
Ten Years of the International Parkinson Disease Genomics Consortium: Progress and Next Steps
Abstract
In June 2009 a small group of investigators met at the annual Movement Disorders Society meeting in Paris. The explicit goal of this meeting was to discuss a potential research alliance focused on the genetics of Parkinson disease (PD). The outcome of this informal meeting was the creation of the International Parkinson Disease Genomics Consortium (IPDGC), a group focused on collaborative genetics research, enabled by trust, sharing, and as little paperwork as possible. The IPDGC has grown considerably since its inception, including over 100 scientists from around the World. The focus has also grown, to include clinical and functional investigation of PD at scale. Most recently, the IPDGC has expanded to initiate major research efforts in East Asia and Africa, and has prioritized collaborations with ongoing major efforts in India and South America. Here we summarize the efforts of the IPDGC thus far and place these in the context of a decade of progress in PD genomics. We also discuss the future direction of IPDGC and our stated research priorities for the next decade.
INTRODUCTION
The International Parkinson Disease Genomics Consortium (IPDGC) is a group of highly collaborative researchers focused on understanding the basis of Parkinson disease (PD) and related disorders including Lewy body diseases, progressive supranuclear palsy, and multiple system atrophy.
The initial formation of the IPDGC, in 2009, was borne out of a realization that no single investigator could deliver on the promise of modern human genetics in isolation. To truly leverage the incredible gains in genetic technologies in the PD space, at scale, would require a highly collaborative approach. This notion brought a small group of PD geneticists together, with the common goal of building an effective, transparent, and functional collaboration. This group, like others, had previously collaborated on a smaller scale, and often in competition with each other.
At that time the disease genetics field, perhaps more than most, was a highly competitive one, where huge gains could be made by individual discoveries. Indeed, the identification of novel, disease-causing genetic mechanisms mostly involved small groups leveraging precious clinical resources and traditional linkage and positional cloning approaches. The resulting novel genetic insights formed the basis of the fields ensuing functional and mechanistic work, and thus such discoveries were scientifically seminal. A corollary of this impact was that such discoveries could launch a career, or at the very least secure funding for the winning group. The stakes were high, there was little reward for second place, the work could be performed by individual investigators, and there was little mechanism for shared success; thus the groups that functioned as gene-hunters tended to do so in competition. This was not a wholly negative approach, competition spurred groups to work incredibly quickly and efficiently, and the pace of discovery, when considering the amount of work required, was high.
However, from ∼2005 onward the genetics field began to witness a revolution. The development of methods for highly parallel genotyping and the generation of knowledge resources such as the Human Genome Project and the International HapMap Project provided the tools and blueprint for understanding the influence of common genetic variability on human traits, including disease. Conversely to previous efforts, this work required large sample series, and was particularly amenable to the aggregation of data. This represented a sea change in genetics. Understanding that each center’s individual ability to discover robust association was limited, the groups that were previously in competition with each other, had to devise ways to work together. In many ways the early seeds of the IPDGC were sown by a coordinated effort in genome wide association (GWA) experiments in PD that began in 2008 [1]. This revealed that sharing, of what were at the time large data, across centers was not only feasible, but necessary, and that when this was done effectively, transformative discoveries could be made. The genesis of the IPDGC came from a rather informal meeting held in Paris in 2009, where a small group of ∼10 investigators established loose criteria for working together. Essentially these were centered on unfettered data sharing, no individual restrictions on the work of each scientist, an understanding that there would be no surprises, and a commitment to help each other on projects. The in-person IPDGC meetings have continued in force over the last decade, with at least one meeting every year hosted by members in Berlin and Tubingen (Germany), Vienna (Austria), London (United Kingdom), Paris, Toulouse and Nice (France), Reykjavik (Iceland), San Diego, Miami (United States of America), Amsterdam (The Netherlands) and Lisbon (Portugal). It is notable that until two years ago there was no external monetary support for these meetings, with each attendee paying their own way. In the last two years, attendance of a dozen or more junior scientists has been sponsored by the Parkinson Foundation, something we are truly grateful for. The last IPDGC meeting included ∼90 attendees, PD scientists across disciplines and from academia, the government, PD foundations, and industry. Here we comment on the history of discovery in IPDGC and then extend this to a view toward the future, and our expectations for the next period of IPDGC research.
IDENTIFICATION OF GENETIC RISK LOCI ASSOCIATED WITH PARKINSON DISEASE
The coordinated analysis of GWA data was perhaps the first success for IPDGC and has continued to be a mainstay of our work. The IPDGC has led or been major contributors to the majority of GWA in PD over the last decade [1–7]. This work has centered on available genome wide SNP genotyping of IPDGC members case and control cohorts from the USA, Canada, England, Wales, The Netherlands, France, Germany, Italy, Spain, Austria, Finland, Norway, Estonia, and Australia (Fig. 1). These studies have involved collaboration within IPDGC and with groups from industry, including Genentech and, notably, 23andMe, who have a continued interest in PD. The source diversity and size of these sample series has grown considerably, from the first efforts that centered on ∼1,500 cases and a similar number of controls, to the most recent effort that included dense genotyping in more than 50,000 cases and proxy-cases, and ∼1.4 million controls [7]. The latest effort, published this year, marks another major step forward in our understanding of the genetic architecture of PD. As in other disorders, as sample size has grown, so has power and the number of loci detected. Currently, there are more than 90 known risk loci for PD. The sample sizes described here certainly show an evolution in our capability for testing of genetic association in PD. It should be noted however that sample series in other complex traits continue to evolve also, with extremely large sample series and continued returns. Work assessing the genetic contribution to traits such as lipid levels, type 2 diabetes, or coronary heart disease have each leveraged between ∼70,000 and ∼180,000 cases and provided novel biological insights [8–10].
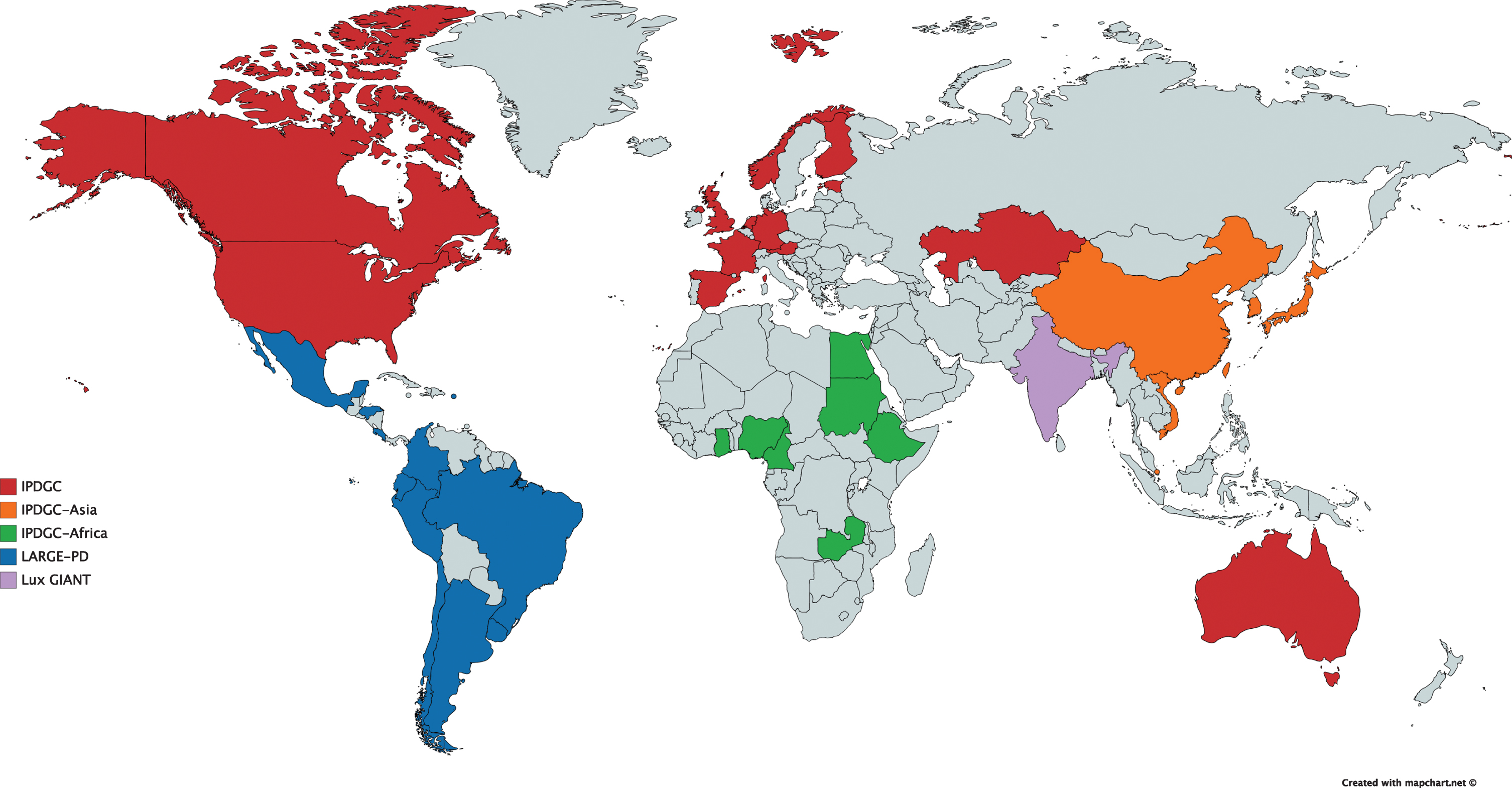
Fig.1
Overview of the currently included countries in the IPDGC and IPDGC-affiliated efforts. IPDGC, International Parkinson Disease Genomics Consortium; LARGE-PD, Latin American Research Consortium on the Genetics of Parkinson Disease; Lux GIANT, Luxembourg-German-Indian Alliance on Neurodegenerative diseases and Therapeutics.
