
Possible Role of Amyloid Cross-Seeding in Evolvability and Neurodegenerative Disease
Abstract
Aging-related neurodegenerative disorders are frequently associated with the aggregation of multiple amyloidogenic proteins (APs), although the reason why such detrimental phenomena have emerged in the post-reproductive human brain across evolution is unclear. Speculatively, APs might provide physiological benefits for the human brain during developmental/reproductive stages. Of relevance, it is noteworthy that cross-seeding (CS) of APs has recently been characterized in cellular and animal models of neurodegenerative disease, and that normal physiological CS of multiple APs has also been observed in lower organisms, including yeast and bacteria. In this context, our main objective is to discuss a possible involvement of the CS of APs in promoting evolvability, a hypothetical view regarding the function of APs as an inheritance of acquired characteristics against human brain stressors, which are transgenerationally transmitted to offspring via germ cells. Mechanistically, the protofibrils formed by the CS of multiple APs might confer hormesis more potently than individual APs. By virtue of greater encoded stress information in parental brains being available, the brains of offspring can cope more efficiently with forth-coming stressors. On the other hand, subsequent neurodegeneration caused by APs in parental brain through the antagonistic pleiotropy mechanism in aging, may suggest that synergistically, multiple APs might be more detrimental compared to singular AP in neurodegeneration. Taken together, we suggest that the CS of multiple APs might be involved in both evolvability and neurodegenerative disease in human brain, which may be mechanistically and therapeutically important.
INTRODUCTION
Aging-related neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD), are pathologically characterized by aggregation of amyloidogenic proteins (APs), the mechanisms of which are incompletely understood. Current prevailing views such as the “amyloid cascade hypothesis” (ACH) postulate that aggregation of APs triggers a toxic cascade of events ultimately resulting in aging-related neurodegenerative diseases [1]. In AD, it is known that familial mutations in various genes, including presenilin (PSEN) 1, PSEN 2 and β-amyloid (Aβ) precursor protein, are associated with early-onset AD [2], while apolipoprotein E4 is the strongest genetic risk factor for late-onset AD [3]. These genetic risk factors promote Aβ and tau oligomer formation, followed by various histopathological features, including neuritic plaque and neurofibrillary tangle formation, synapse loss, neuronal death and widespread neuroinflammation, eventually leading to manifestation of disease symptoms (Fig. 1) [1]. Although the ACH was primarily proposed in AD, it has also been the dominant hypothesis for other related neurodegenerative diseases. For instance, aggregation of α-synuclein (αS) is thought to be upstream of other neuropathological features leading to the disease manifestation in PD [4].
Fig.1
The relationship of ACH with EVH. ACH postulates that protein aggregation triggers a cascade of events ultimately resulting in neurodegenerative diseases. In AD, it is known that familial mutations in various genes and risk factors stimulate the Aβ and tau oligomer formation, eventually leading to various histopathological features, including plaque and neurofibrillary tangles formation, loss of synapses neuronal death and widespread neuroinflammation, and manifestation of disease symptoms (right). In contrast to ACH focusing mainly on the pathology of brain in aging, EVH covers physiological aspects in both development/reproduction and aging stages (left). The protofibrils of APs might confer hormesis in parental brains, which is transgenerationally transmitted to offspring via germ cells so as to cope with the forthcoming diverse stresses in the offspring’s brain [6]. On the other hand, neurodegeneration might later manifest in parental brain through the antagonistic pleiotropy mechanism in aging. It is therefore expected that EVH may play a complementary role for ACH.
![The relationship of ACH with EVH. ACH postulates that protein aggregation triggers a cascade of events ultimately resulting in neurodegenerative diseases. In AD, it is known that familial mutations in various genes and risk factors stimulate the Aβ and tau oligomer formation, eventually leading to various histopathological features, including plaque and neurofibrillary tangles formation, loss of synapses neuronal death and widespread neuroinflammation, and manifestation of disease symptoms (right). In contrast to ACH focusing mainly on the pathology of brain in aging, EVH covers physiological aspects in both development/reproduction and aging stages (left). The protofibrils of APs might confer hormesis in parental brains, which is transgenerationally transmitted to offspring via germ cells so as to cope with the forthcoming diverse stresses in the offspring’s brain [6]. On the other hand, neurodegeneration might later manifest in parental brain through the antagonistic pleiotropy mechanism in aging. It is therefore expected that EVH may play a complementary role for ACH.](https://content.iospress.com:443/media/jpd/2019/9-4/jpd-9-4-jpd191675/jpd-9-jpd191675-g001.jpg)
The recent failure of Aβ immunotherapy for AD, however, raises concerns regarding the validity of the ACH [1, 5]. Since current interpretations of the ACH do not take into account the physiological function of APs, we recently proposed that evolvability of APs in brain might be physiologically important against stressors [6]. More specifically, APs might confer hormesis in parental brains, which is transgenerationally transmitted to offspring via germ cells so as to cope with the forthcoming diverse stresses in the offspring’s brain [6]. Thus, the evolvability of APs could be regarded as an inheritance of acquired characteristics against stressors, which may be beneficial to offspring [6]. The inheritance of acquired characteristics has been a historical controversy [7], yet, this issue has been extensively investigated in the lower organism such as Caenorhabditis elegans [8]. On the other hand, neurodegeneration might later manifest in parental brain through the antagonistic pleiotropy mechanism in aging [9]. Thus, our view, referred to as the “evolvability hypothesis” (EVH), might explain why such a detrimental phenomenon has not been selected out during evolution. Such an idea may be consistent with the concept of Darwinian medicine, the emerging field of study devoted to applying evolutionary biology principles to medicine [10]. In contrast to the ACH, which focuses mainly on brain pathology in aging, our EVH applies not only to aging, but also to developmental/reproductive stages (Fig. 1). Thus, EVH, as a theoretical framework for the role of APs, may complement ACH to elucidate issues that are currently unresolved in the field of neurodegenerative diseases.
