
Is It Possible to Conduct a Multi-Arm Multi-Stage Platform Trial in Parkinson’s Disease: Lessons Learned from Other Neurodegenerative Disorders and Cancer
Abstract
Many potential disease modifying therapies have been identified as suitable for clinical evaluation in Parkinson’s disease (PD). Currently, the evaluation of compounds in phase II and phase III clinical trials in PD are set up in isolation, a process that is lengthy, costly and lacks efficiency. This review will introduce the concept of a multi-arm, multi-stage (MAMS) trial platform which allows for the assessment of several potential therapies at once, transitioning seamlessly from a phase II safety and efficacy study to a phase III trial by means of an interim analysis. At the interim checkpoint, ineffective arms are dropped and replaced by new treatment arms, thereby allowing for the continuous evaluation of interventions. MAMS trial platforms already exist for prostate, renal and oropharyngeal cancer and are currently being developed for progressive multiple sclerosis (PMS) and motor neuron disease (MND) within the UK. As a MAMS trial will evaluate many potential treatments it is of critical importance that a widely endorsed core protocol is developed which will investigate outcomes and objectives meaningful to patients. This review will discuss the challenges of drug selection, trial design, stratification and outcome measures and will share strategies implemented in the planned MAMS trials for MND and PMS that may be of interest to the PD field.
INTRODUCTION
Parkinson’s disease (PD) is a neurodegenerative disease for which there are currently no therapies that delay or stop progression. The disease encompasses an array of motor and non-motor symptoms which have an increasing impact on patients’ and carers’ quality of life.
An estimated 0.85% of the population over 60 years of age is affected by PD in the UK [1, 2]. This amounted to an estimated annual care cost (direct and indirect) of £3.6 billion in 2018 [3, 4]. As both prevalence and incidence are projected to increase, it is imperative to develop disease-modifying therapies.
With the ever increasing pace of research, including in vitro and in vivo screening systems, advances in virtual drug modelling and bioinformatics approaches, the number of suitable drug candidates for clinical evaluation as potential disease modifying therapies is on the rise [5–8].
The development, financing and conducting of a clinical trial can take many years, creating a bottleneck for the clinical evaluation of potential therapies, particularly in the phase III setting. There is therefore a need to adopt an innovative and adaptive approach that allows for the seamless streamlining of trials and the testing of multiple hypotheses at once. One such example is the multi-arm, multi-stage (MAMS) platform trial design. Most MAMS trials are phase III trials that allow the evaluation of multiple therapeutic interventions against a common placebo arm (multi-arm), with pre-determined interim assessments that will evaluate safety and efficacy of all interventions (multi-stage). This allows for the early detection of ineffective treatment arms, which can then be replaced in favour of other interventions (Fig. 1). Most importantly, the multi-stage approach allows for the incorporation of phase II equivalent findings to be carried forward to the final phase III results without requiring the initiation of a separate trial. A MAMS trial is not only quicker and cheaper than a conventional approach, but also more efficient as fewer patients are required to come to a phase III powered conclusion regarding a particular intervention.
Fig.1
MAMS trial schematic. In this example, a five year phase III set up is shown that assumes one out of four tested interventions (D) show a positive signal at an interim analysis after 18 months. By the time one phase III conclusion has been reached, 12 study drugs will have been initiated into the trial, eight will have been terminated at the interim analysis stage, two will have shown sufficient evidence to be carried forward to phase III and two will be pending interim results in year six. (—analysis).
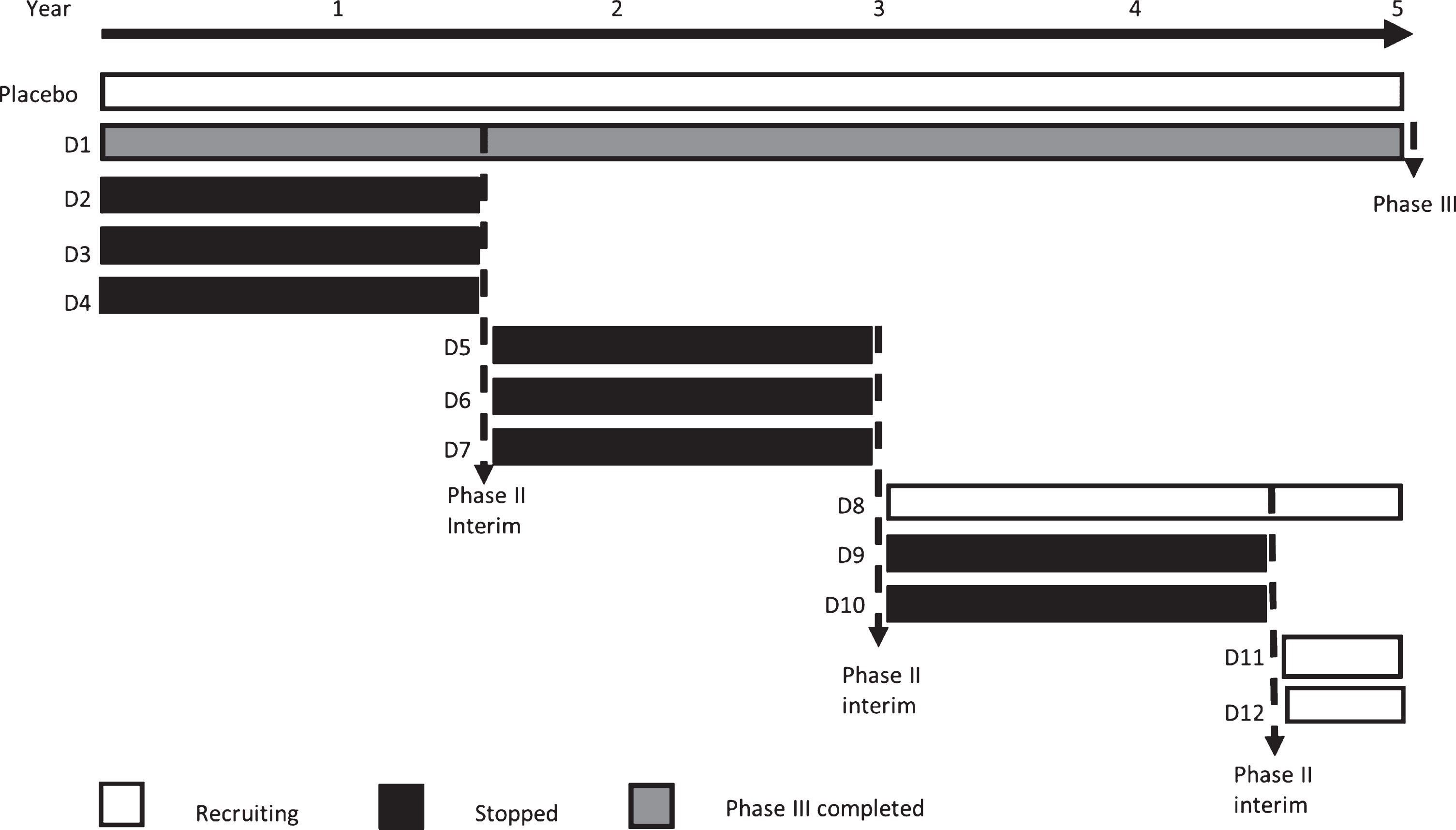
There are many challenges inherent to the implementation of a platform trial. In PD, eight phase II trials have provided sufficient evidence of efficacy for the initiation of eight phase III trials within the last 10 years. Whilst all phase II studies exhibited at least a trend towards activity, only one phase III trial (the ADAGIO study) successfully showed evidence of benefit on its efficacy endpoints (Table 1). Although it is possible that these drugs were not neuroprotective, current outcome measures, trial design features and patient population heterogeneity may play a significant role in the ability of trials to show efficacy of a new treatment.
Table 1
The outcome of phase III studies and the phase II studies that preceded them
| Phase III studies | Clinical Trial Identifier | Endpoint met | Was there evidence of disease improvement in phase II |
| Isradipine | NCT02168842 | Unpublished | No, but trend towards efficacy |
| Inosine | NCT02642393 | No | Yes |
| Creatine | NCT00449865 | No | Yes |
| Coenzyme Q10 | NCT00740714 | No | Yes (n = 2) |
| Yes in advanced disease (n = 1) | |||
| Rasagiline | NCT00256204 | Yes | No neuroprotective trials conducted |
| Ganoderma | NCT03594656 | Unpublished | Unpublished |
| Pramipexole | NCT00321854 | No | No neuroprotective trials conducted |
| Levodopa+Carbidopa | ISRCTN30518857 | No | No neuroprotective trials conducted |
This review will focus on the challenges ahead for the implementation of a MAMS platform in PD including drug selection, trial design, stratification and outcome measures. Many of these challenges are not unique to PD and therefore insights from other diseases that have embarked on this process will be shared. Here we will particularly focus on current efforts made in the development for MAMS trial platforms in motor-neuron disease (MND) and progressive multiple sclerosis (PMS) as well as one well established MAMS trial for the investigation of metastatic prostate cancer (STAMPEDE). Furthermore, we will discuss why common design decisions such as restriction of study populations to early disease and lack of stratification should be reconsidered.
