada T , Tooyama I , Moroo I , Kimura H , Yamamoto T , Okada H ((1996) ) Insulin receptor mRNA in the substantia nigra in Parkinson’s disease, Neurosci Lett 204: , 201–204. |
[66] | Jaikaran ETAS , Nilsson MR , Clark A ((2004) ) Pancreatic beta-cell granule peptides form heteromolecular complexes which inhibit islet amyloid polypeptide fibril formation, Biochem J 377: , 709–716. |
[67] | Wang H , Raleigh DP ((2014) ) The ability of insulin to inhibit the formation of amyloid by pro-islet amyloid polypeptide processing intermediates is significantly reduced in the presence of sulfated glycosaminoglycans, Biochemistry 53: , 2605–2614. |
[68] | Spillantini MG , Schmidt ML , Lee VMY , Trojanowski JQ , Jakes R , Goedert M ((1997) ) α-synuclein in Lewy bodies, Nature 388: , 839–840. |
[69] | Horvath I , Wittung-Stafshede P ((2016) ) Cross-talk between amyloidogenic proteins in type-2 diabetes and Parkinson’s disease, Proc Natl Acad Sci U S A 113: , 12473–12477. |
[70] | Martinez-Valbuena I , Amat-Villegas I , Valenti-Azcarate R , Del Mar Carmona-Abellan M , Marcilla I , Tuñon M-T , Luquin MR ((2018) ) Interaction of amyloidogenic proteins in pancreatic β cells from subjects with synucleinopathies, Acta Neuropathol 135: , 877–886. |
[71] | Vidal-Martinez G , Yang B , Vargas-Medrano J , Perez RG ((2018) ) Could α-synuclein modulation of insulin and dopamine identify a novel link between parkinson’s disease and diabetes as well as potential therapies? Front Mol Neurosci 11: , 465. |
[72] | Patel J , Witkovsky P , Coetzee W , Rice ME ((2011) ) Subsecond regulation of striatal dopamine release by pre-synaptic KATP channels, J Neurochem 119: , 721–736. |
[73] | Duka T , Duka V , Joyce JN , Sidhu A ((2009) ) α-synuclein contributes to GSK-3β-catalysed Tau phosphorylation in Parkinson’s disease models, J Fed Am Soc Exp Biol 23: , 2820–2830. |
[74] | Wills J , Jones J , Haggerty T , Duka V , Joyce JN , Sidhu A ((2010) ) Elevated tauopathy and alpha-synuclein pathology in postmortem Parkinson’s disease brains with and without dementia, Exp Neurol 225: , 210–218. |
[75] | Imamura K , Hishikawa N , Sawada M , Nagatsu T , Yoshida M , Hashizume Y ((2003) ) Distribution of major histocompatibility complex class II-positive microglia and cytokines profile of Parkinson’s disease brains, Acta Neuropathol 106: , 518–526. |
[76] | Kim DS , Choi H , Wang Y , Luo Y , Hoffer BJ , Greig NH ((2017) ) A new treatment strategy for Parkinson’s disease through the gut-brain axis: The glucagon-like peptide-1 receptor pathway, Cell Transplant 26: , 1560–1571. |
[77] | Bartels AL , Willemsen ATM , Doorduin J , de Vries EFJ , Dierckx RA , Leenders KL ((2010) ) [11C]-PK11195 PET: Quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease?, Parkinsonism Relat Disord 16: , 57–59. |
[78] | Chen H , O’Reilly EJ , Schwarzschild MA , Ascherio A ((2008) ) Peripheral inflammatory biomarkers and risk of Parkinson’s disease, Am J Epidemiol 167: , 90–95. |
[79] | Chen Z , Jalabi W , Hu W , Park HJ , Gale JT , Kidd GJ , Bernatowicz R , Gossman ZC , Chen JT , Dutta R , Trapp BD ((2014) ) Microglial displacement of inhibitory synapses provides neuroprotection in the adult brain, Nat Commun 5: , 4486. |