In this paper, we discuss the potential role of cross-seeding (CS) of APs in evolvability and neurodegenerative diseases. Based on the EVH, the CS of multiple APs may stimulate formation of the protofibrils of APs, leading to increased efficiency of evolvability which may be beneficial for offspring, while neurodegeneration caused by APs through the antagonistic pleiotropy mechanism might become more damaging to brain during parental aging. Thus, neurodegenerative diseases associated with multiple APs might have emerged during evolution due to the potential beneficial effect of the CS of multiple APs on evolvability. Given this, the CS of multiple APs may be a therapeutic target against neurodegenerative disorders.
POSSIBLE ROLE OF THE CS OF APs IN NEURODEGENERATIVE DISEASES
Aggregation of multiple APs in neurodegenerative disorders
One of the major features in the pathogenesis of neurodegenerative diseases is the aggregation of multiple APs [11]. As an example, the co-presence of aggregated Aβ and tau is a defining pathological feature of AD. Furthermore, in dementia with Lewy bodies (DLB) as well as familial AD, αS aggregates frequently co-localize with those of Aβ [12, 13]. Moreover, in amyotrophic lateral sclerosis (ALS), SOD-1 (Cu/Zn superoxide dismutase) and TAR DNA-binding protein of 43 kDa (TDP-43) may also be co-aggregated [14]. Perhaps the most prominent disorder featuring aggregation of multiple APs is the ALS/parkinsonism-dementia complex (PDC) of Guam that is associated with aggregation of Aβ, tau, αS, and TDP-43 [15–17]. Because the etiology of ALS/PDC complex is unknown, several hypotheses have been provided, including cycad toxicity and flying fox consumption [18]. Parallel pathologic mechanisms may also apply to the ALS/PDC compex found in Kii peninsula in Japan [19]. Collectively, multiple combinations of APs may be general phenomena in neurodegeneration.
The CS of APs in experimental models of neurodegeneration
The mechanism by which the aggregation of multiple APs occurs in neurodegenerative diseases is obscure. In this regard, recent study shows that the CS of APs may play a major role in a wide variety of experimental models of neurodegeneration, including AD, PD, DLB, ALS and transmissible spongiform encephalopathy [20]. In AD, the CS of Aβ with tau might be caused by various mechanisms. Supporting this, Aβ directly associated with tau in vitro [21], while phosphorylation of tau by Aβ was essential to pathogenesis in both cell-based and animal studies in AD [22]. Similarly, aggregation of αS was increased by Aβ in vitro [23] as well as in transgenic mouse model of α-synucleinopathies [24]. Moreover, Aβ promotes the aggregation of other APs, such as prion protein (PrP) and TDP-43 [11, 25]. Collectively, Aβ may play a central role in the CS of APs, yet other APs may also fulfill a similar role in the CS of APs. For instance, stimulation of αS aggregation in the presence of tau and PrP in α-synucleinopathies may indicate the CS of APs other than Aβ [26, 27]. In the similar context, SOD-1 and TDP-43 may co-aggregate in ALS. In ALS/PDC complex, it is probable that environmental factors might cause neurotoxicity, ultimately promoting the CS of APs. Taken together, the CS of APs may underlie a broad range of neurodegenerative conditions, which prompts the question as to why the aggregation of multiple APs, apparently injurious to the brain, has emerged and persisted against the pressures of natural selection.
THE CS OF APs AS A PHYSIOLOGICAL FUNCTION IN THE LOWER ORGANISMS
Accumulating evidence suggests that amyloids are functionally significant in bacteria and fungi [28]. Notably, the CS of APs is normally observed in the physiology of lower biological systems, such as bacteria and yeast, suggesting that the CR of APs may be physiological and appears early in evolution.
Bacteria
Specifically, bacterial APs are highly conserved, being involved in biofilm formation, which benefit bacteria during invasion, host adhesion, and resistance to destruction [29]. Curli, the best studied bacterial AP, is made by enteric bacteria such as Escherichia coli and Salmonella spp, and its key element, CsgA, has been found to contain amyloidogenic peptide repeat motifs shared by prions and αS that assemble into amyloid fibers. In E. coli, the polymerization of the major curli fiber subunit protein CsgA into an amyloid fiber depends on the minor curli subunit protein, CsgB. The outer membrane-localized CsgB protein shares approximately 30% sequence identity with the amyloid-forming protein CsgA, suggesting that CsgB might also have amyloidogenic properties. Also described, curli promotes the conversion of PAP248–286 into the amyloid SEVI, exemplifying the CS of dissimilar amyloid sequences (Fig. 2a) [30]. Thus, the CS of CsgA and CsgB may play an important role in bacterial physiology.
Fig.2
Physiological roles of the CR of APs in microorganisms. a) Bacterial curli protein promotes the conversion of PAP248–286 into the amyloid SEVI. Electron microscopic images of SEVI fibers formed in the absence of curli (left) and in the presence of 5 mol% CsgA (middle) and CsgB (right) fibers. Fibers were grown at a concentration of 440μM PAP248–286 at 37°C under 1400 rpm orbital shaking for 7 days. Bars = 500 nm. Quantification of the fibers are shown. b) Heterologous prion-forming proteins interact to cross-seed aggregation in Saccharomyces cerevisiae. Modified from Hartman et al. [30] (a) and Keefer et al. [31] (b) with permission.