As the complexity of these data sets has grown, so has the sophistication of downstream analyses. Initial work centered solely on the identification of risk alleles. However, understanding that genetic association testing could be applied to expression or DNA methylation, a natural extension was to examine whether disease risk loci had a detectable association with changes in expression of nearby transcripts. As these data sets have become larger, and available in disease relevant tissues, the results of these analyses have become more robust. Another step forward has come with the availability of single cell expression data. By examining whether the genes near GWA loci are enriched in certain tissues and cell types, one can begin to make inferences about the major tissues and cells through which genetic risk is mediated. In the context of PD, the latest work has revealed that brain tissue generally, and nigral neurons specifically, are critical in this aspect of the disease process. While this is perhaps to be expected, it contrasts quite strikingly to what has been observed in Alzheimer disease, where immune cells are a key effector of genetic risk.
Our search for genetic risk began without a realistic understanding of the contribution of common variability to disease. Again, methodology and data have combined to help clarify this position. The application of heritability estimation methods such as Genome-wide Complex Trait Analysis (GCTA) and Linkage Disequilibrium Score Regression (LDSC) have provided and refined our estimates of common variant heritability in PD, suggesting that 16–36% of the liability of disease is driven by common genetic variability [11]. In effect this tells us the upper bounds of genetic discovery and, importantly genetic risk prediction.
Another increasingly popular extension of genetic risk, is through the application of Mendelian Randomization (MR). This method aims to examine the relationship between a modifiable trait and disease (in our case PD), using genetic variability for the trait as an instrumental variable (a more complete description here [12]. Using this method it is possible to test whether a trait is causally associated with PD. MR has become increasingly popular over the last few years, in large part due to the growing amount of robust genetic association results and data [13].
BEYOND RISK LOCI DETECTION
Risk scores, prediction, pathways, progression, modifiers, and age at onset
The IPDGC also made substantial progress beyond standard risk loci detection. Using as a basis the large IPDGC genetic datasets (currently including over 20,000 cases and 20,000 controls) multiple advanced analyses are performed to predict disease, assign more “function” or biology to GWAS loci and identifying potential disease relevant pathways. Shortly after the larger GWAS efforts a genetic risk score was generated based on the GWAS loci effect sizes. This genetic risk score has been identified to be useful in potential prediction of PD and including simple clinical tests such as a smell test high accuracy can be achieved [14]. This genetic risk score is also negatively correlated with the age of onset of PD [15] and interestingly certain variants appear to have a large effect on age of onset while others do not seem to influence age of onset [16]. Similarly, some variants are associated with certain clinical features of PD [17, 18]. More large-scale pathway approaches have identified important roles of the lysosome [19], endocytic membrane trafficking [20] and the mitochondrial pathway [21]. Another recent topic is penetrance or age of onset modifiers in carriers of LRRK2 and GBA damaging variants. Where for LRRK2 the GRS was shown to play a role in penetrance and age of onset [22, 23] and for GBA besides the GRS affecting penetrance and age of onset variants in close proximity to CTSB and SNCA were affecting penetrance [24].
Causal rare variants
As we gained confidence in our ability to share large data, and as new methods became available, we began to explore sharing of data beyond array-based genotyping, to include targeted resequencing, whole exome sequencing, and genome sequencing. A principal component of this work has centered on new discovery, with the sharing of data leading to the identification of new genes or pathways associated with PD. In the context of rare, novel, disease causing mutations, the discovery of VPS13C mutations as a cause of young onset PD is the cardinal example of how collaboration within IPDGC, led by a founding member, Alexis Brice, facilitates discovery [25]. Moreover, using IPDGC resources we were able to replicate findings such as the association between 22q11.2 and idiopathic PD disease [26], RAB39B and PD [27] and GBA variants and PD [19, 28]. Multiple projects inside the IPDGC are currently in progress on identifying novel causal rare variants in known and novel genes using aggregated exome sequencing and genome sequencing data.
Replicating emerging associations and mutations
Because the data within IPDGC is both broad and deep an important role we play is to assess emerging mutations or variants associated with disease. Clearly, given the investment in time and resources for functional investigation of individual loci or variants, it is critical that these are investigated rigorously. The IPDGC supports this effort in three ways, through the provision of publicly available data, through the assessment of genes/variants upon request (>10 such requests have been addressed by IPDGC), or by assessing variants after they are published. Rare variants in ADORA1 [29], SNCA (p.H50Q) [30], TMEM230 [31], EIF4G1 [32], LRRK2 (loss of function variants) [33], MC1R (p.R160W) [34], CHCHD2 [35], ADH1C [36], ARSA [37], and LRP10 [38] were all previously identified to be associated with “monogenic” forms of PD or high risk variants, however none of these results could be replicated in IPDGC data. Additionally common variants in PARK10 [39] and multiple other associations with age of onset of PD [16] were not replicated in IPDGC efforts. Similarly we have not been able to replicate the associations between vitamin D and PD [40] and the causal relationship between urate levels and PD [41]. While false negative replication can be a result of testing extremely rare variants (private mutations or single family) or may reflect population specific alterations, the relatively large sample series available through IPDGC does provide a frame of reference for such associations and suggest that these associations may represent false positives or very rare family-based or population specific effects.
Genetics based functional work
More recently the IPDGC also has expanded its efforts to more functional follow-up of certain variants and genes of interest. One area that has been proven to be a complex issue is the functional follow-up and validation of GWAS loci. To better understand GWAS loci protein–protein interactions can help determine the most likely candidate for several GWAS loci. Using protein–protein interaction arrays, RAB29 (RAB7L1) was identified as a binding partner of LRRK2 therefore nominating RAB29 as primary candidate at the RAB29 locus [42]. The previously mentioned discovery of the VPS13C mutations included extensive functional work [25]. More recently an effort to identify novel autosomal recessive genes tested 27 genes that have homozygous or compound heterozygous loss-of-function variants in PD cases only. After extensive functional work 5 potential novel genes were nominated including again VPS13C [43].
