
Increasing Comparability and Utility of Gut Microbiome Studies in Parkinson’s Disease: A Systematic Review
Abstract
Gut microbiota have been studied in relation to the pathophysiology of Parkinson’s disease (PD) due to the early gastrointestinal symptomatology and presence of alpha-synuclein pathology in the enteric nervous system, hypothesized to ascend via the vagal nerve to the central nervous system. Accordingly, sixteen human case-control studies have published gut microbiome composition changes in PD and reported over 100 differentially abundant taxa covering all taxonomic levels from phylum to genus or species, depending on methodology. While certain findings were replicated across several studies, various contradictory findings were reported. Here, differences in methodologies and the presence of possible confounders in the study populations are assessed for their potential to confound the results of gut microbiome studies in PD. Gut microbiome studies in PD exhibited considerable variability with respect to the study population, sample transport conditions, laboratory protocols and sequencing, bioinformatics pipelines, and biostatistical methods. To move from the current heterogeneous dataset towards clinically relevant biomarkers and the identification of putative therapeutic targets, recommendations are derived from the limitations of the available studies to increase the future comparability of microbiome studies in PD. In addition, integration of currently available data on the gut microbiome in PD is proposed to identify robust gut microbiome profiles in PD. Furthermore, expansion of the current dataset with atypical parkinsonism cohorts, prodromal and treatment-naïve de novo PD subjects, measurements of fecal microbial concentrations and multi-omics assessments are required to provide clinically relevant biomarkers and reveal therapeutic targets within the gut microbiome of PD.
BACKGROUND
Parkinson’s disease (PD) is clinically defined as a movement disorder characterized by motor symptoms related to dopaminergic degeneration of the substantia nigra [1]. Non-motor symptoms precede the motor symptoms by years in a majority of cases [2]. Different non-motor symptoms such as idiopathic REM sleep behavior disorder (iRBD), hyposmia, depression and constipation, are associated with an increased risk of PD [3]. Combined, they can be used to identify probable prodromal PD subjects with over 80% certainty [3]. The longest prodromal period, up to 20 years, has been suggested for constipation [4].
In concordance with the gastrointestinal symptomatology, low grade inflammation and intestinal barrier dysfunction has been reported in PD [5, 6]. Moreover, alpha synuclein (aSyn) deposition in Lewy bodies and neurites, the pathological hallmark of PD, is also found in the enteric nervous system (ENS) of established and prodromal PD cases [7]. The synucleinopathy is hypothesized to spread in a prion-like manner via the vagal nerve. Accordingly, a protective effect of truncal vagotomy for PD was established in large epidemiological studies [8, 9]. Given the early gastrointestinal dysfunction and pathology, the gut could be a source organ for disease initiation in PD.
In the last decade, non-targeted sequencing of the gut microbiome or the bacterial and archaeal community structure, using shotgun metagenomics or 16S rRNA gene amplicon surveys respectively, has become accessible to clinical researchers [10]. Changes in gut microbiome composition have been described in various neurodegenerative disorders, of which PD has been studied most extensively [11]. Besides the associative evidence from human case-control studies, there is a substantial body of evidence from animal models for a potential role of gut microbiota in the pathophysiology of PD [12, 13]. Notably, Sampson et al. showed gut microbiota to regulate the synucleinopathy, neuroinflammation and motor impairments in a rodent PD model [12].
Despite the large societal impact of PD, the cause of PD remains elusive and only symptomatic treatments exists. In addition, the clinical diagnosis of PD poses a clear diagnostic challenge and is rejected in 20% of the cases [14]. There is furthermore a need for biomarkers that would allow identification of PD already in the prodromal period [15]. To further elucidate the pathophysiological significance of gut microbiome composition changes in PD and to assess their potential as a marker for PD risk, diagnosis, stratification and prognosis, it is essential to assess whether microbiome composition changes specific for PD can be discerned. Here, a systematic review of the currently available fecal gut microbiome composition studies in PD is provided, which are compared based on their methodologies and results. To overcome the current inconsistencies and move towards clinical applications, recommendations are made to increase the quality and comparability of future microbiome studies in PD.
METHODS
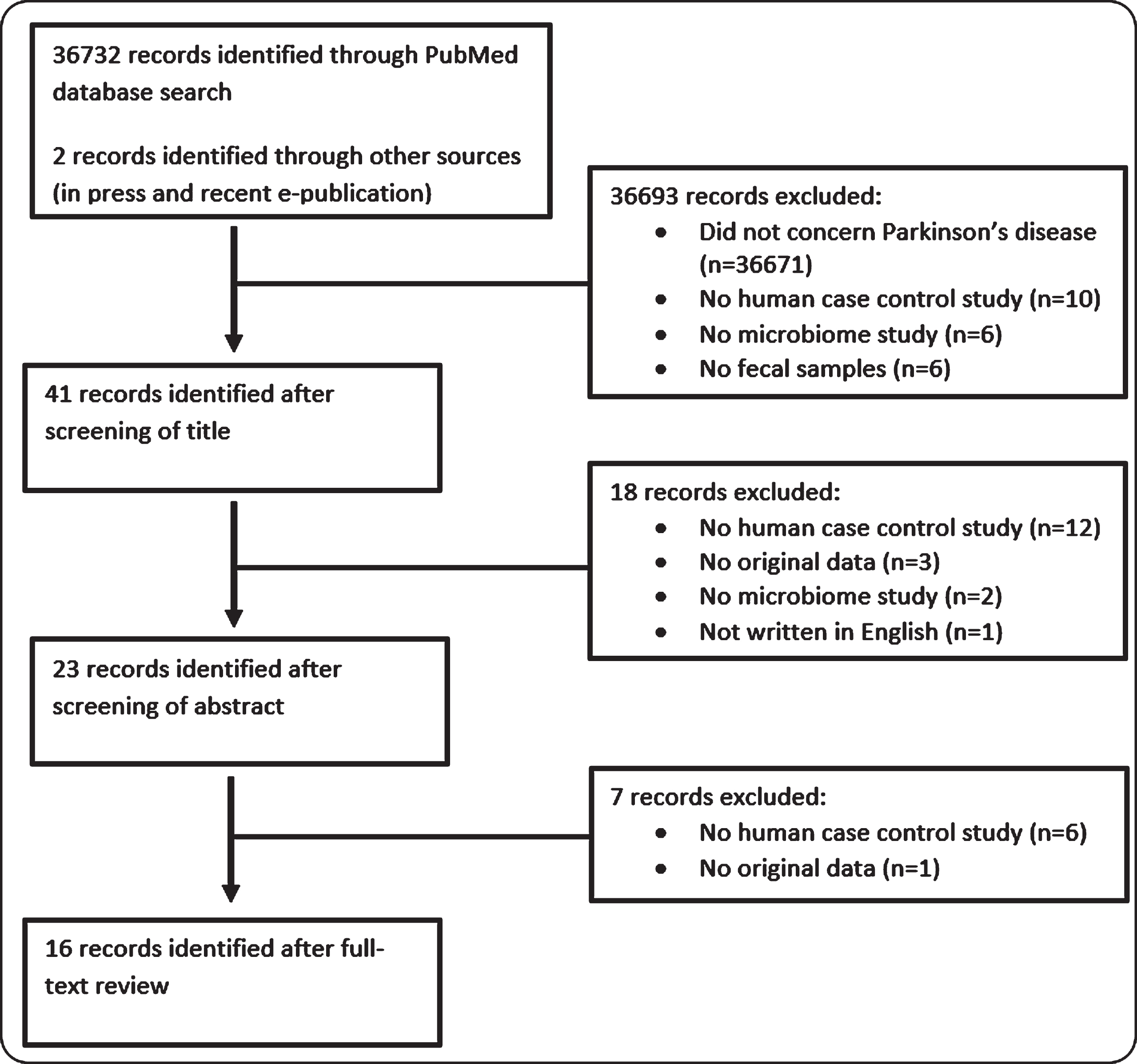
Original studies published in peer-reviewed journals were searched in the PubMed database using the search string “Parkinson” AND “Microbiome” OR “Microbiota” OR “Dysbiosis” NOT “Review”. Publications were included based on the following criteria: Parkinson’s disease is the phenotype of interest; Human case-control studies; Analysis of gut microbiome composition; Use of fecal samples; Original microbiome data; Written in English. Sixteen case-control studies concerning original gut (fecal) microbiome data were identified, including the recently accepted study by Aho et al., a follow-up study on the first gut microbiome case-control cohort in PD of Scheperjans et al. [16–31]. An overview of the selection process of eligible publications is provided in Fig. 1.
Fig.1
Overview of screening procedure to identify case-control gut microbiome studies in Parkinson’s disease.
