
Sleep Disturbance as Potential Risk and Progression Factor for Parkinson’s Disease
Abstract
Sleep disturbances are common and a major source of disability in Parkinson’s disease (PD). Primary and secondary insomnia, rapid eye movement sleep behavior disorder (RDB), central sleep apnea, restless legs, and nocturnal akinesia are common sleep disturbances in PD. Prodromal presence of RBD is associated with a more severe motor and non-motor PD subtype implying a significant disease-modifying effect of this parasomnia. Other disease-modifying mechanisms of sleep disturbances in PD include impaired glymphatic clearance, endoplasmic reticulum stress, nocturnal brain deoxygenation and inflammatory processes among others. Impairments of neural circuit switching and imbalance between inhibitory and excitatory neuronal populations are likely responsible for episodic sleep disturbances, in particular RBD. As neural circuits may predict patterns of α-synuclein propagation in the nervous system, impairments of such circuits are of high relevance for PD pathophysiology. Future research is needed to determine whether appropriate treatment for disturbed sleep might slow progression of PD.
INTRODUCTION
Parkinson’s disease (PD) is believed to be a multifactorial disease caused by a complex interaction of environmental factors against a predisposed genetic background [1]. Although defining pathological features of PD are loss of dopaminergic neurons in the substantia nigra accompanied by accumulation of α-synuclein, current pathophysiological conceptions suggest a multi-system model of the disease. The multi-system involvement is not only present at the organ but also at cellular and diverse sub-cellular molecular levels. A current popular paradigm to explain disease progression is trans-synaptic propagation of α-synuclein.
Emerging evidence from the field of Alzheimer’s disease (AD) research implicates sleep quality as an important factor underlying risk and disease progression in this disorder [2, 3]. There is also evidence that the presence of sleep disturbances may increase the risk of developing PD. For example, a large population-based retrospective cohort study in over 91,000 Taiwanese patients with non-apnea sleep disorders and without evidence of pre-existing PD found that the presence of sleep disorders was an independent risk factor for incident PD compared to matched control subjects without reported sleep disorders [4]. Notably, subgroup analysis showed that patients with chronic insomnia (lasting more than 3 months) were at greatest risk. A longitudinal study of nurse shift workers also point to increased risk of PD compared to nurses without night shift jobs [5]. Prodromal presence of REM sleep behavior disorder (RBD) is associated with a more severe motor and non-motor PD subtype implying a significant disease-modifying effect of this parasomnia [6, 7]. A disease-modifying effect may explain why sleep disturbances are more common in the motor symptomatic stage of PD and further increase with greater severity of disease [8, 9]. Although some of causes of disturbed sleep could be due to other PD-related symptoms, such as nocturia, it is clear from these observations that abnormal sleep is intricately connected to mechanisms of risk, prodromal manifestations and symptomatic disease progression in PD. The purpose of this ‘hot topic’ review is to explore associations between disturbed sleep and PD, and discuss whether sleep disturbance may be an early symptomatic manifestation of prodromal PD or could act as an important disease-modifying factor in those susceptible to developing PD because of genetic, environmental or other etiologies. PubMed literature search was focused on sleep and PD pathophysiology α-synuclein, amyloid-β, tau, inflammation, disease progression and risk factors.
THE PHYSIOLOGICAL BASIS OF SLEEP: SLEEP NEUROBIOLOGY AND NEUROPHYSIOLOGY
In humans, as in most mammals, three vigilance states characterized by differences in electro-encephalogram (EEG), electromyogram (EMG), and electro-oculogram (EOG) recordings exist. Different neurochemical mechanisms acting in fine balance are responsible for the transition between the three vigilance states across the 24 hour day.
(i) Waking state typified by high-frequency (40– 300 Hz), low-amplitude desynchronized EEG activity, sustained EMG24-hourty and ocular movements on EOG.
(ii) Non-REM (NREM) sleep (also called slow-wave sleep (SWS)) typified by low-frequency (0.5– 4 Hz), high-amplitude delta oscillations on the EEG, low EMG muscular activity and no ocular movement. NREM sleep is currently subdivided into three stages according to the American Academy of Sleep Medicine scoring rules, defined mainly on the EEG. The EEG pattern in NREM sleep is described as synchronous, with characteristic wave forms such as sleep spindles, K-complexes and high-voltage slow waves. NREM stages 1 to 3 represent a depth-of-sleep continuum where the arousal threshold is generally lowest in stage 1 and highest in stage 3 sleep. NREM sleep is a stage of minimal energy expenditure and motor activity.
(iii) REM sleep (also called paradoxical sleep (PS) typified by predominant theta (6– 9 Hz) and gamma (30– 300 Hz) rhythms similar to waking state EEG, but with complete disappearance of postural muscle tone and occurrence of REMs and muscle twitches. Conversely to NREM sleep, REM sleep is defined by rapid desynchronixed, low amplitude EEG activity, muscle atonia and episodic bursts of REM’s. Usually, REM sleep is not divided into stages although for research purposes can be divided into tonic and phasic components. REM sleep incurs high energy expenditure and motor activity, due to loss of posture control, and highly unstable autonomic activity.
During wakefulness, multiple neurotransmitter systems, some of which belong to the ascending reticular activating system control wakefulness and arousal through projections to the thalamus and/or neocortex. These include serotonergic systems neurons (dorsal raphe nucleus), noradrenergic neurons (locus ceruleus), cholinergic neurons (in the pontine brainstem and basal forebrain), histaminergic neurons (tuberomamillary nucleus), and the orexin (hypocretin) system in the hypothalamus [10]. During sleep wakefulness systems are progressively inhibited, likely through the effects of γ-aminobutyric acid (GABA) leading to non-REM sleep. The balance between wakefulness and SWS is due to reciprocal inhibitory projections between GABA neurons in the forebrain preoptic nucleus and a number of waking arousal systems.
Non-REM sleep is driven by slow-wave sleep-promoting neurons in forebrain preoptic sleep center, in particular the median preoptic nucleus (MnPn) and the ventrolateral preoptic nucleus (VLPO). Tract studies suggest that the VLPO and MnPn neurons are reciprocally connected with the above wake-active neurons. This led to the current hypothesis that during wakefulness VLPO/MnPn GABAergic neurons are inhibited by multiple noradrenergic and cholinergic arousal circuits. At sleep onset, these neurons start to fire in response to unidentified excitatory inputs, as well as homeostatic (adeonosine and serotonin) and circadian drives through suprachiasmatic input, causing sleep onset and maintaining slow wave sleep [10].
REM sleep (also called paradoxical sleep) is caused by the activation of glutamatergic neurons localized in rats to the sublaterodorsal tegmental nucleus (SLD) of the pontine brainstem [11]. In humans, the corresponding areas are likely to be the subceruleus nucleus and magnocellularis nucleus in the brainstem. Disinhibited ascending glutamatergic neurons generate cortical activation via projections to thalamic relay neurons. Descending glutamatergic neurons induce the muscle atonia seen in REM sleep by excitatory projections to GABA and glycinergic pre-motoneurons located in the medulla. The onset of paradoxical REM sleep is controlled by the activation of GABA and melanin-concentrating hormone neurons in the lateral hypothalamus, removing the tonic inhibition of the glutamatergic SLD neurons during wakefulness and slow wave sleep.