Creation of resources
Besides the genomic work the IPDGC has also been actively creating resources including the development genotyping array and browsable result pages. A potential confounding factor in GWAS and genetics is the introduction of multiple genotyping arrays with different “backbones”. In order to standardize the genotyping arrays used in genetics research, the IPDGC started a collaboration with Illumina in 2014 to develop a neurodegenerative focused genotyping array with a large custom content named NeuroX [44]. This array (and the custom content on other arrays) was widely used for neurodegenerative focused research. In 2017 this array was updated and was named NeuroChip (or Neuro Array) [45]. The next version of our neurodegenerative focused array will be finalized in winter 2019, this array content will more thoroughly catalog risk variants both common and rare, as well as facilitate expanded studies across diverse ancestry groups often underrepresented in the literature.
Another important role the IPDGC has, is providing the research community with easy access result summaries that are usable and understandable to basic researchers, such as results from GWAS summary stats, from the MR studies [13] (https://pdgenetics.shinyapps.io/MRportal/) and PD progression studies [17] (https://pdgenetics.shinyapps.io/pdprogmetagwasbrowser/). Currently under development are the IPDGC sequencing browser and the IPDGC GWAS browser. In the IPDGC sequencing browser researchers can browse all variants identified in IPDGC derived sequencing data to allow quick lookup, scrutinize and assess potential pathogenic variants. The IPDGC GWAS browser is a community driven effort to nominate causal genes in GWAS loci based on public data to assist functional study design.
OUR FUTURE CHALLENGES
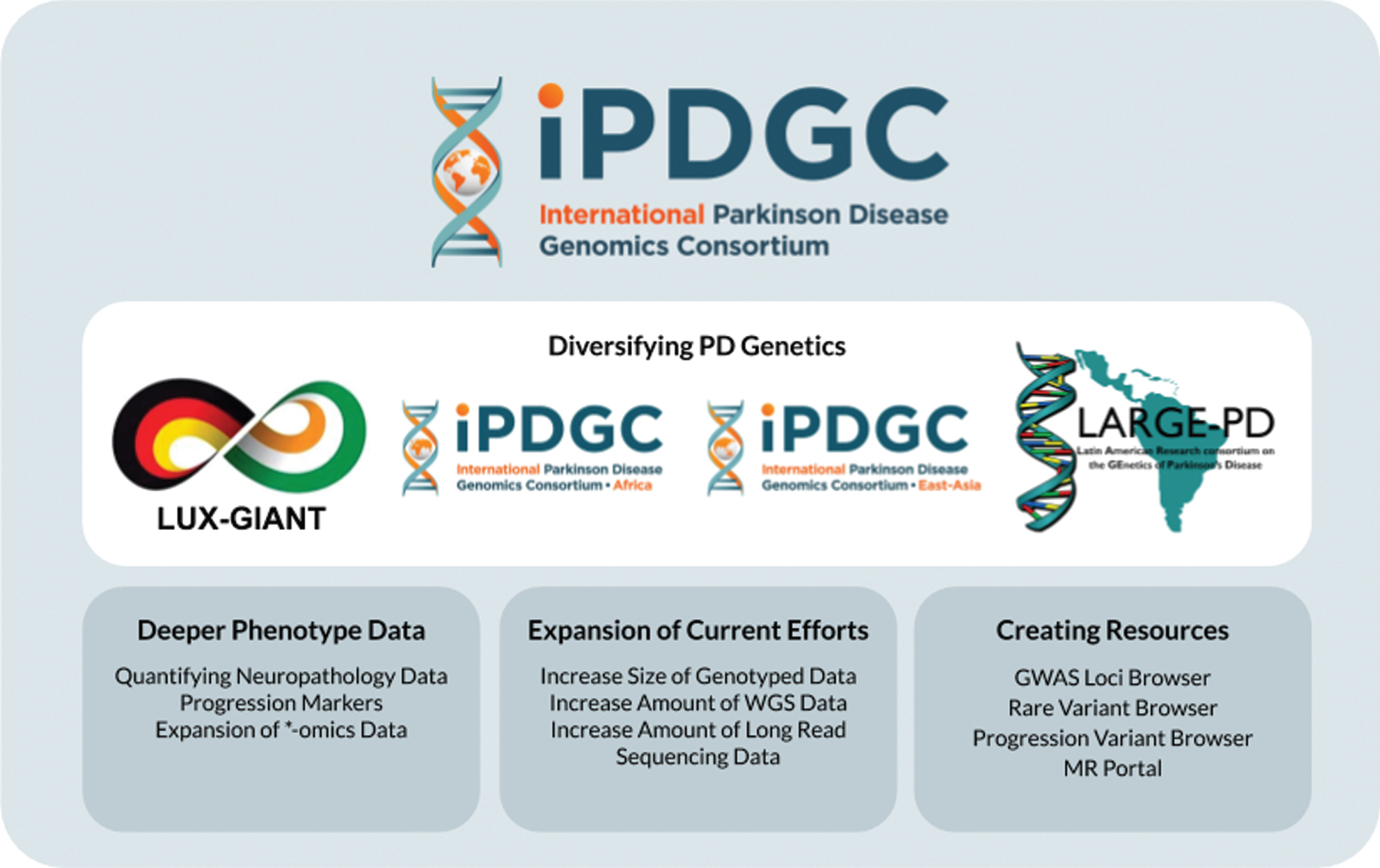
At the end of each IPDGC meeting we typically spent the last couple of hours comprising a list of action items. These points range from short-term action points that are relatively easy to address in the following months to long-term, broad conceptual goals for the IPDGC and the field in general. Our current areas of focus stretch across four major themes: 1) expansion of the known genetic architecture of PD, 2) enabling and supporting genetic investigation in ancestrally diverse populations, 3) an investment in the collection of deeper phenotypic data, and 4) the creation of foundational resources to move from gene to function (Fig. 2). Critically any new aims remain faithful to our original goals, to provide tools with which to develop treatments for this disease. Clearly these can be targets, precision patient stratification, trial ready cohorts, predictors of disease and progression, and leads for biomarkers. As discussed briefly above, we continue to engage our industry colleagues to understand their needs to move PD therapeutics forward.
Fig.2
Overview of the past and future work of the IPDGC. WGS, whole-genome sequencing; MR, Mendelian Randomization.
Expanding the known genetic architecture
Based on our work so far, we know that there is a considerable genetic component underlying PD that remains unidentified. In northern European ancestry individuals, there are ∼90 known risk loci, but these only represent 16–36% of the heritable component of disease. Experience from other disorders has shown that there continue to be significant returns on GWA as case numbers increase into the range of hundreds of thousands. These continued discoveries offer novel biological insights, directly highlighting new biological mechanisms and in this regard, we have clearly not yet reached a point at which we have mechanistic saturation (i.e. in the context of biology, nothing new is being learned). As sample sizes grow, we also increase our ability to define etiologic subtypes of disease, should they exist. By testing for genetic clustering of disease we can define whether there are etiologic/mechanistic differences among patients (as a simplistic example, do we see a lysosomal subgroup, or a mitochondrial subgroup). Clearly, such data also contributes to models of prediction and progression, which we will discuss below, however, we believe that these outcomes alone justify the continued investment in case ascertainment, assessment, and genotyping. Thus an action point for the IPDGC over the next period is to support an expansion of our current genome wide association work to include more than 100,000 patients.
Genetics in diverse ancestries
As with the majority of diseases, most investment in human genetics research has been placed in studies of individuals of European Ancestry. There is a paucity of genetic data in patients outside of this ancestral group. Clearly, this must change. We have an obligation as a field to understand the basis of disease in different ancestral groups and genetics should be the beginning of that understanding. Knowing what differences exist between PD in different ancestral groups has the potential to inform us regarding individualized risk, the predominant underlying disease processes in different populations, and how treatment effectiveness may vary between individuals and ancestral groups. From a purely genetic perspective, combining information across populations facilitates a finer scale dissection of risk loci through trans-ethnic fine mapping, providing more certainty around the underlying biologically important variants and genes.
