
EASE LID 2: A 2-Year Open-Label Trial of Gocovri (Amantadine) Extended Release for Dyskinesia in Parkinson’s Disease
Abstract
Background:
Gocovri® (amantadine) extended release capsules are approved for the treatment of dyskinesia in patients with Parkinson’s disease (PD) receiving levodopa-based therapy.
Objective:
To evaluate the long-term safety, tolerability, and efficacy of Gocovri in patients with PD experiencing levodopa-induced dyskinesia.
Methods:
In this 2-year open-label trial, patients completing double-blind Gocovri clinical trials or excluded from prior trials because of deep-brain stimulation (DBS) received Gocovri 274 mg once daily at bedtime. The primary objective was to evaluate long-term safety and tolerability. In addition, dyskinesia and OFF time were assessed using Part IV (Motor Complications) scores on the Movement Disorder Society–Unified Parkinson’s Disease Rating Scale (MDS-UPDRS).
Results:
Among 223 enrolled patients (mean PD duration, 11.7 years; mean levodopa use, 9.3 years), 75.8% completed 1 year of treatment and 57.8% completed the trial, with a median treatment duration of 1.9 years. Common adverse events were fall (32.7%), hallucination (24.2%), peripheral edema (16.1%), constipation (13.5%), and urinary tract infection (10.3%); 31 patients (13.9%) discontinued because of adverse events considered related to study drug. At baseline, MDS-UPDRS Part IV scores were lower for patients continuing Gocovri (mean, 6.5 points) than for previous placebo (9.4) or DBS groups (10.5) but were similar for all groups by week 8 (6.3, 6.2, 6.4, respectively), and remained low for the duration of the trial (at week 100: 6.9, 7.3, 7.0, respectively).
Conclusions:
In patients with PD, Gocovri showed long-term safety and tolerability consistent with double-blind trial findings, and durable reduction in motor complications (dyskinesia and OFF time).
INTRODUCTION
Levodopa-induced dyskinesias (LID) and OFF episodes most frequently occur in the morning and throughout the daytime in patients with Parkinson’s disease (PD) [1]. Gocovri® (Adamas Pharmaceuticals, Inc., Emeryville, CA) is an extended release amantadine that was specifically designed to provide high drug concentrations when motor complications are most prevalent [2, 3]. With bedtime Gocovri administration, amantadine concentrations rise slowly overnight and near their peak upon awakening (before the patient’s first levodopa dose); high concentrations are then maintained throughout the day and begin to decline in the evening, with the intent of minimizing effects on sleep [3]. This drug concentration–time profile is a unique feature of the Gocovri product.
In phase 3 placebo-controlled trials lasting up to 25 weeks and enrolling approximately 200 PD patients, Gocovri 274 mg (corresponding to 340 mg of amantadine HCl) once daily at bedtime significantly reduced dyskinesia, with the additional benefit of significantly reducing OFF time. The Gocovri safety profile was consistent with the types of adverse events (AEs) known to occur with amantadine immediate-release (amantadine IR) treatment [4–7]. Based on these data, Gocovri was approved by the United States Food and Drug Administration in 2017, and is the only medication indicated for the treatment of dyskinesia in patients with PD receiving levodopa-based therapy, with or without concomitant dopaminergic medications.
This open-label trial was undertaken to assess long-term safety and durability of Gocovri effects, in a broader patient population, under circumstances more relevant to a “real-world” clinical setting. Interim analyses have been published [8, 9]. Here, we report the final results of this 2-year trial.
METHODS
Trial participants
Patients were recruited after completing participation in any of 3 double-blind Gocovri clinical trials: EASED, EASE LID, or EASE LID 3 [4, 5, 12]. Patients ineligible for previous Gocovri trials because they were receiving deep brain stimulation (DBS) were also recruited. All patients were required to have a diagnosis of PD by United Kingdom Parkinson’s Disease Society Brain Bank criteria [10] and a history of peak-dose LID that the patient and investigator felt might benefit from specific treatment. Patients were also required to be on a stable regimen of antiparkinson medications including a levodopa preparation administered at least 3 times daily for at least the previous 30 days. Other key inclusion criteria required all patients to be ambulatory while ON, with or without an aid such as a walker or cane. Key exclusion criteria included orthostatic hypotension, major psychiatric disorders, and renal impairment (an estimated glomerular filtration rate [eGFR] less than 50 mL/min/1.73 m2), as amantadine is renally eliminated.
Enrolled patients were assigned to the following protocol-defined groups:
• Gocovri-randomized patients from a double-blind clinical trial (EASE LID or EASE LID 3) who entered the present trial without interruption (Continuing-Gocovri group).
• Placebo-randomized patients from a double-blind clinical trial (EASE LID or EASE LID 3) who entered the present trial without interruption (Previous-Placebo group).
• Patients who completed double-blind treatment in a Gocovri clinical trial (EASED, EASE LID, or EASE LID 3) but did not enroll directly in the present trial (Participation-Gap group).
• Patients who were previously ineligible for participation in double-blind Gocovri trials because of DBS surgery (DBS group).
For patients who entered this open-label study directly from a double-blind trial, previous study drug assignment was not unblinded at enrollment. Safety and efficacy analyses were also completed for a subgroup of 32 patients who were being treated with amantadine IR at the time of enrollment in the present trial and switched directly to open-label Gocovri (Previous-Amantadine IR subgroup) [9]. These patients came from either the Participation-Gap (n = 8) or the DBS (n = 24) group.
Trial design
For patients transitioning without interruption from a prior double-blind trial the final double-blind Movement Disorder Society-Unified Parkinson Disease Rating Scale (MDS-UPDRS) served as the prestudy efficacy assessment. These patients then began the prescribed 1-week double-blind study drug taper from 274 mg to 137 mg Gocovri (or remained on placebo), before starting this trial. In the present trial, patients were treated for up to 101 weeks with open-label Gocovri, which was taken once daily at bedtime. For all patients, including those transitioning immediately from a previous trial, the Gocovri daily dosage was started at 137 mg (1 capsule) during week 1, and was increased to the maintenance dosage of 274 mg (2 capsules) starting at week 2 through week 100, and tapered back to 137 mg (1 capsule) for the final week (week 101). Patients were allowed to change their PD medications (including levodopa dosage) as needed during the trial. Use of other amantadine formulations (other than study medication) was not permitted. Safety and efficacy assessments were conducted pre-study (1 to 14 days prior to baseline) and at weeks 8, 16, 28, 40, 52, 64, 76, 88, and 100 (or early termination), with additional safety assessments at baseline (week 0), week 4, and week 103 (or 2 weeks following early termination).
Safety measures and analyses
Throughout the trial, investigators were responsible for recording all directly observed and patient-reported AEs, including serious AEs (SAEs). AEs were coded according to the Medical Dictionary for Regulatory Activities (MedDRA) version 17.0. For each AE, the severity (mild, moderate, or severe) and study-drug relationship (related or not related) of the event were categorized by the investigator.
Hematology, serum chemistry, and urinalysis tests were performed at screening and all follow-up visits except the post-treatment safety follow-up visit. A complete physical examination was performed at screening and weeks 16, 52, and 100 (or early termination), with a targeted physical examination at other visits. Vital signs were measured at all visits.
All patients who received at least 1 dose of study drug were included in safety analyses. AE incidence patterns were summarized descriptively by analysis group and also by age (≥65 vs <65 years) and renal function (eGFR 50–59, 60–89, and ≥90). Additional analyses, including time to onset, were conducted for events of special interest, including hallucinations (all MedDRA preferred terms containing hallucination) and neuropsychiatric events (combining all MedDRA System Organ Class nervous system and psychiatric disorder events).
Efficacy measures and analyses
Efficacy was assessed using the MDS-UPDRS [11]. Changes in dyskinesia and OFF signs or symptoms were captured as efficacy outcomes by MDS-UPDRS Part IV scores and its six items (each item is rated by increasing severity from 0 [none] to 4 [severe], for a maximum total score of 24). MDS-UPDRS Parts I, II, and III were also assessed as a measure of Parkinson’s disease status.
All observed cases were included in efficacy analyses. Data were analyzed descriptively.
Protocol approvals, registrations, and patient consents
The trial protocol and patient-consent forms were approved by each site’s institutional review board or research ethics board, or by an independent ethics committee. All patients provided written informed consent prior to any trial procedures. The trial’s ClinicalTrials.gov registry number is NCT02202551.
Data availability
Results from this trial are publicly available at ClinicalTrials.gov. Individual participant data will not be shared.