AGING, SLEEP PATTERN, AND SLEEP STAGES
General statements about sleep can only be made regarding sleep in the normal young adult without sleep complaints, living on a conventional sleep-wake cycle. This is because in elderly people, there is a profound increase in inter-individual sleep variability that precludes any generalizations other than those in young adults [12]. Aging is accompanied by a reduction in sleep quality and quantity, with a progressive reduction in sleep slow wave activity from the age of thirty years. Increasing evidence suggests that slow wave activity reflects synaptic density or the strength of cortical synapses, and reduced SWS in the elderly is associated with grey matter medial prefrontal atrophy as well as an age-related decline in memory performance. With aging, time spent in bed reduces due to earlier wake-up times compared to younger adults. The circadian rhythm change with increasing age is characterized by a phase advance of around 1 hour, with earlier bed times and wake up times. Aging is also associated with a weakening of circadian regulation, with the elderly having reduced 24 hour melatonin secretion and circadian modulation of REM sleep.
The normal young human adult enters sleep through non-REM sleep. REM sleep does not occur until 80 minutes or longer following; and non REM sleep and REM sleep alternate throughout the night with an approximate 90 minute cycle [12]. In young adults, slow wave sleep dominates the non-REM portion of the first one-third of the night, while REM sleep episodes are longest in the last one-third of the night. Brief episodes of wakefulness occur later in the night, usually near REM sleep transitions, but are usually too brief to be remembered in the morning. The preferential delineation of REM sleep in the later portion of the night is likely linked to the circadian oscillator, gauged by the oscillation of body temperature. Slow wave sleep, on the other hand, is not mediated by circadian processes but by sleep timing and previous wake duration.
In general non REM sleep constitutes around 75– 80% of total sleep, while REM sleep is usually 20– 25% of sleep occurring in four to six discrete episodes during the night. During sleep, an individual may switch back and forth from non REM to REM sleep, with occasional transitions to periods of wakefulness. The characteristic EEG pattern in non REM sleep is described as synchronous, with characteristic wave forms including sleep spindles, K-complexes and high-voltage slow waves.
Non-REM sleep is divided into three stages according to the American Academy of Sleep Medicine scoring rules, defined mainly on the EEG. Non-REM stages 1 to 3 represent a depth of sleep continuum, where the arousal threshold is generally lowest in stage 1 and highest in stage 3 sleep. There are strong associations between non-REM sleep and memory consolidation processes. Non-REM sleep is a state of minimal energy expenditure and motor activity during which cardiorespiratory and thermoregulatory variables are driven by the autonomic nervous system and a lower and more stable level compared to wakefulness.
During REM sleep in contrast, posture control is lost, autonomic activity is highly unstable with centrally driven surges in heart rate and blood pressure, breathing is irregular and thermoregulation is suspended. REM sleep is defined by rapid desynchronized, low amplitude EEG activity, muscle atonia and episodic bursts of rapid eye movements.
The link between sleep and body temperature regulation and corresponding energy expenditure is well recognized. The most favorable moment of the day for sleep is the rest period when the circadian system drives a reduction in core body temperature and energy expenditure. The rest period is also when the human or animal is least likely to actively interact with its external environment. Overall, body energy expenditure decreases during sleep. Brain energy metabolism measured using brain glucose utilization or oxygen uptake imaging techniques, progressively decreases during successive non REM sleep stages. In contrast, during REM sleep brain energy metabolism increases to levels similar or even in excess of wakefulness.
SLEEP-WAKE DISTURBANCE IN PD: MULTIFACTORIAL ETIOLOGIES
Sleep-wake disturbance is particularly common in PD occurring in up to 75– 80% of patients over the course of disease [9, 13]. Sleep problems are among the most distressing non-motor symptoms of the disease, impacting the quality of life of patients and caregivers [14]. In the largest survey of non-motor symptoms of PD, 64% of newly diagnosed patients reported sleep problems making it the second most common non-motor complaint [15]. The most common problems are sleep fragmentation, sleep-related breathing disorders, restless legs/periodic leg movements, RBD, nocturnal hallucinations and altered sleep-wake cycle [16]. Sleep efficiency, which is the percentage of time spent asleep in bed is reduced in PD. PD patients also experience sleep attacks and excessive daytime sleepiness [17]. Lastly, patients with PD also experience both primary insomnia and secondary insomnia commonly due to depression, or nocturnal worsening of motoric/non-motor symptoms including nocturia, tremor, nocturnal akinesia, and spasms.
Subjective and objective measures of sleep often show contradictory findings. In particular middle-aged and older adults with insomnia tend to overestimate their lack of sleep, and consistently report shorter sleep durations than those measured with actigraphy [18]. Few studies have compared subjective and objective measures of sleep in patients with chronic disease. Actigraphy has emerged as an objective means of measuring sleep in PD, including sleep quality [19]. Actigraphically-defined sleep disturbance in PD has been shown to be associated with differential aspects of cognitive functioning [20]. Rather than a global pattern of cognitive dysfunction, reduced sleep efficiency in PD versus control subjects was significantly associated with working memory and verbal memory consolidation, while verbal fluency and attentional set-shifting ability was spared.
Few polysomnography (PSG) studies have been performed in PD. A study of 30 newly diagnosed PD subjects versus 15 age/gender matched controls admitted for 2 consecutive nights of PSG recording found PD patients exhibited increased sleep latency (time to sleep onset), reduced sleep efficiency, increased non-REM stage I sleep, and reduced REM sleep [21]. Two PD patients exhibited no REM sleep whatsoever, and aspects of sleep architecture were similarly affected in patients with and without obstructive sleep apnea. PD patients also showed a sustained elevation in serum cortisol levels and reduced circulating melatonin levels. Eight out of 30 PD patients (27%) had polysomnographic evidence of RBD. Other PSG studies have confirmed RBD in 46% of 457 PD cases with subjective sleep complaints and average disease duration of 7.5 years [22]. Similar findings have been shown in early, untreated PD [23], with progressive ‘destructuring of sleep architecture’ found in patients with more advanced PD [8].
REM SLEEP BEHAVIOR DISORDER AND DISEASE MODIFICATION IN PD
Impairments of neural circuit switching and imbalance between the inhibitory and excitatory neuronal populations described above are likely responsible for episodic sleep disturbances, in particular found in RBD [24, 25]. This might occur through malfunction of a putative ‘flip-flop’ switch for REM control, or through a breakdown of the underlying REM sleep circuitry [24]. The majority of idiopathic RBD cases will eventually be diagnosed with PD, dementia with Lewy bodies (DLB) or multiple system atrophy (MSA), with clinicopathological correlations in 172 RBD cases showing that the vast majority has a α-synucleinopathy [26]. RBD is common among the α-synucleinopathy disorders of PD, DLB and MSA probably because in these diseases cell loss is common within neuronal structures regulating REM sleep atonia, namely the subceruleus nucleus and magnocellularis nucleus in the brainstem, and the amygdala, which is linked to the emotional content of dreams.
RBD is characterized by dream-enacting behaviors and nightmares linked to REM sleep without muscle atonia. RBD can be classified into an idiopathic form (iRBD) and a secondary form, which occurs in patients already diagnosed with PD (25– 58% of cases), DLB (70– 80%), MSA (90– 100%), autoimmune disease (e.g., narcoplepsy, paraneoplastic syndromes, limbic encephalitis, IgLON5 parasomnia, multiple sclerosis) and focal brainstem lesions (e.g., tumor, stroke, demyelinating plaques). The use of some medications (e.g., antidepressants and β-adrenergic blockers) can also trigger RBD.