MAMS PLATFORM TRIALS: STAMPEDE AND NEURODEGENERATIVE DISEASES
The STAMPEDE trial is perhaps one of the most successful examples of a MAMS platform. The protocol was initiated in October 2005 for men with poor prognosis prostate cancer who were starting long-term hormone therapy for the first time. Long-term hormone therapy was discovered in the 1950s and since then there had been no new treatments for men with this stage of disease. In the first version of the protocol, 5 new treatments (added to long term hormone therapy) were evaluated against a control arm of long-term hormone therapy alone. At the time of writing (October 2019), more than 11,000 patients had been recruited to the study, and version number 20 of the protocol had been launched. Over the last 14 years, the control (standard of care) arm of STAMPEDE has been adapted and improved 4 times – 3 times due to results emerging from research arms within STAMPEDE and once from results from another trial. Over a period of 20 years from the start of the protocol, STAMPEDE will have evaluated 10 different new treatments. The aim is to evaluate further treatments within STAMPEDE until at least 2030 and continue to improve the survival of men with advanced prostate cancer.
Within the UK, MAMS adaptive platform trials are being developed for MND and PMS. As with PD, these initiatives have arisen to address the unmet need for disease modifying therapies with strong backing from patients, carers and charitable groups. A MAMS platform design was selected in both conditions to enable more efficient testing of potential therapies.
The MND Systematic Multi-Arm Adaptive Randomised Trial (MND SMART) is being sponsored by the University of Edinburgh with funding from a number of charitable organisations and has recently gained regulatory approval. It takes advantage of Scotland’s CARE-MND integrated care and research platform.
For PMS, the UK MS Society Clinical Trials Network was initiated in 2007 to develop an efficient, multi-arm trial of repurposed drugs. This led to the development of MS-Secondary Progressive Multi-Arm Randomisation Trial (MS SMART), a multi-arm trial evaluating three candidate neuroprotective drugs with distinct mechanistic actions [11]. The trial was completed in 2018 with 445 patients recruited. Although none of the research arms showed a benefit on the primary phase II outcome (MRI brain atrophy) over placebo, this trial did demonstrate the feasibility of conducting a multi-arm trial of this scale.
The UK MS Society’s 2018-2022 Research Strategy has focused on designing and delivering a clinical MAMS trial platform to efficiently identify and evaluate potential treatments for PMS. It will incorporate both phase II (interim) and phase III (final) evaluations, with the possibility of dropping arms at interim stages where there is insufficient signal for efficacy. Building on the work of treatment selection, trial design, outcomes and infrastructure strategic working groups, a programme grant has been awarded and the trial will commence setup from early 2020 with recruitment beginning in 2021.
CHALLENGES TO INITIATING A MAMS PLATFORM IN PD
The continued failure to discover new medicines for neurodegenerative diseases reflects bottlenecks at two pivotal stages of drug development (i) discovery/identification of promising candidate interventions and (ii) translation of these agents into treatments that are available to patients through cost and time-efficient testing in clinical trials. To maximise the potential of a MAMS platform trial which will run over many years and interrogate many research questions, it is crucial that there is a pipeline in place that will continuously identify and evaluate suitable drug candidates. Furthermore, outcome measures have to be chosen that are sensitive enough to changes in disease progression over interim stages as well as the full duration of the trial. Decisions on outcome measures, trial population and design need to be widely endorsed not only in the scientific community to encourage widespread collaboration on the programme, but also by patients as their support is crucial for success.
Challenge 1: Drug selection
Many diseases have common etiopathological processes that are echoed in PD such as inflammation, protein trafficking and accumulation, oxidative stress and mitochondrial dysfunction. This is particularly true for neurodegenerative diseases (Supplementary Table 1) but can be found across many medical conditions including cancer [9] and diabetes [10]. It is therefore conceivable that agents targeting these mechanisms in other diseases may also be of value in PD.
Drug repurposing is an effective way to identify new therapeutic strategies as it uses existing clinical efficacy, safety and regulatory data and if off-patent, drugs are likely to be quicker and cheaper to get to licensed approval status.
For the successful implementation of a MAMS trial platform, a process for the selection of suitable drugs needs to be in place. In PD, a process has been devised through the linked clinical trials initiative (LCT) [11, 12]. Similar to its predecessor, the National Institute of Neurological Disorders and Stroke (NINDS) Committee to Identify Neuroprotective Agents for Parkinson’s (CINAPS) [13, 14], the identification and prioritisation of candidates to be entered into pilot clinical trials for PD is the main objective of the LCT committee. In short, the process involves the screening and selection of candidates on the basis of their potential efficacy in PD, evidence for safety, ability to penetrate the blood-brain-barrier, evidence of efficacy in PD animal models, the possibility for measuring target engagement and their commercial/patent status. This results in the compilation of detailed dossiers for a subset of drugs that meet basic pharmacological and safety criteria which are then scored and presented at an annual meeting of international experts where prioritisation for clinical drug testing is finalised [11, 12].
In the MS SMART study, a similar process of systematic review, commissioned by the MS- Clinical Trials Network was undertaken of animal and human trials of putative neuroprotective drugs. This process included a three-part systematic review and meta-analysis assessing the neurodegenerative disease literature including clinical and pre-clinical (animal and human induced pluripotent stem cell) publications to identify drugs ranked by a product score that takes into account efficacy, safety, study size and quality. The human analysis was extended to other neurodegenerative diseases including amyotrophic lateral sclerosis (ALS), Huntington’s, Alzheimer’s and PD, because of the existence of shared pivotal pathways in neurodegeneration. The focus of the upcoming PMS MAMS adaptive platform trial is on repurposed (already approved drugs) and rescued drugs (drugs at advanced stage of development but abandoned before approval) that act on mechanistic pathways in PMS [15].
MND SMART is utilising the infrastructure established by MS SMART for drug selection. Additionally, late-breaking drugs that may not have been tested previously in neurodegenerative diseases but show promise from preclinical data or from clinical trials in other diseases with similar underlying pathological processes are being considered.
Beyond the selection and prioritisation of candidate drugs there are other practical considerations that pose unique challenges to a MAMS trial protocol, such as the use of a common placebo. Traditionally, the MAMS trial design allows for the comparison of multiple therapies against a common control arm. Whilst this is a useful approach to reduce trial cost, it makes it difficult to incorporate and compare drugs that have different routes of administration. Out of the drug candidates initially shortlisted and published by the LCT in 2013, 11 are available as tablets, 5 as capsules, 3 as suspension, 3 as injection and 1 as oromucosal spray [12]. Not all placebos are equal and therefore treatments would either have to be standardised or an additional placebo arm may have to be introduced together with drugs using different administration routes. The latter may negate some of the efficiency savings anticipated through a MAMS trial [16].
Challenge 2: Outcome measures
Motor progression
Currently, the Movement Disorder Society - Unified PD rating scale (MDS-UPDRS) is used worldwide for the assessment of the severity of PD symptoms and is endorsed by regulators such as the FDA. The scale consists of four parts: Parts I and II – Non-Motor and Motor Experience of Daily Living; Part III – Motor Examination; Part IV – Motor Complications. This revision of the UPDRS rating scale was introduced in 2008 and aimed to allow finer differentiation at earlier stages of the disease [17].
Clinical trials have generally utilised either the clinically assessed part III motor score as their outcome measure or a composite score of several parts of the scale. A search of clinicaltrials.gov looking at neuroprotective trials within the last 10 years shows that out of 31 phase II studies with a motor outcome, 42% used the part I-III or I-IV total score and 55% assessed part III only. In contrast, a composite score is more likely to be used in the phase III setting (out of eight phase III trials, six utilised a change in I-III or I-IV composite score as their primary endpoint, one assessed part III only and one trial did not use the UPDRS as an outcome measure) [18–25].
However, changes in the MDS-UPDRS total score (within the first five years of disease) are largely driven by changes in the MDS-UPDRS Part III score; as part I and II score changes are not as pronounced over time [26] their addition into a total score may reduce sensitivity to change [27]. This is in line with findings examining the old UPDRS scale [28]. Although several studies have suggested that composite scores are able to detect clinically relevant differences over time [26, 27, 29], the authors of the new scale clearly recommended against the use of I-III composite scores as their unequal factorial loading reduces sensitivity to change [17].
Assessing part III only has its own limitations: it provides only a snapshot of the patients’ condition and is subject to significant inter- and intra- rater variability, which can undermine score accuracy. This is especially relevant for large multi-centre trials were many different raters are assessing patients throughout the study. Part III results are also dependent on the medication state of the patient [26, 28, 30] with OFF state results being more reliable compared with the ON state [30].
Longitudinal observations of clinical progression on the rating scale are important in determining the sensitivity of the scale in detecting differences at various stages of disease. Whilst some data indicate that overall disease progression up to at least five years from diagnosis is linear [26, 31, 32], it is generally accepted that the rate of motor symptom decline decelerates beyond five years [28, 32–34]. Progression studies using the revised scale are only just emerging [26, 27, 35]; to date these studies do not report data of patients beyond five years of disease, representing an important knowledge gap.
Cognition
Mild cognitive impairment (MCI) affects 20%–33% of patients at diagnosis and 80% of PD patients will go on to develop PD associated dementia within 12 years of disease, making cognitive dysfunction one of the most prevalent non-motor symptoms of PD [36, 37]. Cognitive function is thus often monitored in neuroprotective trials. Progression of cognitive impairment could be described as inverse to that of motor symptoms, with slow initial progression for up to 10 years followed by a rapid decline thereafter [32, 38, 39].