RESULTS
Trial participants
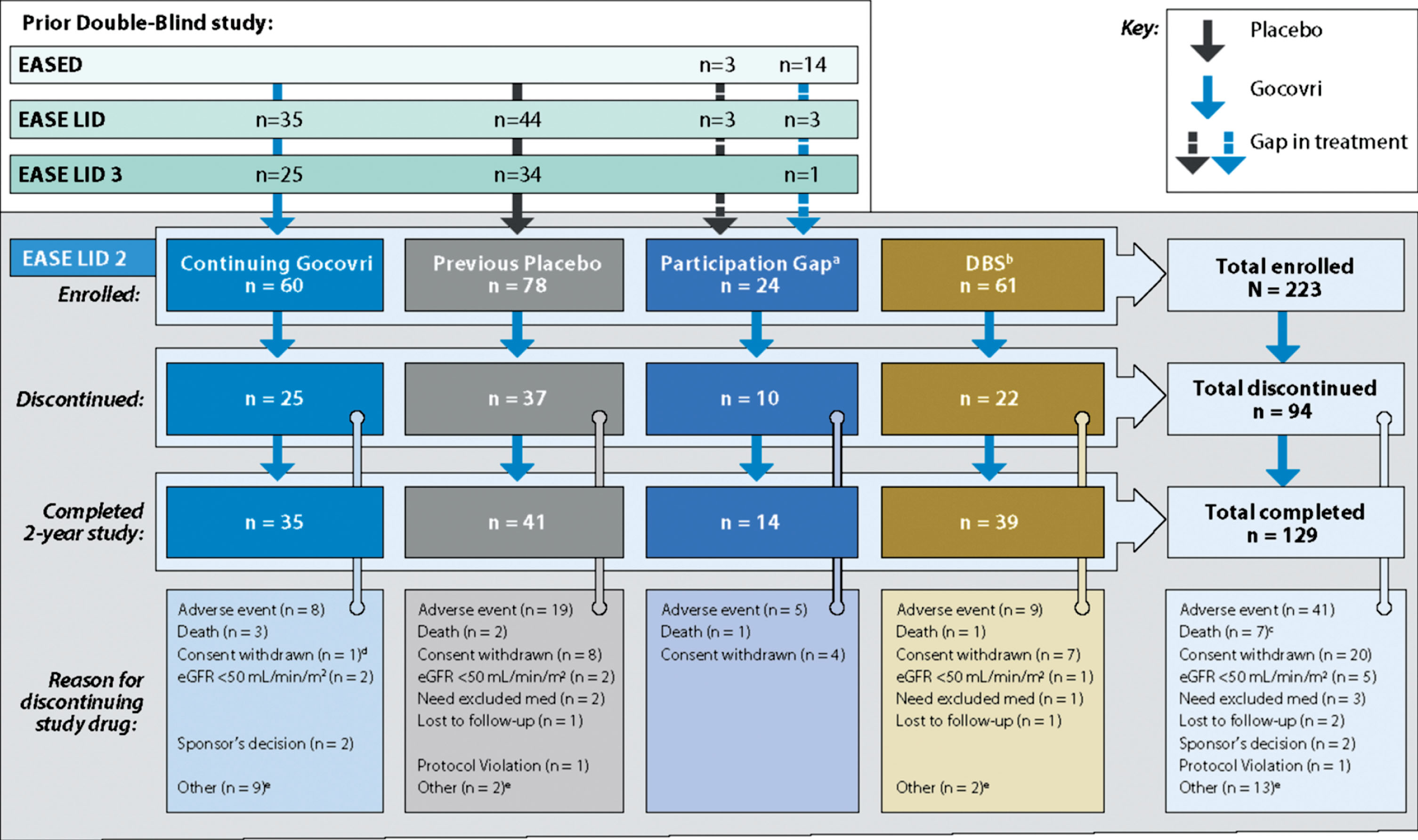
Between July 28, 2014, and March 10, 2016, 243 patients were screened at 67 sites, and 223 were enrolled, including 138 who transitioned directly from double-blind treatment (60 with Gocovri and 78 with placebo), 61 with prior DBS surgery, and 24 with time gaps after their prior trial [4, 5, 12].
The characteristics of enrolled patients are summarized by group in Table 1. Most patients were male (58.7%) and white (93.3%), with a mean (SD) age of 63.7 (9.3) years. Patients had a mean (SD) PD duration of 11.8 (5.3) years, had been receiving levodopa treatment for 9.3 (4.7) years, and had reported dyskinesia for 5.3 (3.7) years. Patients were taking a median of 7 non-study-drug medications at baseline; 84% were taking at least 2 PD medications (including levodopa), and 55.6% were taking 3 or more PD medications. All patients were receiving levodopa, with a mean (SD) dosage of 756 (456.5) mg/day (median, 650 mg/day). Other concomitant PD medications are listed in Table 1. The mean duration of amantadine IR treatment for the 32 patients using it immediately prior to enrollment was 2.5 years, at a mean dose equivalent of 221 mg/day (median; 242 mg/day) of amantadine (corresponding to amantadine HCl ∼275 mg/day; median 300 mg/day). Demographic characteristics were similar among groups, except that patients in the DBS group were younger than patients in the other groups but had longer durations of PD, levodopa treatment, and dyskinesia.
Table 1
Patients’ baseline characteristics, by group (safety population)
| Variable | Continuing-Gocovri group (N = 60) | Previous-Placebo group (N = 78) | Participation-Gap group (N = 24) | DBS group (N = 61) | All patients (N = 223) |
| Age, mean (SD), years | 64.0 (9.8) | 65.9 (8.8) | 64.4 (7.8) | 60.2 (9.2) | 63.7 (9.3) |
| ≥65, n (%) | 32 (53.3) | 48 (61.5) | 12 (50.0) | 22 (36.1) | 114 (51.1) |
| Gender, males, n (%) | 34 (56.7) | 45 (57.7) | 11 (45.8) | 41 (67.2) | 131 (58.7) |
| Time since PD diagnosis, mean (SD), years | 11.3 (4.5) | 10.1 (4.1) | 11.0 (4.7) | 14.7 (6.3) | 11.8 (5.3) |
| Duration of dyskinesia, mean (SD), years | 4.6 (3.4) | 4.0 (2.5) | 4.8 (2.7) | 8.0 (4.4) | 5.3 (3.7) |
| Duration of levodopa treatment, mean (SD), years | 8.7 (3.8) | 7.9 (4.0) | 8.5 (5.0) | 11.9 (5.4) | 9.3 (4.7) |
| Levodopa dosage, mean (SD), mg/day | 805 (418) | 750 (558) | 685 (309) | 742 (399) | 756 (457) |
| Levodopa equivalent dosagea (mg/d), mean (SD) | 1049 (459) | 1015 (600) | 913 (444) | 1012 (510) | 1012 (522) |
| Use of other PD drugs, n (%) | |||||
| Dopamine agonists | 32 (53.3) | 46 (59.0) | 14 (58.3) | 41 (67.2) | 133 (59.6) |
| MAO-B inhibitors | 28 (46.7) | 37 (47.4) | 16 (66.7) | 16 (26.2) | 97 (43.5) |
| COMT inhibitorsb | 25 (41.7) | 31 (39.7) | 7 (29.2) | 27 (44.3) | 90 (40.4) |
| Anticholinergics | 0 | 3 (3.8) | 0 | 1 (1.6) | 4 (1.8) |
| MDS-UPDRS Part IV Total Score, mean (SD)c | 6.5 (3.4) | 9.4 (3.2) | 9.8 (3.9) | 10.5 (2.8) | 8.9 (3.5) |
| Item 4.1 (Time with dyskinesia) | 1.6 (1.1) | 2.0 (1.1) | 1.9 (1.0) | 1.9 (0.9) | 1.8 (1.0) |
| Item 4.2 (Impact of dyskinesia) | 0.9 (1.0) | 1.8 (0.9) | 1.8 (1.1) | 2.4 (1.0) | 1.7 (1.0) |
| Item 4.3 (Time in OFF state) | 0.9 (0.6) | 1.3 (0.6) | 1.3 (0.7) | 1.3 (0.6) | 1.2 (0.6) |
| Item 4.4 (Impact of fluctuations) | 1.4 (1.2) | 1.9 (1.3) | 2.1 (1.1) | 2.4 (1.1) | 1.9 (1.3) |
| Item 4.5 (Complexity of fluctuations) | 1.2 (1.0) | 1.5 (1.1) | 1.6 (1.2) | 1.6 (1.0) | 1.4 (1.0) |
| Item 4.6 (Time with painful dystonia) | 0.6 (1.0) | 0.9 (1.1) | 1.1 (1.6) | 1.0 (1.3) | 0.8 (1.2) |
| eGFR, mean (SD), (mL/min/1.73m2) | 88.7 (24.3) | 92.2 (19.6)d | 96.3 (20.5)e | 92.1 (20.0) | 91.6 (21.2) |
| 50–59, n (%) | 4 (6.7) | 2 (2.6) | 0 | 1 (1.6) | 7 (3.1) |
| 60–89, n (%) | 31 (51.7) | 37 (47.4) | 10 (41.7) | 27 (44.3) | 105 (47.1) |
| ≥90, n (%) | 25 (41.7) | 37 (47.4) | 13 (54.2) | 33 (54.1) | 108 (48.4) |
| Missing, n (%) | 0 | 2 (2.6) | 1 (4.2) | 0 | 3 (1.3) |
aAn estimate of the overall strength of a patient’s PD pharmacotherapy, calculated as the amount of levodopa (taken with carbidopa) expected to have a similar effect. bIncluding entacapone taken in combination with levodopa/carbidopa. cSample sizes are: Continuing-Gocovri n = 60; Previous Placebo n = 78; Participation Gap n = 16; DBS n = 60; Total n = 214. Items are scored on an anchored, rank-order scale from 0 (none), to 1 (slight), 2 (mild), 3 (moderate), or 4 (severe). dn = 76. en = 23. COMT, catechol-O-methyltransferase; DBS, deep brain stimulation; eGFR, estimated glomerular filtration rate; MAO-B, monoamine oxidase B; MDS-UPDRS, Movement Disorder Society–Unified Parkinson’s Disease Rating Scale; PD, Parkinson’s disease; SD, standard deviation.
Patient disposition is presented in Fig. 1. Among the 223 enrolled patients, 75.8% of patients completed at least 1 year (52 weeks) of treatment and 129 (57.8%) completed the 2-year trial. The median duration of patients’ exposure to open-label Gocovri was 704 days (1.9 years).
Fig.1
Study design and patient disposition. aEight patients in the Participation-Gap group switched directly from amantadine IR to open-label Gocovri treatment at enrollment. bTwenty-four patients in the DBS group switched directly from amantadine IR to open-label Gocovri treatment at enrollment. cTwo of the 9 patients who died during the trial discontinued study drug due to AEs and died subsequently (4 and 15 days later). dThis patient who discontinued for unwilling to proceed/consent withdrawn was noted to have an AE leading to withdrawal in safety reporting. eDiscontinuations for “other” reasons were mostly administrative or resulted from other medical issues. In the Continuing-Gocovri group the discontinuations were: site closure (n = 3), underwent DBS surgery (n = 3), dyskinesia upon taper (n = 1), physician decision (n = 1), unable to complete visits per protocol (n = 1). In the Placebo Group they were: lack of efficacy (n = 1), patient relocation (n = 1), DBS group: site closure (n = 1), worsening dyskinesia (n = 1). AE, adverse events; DBS, deep brain stimulation; eGFR, estimated glomerular filtration rate; IR, immediate-release.