[80] | Sekiyama K , Sugama S , Fujita M , Sekigawa A , Takamatsu Y , Waragai M , Takenouchi T , Hashimoto M ((2012) ) Neuroinflammation in Parkinson’s disease and related disorders: A lesson from genetically manipulated mouse models of α-synucleinopathies, Parkinsons Dis 2012: , 271732. |
[81] | Vicente Miranda H , El-Agnaf OMA , Outeiro TF ((2016) ) Glycation in Parkinson’s disease and Alzheimer’s disease, Mov Disord 31: , 782–790. |
[82] | Schapira AH ((2008) ) Mitochondria in the aetiology and pathogenesis of Parkinson’s disease, Lancet Neurol 7: , 97–109. |
[83] | Kleinridders A , Cai W , Cappellucci L , Ghazarian A , Collins WR , Vienberg SG , Pothos EN , Kahn CR ((2015) ) Insulin resistance in brain alters dopamine turnover and causes behavioral disorders, Proc Natl Acad Sci U S A 112: , 3463–3468. |
[84] | Wang S , Zhang C , Sheng X , Zhang X , Wang B , Zhang G ((2014) ) Peripheral expression of MAPK pathways in Alzheimer’s and Parkinson’s diseases, J Clin Neurosci 21: , 810–814. |
[85] | Guo S , Copps KD , Dong X , Park S , Cheng Z , Pocai A , Rossetti L , Sajan M , Farese RV , White MF ((2009) ) The Irs1 branch of the insulin signaling cascade plays a dominant role in hepatic nutrient homeostasis, Mol Cell Biol 29: , 5070–5083. |
[86] | Khang R , Park C , Shin JH ((2015) ) Dysregulation of parkin in the substantia nigra of db/db and high-fat diet mice, Neuroscience 294: , 182–192. |
[87] | Dong XC , Copps KD , Guo S , Li Y , Kollipara R , Depinho A , White MF ((2008) ) Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation, Cell Metab 8: , 65–76. |
[88] | Zhai S , Tanimura A , Graves SM , Shen W , Surmeier DJ ((2018) ) Striatal synapses, circuits, and Parkinson’s disease, Curr Opin Neurobiol 48: , 9–16. |
[89] | Calabresi P , Mercuri NB , Di Filippo M ((2009) ) Synaptic plasticity, dopamine and Parkinson’s disease: One step ahead, Brain 132: , 285–287. |
[90] | Thomanetz V , Angliker N , Cloëtta D , Lustenberger RM , Schweighauser M , Oliveri F , Suzuki N , Rüegg MA ((2013) ) Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology, J Cell Biol 201: , 293–308. |
[91] | Passafaro M , Piãch V , Sheng M ((2001) ) Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons, Nat Neurosci 4: , 917–926. |
[92] | Grillo CA , Piroli GG , Lawrence RC , Wrighten SA , Green AJ , Wilson SP , Mott DD , Reagan LP ((2015) ) Hippocampal insulin resistance impairs spatial learning and synaptic plasticity, Diabetes 64: , 3927–3936. |
[93] | Di Luca M , Ruts L , Gardoni F , Cattabeni F , Biessels GJ , Gispen WH ((1999) ) NMDA receptor subunits are modified transcriptionally and post- translationally in the brain of streptozotocin-diabetic rats, Diabetologia 42: , 693–701. |
[94] | Cacabelos R ((2017) ) Parkinson’s disease: From pathogenesis to pharmacogenomics, Int J Mol Sci 18: , E551. |
[95] | Hölscher C ((2014) ) First clinical data of the neuroprotective effects of nasal insulin application in patients with Alzheimer’s disease, Alzheimers Dement 10: , S33–537. |
[96] | Craft S , Baker L , Montine T , Minoshima S , Watson S , Claxton A , Arbuckle M , Callaghan M , Tsai E , Plymate SR , Green PS , Leverenz J , Cross D ((2012) ) Intranasal Insulin Therapy for Alzheimer Disease and Amnestic Mild Cognitive Impairment, Arch Neurol 69: , 29–38. |
[97] | Baggio LL , Drucker DJ ((2007) ) Biology of incretins: GLP-1 and GIP, Gastroenterology 132: , 2131–2157. |