Recent evidence suggests that progression on the MoCA, MMSE, and Scales for Outcomes in Parkinson’s disease-Cognition (SCOPA-Cog) in non-demented PD patients is not reliably detectable over the typical timeframe of a clinical trial (1 year) [36, 40] with the MoCA being potentially more sensitive than the MMSE at detecting MCI [37].
Time to event measures
Historically, the most common time to event measure employed in neuroprotective trials of PD has been time to initiation of levodopa therapy. A study published in 2016 showed that 50% of neuroprotective trials identified at the time investigated time to initiation of dopaminergic treatment [41]. However, this measure is highly dependent on the judgement of the site investigator. In the case of studies where study drug is withdrawn after commencement of symptomatic therapy (ST), differing motivation of patients to continue the study may also play a role. Time to commencement of ST is therefore prone to bias. Other events that signify milestones for disease progression are onset of dementia, falls, nursing home placement and death.
Time to falls is of particular interest here as a recent study suggests that approximately 70% of PD patients fall within the first 5 years of diagnosis [42] and thus recording time to first fall may be a relatively rapidly evolving event in PD in comparison to events that occur much later such as dementia, nursing home placement or death.
The choice and feasibility of time to event measures employed in a clinical trial largely depend on the patient population being investigated. For example, time to death is dependent on the demographic of patients included in the study [43] with younger onset patients generally having a lower life expectancy [44–48], although some studies have found no difference [49].
Delay of these disease progression milestones are highly meaningful to patients and may therefore represent an alternative to conventional primary outcomes used in phase III trials.
Non-motor symptoms
Progression of non-motor symptoms (NMS) such as sleep, anxiety, apathy, depression, autonomic dysfunction and cognition are less influenced by dopamine therapy and significantly progress over short periods of 1-2 years even in an early disease population [50]. This makes measurement of NMS an attractive tool for evaluating disease progression in patients that require symptomatic treatment. NMS in PD can be measured by part I of the MDS-UPDRS scale or the NMS scale for PD. Both are suitable for detecting prevalence of NMS within PD populations [51] and have been used as secondary outcomes in PD trials. The MDS-NMS has recently been published [52].
Quality of life measures
Quality of life (QoL) measures are patient reported outcomes that attempt to measure physical, mental and social wellbeing that can either be related to general health or disease specific depending on the instrument. There are many generic and PD specific tools available although the most common QoL scale used in clinical trials of PD is the Parkinson’s Disease Questionnaire (PDQ-39) which captures the impact of both motor and non-motor symptoms on quality of life [53]. The impact of PD on QoL has been reviewed by others. In summary, PD has a profound impact on quality of life, has been shown to correlate with disease severity and has been useful at determining the impact of symptomatic therapy on patients in early as well as late disease [53, 54].
Biomarkers
A biomarker is an objectively measured characteristic that serves as an indicator of normal biological processes, pathogenic processes or pharmacological responses to a therapeutic intervention [55]. There is currently no biomarker that can reliably and sensitively track PD severity [32, 56, 57]. Consequently, biomarkers are not always measured and if so, they are generally ancillary exploratory outcomes in PD trials. Only 11 of 29 published phase II studies included at least one biomarker measurement in their protocol. These included 11 studies with brain imaging assessments [24, 58–64], one assessment of 8-hydroxy-2’ -deoxyguanosine measurement in plasma and one in urine [65], as well as two studies with cerebrospinal fluid (CSF) analysis of protein aggregation [66, 67]. Progress on the usefulness of imaging techniques and fluid or tissue biomarkers has been reviewed extensively by others [56, 57, 68]. Importantly, evidence regarding their relevance to clinical worsening of the disease is often contradictory.
Digital health technologies (DHT)
DHTs can be broadly categorised into passive (such as wearables) and active (apps that require data input or task completion) data capture. Where active measures are used, compliance may create problems with accuracy and reliability of data sets over long study periods [69–72]. Uploading of data collected by apps and sensors represents an additional challenge, making studies liable to data gaps due to patients living in poor network areas or outdated hospital IT systems. Mobilise-D is an EU funded project that has set out to improve the accurate assessment of daily life mobility by developing standardised tools [73]. A MAMS trial could provide a platform for integrated assessment of different digital measures that can be tied in to routinely collected clinical data of disease progression thereby validating their use.
Shaping future outcomes
The challenge of assessing the neuroprotective effect of drugs is not unique to PD. Measurement of worsening neurological function in a slowly progressive condition with relatively short (2–3 years) trials is inherently difficult. In MS there are also significant variations in progression between patients and changes in rate over time across several domains of neurological function. MRI brain atrophy measurements can be used to quantify aspects of neurodegeneration [74] and have been employed as the primary outcome in the phase II MS-STAT [75] and MS-SMART trials, as well as a secondary outcome in phase III trials of PMS. However, clinical outcome measures of relevance to patients are critically important to determine the functional impact of treatments on patients with PMS and expected by regulators in phase III trials.
The Expanded Disability Status Scale (EDSS) is currently the most widely used scale in phase III PMS clinical trials. Similar to the UPDRS scale in PD, where motor function is the main driver of progression [26], the EDSS overemphasises walking ability, whilst minimising the contribution of cognition and arm function [76]. Considering the range of disability typical of PMS patients, this is a major shortcoming.
To overcome this limitation, the Multiple Sclerosis Functional Composite (MSFC) was designed and includes measures of lower limb function (timed 25-foot walk), upper limb function and hand dexterity (9-hole peg test) and cognitive function (Paced Auditory Serial Addition Test) [77]. Subsequent amendments replaced the Paced Auditory Serial Addition Test by the Symbol Digit Modalities Test, a test of processing speed, and low-contrast letter acuity as a measurement of visual function [78]. A composite outcome measure has been successfully used in the INFORMS trial [79] and was an exploratory outcome in the ORATORIO trial (phase III trial of fingolimod and Ocrelizumab respectively) [80].
In the design of the UK MS Society’s PMS adaptive trial platform, a composite clinical endpoint, based on EDSS, 9-hole peg test and timed 25-foot walk, was decided as the primary phase III outcome based on expert consensus from the outcomes working group. As a composite endpoint is reached by confirmed disability progression on just one of the scales included in the composite measure, more progression events are recorded than if just assessing one scale alone, thereby reducing the time required to reach a progression endpoint as well as the number of participants required. It was also favoured by patient and public interest groups as it encompassed a broader range of meaningful measurements, particularly in non-ambulant patients in whom walking tests may not be possible or relevant.
Composite outcomes have also been successfully developed for MND. Given the typical trajectory of the disease, mortality is an important outcome for MND clinical trials. However, the large sample sizes and duration required to power such a study makes this practically challenging.
The most commonly used and best validated tool is the Amyotrophic Lateral Sclerosis-Functional Rating Score ALS-FRS(R). The ALS-FRS(R) has the advantage of assessing a wide range of neurological functions including communication, activities of daily living and mobility [81, 82]. Thus, the combined assessment of function and survival (CAFS) ranks outcomes based on survival time and change in ALS-FRS(R) score was developed [83].
Combining endpoints might be a useful strategy in PD to enable the simultaneous analysis of multiple equally important endpoints and may serve to give aspects of the disease, such as cognition, that become more pronounced at later stages of disease, a greater weight.
To date only one phase III trial (The NET-PD LS-1 trial) has attempted to measure a one year delay in cumulative disability using a global statistic measure combining the Modified Rankin Scale, Symbol Digit Modalities Test, PDQ-39 Summary Index, Schwab and England Activities of Daily Living scale, and ambulatory capacity over a five year period as primary efficacy endpoint. In addition to the necessity for long term monitoring of these measures, which has implications on cost and attrition rates, the tests and scales employed exhibited slower progression and higher variability across the study population than originally anticipated [23].
Challenge 3: Trial design
Inclusion criteria
It is important that the trial design for a MAMS platform has strategies in place to minimise or account for patient heterogeneity in terms of disease severity and rate of progression as this will affect variance of efficacy outcomes. Almost all phase III clinical trials conducted within the last 10 years have restricted recruitment to early, untreated disease for this reason (Supplementary Table 2). This patient group is more likely to follow a linear progression trajectory which makes it easier and more reliable to compute efficacy endpoints [31] and drug naïve PD can be modelled more accurately [35]. It is also considered that this restrictive recruitment strategy will capture a population where pathological deterioration within affected brain regions may still be at a redeemable stage [84]. It should be noted here that the assessment of a drug naïve PD population requires adjustments in order to account for patients that initiate dopamine therapy during the course of the trial. A common strategy is to define the endpoint as change from baseline to the end of the study duration or until initiation of dopaminergic therapy. However, this method treats differential progression as equal and does not take into account the potential of these groups responding differently to the study drug.
Limitation of recruitment to early disease has other disadvantages. Most importantly, unless the treatment effect of a neuroprotective drug is comparable to symptomatic benefit of levodopa, patients are likely to require and receive adjunctive dopamine therapy. Therefore, if no benefit can be detected whilst patients receive symptomatic therapy, the agent is not going to have an impact on the patient’s quality of life. Furthermore, early disease trials generally preclude data collection on efficacy of treatments in later disease stages and therefore there is no data to support efficacy or safety as disease advances, which will require further costly trials. Any beneficial effect at later disease stages, where different neurological systems are starting to be involved, would be missed by such a design and exploration of these drugs for PD are most likely going to be stopped after a negative result in an early disease group. In fact, one phase II trial of co-enzyme Q10 stratified patients into early (not treated with levodopa) and more advanced (PD with wearing off) arms and found a treatment benefit (change in total UPDRS score) only in more advanced disease [85]. Although this result came from a very small study (average of 12 patients per arm) and may represent the differential sensitivity of the scale at these two disease stages, a potential beneficial effect in this patient group could not have been (and was not) captured in the later phase III trial that focused on early, untreated disease [19]. These are important considerations that are particularly relevant in the context of establishing a continuous platform trial where there is a high throughput of interventions.