Adverse events
Most patients reported at least one AE (91.9%), with fall (32.7%), hallucination (24.2%), peripheral edema (16.1%), constipation (13.5%), and urinary tract infection (10.3%) being most commonly reported (Table 2). For 55.6% of patients, one or more AEs were considered study drug-related. The study drug-related AEs with an incidence ≥5% were visual hallucination (18.4%), peripheral edema (10.8%), livedo reticularis (7.6%), dry mouth (7.2%), and fall (6.3%).
Table 2
Adverse events and discontinuations due to adverse events (safety population)
| Adverse events, n (%) | |||||
| Continuing-Gocovri group (N = 60) | Previous-Placebo group (N = 78) | Participation-Gap group (N = 24) | DBS group (N = 61) | All patients (N = 223) | |
| Summary | |||||
| Any AE | 57 (95.0%) | 70 (89.7%) | 23 (95.8%) | 55 (90.2%) | 205 (91.9%) |
| Study-drug related | 31 (51.7%) | 45 (57.7%) | 16 (66.7%) | 32 (52.5%) | 124 (55.6%) |
| Any SAE | 16 (26.7%) | 21 (26.9%) | 6 (25.0%) | 17 (27.9%) | 60 (26.9%) |
| Study-drug related | 1 (1.7%) | 3 (3.8%) | 0 | 1 (1.6%) | 5 (2.2%) |
| Any leading to study-drug discontinuation or death | 12 (20.0%) | 21 (26.9%) | 6 (25.0%) | 10 (16.4%) | 49 (22.0%) |
| Study-drug related | 4 (6.7%) | 15 (19.2%) | 4 (16.7%) | 8 (13.1%) | 31 (13.9%) |
| By preferred terma | |||||
| Fall | 13 (21.7%) | 29 (37.2%) | 8 (33.3%) | 23 (37.7%) | 73 (32.7%) |
| Hallucination | 15 (25.0%) | 24 (30.8%) | 5 (20.8%) | 10 (16.4%) | 54 (24.2%) |
| Visual | 14 (23.3%) | 24 (30.8%) | 4 (16.7%) | 10 (16.4%) | 52 (23.3%) |
| Auditory | 1 (1.7%) | 1 (1.3%) | 1 (4.2%) | 2 (3.3%) | 5 (2.2%) |
| Peripheral edema | 10 (16.7%) | 12 (15.4%) | 2 (8.3%) | 12 (19.7%) | 36 (16.1%) |
| Constipation | 9 (15.0%) | 12 (15.4%) | 6 (25%) | 3 (4.9%) | 30 (13.5%) |
| Urinary tract infection | 7 (11.7%) | 8 (10.3%) | 3 (12.5%) | 5 (8.2%) | 23 (10.3%) |
| Dizziness | 4 (6.7%) | 10 (12.8%) | 5 (20.8%) | 3 (4.9%) | 22 (9.9%) |
| Nausea | 7 (11.7%) | 8 (10.3%) | 6 (25.0%) | 1 (1.6%) | 22 (9.9%) |
| Insomnia | 8 (13.3%) | 5 (6.4%) | 4 (16.7%) | 4 (6.6%) | 21 (9.4%) |
| Livedo reticularis | 6 (10.0%) | 5 (6.4%) | 3 (12.5%) | 6 (9.8%) | 20 (9.0%) |
| ON and OFF phenomenon | 7 (11.7%) | 4 (5.1%) | 4 (16.7%) | 3 (4.9%) | 18 (8.1%) |
| Dry mouth | 4 (6.7%) | 6 (7.7%) | 4 (16.7%) | 3 (4.9%) | 17 (7.6%) |
aIncludes all preferred terms with an incidence ≥7.5% among all patients. AE, adverse event; DBS, deep brain stimulation; SAE, serious adverse event.
Most AEs were rated as mild or moderate in intensity. Sixty-six patients (29.6%) reported at least one AE rated as severe in intensity. AEs rated as severe in more than 2 patients were fall (9 patients, 4.0%), visual hallucinations (6 patients, 2.7%), and worsening PD symptoms (3 patients, 1.3%).
Sixty patients (26.9%) had one or more recorded SAEs, most commonly fractures, typically subsequent to falls (5.8%), none of which were considered treatment-related. Five patients (2.2%) had treatment-related SAEs, including confusional state, urinary tract infection, worsening PD symptoms, postoperative confusion, and suicidal ideation. A listing of SAEs occurring in at least 2 patients is provided in Table 3.
Table 3
Serious adverse events (safety population)
| Preferred terma | Adverse event, n (%) | ||||
| Continuing-Gocovri group (N = 60) | Previous-Placebo group (N = 78) | Participation-Gap group (N = 24) | DBS group (N = 61) | All patients (N = 223) | |
| Fractures (pooled) | 2 (3.3%) | 7 (9.0%) | 1 (4.2%) | 3 (4.9%) | 13 (5.8%) |
| PD symptoms worse | 1 (1.7%) | 1 (1.3%) | 0 | 3 (4.9%) | 5 (2.2%) |
| Confusional state | 1 (1.7%) | 1 (1.3%) | 0 | 1 (1.6%) | 3 (1.3%) |
| Acute MI | 1 (1.7%) | 0 | 0 | 1 (1.6%) | 2 (0.9%) |
| Constipation | 1 (1.7%) | 1 (1.3%) | 0 | 0 | 2 (0.9%) |
| Intestinal obstruction | 1 (1.7%) | 1 (1.3%) | 0 | 0 | 2 (0.9%) |
| MI | 0 | 1 (1.3%) | 0 | 1 (1.6%) | 2 (0.9%) |
| Rhabdomyolysis | 0 | 1 (1.3%) | 0 | 1 (1.6%) | 2 (0.9%) |
| Sepsis | 0 | 0 | 1 (4.2%) | 1 (1.6%) | 2 (0.9%) |
| Volvulus | 1 (1.7%) | 0 | 0 | 1 (1.6%) | 2 (0.9%) |
aThe list includes all preferred terms reported in more than 1 patient. Note: The reasons provided for the 9 deaths that occurred during the trial were: “natural causes” (study day 661), cardiac arrest (day 190), myocardial infarction (day 55), arrhythmia (day 473), pneumonia (day 99), Clostridium difficile sepsis (day 425), Pseudomonal sepsis (day 570), septic shock (day 508), and sepsis (day 414). None were considered related to study drug by the investigators. DBS, deep brain stimulation; MI, myocardial infarction; PD, Parkinson’s disease.
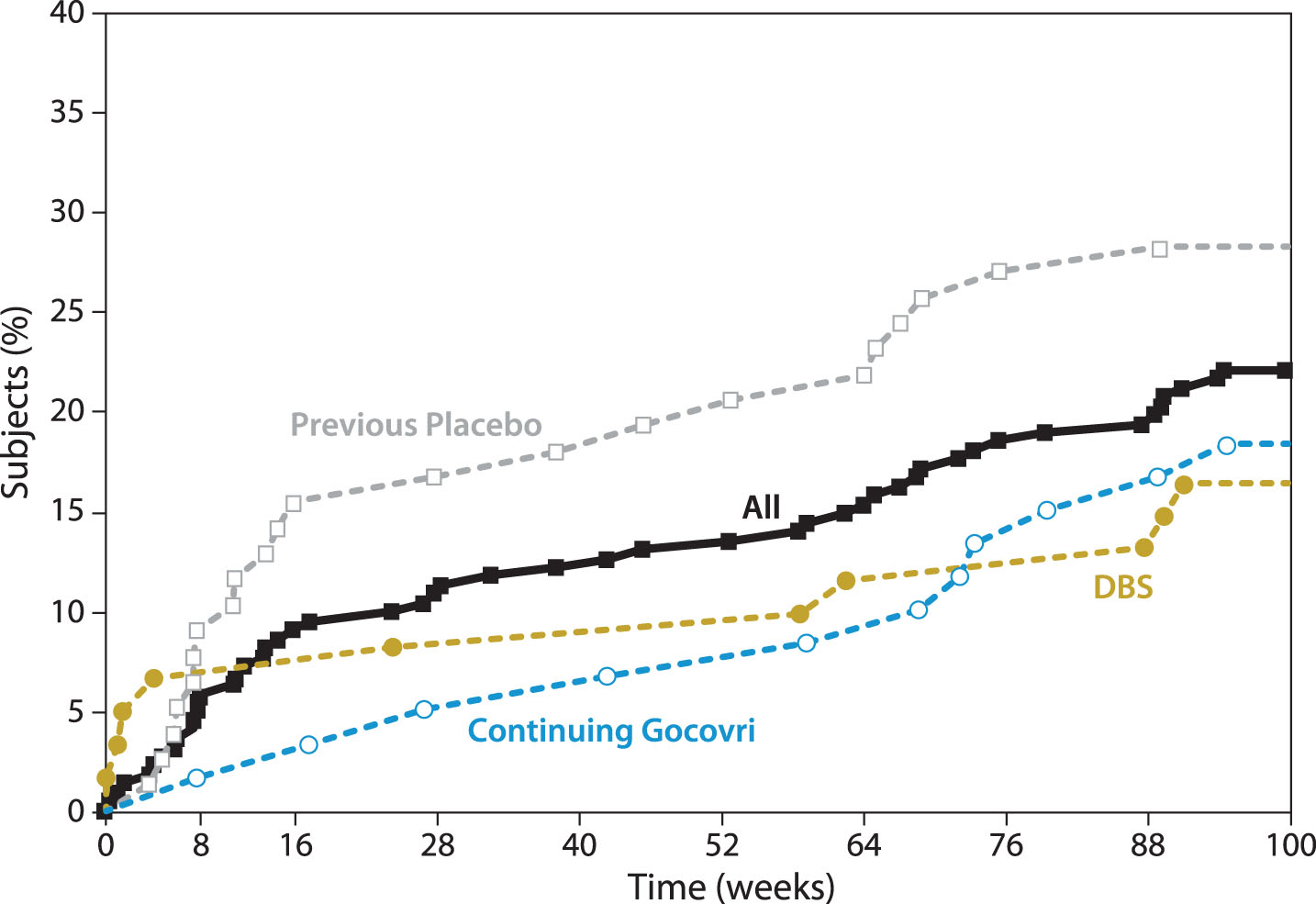
Forty-nine patients (22%) had AEs that eventually led to trial withdrawal or death (Table 4), with an additional 5 discontinuing due to low eGFR (protocol-mandated withdrawal). Nine patients (4%) died during the study; none (0%) of the deaths were considered by the investigator as related to study drug. Figure 2 graphs the timing of withdrawals due to AEs. Consistent with findings of the published interim analysis [8], in the first months of the trial, discontinuation for AEs occurred more frequently among patients who initiated Gocovri in this trial compared with those who continued Gocovri treatment from the double-blind trials. Hallucinations were also more common early in the trial among patients naïve to Gocovri at enrollment. Among the 54 patients who experienced hallucinations during EASE LID 2, the median time to onset was 91 days (range 7–663). For those patients who experienced hallucinations in the Continuing-Gocovri group (n = 15), the median time to onset was 235 days, compared with 51 days among the Previous-Placebo group (n = 24). The median time to onset of hallucination for patients in the DBS group and Previous-Amantadine IR subgroup were 158 and 154 days, respectively.