RESULTS
Study characteristics
Study populations varied in sample size from 10 to 197 PD subjects and 10 to 130 healthy control subjects (HC). All studies represented a total of seven different countries. The mean age was similar between the PD and HC groups for all studies except for one study with a control group that was on average fifteen years younger [25]. The proportion of females, however, varied considerably between the PD and HC groups in nine studies with at least ten percent difference, the highest difference being 34%. In three studies, the samples were frozen immediately after collection and remained frozen until DNA extraction. Other studies used either a nucleotide stabilizer before freezing (n = 4), shipped the samples at ambient temperatures within a certain timeframe (n = 3), shipped the samples on ice (n = 1) or did not report on the sample shipment conditions before freezing (n = 5). Almost all studies used different DNA extraction kits with only the PSP Stool DNA Plus Kit and the QIAamp DNA Stool Mini Kit being used in multiple studies: four and two, respectively. Two studies used targeted measurements of the abundance of selected taxa, whereas all other studies used amplicon sequencing. Of these, thirteen characterized the bacterial community structure, possibly supplemented with the archaeal community structure, through 16S rRNA gene sequencing, whereas one used shotgun metagenomic sequencing to also include microbial eukaryotes, viruses and bacteriophages. An overview of the study characteristics, including demographic data and the procedures for sample collection, DNA extraction and sequencing, is provided in Table 1.
Table 1
Demographic data and methodology of fecal gut microbiome studies in Parkinson’s disease patients (PD) and healthy controls (HC) ordered by sample size from highest to lowest
| Reference | Hill-Burns | Barichella | Petrov | Pietrucci | Scheperjans | Aho1 | Lin | Qian | Hasegawa | Unger | Keshavarzian | Heintz-Buschart | Bedarf | Hopfner | Li | Li |
| 2017 | 2019 | 2017 | 2019 | 2015 | 2019 | 2018 | 2018 | 2015 | 2016 | 2015 | 2018 | 2017 | 2017 | 2017 | 2019 | |
| n (PD/HC) | 197/130 | 193/113 | 89/66 | 80/72 | 72/72 | 64/64 | 75/45 | 45/45 | 45/35 | 34/34 | 34/31 | 26/38 | 31/28 | 29/29 | 24/14 | 10/10 |
| Mean age | 68.4/70.3 | 67.6/65.9 | 67/632 | 66.2/62.6 | 65.3/64.5 | 65.2/64.53 | 60.5/63.2 | 68.1/67.9 | 68.9/68.44 | 67.7/64.6 | 61.5/45.1 | 66.0/69.8 | 64.8/65.6 | 69.2/69.4 | 73.8/74.6 | 79.5/76.5 |
| Sex % female | 33/39 | 40/58 | NA | 35/50 | 49/50 | 48/50 | 35/49 | 51/49 | 60/424 | 29/47 | 32/45 | 34/27 | 0/0 | 21/55 | 33/43 | 30/50 |
| Country | United States | Italy | Russia | Italy | Finland | Finland | China | China | Japan | Germany | United States | Germany | Germany | Germany | China | China |
| Methodology | ||||||||||||||||
| Sample transport | Ambient temp (no stabilizer) | Immediate freezing | Transport not reported | DNA stabilizer (PSP®) | DNA stabilizer (PSP®) | DNA stabilizer (PSP®) | Ambient temp (no stabilizer) | Transport on ice (no stabilizer) | RNA stabilizer (RNAlater) at 4°C | No stabilizer (no temp reported) | BD Gaspak (no temp reported) | Immediate freezing | Transport not reported | Ambient temp (no stabilizer) | Transport not reported | Immediate freezing |
| DNA extraction | Earth microbiome project protocol | QIAamp DNA Stool Mini Kit | Custom protocol | PSP Spin Stool DNA Plus Kit | PSP Spin Stool DNA Plus Kit | PSP Spin Stool DNA Plus Kit | Huirui DNA kit | QIAamp DNA Stool Mini Kit | Custom protocol | QIAsym-phony® DSP Virus/Pathogen Mini-Kit | FastDNA Spin Kit for Soil | Qiagen AllPrep | NA | PowerSoil Kit | TIANamp stool DNA kit | PSP Spin Stool DNA Plus Kit |
| Sequencing target | 16S (V4) | 16S (V3-V4) | 16S (V3-V4) | 16S (V3-V4) | 16S (V1-V3) | 16S (V3-V4) | 16S (V4) | 16S (V3-V4) | qPCR of selected taxa | qPCR of selected taxa | 16S (V4) | 16S (V4)5 | Shotgun meta-genomics | 16S (V1-V2) | 16S (V3-V5) | 16S (V3-V4) |
1Follow-up study of Scheperjans et al. 2015 PD and HC subjects, including re-analysis of baseline samples and collection of new samples with a mean follow-up time of 2.25 years. 2Median age. 3Mean age at baseline. 4Data on the entire study cohort is provided (n = 52/36, as no demographic data is reported on the subcohort of whom fecal samples were available. 5Also performed 18S sequencing. n, number of participants; NA, not available; qPCR, quantitative polymerase chain reaction; 16S, 16S rRNA gene sequencing.
Microbiome composition changes in PD
Table 2
Microbial load, alpha diversity and beta diversity of fecal gut microbiome in Parkinson’s disease (PD) ordered by sample size from highest to lowest
| Reference | Hill-Burns | Barichella | Petrov | Pietrucci | Scheperjans | Lin | Qian | Hasegawa | Unger | Keshavarzian | Heintz-Buschart | Bedarf | Hopfner | Li | Li |
| 2017 | 2019 | 2017 | 2019 | 2015 | 2018 | 2018 | 2015 | 2016 | 2015 | 2018 | 2017 | 2017 | 2017 | 2019 | |
| Microbial load | NA | NA | NA | NA | NA | NA | NA | ↓ | NA | NA | n.d.1,2 | NA | NA | NA | NA |
| Alpha diversity | NA | ↑ | ↓ | n.d. | n.d. | n.d.2 | ↑ | NA | NA | ↑ | n.d. | n.d. | n.d. | n.d. | NA |
| Beta diversity significantly differed between PD and HC (weighted) | Yes | Yes | Yes | Yes | Yes | Yes3 | Yes3 | NA | NA | Yes1 | Yes4 | Yes | Yes3 | Yes1 | Yes1,4 |
1No uncertainty estimate reported. 2Direction of difference not reported. 3Unweighted UniFrac. 4Not specified whether a weighted or unweighted method was used. NA, not available; ↑ increased in PD; ↓, decreased in PD; n.d., no difference between PD and HC.
Differences in microbial community composition can be described in terms of fecal microbial concentration, alpha and beta diversity, and relative abundances in the form of sequence counts or proportions. Microbial concentrations were reported in one study, showing a decreased abundance of microbes in the feces of PD compared to HC. Out of thirteen studies using amplicon sequencing methods in original study populations, alpha diversity indices were reported in eleven studies of which three reported higher alpha diversity in PD, six reported no difference, one reported lower alpha diversity in PD and one did not report on the direction of the found difference. Beta diversity was reported in all studies that performed 16S rRNA gene sequencing or metagenomic sequencing, all of which reported a difference between PD and HC. An overview of the microbial concentrations, alpha- and beta-diversity results in fifteen original study populations is provided in Table 2.
All studies found taxa that were differentially abundant between PD and HC. Combined, over 110 differentially abundant taxa are described that cover all taxonomic levels from phylum to species. In particular, an increase in the relative abundances of Verrucomicrobiaceae and Akkermansia, as well as a decrease of Prevotellaceae were consistently reported in nine, six and eight studies respectively. An overview of the most frequently reported differentially abundant taxa, mentioned in at least four independent populations, is provided in Table 3.
In addition to case-control comparisons, eleven studies assessed gut microbiome differences in relation to PD clinical variables, including clinical subtypes, disease duration and motor symptom burden, of which disease duration had the most consistent association with gut microbiome composition. Notably, in the only follow-up study using amplicon sequencing in PD, beta diversity did not differ between timepoints, but did differ between patients and controls at both timepoints [30]. PD-associated changes in microbiome composition therefore outweighed temporal intra-subject variability.
DISCUSSION
Since the first publication on gut microbiome differences in PD by Scheperjans et al. [16], several findings, such as an increase of Verrucomicrobiaceae and Akkermansia, and a decrease of Prevotellaceae were robustly replicated. Nonetheless, most findings could not be replicated and various contradictory findings were reported, hampering the extrapolation to PD as a whole or to specific subtypes. Examples of frequently reported taxa with contradictory results are Lactobacillaceae and Bacteroidetes. Four studies reported a statistically significant increase of Lactobacillaceae in PD, whereas two other report a statistically significant decrease. Bacteroidetes were significantly increased in one study, whereas two other reported a significant decrease.
Though, to a certain degree, different findings can be attributed to the inherent intra- and inter-subject variability of gut microbiome composition [32], methodological inconsistencies between gut microbiome case-control studies in PD might contribute to the heterogeneity of the results. Here, differences in study population, sample collection, laboratory procedures and sequencing of the fifteen studies concerning original study populations are discussed and assessed for their potential to confound the results of gut microbiome studies in PD.