The importance of the dissection of genetic risk in non-European ancestry populations has led us to invest more in establishing research in underrepresented groups. With the support of the Michael J Fox Foundation for Parkinson’s Research, the IPDGC has initiated large efforts in South East Asia and China, and across Africa. We are also working closely with collections centered in India (LUX-GIANT; Luxembourg-German-Indian Alliance on Neurodegenerative diseases and Therapeutics) and the well-established effort LARGE-PD (Latin American Research Consortium on the Genetics of Parkinson’s Disease; http://large-pd.org/), led by Dr. Ignacio Mata (Fig. 1). To facilitate research across populations we have been working with Illumina Inc. to create an affordable genotyping array that is capable of maximizing information across different ancestral groups. As with the previous arrays developed by us this commercial array will include a large number of neurological disease relevant variants; however, this time the array will be placed on the backbone of popular genotyping platforms such as the Multi-Ethnic Genotyping Array (MEGA) or the Global Screening Array (GSA) creating the power to assess both common and rare variants in diverse populations. Thus, in the first instance our approach will be to facilitate the generation of large-scale and unified genotyping data across diverse populations, with the explicit goals of 1) identifying genetic risk for PD in each population, 2) determining the differences in genetic architecture of PD between populations, and 3) combining data to fine map genetic risk loci. A limitation in this space is the lack of reference whole genome sequence data in certain groups, thus limiting the capacity to capture the genetic diversity in these underserved groups; we will contribute to filling this need by generating whole genome sequence in select ancestral groups.
Our view is that critical to the success of these global initiatives is the involvement of local stakeholders in the leadership and management of research efforts, in addition to providing training opportunities for local scientists and clinicians.
Advanced cohort building
Data generation within IPDGC is ongoing and accelerating. With this massive growth planned in both depth and breadth of data, new models of sharing and analyses become necessary. It is our goal to make as much participant level data, code and summary statistics available in as close to real time as possible. Additionally, supporting these centralized repositories with enough compute infrastructure to facilitate federated analysis methods from the large central data pool to integrate many smaller silos of data isolated by various logistic constraints will be necessary.
Our next phase of analyses will focus not only on just risk relating to case-control status but also on disease course, analyzing markers of progression on both the clinical and molecular levels. Precedence for this has been set by reports such as Iwaki et al 2019 [17], illustrating the utility of large-scale data harmonization across over a dozen longitudinal studies of PD progression. This type of detailed clinical data coupled with multi-modal biomarker studies and various *-omics assays on the same study participants will exponentially increase our data complexity and hopefully yield new insights.
To maximize the utility of these complex datasets in our investigations of disease etiology, we will likely need to push past relying on current GWAS derived estimates of risk. While polygenic risk scoring works well for SNP based studies, integrating multiple data substrates and including various data silos in risk predictions will necessitate robust infrastructure and adoption of methods such as federated learning via neural networks to predict various aspects of disease course. Methods such as decomposition trees and other graph-based methods will help us identify subsets of disease that are more similar or more simply put, “predicting the flavors of parkinsonism.” All of these methods are relatively new to biology but are well established in the business and/or technology sectors, with these new tools possibly helping us move down a path toward more efficient clinical trials and better therapeutic targets.
As these tools develop, we are likely to improve PD risk prediction in the general population, particularly when combined with pre-motor measures such as changes in olfaction and sleep, other measures including imaging, and, likely, biological markers. However, currently patients present to clinic with early PD and our challenge is to make an accurate diagnosis, to predict the future disease course and to modulate disease progress. The integration of clinical data with genetics is key to developing these approaches, with some progress already made in this regard [14]. It is also likely that collaboration with industry and with large investigator led cohort studies will be central to success, and this has been a central theme of the last three IPDGC meetings.
The work on GBA variants and clinical phenotype serves to highlight the effect of variants in a single gene on age at onset, disease progression and disease phenotype, in this case the early development of cognitive impairment. It is likely that there are a series of genetically determined pathways, not yet discovered that will provide further insights into disease phenotype and may mandate different treatment approaches. Progression is of particular interest. There is a huge range in PD progression. It is likely that patients with rapid PD progression will have genetic determinants related to the spread of protein pathology from cell to cell, or to modulating factors such as neuro-inflammation and these may represent new therapeutic targets. Furthermore, predicting rate of progression will be important in balancing treatment/placebo arms in therapeutic trials and may reduce sample sizes needed in future clinical trials. We have modelled the effects of genetic bias in treatment arms in clinical trials [46]. A further initiative is to collate genetic resources from ongoing drug trials to leverage high quality longitudinal data form clinical trials and to enable the study of genetic determinants of drug response and side effects.
Creating Parkinson disease resources for the research community
Relative to other branches of research, the genetics of disease has become relatively straightforward. There is a clear and increasingly common path that yields predictable progress. Traveling this path and realizing the potential of the understanding you arrive at can be most efficiently supported through the creation of foundational resources for the research community. Most obviously the sharing of genetic data enables rapid progress and democratizes access to multimillion-dollar datasets. The discovery and dissemination of genetic findings is no longer the rate limiting step in our understanding of biology; however, the next step, translating maps to mechanisms to medicine (M2M2M), remains a significant challenge and one collectively recognized and prioritized by the global genetics community (https://www.icda.bio/). Another promising initiative is the Aligning Science Across Parkinson’s (ASAP, https://parkinsonsroadmap.org/) [47]. ASAP is aiming to build an international network of researchers to improve our understanding of the biology underlying Parkinson’s disease and has a particular interest in the “Biology Of PD-associated Genetics”.
The work of translating variant to function has been limited by methodological constraints that have necessitated traditional low-throughput dissection of the biology of genetic variation. Like genetics 15 years ago, there are a series of technological developments that mean a more systematic and unbiased effort can be undertaken to understand consequences of genetic risk; the development of induced pluripotent stem cells, gene editing, and single cell -omics, applied at scale, mean that variant to function is the likely space for the next transformative efforts in disease research. We believe that a critical need in this space is the development of foundation reference datasets and have therefore prioritized two projects, FOUNDIN-PD and the PD Single Cell Reference set.
The integration of genetics and single cell transcriptomics from human brain is beginning to reveal the biological context of risk variants. In PD, these data show a clear enrichment of genes from risk loci in dopaminergic neurons as well as in oligodendroglia [7]; while this would suggest that modeling the biological effects of risk variants should be prioritized in these cell types, an important prerequisite is an understanding of the biological landscape of such cells, so that we can understand the biological fidelity of our models.
The FOUNDIN-PD project represents a pilot for translating PD risk variants to function. Within this study we have leveraged approximately 100 induced pluripotent stem cells generated from subjects within the longitudinal PD study Parkinson’s Progression Markers Initiative (PPMI; https://www.ppmi-info.org/). Because there is extensive clinical, molecular, and genetic information on these lines, we are able to assess these lines in the context of diagnosis, simple genetic load (LRRK2, SNCA, GBA mutation) and also complex genetics through an assessment of genetic risk load. In FOUNDIN-PD, these lines are driven to a dopaminergic lineage, and then a series of molecular readouts generated assessing transcription, chromatin accessibility, DNA methylation, and the three-dimensional architecture of the genome. Importantly, these assays include single cell RNA sequencing and single cell ATAC sequencing, reducing the impact of cellular heterogeneity in our readouts. The output of this work will be a pilot reference dataset, that will ultimately allow the rapid lookup of the immediate effects of genetic variability (including disease linked variability) to biological function; clearly an extension of this capacity will be the revealing of the molecular networks that are at their core, the disease process.