Stratification
A trial that allows for the recruitment of de novo and more advanced PD will require stratification of patients based on disease severity. However, it might be possible to further categorise patients into subtypes that have differential predicted disease progression. Patients with baseline characteristics of increased age, male sex, mild cognitive impairment, REM sleep behaviour disorder and orthostatic hypotension are more likely to progress faster, whereas tremor-dominant patients follow a milder disease course [86–90]. The differences in disease progression between these clinical subtypes can be very significant [86]. Progress in practically distinguishing between PD subtypes has been made by Fereshtehnejad et al and Velseboer et al, who have developed subtype calculators for newly diagnosed patients [87, 91]. Unless these groups are treated as clinically distinct in trial design, differences in their treatment response will never be detected and thus their importance as etiologically distinct groups cannot be verified. However, whether predictions of these subtype calculators hold true in longitudinal data sets remains to be investigated and previous attempts to verify clinical subtypes in additional cohorts have pointed towards a lack of reproducibility as well as instability of identified phenotypes in early disease populations [92, 93].
Although the reproducibility of clinical subtypes in additional cohorts is often inconsistent [92], evidence suggests that tremor dominance in particular, is highly predictive of slower progression on the UPDRS scale [86–90, 94–96]. This may be due to the fact that the UPDRS and MDS-UPDRS scale is weighted towards tremor and an early presentation of high tremor scores may reduce the probability of tremor dominant individuals to progress.
PD is complex and multi-factorial and therefore it might be wise to step back from the single endpoint paradigm of trial design that is based on the assumption that a population as a whole will respond and show improvement homogeneously. One way to acknowledge the complex multifactorial nature of PD in a MAMS trial without tightening inclusion criteria, could be to measure and power interim progression on individuals with low tremor scores only, whilst ultimate progression for phase III analysis may be a non- UPDRS based measure. Whether this is a feasible approach remains to be determined. In the MND-SMART trial, interim analysis excludes individuals classified as long survivors in order to reduce heterogeneity at that point without excluding long survivors from the trial. The planned inclusion EDSS range of 3.0–8.0 for the PMS MAMS trial is also broader than in previous phase III trials (typically 3.5 or 4.0–6.5). Making clinical trials more inclusive has been emphasised by patient and public involvement groups. Baseline EDSS will be one of the minimisation factors in randomisation to ensure comparability of study arms.
Methodology
There are two distinct trial designs that aim to distinguish between symptomatic and disease modifying effects of a study drug. These are the delayed start and wash-out design (Fig. 2). Within the past 10 years, four phase III trials have used a delayed start design, one has employed a wash out design and three chose to assess beneficial effects without attempting to distinguish between symptomatic and neuroprotective benefits (Table 2). The delayed start design has been successfully employed by the ADAGIO study that investigated the MAO inhibitor rasagiline, which is currently used as a symptomatic treatment of PD. However, the design has been criticised as it assumes linearity of disease progression over the trial period and whilst it may be reasonable to make such an assumption in a de novo PD patient group, its feasibility in more advanced disease is questionable. Furthermore, differential dropout rates between the two arms have to be imputed. This is particularly relevant in trials that examine therapies with known symptomatic effect on a drug naïve patient population (such as the ADAGIO study), as the need for symptomatic therapy will occur earlier in the delayed start group resulting in increased drop out.
Fig.2
Methodology to investigate disease modification. The delayed start design is a two period trial design. In period one, patients are initially randomised into placebo (delayed start) and treatment arms (early start) at the end of which, the placebo group is switched to the study drug (period two). Both groups will receive the study drug for the remaining duration of the trial. Should a treatment be neuroprotective rather than symptomatic, the early start group should have a reduced progression rate as compared to placebo in period one, a significantly improved MDS-UPDRS score from baseline at the end of period two and an equal or reduced rate of disease progression in the early start group compared to the delayed start group in period two. Thus, such a design has three endpoints [31]. The wash out design is a two period design that evaluates a global change in the outcome measure of choice from baseline over a drug administration period and the maintenance of this change after the study drug is withdrawn (washed out).
![Methodology to investigate disease modification. The delayed start design is a two period trial design. In period one, patients are initially randomised into placebo (delayed start) and treatment arms (early start) at the end of which, the placebo group is switched to the study drug (period two). Both groups will receive the study drug for the remaining duration of the trial. Should a treatment be neuroprotective rather than symptomatic, the early start group should have a reduced progression rate as compared to placebo in period one, a significantly improved MDS-UPDRS score from baseline at the end of period two and an equal or reduced rate of disease progression in the early start group compared to the delayed start group in period two. Thus, such a design has three endpoints [31]. The wash out design is a two period design that evaluates a global change in the outcome measure of choice from baseline over a drug administration period and the maintenance of this change after the study drug is withdrawn (washed out).](https://content.iospress.com:443/media/jpd/2020/10-2/jpd-10-2-jpd191856/jpd-10-jpd191856-g002.jpg)
Table 2
Trial methodology and endpoints of phase III neuroprotective trials and their preceding phase II trials within the last 10 years; ST, symptomatic therapy
| Methodology | Endpoint | Intervention | Phase | Clinical trial identifier | Treatment duration | Comments |
| Delayed start | 1) Difference in rate of progression (period 1) | Rasagiline | 3 | NCT00256204 | Period 1: 9 months; Period 2: 9 months | Allowed 3 months wash in for slope analyses |
| 2) non-inferiority of slopes (period 2) | Ganoderma | 3 | NCT03594656 | Period 1: 6 months; Period 2: 12months | ||
| 3) difference in total UPDRS score between early and delayed (period 1 + 2) | Pramipexole | 3 | NCT00321854 | Period 1 : 6–9 months;Period 2 : 6 months | ||
| Levodopa+Carbidopa | 3 | ISRCTN30518857 | Period 1 : 9 months;Period 2 : 9 months | |||
| Wash out | Difference in rate of change from shared baseline | Inosine | 3 | NCT02642393 | Period 1: 24 months; Period 2: 3 months | |
| Change from baseline vs each arm | Isradipine | 2 | NCT00909545 | Period 1: 12 months; Period 2: 0.5 months | ||
| Change from baseline | Ubiquinol-10 | 2 | N/A | Period 1: 12 months (advanced PD arm), 24 months (early PD arm); Period 2: 2 months | ||
| None | Change from baseline | Creatine | 3 | NCT00449865 | 60 months | |
| Change from baseline | Coenzyme Q10 with Vitamin E | 3 | NCT00740714 | 16 months or initiation of ST | ||
| Change from baseline for those receiving ST vs placebo | Isradipine | 3 | NCT02168842 | 36 months | ||
| Futility boundary 70% of placebo group progression | Creatine and Minocycline | 2 | NCT00063193 | 12 months or initiation of ST | DATATOP placebo + trial placebo arm | |
| Change from baseline | Coenzyme Q10 | 2 | NCT00004731 | 16 months or initiation of ST | ||
| Futility boundary 70% of placebo group progression | Coenzyme Q10 | 2 | NCT00076492 | 12 months or initiation of ST | DATATOP placebo + trial placebo arm | |
| Futility boundary 70% of placebo group progression | Inosine | 2 | NCT00833690 | 23 months |
The delayed start design is an interesting consideration for a MAMS trial design as it already allows for an interim analysis after period one which could serve as an interim checkpoint in a multi-arm trial. A primary outcome measure that is sensitive enough to detect changes within the relatively short time scales of period one and two would have to be chosen.
The wash-out design requires knowledge of the pharmacology of the drug and thus evaluation of blood and plasma levels would have to be incorporated into the initial phase II component of the MAMS trial. This ensures that the wash-out period is long enough to allow for clearance of the drug from the body so that a persisting benefit thereafter, indicative of its influence on underlying disease pathology, can be confirmed. The advantage of this design is that period one is generally longer than period one of a delayed start design allowing for the evaluation of potentially slower evolving outcome measures. However, long duration effects are often poorly understood and therefore it is hard to conclude whether a given wash-out period will be sufficient to preclude long lasting symptomatic benefits. For example, despite the short half-life of levodopa in plasma [97, 98] clinical benefits persist much longer [99, 100]. Thus the results of the ELLDOPA study, where levodopa treatment led to sustained improvement of the total UPDRS score over a two week wash out period (indicative of slowed disease progression) remained inconclusive and were further confounded by its imaging substudy that showed a decline in dopamine transporter (DAT) activity ([123I] β-CIT uptake) [99, 101].
Distinguishing between a symptomatic and a disease modifying therapy at the interim check point may be challenging. A MAMS trial is only feasible if an interim analysis is conducted before the recruitment target for the phase III element is reached. Introducing a wash out or delayed start design prior to the interim analysis may increase the trial duration beyond the feasibility threshold for a MAMS trial. Therefore, a MAMS trial design that postpones distinguishing between disease modification and symptomatic effects until the phase III analysis may be more practical.