![Physiological roles of the CR of APs in microorganisms. a) Bacterial curli protein promotes the conversion of PAP248–286 into the amyloid SEVI. Electron microscopic images of SEVI fibers formed in the absence of curli (left) and in the presence of 5 mol% CsgA (middle) and CsgB (right) fibers. Fibers were grown at a concentration of 440μM PAP248–286 at 37°C under 1400 rpm orbital shaking for 7 days. Bars = 500 nm. Quantification of the fibers are shown. b) Heterologous prion-forming proteins interact to cross-seed aggregation in Saccharomyces cerevisiae. Modified from Hartman et al. [30] (a) and Keefer et al. [31] (b) with permission.](https://content.iospress.com:443/media/jpd/2019/9-4/jpd-9-4-jpd191675/jpd-9-jpd191675-g002.jpg)
Yeast
In addition, it was recently shown that the CS of yeast prions, including Rnq1 and Sup35, is a predominant mechanism leading to self-propagating and aggregation of the translation termination factor sup35 for formation of the yeast prion [PSI+], suggesting that CS of APs may be physiological in yeast (Fig. 2b) [31]. Collectively, it is predicted that the CS of APs might evolve as a physiological phenomena in the lower organisms.
PERSPECTIVE: INCREASED EVOLVABILITY THROUGH THE CS OF MULTIPLE APs
Similar to the lower organisms, it is tempting to speculate that CS of APs might be similarly functional under normal conditions in humans. In this context, the CS of multiple APs during aging might be beneficial for evolvability, a possible physiological function of APs during developmental/reproductive stages in humans.
Hormesis
A recent study suggests that APs are composed of structurally heterogeneous populations in part due to the ‘intrinsically disordered structure’ of APs that lack fixed or ordered three-dimensional structures [32, 33]. As such, it is conceivable that AP heterogeneity might correspond to diverse stressors and that APs might retain information regarding a variety of stressors through structural changes. Given that AP evolvability is beneficial, one might further predict that the CS of multiple APs may synergistically increase the heterogeneity of APs aggregates, allowing greater capacity to cope with increasingly diverse stressors (Fig. 3). Thus, the CS of APs may be physiologically regulated to acquire the resistance of stresses, namely hormesis.
Fig.3
Role of the CS of APs in evolvability and neurodegenerative diseases. The CS of multiple APs, such as Aβ, αS, tau, SOD-1, TDP-43, and PrP, may stimulate protein aggregation, resulting in increased evolvability for offspring to cope with forth-coming stressors. Instead, antagonistic pleiotropy in aging might promote neurodegeneration in parental brain. Therefore, the CS of multiple APs may be beneficial for offspring, but detrimental for parents.
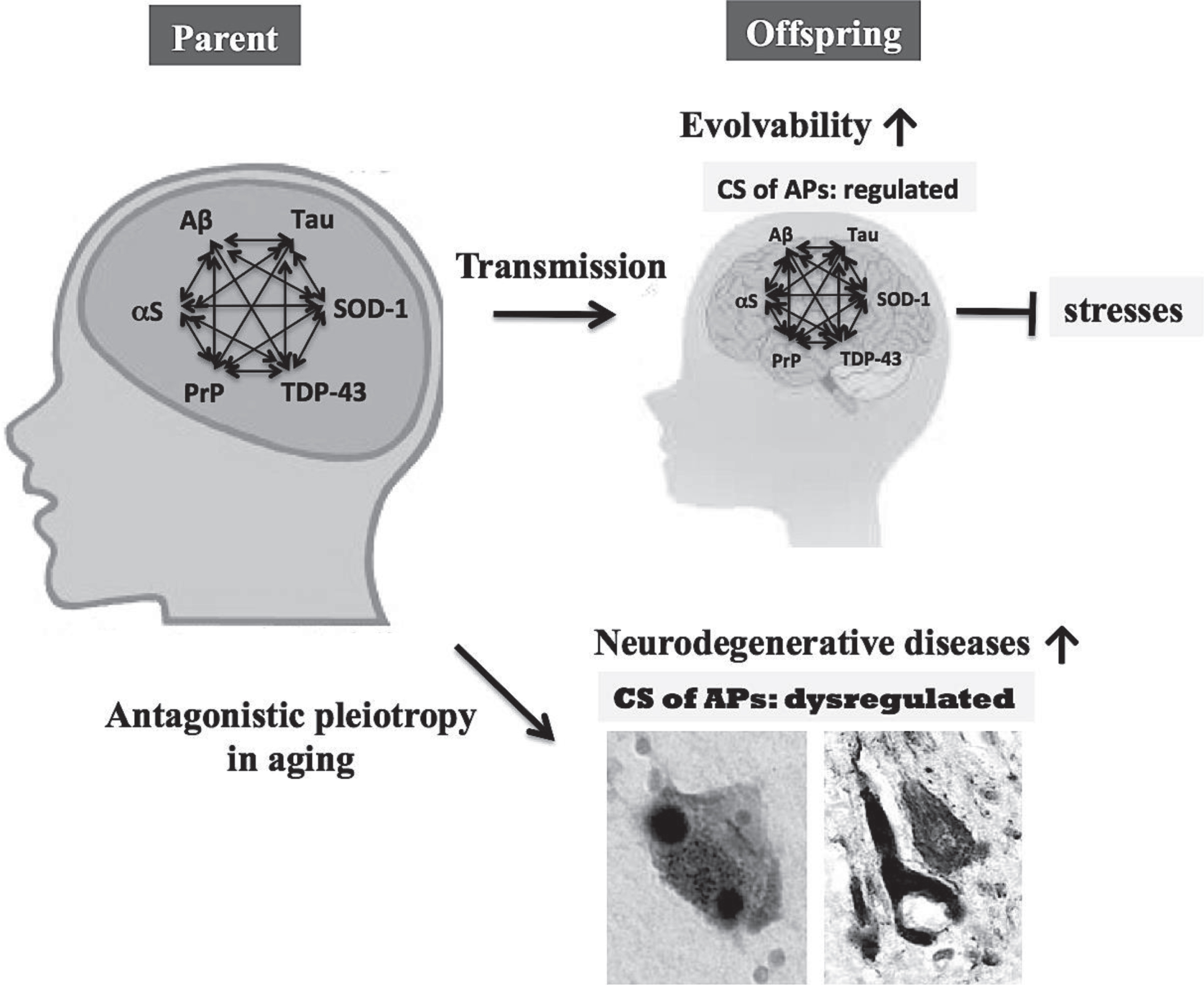
Transgenerational transmission
Since the requirements for evolvability include not only hormesis, but also heredity to generate adaptive genetic diversity [34], it follows that transgenerational transmission is a critical component. In case of unicellular organisms, such as yeast and bacteria, protofibrillar APs may be easily transmitted from parental to progeny cells in concert with cell division. By contrast, the transgeneration of AP protofibrils in multicellular organisms may be more complex. In humans, protofibrillar APs may be transmitted to offspring via germ lines [6, 35]. As far we are aware, few reports demonstrate the presence of APs protofibrils in vivo, including germ cells, however, further investigations are warranted since amyloid fibrils are abundantly present in semen [36].
