
Urate and Homocysteine: Predicting Motor and Cognitive Changes in Newly Diagnosed Parkinson’s Disease
Abstract
Background:
Urate and homocysteine are potential biomarkers for disease progression in Parkinson’s disease (PD). Baseline serum urate concentration has been shown to predict motor but not cognitive decline. The relationship between serum homocysteine concentration and cognitive and motor impairment is unknown.
Objectives:
The aim of this study was to examine the association between baseline serum urate and homocysteine, and prospective measures of disease progression and cognition over 54 months in early PD.
Methods:
154 newly diagnosed PD participants and 99 age-matched controls completed a schedule of assessments at baseline, 18, 36 and 54 months. The Movement Disorders Society Unified Parkinson’s Disease Scale Part III (MDS-UPDRS III) was used to assess motor severity. The Montreal Cognitive Assessment (MoCA) was used to assess global cognition. Serum samples drawn at baseline were analysed for urate, homocysteine, red cell folate and vitamin B12 concentrations.
Results:
Baseline urate was 331.4±83.8 and 302.7±78.0μmol/L for control and PD participants, respectively (p = 0.015). Baseline homocysteine was 9.6±3.3 and 11.1±3.8μmol/L for controls and PD participants, respectively (p < 0.01). Linear mixed effects modelling showed that lower baseline urate (β= 0.02, p < 0.001) and higher homocysteine (β= 0.29, p < 0.05) predicted decline in motor function. Only higher homocysteine concentrations at baseline, however, predicted declining MoCA scores over 54 months (β= 0.11, p < 0.01).
Conclusions:
Lower serum urate concentration is associated with worsening motor function; while higher homocysteine concentration is associated with change in motor function and cognitive decline. Therefore, urate and homocysteine may be suitable biomarkers for predicting motor and cognitive decline in early PD.
INTRODUCTION
Parkinson’s disease (PD) is a progressive neurodegenerative disorder encompassing motor, cognitive and behavioural and autonomic features. There is growing evidence that oxidative stress is involved in the pathophysiology, disease progression and development of cognitive impairment in PD. Dopaminergic neurons in the basal ganglia are particularly susceptible to oxidative stress [1] and the post-mortem brains of PD subjects have higher concentrations of the products of oxidative stress than controls [2].
Both urate and homocysteine may have a role in regulating oxidative stress in PD. Urate is a product of purine breakdown, which accounts for 60% of serum antioxidant capacity [3]. Prospective studies have shown that low serum urate in middle age is associated with an increased risk of developing PD over the subsequent 15–20 years, with participants in the lowest quartile 2.3–2.5 times as likely to develop the condition than those in the highest quartile [4, 5]. Furthermore, amongst patients with newly diagnosed PD who had not yet started treatment, those with the highest quintile of urate were half as likely to require treatment than those in the lowest quintile over the following two years [6]. Low plasma urate in PD patients has also been associated with poorer performance in neuropsychological tests sensitive to attention, executive function and visuospatial function [7], suggesting it may be associated with cognitive impairment.
The association between hyperhomocysteinaemia and risk of dementia in community dwelling older adults has been previously reported [8], raising the possibility that it may also have a role in the development of PD dementia (PDD). In established PD, cross-sectional studies have reported that elevated plasma homocysteine levels are associated with dementia [9].
The aim of this study was to examine the association between baseline serum urate and homocysteine and measures of disease progression in early PD using participants in an incident cohort study with prospective follow up. We hypothesised that PD patients with low urate and/or high homocysteine at baseline would display a more rapid decline in motor and cognitive function over the 54 month follow up.
PARTICIPANTS AND METHODS
Subjects
Participants were recruited prospectively as part of the Incidence of Cognitive Impairment in Cohorts with Longitudinal Evaluation in Parkinson’s disease (ICICLE-PD) study [10] Inclusion criteria comprised a diagnosis of idiopathic PD according to UK Brain Bank Criteria [11] in the North East of England between 1 June 2009 and 31 December 2011. Exclusion criteria were: significant cognitive impairment at presentation (Mini Mental State Examination (MMSE) score <24) or a diagnosis of dementia, parkinsonism due to another cause, insufficient working knowledge of English, major psychiatric illness or treatment with COMT inhibitors.
To generate normative values, age-matched healthy control subjects were recruited from local advertising and community sources. Carers and spouses of patients with PD were not approached to act as controls to limit bias. Exclusion criteria for controls were: significant cognitive impairment at presentation (MMSE <24) or diagnosis of dementia, major psychiatric illness or movement disorder.