SUMMARY
The field of PD genetics is one that has changed dramatically over the last 10 years. There has been an exponential growth in our appreciation of the genetic architecture of the disease, and a greater understanding of how to proceed with genetic prosecution of PD. Our future path promises to expand this work and leverage its clinical, mechanistic, and biological potential. Thus, while we believe the work of the IPDGC has had a significant and lasting impact on our field over the last ten years, we are even more excited by the course we have charted for the next decade.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
We would like to thank all of the subjects who donated their time and biological samples to be part of all IPDGC studies without them we could not have made such progress. We would also like to thank all members of the International Parkinson Disease Genomics Consortium (IPDGC) for their continuous support and collaboration. For a complete overview of members, acknowledgements and funding, please see: http://pdgenetics.org/partners. The work of the IPDGC has been supported either directly or indirectly by a number of bodies. These are at http://pdgenetics.org/partners and noted below, but it is also worth explicitly mentioning the support of the Parkinson’s Foundation and Illumina, who have both provided generous support for IPDGC meetings.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JPD-191854.
REFERENCES
[1] | Simón-Sánchez J , Schulte C , Bras JM , Sharma M , Gibbs JR , Berg D , Paisan-Ruiz C , Lichtner P , Scholz SW , Hernandez DG , Krüger R , Federoff M , Klein C , Goate A , Perlmutter J , Bonin M , Nalls MA , Illig T , Gieger C , Houlden H , Steffens M , Okun MS , Racette BA , Cookson MR , Foote KD , Fernandez HH , Traynor BJ , Schreiber S , Arepalli S , Zonozi R , Gwinn K , van der Brug M , Lopez G , Chanock SJ , Schatzkin A , Park Y , Hollenbeck A , Gao J , Huang X , Wood NW , Lorenz D , Deuschl G , Chen H , Riess O , Hardy JA , Singleton AB , Gasser T ((2009) ) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41: , 1308–1312. |
[2] | Bandres-Ciga S , Ahmed S , Sabir MS , Blauwendraat C , Adarmes-Gómez AD , Bernal-Bernal I , Toribio MB , Buiza-Rueda D , Carrillo F , Carrión-Claro M , Gómez-Garre P , Jesús S , Labrador-Espinosa MA , Macias D , Méndez-del-Barrio C , Periñán-Tocino T , Tejera-Parrado C , Vargas-González L , Diez-Fairen M , Alvarez I , Tartari JP , Buongiorno MT , Aguilar M , Gorostidi A , Bergareche JA , Mondragon E , Ruiz-Martínez J , Dols-Icardo O , Kulisevsky J , Marín-Lahoz J , Pagonabarraga J , Pascual-Sedano B , Ezquerra M , Cámara A , Compta Y , Fernández M , Fernández-Santiago R , Muñoz E , Tolosa E , Valldeoriola F , Gonzalez-Aramburu I , Rodriguez AS , Sierra M , Menéndez-González M , Blazquez M , Garcia C , Martin ES-S , García-Ruiz P , Martínez-Castrillo JC , Vela-Desojo L , Ruz C , Barrero FJ , Escamilla-Sevilla F , Mínguez-Castellanos A , Cerdan D , Tabernero C , Heredia MJG , Errazquin FP , Romero-Acebal M , Feliz C , Lopez-Sendon JL , Mata M , Torres IM , Kim JJ , Brooks J , Saez-Atienzar S , Raphael Gibbs J , Jorda R , Botia JA , Bonet-Ponce L , Morrison KE , Clarke C , Tan M , Morris H , Edsall C , Hernandez D , Simon-Sanchez J , Nalls MA , Scholz SW , Jimenez-Escrig A , Duarte J , Vives F , Duran R , Hoenicka J , Alvarez V , Infante J , Marti MJ , Clarimón J , de Munain AL , Pastor P , Mir P , Singleton A , on behalf of the International Parkinson Disease Genomics Consortium ((2019) ) The genetic architecture of Parkinson disease in Spain: Characterizing population-specific risk, differential haplotype structures, and providing etiologic insight. Mov Disord, doi: 10.1002/mds.27864 |
[3] | International Parkinson Disease Genomics Consortium, Nalls MA , Plagnol V , Hernandez DG , Sharma M , Sheerin U-M , Saad M , Simón-Sánchez J , Schulte C , Lesage S , Sveinbjörnsdóttir S , Stefánsson K , Martinez M , Hardy J , Heutink P , Brice A , Gasser T , Singleton AB , Wood NW ((2011) ) Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet 377: , 641–649. |
[4] | Lill CM , Roehr JT , McQueen MB , Kavvoura FK , Bagade S , Schjeide B-MM , Schjeide LM , Meissner E , Zauft U , Allen NC , Liu T , Schilling M , Anderson KJ , Beecham G , Berg D , Biernacka JM , Brice A , DeStefano AL , Do CB , Eriksson N , Factor SA , Farrer MJ , Foroud T , Gasser T , Hamza T , Hardy JA , Heutink P , Hill-Burns EM , Klein C , Latourelle JC , Maraganore DM , Martin ER , Martinez M , Myers RH , Nalls MA , Pankratz N , Payami H , Satake W , Scott WK , Sharma M , Singleton AB , Stefansson K , Toda T , Tung JY , Vance J , Wood NW , Zabetian CP , 23andMe Genetic Epidemiology of Parkinson’s Disease Consortium; International Parkinson’s Disease Genomics Consortium; Parkinson’s Disease GWAS Consortium; Wellcome Trust Case Control Consortium 2), Young P , Tanzi RE , Khoury MJ , Zipp F , Lehrach H , Ioannidis JP , Bertram L ((2012) ) Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: The PDGene database. PLoS Genet 8: , e1002548. |
[5] | Nalls MA , Pankratz N , Lill CM , Do CB , Hernandez DG , Saad M , DeStefano AL , Kara E , Bras J , Sharma M , Schulte C , Keller MF , Arepalli S , Letson C , Edsall C , Stefansson H , Liu X , Pliner H , Lee JH , Cheng R ; International Parkinson’s Disease Genomics Consortium (IPDGC); Parkinson’s Study Group (PSG) Parkinson’s Research: The Organized GENetics Initiative (PROGENI); 23andMe; GenePD; NeuroGenetics Research Consortium (NGRC); Hussman Institute of Human Genomics (HIHG); Ashkenazi Jewish Dataset Investigator; Cohorts for Health and Aging Research in Genetic Epidemiology (CHARGE); North American Brain Expression Consortium (NABEC); United Kingdom Brain Expression Consortium (UKBEC); Greek Parkinson’s Disease Consortium; Alzheimer Genetic Analysis Group, Ikram MA , Ioannidis JP , Hadjigeorgiou GM , Bis JC , Martinez M , Perlmutter JS , Goate A , Marder K , Fiske B , Sutherland M , Xiromerisiou G , Myers RH , Clark LN , Stefansson K , Hardy JA , Heutink P , Chen H , Wood NW , Houlden H , Payami H , Brice A , Scott WK , Gasser T , Bertram L , Eriksson N , Foroud T , Singleton AB ((2014) ) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46: , 989–993. |
[6] | Chang D , Nalls MA , Hallgrímsdóttir IB , Hunkapiller J , van der Brug M , Cai F , International Parkinson’s Disease Genomics Consortium; 23andMe Research Team, Kerchner GA , Ayalon G , Bingol B , Sheng M , Hinds D , Behrens TW , Singleton AB , Bhangale TR1 , Graham RR1 ((2017) ) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49: , 1511–1516. |