Table 4
Adverse events leading to study-drug discontinuation (safety population)
| Preferred terma | Adverse event, n (%) | ||||
| Continuing-Gocovri group (N = 60) | Previous-Placebo group (N = 78) | Participation-Gap group (N = 24) | DBS group (N = 61) | All patients (N = 223) | |
| Hallucination, visual | 1 (1.7%) | 4 (5.1%) | 0 | 1 (1.6%) | 6 (2.7%) |
| Fall | 1 (1.7%) | 1 (1.3%) | 1 (4.2%) | 2 (3.3%) | 5 (2.2%) |
| Confusional state | 1 (1.7%) | 0 | 1 (4.2%) | 1 (1.6%) | 3 (1.3%) |
| PD symptoms worse | 1 (1.7%) | 1 (1.3%) | 0 | 0 | 2 (0.9%) |
| Peripheral edema | 0 | 2 (2.6%) | 0 | 0 | 2 (0.9%) |
| Psychotic disorder | 1 (1.7%) | 0 | 0 | 1 (1.6%) | 2 (0.9%) |
| Sleep disorder | 0 | 2 (2.6%) | 0 | 0 | 2 (0.9%) |
aThe list includes all MedDRA preferred terms reported in more than 1 patient. PD, Parkinson’s disease.
Fig.2
Cumulative withdrawal for AEs over 2-year trial (includes patients who died during the study). AEs, adverse events; DBS, deep brain stimulation. Note that Participation-Gap patients were not summarized here due to small sample size.
Among other AEs of specific interest for PD patients, orthostatic hypotension and/or hypotension were reported in 6 patients (2.7%), and impulse control disorders in 5 patients (2.2%), 4 of whom were concomitantly using dopamine agonists. One patient in the Previous-Placebo group attempted suicide, and 1 patient in the DBS group reported suicidal ideation with intent for self-harm. Both patients had a history of anxiety and depression and subsequently withdrew from the trial.
Adverse event risk factors
The incidence of AEs was evaluated by age, per protocol, and renal function as an additional analysis. AEs were more common in older than younger patients, occurring in 96.5% of the 114 patients ≥65 years of age and 87.2% of the 109 patients <65 years of age. All AEs occurring in ≥10% of overall patients occurred more frequently in those ≥65 years of age vs <65 years of age, including fall (42.1% vs 22.9%), hallucinations (35.1% vs 12.8%), peripheral edema (21.9% vs 10.1%), constipation (15.8% vs 11.0%), and urinary tract infection (14.0% vs 6.4%). Other AEs with incidence ≥10% in one age group were dizziness (12.3%) and nausea (11.4%) in patients ≥65 years of age; and insomnia (11.0%), ON and OFF phenomenon (11.0%), livedo reticularis (11.0%), and depression (10.1%) in patients <65 years of age. Patients who completed the trial were generally younger at baseline (mean age 63.5 years; 49.6% ≥65 years) than those who withdrew due to AEs (mean age 66.6 years; 69.4% age ≥65).
For patients with baseline eGFR ≥90, 60–89, or 50–59 mL/min/m2, AEs occurred in 98/108 (90.7%), 97/105 (92.4%), and 7/7 (100%) patients, respectively (53.7%, 57.1%, and 57.1%, respectively, had AEs that were considered study-drug-related). Patients completing the trial had higher baseline eGFR, compared with those who discontinued due to an AE (mean 94.4 vs 84.1 mL/min/1.73m2; with 53.5% vs 38.8% having eGFR ≥90). 16.6% of patients shifted from normal to low eGFR (where low is <75 mL/min/1.73m2 for females and <85 mL/min/1.73m2 for males) during the trial, and 5 patients (2.2%) were withdrawn per protocol because their eGFR fell below 50 mL/min/1.73 m2.
Gocovri dose modification following adverse events
In all, 29 patients (13.0%) had their Gocovri dose reduced to 137 mg because of AEs (3 patients in the Continuing-Gocovri, 14 in the Previous-Placebo, 4 in the Participation-Gap, and 8 in the DBS group). Seventeen (58.6%) of the 29 patients subsequently completed the trial; the other 12 (41.4%) discontinued. The AE most commonly associated with Gocovri dose reduction was hallucination (n = 14).
Efficacy
Mean MDS-UPDRS Part IV total scores are displayed by visit for each patient group in Fig. 3. For those enrolling directly from a double-blind trial, scores from the prior trial are also shown. As previously described in an interim analysis [8], the Continuing-Gocovri group had already experienced a marked mean (SD) improvement of –4.7 (3.8) points in MDS-UPDRS Part IV total score, from a double-blind baseline of 11.2, and therefore exhibited a lower mean score at baseline of the present trial. The changes from open-label baseline for all time points are shown in Table 5.
Fig.3
Time course of mean MDS-UPDRS Part IV total score by group (observed cases). Error bars are standard errors of the mean. Participation-Gap patients were not summarized here due to small sample size. *Data from weeks 12 and 18 of EASE LID are not shown here. **For participants enrolling directly from double-blind Gocovri trials, their final efficacy visit in EASE LID 3 or EASE LID (week 12 or up to week 24) was also their baseline MDS-UPDRS assessment for the open-label trial. For all patients, the baseline MDS-UPDRS assessment was performed between study days –14 and –1. *** The Previous-Amantadine IR subgroup and the prespecified patient groups are not mutually exclusive; the Previous-Amantadine IR subgroup contains 32 patients who were switched directly from amantadine IR to Gocovri at enrollment in EASE LID 2 (24 of whom are from the DBS group and 8 from the Participation-Gap group). At baseline, the patients in this group had a mean 221 mg daily dose of amantadine (corresponding to ~275 mg amantadine HCl) and mean treatment duration of 2.5 years. †Charts show change from Gocovri initiation (ie, change from double-blind baseline for the Continuing-Gocovri group and change from open-label baseline for Previous-Placebo, DBS, and Previous-Amantadine IR groups). AMT, amantadine; BSL, baseline; DBS, deep brain stimulation; IR, immediate-release; MDS-UPDRS, Movement Disorder Society–Unified Parkinson’s Disease Rating Scale.