The study was approved by Newcastle and North Tyneside Research Ethics Committee. All participants provided written informed consent at each review.
Assessments
Demographic information including age, sex, and education were collected. All subjects underwent a medical assessment by a movement disorders specialist; we sought details of disease duration, co-morbid diseases, medication use, smoking history and alcohol consumption. Patients were evaluated in the “on” motor state. Motor severity was rated using the Movement Disorders Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) Part III [12] and Hoehn and Yahr scale [13]. MDS-UPDRS scores range from 0–132, with higher scores indicating more severe motor symptoms. Levodopa equivalent daily dose (LEDD) was calculated for all dopaminergic medications [14]. Participants completed the National Adult Reading Test (NART) [15] as a measure of pre-morbid IQ and the Geriatric Depression Scale (GDS) [16] was used to measure depressive symptoms. Global cognitive function was assessed with the Mini-Mental State Examination (MMSE) [17] and Montreal Cognitive Assessment (MoCA). Both tests have a maximum score of 30 with a score of <26 suggesting cognitive impairment. The MoCA has been validated in patients with PD [18]. Assessments were repeated at 18-month intervals.
Laboratory analyses
A single baseline non-fasting blood sample was obtained for measurements of total serum urate, plasma homocysteine, vitamin B12 and red cell folate concentrations. A serum separate tube (SST) was collected for urate analysis, which was later determined using an enzymatic (Uricase) method on the Roche Modular P. Homocysteine samples were collected into citrate plasma tubes and put on ice immediately. Total plasma homocysteine was measured using an automated latex immunoassay (Hemosil, Homocysteine, Instrumentation Laboratory) on the IL-TOP Coagulation analyser. Vitamin B12 and red cell folate samples were collected into EDTA plasma tubes; levels were determined using a chemiluminescent microparticle assay (Architect, Abbott).
Statistical analyses
Data were analysed using SPSS (Version 21, Armonk, NY: IBM Corp). Data were examined for normality by visual inspection of histograms and Kolmogorov-Smirnov testing. Comparison of means between PD and control subjects at baseline were performed using independent t-tests or Mann-Whitney U tests as appropriate. For comparisons of repeated measures between baseline and 54 months, repeated measures ANOVA or Friedman tests were used as appropriate. Chi-squared testing was used to assess binary outcome variables. The significance level for all statistical tests was set at α= 0.05 (two-tailed).
R [19] and lme4 [20] were used to perform linear mixed effects analysis. A random intercept model was used to determine baseline predictors of change in motor severity and global cognition from baseline to 54 months in PD subjects. The intercept varied at the participant and time level, accounting for by-subject and by-time variability. A reduced model was produced by excluding non-significant predictors to which baseline serum urate or baseline plasma homocysteine were then added, in addition to baseline folate and vitamin B12 and their interactions with time as covariates. Baseline age, sex, LEDD, time, age×time and LEDD×time were entered into the model as fixed factors predicting change in PD motor severity. Baseline age, sex, years of education, LEDD, MDS-UPDRS III, GDS-15 and time were added as fixed factors predicting change in MoCA score, as well as interactions of time with baseline age (age×time), motor severity (MDS-UPDRS III×time), LEDD (LEDD×time) and GDS-15 (GDS-15×time). LEDD was controlled for in all models that included total plasma homocysteine (tHyc). To account for confounders that may mediate the relationship between urate and/or tHyc, we repeated the above analysis adjusting for the presence of baseline smoking status, type II diabetes, hypertension, ischemic heart disease, previous stoke or transient ischemic attack (TIA), hypercholesterolemia and gout. Visual inspection of residual plots did not reveal any obvious deviations from homoscedasticity or normality. Fit of the models was assessed by likelihood ratio tests.