Because monomeric APs may be unstable due to its intrinsically disordered nature [37], it is possible that oligomers and protofibrils of APs might be more stable. Thus, it is interesting to determine whether the protofibrils composed of heterogeneous APs might be more stable compared to the homogeneous protofibrils of APs (Fig. 3). There is also great interest in the role of exosomal RNAs, including ncRNAs and miRNA, transmitted transgenerationally in a non-genetic manner through germ cells. [38]. Considering that prefibrillar Aβ aggregates preferentially bind to exosomes [39], it is possible that these biologic processes might in some way be relavant to the transgenerational transmission of Aβ prefibrils. Taken together, further studies may reveal that multiple APs may cooperate to increase the efficiency of evolvability, including the CS of APs.
Antagonistic pleiotropy
On the other hand, neurodegenerative diseases may manifest in parental brain through the antagonistic pleiotropy mechanism in aging [9, 40], whereby in neurodegeneration multiple APs might be more detrimental compared to singular AP. Although the neurotoxicity associated with the evolvability of APs protofibrils may be well regulated during the reproductive stage, the same might be reduced or absent in post-reproductive senescence, leading to neuropathological phenotypes characterized by mature fibrils and the CS of APs. Thus, the CS of APs is primarily beneficial in development/reproduction, but is detrimental in aging. This viewpoint might explain why the CS of APs, a deleterious pathological phenomenon during aging has not been selected out during the evolutionary process.
Role of the CS of APs to promote evolvability in non-neuronal tissues
Given that type 2 diabetes mellitus (T2DM) and neurodegenerative disorders, such as AD and PD, are analogous in terms of amyloidosis associated with stressors [41], there might be common mechanisms underlying these two diseases. Similar to AD, the aggregation of amylin in pancreatic β cells during T2DM pathogenesis could represent antagonistic pleiotropy of amylin evolvability. Hypothetically, gestational DM might promote transmission of the amylin protofibrils to deliver information about maternal stressors, which if deficient, might promote the risk of type 1 DM in offspring. T2DM, however, might manifest later through antagonistic pleiotropy in parental aging. Furthermore, it is interesting to note that amylin and αS co-aggregate in pancreatic β cells, suggesting that the CS of these APs might be pathologically important [42].
Experimental approach
The effect of the CS of multiple APs on evolvability would be assessed experimentally using transgenic (tg) mice model of neurodegenerative diseases. For instance, it was shown that tau and αS synergistically promoted fibrillization each other both in vitro and in the bigenic (αS/tau) mice expressing wild-type human αS plus P301L mutant tau [43]. According to our EVH, it is anticipated that the offspring born from the tg mice with administration to neurotoxins, such as 1-methyl–4-phenyl-1,2,3,6-tetrahydropyridine and 6-hydroxydopamine, might be more resistant compared to those born from the same parent without treatment. Such effects might be more stronger if the parents are the bigenic (αS/tau) mice compared to the single tg mice. Essentially similar results could be obtained using tg mice for αS and amyloid precursor protein [24]. In both cases, it should be considered that the fertility of the bigeic mice might be compromised compared to those of the single tg mice.
VIRAL INFECTION AND AGGREGATION OF APS: INVOLVEMENT OF THE CS OF APs?
Increasing evidence suggests that some APs are functionally involved in various biological processes, such as melanosome biogenesis, long-term memory formation and the release of peptide hormones in higher organisms [44]. However, the role of amyloid-fibril formation has been unclear.
Aβ aggregation by viral infection
Notably, it was recently shown that the seeding of Aβ aggregation was stimulated by infection of Herpes simplex virus type 1 (HSV1) in cellular and tg mice models of AD (Fig. 4a) [45]. Mechanistically, it was suggested that Aβ aggregates entrap virus at the cell membrane and protect the brain against viral infection [45]. Based on a series of experimental results, the authors proposed the “antimicrobial protection model” (APM) of AD [46], suggesting that Aβ fibrillization may drive neuroinflammatory pathways that help fight the infection and clear Aβ/pathogen deposits.