[98] | Hamilton A , Holscher C ((2009) ) Receptors for the incretin glucagon-like peptide-1 are expressed on neurons in the central nervous system, Neuroreport 20: , 1161–1166. |
[99] | Fan R , Li X , Gu X , Chan JCN , Xu G ((2010) ) Exendin-4 protects pancreatic beta cells from human islet amyloid polypeptide-induced cell damage: Potential involvement of AKT and mitochondria biogenesis, Diabetes Obes Metab 12: , 815–824. |
[100] | Liu W , Jalewa J , Sharma M , Li G , Li L , Hölscher C ((2015) ) Neuroprotective effects of lixisenatide and liraglutide in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease, Neuroscience 303: , 42–50. |
[101] | Bertilsson G , Patrone C , Zachrisson O , Andersson A , Danneus K , Heidrich J , Kortesmaa J , Mercer A , Nielsen E , Ronnholm H , Wikstrom L ((2008) ) Distributions of heavy metal concentrations in different tissues of the mangrove snail nerita lineata, J Neurosci Res 86: , 326–338. |
[102] | Elbassuoni EA , Ahmed RF ((2019) ) Mechanism of the neuroprotective effect of GLP-1 in a rat model of Parkinson’s with pre-existing diabetes, Neurochem Int 131: , 104583. |
[103] | Aviles-Olmos I , Dickson J , Kefalopoulou Zi , Djamshidian A , Ell P , Soderlund T , Whitton P , Wyse R , Isaacs T , Lees A , Limousin P , Foltynie T ((2013) ) Exenatide and the treatment of patients with Parkinson’s disease, J Clin Invest 123: , 2730–2736. |
[104] | Aviles-Olmos I , Dickson J , Kefalopoulou Z , Djamshidian A , Kahan J , Ell P , Whitton P , Wyse R , Isaacs T , Lees A , Limousin P , Foltynie T ((2014) ) Motor and cognitive advantages persist 12 months after Exenatide exposure in Parkinson’s disease, J Parkinsons Dis 4: , 337–344. |
[105] | Athauda D , Maclagan K , Skene SS , Bajwa-joseph M , Letchford D , Chowdhury K , Hibbert S , Budnik N , Wolfson L , Zampedri L , Dickson J ((2017) ) Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial, Lancet 390: , 1664–1675. |
[106] | Athauda D , Gulyani S , Karnati HK , Li Y , Tweedie D , Mustapic M , Chawla S , Chowdhury K , Skene SS , Greig NH , Kapogiannis D , Foltynie T ((2019) ) Utility of neuronal-derived exosomes to examine molecular mechanisms that affect motor function in patients with Parkinson disease: A secondary analysis of the Exenatide-PD Trial, JAMA Neurol 76: , 420–429. |
[107] | Swanson C , Emborg M ((2014) ) Expression of peroxisome proliferator-activated receptor-gamma in the substantia nigra of hemiparkinsonian nonhuman primates, Neurol Res 36: , 634–646. |
[108] | Ghosh S , Patel N , Rahn D , McAllister J , Sadeghi S , Horwitz G , Berry D , Wang KX , Swerdlow RH ((2007) ) The thiazolidinedione pioglitazone alters mitochondrial function in human neuron-like cells, Mol Pharmacol 71: , 1695–1702. |
[109] | Connolly JG , Bykov K , Gagne JJ ((2015) ) Thiazolidinediones and Parkinson disease: A cohort study, Am J Epidemiol 182: , 936–944. |
[110] | NINDS Exploratory Trials in Parkinson Disease (NET-PD) FS-ZONE Investigators ((2015) ) Pioglitazone in early Parkinson’s disease: A phase 2 multicentre, double-blind, randomised trial, Lancet Neurol 14: , 795–803. |
[111] | Yu Y , Hsueh S , Lai J , Chen Y , Kang S , Chen K , Kang SJ , Chen KY , Hsieh TH , Hoffer BJ , Li Y , Greig NH , Chiang YH ((2018) ) Glucose-dependent insulinotropic polypeptide mitigates 6-OHDA-induced behavioral impairments in parkinsonian rats, Int J Mol Sci 19: , E1153. |