RESULTS
After exclusions, at baseline 154 patients with PD and 99 healthy control subjects completed assessments (Fig. 1). The demographic and clinical characteristics of the PD participants and control subjects at baseline were compared (Table 1). Mean time from PD diagnosis was 6.3±6.1 months. There were no significant differences between PD participants and controls with regard to age, sex, years of education or smoking status (p > 0.05 for all). Previous stroke or TIAs were more common in those with PD than controls (10 vs. 0 patients, p < 0.01), but there were no differences in rates of ischaemic heart disease, hypertension, hypercholesterolaemia, gout or type 2 diabetes. PD participants scored significantly poorer on both the MoCA and MMSE as measures of global cognition (p < 0.01 for both). At 54 month evaluation n = 91 PD participants (58%) and 69 control participants (68%) returned (Fig. 1). In total, 18 PD participants declined further follow up, 35 were lost to follow up and 10 were deceased. There were no significant differences in either PD or control subjects who completed all assessments compared those who did not, in terms of baseline age, NART scores, GDS-15, LEDD, MMSE, urate or homocysteine concentrations (p > 0.05 for all). At baseline completers had completed more years of education (13.3±3.8 vs. 12.0±3.7, Z = –2.3, p = 0.024), a lower MDS-UPDRS III score (24.2±10.5 vs. 30.8±13.2, Z = –3.1, p = 0.002) and higher MoCA score (25.8±3.3 vs. 24.3±3.9, Z = –2.4, p = 0.018).
Fig. 1.
Consort diagram of Parkinson’s disease and control participants.
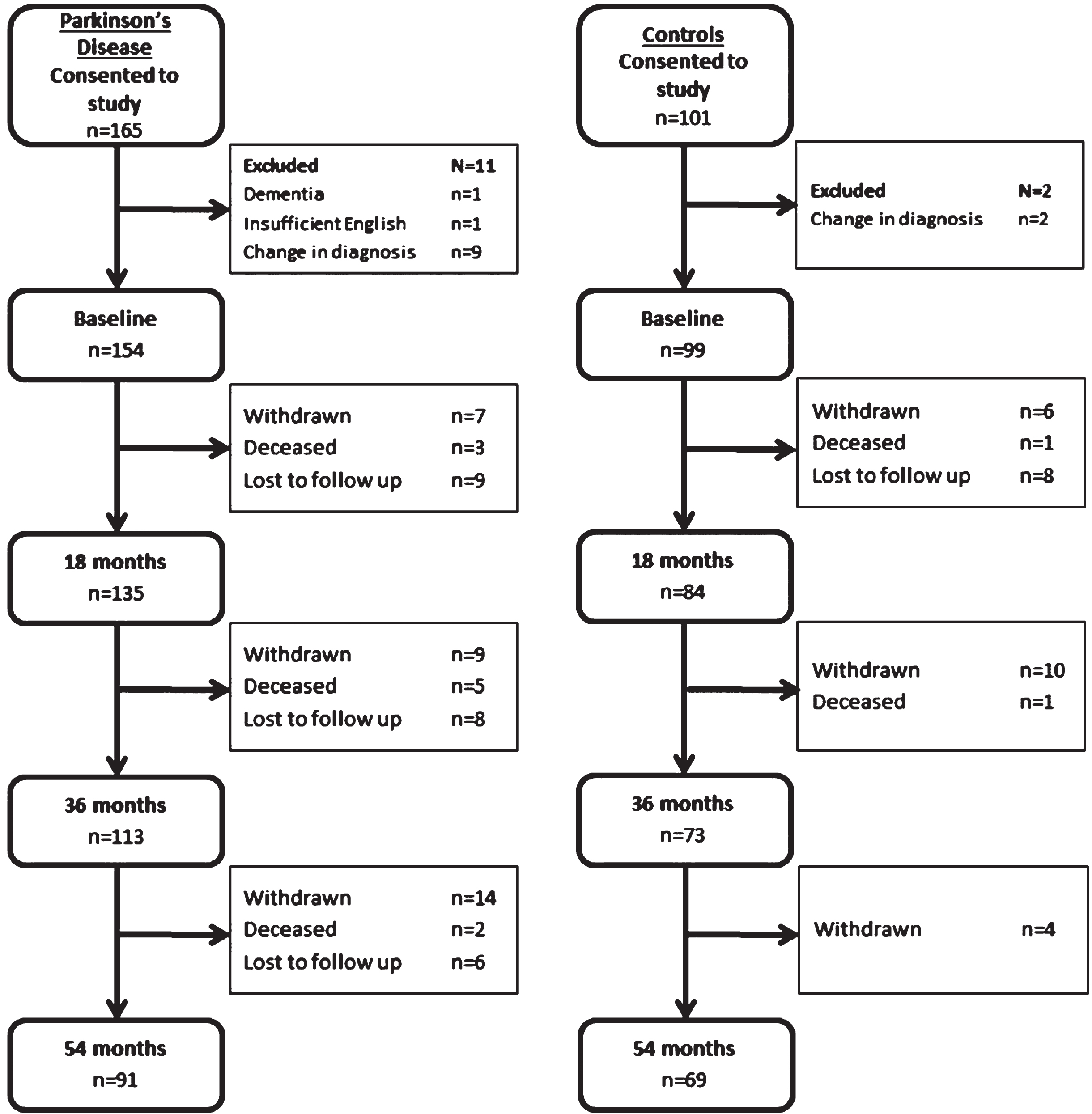
Table 1
Baseline demographic and clinical characteristics of the study participants
| Control | PD | t/z | p | |||
| Mean | SD | Mean | SD | |||
| Age (years) | 67.9 | 8.2 | 66.4 | 10.4 | 1.2 | 0.214 |
| Education (years) | 13.1 | 3.4 | 12.8 | 3.8 | –1.1 | 0.251 |
| NART | 115.9 | 8.7 | 114.7 | 10.7 | –0.3 | 0.770 |
| PD duration (months) | – | – | 6.4 | 6.0 | – | – |
| MDS-UPDRS III | – | – | 26.9 | 12.1 | – | – |
| Hoehn and Yahr | – | – | 2.0 | 0.7 | – | – |
| LEDD (mg/d) | – | – | 178.0 | 148.2 | – | – |