Study population and assessments
Table 3
Differentially abundant taxa between Parkinson’s disease patients (PD) and healthy controls (HC)
| Phylum | Family | Genus | Number of citations (statistically significant) | Increased abundance (statistically significant) | Decreased abundance (statistically significant) |
| Verrucomicrobia | Verrucomicrobiaceae | 9 (6) | 9 (6) | 0 (0) | |
| Bacteroidetes | Prevotellaceae | 8 (3) | 0 (0) | 8 (3) | |
| Firmicutes | Lactobacillaceae | 7 (6) | 5 (4) | 2 (2) | |
| Verrucomicrobia | Verrucomicrobiaceae1 | Akkermansia | 6 (6) | 6 (6) | 0 (0) |
| Actinobacteria | Bifidobacteriaceae | Bifidobacterium | 6 (4) | 5 (4) | 1 (0) |
| Firmicutes | Lachnospiraceae | 6 (5) | 1 (1) | 5 (4) | |
| Verrucomicrobia | 5 (4) | 5 (4) | 0 (0) | ||
| Firmicutes | Clostridiaceae | Faecalibacterium | 5 (3) | 0 (0) | 5 (3) |
| Bacteroidetes | Prevotellaceae | Prevotella | 5 (1) | 0 (0) | 5 (1) |
| Proteobacteria | Enterobacteriaceae | 5 (4) | 4 (4) | 1 (0) | |
| Firmicutes | Lactobacillaceae | Lactobacillus | 5 (5) | 4 (4) | 1 (1) |
| Actinobacteria | Bifidobacteriaceae | 5 (3) | 4 (3) | 1 (0) | |
| Firmicutes | Lachnospiraceae | Roseburia | 4 (4) | 0 (0) | 4 (4) |
| Firmicutes | Enterococcaceae | 4 (4) | 3 (3) | 1 (1) | |
| Firmicutes | 4 (3) | 1 (1) | 3 (2) | ||
| Bacteroidetes | 4 (3) | 2 (1) | 2 (2) | ||
| Firmicutes | Erysipelotrichaceae | 4 (3) | 2 (2) | 2 (1) | |
| Firmicutes | Ruminococcaceae | 4 (1) | 2 (1) | 2 (0) |
Overview of the most frequently reported differentially abundant fecal microbiota between PD and HC, found in at least four independent populations. Reported taxa are mentioned in bold, with higher taxonomic levels (phylum and/or family) provided when applicable. Total number of citations and the number of citations indicating an increased or decreased relative abundance in PD are mentioned respectively. Number of publications reporting a statistically significant difference in relative abundance (P < 0.05) are mentioned between parentheses. 1Depending on the taxonomic nomenclature, Akkermansia can also be classified as a genus of the family Akkermansiaceae.
Despite large differences in sample size, ranging from 10 to 197 PD subjects, all studies reported statistically significant differences between PD and HC. Nonetheless, the inability to replicate previous findings could be attributed in some cases to the low sample size of less than 50 PD subjects and controls found in nine studies. On the contrary, the two largest studies, with 193 and 197 PD subjects, also did not replicate findings from previous studies. Other variables within the study population should therefore be assessed for their confounding influence, including differences in geographical background, age and sex distributions, PD disease status, medication use and the occurrence of gastrointestinal determinants of gut microbiome composition.
Geographical gut microbiome signatures may prevent the extrapolation of phenotype associated microbiome changes between populations from different geographical background [33, 34]. They can therefore account for inter-study differences in the microbiome studies in PD, as seven different countries are represented in the current dataset. Intra-study geographical distributions, however, were adequately addressed as each study recruited a control group through either the same medical center or institutes within the same area. Hill-Burns et al., the only study that recruited from three distinct locations, reported a statistically significant contribution of geographical site to gut microbiome composition changes in PD that was adjusted for during modeling.
Age and sex are well-known determinants of gut microbiome composition which are classical matching criteria between case and control groups [35, 36]. In nine studies more than ten percent difference in the proportion of females between the PD and HC group is observed, with one study also reporting a difference in mean age of 15 years. A possible explanation for these differences could be the inclusion of cohabitants or spouses as HC in at least six studies. Though cohabitant or spousal controls might be more suitable to adjust for environmental confounders, differences in age and sex distribution should be accounted for as potential confounders in all case-control gut microbiome studies. Age and sex differences were only incorporated as covariates in seven studies. Due to inadequate matching in various studies and the lack of post-hoc correction, age and sex effects could therefore possibly contribute to the heterogeneity of results between microbiome studies in PD.
PD is a heterogeneous disorder with varying clinical presentations that might represent different etiologies [37]. The clinical variability of PD was addressed in nine studies by adding disease duration or symptom severity as a covariate, or by analyzing different PD subtypes within the PD cohort. Eight studies could correlate microbiome signatures to PD-related clinical variables, most often disease duration, despite a further reduction of statistical power. In particular, Barichella et al. reported beta diversities to become more distant between PD and HC as the disease progresses. To date, two longitudinal follow-up microbiome studies in PD have been performed [30, 38]. Minato et al., a two year follow-up study of the subjects from the Hasegawa et al. study, reported differences in selected taxa over time, suggesting that the gut microbiome composition of PD subjects changes with disease duration [38]. On the contrary, Aho et al. found no differences in microbial community structure in both cases and controls after a similar follow-up period [30]. Possibly, the follow-up period of a little over two years in Aho et al. is too short to reveal temporal changes as the disease progresses, whereas Barichella et al. describe microbiome composition in relation to the large variation in disease duration observed in their cross-sectional sample of zero to 25 years. Therefore, differences in PD clinical status could explain part of the non-replicated results in PD microbiome studies, as microbiome shifts in PD might become more distinct as the disease progresses.
Besides clinical features, various genetic variants have been described to increase the risk of PD, possibly accentuating different parts of PD etiology with varying degrees of gastrointestinal involvement [39]. Also, different genome wide association studies (GWAS) have identified genetic variants as determinants of microbiome composition, regardless of PD status [40–42]. Genetic drift and founder effects can lead to different genetic backgrounds in different populations [43]. Various microbiome studies in PD have attempted to limit the influence of the genetic background through the exclusion of subjects with a positive family history and/or age of onset below 50 years. However, most PD associated variants only increase the disease risk by a few percent and can be present without a positive family history. Only Barichella et al. screened for mutations in one PD associated gene (Parkin), found in two subjects. No microbiome study in PD assessed the host genome on a genome wide scale, including possible genetic determinants of gut microbiome composition. Therefore, differences in genetic background could potentially explain inter-study differences of the current PD microbiome results.
To date, the effect of dopaminergic medication on gut microbiome composition has not been studied directly. Nonetheless, the effect of dopaminergic medication can be hypothesized to be substantial, as effects of various medications on gut microbiota composition have been described [17, 35, 36]. Most microbiome case-control studies in PD concern a cross-sectional sample of already treated PD subjects. Although five studies adjusted for the effects of medications that were differentially prescribed within their PD cohort, or tried to link microbial effects to the Levodopa Equivalent Daily Dose (LEDD), the one-sided introduction of dopaminergic medication as a possible confounder hinders the interpretation of the current microbiome data in PD. Bedarf et al. introduced a cohort that is Levodopa naïve. However, all PD subjects were treated using other dopaminergic drugs. Contrary to other publications, no effect from differentially prescribed dopaminergic drugs was reported by Bedarf et al. which the authors attribute to the small sample size of the study. Keshavarzian et al. and Barichella et al. included 12 and 39 treatment-naïve PD subjects respectively. Keshavarzian et al. found a difference in microbiome composition between untreated and treated PD subjects, but attributed this to differences in disease duration. Barichella et al. report a lower Lachnospiraceae abundance in untreated PD compared to HC, which was not found in treated subjects. Another treatment-naïve sample can be derived from Heintz-Buschart et al. who included 20 polysomnography (PSG) proven iRBD patients as prodromal PD [2]. Compared to HC, 41 operational taxonomic units (OTUs) were differentially abundant in iRBD of which over 75% showed a similar change as was found in their PD cohort compared to HC. Nonetheless, the extent to which the remaining 25% can be attributed to medication effects, the temporal changes as the disease progresses or a distinct RBD positive clinical subtype, remains to be seen. Larger gut microbiome studies in treatment-naïve PD subjects are therefore required to distinguish medication effects from disease specific changes in gut microbiome composition.
A variety of gastrointestinal determinants of gut microbiome composition have been identified in population-based cohort studies, including gastrointestinal disease and/or surgery, constipation, and dietary influences [35, 36]. First, gastrointestinal disease and/or surgery were used as exclusion criteria in the microbiome studies in PD with varying stringency. Second, constipation is a known determinant of gut microbiome composition and a common non-motor symptom in PD, with objective measures revealing a prolonged stool transit time in up to 80% of patients [35, 36, 44, 45]. No direct comparison with idiopathically constipated subjects was performed. However, constipation was assessed in five microbiome studies in PD using questionnaires or an interview, which can underestimate its prevalence [44]. Where the most objective measurement, using radio-opaque markers [44], can be unattainable for large-scale microbiome studies, more objective measurements of stool frequency and consistency can be obtained through a stool diary and the Bristol Stool Chart [45, 46]. Also, moist content of the fecal sample can be used as a marker for stool transit time and consistency [47]. Last, various nutrients exert an influence on gut microbiome composition [35, 36]. Dietary habits were included as covariate in five studies, with different emphasis such as protein consumption and fruit-vegetable consumption. To minimize recall bias and provide a complete assessment of nutrient intake, food diaries can be used in combination with food frequency questionnaires to obtain more reliable and complete information on dietary habits [48].