Other than α-synucleinopathies, RBD has also been described in small series of other neurodegenerative disorders including progressive supranuclear palsy and Machado-Joseph disease, but is rare in AD, Huntington’s disease, amyotrophic lateral sclerosis, corticobasal degeneration and frontotemporal dementias. In these disorders, combined brainstem and limbic neuronal damage is rare. RBD is also rare among tauopathies, amyloidopathies and ubiquitinopathies, thereby representing the archetypal synuclein disorder.
As over 80% of idiopathic RBD patients will eventually develop an α-synuclein disorder, the dichotomy between idiopathic RBD and secondary RBD could be seen as a false one, as both disorders might represent different ends of the clinicopathological disease continuum. Careful phenotyping of large iRBD cohorts demonstrates these individuals already have a virtually identical non-motor phenotype as those with early PD, with similar rates of depression, anxiety, apathy, cognitive impairment, and postural hypotension [7, 27]. Motor deficits in idiopathic RBD at a group level are already sufficient to distinguish them from age and gender-matched control subjects using standard rating scales or digital motor testing, however the degree of motor impairment is not yet as severe as that encountered in established early PD patients [28].
To date the largest ever study of 1,280 polysomnographically-diagnosed iRBD subjects from 24 international RBD Study Group sleep centers, found an overall conversion rate from iRBD to an overt neurodegenerative syndrome of 6.3% per year, with 73.5% converting after 12-year follow up. The rate of conversion was significantly increased with abnormal quantitative motor testing (HR-3.16), objective motor examination (HR = 2.13), motor symptoms (HR = 2.11), an abnormal dopamine nerve terminal scan (HR = 1.98), color vision abnormalities (HR = 1.69), constipation (HR = 1.67), REM atonia loss (HR = 1.54) and age (HR = 1.54) [29].
Interestingly, both video-PSG analysis and bed partner’s reports demonstrate alleviation of parkinsonism during complex RBD episodes; movements seen during RBD episodes were faster, stronger and smoother than movements made while the PD patient was awake. Similarly, speech during RBD episodes was better articulated and louder than during wakefulness, implying restoration of normal muscle control and movement in PD during REM sleep [30].
An ‘RBD-PD phenotype’ has been described in some studies, where patients have more symmetric, akinetic-rigid dominant motor disease, older age, male gender, increased falls, autonomic dysfunction, excessive daytime somnolence, visual hallucinations and increased risk of developing future dementia [31]. A longitudinal study of 61 PD patients with and without PSG-determined RBD at baseline found that by 4 years, 48% of PD patients with RBD had developed dementia, compared to 0% without [6]. This suggests that the presence of RBD in PD is related to widespread neuropathological changes in the brain. Our group have used data-driven approaches to delineate PD subtypes in >2,500 early patients, finding the most severe subtype was delineated by severe baseline motor disease, poor psychological well being and poor sleep including worse RBD on screening questionnaires [32].
SLEEP APNEA AND INCIDENT PD: INTERMITTENT HYPO-OXYGENATION AND INFLAMMATORY MECHANISMS
Sleep apnea is characterized by repeated periods of hypoxia and reoxygenation during sleep. Sleep apnea may have central, obstructive/peripheral or mixed causes. It has been hypothesized that sleep apnea-induced chronic intermittent hypoxia may increase oxidative stress and inflammation [33], which may contribute to the pathophysiology of PD [34]. A recent 3-year nationwide retrospective population-based study of 1,944 Taiwanese patients with sleep apnea and 9,720 matched control subjects showed a higher risk of incident PD diagnosis (0.9%) in the apneic patients compared to 0.4% in the controls [35]. The risk of PD remained significant after adjustments for potential confounders, including sex, age, hypertension, lung disease, head injury, rural vs. urban living, geographic region, income, diabetes, hypertension and hyperlipidemia. After age and sex stratification, PD development was independently associated with sleep apnea only in men (adjusted hazard ratio 2.26; 95% CI 1.11– 4.63) and in patients aged 60 years and over (adjusted hazard ratio 0.93; 95% CI 1.01– 3.71). In contrast, women did not exhibit a significantly increased risk of PD in the sleep apnea cohort compared to those in the non-apnea cohort during the 3-year follow-up period. Whether sleep is an independent risk factor for PD or merely an early symptom during the prodromal phase of PD remains unclear [35].
Sleep apnea-associated intermittent brain hypo-oxygenation and inflammation may potentially accelerate the degenerative process in already vulnerable or affected nigral dopaminergic neurons. As such the presence of sleep apnea may exacerbate or accelerate the clinical manifestation of prodromal PD rather than a primary triggering event. Furthermore, the high prevalence of sleep apnea in the general population widely exceeds the prevalence of PD suggesting the presence of either prodromal PD or other PD-specific susceptibility or vulnerability factors in at risk patients. Women, at least at pre-menopausal age, have higher binding levels of nigostriatal nerve terminals compared to males [36]. Given the prominent age-associated declines of striatal dopamine transporters in both genders [37], the extra dopaminergic buffer may provide a delayed phase protection in women compared to men. It is clear, however, that age-associated losses in nigrostriatal nerve terminals and longer duration of untreated apnea may explain why older age is an important risk factor for incident PD in patients with sleep apnea. Sleep apnea is also associated with substantial cardiovascular morbidity. Therefore, it is possible that at least some of the sleep apnea patients with incident motor parkinsonism may have vascular parkinsonism rather than idiopathic PD. This notion is supported by postmortem data from the Honolulu aging study where the presence of hypo-oxygenation during life was found to be associated with the development of micro-infarcts and brain atrophy but not AD or Lewy body pathology [38].
Neuroinflammation is now being recognized as playing an important role in neurodegeneration. Recent studies from the AD field have shed further light on this intriguing area of research where inflammation caused by poor sleep is believed to increase amyloid-β burden. Furthermore, a bidirectional relationship between sleep and AD is supported by advances in our understanding of sleep disturbance-induced increases in systemic inflammation (for review, see [2], see also Fig. 1). More specifically, sleep disturbance may lead to increases in systemic inflammation (as measured by proinflammatory cytokines and C-reactive protein), which in turn is hypothesized to ‘prime’ microglial cells in the brain. Priming of microglia cells will compromise these cells’ abilities to clear amyloid-β, which in turn may result in more increased production of pro-inflammatory cytokines and local inflammation in the central nervous system (CNS). In other words, initiation of this cycle may result in a forward feedback loop, in which greater increases in local inflammation in the brain results in more priming of activated microglial cells with greater production of AD pathology, including neuronal damage, etc [2]. Although this theory is attractive and of high relevance for the field of AD, it lacks specificity to explain the increased risk of PD rather than AD unless one assumes the presence of pre-existing individual genetic or other vulnerability to develop PD. The AD model, however, may explain the emergence of AD pathology with disturbed sleep in patients with later stage PD at risk of developing dementia. As expected, genetic predisposition appears to play an important role in systemic and neuroinflammation and risk of neurodegeneration. For example, a recent prospective analysis of the Framingham cohort study found evidence of an interaction between APOE4 status and systemic inflammation that dramatically increases the risk of AD [39].