[7] | Nalls MA , Blauwendraat C , Vallerga CL , Heilbron K , Bandres-Ciga S , Chang D , Tan M , Kia DA , Noyce AJ , Xue A , Bras J , Young E , von Coelln , Simon-Sanchez J , Schulte C , Sharma M , Korhn L , Pihlstrom L , Siitonen A , Iwaki H , Leonard H , Faghri F , Gibbs JR , Hernandez DG , Scholz SW , Botia JA , Martinez M , Corvol JC , Lesage S , Jankovic J , Shulman LM , The 23andMe Research Team, System Genomics of Parkinson’s Disease (SGPD) Consortium, Sutherland M , Tienari P , Majamaa K , Toft M , Brice A , Yang J , Gan-Orr Z , Gasser TM , Heutink PM , Shulman JM , Wood NA , Hinds DA , Hardy JR , Morris HR , Gratten JM , Viccher PM , Graham RR , Singleton AB , International Parkinson’s Disease Genomics Consortium ((2019) ) Expanding Parkinson’s disease genetics: Novel risk loci, genomic context, causal insights and heritable risk. BioRxiv 388165. |
[8] | Mahajan A , Taliun D , Thurner M , Robertson NR , Torres JM , Rayner NW , Payne AJ , Steinthorsdottir V , Scott RA , Grarup N , Cook JP , Schmidt EM , Wuttke M , Sarnowski C , Magi R , Nano J , Gieger C , Trompet S , Lecoeur C , Preuss MH , Prins BP , Guo X , Bielak LF , Below JE , Bowden DW , Chambers JC , Kim YJ , Ng MCY , Petty LE , Sim X , Zhang W , Bennett AJ , Bork-Jensen J , Brummett CM , Canouil M , Ec Kardt KU , Fischer K , Kardia SLR , Kronenberg F , Lall K , Liu CT , Locke AE , Luan J , Ntalla I , Nylander V , Schonherr S , Schurmann C , Yengo L , Bottinger EP , Brandslund I , Christensen C , Dedoussis G , Florez JC , Ford I , Franco OH , Frayling TM , Giedraitis V , Hackinger S , Hattersley AT , Herder C , Ikram MA , Ingelsson M , Jorgensen ME , Jorgensen T , Kriebel J , Kuusisto J , Ligthart S , Lindgren CM , Linneberg A , Lyssenko V , Mamakou V , Meitinger T , Mohlke KL , Morris AD , Nadkarni G , Pankow JS , Peters A , Sattar N , Stancakova A , Strauch K , Taylor KD , Thorand B , Thorleifsson G , Thorsteinsdottir U , Tuomilehto J , Witte DR , Dupuis J , Peyser PA , Zeggini E , Loos RJF , Froguel P , Ingelsson E , Lind L , Groop L , Laakso M , Collins FS , Jukema JW , Palmer CNA , Grallert H , Metspalu A , Dehghan A , Kottgen A , Abecasis GR , Meigs JB , Rotter JI , Marchini J , Pedersen O , Hansen T , Langenberg C , Wareham NJ , Stefansson K , Gloyn AL , Morris AP , Boehnke M , McCarthy MI ((2018) ) Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet 50: , 1505–1513. |
[9] | Allara E , Morani G , Carter P , Gkatzionis A , Zuber V , Foley CN , Rees JM , Mason AM , Bell S , Gill D , Lindstroem S , Butterworth AS , Di Angelantonio E , Peters J , Burgess S ((2019) ) Genetic determinants of lipids and cardiovascular disease outcomes: A wide-angled Mendelian randomization investigation. Circ Genom Precis Med, doi: 10.1161/CIRCGEN.119.002711 |
[10] | van der Harst P , Verweij N ((2018) ) Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res 122: , 433–443. |
[11] | Keller MF , Saad M , Bras J , Bettella F , Nicolaou N , Simón-Sánchez J , Mittag F , Büchel F , Sharma M , Gibbs JR , Schulte C , Moskvina V , Durr A , Holmans P , Kilarski LL , Guerreiro R , Hernandez DG , Brice A , Ylikotila P , Stefánsson H , Majamaa K , Morris HR , Williams N , Gasser T , Heutink P , Wood NW , Hardy J , Martinez M , Singleton AB , Nalls MA , International Parkinson’s Disease Genomics Consortium (IPDGC); Wellcome Trust Case Control Consortium 2 (WTCCC2) ((2012) ) Using genome-wide complex trait analysis to quantify ‘missing heritability’ in Parkinson’s disease. Hum Mol Genet 21: , 4996–5009. |
[12] | Bandres-Ciga S , Noyce AJ , Traynor BJ ((2019) ) Mendelian randomization—a journey from obscurity to center stage with a few potholes along the way. JAMA Neurol, doi: 10.1001/jamaneurol.2019.3419 |
[13] | Noyce AJ , Bandres-Ciga S , Kim J , Heilbron K , Kia D , Hemani G , Xue A , Lawlor DA , Smith GD , Duran R , Gan-Or Z , Blauwendraat C , Gibbs JR ; 23andMe Research Team, International Parkinson’s Disease Genomics Consortium (IPDGC), Hinds DA , Yang J , Visscher P , Cuzick J , Morris H , Hardy J , Wood NW , Nalls MA , Singleton AB ((2019) ) The Parkinson’s Disease Mendelian Randomization Research Portal. Mov Disord, doi: 10.1002/mds.27873 |
[14] | Nalls MA , McLean CY , Rick J , Eberly S , Hutten SJ , Gwinn K , Sutherland M , Martinez M , Heutink P , Williams NM , Hardy J , Gasser T , Brice A , Price TR , Nicolas A , Keller MF , Molony C , Gibbs JR , Chen-Plotkin A , Suh E , Letson C , Fiandaca MS , Mapstone M , Federoff HJ , Noyce AJ , Morris H , Van Deerlin VM , Weintraub D , Zabetian C , Hernandez DG , Lesage S , Mullins M , Conley ED , Northover CAM , Frasier M , Marek K , Day-Williams AG , Stone DJ , Ioannidis JPA , Singleton AB , Parkinson’s Disease Biomarkers Program and Parkinson’s Progression Marker Initiative investigators ((2015) ) Diagnosis of Parkinson’s disease on the basis of clinical and genetic classification: A population-based modelling study. Lancet Neurol 14: , 1002–1009. |
[15] | Nalls MA , Escott-Price V , Williams NM , Lubbe S , Keller MF , Morris HR , Singleton AB , International Parkinson’s Disease Genomics Consortium ((2015) ) Genetic risk and age in Parkinson’s disease: Continuum not stratum. Mov Disord 30: , 850–854. |
[16] | Blauwendraat C , Heilbron K , Vallerga CL , Bandres-Ciga S , von Coelln R , Pihlstrøm L , Simón-Sánchez J , Schulte C , Sharma M , Krohn L , Siitonen A , Iwaki H , Leonard H , Noyce AJ , Tan M , Gibbs JR , Hernandez DG , Scholz SW , Jankovic J , Shulman LM , Lesage S , Corvol J-C , Brice A , van Hilten JJ , Marinus J , andMe Research T , Eerola-Rautio J , Tienari P , Majamaa K , Toft M , Grosset DG , Gasser T , Heutink P , Shulman JM , Wood N , Hardy J , Morris HR , Hinds DA , Gratten J , Visscher PM , Gan-Or Z , Nalls MA , Singleton AB , International Parkinson’s Disease Genomics Consortium ((2019) ) Parkinson’s disease age at onset genome-wide association study: Defining heritability, genetic loci, and α-synuclein mechanisms. Mov Disord 34: , 866–875. |