| GDS-15 | 1.0 | 1.5 | 2.8 | 2.5 | –6.87 | <0.001 |
| MoCA | 27.0 | 2.5 | 25.2 | 3.7 | –3.7 | <0.001 |
| MMSE | 29.0 | 1.2 | 28.6 | 1.3 | –2.7 | 0.006 |
| BMI | 27.7 | 3.9 | 27.2 | 4.8 | 0.8 | 0.411 |
| Urate (μmol/L) | 331.4 | 83.8 | 302.7 | 77.9 | –2.4 | 0.015 |
| Homocysteine (μmol/L) | 9.6 | 3.3 | 11.1 | 3.8 | –3.8 | <0.001 |
| Vitamin B12 (pg/mL) | 472.6 | 171.1 | 391.5 | 170.0 | –4.1 | <0.001 |
| Red cell folate (ng/ml) | 324.4 | 162.0 | 277.2 | 148.5 | –3.0 | 0.003 |
| n | % | n | % | χ2 | p | |
| Sex (male) | 54 | 54.5 | 100 | 64.9 | 2.7 | 0.098 |
| Smoker | 7 | 7.1 | 9 | 5.8 | 0.4 | 0.725 |
| Hypertension | 35 | 35.4 | 48 | 31.2 | 0.5 | 0.989 |
| Type 2 diabetes mellitus | 5 | 5.1 | 12 | 7.8 | 0.7 | 0.395 |
| Ischaemic heart disease | 9 | 9.1 | 18 | 11.7 | 0.4 | 0.514 |
| Previous stroke or transient ischaemic attack | 0 | 0.0 | 10 | 6.5 | 6.7 | 0.010 |
| Hypercholesterolaemia | 17 | 17.2 | 22 | 14.3 | 0.4 | 0.535 |
| Gout | 3 | 3.0 | 5 | 3.2 | 0 | 0.924 |
PD, Parkinson’s disease; SD, Standard deviation; NART, National Adult Reading Test; MDS UPDRS III, Movement Disorders Society Unified Parkinson’s Disease Rating Scale Part III; LEDD, Levodopa equivalent daily dose; GDS-15, Geriatric Depression Scale; MoCA, Montreal Cognitive Assessment; MMSE, Mini Mental State Examination. Significant results are highlighted in bold.
Changes in serum urate and homocysteine
Total plasma urate concentration was significantly lower in those with PD than in controls at baseline (302.7±77.9 vs. 331.4±77.9μmol/L, respectively, p < 0.01), 18 months (301.9±77.0 vs. 339.4±90.7μmol/L, respectively, p < 0.01) and 36 months (305.0±76.4 vs. 331.±79.7μmol/L, respectively, p < 0.05). Within groups, there was no significant change over time in urate concentrations for either PD or control participants using repeated measures analysis (p > 0.05). Four participants with PD and one control were taking allopurinol at baseline. For both PD and control participants, urate concentrations were significantly higher in males compared to females at each time point (p < 0.001 for all, Supplementary Table 1).