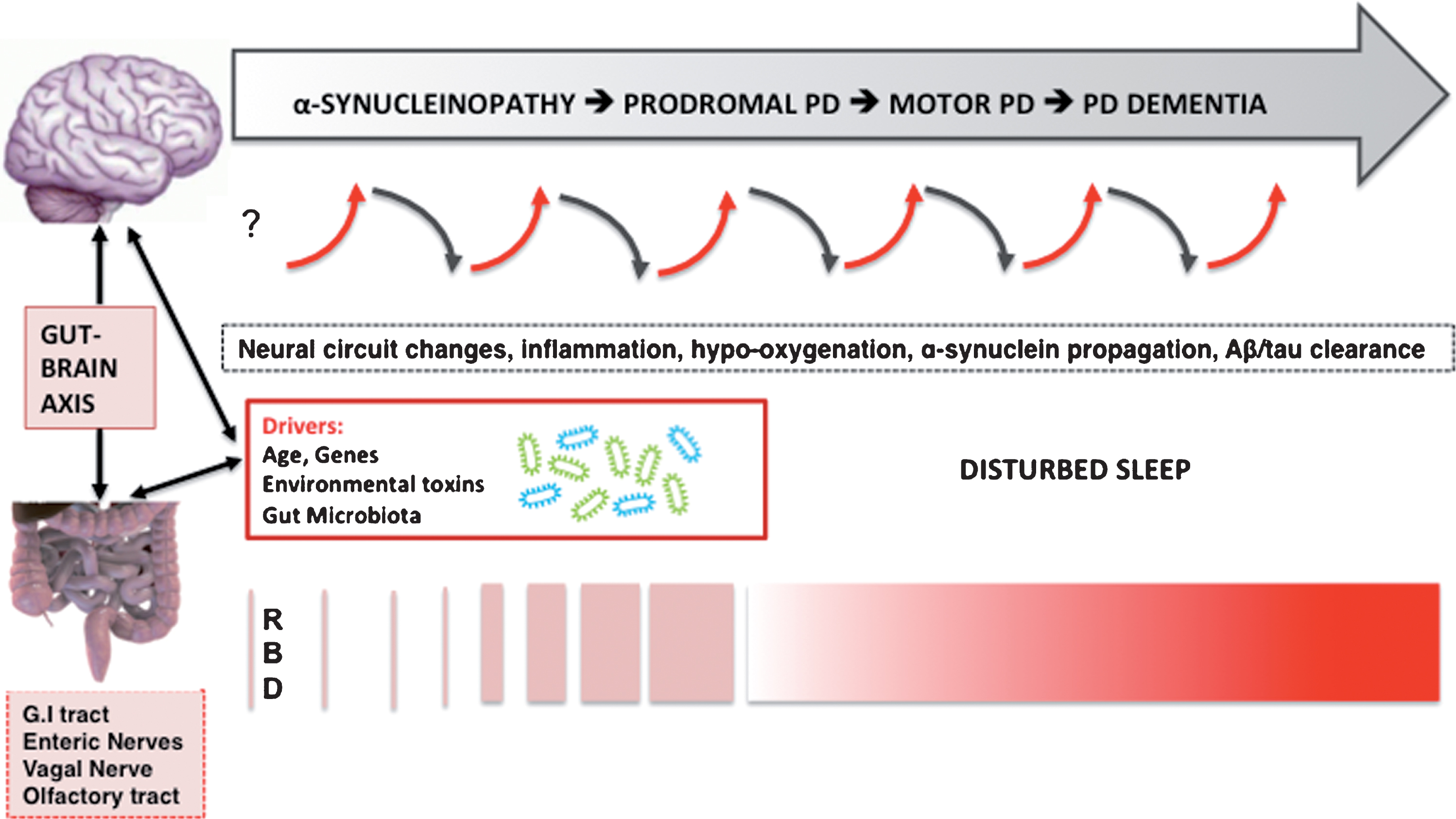
Fig.1
Hypothesized bidirectional changes between the presence of disturbed sleep, risk, and progression of PD. The selected neurodegenerative mechanisms do not follow a pre-specified order and likely overlap.
MODIFYING THE ACCUMULATION OF ABNORMAL PROTEINS AND DISTURBED SLEEP: THE GLYMPHATIC SYSTEM
Abnormal build-up of proteins, such as α-synuclein, amyloid-β, TDP-43, or phosphorylated tau may occur in PD. The ‘glymphatic’ system is a paravascular pathway that promotes clearance of waste products from the brain [40]. The glymphatic system has been implicated in clearing soluble species of amyloid-β in patients with AD allowing potentially toxic proteins to clear the brain and prevent the build-up of pathological deposits.
Studies in live mice using real-time assessment of tetra methylammonium diffusion and two-photon imaging have shown that natural sleep is associated with a 60% increase in the interstitial space, resulting in increased convective change of cerebrospinal fluid with interstitial fluid. This in turn leads to increased rates of amyloid-β clearance during sleep via glymphatic clearance. Thus the restorative function of sleep may be through enhanced removal of potentially neurotoxic waste products that accumulate during wakefulness as a consequence of neural activity across the central nervous system [41]. Hypothetically, a similar case may be made for soluble phase α-synuclein species in the brain but remains unproven at the present time. If true in humans, any future glymphatic clearance or sleep enhancing therapies should be given as early as possible into the course of disease, preferably in the presymptomatic or prodromal phase. Medications such as gamma hydroxybutyrate have been shown to reliably stimulate slow wave sleep in normal subjects and narcoleptic patients. Gamma hydroxybutyrate has also been shown on polysomnography to remarkably increase slow wave sleep and improve insomnia in a patient with fatal familial insomnia [42]; however, it does not work for primary insomnia patients.
Nonetheless, glymphatic clearance promoting intervention still may be of benefit in the early motor symptomatic phase of PD as α-synuclein spreading may extend beyond mesencephalic and limbic regions. Furthermore, subsequent development of dementia in PD is also associated with co-occurring AD pathology. Therefore, sleep or glymphatic clearance enhancing strategies may also lower the build-up of non-α-synuclein protein deposits in advancing PD at risk of dementia. Sleep enhancing interventions that promote glymphatic clearance may therefore have the potential to serve important disease-modifying goals in PD.