Fig.4
Seeding of Aβ fibrils by HSV1 infection. a) Ultrstractural analysis of immunoelectron micrography shows Aβ fibrillization seeded by HSV1 in cell culture leading to virus capture and entrapment. Please see the experimental conditions in Eimer et al. (2018) [45]. Reprinted from Eimer et al. (2018) [45] with permission. b) Schematic of Aβ evolvability of and disease manifestation related to HSV1 infection. Aβ evolvability might be an epigenetic phenomenon transmitted transgenerationally to confer resistance against the HSV1 infection in offspring during reproduction, which may be beneficial in evolution. However, evolvability might lead to T2DM during parental aging through the antagonistic pleiotropy mechanism. The Aβ evolvability is increased by various causes, such as the CS of APs, may result in an efficient delivery of information of stresses associated with the HSV1 infection for offspring, but increased frequency of AD in parents aging. The CS of APs and the antagonistic pleiotropy may be targets of therapy strategy (Tx).
![Seeding of Aβ fibrils by HSV1 infection. a) Ultrstractural analysis of immunoelectron micrography shows Aβ fibrillization seeded by HSV1 in cell culture leading to virus capture and entrapment. Please see the experimental conditions in Eimer et al. (2018) [45]. Reprinted from Eimer et al. (2018) [45] with permission. b) Schematic of Aβ evolvability of and disease manifestation related to HSV1 infection. Aβ evolvability might be an epigenetic phenomenon transmitted transgenerationally to confer resistance against the HSV1 infection in offspring during reproduction, which may be beneficial in evolution. However, evolvability might lead to T2DM during parental aging through the antagonistic pleiotropy mechanism. The Aβ evolvability is increased by various causes, such as the CS of APs, may result in an efficient delivery of information of stresses associated with the HSV1 infection for offspring, but increased frequency of AD in parents aging. The CS of APs and the antagonistic pleiotropy may be targets of therapy strategy (Tx).](https://content.iospress.com:443/media/jpd/2019/9-4/jpd-9-4-jpd191675/jpd-9-jpd191675-g004.jpg)
Given that amyloid-fibril structures are shared among APs, it is likely that APs other than Aβ may similarly entrap viral particles and neutralize viral infection. Indeed, many studies recently have described an association between virus infection and neurodegenerative as another important common feature of these disorders, not only HSV1 in AD, but also H1N1 influenza virus in PD and retroviruses in ALS [47]. As Aβ is protective against HSV1, it is possible that αS and TDP-43 might also be protective against some other viruses. In particular, it is of interest to determine whether the CS of APs might further enhance protection against various viral infections. It would be great interest to experimentally validate such hypotheses using tg mouse models for the various APs.
Comparison of APM with other hypotheses
There are at the present, at least three hypotheses which are relevant to the aggregation of APs, namely APM, EVH and ACH (Table 1). The ACH focuses on neurodegeneration, a pathological phenomenon in aging, whereas the EVH covers evolvability, a physiological phenomenon mainly in the developmental/reproduction stage in offspring/parents, as well as aging by antagonistic pleiotropy in parent. In contrast to these two hypotheses, the APM addresses a singular factor, namely viral infection, which may occur during any stage of life.
Table 1
Comparison of the APM with other hypotheses; the EVH and the ACH. The APM was compared with two other hypotheses, the ACH and the EVH, in terms of ‘biological significance’, ‘life-stage’, ‘inheritance’, ‘role of the CS of APs’, ‘role of Aβ’, and ‘experimental approach’
| ACH | EVH | APM | |
| Role of the CS of APs | Pathological | Physiological | Physiological |
| Life-stage | Aging | Development/Reproduction | Development/Reproduction, Aging |
| Inheritance | Not addressed | Inheritance of acquired characteristic | Not addressed |
| Experimental approach | Reccombinant proteins, Cells, and Animals | To be addressed | Cells and Animals |
| Role of Aβ | Dominant | Similar to other APs | Exclusive |
Because aging-associated neurodegeneration and viral encephalitis are commonly associated with various stresses, including neurotoxins, inflammation, oxidative stress and hyperthermia, both the APM and the ACH may correspond to hormesis in the EVH, but fails to encompass the relevance to transgenerational transmission from parents to offspring of the information regarding stressors (Fig. 4b). Although the APM is physiological, whereas the ACH is pathological, both are similar in that Aβ is dominant compared to other APs, and in that both hypotheses are based on experimental results (Table 1). On the other hand, the EVH presumes the presence of the APs protofibrils, but is attractive due to a quality of inheritance of APs (Table 1). At this point, the three hypotheses have both advantages and disadvantages, and the biological significance of the CS of APs may depend on all these hypotheses.
IMPLICATION FOR THERAPY STRATEGY
Since a disease-modifying therapy for neurodegenerative disorders is presently unavailable, the many alternative theories of neurodegeneration must be seriously considered and perhaps leveraged to increase the likelihood that such as therapy will be identified. Thus, the EVH may provide novel insights into neurodegenerative etiologies and also open the door to potential therapies.