Sample collection, transport, and storage
Sampling procedures can introduce bias during sampling itself or during transportation and storage [49, 50]. During sampling, contact with urine and toilet water should be avoided to not dilute and contaminate the sample. Commercially available paper fecal collectors minimize the risk of contact of urine and water with the stool. Not all studies reported the usage of a fecal collector, but it can be assumed that all studies used similar collectors, as a fecal microbiome study would otherwise become too impractical for most participants, in particular PD subjects who might have impaired fine motor skills. Correct usage of the collector was not assessed by means of, for instance, questionnaires, nor were samples assessed for moist content in relation to stool type to red flag possibly contaminated samples.
Continued differential growth of microbes during transportation and storage can introduce artificial differences [51]. When frozen, microbiome composition remains stable for a few months at –20 C and up to two years at –80 C [51]. Ideally, samples should be frozen within 48 hours when stored at 4°C or within four hours when stored at ambient temperatures [51]. Compared to the “gold standard” of immediate freezing, various preservatives have been assessed, most of which adequately prevent microbial composition shifts for one week [50]. However, RNAlater is an example of a preservative that less adequately prevents microbiome composition shifts, which in particular hampers the analysis of rare taxa [52]. An additional issue is the ease with which the preservative can penetrate the sample, which is dependent on the size of the sample and the stool consistency [51], the latter being firmer in PD subjects who often suffer from constipation [44]. The microbial shifts found when using a preservative are, however, small compared to the temporal intra-subject microbiome variation and inter-subject variability [32]. Nonetheless, not immediately freezing the samples can potentially introduce biases.
From the fifteen PD microbiome studies in original populations, only three immediately froze the sample, whereas others transported the sample on ice, used a preservative or shipped the sample at ambient temperatures. Five studies did not report the use of a preservative and/or the transportation conditions. Only Hill-Burns et al., who shipped samples without preservative at ambient temperatures, reported on sample shipping time [17]. Shipping time was identified as a significant contributor to microbiome composition changes, which was adjusted for during modeling [17]. Inadequate adjustment for microbial shifts due to sample transportation conditions can therefore be a potential source for both intra- and inter-study differences.
Laboratory procedures
All phases during the library preparation, from DNA extraction to sequencing, can potentially introduce bias through differences in reagent quality, usage of different kit production batches during the same project, changes to protocols and/or lab personnel during the project, and contamination between batches [51, 53]. Various DNA extraction kits are available for fecal samples that rely on chemical lysis, possibly in combination with mechanical lysis [51]. Different lysis methods yield different microbial DNA based on the effectiveness with which the cell wall can be broken down [51]. Besides the method of lysis, which will be the same for case and control samples in studies, contamination of reagents can introduce batch effects that might drive artificial differences between PD and HC [53]. Most notably in this respect is the paper by Salter et al. who analyzed a blank control parallel to a pure culture of Salmonella bongori using four widely used DNA extraction kits, three of which were used in microbiome studies in PD [53]. All kits showed contamination, which led to the notion of a “kitome” that should be corrected for [51]. This kitome might also differ between kits from the same manufacturer depending on production batch and laboratory procedures. As DNA extraction of all case and control samples together in a single batch using the same DNA extraction kit and manufacturing batch is often not feasible, Salter et al. provide concrete recommendations to mitigate the risk and effects of reagent contamination. They suggest the use of high biomass samples to avoid a predominant effect of the contaminants; to use technical controls for each DNA extraction and PCR kit; and to randomize samples both before DNA extraction and library preparation for sequencing, to avoid batch effects [53]. Failure to do so might lead to misleading results that represent technical batch effects that can exceed a biological effect and should be corrected for in silico [53].
At least ten different DNA extraction kits were used in the microbiome studies in PD, that deployed different methods of chemical and mechanical lysis, with some only relying on chemical lysis. Different yields for different microbes might contribute to the heterogeneity of results between studies. In addition, only Heintz-Buchart and the follow-up of Scheperjans et al. by Aho et al. reported the implementation of the recommendations made by Salter et al. [26, 30] Instead of mere inter-study differences due to a different scope resulting from lysis efficiency, this might introduce artificial results that do not reflect any PD related effect and can potentially lead to contradictory results between studies.
Sequencing
The scope of the microbiome studies in PD is primarily determined by the sequencing method and can be divided into three groups. Hasegawa et al. and Unger et al. resorted to direct quantification of selected microbes which obviously limits the scope to the selected taxa. Only Bedarf et al. used shotgun metagenomic sequencing which is based on all available DNA fragments and therefore includes microbial eukaryotes and viruses besides bacteria and archaea [10]. All other studies sequenced one or more of the variable regions of the 16S rRNA gene which evolutionary conservation is often used for the taxonomic classification of bacteria and can also be used to classify archaea if dedicated primers are used [10]. Minor variations in the discriminative power of the variable regions exist, but do not exceed inter-individual variance [54]. The possibility of bias introduced by sequencing different variable regions is further reduced as only two out of thirteen studies using 16S sequencing did not include the V4 variable region. Nonetheless, different discriminative abilities between the variable regions and PCR primers could account for failed replication of specific taxa, especially on lower taxonomic levels [55]. Non-replication of differences in the abundance of Bifidobacteria can for instance be attributed to the different variable regions used and primer choice [55]. Notably, Aho et al. picked up differences in Bifidobacteria abundance when resequencing the baseline samples of Scheperjans et al. using the V3-V4 variable regions rather than V1-V3 [30].
With the exception of Hasegawa et al. and Unger et al., all microbiome studies in PD report on microbial abundances relative to the abundance of other microbes. Though relative microbiome profiling (RMP) is common practice in microbiome studies across phenotypes, the biological interpretation is limited without information on the fecal concentration of microbes [47]. For instance, a direct influence of fecal microbial concentration on the phenotype of interest is neglected and no accurate estimates of metabolite concentrations can be made [47]. Moreover, Vandeputte et al. showed how fecal microbiome concentrations are a determinant of microbiome diversity and suggest the use of flowcytometry and/or qPCR of the 16S rRNA gene to adjust for the confounding effect of fecal microbiome concentrations [47]. Only one microbiome study in PD reported on the fecal microbial concentration, showing a decreased concentration of microbes in the feces. Fecal microbiome concentrations could therefore introduce an unexplored bias between PD and HC.
Increasing comparability of microbiome studies in PD
Microbiome studies are notorious for the large variety of methodologies [51]. Gut microbiome studies inPDare no exception, as different methodologies were found in all phases, including sample collection, laboratory procedures, sequencing and adjustment for confounders within the study populations. An overview of the differences in study populations and methodologies between gut microbiome studies in PD is provided in Fig. 2. Although technical differences might introduce bias, practical considerations to obtain sufficient sample size can justify the deviation from “gold standard” practices. Here, the potential confounding effects of various technical, phenotypical and environmental determinants of gut microbiome composition were assessed. From this, recommendations can be derived to standardize and increase the utility of future microbiome studies in PD, which are listed in Box 1.
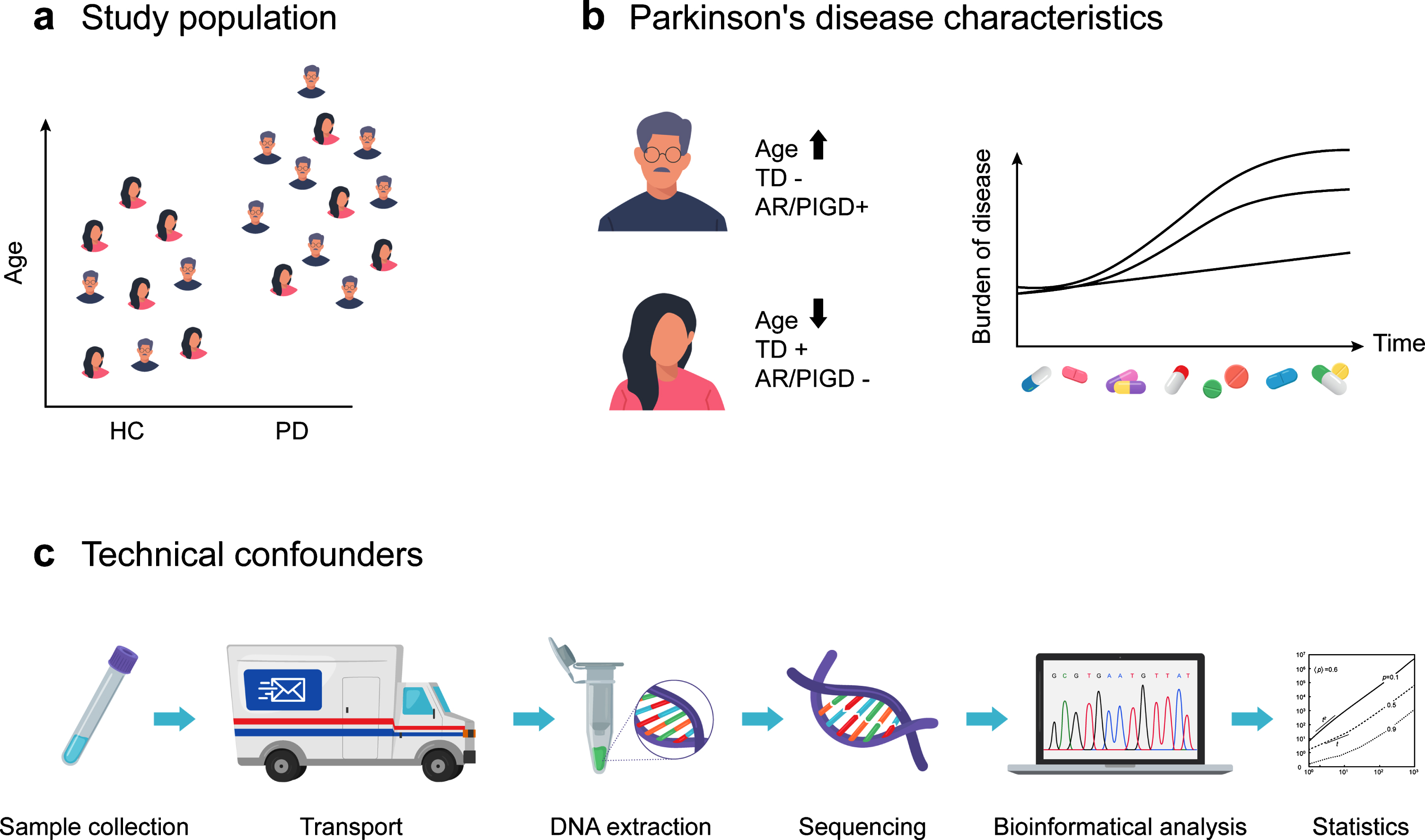
Fig.2
Differences in study populations and methodologies between gut microbiome studies in Parkinson’s disease (PD). (a) Study populations differed in age and sex distributions, sample size, geographical background, and the extent to which gastrointestinal confounders of gut microbiome composition were assessed. (b) Differences in PD subtypes, disease duration and PD medication regimen were linked to gut microbiome composition changes. (c) Sample collection procedures, transport conditions, DNA extraction, sequencing and the analytical and bio-informatics pipelines are known technical confounders of gut microbiome composition studies and differed across gut microbiome studies in PD. TD, tremor dominant subtype; AR/PIGD, akinetic rigid and/or postural instability and gait disorders subtypes; HC, healthy controls.