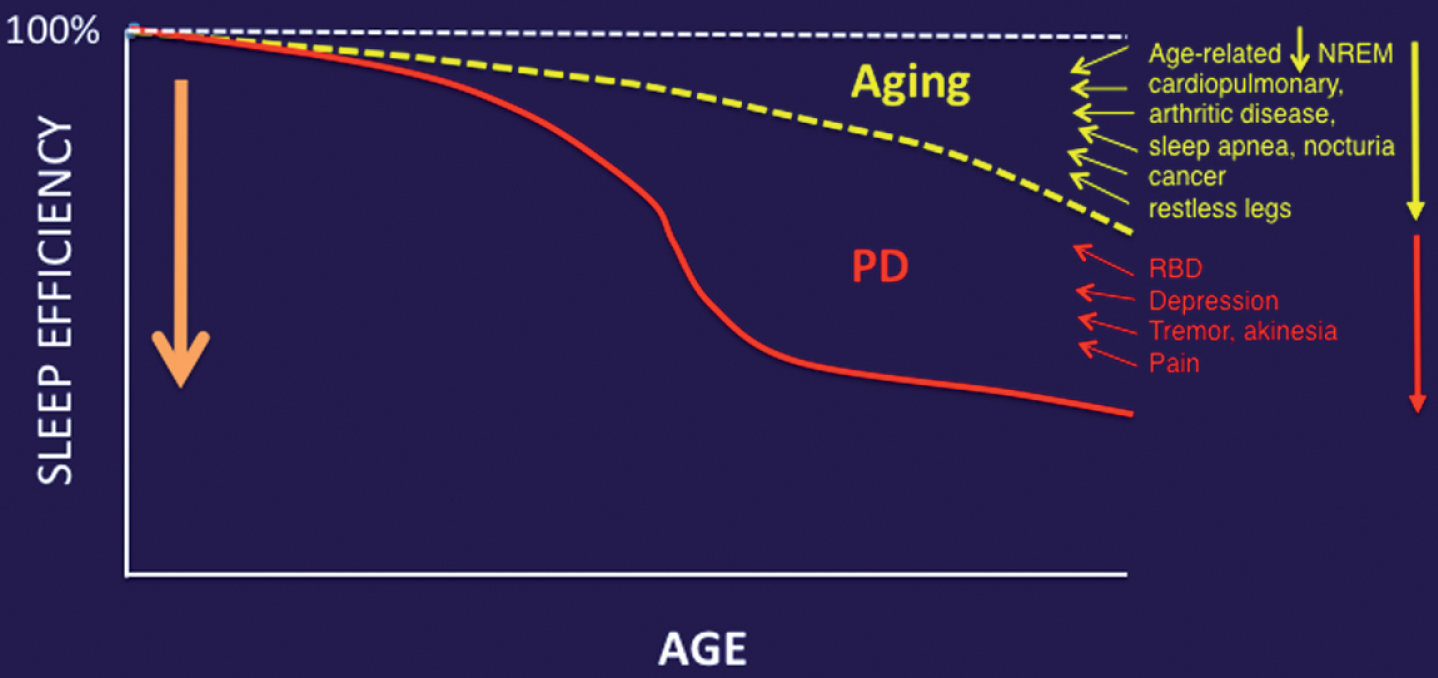
Aging is an important contributor to the multifactorial etiology of impaired sleep in PD. For example, studies in mice have shown a substantial age-associated decrease in the capacity of the glymphatic clearance system [43]. Furthermore, another mouse study showed an inverse relationship between EEG delta power and glymphatic clearance [44]. Aging is associated with reduced EEG delta power [45]. Therefore, the reliance of glymphatic function on slow wave activity at night illustrates the significant impact of aging on glymphatic clearance. Given the fact that age is a primary risk factor for PD, it is plausible that age-related compromises in sleep quality will not only augment but also may lower the manifesting threshold for those impaired sleep mechanisms specifically related to PD pathophysiology (Fig. 2).
Fig.2
Aging and medical comorbidities of older age are important contributors to impaired sleep in PD.
MODIFYING THE ACCUMULATION OF ABNORMAL PROTEINS IN ABNORMAL SLEEP: IMPAIRED CELLULAR CLEARANCE
Age-associated decline in cellular functions are due in part to a progressive failure of the chaperoning systems. Protein misfolding, accumulation and aggregation characterize many aging related diseases, which typically occur later in life including AD, PD, type 2 diabetes and fatal familial insomnia. That protein aggregates do not accumulate in unstressed cells is in part due to the existence of cellular ‘quality control machinery’, of which the endoplasmic reticulum (ER) plays a major role [46]. Accumulation of misfolded proteins in the ER causes ER stress and activates a signaling pathway called the unfolded protein response (UPR). The UPR limits protein load by up-regulating ER chaperones (e.g., BiP/GRP78) and reducing protein translation through phosphorylation.
Acute sleep deprivation in young mice leads to normal induction of the UPR response. However, studies in aged mice have shown that aging impairs this adaptive response to sleep deprivation. Aged mice do not display an increase in BiP expression with sleep deprivation, and have higher cortical levels of pro-apoptic proteins [46], leading to cell injury and death. As in humans, elderly healthy mice display fragmented sleep patterns. Given that patients with neurodegenerative disorders such as AD and PD have well documented sleep disorders, it is not unreasonable to speculate that the sleep disturbances place an additional burden on an already stressed protein folding and degradation system, further exacerbating protein aggregation in an accelerating cascade.
Evidence from human studies to support this hypothesis is now starting to emerge. Sleep and circadian problems are very common in AD, with animal studies suggesting a bidirectional relationship between sleep and amyloid-β, a key player in AD pathogenesis. Amyloid deposition was assessed with cerebrospinal fluid Aβ42 levels in 145 cognitively normal individuals with 2 weeks actigraphy to objectively measure sleep parameters [47]. Amyloid deposition was present in 32 participants (22.5%) who had worse sleep quality measured by sleep efficiency after correcting for age, sex, and APOE4 carrier status. This study suggests that amyloid deposition in the preclinical stage of AD appears to be associated with poor sleep quality. Amyloid deposition could itself cause sleep-wake fragmentation and reduced sleep efficiency by directly interfering with neuronal function in brain regions key to sleep and wake promotion. Amyloid accumulation could negatively affect sleep behavior, while conversely poor sleep could increase Aβ aggregation in a positive feedback loop [47].
While there is growing evidence that amyloid-β aggregation initiates AD pathogenesis it is tau not Aβ accumulation that appears to drive the neurodegenerative process in AD [48]. Tau aggregation in the brain of AD, progressive supranuclear palsy and other tauopathies significantly correlates with neuronal and synaptic loss. Once tau aggregation occurs, there is evidence that it can spread from one synaptically connected region to another [48]. While tau is predominantly cytoplasmic it is normally released by neurons into the extracellular space, with release being increased by excitatory activity. Tau is also likely to be highly relevant to PD pathogenesis, as variants in the microtubule-associated protein tau (MAPT) gene locus remain the second strongest genetic risk factor for PD following α-synuclein gene variants [49]. Furthermore, emerging evidence suggests a link between tau and α-synuclein pathology in Lewy body dementia (LBD). Postmortem studies suggest clinically significant AD co-pathology is present in 50% of all cases of LBD, and that these cases are associated with greater overall α-synuclein pathology, and faster progression rates to dementia. Emerging work has now found that the topology of neocortical tau pathology in LBD maps most closely to the distribution of α-synuclein pathology, and that the severity of tau pathology in LBD correlates strongly with cognitive performance [50]. Studies in mice and humans aged 30– 60 years have shown a diurnal fluctuation in tau levels, with increases in dark compared to light conditions. Furthermore, a 50% increase in interstitial fluid (ISF) or cerebrospinal fluid (CSF) tau levels were seen when mice or humans were subjected to sleep deprivation. Interestingly, CSF α-synuclein levels in humans showed a similar diurnal variation, and increased during sleep deprivation. Chronic sleep deprivation in a mouse model also increased tau pathology spreading and may have relevance for PD pathophysiology.
CAN DISTURBED SLEEP TRIGGER PD?
Current evidence suggests that the mechanistic link between disturbed sleep and neurodegeneration may involve intermittent hypo-oxygenation, inflammatory, or protein homeostatic changes and that the sleep-neurodegeneration relationship is likely bidirectional in nature (Fig. 1). A more rudimentary question is whether disturbed sleep may be able to play a primary function triggering the cascade of upstream events of PD pathophysiology?
The unique intermittent and sometimes very long duration nature of idiopathic RBD prior to the disease-defining motor features of PD raises questions about the nature of this parasomnia and what may trigger PD in this subset of patients. Although the pathophysiology of idiopathic RBD may start in peripheral autonomic nerves and then spread to the locus ceruleus there can be a very long lag time before nigral pars compacta dopaminergic neurons become affected. According to the prevailing Braak hypothesis RBD emerges when α-synuclein pathology affect key brainstem areas involved in sleep regulation [51]. A recent in vivo multi-modal neuroinflammatory, cholinergic intestinal, cardiac sympathetic and brain monoaminergic imaging study in patients with idiopathic RBD provide support for the Braak hypothesis [52]. Findings showed that idiopathic RBD had fully developed pathology in the peripheral autonomic nervous system and the locus ceruleus, equal to that in diagnosed PD. The RBD patients also showed noradrenergic thalamic denervation, but most had normal putaminal dopaminergic storage capacity. This caudo-rostral gradient of dysfunction supports the hypothesis that α-synuclein pathology in PD initially targets peripheral autonomic nerves and then spreads rostrally to the brainstem. A limitation of this multi-modal imaging study is that information about sequential pathophysiological events cannot be inferred. Therefore, a key question that remains is whether brainstem α-synucleinopathy is an essential primary component of RBD in prodromal PD?.