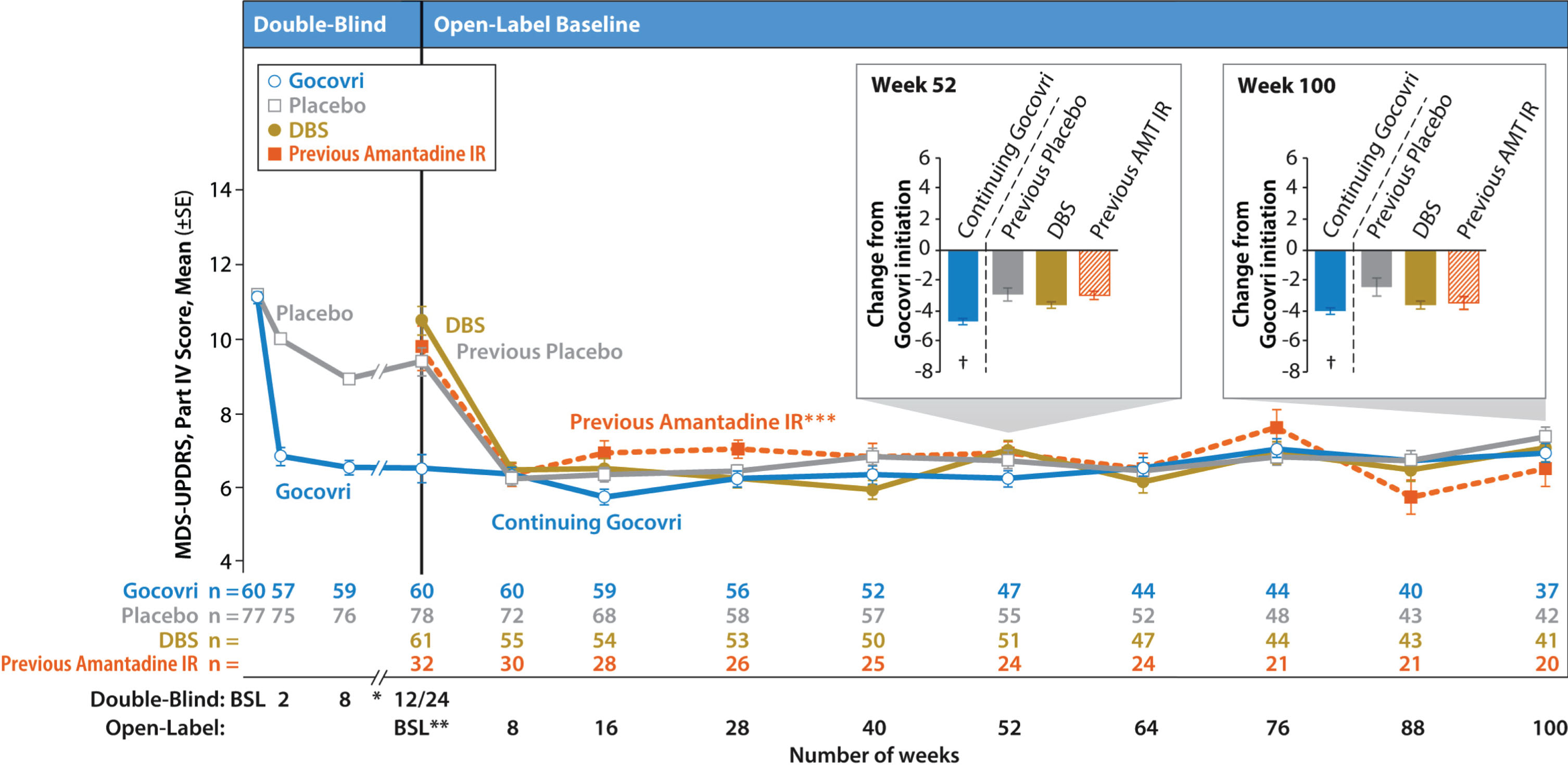
Table 5
Mean changes from open-label baseline in MDS-UPDRS Part IV total score at all observation time points, by group (observed cases)
| Time point/variable | Continuing-Gocovri group | Previous-Placebo group | Participation-Gap group | DBS group |
| Baseline | ||||
| n | 60 | 78 | 16 | 60 |
| Total score, mean (SD) | 6.5 (3.4) | 9.4 (3.2) | 9.8 (3.9) | 10.5 (2.8) |
| Week 8 | ||||
| n | 60 | 72 | 16 | 55 |
| Total score, mean (SD) | 6.3 (3.1) | 6.2 (3.3) | 6.3 (3.7) | 6.4 (3.6) |
| Mean change (SD) | –0.2 (3.2) | –3.4 (3.3) | –3.6 (4.2) | –4.0 (4.2) |
| Week 16 | ||||
| n | 59 | 68 | 15 | 54 |
| Total score, mean (SD) | 5.7 (3.3) | 6.3 (3.3) | 8.1 (4.9) | 6.5 (3.7) |
| Mean change (SD) | –0.8 (3.6) | –3.2 (3.6) | –1.1 (5.0) | –3.9 (3.7) |
| Week 28 | ||||
| n | 56 | 58 | 14 | 53 |
| Total score, mean (SD) | 6.2 (3.4) | 6.4 (3.1) | 7.9 (3.4) | 6.2 (3.6) |
| Mean change (SD) | –0.3 (3.6) | –3.3 (3.1) | –1.4 (3.8) | –4.4 (3.8) |
| Week 40 | ||||
| n | 52 | 57 | 14 | 50 |
| Total score, mean (SD) | 6.3 (3.7) | 6.8 (3.4) | 6.4 (3.6) | 5.9 (3.6) |
| Mean change (SD) | 0 (4.0) | –2.8 (3.4) | –2.9 (4.8) | –4.7 (4.1) |
| Week 52 | ||||
| n | 47 | 55 | 12 | 51 |
| Total score, mean (SD) | 6.2 (3.2) | 6.7 (3.3) | 7.4 (3.5) | 7.0 (3.3) |
| Mean change (SD) | 0.2 (3.6) | –2.9 (3.6) | –2.5 (4.4) | –3.6 (3.6) |
| Week 64 | ||||
| n | 44 | 52 | 12 | 47 |
| Total score, mean (SD) | 6.5 (3.0) | 6.4 (3.0) | 8.0 (3.1) | 6.1 (4.0) |
| Mean change (SD) | 0.4 (3.8) | –3.3 (3.4) | –1.9 (4.3) | –4.5 (4.0) |
| Week 76 | ||||
| n | 44 | 48 | 11 | 44 |
| Total score, mean (SD) | 7.0 (3.8) | 6.8 (3.5) | 6.3 (4.6) | 6.9 (4.0) |
| Mean change (SD) | 0.9 (4.2) | –2.9 (3.7) | –2.7 (3.9) | –3.7 (3.7) |
| Week 88 | ||||
| n | 40 | 43 | 11 | 43 |
| Total score, mean (SD) | 6.7 (3.4) | 6.7 (3.4) | 5.3 (4.0) | 6.4 (3.6) |
| Mean change (SD) | 0.4 (3.8) | –2.8 (3.6) | –3.7 (4.7) | –4.3 (3.6) |
| Week 100 | ||||
| n | 37 | 42 | 10 | 41 |
| Total score, mean (SD) | 6.9 (3.1) | 7.3 (4.0) | 5.6 (3.7) | 7.0 (4.4) |
| Mean change (SD) | 0.4 (3.3) | –2.4 (4.2) | –3.6 (3.7) | –3.6 (3.9) |
| Last on-study measurement | ||||
| n | 60 | 77 | 16 | 58 |
| Total score, mean (SD) | 7.2 (4.1) | 7.3 (3.7) | 7.5 (4.0) | 7.0 (4.2) |
| Mean change (SD) | 0.7 (4.3) | –2.1 (3.9) | –2.3 (4.4) | –3.5 (3.9) |
DBS, deep brain stimulation; MDS-UPDRS, Movement Disorder Society–Unified Parkinson’s Disease Rating Scale; SD, standard deviation.
For the Continuing-Gocovri group, the efficacy achieved during double-blind treatment appeared sustained throughout the present trial, with mean MDS-UPDRS Part IV decreases from original double-blind baseline of –4.7 (3.5) at week 52 of this trial and –4.0 (3.2) at week 100 (total treatment duration from double-blind entry to open-label completion was up to 128 weeks); similar changes in scores were seen at other visits (Fig. 3). By contrast, the groups initiating Gocovri in this trial showed marked improvement in MDS-UPDRS Part IV scores, achieving a level similar to patients in the Continuing-Gocovri group, by Week 8 (the first scheduled efficacy assessment). These improvements were maintained at subsequent visits throughout the 2-year trial (Fig. 3, Table 5).
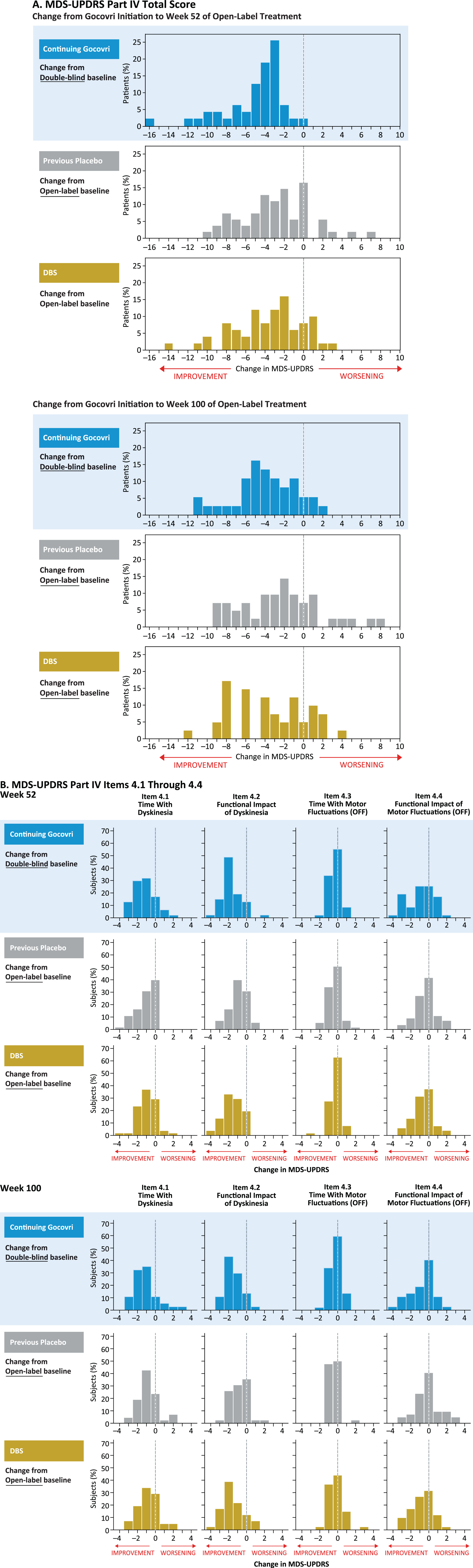
A categorical distribution of patients experiencing improvement, no change, or worsening in MDS-UPDRS Part IV total score is shown in Fig. 4A; for the Continuing-Gocovri group, changes are shown from original double-blind baseline. At week 100 for the Continuing-Gocovri, Previous-Placebo, Participation-Gap, and DBS groups, respectively, the proportions with at least a 1-point improvement from pre-Gocovri baseline were 86.5%, 71.4%, 80.0%, and 75.6%. The proportions with at least a 2-point improvement were 75.7%, 61.9%, 50.0%, and 63.4%, respectively. Evaluation of the individual MDS-UPDRS Part IV subscales 4.1, 4.2, 4.3, and 4.4, measuring time spent with dyskinesia, functional impact of dyskinesia, time spent with OFF, and functional impact of OFF, respectively, showed the observed improvements derived from reductions in both dyskinesia and OFF time (Fig. 4B). At week 100, the overall change in these subscores remained below pre-Gocovri baseline, with the largest reductions seen for functional impact of dyskinesia. Evaluation of changes from pre-Gocovri baseline to last trial visit (including patients who did not complete the trial) showed a similar result. For patients in the Continuing-Gocovri group, this improvement represents maintenance of effect through 113 (EASE LID 3) or 125 weeks (EASE LID) of treatment.