Box 1
Recommendations to increase the comparability and utility of (gut) microbiome studies in PD
Technical aspects:
Include information on transport and storage conditions (temperatures, time and use of preservatives) if samples are not immediately frozen after collection.
DNA extraction, library preparation and sequencing should ideally be performed for all samples in one run. Randomization of samples before each step and batch annotation should be performed to correct for batch effects.
Multiple sequencing runs can be performed with all biological samples included per run, after which the total sequence data can be merged per sample.
Negative controls should be analyzed for each batch during DNA extraction, library preparation and sequencing to adjust for contamination. Ideally, replicates of samples should be present in different batches.
16S sequencing should include the V4 variable region to assure maximum compatibility with previous studies.
Microbial concentrations of samples should be reported to allow for quantitative microbiome profiling.
Age and sex
PD subtype and disease duration
Use of medication
Host genome
Gastrointestinal disease and/or surgery
Stool frequency and consistency, possibly supplemented with moist content of the fecal sample
Dietary habits by means of a food diary
Treatment-naïve de novo PD subjects
Atypical parkinsonism and other differential diagnoses of PD as control group
Prodromal syndromes (e.g., iRBD)
In parallel, integration of the currently available microbiome data in PD would allow for the discovery of robust microbial associations and confounder adjustment necessary to address the current variability of the results. Currently, a meta-analysis of differentially abundant taxa in PD is hampered by the lack of descriptive statistics and an overemphasis on statistically significant results, ignoring the effect estimates of previously reported taxa. To adequately assess effect sizes and their respective degree of uncertainty, results on differentially abundant taxa should include all previously reported taxa, regardless of effect size or statistical significance. Nonetheless, the variety of bioinformatic and analytical tools used in microbiome studies can still impact inter-study comparability due to different annotation pipelines, clustering cut-offs and compositional metrics, as well as statistical methods. Ideally, raw sequencing data should be made publicly available to aid meta-analyses using standardized analytical pipelines. A meta-analysis of sequencing data from different variable regions of the 16S rRNA gene is possible through closed-reference operational taxonomic unit (OTU) picking [54]. Even though this will not resolve differences in the resolution of the variable regions, the predominant use of the V4 region in twelve out of fourteen 16S studies in PD, including the re-sequenced baseline samples of Aho et al., increases the inter-study comparability. More importantly, the increase in sample size would allow for more robust conclusions on the gut microbiome composition of PD subjects: a total sample size of 874 PD and 665 HC subjects could be achieved through integration of the raw sequencing data of all PD microbiome studies using 16S sequencing data. In addition, disclosure of per sample technical, phenotypical and environmental data would aid adjusting for confounders that could possibly not be adjusted for within a given study due to low sample size. As public availability of subject information might be restricted due to privacy and confidentiality legislation, sample metadata can still be made available upon request. Currently, seven studies have made their raw sequencing data publicly available.
Towards gut microbial biomarkers for PD
Exploitation of gut microbiome changes for early PD diagnosis or as a possible prognostic marker is not only dependent on standardization of case-control studies and the integration of data to achieve sufficient sample sizes to identify robust markers of disease. Different practical applications require different control settings and study cohorts. Moreover, clinical applications will likely depend on rapid quantification of selected taxa.
The clinical diagnosis of PD poses a diagnostic challenge with a rejection rate of up to 20% [14]. In particular, multiple system atrophy (MSA), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), secondary parkinsonism, vascular parkinsonism, essential tremor and dystonic tremor are differential diagnoses that can be difficult to differentiate from idiopathic PD [56]. To increase diagnostic accuracy, physicians can resort to MRI to exclude other diagnoses (e.g., PSP, vascular parkinsonism and ferrinopathies) or use FDOPA-PET or DAT-SPECT to exclude patients with scans without evidence of dopaminergic deficit (SWEDDs) such as dystonic tremor [56]. Nonetheless, PD diagnosis is still rejected in up to 10% of cases in specialized movement disorder clinics based on insufficient response to chronic dopaminergic therapy, rate of progression or autopsy [14]. Especially, atypical parkinsonisms that still exhibit some dopa-responsiveness (e.g., MSA) can be misclassified as idiopathic PD despite a positive FDOPA-PET or DAT-SPECT [56]. To avoid unnecessary treatment and adequately inform patients about their disorder and the associated prognosis, there is still a large need for early diagnostic markers of PD. To assess the diagnostic potential of gut microbiome changes in PD, the comparison with healthy age- and sex-matched controls is insufficient. Ideally, gut microbiome changes should be compared to imaging modalities for their ability to differentiate between PD and related disorders.
To date, two studies reported on microbiome changes in atypical parkinsonisms. Engen et al. compared the fecal microbiome of 6 MSA patients to 11 HCs and reported increased relative abundances in MSA of Bacteroidetes, Clostridiaceae and Rikenellaceae, whereas the relative abundances of Firmicutes and Coprobacillaceae were reduced [57]. Barichella et al. included 22 MSA patients and 22 PSP patients in their study next to 193 PD subjects and 113 HCs, allowing for a first direct comparison between PD and other neurodegenerative disorders [18]. Contrary to PD, MSA and PSP subjects were not characterized by higher alpha-diversity. Microbiome compositions were also different between PD on the one hand and PSP and MSA on the other hand in terms of beta-diversity. Due to the small sample size, the authors did not report extensively on taxonomic differences that were directly assessed between the disorders. However, the increased levels of Lactobacillaceae in PD compared to HC were also observed in MSA, but not in PSP. Given the lower prevalence of atypical parkinsonisms and other PD mimicking syndromes [58], the public availability of data becomes even more important to obtain a large enough sample size to allow robust adjustment for various potential confounders that could not be addressed in the current studies. A meta-analysis comparing the various disorders, as has been performed for other phenotypes [59], would allow for an assessment of the discriminative power of gut microbial signatures between PD and associated disorders.
Next to the need for relevant control cohorts, treatment-naïve de novo PD subjects are required to validate gut microbial biomarkers for the diagnosis of PD. Barichella et al. showed less pronounced taxonomic differences between treatment-naïve de novo PD subjects and HC compared to already treated subjects. Only a statistically significant reduction in Lachnospiraceae was reported, whereas various decreased an increased taxa were reported in already treated subjects. Besides the lower sample size of untreated subjects, this could be attributed to the unexplored effect of dopaminergic medication on gut microbiome composition. Disease duration, as a known determinant of microbiome composition in PD, obviously varied considerably between de novo and already treated subjects. To adequately reflect the context of PD diagnostics, gut microbiome studies in untreated or recently treated patients in the early phase of the disease are required.
Microbial changes to differentiate PD and its subtypes from other clinical entities can vary in complexity from different quantities of specific taxa or metabolites to complex microbial signatures. Current microbiome study procedures are time consuming and require extensive and specialized bioinformatic and statistical expertise [10]. Clinical applications of microbiome signatures will therefore ideally rely on rapid quantification of selected taxa using established methods such as qPCR. To facilitate the shift from microbiome studies based on relative abundance to clinically applicable quantification methods, microbiome studies should report on the fecal microbial concentration of samples through for instance flow cytometry or qPCR of the 16S rRNA gene [47]. From this, taxa of interest can be quantified using the already available data [47].