The gut microbiome seems to influence development and diseases of the enteric and central nervous system, including PD. By leveraging animal models, several different pathways of communication have been identified along the ‘gut-brain-axis’ including those driven by the immune system, the vagus nerve, or by modulation of neuroactive compounds by the microbiota [53]. For example, GABA producing vs. consuming gut bacteria may modulate the microbiome. Strandwitz and colleagues studied a rare type of gut bacteria called KLE178 that eat GABA. A different type of bacteria, Bacteroides, produces GABA, keeping the KLE1738 bacteria alive by feeding them [54]. These findings underscore how different types of bacteria work together in the gut. This is relevant as GABA is a CNS-active chemical and may affect brain functions that could be of relevance for PD pathophysiology.
Clinical and basic neuroscience evidence indicates that RBD results from breakdown of the network underlying REM sleep atonia [25]. It is also clear that GABA/glutamatergic imbalance in neural brainstem-subcortical circuits can induce RBD-like behaviors in animals. For example, genetic inactivation of glutamate neurons in the rat sublaterodorsal tegmental nucleus display symptoms and behaviors during paradoxical sleep that closely mimic human REM sleep behavior disorder [55]. If enteric α-synucleinopathy and/or change in the gut microbiome may be one of the earliest events in some patients with PD, then the observations of GABA producing/consuming bacteria in the gut [54], and evidence of vagal nerve mediated GABA-ergic influences on the brain may have the potential to disrupt the delicate GABA/glutamatergic balances of neural sleep circuits resulting in oneiric motor behaviors in the absence of primary brainstem α-synucleinopathy. The speculation that very early and more isolated and sporadic RBD behaviors may potentially occur in the absence of α-synucleinopathy in brainstem vulnerable cell populations may also explain the very long interval between first occurrence of dream enactment behavior and emergence of prototypical parkinsonian impairment up to 5 decades in a small subset of patients [56]. Alternatively, it may be possible that naturally occurring α-synuclein in the setting of primary ER stress in vulnerable neuronal cell population of RBD sleep circuits start to misfold and subsequently spread caudally, but this remains speculative at the present time. However, the existence of the olfactory/nasal lymphatic route where CSF passes through the cribriform plate along perineural spaces near the olfactory nerves to a lymphatic network at the nasal mucosa, and finally reaches to cervical lymph nodes [57, 58] raises the possibility that changes in the integrity of nasal mucosal or cervical lymph nodes could be another source for abnormal proteinopathy. For example, a recent study showed that ligation of deep cervical lymph nodes exacerbated AD-like phenotypes of APP/PS1 mice, showing more severe brain Aβ accumulation, neuroinflammation, synaptic protein loss, impaired polarization of aquaporin-4 and deficits in cognitive and exploratory behaviors [59]. Further research into these speculative and hypothetical mechanisms is important as positive findings may provide novel clues for therapeutic interventions. As neural circuits may predict patterns of α-synuclein propagation in the nervous system [60, 61], impairments of such circuits involved in sleep may be of high relevance for PD pathophysiology.
CONCLUSION
Emerging evidence suggests that sleep can serve an important disease-modifying role in PD and may be best explained by a bidirectional nature between sleep disturbance and neurodegeneration. Such disease-modifying mechanisms may include activation of inflammatory pathways, impaired nocturnal brain oxygenation, abnormal proteostasis, changes in glymphatic clearance, and altered modulation of specific sleep neural circuits that may prime further propagation of α-synucleinopathy in the brain. Epidemiological studies also suggest that disturbed sleep may increase the risk of developing PD. Further research is needed to determine whether sleep may have a more primary disease-triggering function in PD, at least in the setting of genetic or other vulnerabilities. Early management of impaired sleep in at risk persons and symptomatic PD may have the potential to modify the natural history of the disease.
CONFLICT OF INTEREST
The authors declare no relevant conflict of interests.
ACKNOWLEDGMENTS
Nicolaas Bohnen has received research support from the NIH, Michael J. Fox Foundation, the Department of Veteran Affairs, Chase Pharmaceuticals and Axovant Sciences. Michele Hu has received research support from Parkinson’s UK, Oxford Biomedical Research centre, Michael J. Fox Foundation and Cure Parkinson’s Trust. She has acted as an advisor to Roche and Biogen.
REFERENCES
[1] | Kieburtz K , Wunderle KB ((2013) ) Parkinson’s disease: Evidence for environmental risk factors. Mov Disord 28: , 8–13. |
[2] | Irwin MR , Vitiello MV ((2019) ) Implications of sleep disturbance and inflammation for Alzheimer’s disease dementia. Lancet Neurol 18: , 296–306. |
[3] | Leng Y , Musiek ES , Hu K , Cappuccio FP , Yaffe K ((2019) ) Association between circadian rhythms and neurodegenerative diseases. Lancet Neurol 18: , 307–318. |
[4] | Hsiao YH , Chen YT , Tseng CM , Wu LA , Perng DW , Chen YM , Chen TJ , Chang SC , Chou KT ((2017) ) Sleep disorders and an increased risk of Parkinson’s disease in individuals with non-apnea sleep disorders: A population-based cohort study. J Sleep Res 26: , 623–628. |
[5] | Chen H , Schernhammer E , Schwarzschild MA , Ascherio A ((2006) ) A prospective study of night shift work, sleep duration, and risk of Parkinson’s disease. Am J Epidemiol 163: , 726–730. |
[6] | Postuma RB , Bertrand JA , Monplaisir J , Desjardins C , Vendette M , Rios Romenets S , Panisset M , Gagnon JF ((2012) ) Rapid eye movement sleep behaviour disorder and risk of dementia in Parkinson’s disease: A prospective study. Mov Disord 27: , 720–726. |
[7] | Barber TR , Muhammed K , Drew D , Lawton M , Crabbe M , Rolinski M , Quinnell T , Zaiwalla Z , Ben-Shlomo Y , Husain M , Hu MTM ((2018) ) Apathy in rapid eye movement sleep behaviour disorder is common and under-recognized. Eur J Neurol 25: , 469–e-432. |