Current strategy: Anti-aggregation
In the past decade or more, the ACH has been the predominant theory in the field of AD, which also has strongly influenced views on other related neurodegenerative conditions. As a result, the current experimental therapeutic paradigm is directed at mitigating protein aggregation to reduce the neurotoxicity of AP aggregates. Based on this, clinical trials using various therapeutic modalities, including antioxidants, anti-inflammatory agents and immunotherapies, have been evaluated extensively and/or are still in progress [48–50]. Despite numerous studies, none has proven efficacious, failing to meet established target endpoints. To be effective, it is highly possible that therapeutic interventions must be initiated early in disease pathogenesis. As a result, a phase 3 study of Aβ immunotherapy was conducted in dominantly inherited AD to assess the effect of therapeutic intervention in the pre-symptomatic stage [51]. Recently, however, it was revealed that the trial in early-onset AD was unsuccessful and thus terminated [52]. As for the failure of such clinical trials, at least two possibilities should be considered. First, immunotherapy against Aβ was not sufficiently efficacious. More specifically, although it was previously shown that there were significant decreases in amyloid plaque load in autopsy brain from Aβ immunotherapy patients, it is possible that residual neurotoxic APs protofibrils were still present. Indeed, neuropathologic examination of participants in previous clinical trials (e.g., AN1792: an active Aβ42 immunization by Elan Pharmaceuticals) may support of this notion [53]. Despite efficient removal of senile plaques in AD brain, most patients had progressed to severe dementia, possibly due to continued tau propagation [53], suggesting that the cross-seeding of Aβ with tau might be important in the pathogenic mechanisms of AD and the simultaneous targeting of Aβ and tau might be therapeutically more effective in AD. Since the current therapy strategy, especially immunotherapy, is directed at a single molecule among various APs, to improve efficacy, it may be necessary to consider collectively, protein aggregation involving combinations of multiple APs (Fig. 4b).
Antagonistic pleiotropy as a therapy target
Alternatively, the protein aggregation of Aβ might be unrelated to neurotoxicity during neurodegeneration, and in this case, it may be necessary to devise a novel therapy strategy. Presuming that neurodegeneration might be a manifestation of evolvability through antagonistic pleiotropy in aging, it follows that an attractive alternate therapy strategy might focus on the antagonistic pleiotropy mechanism (Fig. 4b). In this regard, a recent study suggests that a 2q22 region corresponding to the TGFβ/activin receptor-signaling pathways might be linked to the risk of major human diseases, including neurodegenerative disorders [54]. It is therefore possible that targeting and modifying the TGFβ/activin receptor-signaling pathways could be therapeutically beneficial for neurodegeneration [37]. This might be assessed by applying various treatment strategies such as receptor antagonists and antisense RNA, to tg mouse models of neurogeneration.
CONCLUSION
Considering that the CS of APs is normally observed in microorganisms, it is predicted that the CS of APs may not only be pathological, but also physiologically relevant to human biology. In this context, the CS of APs in neurodegenerative diseases might reflect an antagonistic phenomenon of the CS of APs in evolvability, a potentially beneficial physiological function. Furthermore, much attention has recently been paid to the role of viral infection in stimulating Aβ aggregation. Since this phenomenon is consistent with the concept of amyloidogenic evolvability, it is intriguing to determine whether the CS of APs is observed in this context.
With this in mind, therapeutic strategies for neurodegeneration might require restructuring. Generally considered as the reason for the failure of clinical trials in AD and PD, excessively late therapeutic intervention may not be the only issue, whereas simultaneously, the importance of the CS of APs might be overlooked. Based on the ACH, current therapeutic strategies, including immunotherapy targets anti-protein aggregation, yet if this concept is carefully examined, directing efforts at protein aggregation by multiple APs rather than individual AP molecules might be more effective. Finally, it should be remembered that there are currently few reports as to the CS of multiple APs in human physiology. Because the CS of APs is a phenomenon in post-reproductive senescence that has specifically evolved in humans, the findings observed in other neurodegeneration models, such as tg mice, may be interpreted with caution in terms of their application to human brain.
CONFLICT OF INTEREST
The authors declare that they have no competing interests.
ACKNOWLEDGMENTS
We are grateful for the continuous encouragement of Drs. Kaori Hashimoto (Tokyo Metropolitan Institute of Medical Science) and Maria del Carmen Ruiz de la Cruz (University of Chicago).
REFERENCES
[1] | Morris GP , Clark IA , Vissel B ((2014) ) Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s disease. Acta Neuropathol Commun 2: , 135. |