In comparison, the total plasma homocysteine concentration was significantly higher in those with PD than in controls at baseline (11.1±3.8 vs. 9.6±3.3μmol/L, respectively, p < 0.01), 18 months (12.3±3.8 vs. 11.1±3.7μmol/L, respectively, p < 0.05) and 36 months 13.9±4.9 vs. 12.3±4.2μmol/L, respectively, p < 0.05). However, repeated measures analysis showed plasma homocysteine significantly increased in both groups over 36 months (p < 0.01 for both). At baseline, females control participants had significantly lower plasma homocysteine concentrations compared to males (8.8±2.9 vs. 10.2±3.5, respectively, p < 0.05) but at no other time point. In PD participants, there were no significant differences in males and females at baseline, 18 and 36 months, but females had significantly lower plasma homocysteine concentrations compared to males at 54 months (11.4±2.3 vs. 14.6±5.0μmol/L, respectively, p < 0.05).
At baseline, 73% of PD participants were levodopa naïve. Comparison of mean tests found no significant differences between levodopa treated and levodopa naïve participants for serum urate concentrations (307.5±71.2 vs. 301.3±73.9μmol/L, respectively, p > 0.05) or serum homocysteine concentrations (11.8±3.4 vs. 10.9±4.0μmol/L, respectively, p > 0.05). However, urate concentration was significantly lower in levodopa naïve PD participants compared to healthy controls (301.3±73.9 vs. 331.4±83.8μmol/L, respectively, p < 0.05), while homocysteine concentration was significantly higher (10.9±4.0 vs. 9.6±3.3μmol/L, respectively <0.05).
Baseline predictors of motor severity and cognition over 54 months
Multivariate modelling was used to determine the effects of baseline homocysteine and urate on change in motor severity and change in cognition over 54 months, respectively. Linear mixed effects modelling was used to predict motor severity (MDS-UPDRS III). After excluding non-significant predictors, age (β= –4.0, p < 0.05), sex (β= 0.2, p < 0.01) and time (β= 5.2, p < 0.01) were controlled for in the basic model.
The interaction of baseline urate with time was a significant predictor of change in MDS-UPDRS III score (p < 0.001), after controlling for B12 and folate concentrations, indicating that low urate levels at diagnosis were associated with declining motor severity (Table 2). Adding urate, urate×time and covariates significantly improved the fit of the model (χ2 = 437.1, p < 0.001). The basic model plus tHyc and tHyc×time, controlling for LEDD, B12 and folate, was also a significantly stronger model than the basic model (χ2 = 405.1 p < 0.001). However, only tHyc×time was significant predictor. There was no significant difference in fit between these two models (p > 0.05). Including both urate×time and tHyc×time, controlling for LEDD, B12 and folate, showed that both low urate and high tHyc predicted declining motor severity (Table 2). However, the model suggested that when taking into account both urate and tHyc, baseline urate was the stronger predictor of change in motor severity (t = 3.5, p < 0.001 vs. t = 2.4, p < 0.05, respectively). The basic model plus urate and homocysteine had stronger predictive power than urate alone (χ2 = 77.2, p < 0.001) or homocysteine alone (χ2 = 109.3, p < 0.001).
Table 2
Baseline predictors of motor severity over 54 months using linear mixed effects modelling
| β | SE | t | p-value | |
| Basic modela + urate | ||||
| Urate | –0.01 | 0.0 | –0.8 | 0.422 |
| Urate×Time | 0.02 | 0.0 | 4.5 | <0.001 |
| Basic modelb + homocysteine | ||||
| tHyc | 0.48 | 0.3 | 1.7 | 0.094 |
| tHyc×Time | 0.44 | 0.1 | 3.8 | <0.001 |
| Basic modelb + urate + homocysteine | ||||
| Urate | –0.02 | 0.0 | –1.0 | 0.313 |
| tHyc | 0.57 | 0.3 | 2.0 | 0.052 |
| Urate×Time | 0.02 | 0.0 | 3.5 | <0.001 |
| tHyc×Time | 0.29 | 0.1 | 2.4 | 0.016 |
Motor severity determined by MDS-UPDRS III. Significant predictors highlighted in bold. aCovariates included in model: age, sex, folate, B12, time, folate×time and B12×time. bCovariates included in model: age, sex, LEDD, folate, B12, time, LEDD×time, folate×time and B12×time. MDS-UPDRS III, Movement Disorder Society Unified Parkinson’s Disease Rating Scale Part III; LEDD, Levodopa equivalent daily dose; tHyc, serum homocysteine.