Fig.4
MDS-UPDRS Part IV: change from Gocovri initiation to weeks 52 and 100 of open-label treatment. DBS, deep-brain stimulation; MDS-UPDRS, Movement Disorder Society–Unified Parkinson’s Disease Rating Scale. Participation-Gap group not shown due to small size of group.
Prior amantadine IR users
Among the previous amantadine IR users, 24 (75.0%) completed Week 52 and 20 (62.5%) completed the Week 100 visit. Improvement in these patients was sustained throughout the duration of Gocovri use; mean (SD) MDS-UPDRS Part IV changes from baseline at these visits were –3.0 (3.2) and –3.5 (4.2), respectively. Including patients who dropped out, the overall mean change from baseline to last on-study assessment was –2.6 (4.3); n = 32.
Levodopa dose adjustments
Trial investigators could adjust their patients’ levodopa dosages based on clinical judgment. The mean levodopa daily dose among all enrolled patients rose from 756 mg/day at baseline to 840 mg/day at last on-study measurement. Among 134 patients who completed 100 weeks in the study, 44 (32.8%) were taking a higher levodopa dose, 69 (51.5%) the same, and 21 (15.6%) a lower dose than at baseline (Fig. 5). Analysis at weeks 52 and 100 showed that mean MDS-UPDRS Part IV scores were improved vs baseline for all 3 of these groups, regardless of the directionality of levodopa dose adjustment.
Fig.5
Changes in levodopa usage status at weeks 52 and 100. Levodopa dose data were available for 168 patients completing the week 52 visit and 134 patients completing the week 100 visit. Recorded levodopa doses were the same as baseline for 109 patients (64.9%) at week 52 and 69 patients (51.5%) at week 100 (not shown on graph). Mean (SD) changes from baseline in MDS-UPDRS Part IV total score to weeks 52 and 100, respectively, were –0.9 (4.0) and –1.4 (4.6) for those who had an increased levodopa dose (n = 39 and n = 44), –2.8 (4.0) and –2.6 (4.2) for those who had no change (n = 105 and n = 66), and –2.3 (2.9) and –1.8 (2.8) for those who had a decreased levodopa dose (n = 19 and n = 20) compared to baseline. MDS-UPDRS assessments were not available for 5 patients at week 52 and for 4 patients at week 100.
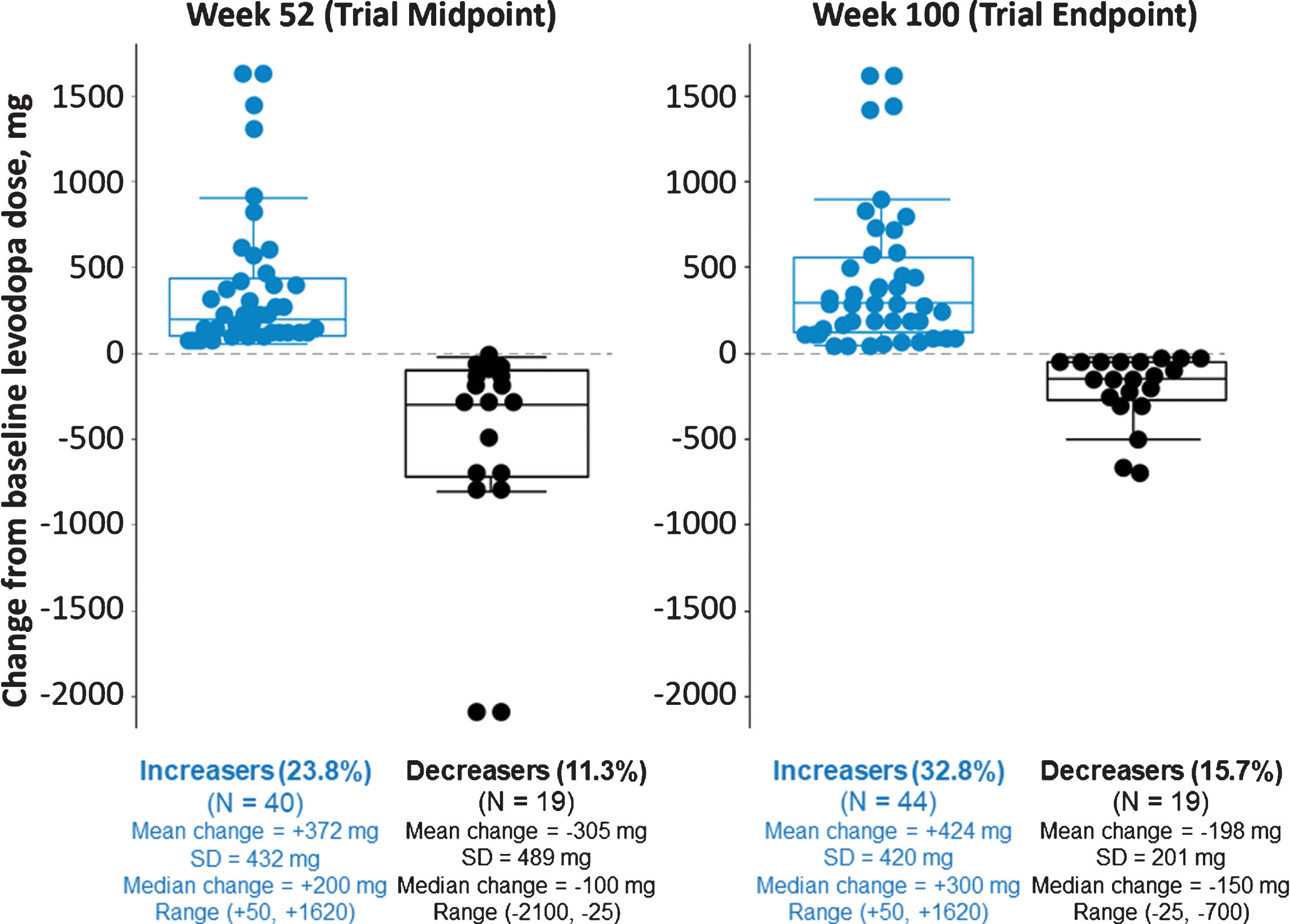
With respect to changes in other PD medications, week 100 analysis did not show large shifts, but overall, more patients discontinued than added PD medications. Sixty patients (44.8%) completed week 100 with no recorded changes in levodopa or any other PD medications.
Changes in MDS Parts I-III scores
MDS-UPDRS Parts I–III individual and combined scores are presented in Table 6. Because patients were not consistently evaluated in the OFF or ON state, all groups showed fluctuations across study visits. By the end of the trial, mean scores had increased in all groups.
Table 6
MDS-UPDRS Parts I-III scores at baseline, and weeks 52 and 100 by group (observed cases)
| MDS-UPDRS score, mean (SD) | Continuing-Gocovri group | Previous-Placebo group | Participation-Gap group | DBS group |
| Baseline | ||||
| N | 60 | 78 | 16 | 60 |
| Part I | 9.1 (5.0) | 9.9 (5.4) | 10.4 (4.8) | 11.0 (5.0) |
| Part II | 11.3 (6.9) | 13.9 (6.5) | 15.6 (8.4) | 16.2 (5.9) |
| Part III | 21.4 (11.4) | 21.3 (13.0) | 26.8 (12.5) | 26.7 (13.5) |
| Totala | 41.8 (18.4) | 45.1 (18.8) | 52.8 (23.1) | 53.8 (17.2) |
| Week 52 | ||||
| N | 47 | 55 | 12 | 51 |
| Part I | 11.5 (6.6)b | 10.0 (5.9)c | 9.9 (4.4) | 9.9 (5.8) |
| Part II | 14.2 (7.6) | 13.4 (7.3) | 16.1 (10.9) | 14.6 (6.9) |
| Part III | 27.2 (13.9) | 22.9 (12.0) | 30.3 (20.2) | 23.5 (12.6) |
| Totala | 52.7 (23.0)b | 46.5 (18.7)c | 56.3 (31.2) | 48.0 (20.9) |
| Week 100 | ||||
| N | 37 | 42 | 10 | 41 |
| Part I | 12.0 (7.1) | 9.6 (4.8) | 8.8 (4.7) | 12.1 (6.5) |
| Part II | 15.3 (6.7) | 14.3 (6.5) | 16.5 (6.0) | 16.6 (8.1) |
| Part III | 22.5 (12.0) | 23.9 (10.7) | 27.6 (13.5) | 27.4 (14.6) |
| Totala | 49.8 (18.6) | 47.8 (15.2) | 52.9 (19.7) | 56.1 (24.9) |
aSum of scores for Parts I, II, and III; summation of individual Parts may differ slightly from Total due to differences in number of patients completing individual MDS-UPDRS sections. bn = 46. cn = 54. DBS, deep brain stimulation; MDS-UPDRS, Movement Disorder Society–Unified Parkinson’s Disease Rating Scale; SD, standard deviation.
DISCUSSION
In the present open-label trial, PD patients completing double-blind, placebo-controlled Gocovri trials, and patients excluded from those trials because of DBS, began or continued once-daily Gocovri under conditions that more closely resemble real-world use. Over the 2 years of open-label treatment, Gocovri exhibited long-term safety, tolerability, and efficacy, consistent with the findings of double-blind trials lasting up to 25 weeks [4–7, 12]. To our knowledge, this is now the longest-running trial to evaluate amantadine in PD patients with LID.