[17] | Iwaki H , Blauwendraat C , Leonard HL , Kim JJ , Liu G , Maple-Grødem J , Corvol J-C , Pihlstrøm L , van Nimwegen M , Hutten SJ , Nguyen K-DH , Rick J , Eberly S , Faghri F , Auinger P , Scott KM , Wijeyekoon R , Van Deerlin VM , Hernandez DG , Gibbs JR , International Parkinson’s Disease Genomics Consortium, Chitrala KN , Day-Williams AG , Brice A , Alves G , Noyce AJ , Tysnes O-B , Evans JR , Breen DP , Estrada K , Wegel CE , Danjou F , Simon DK , Andreassen O , Ravina B , Toft M , Heutink P , Bloem BR , Weintraub D , Barker RA , Williams-Gray CH , van de Warrenburg BP , Van Hilten JJ , Scherzer CR , Singleton AB , Nalls MA ((2019) ) Genomewide association study of Parkinson’s disease clinical biomarkers in 12 longitudinal patients’ cohorts. Mov Disord, doi: 10.1002/mds.27845 |
[18] | Iwaki H , Blauwendraat C , Leonard HL , Liu G , Maple-Grødem J , Corvol J-C , Pihlstrøm L , van Nimwegen M , Hutten SJ , Nguyen K-DH , Rick J , Eberly S , Faghri F , Auinger P , Scott KM , Wijeyekoon R , Van Deerlin VM , Hernandez DG , Day-Williams AG , Brice A , Alves G , Noyce AJ , Tysnes O-B , Evans JR , Breen DP , Estrada K , Wegel CE , Danjou F , Simon DK , Ravina B , Toft M , Heutink P , Bloem BR , Weintraub D , Barker RA , Williams-Gray CH , van de Warrenburg BP , Van Hilten JJ , Scherzer CR , Singleton AB , Nalls MA ((2019) ) Genetic risk of Parkinson disease and progression: An analysis of 13 longitudinal cohorts. Neurol Genet 5: , e348. |
[19] | Robak LA , Jansen IE , van Rooij J , Uitterlinden AG , Kraaij R , Jankovic J , International Parkinson’s Disease Genomics C, Heutink P , Shulman JM ((2017) ) Excessive burden of lysosomal storage disorder gene variants in Parkinson’s disease. Brain 140: , 3191–3203. |
[20] | Bandres-Ciga S , Saez-Atienzar S , Bonet-Ponce L , Billingsley K , Vitale D , Blauwendraat C , Gibbs JR , Pihlstrøm L , Gan-Or Z , International Parkinson’s Disease Genomics Consortium, Cookson MR , Nalls MA , Singleton AB ((2019) ) The endocytic membrane trafficking pathway plays a major role in the risk of Parkinson’s disease. Mov Disord 34: , 460–468. |
[21] | Billingsley KJ , Barbosa IA , Bandrés-Ciga S , Quinn JP , Bubb VJ , Deshpande C , Botia JA , Reynolds RH , Zhang D , Simpson MA , Blauwendraat C , Gan-Or Z , Gibbs JR , Nalls MA , Singleton A , International Parkinson’s Disease Genomics Consortium, Ryten M , Koks S ((2019) ) Mitochondria function associated genes contribute to Parkinson’s Disease risk and later age at onset. NPJ Parkinsons Dis 5: , 8. |
[22] | Iwaki H ((2019) ) Penetrance of Parkinson’s disease in LRRK2 p.G2019S carriers is modified by a polygenic risk score. BioRxiv, https://www.biorxiv.org/content/10.1101/738260v2. |
[23] | Brown E ((2019) ) Analysis of DNM3 and VAMP4 as genetic modifiers of LRRK2 Parkinson’s disease. BioRxiv, https://www.biorxiv.org/content/10.1101/686550v2. |
[24] | Blauwendraat C , Reed X , Krohn L , Heilbron K , Bandres-Ciga S , Tan M , Gibbs JR , Hernandez DG , Kumaran R , Langston R , Bonet-Ponce L , Alcalay RN , Hassin-Baer S , Greenbaum L , Iwaki H , Leonard HL , Grenn FP , Ruskey JA , Sabir M , Ahmed S , Makarious MB , Pihlstrom L , Toft M , van Hilten JJ , Marinus J , Schulte C , Brockmann K , Sharma M , Siitonen A , Majamaa K , Eerola-Rautio J , Tienari PJ , Team TAR , Pantelyat A , Hillis AE , Dawson TM , Rosenthal LS , Albert MS , Resnick SM , Ferrucci L , Morris CM , Pletnikova O , Troncoso J , Grosset D , Lesage S , Corvol JC , Brice A , Noyce AJ , Masliah E , Wood N , Hardy J , Shulman LM , Jankovic J , Shulman JM , Heutink P , Gasser T , Cannon P , Scholz SW , Morris H , Cookson MR , Nalls MA , Gan-Or Z , Singleton AB ((2019) ) Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain, doi: 10.1093/brain/awz350 |
[25] | Lesage S , Drouet V , Majounie E , Deramecourt V , Jacoupy M , Nicolas A , Cormier-Dequaire F , Hassoun SM , Pujol C , Ciura S , Erpapazoglou Z , Usenko T , Maurage C-A , Sahbatou M , Liebau S , Ding J , Bilgic B , Emre M , Erginel-Unaltuna N , Guven G , Tison F , Tranchant C , Vidailhet M , Corvol J-C , Krack P , Leutenegger A-L , Nalls MA , Hernandez DG , Heutink P , Gibbs JR , Hardy J , Wood NW , Gasser T , Durr A , Deleuze J-F , Tazir M , Destée A , Lohmann E , Kabashi E , Singleton A , Corti O , Brice A , French Parkinson’s Disease Genetics Study (PDG); International Parkinson’s Disease Genomics Consortium (IPDGC) ((2016) ) Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am J Hum Genet 98: , 500–513. |
[26] | Mok KY , Sheerin U , Simón-Sánchez J , Salaka A , Chester L , Escott-Price V , Mantripragada K , Doherty KM , Noyce AJ , Mencacci NE , Lubbe SJ , International Parkinson’s Disease Genomics C, Williams-Gray CH , Barker RA , van Dijk KD , Berendse HW , Heutink P , Corvol J-C , Cormier F , Lesage S , Brice A , Brockmann K , Schulte C , Gasser T , Foltynie T , Limousin P , Morrison KE , Clarke CE , Sawcer S , Warner TT , Lees AJ , Morris HR , Nalls MA , Singleton AB , Hardy J , Abramov AY , Plagnol V , Williams NM , Wood NW ((2016) ) Deletions at 22q11.2 in idiopathic Parkinson’s disease: A combined analysis of genome-wide association data. Lancet Neurol 15: , 585–596. |
[27] | Lesage S , Bras J , Cormier-Dequaire F , Condroyer C , Nicolas A , Darwent L , Guerreiro R , Majounie E , Federoff M , Heutink P , Wood NW , Gasser T , Hardy J , Tison F , Singleton A , Brice A , French Parkinson’s Disease Genetics Study Group (PDG) and the International Parkinson’s Disease Genomics Consortium (IPDGC) ((2015) ) Loss-of-function mutations in RAB39B are associated with typical early-onset Parkinson disease. Neurol Genet 1: , e9. |
[28] | Blauwendraat C , Bras JM , Nalls MA , Lewis PA , Hernandez DG , Singleton AB , International Parkinson’s Disease Genomics Consortium ((2018) ) Coding variation in GBA explains the majority of the SYT11-GBA Parkinson’s disease GWAS locus. Mov Disord 33: , 1821–1823. |
[29] | Blauwendraat C , Nalls MA , Federoff M , Pletnikova O , Ding J , Letson C , Geiger JT , Gibbs JR , Hernandez DG , Troncoso JC , Simón-Sánchez J , Scholz SW , International Parkinson’s Disease Genomics Consortium ((2017) ) ADORA1 mutations are not a common cause of Parkinson’s disease and dementia with Lewy bodies. Mov Disord 32: , 298–299. |
[30] | Blauwendraat C , Kia DA , Pihlstrøm L , Gan-Or Z , Lesage S , Gibbs JR , Ding J , Alcalay RN , Hassin-Baer S , Pittman AM , Brooks J , Edsall C , Chung SJ , Goldwurm S , Toft M , Schulte C , International Parkinson’s Disease Genomics Consortium (IPDGC), COURAGE-PD Consortium, Hernandez D , Singleton AB , Nalls MA , Brice A , Scholz SW , Wood NW ((2018) ) Insufficient evidence for pathogenicity of SNCA His50Gln (H50Q) in Parkinson’s disease. Neurobiol Aging 64: , 159.e155–159.e158. |