Baseline predictors of MoCA score over 54 months were age (β= –0.16, p < 0.001), MDS-UPDRS III score (β= –0.05, p < 0.01), time (β= 1.14, p < 0.01) and MDS-UPDRS×time (β= –0.03, p < 0.001) and were included in the basic model. Due to sex differences in baseline urate concentration, sex was controlled for in the basic model. Urate and urate×time were not significant predictors of change in MoCA score (p > 0.05, Table 3). When tHyc and tHyc×time were added to the basic model, controlling for LEDD, B12 and folate, tHyc×time was a significant predictor of MoCA score over time (Table 3) and the model significantly improved (χ2 = 230.0, p < 0.001).
Table 3
Baseline predictors of global cognition over 54 months using linear mixed effects modelling
| β | SE | t | p-value | |
| Basic modela + urate | ||||
| Urate | 0.00 | 0.00 | –1.03 | 0.305 |
| Urate×Time | 0.00 | 0.00 | 1.38 | 0.170 |
| Basic modelb + homocysteine | ||||
| tHyc | –0.05 | 0.09 | –0.53 | 0.598 |
| tHyc×Time | 0.12 | 0.04 | 2.89 | 0.004 |
| Basic modelb + urate + homocysteine | ||||
| Urate | –0.01 | 0.00 | –1.13 | 0.261 |
| tHyc | –0.04 | 0.09 | –0.42 | 0.678 |
| Urate×Time | 0.00 | 0.00 | 0.94 | 0.346 |
| tHyc×Time | 0.11 | 0.04 | 2.70 | 0.007 |
Global cognition determined by MoCA. Significant predictors highlighted in bold. aCovariates included in model: sex, age, MDS-UPDRS III, folate, B12, time, folate×time and B12×time. bCovariates included in model: sex, age, MDS-UPDRS III, LEDD, folate, B12, time, LEDD×time, folate×time and B12×time. MoCA, Montreal Cognitive Assessment; MDS-UPDRS III, Movement Disorder Society Unified Parkinson’s Disease Rating Scale Part III; LEDD, Levodopa equivalent daily dose; tHyc, serum homocysteine.
DISCUSSION
We found that participants with early PD had lower serum urate and higher serum homocysteine than age-matched controls. Mixed effect modelling revealed that lower urate and higher homocysteine at baseline predicted worse motor scores over 54 months, while only high baseline homocysteine predicted declining MoCA scores.
In keeping with previous studies, our cohort of PD participants had significantly lower serum urate concentrations than controls [6, 21]. These studies also found a correlation between lower baseline urate and worsening motor function; however, our study design differed in a number of ways. While we recruited patients with mean disease duration of six months, the other studies recruited PD patients, who did not require levodopa, with disease duration of up to five years. Our end-point was a motor progression over 54 months using the MDS-UPDRS III, whilst the end-point in others studies was the initiation of dopaminergic therapy over the two-year follow up period.
Low serum urate concentration predicted worsening motor symptoms over 54 months. The protective effect of urate is thought to relate to its ability to act as a physiological buffer to oxidative stress [3]. Evidence from human post-mortem studies suggests dopaminergic neurons in the substantia nigra of PD patients are subject to higher levels of oxidative stress than controls [1]. Rodents have an enzyme, urate oxidase, which breaks down urate; urate oxidase is not present in humans. Transgenic urate oxidase knockout mice, who consequently have higher levels of circulating urate, were shown to lose fewer nigral dopamine neurons in response to an intrastriatal injection of the dopaminergic toxin 6-hydroxydopamine, than wild-type mice [22]. Our findings provide further evidence of the effects of urate on progression of PD even in early disease, and therefore, its role as a possible therapeutic target. A recent placebo controlled trial has shown that inosine administration increases urate concentration and oxidative capacity in patients with early PD [23]. The increased oxidative capacity, as measured by the ferric reducing property of plasma assay, was associated with reduced change in the UPDRS III over six months. This result provides evidence of the therapeutic potential of inosine to increase urate concentration, although further longitudinal evaluation is needed.
Homocysteine was significantly higher in newly diagnosed PD compared to controls. This is consistent with a previous study in established PD [24]. Homocysteine was also associated with increasing motor severity and cognitive impairment. This is consistent with the findings of O’Suilleabhain and colleagues [25] who reported that in patients with established PD, those with hyperhomocysteinaemia performed worse on a range of neuropsychometric tests despite comparable motor severity. 20% of patients with hyperhomocysteinaemia in the original cohort were not included in the longitudinal analyses, however, because of excess mortality in this group.