PROPELLING MAMS INTO ACTION
A MAMS trial is a substantial logistical undertaking and thus requires significant investment to ensure trial delivery. STAMPEDE, which has been running for more than 10 years currently employs tens of core staff that are responsible for continued data collection, management, analysis and biobanking, amongst a range of other roles as well as facilitating the seamless introduction of new study arms. Beyond the core staff required for the trial, which is generally funded through disease specific charities, funding for individual research arms is sourced separately through standard clinical research grants or industry sponsorship. MAMS trial arms should
1) investigate drugs that are as divergent as possible (a practical consideration that also retains the possibility of combining effective treatments at a later stage) and
2) be powered to compare active arms to placebo only
thereby avoiding direct competition between drugs making it more appealing to industry and has attracted industry to the STAMPEDE trial.
Gaining regulatory approval for a seamless phase II/III design may be perceived as a barrier to the implementation of an adaptive trial platform. Although this approach is novel in PD, the Medical Research Council Clinical Trials Unit for the STAMPEDE trial has a proven track record for the implementation of adaptive trials within the UK and have experienced regulators to be open minded and helpful in the development of adaptive trial protocols. The MND SMART trial has obtained regulatory approval to commence recruitment in early 2020. This represents a growing recognition that in situations where there is an unmet need for effective therapies, and/or patient populations are limited, such designs have the potential to facilitate ongoing evaluation of drugs over time and are thus worthy of the considerable effort required for their implementation and continued adaptation. Selection of the primary outcomes would need to be made with regulatory agencies in mind and any adaptations made during the course of the trial would also need to be aligned with regulatory requirements.
In order to establish a MAMS trial platform consensus will have to be reached on 1) drug selection, 2) an appropriate patient population for study, 3) methodology for identifying disease modification and whether this is necessary as well as 4) effective and relevant outcome measures. Current phase III studies have been focused on an early PD patient population and whilst practical considerations have played a role in dictating this, recent development in identification of progression subgroups as well as the development of composite measures may open the gate to more inclusive recruitment strategies.
To reach consensus on these issues in PD, a Delphi process is currently being developed to inform the design of a MAMS platform. This method is an iterative approach whereby experts from different backgrounds such as clinicians, funders, industry, academics, regulators and patients will be invited to complete a series of questionnaires reaching consensus over multiple survey rounds.
We propose that the Delphi process should inform the remit for focused working groups to develop trial design, outcome measures and trial infrastructure, thereby ensuring that the wider research community, as well as patients have the opportunity to engage in the development of the MAMS platform.
CONCLUSION
A MAMS trial is an excellent platform that gives the opportunity to simultaneously test the benefit of multiple agents with the added benefit of integrated interim analyses allowing for the removal of arms that do not show sufficient evidence of efficacy. To date, clinical investigation of neuroprotective therapies is hampered by a lack of availability and consensus around methodology and outcome measures. By including a broad field of clinical research stakeholders, as well as patients, it is hoped that a strategy for implementing a MAMS trial will be formulated that is internationally endorsed and of relevance to patients, thereby giving it the best chance for success.
Dedicated effort over two years in the MS field, which has faced similarly complex issues, especially with regards to outcome measures, has led to a research grant award for the implementation of their MAMS trial. This is encouraging and inspires confidence that a similar approach will make a MAMS trial for PD possible in the near future.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
This work is supported by the Cure Parkinson’s Trust
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JPD-191856.
REFERENCES
[1] | Weir S , Samnaliev M , Kuo TC , Tierney TS , Walleser Autiero S , Taylor RS , Schrag A ((2018) ) Short- and long-term cost and utilization of health care resources in Parkinson’s disease in the UK. Mov Disord 33: , 974–981. |
[2] | Rossi A , Berger K , Chen H , Leslie D , Mailman RB , Huang X ((2018) ) Projection of the prevalence of Parkinson’s disease in coming decades: Revisited HHS Public Access. Mov Disord 33: , 156–159. |
[3] | Statistics for journalists, Parkinson’s UK, https://www.parkinsons.org.uk/about-us/statistics-journalists, Last updated 2018, Accessed on, 2018. |
[4] | Mid-2018:2019 LA boundaries document, Estimates of the population for the UK, England and Wales, Scotland and Northern Ireland – Office for National Statistics, https://www.ons.gov.uk/peoplepopulationandcommunity/populationandmigration/populationestimates/datasets/populationestimatesforukenglandandwalesscotlandandnorthernireland, Last updated 2019, Accessed on, 2019. |
[5] | Wooller SK , Benstead-Hume G , Chen X , Ali Y , Pearl FMG ((2017) ) Bioinformatics in translational drug discovery. Biosci Rep 37: , 20160180. |
[6] | Kesik-Brodacka M ((2018) ) Progress in biopharmaceutical development. Biotechnol Appl Biochem 65: , 306–322. |
[7] | Hollingsworth SA , Dror RO ((2018) ) Molecular dynamics simulation for all. Neuron 99: , 1129–1143. |
[8] | Tresadern G , Rombouts FJR , Oehlrich D , Macdonald G , Trabanco AA ((2017) ) Industrial medicinal chemistry insights: Neuroscience hit generation at Janssen. Drug Discov Today 22: , 1478–1488. |
[9] | Ariga H ((2015) ) Common mechanisms of onset of cancer and neurodegenerative diseases. Biol Pharm Bull 38: , 795–808. |
[10] | Ashraghi MR , Pagano G , Polychronis S , Niccolini F , Politis M ((2016) ) Parkinson’s disease, diabetes and cognitive impairment. Recent Pat Endocr Metab Immune Drug Discov 10: , 11–21. |
[11] | Brundin P , Wyse RK ((2019) ) The Linked Clinical Trials initiative (LCT) for Parkinson’s disease. Eur J Neurosci 49: , 307–315. |
[12] | Brundin P , Barker RA , Conn PJ , Dawson TM , Kieburtz K ((2013) ) Linked clinical trials – the development of new clinical learning studies in Parkinson’s disease using screening of multiple prospective new treatments. J Parkinsons Dis 3: , 231–239. |
[13] | Tilley BC , Galpern WR ((2007) ) Screening potential therapies: Lessons learned from new paradigms used in Parkinson disease. Stroke 38: , 800–803. |
[14] | Ravina BM , Fagan SC , Hart RG , Hovinga CA , Murphy DD , Dawson TM , Marler JR ((2003) ) Neuroprotective agents for clinical trials in Parkinson’s disease: A systematic assessment. Neurology 60: , 1234–1240. |
[15] | Connick P , Angelis F De , Parker RA , Plantone D , Doshi A , John N , Stutters J , MacManus D , Carrasco FP , Barkhof F , Ourselin S , Braisher M , Ross M , Cranswick G , Pavitt SH , Giovannoni G , Wheeler-Kingshott CAG , Hawkins C , Sharrack B , Bastow R , Weir CJ , Stallard N , Chandran S , Chataway J , UK Multiple Sclerosis Society Clinical Trials Network ((2018) ) Multiple Sclerosis-Secondary Progressive Multi-Arm Randomisation Trial (MS-SMART): A multiarm phase IIb randomised, double-blind, placebo-controlled clinical trial comparing the efficacy of three neuroprotective drugs in secondary progressive multiple sclerosis. BMJ Open 8: , e021944. |
[16] | de Craen AJ , Tijssen JG , de Gans J , Kleijnen J ((2000) ) Placebo effect in the acute treatment of migraine: Subcutaneous placebos are better than oral placebos. J Neurol 247: , 183–188. |