Towards gut microbial interventions in PD
To move towards gut microbial interventions in PD, integration of gut microbiome data in PD can aid the identification of putative causative or protective microbes by means of mendelian randomization (MR) [60]. Moreover, expansion of microbiome data with a multi-omics assessment will provide insight into microbiome-host interactions in PD [61].
MR leverages genetically determined variance of a given exposure to establish causality between a phenotype and the epidemiologically associated exposure [60]. Using a two-sample MR study, causal relationships between the abundance of microbial taxa (exposure) and PD (phenotype) can be inferred given two prerequisites. First, a strong epidemiological association between specific taxa and PD should be established, possibly through integration of currently available microbiome data in PD. Second, the influence of host genetics on the abundance of specific taxa should be established in genome wide association studies (GWAS). Three GWAS studies have identified genetic variants in the lactase gene (LCT) locus associated with gut microbiome composition changes [40–42]. Currently, the MiBioGen consortium is an international initiative to expand the current genome-microbiome associations by means of a meta-analysis of 18 genome-microbiome studies that combined have around 19.000 participants [54]. The establishment of well-powered genome-microbiome associations and the integration of gut microbiome studies in PD could be leveraged to indicate putative causal microbiota in PD.
Expansion of the current data-set with multi-omics assessments allows for a functional interrogation of microbiome-host interactions [61]. These could include metagenomic, metatranscriptomic, metaproteomic and metabolomic assessments [61], as well as parallel sampling using glycerol embedded tubes for large-scale culturing of microbes [62]. Currently, only Bedarf et al. preformed shotgun metagenomics sequencing in PD subjects. Contrary to 16S sequencing, where pathway annotation can only be performed indirectly based on the putative metabolic arsenal of a given taxon, shotgun metagenomic sequencing data can be correlated directly to genes and their biological function next to taxonomic correlation. Interestingly, Wang et al. derived biosynthetic gene clusters (BGCs) from the Bedarf et al. metagenomics data and found 43 enriched BGCs of which 14 could not be correlated to differences in source taxa between PD and HC [63]. Metagenomic sequencing therefore transcends 16S sequencing in its potential to identify biological pathways that might be of relevance for PD pathophysiology. Nonetheless, the lack of expression data limits the interpretation of metagenomic data, as no fecal metatranscriptomics or metabolomics study in PD has been performed to date. Augmentation of metagenomic sequencing with metatranscriptomics and/or metabolomics in a multi-omics approach would further improve insight in the metabolic consequences of microbiome composition shift in the context of PD pathophysiology.
Putative causal relationships, derived from data mining efforts of gut microbiome studies in PD, should still be assessed for their pathophysiological implications in relation to other determinants of the enteric pathology in PD, including intestinal wall permeability, inflammation, alpha-synuclein aggregation and gastrointestinal dysfunction. Mucosal microbiome composition was assessed by Keshavarzian et al. using sigmoid biopsies and revealed less marked differences compared to fecal samples, indicating not all differently abundant taxa in feces to interact with the intestinal mucosa [25]. To reveal relevant interactions in the multifactorial pathogenesis of PD, integration of various determinants of the enteric pathology in PD can be achieved using human intestinal organoids (HIOs), besides regularly used in vitro and preclinical models [64]. HIOs are 3D cellular models of the human intestine established through differentiation of induced pluripotent stem cells, which can be reprogrammed from patient-derived material [64]. Co-culture with enteric nervous system tissue and gut microbiota allows for a patient specific integrative assessment to elucidate significant contributions of gut microbiota to the pathophysiology of PD and reveal putative therapeutic targets [65, 66].
Conclusion
In light of the possible etiological implications of gut microbiota in PD, microbiome data in PD has rapidly accumulated in the past few years. Several findings could be replicated in various studies, such as an increase of Verrucomicrobiaceae and Akkermansia, and a decrease of Prevotellaceae. However, there is currently no consensus on PD specific changes in microbiome composition and their pathophysiological implications due to inconsistent results, differences in methodologies and unaddressed confounders. Standardization of study protocols and the collection and publication of possible confounders and results, regardless of statistical significance within one’s own dataset, could lead to the homogenization of results needed to identify relevant taxa. In this respect, public availability of raw sequencing data and sample metadata would allow for an integrative dataset of PD microbiome studies that could address various possible confounders. Expansion of the current dataset with atypical parkinsonisms, prodromal and treatment-naïve de novo PD subjects, quantitative data on fecal microbial concentration and multi-omics assessments are required to provide clinically relevant biomarkers and reveal therapeutic targets within the gut microbiome of PD.
CONFLICTS OF INTEREST
JMB has no conflicts of interest to report. PABP, VTEA, and FS have a patent FI127671B issued, a patent US10139408B2 issued, a patent US16/186,663 pending, and a patent EP3149205 pending.
FS is founder and CEO of NeuroInnovation Oy, has received a grant from Renishaw, consulting fees from Herantis Pharma and Orion, lecture fees from Abbvie, UCB, Zambon, and Orion, travel support from Abbvie, Herantis Pharma, Global Kinetics, UCB, NordicInfu Care, Zambon, and Medtronic. He is member of the scientific advisory boards of Axial Biotherapeutics and LivaNova.
ACKNOWLEDGMENTS
PABP, VTEA, and FS received funding from the Academy of Finland (295724, 310835)
REFERENCES
[1] | Postuma RB , Berg D , Stern M , Poewe W , Olanow CW , Oertel W , Obeso J , Marek K , Litvan I , Lang AE , Halliday G , Goetz CG , Gasser T , Dubois B , Chan P , Bloem BR , Adler CH , Deuschl G ((2015) ) MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 30: , 1591–1601. |
[2] | Postuma RB , Gagnon JF , Bertrand JA , Génier Marchand D , Montplaisir JY ((2015) ) Parkinson risk in idiopathic REM sleep behavior disorder: Preparing for neuroprotective trials. Neurology 84: , 1104–1113. |
[3] | Berg D , Postuma RB , Adler CH , Bloem BR , Chan P , Dubois B , Gasser T , Goetz CG , Halliday G , Joseph L , Lang AE , Liepelt-Scarfone I , Litvan I , Marek K , Obeso J , Oertel W , Olanow CW , Poewe W , Stern M , Deuschl G ((2015) ) MDS research criteria for prodromal Parkinson’s disease. Mov Disord 30: , 1600–1611. |
[4] | Postuma RB , Gagnon J-F , Pelletier A , Montplaisir J ((2013) ) Prodromal autonomic symptoms and signs in Parkinson’s disease and dementia with Lewy bodies. Mov Disord 28: , 597–604. |
[5] | Forsyth CB , Shannon KM , Kordower JH , Voigt RM , Shaikh M , Jaglin JA , Estes JD , Dodiya HB , Keshavarzian A ((2011) ) Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLoS One 6: , e28032. |
[6] | Mulak A , Budrewicz S , Panek-Jeziorna M , Koszewicz M , Jasinska M , Marczak-Karpina B , Slotwinski K , Podemski R , Paradowski L ((2017) ) Fecal biomarkers of gut inflammation and intestinal barrier dysfunction in Parkinson’s disease.S. Gastroenterology 152: , 924. |
[7] | Stokholm MG , Danielsen EH , Hamilton-Dutoit SJ , Borghammer P ((2016) ) Pathological α-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann Neurol 79: , 940–949. |
[8] | Svensson E , Horváth-Puhó E , Thomsen RW , Djurhuus JC , Pedersen L , Borghammer P , Sørensen HT ((2015) ) Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol 78: , 522–529. |