[8] | Diederich NJ , Vaillant M , Mancuso G , Lyen P , Tiete J ((2005) ) Progressive sleep ‘destructuring’ in Parkinson’s disease. A polysomnographic study in 46 patients. Sleep Med 6: , 313–318. |
[9] | Chahine LM , Amara AW , Videnovic A ((2017) ) A systematic review of the literature on disorders of sleep and wakefulness in Parkinson’s disease from 2005 to 2015. Sleep Med Rev 35: , 33–50. |
[10] | Fort P , Bassetti CL , Luppi PH ((2009) ) Alternating vigilance states: New insights regarding neuronal networks and mechanisms. Eur J Neurosci 29: , 1741–1753. |
[11] | Boissard R , Gervasoni D , Schmidt MH , Barbagli B , Fort P , Luppi PH ((2002) ) The rat ponto-medullary network responsible for paradoxical sleep onset and maintenance: A combined microinjection and functional neuroanatomical study. Eur J Neurosci 16: , 1959–1973. |
[12] | Carskadon MA , Dement WC ((2011) ) Normal human sleep: An overview. In Kryger MH, Roth T, Dement WC, eds., Principles and Practice of Sleep Medicine, Elsevier Saunders, Philedelphia, PA, pp. 16–26. |
[13] | Goetz CG , Wuu J , Curgian LM , Leurgans S ((2005) ) Hallucinations and sleep disorders in PD: Six-year prospective longitudinal study. Neurology 64: , 81–86. |
[14] | Comella CL ((2006) ) Sleep disturbances and excessive daytime sleepiness in Parkinson disease: An overview. J Neural Transm Suppl 349–355. |
[15] | Barone P , Antonini A , Colosimo C , Marconi R , Morgante L , Avarello TP , Bottacchi E , Cannas A , Ceravolo G , Ceravolo R , Cicarelli G , Gaglio RM , Giglia RM , Iemolo F , Manfredi M , Meco G , Nicoletti A , Pederzoli M , Petrone A , Pisani A , Pontieri FE , Quatrale R , Ramat S , Scala R , Volpe G , Zappulla S , Bentivoglio AR , Stocchi F , Trianni G , Dotto PD ((2009) ) The PRIAMO study: A multicenter assessment of nonmotor symptoms and their impact on quality of life in Parkinson’s disease. Mov Disord 24: , 1641–1649. |
[16] | Fahn S ((2003) ) Description of Parkinson’s disease as a clinical syndrome. Ann N Y Acad Sci 991: , 1–14. |
[17] | Askenasy JJ ((2003) ) Sleep disturbances in Parkinsonism. J Neural Transm (Vienna) 110: , 125–150. |
[18] | Van Den Berg JF , Van Rooij FJ , Vos H , Tulen JH , Hofman A , Miedema HM , Neven AK , Tiemeier H ((2008) ) Disagreement between subjective and actigraphic measures of sleep duration in a population-based study of elderly persons. J Sleep Res 17: , 295–302. |
[19] | Stavitsky K , Saurman JL , McNamara P , Cronin-Golomb A ((2010) ) Sleep in Parkinson’s disease: A comparison of actigraphy and subjective measures. Parkinsonism Relat Disord 16: , 280–283. |
[20] | Gunn DG , Naismith SL , Bolitho SJ , Lewis SJ ((2014) ) Actigraphically-defined sleep disturbance in Parkinson’s disease is associated with differential aspects of cognitive functioning. J Clin Neurosci 21: , 1112–1115. |
[21] | Breen DP , Vuono R , Nawarathna U , Fisher K , Shneerson JM , Reddy AB , Barker RA ((2014) ) Sleep and circadian rhythm regulation in early Parkinson disease. JAMA Neurol 71: , 589–595. |
[22] | Sixel-Doring F , Trautmann E , Mollenhauer B , Trenkwalder C ((2011) ) Associated factors for REM sleep behavior disorder in Parkinson disease. Neurology 77: , 1048–1054. |
[23] | Buskova J , Klempir J , Majerova V , Picmausova J , Sonka K , Jech R , Roth J , Ruzicka E ((2011) ) Sleep disturbances in untreated Parkinson’s disease. J Neurol 258: , 2254–2259. |
[24] | Lu J , Sherman D , Devor M , Saper CB ((2006) ) A putative flip-flop switch for control of REM sleep. Nature 441: , 589–594. |
[25] | Peever J , Luppi PH , Montplaisir J ((2014) ) Breakdown in REM sleep circuitry underlies REM sleep behavior disorder. Trends Neurosci 37: , 279–288. |
[26] | Boeve BF , Silber MH , Ferman TJ , Lin SC , Benarroch EE , Schmeichel AM , Ahlskog JE , Caselli RJ , Jacobson S , Sabbagh M , Adler C , Woodruff B , Beach TG , Iranzo A , Gelpi E , Santamaria J , Tolosa E , Singer C , Mash DC , Luca C , Arnulf I , Duyckaerts C , Schenck CH , Mahowald MW , Dauvilliers Y , Graff-Radford NR , Wszolek ZK , Parisi JE , Dugger B , Murray ME , Dickson DW ((2013) ) Clinicopathologic correlations in 172 cases of rapid eye movement sleep behavior disorder with or without a coexisting neurologic disorder. Sleep Med 14: , 754–762. |
[27] | Barber TR , Lawton M , Rolinski M , Evetts S , Baig F , Ruffmann C , Gornall A , Klein JC , Lo C , Dennis G , Bandmann O , Quinnell T , Zaiwalla Z , Ben-Shlomo Y , Hu MTM ((2017) ) Prodromal parkinsonism and neurodegenerative risk stratification in REM sleep behavior disorder. Sleep 40: , 10.1093/sleep/zsx071 |
[28] | Arora S , Baig F , Lo C , Barber TR , Lawton MA , Zhan A , Rolinski M , Ruffmann C , Klein JC , Rumbold J , Louvel A , Zaiwalla Z , Lennox G , Quinnell T , Dennis G , Wade-Martins R , Ben-Shlomo Y , Little MA , Hu MT ((2018) ) Smartphone motor testing to distinguish idiopathic REM sleep behavior disorder, controls, and PD. Neurology 91: , e1528–e-1538. |
[29] | Postuma RB , Iranzo A , Hu M , Hogl B , Boeve BF , Manni R , Oertel WH , Arnulf I , Ferini-Strambi L , Puligheddu M , Antelmi E , Cochen De Cock V , Arnaldi D , Mollenhauer B , Videnovic A , Sonka K , Jung KY , Kunz D , Dauvilliers Y , Provini F , Lewis SJ , Buskova J , Pavlova M , Heidbreder A , Montplaisir JY , Santamaria J , Barber TR , Stefani A , St Louis EK , Terzaghi M , Janzen A , Leu-Semenescu S , Plazzi G , Nobili F , Sixel-Doering F , Dusek P , Bes F , Cortelli P , Ehgoetz Martens K , Gagnon JF , Gaig C , Zucconi M , Trenkwalder C , Gan-Or Z , Lo C , Rolinski M , Mahlknecht P , Holzknecht E , Boeve AR , Teigen LN , Toscano G , Mayer G , Morbelli S , Dawson B , Pelletier A ((2019) ) Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: A multicentre study. Brain 142: , 744–759. |
[30] | De Cock VC , Vidailhet M , Leu S , Texeira A , Apartis E , Elbaz A , Roze E , Willer JC , Derenne JP , Agid Y , Arnulf I ((2007) ) Restoration of normal motor control in Parkinson’s disease during REM sleep. Brain 130: , 450–456. |
[31] | Burn DJ , Anderson K ((2012) ) To sleep, perchance to dement: RBD and cognitive decline in Parkinson’s disease. Mov Disord 27: , 671–673. |
[32] | Lawton M , Ben-Shlomo Y , May MT , Baig F , Barber TA , Klein JC , Swallow DMA , Malek N , Grosset KA , Bajaj N , Barker RA , Williams N , Burn DJ , Foltynie T , Morris HR , Wood NW , Grosset DG , Hu MTM ((2018) ) Developing and validating Parkinson’s disease subtypes and their motor and cognitive progression. J Neurol Neurosurg Psychiatry 89: , 1279–1287. |