In this study, Gocovri showed persistent benefits in patients continuing therapy after double-blind use, with MDS-UPDRS Part IV scores showing improvement achieved during the double-blind phase to be maintained throughout 2 years of open-label treatment. In patients naïve to Gocovri, including those with a history of DBS and/or who switched to Gocovri from amantadine IR, MDS-UPDRS Part IV scores decreased 3.4 to 4.0 points by the first clinical efficacy assessment and were maintained for up to 2 years of open-label Gocovri treatment. The observed improvements in MDS-UPDRS Part IV scores were maintained regardless of levodopa dosage increases or decreases during the study. These results for Gocovri are similar to those reported in the double-blind trials [4, 5, 12]. The observed improvements were greater than the estimated minimum clinically important change of approximately 1 point in the MDS-UPDRS Part IV score [13].
The observed MDS-UPDRS Part IV improvements resulted primarily from decreases in scores for items 4.1, 4.2, and 4.4, representing improvement in total time with dyskinesias as well as the functional impact of both dyskinesias and motor fluctuations (OFF). Although changes in the amount and predictability of OFF time (item 4.3) were small, the results remained numerically favorable at 2 years, consistent with phase 3 trial findings of decreased OFF time in patient diaries. Results at 2 years were also consistent with Part IV scores for all patients, analyzed using a last observation carried forward approach (including the last on-study score for patients who dropped out), suggesting that long-term improvement was not solely attributable to drop-out of poorly responding patients.
In general, Gocovri was well tolerated. Approximately 60% of trial participants completed 100 weeks of Gocovri treatment (the start of the planned taper), despite a mean PD duration of 11.8 years, a median of 7 concomitant medications (with a median 3 other PD medications), and a majority being ≥65 years of age (51% of patients) (Table 1). Moreover, the incidence of AEs leading to trial withdrawal (22%) was similar to that seen in shorter, double-blind Gocovri trials lasting 12–25 weeks. Not surprisingly, discontinuation due to AEs considered to be related to study drug was lower in patients continuing Gocovri (7%) than in patients initiating treatment (17%), including patients with DBS.
In the present trial, the most common AE was falls. Most of the reported falls (94%) were judged by investigators to be unrelated to study drug. In the Gocovri prescribing information, falls are listed as a potential adverse reaction. However, falls are commonly reported in PD studies, and are consistent with the motor impairments characteristic of advanced PD [14–17]. In the general population and in PD specifically, falls are also related to aging. Consistently, the frequency of falls in this trial was higher in patients ≥65 years of age (42%) than in patients <65 years of age (23%). In a cross-sectional study of 160 consecutive PD patients attending a movement disorder clinic, 39% of patients had fallen at least once since the onset of their PD [18]. In that study, the risk of falls increased dramatically with age, especially among participants over the age of 70 years. In a long-term prospective study of 211 PD patients initially able to walk, the prevalence of falls increased from 41% at baseline to 72% at 8 years, at which time age at baseline was the only identified risk factor for falling.
Hallucinations are also common in PD. In general, they are thought to be caused by neurotransmitter signaling abnormalities resulting from disease-related neurodegeneration, and may be exacerbated by dopaminergic, serotonergic, antiglutamatergic (N-methyl-D-aspartate [NMDA] receptor antagonist), or anticholinergic medications [19–21]. In patients taking Gocovri, hallucinations are likely to reflect a combination of drug activity, dosage, and disease-state factors. While hallucinations are often attributed to the activity of amantadine as a weak, uncompetitive NMDA-receptor antagonist, they could also be related to the drug’s described anticholinergic properties. Although amantadine has not been shown to possess direct anticholinergic activity in animal studies, in humans its use is often associated with anticholinergic-like side effects such as dry mouth, urinary retention, and constipation [21–23]. In the present trial, the 2-year incidence of hallucinations (24%) was similar to that in pooled double-blind phase 3 Gocovri trials lasting up to 25 weeks (21%) [6]. The reported events were predominately visual, mostly mild to moderate in intensity, and occurred most frequently in patients ≥65 years of age (40/54 cases, 74%). Hallucinations occurred within the first 3 months of open-label treatment in approximately half of patients who reported them (median 91 days), and generally occurred earlier in patients starting Gocovri treatment in this trial (51 days for Prior-Placebo group), when compared to those in the Continuing-Gocovri group (median 235 days). Nevertheless, a potential relationship to Gocovri treatment, or a need for medication regimen adjustment, should not be overlooked in patients who develop hallucinations, even after long-standing treatment. Hallucinations were managed by dose reduction in approximately 26% of cases, and by drug discontinuation in approximately 13%. The remainder did not require intervention. Dose reduction could therefore be considered for patients in whom Gocovri is considered efficacious.
Gocovri is primarily renally eliminated, and older individuals are more likely to have renal impairment [24]. Therefore, we analyzed the incidence of AEs by baseline eGFR, and no clear relationship between baseline eGFR and AEs considered study drug-related was observed. In previously reported double-blind trials, there was a greater incidence of AEs such as hallucinations in patients with lower baseline eGFR [6, 7]. Lack of observed relationship in this study may be explained in part by the long duration of EASE LID 2, during which many patients experienced some degree of reduction in eGFR from baseline. For patients with moderate or severe renal impairment, the Gocovri prescribing information recommends a lower initial and maintenance dose be administered daily at bedtime [2]. This dosing strategy should provide for drug exposure similar to patients with no or mild renal impairment, and differs from the alternate-day renal dosing recommendations of other amantadine products [25, 26]. Alternate-day dosing could result in wider peak-to-trough (or dosing to non-dosing day) exposure fluctuations of amantadine, and may not be well-suited to the release characteristics of Gocovri.
Strengths of this trial include a 2-year duration, large patient population (including follow-up from double-blind treatment), and rigorous safety monitoring. It also employed a more “real-world” design that permitted adjustment of levodopa and other concomitant PD medications, the inclusion of patients with DBS, and patients who had been using amantadine IR. The major limitation of this trial is the lack of a blinded control group, which reduces certainty that study findings are due to Gocovri. Alterations in DBS settings were not prohibited in the trial, consistent with the “real-world” design; however, information on DBS settings was not collected as part of the trial record, which is also a potential study limitation. Nevertheless, the Gocovri treatment effect appeared similar among participants with and without DBS, suggesting that DBS settings did not disproportionately influence trial results. Also, with respect to the MDS-UPDRS Part III data, there were no stated requirements in the protocol for this to be completed consistently in either the ON or OFF state, thereby limiting the interpretability of the MDS-UPDRS Part III data.
The Gocovri phase 3 clinical trial program consistently demonstrated important reductions in troublesome dyskinesia and OFF time in PD patients on levodopa therapy [4–6]. The present long-term trial provides supportive evidence that 274 mg Gocovri, taken once daily at bedtime, durably reduces the daily duration, severity, and functional impact of dyskinesia as well as OFF episodes throughout 100 weeks of continued use. Safety and tolerability outcomes were consistent with those in the phase 3 trials, with no new or unexpected findings. Onset of AEs (including hallucination), as well as discontinuation due to AEs, tended to occur earlier among those naïve to Gocovri at enrollment than among those continuing Gocovri from a prior trial (Fig. 2). Bedtime dosing of Gocovri and the resulting plasma-concentration time curve are unique product features; therefore, these trial findings may not be generalizable to different amantadine products with dissimilar doses or pharmacokinetics. Overall, the results support the safety and efficacy of Gocovri for long-term treatment of dyskinesia and OFF in PD.
CONFLICT OF INTEREST
Caroline M. Tanner: an employee of the University of California–San Francisco and the San Francisco Veterans Affairs Medical Center; serving on the scientific advisory boards of the Michael J. Fox Foundation and the National Spasmodic Dysphonia Association as a voluntary consultant; providing paid consulting services to Neurocrine Biosciences, Acorda Therapeutics, Acadia Pharmaceuticals, Amneal Pharmaceuticals, Grey Matter, and Adamas Pharmaceuticals; receiving compensation for serving on data monitoring committees from Biotie Therapeutics, Voyager Therapeutics, and Intec Pharma; and receiving grant support from the Michael J. Fox Foundation, the Parkinson’s Foundation, the Department of Defense, the National Institutes of Health, Gateway, Roche/Genentech, Biogen, and BioElectron.
Rajesh Pahwa: compensated advisory services, consulting, research grant support, or speaker honoraria: Abbott, AbbVie, Acadia, Acorda, Adamas, Biogen, Boston Scientific, CalaHealth, Cavion, Global Kinetics, Intec, Kyowa, Lilly, Lundbeck, Neurocrine, NIH/NINDS, Parkinson Foundation, Photopharmics, Prilenia, Sunovion, Teva Neuroscience, Theranexus, Theravance, US WorldMeds, Voyager.
Robert A. Hauser: is supported in part by a Center of Excellence grant from the Parkinson Foundation and is employed by the University of South Florida (Florida). He received payment from Adamas Pharmaceuticals for participating as a Steering Committee member and reports receiving fees from Adamas Pharmaceuticals, Teva Pharmaceuticals, UCB BioSciences, AbbVie, Novartis, Biotie Therapies, Lundbeck, Pfizer, Allergan Neuroscience, Neurocrine Biosciences, Chelsea Therapeutics, Auspex Pharmaceuticals, Acadia Pharmaceuticals, Michael J. Fox Foundation, GLG, AstraZeneca, Acorda Therapeutics, Impax Pharmaceuticals, Cynapsus Therapeutics, US WorldMeds, Neuropore, and Prexton Therapeutics.
Wolfgang H. Oertel: Adamas LID Steering Committee and received compensation for this service.
Stuart H. Isaacson: honoraria for CME, consultant, research grants, and/or promotional speaker on behalf of AbbVie, Acadia, Acorda, Adamas Pharmaceuticals, Addex, Allergan, Amarantus, Auspex, Avid, Axovant, AZ Therapies, Biogen, Biotie, Britannia, Cynapsus, Eisai, Eli Lilly, GE Healthcare, Impax, Intec Pharma, Ipsen, Kyowa, Lundbeck, Medtronics, Merz, The Michael J. Fox Foundation, Neurocrine, Neuroderm, NINDS/NIH, Parkinson Study Group, Pfizer, Pharma2B, Prothena, Roche, Sanofi, Shire, Sunovion, Teva, UCB, US WorldMeds, and XenoPort.