[2] | Kowalska A ((2004) ) Genetic basis of neurodegeneration in familial Alzheimer’s disease. Pol J Pharmacol 56: , 171–178. |
[3] | Roda AR , Montoliu-Gaya L , Villegas S ((2019) ) The role of apolipoprotein E isoforms in Alzheimer’s disease. J Alzheimers Dis 68: , 459–471. |
[4] | Nussbaum RL , Ellis CE ((2003) ) Alzheimer’s disease and Parkinson’s disease. N Engl J Med 348: , 1356–1364. |
[5] | Ricciarelli R , Fedele E ((2017) ) The amyloid cascade hypothesis in Alzheimer’s disease: It’s time to change our mind. Curr Neuropharmacol 15: , 926–935. |
[6] | Hashimoto M , Ho G , Sugama S , Takamatsu Y , Shimizu Y , Takenouchi T , Waragai M , Masliah E ((2018) ) Evolvability of amyloidogenic proteins in human brain. J Alzheimers Dis 62: , 73–83. |
[7] | Liu Y ((2007) ) Like father like son. A fresh review of the inheritance of acquired characteristics. EMBO Rep 8: , 798–803. |
[8] | Kishimoto S , Uno M , Okabe E , Nono M , Nishida E ((2017) ) Environmental stresses induce transgenerationally inheritable survival advantages via germline-to-soma communication in Caenorhabditis elegans. Nat Commun 8: , 14031. |
[9] | Williams GC ((1957) ) Pleiotropy, natural selection, and the evolution of senescence. Evolution 11: , 398–411. |
[10] | Williams GC , Nesse RM ((1991) ) The dawn of Darwinian medicine. Q Rev Biol 66: , 1–22. |
[11] | Spires-Jones TL , Attems J , Thal DR ((2017) ) Interactions of pathological proteins in neurodegenerative diseases. Acta Neuropathol 134: , 187–205. |
[12] | Lippa CF , Fujiwara H , Mann DM , Giasson B , Baba M , Schmidt ML , Nee LE , O’Connell B , Pollen DA , St George-Hyslop P , Ghetti B , Nochlin D , Bird TD , Cairns NJ , Lee VM , Iwatsubo T , Trojanowski JQ ((1998) ) Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol 153: , 1365–1370. |
[13] | Hashimoto M , Masliah E ((2003) ) Cycles of aberrant synaptic sprouting and neurodegeneration in Alzheimer’s and dementia with Lewy bodies. Neurochem Res 28: , 1743–1756. |
[14] | Liscic RM , Breljak D ((2011) ) Molecular basis of amyotrophic lateral sclerosis. Prog Neuropsychopharmacol Biol Psychiatry 35: , 370–372. |
[15] | Forman MS , Schmidt ML , Kasturi S , Perl DP , Lee VM , Trojanowski JQ ((2002) ) Tau and alpha-synuclein pathology in amygdala of Parkinsonism-dementia complex patients of Guam. Am J Pathol 160: , 1725–1731. |
[16] | Schmidt ML , Lee VM , Saido T , Perl D , Schuck T , Iwatsubo T , Trojanowski JQ ((1998) ) Amyloid plaques in Guam amyotrophic lateral sclerosis/parkinsonism-dementia complex contain species of A beta similar to those found in the amyloid plaques of Alzheimer’s disease and pathological aging. Acta Neuropathol 95: , 117–122. |
[17] | Hasegawa M , Arai T , Akiyama H , Nonaka T , Mori H , Hashimoto T , Yamazaki M , Oyanagi K ((2007) ) TDP-43 is deposited in the Guam parkinsonism-dementia complex brains. Brain 130: , 1386–1394. |
[18] | Cox PA , Sacks OW ((2002) ) Cycad neurotoxins, consumption of flying foxes, and ALS-PDC disease in Guam. Neurology 58: , 956–959. |
[19] | Kuzuhara S , Kokubo Y ((2005) ) Atypical parkinsonism of Japan: Amyotrophic lateral sclerosis-parkinsonism-dementia complex of the Kii peninsula of Japan (Muro disease): An update. Mov Disord 20: (Suppl 12), S108–113. |
[20] | Morales R , Moreno-Gonzalez I , Soto C ((2013) ) Cross-seeding of misfolded proteins: Implications for etiology and pathogenesis of protein misfolding diseases. PLoS Pathog 9: , e1003537. |
[21] | Rank KB , Pauley AM , Bhattacharya K , Wang Z , Evans DB , Fleck TJ , Johnston JA , Sharma SK ((2002) ) Direct interaction of soluble human recombinant tau protein with Abeta 1-42 results in tau aggregation and hyperphosphorylation by tau protein kinase II. FEBS Lett 514: , 263–268. |
[22] | Guo JP , Arai T , Miklossy J , McGeer PL ((2006) ) Abeta and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer’s disease. Proc Natl Acad Sci U S A 103: , 1953–1958. |
[23] | Yoshimoto M , Iwai A , Kang D , Otero DA , Xia Y , Saitoh T ((1995) ) NACP, the precursor protein of the non-amyloid beta/A4 protein (A beta) component of Alzheimer disease amyloid, binds A beta and stimulates A beta aggregation. Proc Natl Acad Sci U S A 92: , 9141–9145. |
[24] | Masliah E , Rockenstein E , Veinbergs I , Sagara Y , Mallory M , Hashimoto M , Mucke L ((2001) ) beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc Natl Acad Sci U S A 98: , 12245–12250. |
[25] | Guerrero-Munoz MJ , Castillo-Carranza DL , Krishnamurthy S , Paulucci-Holthauzen AA , Sengupta U , Lasagna-Reeves CA , Ahmad Y , Jackson GR , Kayed R ((2014) ) Amyloid-beta oligomers as a template for secondary amyloidosis in Alzheimer’s disease. Neurobiol Dis 71: , 14–23. |
[26] | Guo JL , Covell DJ , Daniels JP , Iba M , Stieber A , Zhang B , Riddle DM , Kwong LK , Xu Y , Trojanowski JQ , Lee VM ((2013) ) Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 154: , 103–117. |
[27] | Katorcha E , Makarava N , Lee YJ , Lindberg I , Monteiro MJ , Kovacs GG , Baskakov IV ((2017) ) Cross-seeding of prions by aggregated alpha-synuclein leads to transmissible spongiform encephalopathy. PLoS Pathog 13: , e1006563. |
[28] | Van Gerven N , Van der Verren SE , Reiter DM , Remaut H ((2018) ) The role of functional amyloids in bacterial virulence. J Mol Biol 430: , 3657–3684. |