[9] | Liu B , Fang F , Pedersen NL , Tillander A , Ludvigsson JF , Ekbom A , Svenningsson P , Chen H , Karin W ((2017) ) Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study). Neurology 88: , 1996–2002. |
[10] | Fraher MH , O’Toole PW , Quigley EMM ((2012) ) Techniques used to characterize the gut microbiota: A guide for the clinician. Nat Rev Gastroenterol Hepatol 9: , 312–322. |
[11] | Gerhardt S , Mohajeri MH ((2018) ) Changes of colonic bacterial composition in Parkinson’s disease and other neurodegenerative diseases. Nutrients 10: , 708. |
[12] | Sampson TR , Debelius JW , Thron T , Janssen S , Shastri GG , Ilhan ZE , Challis C , Schretter CE , Rocha S , Gradinaru V , Chesselet MF , Keshavarzian A , Shannon KM , Krajmalnik-Brown R , Wittung-Stafshede P , Knight R , Mazmanian SK ((2016) ) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 67: , 1469–1480.e12. |
[13] | Chen SG , Stribinskis V , Rane MJ , Demuth DR , Gozal E , Roberts AM , Jagadapillai R , Liu R , Choe K , Shivakumar B , Son F , Jin S , Kerber R , Adame A , Masliah E , Friedland RP ((2016) ) Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged Fischer 344 rats and Caenorhabditis elegans. Sci Rep 6: , 34477. |
[14] | Jellinger KA ((2016) ) Accuracy of clinical diagnosis of Parkinson disease: A systematic review and meta-analysis. Neurology 87: , 237–238. |
[15] | Heinzel S , Roeben B , Ben-Shlomo Y , Lerche S , Alves G , Barone P , Behnke S , Berendse HW , Bloem BR , Burn D , Dodel R , Grosset DG , Hu M , Kasten M , Krüger R , Moccia M , Mollenhauer B , Oertel W , Suenkel U , Walter U , Wirdefeldt K , Liepelt-Scarfone I , Maetzler W , Berg D ((2016) ) Prodromal markers in Parkinson’s disease: Limitations in longitudinal studies and lessons learned. Front Aging Neurosci 8: , 147. |
[16] | Scheperjans F , Aho V , Pereira PAB , Koskinen K , Paulin L , Pekkonen E , Haapaniemi E , Kaakkola S , Eerola-Rautio J , Pohja M , Kinnunen E , Murros K , Auvinen P ((2015) ) Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord 30: , 350–358. |
[17] | Hill-Burns EM , Debelius JW , Morton JT , Wissemann WT , Lewis MR , Wallen ZD , Peddada SD , Factor SA , Molho E , Zabetian CP , Knight R , Payami H ((2017) ) Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov Disord 32: , 739–749. |
[18] | Barichella M , Severgnini M , Cilia R , Cassani E , Bolliri C , Caronni S , Ferri V , Cancello R , Ceccarani C , Faierman S , Pinelli G , De Bellis G , Zecca L , Cereda E , Consolandi C , Pezzoli G ((2019) ) Unraveling gut microbiota in Parkinson’s disease and atypical parkinsonism. Mov Disord 34: , 396–405. |
[19] | Petrov VA , Saltykova IV , Zhukova IA , Alifirova VM , Zhukova NG , Dorofeeva YB , Tyakht AV , Kovarsky BA , Alekseev DG , Kostryukova ES , Mironova YS , Izhboldina OP , Nikitina MA , Perevozchikova TV , Fait EA , Babenko VV , Vakhitova MT , Govorun VM , Sazonov AE ((2017) ) Analysis of gut microbiota in patients with Parkinson’s disease. Bull Exp Biol Med 162: , 734–737. |
[20] | Pietrucci D , Cerroni R , Unida V , Farcomeni A , Pierantozzi M , Mercuri NB , Biocca S , Stefani A , Desideri A ((2019) ) Dysbiosis of gut microbiota in a selected population of Parkinson’s patients. Parkinsonism Relat Disord–10.1016/j.parkreldis.2019.06.003. |
[21] | Lin A , Zheng W , He Y , Tang W , Wei X , He R , Huang W , Su Y , Huang Y , Zhou H , Xie H ((2018) ) Gut microbiota in patients with Parkinson’s disease in southern China. Parkinsonism Relat Disord 53: , 82–88. |
[22] | Qian Y , Yang X , Xu S , Wu C , Song Y , Qin N , Di Chen S , Xiao Q ((2018) ) Alteration of the fecal microbiota in Chinese patients with Parkinson’s disease. Brain Behav Immun 70: , 194–202. |
[23] | Hasegawa S , Goto S , Tsuji H , Okuno T , Asahara T , Nomoto K , Shibata A , Fujisawa Y , Minato T , Okamoto A , Ohno K , Hirayama M ((2015) ) Intestinal dysbiosis and lowered serum lipopolysaccharide-binding protein in Parkinson’s disease. PLoS One 10: , e0142164. |
[24] | Unger MM , Spiegel J , Dillmann KU , Grundmann D , Philippeit H , Bürmann J , Faßbender K , Schwiertz A , Schäfer KH ((2016) ) Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord 32: , 66–72. |
[25] | Keshavarzian A , Green SJ , Engen PA , Voigt RM , Naqib A , Forsyth CB , Mutlu E , Shannon KM ((2015) ) Colonic bacterial composition in Parkinson’s disease. Mov Disord 30: , 1351–1360. |
[26] | Heintz-Buschart A , Pandey U , Wicke T , Sixel-Döring F , Janzen A , Sittig-Wiegand E , Trenkwalder C , Oertel WH , Mollenhauer B , Wilmes P ((2018) ) The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov Disord 33: , 88–98. |
[27] | Bedarf JR , Hildebrand F , Coelho LP , Sunagawa S , Bahram M , Goeser F , Bork P , Wüllner U ((2017) ) Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med 9: , 39. |
[28] | Hopfner F , Künstner A , Müller SH , Künzel S , Zeuner KE , Margraf NG , Deuschl G , Baines JF , Kuhlenbäumer G ((2017) ) Gut microbiota in Parkinson disease in a northern German cohort. Brain Res 1667: , 41–45. |
[29] | Li W , Wu X , Hu X , Wang T , Liang S , Duan Y , Jin F , Qin B ((2017) ) Structural changes of gut microbiota in Parkinson’s disease and its correlation with clinical features. Sci China Life Sci 60: , 1223–1233. |
[30] | Aho VTEA , Pereira PAB , Voutilainen S , Paulin L , Pekkonen E , Auvinen P , Scheperjans F ((2019) ) Gut microbiota in Parkinson’s disease: Temporal stability and relations to disease progression. EBioMedicine 44: , 691–707. |
[31] | Li F , Wang P , Chen Z , Sui X , Xie X , Zhang J ((2019) ) Alteration of the fecal microbiota in North-Eastern Han Chinese population with sporadic Parkinson’s disease. Neurosci Lett 707: , 134297. |
[32] | Voigt AY , Costea PI , Kultima JR , Li SS , Zeller G , Sunagawa S , Bork P ((2015) ) Temporal and technical variability of human gut metagenomes. Genome Biol 16: , 73. |
[33] | Gaulke CA , Sharpton TJ ((2018) ) The influence of ethnicity and geography on human gut microbiome composition. Nat Med 24: , 1495–1496. |
[34] | He Y , Wu W , Zheng HM , Li P , McDonald D , Sheng HF , Chen MX , Chen ZH , Ji GY , Zheng ZDX , Mujagond P , Chen XJ , Rong ZH , Chen P , Lyu LY , Wang X , Bin Wu C , Yu N , Xu YJ , Yin J , Raes J , Knight R , Ma WJ , Zhou HW ((2018) ) Regional variation limits applications of healthy gut microbiome reference ranges and disease models. Nat Med 24: , 1532–1535. |
[35] | Zhernakova A , Kurilshikov A , Bonder MJ , Tigchelaar EF , Schirmer M , Vatanen T , Mujagic Z , Vila AV , Falony G , Vieira-Silva S , Wang J , Imhann F , Brandsma E , Jankipersadsing SA , Joossens M , Cenit MC , Deelen P , Swertz MA , Weersma RK , Feskens EJMM , Netea MG , Gevers D , Jonkers D , Franke L , Aulchenko YS , Huttenhower C , Raes J , Hofker MH , Xavier RJ , Wijmenga C , Fu J ((2016) ) Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science 352: , 565–569. |
[36] | Falony G , Joossens M , Vieira-Silva S , Wang J , Darzi Y , Faust K , Kurilshikov A , Bonder MJ , Valles-Colomer M , Vandeputte D , Tito RY , Chaffron S , Rymenans L , Verspecht C , De Sutter L , Lima-Mendez G , D’hoe K , Jonckheere K , Homola D , Garcia R , Tigchelaar EF , Eeckhaudt L , Fu J , Henckaerts L , Zhernakova A , Wijmenga C , Raes J ((2016) ) Population-level analysis of gut microbiome variation. Science 352: , 560–564. |
[37] | Van Rooden SM , Colas F , Martínez-Martín P , Visser M , Verbaan D , Marinus J , Chaudhuri RK , Kok JN , Van Hilten JJ ((2011) ) Clinical subtypes of Parkinson’s disease. Mov Disord 26: , 51–58. |
[38] | Minato T , Maeda T , Fujisawa Y , Tsuji H , Nomoto K , Ohno K , Hirayama M ((2017) ) Progression of Parkinson’s disease is associated with gut dysbiosis: Two-year follow-up study. PLoS One 12: , e0187307. |