Joseph Jankovic: has received grants from Adamas Pharmaceuticals, Inc., Allergan, Inc., CHDI Foundation, Civitas/Acorda Therapeutics, Huntington Study Group, Ipsen Limited, Kyowa Haako Kirin Pharma, Inc., Lundbeck Inc., Medtronic, Merz Pharmaceuticals, Michael J. Fox Foundation for Parkinson Research, National Institutes of Health, National Parkinson Foundation, Omeros Corporation, Parkinson Study Group, Pfizer, Prothena Biosciences Inc., Psyadon Pharmaceuticals, Inc., St. Jude Medical, and Teva Pharmaceutical Industries Ltd. Dr. Jankovic receives consulting fees from Adamas Pharmaceuticals, Inc., Allergan, Inc., and Teva Pharmaceutical Industries Ltd. Dr. Jankovic has received royalties from Cambridge, Elsevier, Future Science Group, Hodder Arnold, Lippincott Williams and Wilkins, and Wiley-Blackwell.
Reed Johnson, Dustin Chernick, and Jean Hubble: are employees of and own stock in Adamas Pharmaceuticals, Inc.
ACKNOWLEDGMENTS
We acknowledge and thank the trial participants and the EASE LID 2 trial investigators and their staff. Editorial support was provided by Michael Feirtag and Robin Smith, PhD of The Curry Rockefeller Group, LLC, which was funded by Adamas Pharmaceuticals, Inc. Andrea Formella provided editorial support and assisted with data analysis and interpretation, and is an employee of and owns stock in Adamas Pharmaceuticals, Inc. Rob Howard provided statistical analysis programming for this manuscript as a consultant for Adamas Pharmaceuticals, Inc. Lily Llorens provided statistical analysis for the trial as a consultant for Adamas Pharmaceuticals, Inc. Adam Steinberg generated artwork and figures as a consultant for Adamas Pharmaceuticals, Inc. Larissa Felt was the Clinical Program Director for this trial and was a former employee of and owned stock in Adamas Pharmaceuticals, Inc. Mary Jean Stempien was the Medical Monitor for this trial and was formerly a consultant for Adamas Pharmaceuticals, Inc. Rajiv Patni and Jack Nguyen aided in the interpretation of data and are or were (respectively) employees of and own stock in Adamas Pharmaceuticals, Inc.
The trial was funded by Adamas Pharmaceuticals, Inc.
REFERENCES
[1] | Hauser RA , Kremens DE , Elmer LW , Kreitzman DL , Walsh RR , Johnson R , Howard R , Nguyen JT , Patni R ((2019) ) Prevalence of dyskinesia and OFF by 30-minute intervals through the day and assessment of daily episodes of dyskinesia and OFF: Novel analyses of diary data from Gocovri pivotal trials. J Parkinsons Dis 9: , 591–600. |
[2] | Gocovri (amantadine) extended release capsules for oral use (prescribing information). 2017. Adamas Pharma, LLC. Emeryville, CA. 2017. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208944lbl.pdf. Accessed 19 August, 2019. |
[3] | Hauser RA , Pahwa R , Wargin WA , Souza-Prien CJ , McClure N , Johnson R , Nguyen JT , Patni R , Went GT ((2019) ) Pharmacokinetics of ADS-5102 (amantadine) extended release capsules administered once daily at bedtime for the treatment of dyskinesia. Clin Pharmacokinet 58: , 77–88. |
[4] | Oertel W , Eggert K , Pahwa R , Tanner CM , Hauser RA , Trenkwalder C , Ehret R , Azulay JP , Isaacson S , Felt L , Stempien MJ ((2017) ) Randomized, placebo-controlled trial of ADS-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson’s disease (EASE LID 3). Mov Disord 32: , 1701–1709. |
[5] | Pahwa R , Tanner CM , Hauser RA , Isaacson SH , Nausieda PA , Truong DD , Agarwal P , Hull KL , Lyons KE , Johnson R , Stempien MJ ((2017) ) ADS-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson disease (EASE LID study): A randomized clinical trial. JAMA Neurol 74: , 941–949. |
[6] | Elmer LW , Juncos JL , Singer C , Truong DD , Criswell SR , Parashos S , Felt L , Johnson R , Patni R ((2018) ) Pooled analyses of phase III studies of ADS-5102 (amantadine) extended-release capsules for dyskinesia in Parkinson’s disease. CNS Drugs 32: , 387–398. |
[7] | Elmer LW , Juncos JL , Singer C , Truong DD , Criswell SR , Parashos S , Felt L , Johnson R , Patni R ((2018) ) Author correction to: Pooled analyses of phase III studies of ADS-5102 (amantadine) extended-release capsules for dyskinesia in Parkinson’s disease. CNS Drugs 32: , 399–400. |
[8] | Hauser RA , Pahwa R , Tanner CM , Oertel W , Isaacson SH , Johnson R , Felt L , Stempien MJ ((2017) ) ADS-5102 (amantadine) extended-release capsules for levodopa-induced dyskinesia in Parkinson’s disease (EASE LID 2 Study): Interim results of an open-label safety study. J Parkinsons Dis 7: , 511–522. |
[9] | Isaacson SH , Fahn S , Pahwa R , Tanner CM , Espay AJ , Trenkwalder C , Adler CH , Patni R , Johnson R ((2018) ) Parkinson’s patients with dyskinesia switched from immediate release amantadine to open-label ADS-5102. Mov Disord Clin Pract 5: , 183–190. |
[10] | Hughes AJ , Daniel SE , Kilford L , Lees AJ ((1992) ) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55: , 181–184. |
[11] | Goetz CG , Tilley BC , Shaftman SR , Stebbins GT , Fahn S , Martinez-Martin P , Poewe W , Sampaio C , Stern MB , Dodel R , Dubois B , Holloway R , Jankovic J , Kulisevsky J , Lang AE , Lees A , Leurgans S , LeWitt PA , Nyenhuis D , Olanow CW , Rascol O , Schrag A , Teresi JA , van Hilten JJ , LaPelle N , Movement Disorder Society UPDRS Revision Task Force ((2008) ) Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Scale presentation and clinimetric testing results. Mov Disord 23: , 2129–2170. |
[12] | Pahwa R , Tanner CM , Hauser RA , Sethi K , Isaacson S , Truong D , Struck L , Ruby AE , McClure NL , Went GT , Stempien MJ ((2015) ) Amantadine extended release for levodopa-induced dyskinesia in Parkinson’s disease (EASED Study). Mov Disord 30: , 788–795. |
[13] | Makkos A , Kovacs M , Pinter D , Janszky J , Kovacs N ((2019) ) Minimal clinically important difference for the historic parts of the Unified Dyskinesia Rating Scale. Parkinsonism Relat Disord 58: , 79–82. |
[14] | Koller WC , Glatt S , Vetere-Overfield B , Hassanein R ((1989) ) Falls and Parkinson’s disease. Clin Neuropharmacol 12: , 98–105. |
[15] | Gray P , Hildebrand K ((2000) ) Fall risk factors in Parkinson’s disease. J Neurosci Nurs 32: , 222–228. |
[16] | Hiorth YH , Larsen JP , Lode K , Pedersen KF ((2014) ) Natural history of falls in a population-based cohort of patients with Parkinson’s disease: An 8-year prospective study. Parkinsonism Relat Disord 20: , 1059–1064. |
[17] | Rascol O , Perez-Lloret S , Damier P , Delval A , Derkinderen P , Destee A , Meissner WG , Tison F , Negre-Pages L ((2015) ) Falls in ambulatory non-demented patients with Parkinson’s disease. J Neural Transm (Vienna) 122: , 1447–1455. |
[18] | Contreras A , Grandas F ((2012) ) Risk of falls in Parkinson’s disease: A cross-sectional study of 160 patients. Parkinsons Dis 2012: , 362572. |
[19] | Chang A , Fox SH ((2016) ) Psychosis in Parkinson’s disease: Epidemiology, pathophysiology, and management. Drugs 76: , 1093–1118. |
[20] | Forsaa EB , Larsen JP , Wentzel-Larsen T , Goetz CG , Stebbins GT , Aarsland D , Alves G ((2010) ) A 12-year population-based study of psychosis in Parkinson disease. Arch Neurol 67: , 996–1001. |
[21] | Rolland B , Jardri R , Amad A , Thomas P , Cottencin O , Bordet R ((2014) ) Pharmacology of hallucinations: Several mechanisms for one single symptom? Biomed Res Int 2014: , 307106. |
[22] | Rogawski MA ((2000) ) Low affinity channel blocking (uncompetitive) NMDA receptor antagonists as therapeutic agents–toward an understanding of their favorable tolerability. Amino Acids 19: , 133–149. |
[23] | Merritt K , McGuire P , Egerton A ((2013) ) Relationship between glutamate dysfunction and symptoms and cognitive function in psychosis. Front Psychiatry 4: , 151. |
[24] | Coresh J , Astor BC , Greene T , Eknoyan G , Levey AS ((2003) ) Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis 41: , 1–12. |
[25] | Symmetrel (amantadine hydrochloride, USP) (prescribing information). 2009. Endo Pharmaceuticals Inc. Chadds Ford, Pennsylvania. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/016023s041,018101s016lbl.pdf. Accessed 16 September, 2019. |
[26] | Osmolex ER (amantadine) extended-release tablets (prescribing information). 2018. Vertical Pharmaceuticals, LLC. Bridgewater, New Jersey. Available from: https://www.osmolexhcp.com/images/pdf/Prescribing_Information.pdf. Accessed 16 September, 2019. |