[112] | Long-Smith CM , Manning S , McClean PL , Coakley MF , O’Halloran DJ , Holscher C , O’Neill C ((2013) ) The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-β plaque and glial pathology in a mouse model of alzheimer’s disease, Neuromolecular Med 15: , 102–114. |
[113] | Hunter K , Hölscher C ((2012) ) Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis, BMC Neurosci 13: , 33. |
[114] | Yuan Z , Li D , Feng P , Xue G , Ji C , Li G , Hölscher C ((2017) ) A novel GLP-1/GIP dual agonist is more effective than liraglutide in reducing inflammation and enhancing GDNF release in the MPTP mouse model of Parkinson’s disease, Eur J Pharmacol 812: , 82–90. |
[115] | Finan B , Ma T , Ottaway N , Muller T , Habegger K , Heppner K , Kirchner H , Holland J , Hembree J , Raver C , Lockie SH , Smiley DL , Gelfanov V , Yang B , Hofmann S , Bruemmer D , Drucker DJ , Pfluger PT , Perez-Tilve D , Gidda J , Vignati L , Zhang L , Hauptman JB , Lau M , Brecheisen M , Uhles S , Riboulet W , Hainaut E , Sebokova E , Conde-Knape K , Konkar A , DiMarchi RD , Tschop TH ((2013) ) Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans, Sci Transl Med 5: , 209ra151. |
[116] | Ji C , Xue GF , Lijun C , Feng P , Li D , Li L , Li G , Hölscher C ((2016) ) A novel dual GLP-1 and GIP receptor agonist is neuroprotective in the MPTP mouse model of Parkinson′s disease by increasing expression of BNDF, Brain Res 1634: , 1–11. |
[117] | Sharma MK , Jalewa J , Hölscher C ((2014) ) Neuroprotective and anti-apoptotic effects of liraglutide on SH-SY5Y cells exposed to methylglyoxal stress, J Neurochem 128: , 459–471. |
[118] | Shi L , Zhang Z , Li L , Hölscher C ((2017) ) A novel dual GLP-1/GIP receptor agonist alleviates cognitive decline by re-sensitizing insulin signaling in the Alzheimer icv. STZ rat model, Behav Brain Res 327: , 65–74. |
[119] | Cao L , Li D , Feng P , Li L , Xue GF , Li G , Hölscher C ((2016) ) A novel dual GLP-1 and GIP incretin receptor agonist is neuroprotective in a mouse model of Parkinson’s disease by reducing chronic inflammation in the brain, Neuroreport 27: , 384–391. |
[120] | Abuirmeileh A , Harkavyi A , Rampersaud N , Lever R , Tadross JA , Bloom SR , Whitton P ((2012) ) Exendin-4 treatment enhances L-DOPA evoked release of striatal dopamine and decreases dyskinetic movements in the 6-hydoxydopamine lesioned rat, J Pharm Pharmacol 64: , 637–643. |
[121] | Harkavyi A , Abuirmeileh A , Lever R , Kingsbury AE , Biggs CS , Whitton PS ((2008) ) Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson’s disease, J Neuroinflammation 5: , 1–9. |
[122] | Aksoy D , Solmaz V , Çavuşoğlu T , Meral A , Ateş U , Erbaş O ((2017) ) Neuroprotective effects of Exenatide in a rotenone-induced rat model of Parkinson’s disease, Am J Med Sci 354: , 319–324. |
[123] | Yun SP , Kam TI , Panicker N , Kim S , Oh Y , Park JS , Kwon SH , Park YJ , Karuppagounder SS , Park H , Kim S , Oh N , Kim NA , Lee S , Brahmachari S , Mao X , Lee JH , Kumar M , An D , Kang SU , Lee Y , Lee KC , Na DH , Kim D , Lee SH , Roschke VV , Liddelow SA , Mari Z , Barres BA , Dawson VL , Lee Seulki , Dawson TM , Ko HS ((2018) ) Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease, Nat Med 24: , 931–938. |
[124] | Li Y , Liu WZ , Li L , Hölscher C ((2016) ) Neuroprotective effects of a GIP analogue in the MPTP Parkinson’s disease mouse model, Neuropharmacology 101: , 255–263. |