[39] | Nalls MA , Blauwendraat C , Vallerga CL , Heilbron K , Bandres-Ciga S , Chang D , Tan M , Kia DA , Noyca AJ , Xue A , Bras J , Young E , von Coelln R , Simón-Sánchez J , Schulte C , Sharma M , Krohn L , Pihlstrom L , Siitonen A , Iwaki H , Leonard H , Faghri F , Gibbs JR , Hernandez DG , Scholz SW , Botia JA , Martinez M , Corvol JC , Lesage S , Jankovic J , Shulman LM , The 23andMe Research Team, System Genomics of Parkinson’s Disease (SGPD) Consortium, Sutherland M , Tienari P , Majamaa K , Toft M , Brice A , Yang J , Gan-Or Z , Gasser T , Heutink P , Shulman JM , Wood N , Hinds DA , Hardy J , Morris HR , Gratten J , Visscher PM , Graham RR , Singleton AB , for the International Parkinson’s Disease Genomics Consortium ((2018) ) Parkinson’s disease genetics: Identifying novel risk loci, providing causal insights and improving estimates of heritable risk. bioRxiv, https://doi.org/10.1101/388165. |
[40] | Bonder MJ , Kurilshikov A , Tigchelaar EF , Mujagic Z , Imhann F , Vila AV , Deelen P , Vatanen T , Schirmer M , Smeekens SP , Zhernakova D V , Jankipersadsing SA , Jaeger M , Oosting M , Cenit MC , Masclee AAM , Swertz MA , Li Y , Kumar V , Joosten L , Harmsen H , Weersma RK , Franke L , Hofker MH , Xavier RJ , Jonkers D , Netea MG , Wijmenga C , Fu J , Zhernakova A ((2016) ) The effect of host genetics on the gut microbiome. Nat Genet 48: , 1407–1412. |
[41] | Wang J , Thingholm LB , Skiecevičie J , Rausch P , Kummen M , Hov JR , Degenhardt F , Heinsen FA , Rühlemann MC , Szymczak S , Holm K , Esko T , Sun J , Pricop-Jeckstadt M , Al-Dury S , Bohov P , Bethune J , Sommer F , Ellinghaus D , Berge RK , Hübenthal M , Koch M , Schwarz K , Rimbach G , Hübbe P , Pan WH , Sheibani-Tezerji R , Häsler R , Rosenstiel P , D’Amato M , Cloppenborg-Schmidt K , Künzel S , Laudes M , Marschall HU , Lieb W , Nöthlings U , Karlsen TH , Baines JF , Franke A ((2016) ) Genome-wide association analysis identifies variation in Vitamin D receptor and other host factors influencing the gut microbiota. Nat Genet 48: , 1396–1406. |
[42] | Turpin W , Espin-Garcia O , Xu W , Silverberg MS , Kevans D , Smith MI , Guttman DS , Griffiths A , Panaccione R , Otley A , Xu L , Shestopaloff K , Moreno-Hagelsieb G , Paterson AD , Croitoru K ((2016) ) Association of host genome with intestinal microbial composition in a large healthy cohort. Nat Genet 48: , 1413–1417. |
[43] | Shi L , Webb BD , Birch AH , Elkhoury L , McCarthy J , Cai X , Oishi K , Mehta L , Diaz GA , Edelmann L , Kornreich R ((2017) ) Comprehensive population screening in the Ashkenazi Jewish population for recurrent disease-causing variants. Clin Genet 91: , 599–604. |
[44] | Knudsen K , Fedorova TD , Bekker AC , Iversen P , Østergaard K , Krogh K , Borghammer P ((2017) ) Objective colonic dysfunction is far more prevalent than subjective constipation in Parkinson’s disease: A colon transit and volume study. J Parkinsons Dis 7: , 359–367. |
[45] | Vandeputte D , Falony G , Vieira-Silva S , Tito RY , Joossens M , Raes J ((2016) ) Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 65: , 57–62. |
[46] | Tigchelaar EF , Zhernakova A , Dekens JAM , Hermes G , Baranska A , Mujagic Z , Swertz MA , Muñoz AM , Deelen P , Cénit MC , Franke L , Scholtens S , Stolk RP , Wijmenga C , Feskens EJM ((2015) ) Cohort profile: LifeLines DEEP, a prospective, general population cohort study in the northern Netherlands: Study design and baseline characteristics. BMJ Open 5: , e006772. |
[47] | Vandeputte D , Kathagen G , D’Hoe K , Vieira-Silva S , Valles-Colomer M , Sabino J , Wang J , Tito RY , De Commer L , Darzi Y , Vermeire S , Falony G , Raes J ((2017) ) Quantitative microbiome profiling links gut community variation to microbial load. Nature 551: , 507–511. |
[48] | Kowalkowska J , Slowinska MA , Slowinski D , Dlugosz A , Niedzwiedzka E , Wadolowska L ((2013) ) Comparison of a full food-frequency questionnaire with the three-day unweighted food records in young Polish adult women: Implications for dietary assessment. Nutrients 5: , 2747–2776. |
[49] | Wu GD , Lewis JD , Hoffmann C , Chen YY , Knight R , Bittinger K , Hwang J , Chen J , Berkowsky R , Nessel L , Li H , Bushman FD ((2010) ) Sampling and pyrosequencing methods for characterizing bacterial communities in the human gut using 16S sequence tags. BMC Microbiol 10: , 206. |
[50] | Wang Z , Zolnik CP , Qiu Y , Usyk M , Wang T , Strickler HD , Isasi CR , Kaplan RC , Kurland IJ , Qi Q , Burk RD ((2018) ) Comparison of fecal collection methods for microbiome and metabolomics studies. Front Cell Infect Microbiol 8: , 301. |
[51] | Wu WK , Chen CC , Panyod S , Chen RA , Wu MS , Sheen LY , Chang SC ((2019) ) Optimization of fecal sample processing for microbiome study — The journey from bathroom to bench. J Formos Med Assoc 118: , 545–555. |
[52] | Flores R , Shi J , Yu G , Ma B , Ravel J , Goedert JJ , Sinha R ((2015) ) Collection media and delayed freezing effects on microbial composition of human stool. Microbiome 3: , 33. |
[53] | Salter SJ , Cox MJ , Turek EM , Calus ST , Cookson WO , Moffatt MF , Turner P , Parkhill J , Loman NJ , Walker AW ((2014) ) Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol 12: , 87. |
[54] | Wang J , Kurilshikov A , Radjabzadeh D , Turpin W , Croitoru K , Bonder MJ , Jackson MA , Medina-Gomez C , Frost F , Homuth G , Rühlemann M , Hughes D , Kim HN , MiBioGen Consortium Initiative, Spector TD, Bell JT , Steves CJ , Timpson N , Franke A , Wijmenga C , Meyer K , Kacprowski T , Franke L , Paterson AD , Raes J , Kraaij R , Zhernakova A ((2018) ) Meta-analysis of human genome-microbiome association studies: The MiBioGen consortium initiative. Microbiome 6: , 101. |
[55] | Graspeuntner S , Loeper N , Künzel S , Baines JF , Rupp J ((2018) ) Selection of validated hypervariable regions is crucial in 16S-based microbiota studies of the female genital tract. Sci Rep 8: , 9678. |
[56] | Ibrahim N , Kusmirek J , Struck A , Floberg J , Perlman S , Gallagher C , Hall L ((2016) ) The sensitivity and specificity of F-DOPA PET in a movement disorder clinic. Am J Nucl Med Mol Imaging 6: , 102–109. |
[57] | Engen PA , Dodiya HB , Naqib A , Forsyth CB , Green SJ , Voigt RM , Kordower JH , Mutlu EA , Shannon KM , Keshavarzian A ((2017) ) The potential role of gut-derived inflammation in multiple system atrophy. J Parkinsons Dis 7: , 331–346. |
[58] | Levin J , Kurz A , Arzberger T , Giese A , Höglinger GU ((2016) ) The differential diagnosis and treatment of atypical parkinsonism. Dtsch Arztebl Int 113: , 61–69. |
[59] | Duvallet C , Gibbons SM , Gurry T , Irizarry RA , Alm EJ ((2017) ) Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat Commun 8: , 1784. |
[60] | Burgess S , Foley CN , Zuber V ((2018) ) Inferring causal relationships between risk factors and outcomes from genome-wide association study data. Annu Rev Genomics Hum Genet 19: , 303–327. |
[61] | Pinu FR , Beale DJ , Paten AM , Kouremenos K , Swarup S , Schirra HJ , Wishart D ((2019) ) Systems biology and multi-omics integration: Viewpoints from the metabolomics research community. Metabolites 9: , E76. |
[62] | Browne HP , Forster SC , Anonye BO , Kumar N , Neville BA , Stares MD , Goulding D , Lawley TD ((2016) ) Culturing of “unculturable” human microbiota reveals novel taxa and extensive sporulation. Nature 533: , 543–546. |
[63] | Wang S , Li N , Zou H , Wu M ((2019) ) Gut microbiome-based secondary metabolite biosynthetic gene clusters detection in Parkinson’s disease. Neurosci Lett, 696: , 93–98. |
[64] | Watson CL , Mahe MM , Múnera J , Howell JC , Sundaram N , Poling HM , SchweitzerJI , Vallance JE , Mayhew CN , Sun Y , Grabowski G , Finkbeiner SR , Spence JR , Shroyer NF , Wells JM , Helmrath MA ((2014) ) An in vivo model of human small intestine using pluripotent stem cells. Nat Med 20: , 1310–1314. |
[65] | Schlieve CR , Fowler KL , Thornton M , Huang S , Hajjali I , Hou X , Grubbs B , Spence JR , Grikscheit TC ((2017) ) Neural crest cell implantation restores enteric nervous system function and alters the gastrointestinal transcriptome in human tissue-engineered small intestine. Stem Cell Reports 9: , 883–896. |
[66] | Hill DR , Huang S , Nagy MS , Yadagiri VK , Fields C , Mukherjee D , Bons B , Dedhia PH , Chin AM , Tsai YH , Thodla S , Schmidt TM , Walk S , Young VB , Spence JR ((2017) ) Bacterial colonization stimulates a complex physiological response in the immature human intestinal epithelium. Elife 6: , e29132. |