Increasing evidence suggests that the pathology of PD dementia includes a synergistic effect of α-synuclein and amyloid pathologies. Post-mortem studies of PD subjects have shown that there is a significant correlation between the extent of amyloid deposition and Lewy body pathology [26]. Interestingly, in the EXPRESS trial, patients with PDD and hyperhomocysteinaemia showed a more favourable response to rivastigmine therapy on measures of cognition, functioning and neuropsychiatric symptomatology than those in the normal range [27], suggesting that stratifying patients by homocysteine level may be useful for predicting response to treatment.
While raised plasma homocysteine is an established risk factor for cardiovascular disease [28], its role in diseases of the nervous system if still under investigation. Homocysteine acts as an N-Nitrosodimethylamine (NMDA) receptor agonist on cultured cortical neurons, leading to neuronal death [29]. In the basal ganglia, five days of homocysteine administration into the lateral ventricle of rats led to reduced levels of striatal dopamine and metabolites, and a decreased number of spontaneous movements [30]. One possible mechanism for this is increased oxidative stress; Bhattacharjee et al. has shown that 60 days of systemic administration of homocysteine increases striatal levels of superoxide dismutase [31].
Strengths of the current study include the recruitment of a cohort of patients with early PD, many of whom had low levels of motor impairment and not yet established on levodopa therapy. The inclusion of a matched control group was important to control for the effects of normal ageing. The study also used a range of validated assessments, which were repeated every 18 months.
Potential limitations of this study include the non-inclusion of patients who declined to be part of the ICICLE-PD study. The participants who declined to take part in the study may have been those with more rapid motor and cognitive decline and therefore of particular interest. Our homocysteine and urate levels were based on a single non-fasting measurement and fluctuations in homocysteine levels over time may be important. A single homocysteine measurement may underestimate the risk of the outcome of interest due to regression dilution bias [32]. Although this study is large in comparison to other studies in PD, our sample size is small in comparison to studies of non-PD populations [8, 33]. Nevertheless, we were still able to demonstrate that impaired cognition was associated with increased homocysteine levels.
Only a small proportion of PD participants were drug naive (12.3%) at baseline and our cohort contained a mixture of participants on and off levodopa therapy. This could have implications for our results as previous studies have shown that levodopa acutely increases homocysteine concentration [34]. However, we controlled for LEDD as part of our analysis to minimise the effects. As with many longitudinal studies, missing data was an issue; 58% of PD participants and 68% of controls completed assessments at each time point. This has implications for data interpretation as participants who did not return for further evaluation may have been those with a more rapid decline in PD and cognition, and would therefore have been of particular interest to this study. However, we used mixed effects modelling as part of our analysis. This form of multilevel modelling has the ability to handle missing data points, and so the omission of individual variables at one time point does not result in all of the data for that subject being excluded from analysis.
In conclusion, we have shown that in a longitudinal study of PD subjects, urate and homocysteine predict motor progression over 54 months, while homocysteine predicted cognitive decline. These findings lend support to the hypothesis that oxidative stress may play a role in the pathophysiology of PD and that motor and cognitive aspects of the disease may have overlapping but separate mechanisms. This hypothesis would require further investigation in order to develop robust neuroprotective strategies to alter the disease trajectory. In addition to potential disease modification, our findings suggest that determining urate and homocysteine concentration at the outset may have a role in predicting patients with PD at greater risk of decline in motor and cognitive function.
CONFLICT OF INTEREST
I Sleeman, F Johnston, GW Duncan and TK Khoo have no financial disclosures. RA Lawson is funded by Parkinson’s UK has previously been supported by grants from the Lockhart Parkinson’s Disease Research Fund. AJ Yarnall is funded by the Biomedical Research Centre, Newcastle University, and has previously been supported by grants from the Lockhart Parkinson’s Disease Research Fund and the Michael J. Fox Foundation (MJFF). She has received honoraria from Teva-Lundbeck and sponsorship from Teva-Lundbeck, UCB, GlaxoSmithKline (GSK), Genus, Britannia Pharmaceuticals Ltd. and AbbVie for attending conferences. DJ Burn has received grants from NIHR, Wellcome Trust, and Parkinson’s UK. He has received speaker fees from Acadia Pharmaceuticals.
ACKNOWLEDGMENTS
The ICICLE-PD study group would like to thank all participants for their contribution towards the study. The research was funded by Parkinson’s UK (J-0802, G-1507) and supported by the Lockhart Parkinson’s Disease Research Fund, the National Institute for Health Research (NIHR) Newcastle Biomedical Research Centre in Ageing and Chronic Disease and Biomedical Research Unit in Lewy Body Dementia based at Newcastle upon Tyne Hospitals NHS Foundation Trust and Newcastle University and the NIHR Biomedical Research Centre award to the University of Cambridge/Addenbrooke's Hospital. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JPD-181535.