[17] | Goetz CG , Tilley BC , Shaftman SR , Stebbins GT , Fahn S , Martinez-Martin P , Poewe W , Sampaio C , Stern MB , Dodel R , Dubois B , Holloway R , Jankovic J , Kulisevsky J , Lang AE , Lees A , Leurgans S , LeWitt PA , Nyenhuis D , Olanow CW , Rascol O , Schrag A , Teresi JA , van Hilten JJ , LaPelle N ((2008) ) Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Scale presentation and clinimetric testing results. Mov Disord 23: , 2129–2170. |
[18] | Biglan KM , Oakes D , Lang AE , Hauser RA , Hodgeman K , Greco B , Lowell J , Rockhill R , Shoulson I , Venuto C , Young D , Simuni T ((2017) ) A novel design of a Phase III trial of isradipine in early Parkinson disease (STEADY-PD III). Ann Clin Transl Neurol 4: , 360–368. |
[19] | Beal MF , Oakes D , Shoulson I , Henchcliffe C , Galpern WR , Haas R , Juncos JL , Nutt JG , Voss TS , Ravina B , Shults CM , Helles K , Snively V , Lew MF , Griebner B , Watts A , Gao S , Pourcher E , Bond L , Kompoliti K , Agarwal P , Sia C , Jog M , Cole L , Sultana M , Kurlan R , Richard I , Deeley C , Waters CH , Figueroa A , Arkun A , Brodsky M , Ondo WG , Hunter CB , Jimenez-Shahed J , Palao A , Miyasaki JM , So J , Tetrud J , Reys L , Smith K , Singer C , Blenke A , Russell DS , Cotto C , Friedman JH , Lannon M , Zhang L , Drasby E , Kumar R , Subramanian T , Ford DS , Grimes DA , Cote D , Conway J , Siderowf AD , Evatt ML , Sommerfeld B , Lieberman AN , Okun MS , Rodriguez RL , Merritt S , Swartz CL , Martin WRW , King P , Stover N , Guthrie S , Watts RL , Ahmed A , Fernandez HH , Winters A , Mari Z , Dawson TM , Dunlop B , Feigin AS , Shannon B , Nirenberg MJ , Ogg M , Ellias SA , Thomas C-A , Frei K , Bodis-Wollner I , Glazman S , Mayer T , Hauser RA , Pahwa R , Langhammer A , Ranawaya R , Derwent L , Sethi KD , Farrow B , Prakash R , Litvan I , Robinson A , Sahay A , Gartner M , Hinson VK , Markind S , Pelikan M , Perlmutter JS , Hartlein J , Molho E , Evans S , Adler CH , Duffy A , Lind M , Elmer L , Davis K , Spears J , Wilson S , Leehey MA , Hermanowicz N , Niswonger S , Shill HA , Obradov S , Rajput A , Cowper M , Lessig S , Song D , Fontaine D , Zadikoff C , Williams K , Blindauer KA , Bergholte J , Propsom CS , Stacy MA , Field J , Mihaila D , Chilton M , Uc EY , Sieren J , Simon DK , Kraics L , Silver A , Boyd JT , Hamill RW , Ingvoldstad C , Young J , Thomas K , Kostyk SK , Wojcieszek J , Pfeiffer RF , Panisset M , Beland M , Reich SG , Cines M , Zappala N , Rivest J , Zweig R , Lumina LP , Hilliard CL , Grill S , Kellermann M , Tuite P , Rolandelli S , Kang UJ , Young J , Rao J , Cook MM , Severt L , Boyar K ((2014) ) A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease. JAMA Neurol 71: , 543–552. |
[20] | Duncan RP , Earhart GM ((2012) ) Disease randomized controlled trial of community-based dancing to modify disease progression in Parkinson. Neurorehabil Neural Repair 26: , 132–143. |
[21] | Olanow W , Rascol O , Hauser R , Feigin PD , Jankovic J , Lang A , Langston W , Poewe W , Stocchi F , Tolosa E ((2009) ) A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 361: , 1268–1278. |
[22] | ClinicalTrials.gov, NCT03594656; Effects of Lingzhi on Disease Progression in Patients With Untreated Early Parkinson’s Disease, https://clinicaltrials.gov/ct2/show/NCT03594656?term=NCT03594656&draw=2&rank=1, Last updated 2018, Accessed on, 2018. |
[23] | Kieburtz K , Tilley BC , Elm JJ , Babcock D , Hauser R , Ross GW , Augustine AH , Augustine EU , Aminoff MJ , Bodis-Wollner IG , Boyd J , Cambi F , Chou K , Christine CW , Cines M , Dahodwala N , Derwent L , Dewey RB , Hawthorne K , Houghton DJ , Kamp C , Leehey M , Lew MF , Lin Liang GS , Luo ST , Mari Z , Morgan JC , Parashos S , Pérez A , Petrovitch H , Rajan S , Reichwein S , Roth JT , Schneider JS , Shannon KM , Simon DK , Simuni T , Singer C , Sudarsky L , Tanner CM , Umeh CC , Williams K , Wills AM ((2015) ) Effect of creatine monohydrate on clinical progression in patients with parkinson disease: A randomized clinical trial. JAMA 313: , 584–593. |
[24] | Schapira AH , McDermott MP , Barone P , Comella CL , Albrecht S , Hsu HH , Massey DH , Mizuno Y , Poewe W , Rascol O , Marek K ((2013) ) Pramipexole in patients with early Parkinson’s disease (PROUD): A randomised delayed-start trial. Lancet Neurol 12: , 747–755. |
[25] | Verschuur CVM , Suwijn SR , Boel JA , Post B , Bloem BR , Van Hilten JJ , Van Laar T , Tissingh G , Munts AG , Deuschl G , Lang AE , Dijkgraaf MGW , De Haan RJ , De Bie RMA ((2019) ) Randomized delayed-start trial of levodopa in Parkinson’s disease. N Engl J Med 380: , 315–324. |
[26] | Holden SK , Finseth T , Sillau SH , Berman BD ((2018) ) Progression of MDS-UPDRS scores over five years in de novo Parkinson disease from the Parkinson’s Progression Markers Initiative Cohort. Mov Disord Clin Pract 5: , 47–53. |
[27] | Makkos A , Kovács M , Aschermann Z , Harmat M , Janszky J , Karádi K , Kovács N ((2018) ) Are the MDS-UPDRS-based composite scores clinically applicable? Mov Disord 33: , 835–839. |
[28] | Schrag A , Dodel R , Spottke A , Bornschein B , Siebert U , Quinn NP ((2007) ) Rate of clinical progression in Parkinson’s disease. A prospective study. Mov Disord 22: , 938–945. |
[29] | Horváth K , Aschermann Z , Kovács Márton , Makkos A , Harmat Márk , Janszky J , Komoly S , Karádi K , Kovács N ((2017) ) Minimal clinically important differences for the experiences of daily living parts of movement disorder society-sponsored unified Parkinson’s disease rating scale. Mov Disord 32: , 789–793. |
[30] | Evers LJW , Krijthe JH , Meinders MJ , Bloem BR , Heskes TM ((2019) ) Measuring Parkinson’s disease over time: The real-world within-subject reliability of the MDS-UPDRS. Mov Disord 34: , 1480–1487. |
[31] | Bhattaram VA , Siddiqui O , Kapcala LP , Gobburu JVS ((2009) ) Endpoints and analyses to discern disease-modifying drug effects in early Parkinson’s disease. AAPS J 11: , 456–464. |
[32] | Maetzler W , Liepelt I , Berg D ((2009) ) Progression of Parkinson’s disease in the clinical phase: Potential markers. Lancet Neurol 8: , 1158–1171. |
[33] | Holford NHG , Chan PLS , Nutt JG , Kieburtz K , Shoulson I , Parkinson Study Group ((2006) ) Disease progression and pharmacodynamics in Parkinson disease – evidence for functional protection with levodopa and other treatments. J Pharmacokinet Pharmacodyn 33: , 281–311. |
[34] | Venuto CS , Potter NB , Dorsey ER , Kieburtz K ((2016) ) A review of disease progression models of Parkinson’s disease and applications in clinical trials. Mov Disord 31: , 947–956. |
[35] | Latourelle JC , Beste MT , Hadzi TC , Miller RE , Oppenheim JN , Valko MP , Wuest DM , Church BW , Khalil IG , Hayete B , Venuto CS ((2017) ) Large-scale identification of clinical and genetic predictors of motor progression in patients with newly diagnosed Parkinson’s disease: A longitudinal cohort study and validation. Lancet Neurol 16: , 908–916. |
[36] | Roheger M , Kalbe E , Liepelt-Scarfone I ((2018) ) Progression of cognitive decline in Parkinson’s disease. J Parkinsons Dis 8: , 183–193. |
[37] | Lawson RA , Yarnall AJ , Duncan GW , Breen DP , Khoo TK , Williams-Gray CH , Barker RA , Burn DJ , ICICLE-PD study group ((2017) ) Stability of mild cognitive impairment in newly diagnosed Parkinson’s disease. J Neurol Neurosurg Psychiatry 88: , 648–652. |
[38] | Aarsland D , Muniz G , Matthews F ((2011) ) Nonlinear decline of Mini-Mental State Examination in Parkinson’s disease. Mov Disord 26: , 334–337. |
[39] | Vu TC , Nutt JG , Holford NHG ((2012) ) Progression of motor and nonmotor features of Parkinson’s disease and their response to treatment. Br J Clin Pharmacol 74: , 267–283. |
[40] | Faust-Socher A , Duff-Canning S , Grabovsky A , Armstrong MJ , Rothberg B , Eslinger PJ , Meaney CA , Schneider RB , Tang-Wai DF , Fox SH , Zadikoff C , Kennedy N , Chou KL , Persad C , Litvan I , Mast BT , Gerstenecker AT , Weintraub S , Reginold W , Marras C ((2019) ) Responsiveness to change of the Montreal Cognitive Assessment, Mini-Mental State Examination, and SCOPA-Cog in non-demented patients with Parkinson’s disease. Dement Geriatr Cogn Disord 17: , 1–11. |