[33] | Lurie A ((2011) ) Inflammation, oxidative stress, and procoagulant and thrombotic activity in adults with obstructive sleep apnea. Adv Cardiol 46: , 43–66. |
[34] | Kaminska M , Lafontaine AL , Kimoff RJ ((2015) ) The interaction between obstructive sleep apnea and Parkinson’s disease: Possible mechanisms and implications for cognitive function. Parkinsons Dis 2015: , 849472. |
[35] | Chou PS , Lai CL , Chou YH , Chang WP ((2017) ) Sleep apnea and the subsequent risk of Parkinson’s disease: A 3-year nationwide population-based study. Neuropsychiatr Dis Treat 13: , 959–965. |
[36] | Wong KK , Muller ML , Kuwabara H , Studenski SA , Bohnen NI ((2012) ) Gender differences in nigrostriatal dopaminergic innervation are present at young-to-middle but not at older age in normal adults. J Clin Neurosci 19: , 183–184. |
[37] | Bohnen NI , Muller ML , Kuwabara H , Cham R , Constantine GM , Studenski SA ((2009) ) Age-associated striatal dopaminergic denervation and falls in community-dwelling subjects. J Rehabil Res Dev 46: , 1045–1052. |
[38] | Gelber RP , Redline S , Ross GW , Petrovitch H , Sonnen JA , Zarow C , Uyehara-Lock JH , Masaki KH , Launer LJ , White LR ((2015) ) Associations of brain lesions at autopsy with polysomnography features before death. Neurology 84: , 296–303. |
[39] | Tao Q , Ang TFA , DeCarli C , Auerbach SH , Devine S , Stein TD , Zhang X , Massaro J , Au R , Qiu WQ ((2018) ) Association of chronic low-grade inflammation with risk of Alzheimer disease in ApoE4 carriers. JAMA Netw Open 1: , e183597. |
[40] | Iliff JJ , Wang M , Liao Y , Plogg BA , Peng W , Gundersen GA , Benveniste H , Vates GE , Deane R , Goldman SA , Nagelhus EA , Nedergaard M ((2012) ) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 4: , 147ra111. |
[41] | Xie L , Kang H , Xu Q , Chen MJ , Liao Yea ((2013) ) Sleep drives metabolite clearance from the adult brain. Science 342: , 373–377. |
[42] | Reder AT , Mednick AS , Brown P , Spire JP , Van Caulter E , Wollmann RL , Cervenakova L ((1995) ) Clinical and genetic studies of fatal familial insomnia. Neurology 45: , 1068–1075. |
[43] | Kress BT , Iliff JJ , Xia M , Wang M , Wei HS , Zeppenfeld D , Xie L , Kang H , Xu Q , Liew JA , Plog BA , Ding F , Deane R , Nedergaard M ((2014) ) Impairment of paravascular clearance pathways in the aging brain. Ann Neurol 76: , 845–861. |
[44] | Hablitz LM , Vinitsky HS , Sun Q , Staeger FF , Sigurdsson B , Mortensen KN , Lilius TO , Nedergaard M ((2019) ) Increased glymphatic influx is correlated with high EEG delta power and low heart rate in mice under anesthesia. Sci Adv 5: , eaav5447. |
[45] | Svetnik V , Snyder ES , Ma J , Tao P , Lines C , Herring WJ ((2017) ) EEG spectral analysis of NREM sleep in a large sample of patients with insomnia and good sleepers: Effects of age, sex and part of the night. J Sleep Res 26: , 92–104. |
[46] | Naidoo N , Ferber M , Master M , Zhu Y , Pack AI ((2008) ) Aging impairs the unfolded protein response to sleep deprivation and leads to proapoptotic signaling. J Neurosci 28: , 6539–6548. |
[47] | Ju YE , McLeland JS , Toedebusch CD , Xiong C , Fagan AM , Duntley SP ((2013) ) Sleep quality and preclinical Alzheimer disease. JAMA Neurol 70: , 587–593. |
[48] | Holth JK , Fritschi SK , Wang C , Pedersen NP , Cirrito JR , Mahan TE ((2019) ) The sleep-wake cycle regulates brain interstitial fluid tau in mice and CSF tau in humans. Science 363: , 880–884. |
[49] | Nalls MA , Pankratz N , Lill CM , Do CB , Hernandez DG , Saad M , DeStefano AL , Kara E , Bras J ((2014) ) Large-scale meta-analysis of genome-wide association data identified six new risk loci for Parkinson’s disease. Nat Genetic 46: , 989–993. |
[50] | Coughlin D , Xie SX , Liang M , Williams A , Peterson C , Weintraub D , McMillan CT , Wolk DA ((2019) ) Cognitive and pathological influences of tau pathology in Lewy body disorders. Ann Neurol 85: , 259–271. |
[51] | Braak H , Del Tredici K , Rub U , de Vos RA , Jansen Steur EN , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: , 197–211. |
[52] | Knudsen K , Fedorova TD , Hansen AK , Sommerauer M , Otto M , Svendsen KB , Nahimi A , Stokholm MG , Pavese N , Beier CP , Brooks DJ , Borghammer P ((2018) ) in-vivo staging of pathology in REM sleep behaviour disorder: A multimodality imaging case-control study. Lancet Neurol 17: , 618–628. |
[53] | Strandwitz P ((2018) ) Neurotransmitter modulation by the gut microbiota. Brain Res 1693: , 128–133. |
[54] | Strandwitz P , Kim KH , Terekhova D , Liu JK , Sharma A , Levering J , McDonald D , Dietrich D , Ramadhar TR , Lekbua A , Mroue N , Liston C , Stewart EJ , Dubin MJ , Zengler K , Knight R , Gilbert JA , Clardy J , Lewis K ((2019) ) GABA-modulating bacteria of the human gut microbiota. Nat Microbiol 4: , 396–403. |
[55] | Valencia Garcia S , Libourel PA , Lazarus M , Grassi D , Luppi PH , Fort P ((2017) ) Genetic inactivation of glutamate neurons in the rat sublaterodorsal tegmental nucleus recapitulates REM sleep behaviour disorder. Brain 140: , 414–428. |
[56] | Claassen DO , Josephs KA , Ahlskog JE , Silber MH , Tippmann-Peikert M , Boeve BF ((2010) ) REM sleep behavior disorder preceding other aspects of synucleinopathies by up to half a century. Neurology 75: , 494–499. |
[57] | Bradbury MW , Cserr HF , Westrop RJ ((1981) ) Drainage of cerebral interstitial fluid into deep cervical lymph of the rabbit. Am J Physiol 240: , F329–336. |
[58] | Bradbury MW , Westrop RJ ((1983) ) Factors influencing exit of substances from cerebrospinal fluid into deep cervical lymph of the rabbit. J Physiol 339: , 519–534. |
[59] | Wang L , Zhang Y , Zhao Y , Marshall C , Wu T , Xiao M ((2019) ) Deep cervical lymph node ligation aggravates AD-like pathology of APP/PS1 mice. Brain Pathol 29: , 176–192. |
[60] | Zeighami Y , Ulla M , Iturria-Medina Y , Dadar M , Zhang Y , Larcher KM , Fonov V , Evans AC , Collins DL , Dagher A ((2015) ) Network structure of brain atrophy in de novo Parkinson’s disease. Elife 4: , 10.7554/eLife.08440 |
[61] | Yau Y , Zeighami Y , Baker TE , Larcher K , Vainik U , Dadar M , Fonov VS , Hagmann P , Griffa A , Misic B , Collins DL , Dagher A ((2018) ) Network connectivity determines cortical thinning in early Parkinson’s disease progression. Nat Commun 9: , 12. |