[29] | Kostakioti M , Hadjifrangiskou M , Hultgren SJ ((2013) ) Bacterial biofilms: Development, dispersal, and therapeutic strategies in the dawn of the postantibiotic era. Cold Spring Harb Perspect Med 3: , a010306. |
[30] | Hartman K , Brender JR , Monde K , Ono A , Evans ML , Popovych N , Chapman MR , Ramamoorthy A ((2013) ) Bacterial curli protein promotes the conversion of PAP248-286 into the amyloid SEVI: Cross-seeding of dissimilar amyloid sequences. Peerj 1: , e5. |
[31] | Keefer KM , Stein KC , True HL ((2017) ) Heterologous prion-forming proteins interact to cross-seed aggregation in Saccharomyces cerevisiae. Sci Rep 7: , 5853. |
[32] | Greenwald J , Riek R ((2012) ) On the possible amyloid origin of protein folds. J Mol Biol 421: , 417–426. |
[33] | Wang C , Zhao C , Li D , Tian Z , Lai Y , Diao J , Liu C ((2016) ) Versatile structures of alpha-synuclein. Front Mol Neurosci 9: , 48. |
[34] | Kirschner M , Gerhart J ((1998) ) Evolvability. Proc Natl Acad Sci U S A 95: , 8420–8427. |
[35] | Hashimoto M , Ho G , Takamatsu Y , Wada R , Sugama S , Takenouchi T , Masliah E , Waragai M ((2018) ) Possible role of the polyglutamine elongation in evolution of amyloid-related evolvability. J Huntingtons Dis 7: , 297–307. |
[36] | Roan NR , Sandi-Monroy N , Kohgadai N , Usmani SM , Hamil KG , Neidleman J , Montano M , Standker L , Rocker A , Cavrois M , Rosen J , Marson K , Smith JF , Pilcher CD , Gagsteiger F , Sakk O , O’Rand M , Lishko PV , Kirchhoff F , Munch J , Greene WC ((2017) ) Semen amyloids participate in spermatozoa selection and clearance. Elife 6: , e24888. |
[37] | Takamatsu Y , Fujita M , Ho GJ , Wada R , Sugama S , Takenouchi T , Waragai M , Masliah E , Hashimoto M ((2018) ) Motor and nonmotor symptoms of Parkinson’s disease: Antagonistic pleiotropy phenomena derived from alpha-synuclein evolvability? Parkinsons Dis 2018: , 5789424. |
[38] | Sharma A ((2017) ) Transgenerational epigenetics: Integrating soma to germline communication with gametic inheritance. Mech Ageing Dev 163: , 15–22. |
[39] | Lim CZJ , Zhang Y , Chen Y , Zhao H , Stephenson MC , Ho NRY , Chen Y , Chung J , Reilhac A , Loh TP , Chen CLH , Shao H ((2019) ) Subtyping of circulating exosome-bound amyloid beta reflects brain plaque deposition. Nat Commun 10: , 1144. |
[40] | Hashimoto M , Ho G , Takamatsu Y , Shimizu Y , Sugama S , Takenouchi T , Waragai M , Masliah E ((2018) ) Evolvability and neurodegenerative disease: Antagonistic pleiotropy phenomena derived from amyloid aggregates. J Parkinsons Dis 8: , 405–408. |
[41] | Moreno-Gonzalez I , Edwards Iii G , Salvadores N , Shahnawaz M , Diaz-Espinoza R , Soto C ((2017) ) Molecular interaction between type 2 diabetes and Alzheimer’s disease through cross-seeding of protein misfolding. Mol Psychiatry 22: , 1327–1334. |
[42] | Martinez-Valbuena I , Amat-Villegas I , Valenti-Azcarate R , Carmona-Abellan MDM , Marcilla I , Tunon MT , Luquin MR ((2018) ) Interaction of amyloidogenic proteins in pancreatic beta cells from subjects with synucleinopathies. Acta Neuropathol 135: , 877–886. |
[43] | Giasson BI , Forman MS , Higuchi M , Golbe LI , Graves CL , Kotzbauer PT , Trojanowski JQ , Lee VM ((2003) ) Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300: , 636–640. |
[44] | Chuang E , Hori AM , Hesketh CD , Shorter J ((2018) ) Amyloid assembly and disassembly. J Cell Sci 131: , jcs189928. |
[45] | Eimer WA , Vijaya Kumar DK , Navalpur Shanmugam NK , Rodriguez AS , Mitchell T , Washicosky KJ , Gyorgy B , Breakefield XO , Tanzi RE , Moir RD ((2018) ) Alzheimer’s disease-associated beta-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron 100: , 1527–1532. |
[46] | Moir RD , Lathe R , Tanzi RE ((2018) ) The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement 14: , 1602–1614. |
[47] | Limongi D , Baldelli S ((2016) ) Redox imbalance and viral infections in neurodegenerative diseases. Oxid Med Cell Longev 2016: , 6547248. |
[48] | Polidori MC , Nelles G ((2014) ) Antioxidant clinical trials in mild cognitive impairment and Alzheimer’s disease - challenges and perspectives. Curr Pharm Des 20: , 3083–3092. |
[49] | Wu SS , Frucht SJ ((2005) ) Treatment of Parkinson’s disease: What’s on the horizon? CNS Drugs 19: , 723–743. |
[50] | Hull M , Berger M , Heneka M ((2006) ) Disease-modifying therapies in Alzheimer’s disease: How far have we come? Drugs 66: , 2075–2093. |
[51] | Sperling R , Mormino E , Johnson K ((2014) ) The evolution of preclinical Alzheimer’s disease: Implications for prevention trials. Neuron 84: , 608–622. |
[52] | Fagan T ((2019) ) Biogen/Eisai Halt Phase 3 Aducanumab Trials. AlzForum, http://www.alzforum.org/news/research-news/biogeneisai-halt-phase-3-aducanumab-trials. |
[53] | Nicoll JAR , Buckland GR , Harrison CH , Page A , Harris S , Love S , Neal JW , Holmes C , Boche D ((2019) ) Persistent neuropathological effects 14 years following amyloid-beta immunization in Alzheimer’s disease. Brain 18: , 484–514. |
[54] | Kulminski AM , He L , Culminskaya I , Loika Y , Kernogitski Y , Arbeev KG , Loiko E , Arbeeva L , Bagley O , Duan M , Yashkin A , Fang F , Kovtun M , Ukraintseva SV , Wu D , Yashin AI ((2016) ) Pleiotropic associations of allelic variants in a 2q22 region with risks of major human diseases and mortality. PLoS Genet 12: , e1006314. |