[41] | McGhee DJM , Ritchie CW , Zajicek JP , Counsell CE ((2016) ) A review of clinical trial designs used to detect a disease-modifying effect of drug therapy in Alzheimer’s disease and Parkinson’s disease. BMC Neurol 16: , 92. |
[42] | Lord S , Galna B , Yarnall AJ , Morris R , Coleman S , Burn D , Rochester L ((2017) ) Natural history of falls in an incident cohort of Parkinson’s disease: Early evolution, risk and protective features. J Neurol 264: , 2268–2276. |
[43] | Bäckström D , Granåsen G , Domellöf ME , Linder J , Mo SJ , Riklund K , Zetterberg H , Blennow K , Forsgren L ((2018) ) Early predictors of mortality in parkinsonism and Parkinson disease A population-based study. Neurology 91: , E2045–E2056. |
[44] | Ishihara LS , Cheesbrough A , Brayne C , Schrag A ((2007) ) Estimated life expectancy of Parkinson’s patients compared with the UK population. J Neurol Neurosurg Psychiatry 78: , 1304–1309. |
[45] | Elbaz A , Bower JH , Peterson BJ , Maraganore DM , McDonnell SK , Ahlskog JE , Schaid DJ , Rocca WA ((2003) ) Survival study of Parkinson disease in Olmsted County, Minnesota. Arch Neurol 60: , 91–96. |
[46] | Hely MA , Morris JGL , Traficante R , Reid WGJ , O’sullivan DJ , Williamson PM ((1999) ) The Sydney multicentre study of Parkinson’s disease: Progression and mortality at 10 years. J Neurol Neurosurg Psychiatry 67: , 300–307. |
[47] | Tison F , Letenneur L , Djossou F , Dartigues JF ((1996) ) Further evidence of increased risk of mortality from Parkinson’s disease. J Neurol Neurosurg Psychiatry 60: , 592–593. |
[48] | Herlofson K , Lie SA , Arsland D , Larsen JP ((2004) ) Mortality and Parkinson disease: A community based study. Neurology 62: , 937–942. |
[49] | Marras C , McDermott MP , Rochon PA , Tanner CM , Naglie G , Rudolph A , Lang AE ((2005) ) Survival in Parkinson disease: Thirteen-year follow-up of the DATATOP cohort. Neurology 64: , 87–93. |
[50] | Simuni T , Caspell-Garcia C , Coffey CS , Weintraub D , Mollenhauer B , Lasch S , Tanner CM , Jennings D , Kieburtz K , Chahine LM , Marek K ((2018) ) Baseline prevalence and longitudinal evolution of non-motor symptoms in early Parkinson’s disease: The PPMI cohort. J Neurol Neurosurg Psychiatry 89: , 78–88. |
[51] | Martinez-Martin P , Chaudhuri KR , Rojo-Abuin JM , Rodriguez-Blazquez C , Alvarez-Sanchez M , Arakaki T , Bergareche-Yarza A , Chade A , Garretto N , Gershanik O , Kurtis MM , Martinez-Castrillo JC , Mendoza-Rodriguez A , Moore HP , Rodriguez-Violante M , Singer C , Tilley BC , Huang J , Stebbins GT , Goetz CG ((2015) ) Assessing the non-motor symptoms of Parkinson’s disease: MDS-UPDRS and NMS Scale. Eur J Neurol 22: , 37–43. |
[52] | Martinez-Martin P , Schrag A , Weintraub D , Rizos A , Rodriguez-Blazquez C , Chaudhuri KR ((2019) ) Pilot study of the International Parkinson and Movement Disorder Society-sponsored Non-motor Rating Scale (MDS-NMS). Mov Disord Clin Pract 6: , 227–234. |
[53] | Stocchi F , Martínez-Martin P , Reichmann H ((2014) ) Movement Disorders Parkinson’s disease quality of life in Parkinson’s disease-patient, clinical and research perspectives. Eur Neurol Rev 9: , 8–12. |
[54] | Reichmann H , Martínez-Martin P , Stocchi F ((2014) ) Effect of therapeutic interventions on health-related quality of life in parkinson’s disease. Eur Neurol Rev 9: , 19–26. |
[55] | The Biomarkers Definitions Working Group ((2001) ) Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin Pharmacol Ther 69: , 89–95. |
[56] | Atik A , Stewart T , Zhang J ((2016) ) Alpha-synuclein as a biomarker for Parkinson’s disease. Brain Pathol 26: , 410–418. |
[57] | Miller DB , O’Callaghan JP ((2015) ) Biomarkers of Parkinson’s disease: Present and future. Metabolism 64: , S40–S46. |
[58] | Aviles-Olmos I , Dickson J , Kefalopoulou Z , Djamshidian A , Ell P , Soderlund T , Whitton P , Wyse R , Isaacs T , Lees A , Limousin P , Foltynie T ((2013) ) Exenatide and the treatment of patients with Parkinson’s disease. J Clin Invest 123: , 2730–2736. |
[59] | Whone AL , Boca M , Luz M , Woolley M , Mooney L , Dharia S , Broadfoot J , Cronin D , Schroers C , Barua NU , Longpre L , Lynn Barclay C , Boiko C , Johnson GA , Fibiger HC , Harrison R , Lewis O , Pritchard G , Howell M , Irving C , Johnson D , Kinch S , Marshall C , Lawrence AD , Blinder S , Sossi V , Stoessl AJ , Skinner P , Mohr E , Gill SS ((2019) ) Extended treatment with glial cell line-derived neurotrophic factor in Parkinson’s disease. J Parkinsons Dis 9: , 301–313. |
[60] | Schneider JS , Cambi F , Gollomp SM , Kuwabara H , Brašić JR , Leiby B , Sendek S , Wong DF ((2015) ) GM1 ganglioside in Parkinson’s disease: Pilot study of effects on dopamine transporter binding. J Neurol Sci 356: , 118–123. |
[61] | Marks WJ , Bartus RT , Siffert J , Davis CS , Lozano A , Boulis N , Vitek J , Stacy M , Turner D , Verhagen L , Bakay R , Watts R , Guthrie B , Jankovic J , Simpson R , Tagliati M , Alterman R , Stern M , Baltuch G , Starr PA , Larson PS , Ostrem JL , Nutt J , Kieburtz K , Kordower JH , Olanow CW ((2010) ) Gene delivery of AAV2-neurturin for Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol 9: , 1164–1172. |
[62] | Hartmann A , Müllner J , Meier N , Hesekamp H , van Meerbeeck P , Habert M-O , Kas A , Tanguy M-L , Mazmanian M , Oya H , Abuaf N , Gaouar H , Salhi S , Charbonnier-Beaupel F , Fievet M-H , Galanaud D , Arguillere S , Roze E , Degos B , Grabli D , Lacomblez L , Hubsch C , Vidailhet M , Bonnet A-M , Corvol J-C , Schüpbach M ((2016) ) Bee venom for the treatment of Parkinson disease - a randomized controlled clinical trial. PLoS One 11: , e0158235. |
[63] | Jucaite A , Svenningsson P , Rinne JO , Cselényi Z , Varnäs K , Johnström P , Amini N , Kirjavainen A , Helin S , Minkwitz M , Kugler AR , Posener JA , Budd S , Halldin C , Varrone A , Farde L ((2015) ) Effect of the myeloperoxidase inhibitor AZD3241 on microglia: A PET study in Parkinson’s disease. Brain 138: , 2687–2700. |
[64] | Villafane G , Thiriez C , Audureau E , Straczek C , Kerschen P , Cormier-Dequaire F , Van Der Gucht A , Gurruchaga J-M , Quéré-Carne M , Evangelista E , Paul M , Defer G , Damier P , Remy P , Itti E , Fénelon G ((2018) ) High-dose transdermal nicotine in Parkinson’s disease patients: A randomized, open-label, blinded-endpoint evaluation phase 2 study. Eur J Neurol 25: , 120–127. |
[65] | Simuni T , Kieburtz K , Tilley B , J Elm J , Ravina B , Babcock D , Emborg M , Hauser R , Kamp C , Morgan JC , Webster Ross G , K Simon D , Bainbridge J , Baker L , Bodis-Wollner I , Boyd J , Cambi F , Carter J , Chou K , Dahodwala N , Dewey RB , Dhall R , Fang J , Farrow B , Feigin A , Glazman S , Goudreau J , LeBlanc P , Lee S , Leehey M , Lew MF , Lowenhaupt S , Luo S , Pahwa R , Perez A , Schneider J , Scott B , Shah B , Shannon KM , Sharma S , Singer C , Truong D , Wagner R , Williams K , Marie Wills A , Shieen Wong P , Zadikoff C , Zweig R ((2015) ) Pioglitazone in early Parkinson’s disease: A phase 2, multicentre, double-blind, randomised trial. Lancet Neurol 14: , 795–803. |
[66] | Silveira CRA , MacKinley J , Coleman K , Li Z , Finger E , Bartha R , Morrow SA , Wells J , Borrie M , Tirona RG , Rupar CA , Zou G , Hegele RA , Mahuran D , MacDonald P , Jenkins ME , Jog M , Pasternak SH ((2019) ) Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: Protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 19: , 20. |
[67] | Pagan FL , Hebron ML , Wilmarth B , Torres-Yaghi Y , Lawler A , Mundel EE , Yusuf N , Starr NJ , Arellano J , Howard HH , Peyton M , Matar S , Liu X , Fowler AJ , Schwartz SL , Ahn J , Moussa C ((2019) ) Pharmacokinetics and pharmacodynamics of a single dose Nilotinib in individuals with Parkinson’s disease. Pharmacol Res Perspect 7: , e00470. |
[68] | Saeed U , Compagnone J , Aviv RI , Strafella AP , Black SE , Lang AE , Masellis M ((2017) ) Imaging biomarkers in Parkinson’s disease and Parkinsonian syndromes: Current and emerging concepts. Transl Neurodegener 6: , 8. |