[31] | Giri A , Mok KY , Jansen I , Sharma M , Tesson C , Mangone G , Lesage S , Bras JM , Shulman JM , Sheerin U-M , International Parkinson’s Disease Consortium (IPDGC), Díez-Fairen M , Pastor P , Martí MJ , Ezquerra M , Tolosa E , Correia-Guedes L , Ferreira J , Amin N , van Duijn CM , van Rooij J , Uitterlinden AG , Kraaij R , Nalls M , Simón-Sánchez J ((2017) ) Lack of evidence for a role of genetic variation in TMEM230 in the risk for Parkinson’s disease in the Caucasian population. Neurobiol Aging 50: , 167.e111–167.e113. |
[32] | Nichols N , Bras JM , Hernandez DG , Jansen IE , Lesage S , Lubbe S , Singleton AB , International Parkinson’s Disease Genomics Consortium ((2015) ) EIF4G1 mutations do not cause Parkinson’s disease. Neurobiol Aging 36: , 2444.e2441–2444. |
[33] | Blauwendraat C , Reed X , Kia DA , Gan-Or Z , Lesage S , Pihlstrøm L , Guerreiro R , Gibbs JR , Sabir M , Ahmed S , Ding J , Alcalay RN , Hassin-Baer S , Pittman AM , Brooks J , Edsall C , Hernandez DG , Chung SJ , Goldwurm S , Toft M , Schulte C , Bras J , Wood NW , Brice A , Morris HR , Scholz SW , Nalls MA , Singleton AB , Cookson MR , COURAGE-PD (Comprehensive Unbiased Risk Factor Assessment for Genetics and Environment in Parkinson’s Disease) Consortium, the French Parkinson’s Disease Consortium, and the International Parkinson’s Disease Genomics Consortium (IPDGC) ((2018) ) Frequency of loss of function variants in LRRK2 in Parkinson disease. JAMA Neurol 75: , 1416–1422. |
[34] | Lubbe SJ , Escott-Price V , Brice A , Gasser T , Hardy J , Heutink P , Sharma M , Wood NW , Nalls M , Singleton AB , Williams NM , Morris HR , International Parkinson’s Disease Genomics Consortium ((2016) ) Is the MC1R variant p.R160W associated with Parkinson’s? Ann Neurol 79: , 159–161. |
[35] | Jansen IE , Bras JM , Lesage S , Schulte C , Gibbs JR , Nalls MA , Brice A , Wood NW , Morris H , Hardy JA , Singleton AB , Gasser T , Heutink P , Sharma M , IPDGC ((2015) ) CHCHD2 and Parkinson’s disease. Lancet Neurol 14: , 678–679. |
[36] | Kim JJ ((2019) ) No genetic evidence for involvement of alcohol dehydrogenase genes in risk for Parkinson’s disease. BioRxiv, https://www.biorxiv.org/content/10.1101/784405v1. |
[37] | Makarious MB , Diez-Fairen M , Krohn L , Blauwendraat C , Bandres-Ciga S , Ding J , Pihlstrom L , Houlden H , Scholz SW , Gan-Or Z ((2019) ) ARSA variants in alpha-synucleinopathies. Brain, doi: 10.1093/brain/awz340 |
[38] | Kia DA , Sabir MS , Ahmed S , Trinh J , Bandres-Ciga S , International Parkinson’s Disease Genomics Consortium ((2018) ) LRP10 in α-synucleinopathies. Lancet Neurol 17: , 1032. |
[39] | Simón-Sánchez J , Heutink P , Gasser T , International Parkinson’s Disease Genomics Consortium ((2015) ) Variation in PARK10 is not associated with risk and age at onset of Parkinson’s disease in large clinical cohorts. Neurobiol Aging 36: , 2907.e2913–2907. |
[40] | Larsson SC , Singleton AB , Nalls MA , Richards JB , International Parkinson’s Disease Genomics Consortium ((2017) ) No clear support for a role for vitamin D in Parkinson’s disease: A Mendelian randomization study. Mov Disord 32: , 1249–1252. |
[41] | Kia DA , Noyce AJ , White J , Speed D , Nicolas A , IPDGC collaborators, Burgess S , Lawlor DA , Davey Smith G , Singleton A , Nalls MA , Sofat R , Wood NW ((2018) ) Mendelian randomization study shows no causal relationship between circulating urate levels and Parkinson’s disease. Ann Neurol 84: , 191–199. |
[42] | Beilina A , Rudenko IN , Kaganovich A , Civiero L , Chau H , Kalia SK , Kalia LV , Lobbestael E , Chia R , Ndukwe K , Ding J , Nalls MA , International Parkinson’s Disease Genomics Consortium; North American Brain Expression Consortium, Olszewski M , Hauser DN , Kumaran R , Lozano AM , Baekelandt V , Greene LE , Taymans JM , Greggio E , Cookson MR ((2014) ) Unbiased screen for interactors of leucine-rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc Natl Acad Sci U S A 111: , 2626–2631. |
[43] | Jansen IE , Ye H , Heetveld S , Lechler MC , Michels H , Seinstra RI , Lubbe SJ , Drouet V , Lesage S , Majounie E , Gibbs JR , Nalls MA , Ryten M , Botia JA , Vandrovcova J , Simon-Sanchez J , Castillo-Lizardo M , Rizzu P , Blauwendraat C , Chouhan AK , Li Y , Yogi P , Amin N , van Duijn CM , International Parkinson’s Disease Genetics Consortium, Morris HR , Brice A , Singleton AB , David DC , Nollen EA , Jain S , Shulman JM , Heutink P ((2017) ) Discovery and functional prioritization of Parkinson’s disease candidate genes from large-scale whole exome sequencing. Genome Biol 18: , 22. |
[44] | Nalls MA , Bras J , Hernandez DG , Keller MF , Majounie E , Renton AE , Saad M , Jansen I , Guerreiro R , Lubbe S , Plagnol V , Gibbs JR , Schulte C , Pankratz N , Sutherland M , Bertram L , Lill CM , DeStefano AL , Faroud T , Eriksson N , Tung JY , Edsall C , Nichols N , Brooks J , Arepalli S , Pliner H , Letson C , Heutink P , Martinez M , Gasser T , Traynor BJ , Wood N , Hardy J , Singleton AB , International Parkinson’s Disease Genomics Consortium (IPDGC); Parkinson’s Disease meta-analysis consortium ((2015) ) NeuroX, a fast and efficient genotyping platform for investigation of neurodegenerative diseases. Neurobiol Aging 36: , 1605.e1607–1612. |
[45] | Blauwendraat C , Faghri F , Pihlstrom L , Geiger JT , Elbaz A , Lesage S , Corvol J-C , May P , Nicolas A , Abramzon Y , Murphy NA , Gibbs JR , Ryten M , Ferrari R , Bras J , Guerreiro R , Williams J , Sims R , Lubbe S , Hernandez DG , Mok KY , Robak L , Campbell RH , Rogaeva E , Traynor BJ , Chia R , Chung SJ , International Parkinson’s Disease Genomics Consortium (IPDGC), COURAGE-PD Consortium, Hardy JA , Brice A , Wood NW , Houlden H , Shulman JM , Morris HR , Gasser T , Krüger R , Heutink P , Sharma M , Simón-Sánchez J , Nalls MA , Singleton AB , Scholz SW ((2017) ) NeuroChip, an updated version of the NeuroX genotyping platform to rapidly screen for variants associated with neurological diseases. Neurobiol Aging 57: , 247.e249–247.e213. |
[46] | Leonard H , Blauwendraat C , Krohn L , Faghri F , Iwaki H , Furgeson G , Day-Williams AG , Stone DJ , International Parkinson’s Disease Genomics Consortium (IPDGC), Singleton AB , Nalls MA , Gan-Or Z ((2018) ) Genetic variability and potential effects on clinical trial outcomes: Perspectives in Parkinson’s disease. BioRxiv, doi: 10.1101/427385. |
[47] | Schekman R , Riley EA ((2019) ) Coordinating a new approach to basic research into Parkinson’s disease. Elife 8: , e51167. |