[69] | Lipsmeier F , Taylor KI , Kilchenmann T , Wolf D , Scotland A , Schjodt-Eriksen J , Cheng W-Y , Fernandez-Garcia I , Siebourg-Polster J , Jin L , Soto J , Verselis L , Boess F , Koller M , Grundman M , Monsch AU , Postuma RB , Ghosh A , Kremer T , Czech C , Gossens C , Lindemann M ((2018) ) Evaluation of smartphone-based testing to generate exploratory outcome measures in a phase 1 Parkinson’s disease clinical trial. Mov Disord 33: , 1287–1297. |
[70] | Lígia Silva de Lima A , Hahn T , de Vries NM , Cohen E , Bataille L , Little MA , Baldus H , Bloem BR , Faber MJ ((2016) ) Large-scale wearable sensor deployment in Parkinson’s patients: The Parkinson@Home Study Protocol. JMIR Res Protoc 5: , e172. |
[71] | Bot BM , Suver C , Neto EC , Kellen M , Klein A , Bare C , Doerr M , Pratap A , Wilbanks J , Dorsey ER , Friend SH , Trister AD ((2016) ) The mPower study, Parkinson disease mobile data collected using ResearchKit. Sci. Data 3: , 160011. |
[72] | Lígia Silva de Lima A , Hahn T , W Evers LJ , de Vries NM , Cohen E , Afek M , Bataille L , Daeschler M , Claes K , Boroojerdi B , Terricabras D , Little MA , Baldus H , Bloem BR , Faber MJ ((2017) ) Feasibility of large-scale deployment of multiple wearable sensors in Parkinson’s disease. PLoS One 12: , e0189161. |
[73] | Mobilise-D, https://www.mobilise-d.eu/, Last updated 2019, Accessed on December 13, 2019. |
[74] | Bermel RA , Bakshi R ((2006) ) The measurement and clinical relevance of brain atrophy in multiple sclerosis. Lancet Neurol 5: , 158–170. |
[75] | Chataway J , Schuerer N , Alsanousi A , Chan D , MacManus D , Hunter K , Anderson V , Bangham CRM , Clegg S , Nielsen C , Fox NC , Wilkie D , Nicholas JM , Calder VL , Greenwood J , Frost C , Nicholas R ((2014) ) Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): A randomised, placebo-controlled, phase 2 trial. Lancet 383: , 2213–2221. |
[76] | Ontaneda D , Fox RJ , Chataway J ((2015) ) Clinical trials in progressive multiple sclerosis: Lessons learned and future perspectives. Lancet Neurol 14: , 208–223. |
[77] | Cutter GR , Baier ML , Rudick RA , Cookfair DL , Fischer JS , Petkau J , Syndulko K , Weinshenker BG , Antel JP , Confavreux C , Ellison GW , Lublin F , Miller AE , Rao SM , Reingold S , Thompson A , Willoughby E ((1999) ) Development of a multiple sclerosis functional composite as a clinical trial outcome measure. Brain 122: , 871–882. |
[78] | Plantone D , De Angelis F , Doshi A , Chataway J ((2016) ) Secondary progressive multiple sclerosis: Definition and measurement. CNS Drugs 30: , 517–526. |
[79] | Lublin F , Miller DH , Freedman MS , Cree BAC , Wolinsky JS , Weiner H , Lubetzki C , Hartung H-P , Montalban X , Uitdehaag BMJ , Merschhemke M , Li B , Putzki N , Liu FC , Häring DA , Kappos L , INFORMS study investigators ((2016) ) Oral fingolimod in primary progressive multiple sclerosis (INFORMS): A phase 3, randomised, double-blind, placebo-controlled trial. Lancet 387: , 1075–1084. |
[80] | Montalban X , Hauser SL , Kappos L , Arnold DL , Bar-Or A , Comi G , de Seze J , Giovannoni G , Hartung H-P , Hemmer B , Lublin F , Rammohan KW , Selmaj K , Traboulsee A , Sauter A , Masterman D , Fontoura P , Belachew S , Garren H , Mairon N , Chin P , Wolinsky JS ((2017) ) Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 376: , 209–220. |
[81] | Kaufmann P , Levy G , Montes J , Buchsbaum R , Barsdorf AI , Battista V , Arbing R , Gordon PH , Mitsumoto H , Levin B , Thompson JLP , Kaufmann P , Levy G , Montes J , Buchsbaum R , Barsdorf AI , Battista V , Arbing R , Gordon PH , Mitsumoto H , Levin B , Thompson JLP , Thompson JLP , QALS study group ((2007) ) Excellent inter-rater, intra-rater, and telephone-administered reliability of the ALSFRS-R in a multicenter clinical trial. Amyotroph Lateral Scler 8: , 42–46. |
[82] | The ALS CNTF Study Group ((1996) ) The Amyotrophic Lateral Sclerosis Functional Rating Scale. Arch Neurol 53: , 141. |
[83] | Berry JD , Miller R , Moore DH , Cudkowicz ME , Van Den Berg LH , Kerr DA , Dong Y , Ingersoll EW , Archibald D ((2013) ) The Combined Assessment of Function and Survival (CAFS): A new endpoint for ALS clinical trials. Amyotroph Lateral Scler Front Degener 14: , 162–168. |
[84] | Kordower JH , Olanow CW , Dodiya HB , Chu Y , Beach TG , Adler CH , Halliday GM , Bartus RT ((2013) ) Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136: , 2419–2431. |
[85] | Yoritaka A , Kawajiri S , Yamamoto Y , Nakahara T , Ando M , Hashimoto K , Nagase M , Saito Y , Hattori N ((2015) ) Randomized, double-blind, placebo-controlled pilot trial of reduced coenzyme Q10 for Parkinson’s disease. Parkinsonism Relat Disord 21: , 911–916. |
[86] | Lawton M , Ben-Shlomo Y , May MT , Baig F , Barber TR , Klein JC , Swallow DMA , Malek N , Grosset KA , Bajaj N , Barker RA , Williams N , Burn DJ , Foltynie T , Morris HR , Wood NW , Grosset DG , Hu MTM ((2018) ) Developing and validating Parkinson’s disease subtypes and their motor and cognitive progression. J Neurol Neurosurg Psychiatry 89: , 1279–1287. |
[87] | Fereshtehnejad SM , Zeighami Y , Dagher A , Postuma RB ((2017) ) Clinical criteria for subtyping Parkinson’s disease: Biomarkers and longitudinal progression. Brain 140: , 1959–1976. |
[88] | Fereshtehnejad S-M , Romenets SR , Anang JBM , Latreille V , Gagnon J-F , Postuma RB ((2015) ) New clinical subtypes of Parkinson disease and their longitudinal progression. JAMA Neurol 72: , 863–873. |
[89] | Schrag A , Siddiqui UF , Anastasiou Z , Weintraub D , Schott JM ((2017) ) Clinical variables and biomarkers in prediction of cognitive impairment in patients with newly diagnosed Parkinson’s disease: A cohort study. Lancet Neurol 16: , 66–75. |
[90] | Mollenhauer B , Zimmermann J , Sixel-Döring F , Focke NK , Wicke T , Ebentheuer J , Schaumburg M , Lang E , Friede T , Trenkwalder C ((2019) ) Baseline predictors for progression 4 years after Parkinson’s disease diagnosis in the De Novo Parkinson Cohort (DeNoPa). Mov Disord 34: , 67–77. |
[91] | Velseboer DC , de Bie RMA , Wieske L , Evans JR , Mason SL , Foltynie T , Schmand B , de Haan RJ , Post B , Barker RA , Williams-Gray CH ((2016) ) Development and external validation of a prognostic model in newly diagnosed Parkinson disease. Neurology 86: , 986–993. |
[92] | Mestre TA , Eberly S , Tanner C , Grimes D , Lang AE , Oakes D , Marras C ((2018) ) Reproducibility of data-driven Parkinson’s disease subtypes for clinical research. Parkinsonism Relat Disord 56: , 102–106. |
[93] | Simuni T , Caspell-Garcia C , Coffey C , Lasch S , Tanner C , Marek K ((2016) ) How stable are Parkinson’s disease subtypes in de novo patients: Analysis of the PPMI cohort? Parkinsonism Relat Disord 28: , 62–67. |
[94] | Lewis SJG , Foltynie T , Blackwell AD , Bobbins TW , Owen AM , Barker RA ((2005) ) Heterogeneity of Parkinson’s disease in the early clinical stages using a data driven approach. J Neurol Neurosurg Psychiatry 76: , 343–348. |
[95] | Reijnders JSAM , Ehrt U , Lousberg R , Aarsland D , Leentjens AFG ((2009) ) The association between motor subtypes and psychopathology in Parkinson’s disease. Parkinsonism Relat Disord 15: , 379–382. |
[96] | Liu P , Feng T , Wang YJ , Zhang X , Chen B ((2011) ) Clinical heterogeneity in patients with early-stage Parkinson’s disease: A cluster analysis. J Zhejiang Univ Sci B 12: , 694–703. |
[97] | Kempster PA , Frankel JP , Bovingdon M , Webster R , Lees AJ , Stern GM ((1989) ) Levodopa peripheral pharmacokinetics and duration of motor response in Parkinson’s disease. J Neurol Neurosurg Psychiatry 52: , 718–723. |
[98] | Muenter MD , Tyce GM ((1971) ) L-dopa therapy of Parkinson’s disease: Plasma L-dopa concentration, therapeutic response, and side effects. Mayo Clin Proc 46: , 231–239. |
[99] | The Parkinson Study Group ((2004) ) Levodopa and the progression of Parkinson’s disease. N Engl J Med 351: , 2498–2508. |
[100] | Hauser RA , Holford NHG ((2002) ) Quantitative description of loss of clinical benefit following withdrawal of levodopa-carbidopa and bromocriptine in early Parkinson’s disease. Mov Disord 17: , 961–968. |
[101] | Fahn S ((2006) ) A new look at levodopa based on the ELLDOPA study. J Neural Transm 70: , 419–426. |