REFERENCES
[1] | Yoritaka A , Hattori N , Uchida K , Tanaka M , Stadtman ER , Mizuno Y ((1996) ) Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci USA 93: , 2696–2701. |
[2] | Zhang J , Perry G , Smith MA , Robertson D , Olson SJ , Graham DG , Montine TJ ((1999) ) Parkinson’s disease is associated with oxidative damage to cytoplasmic DNA and RNA in substantia nigra neurons. Am J Pathol 154: , 1423–1429. |
[3] | Ames BN , Cathcart R , Schwiers E , Hochstein P ((1981) ) Uric acid provides an antioxidant defense in humans against oxidant-and radical-caused aging and cancer: A hypothesis. Proc Natl Acad Sci USA 78: , 6858–6862. |
[4] | Chen H , Mosley TH , Alonso A , Huang X ((2009) ) Plasma urate and Parkinson’s disease in the atherosclerosis risk in communities (ARIC) study. Am J Epidemiol 169: , 1064–1069. |
[5] | Weisskopf MG , O'Reilly E , Chen H , Schwarzschild MA , Ascherio A ((2007) ) Plasma urate and risk of Parkinson’s disease. Am J Epidemiol 166: , 561–567. |
[6] | Schwarzschild MA , Schwid SR , Marek K , Watts A , Lang AE , Oakes D , Shoulson I , Ascherio A , Hyson C , Gorbold E , Rudolph A , Kieburtz K , Fahn S , Gauger L , Goetz C , Seibyl J , Forrest M , Ondrasik J ((2008) ) Serum urate as a predictor of clinical and radiographic progression in Parkinson disease. Arch Neurol 65: , 716–723. |
[7] | Annanmaki T , Pessala-Driver A , Hokkanen L , Murros K ((2008) ) Uric acid associates with cognition in Parkinson’s disease. Parkinsonism Relat Disord 14: , 576–578. |
[8] | Seshadri S , Beiser A , Selhub J , Jacques PF , Rosenberg IH , D'Agostino RB , Wilson PWF , Wolf PA ((2002) ) Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N Engl J Med 346: , 476–483. |
[9] | Zoccolella S , dell'Aquila C , Abruzzese G , Antonini A , Bonuccelli U , Canesi M , Cristina S , Marchese R , Pac-chetti C , Zagaglia R , Logroscino G , Defazio G , Lamberti P , Livrea P ((2009) ) Hyperhomocysteinemia in levodopa-treated patients with Parkinson’s disease dementia. Mov Disord 24: , 1028–1033. |
[10] | Duncan GW , Khoo TK , Coleman SY , Brayne C , Yarnall AJ , O’Brien JT , Barker RA , Burn DJ ((2014) ) The incidence of Parkinson’s disease in the North-East of England. Age Ageing 43: , 257–263. |
[11] | Hughes AJ , Daniel SE , Kilford L , Lees AJ ((1992) ) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55: , 181–184. |
[12] | Goetz CG , Tilley BC , Shaftman SR , Stebbins GT , Fahn S , Martinez-Martin P , Poewe W , Sampaio C , Stern MB , Dodel R , Dubois B , Holloway R , Jankovic J , Kulisevsky J , Lang AE , Lees A , Leurgans S , LeWitt PA , Nyenhuis D , Olanow W , Rascol O , Schrag A , Teresi JA , van Hilten JJ , LaPelle N ((2008) ) Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Scale presentation and clinimetric testing results.Mov Disord 23: , 2129–2170. |
[13] | Hoehn MM , Yahr MD ((1967) ) Parkinsonism: Onset, progression and mortality. eurology 17: , 427–442. |
[14] | Tomlinson CL , Stowe R , Patel S , Rick C , Gray R , Clarke CE ((2010) ) Systematic review of levodopa dose equivalency reporting in Parkinson’s disease. Mov Disord 25: , 26492653. |
[15] | Mathias JL , Bowden SC , Barrett-Woodbridge M ((2007) ) Accuracy of the Wechsler Test of Adult Reading (WTAR) and National Adult Reading Test (NART) when estimating IQ in a healthy Australian sample. Aust Psychol 42: , 49–56. |
[16] | Yesavage JA , Brink TL , Rose TL , Lum O , Huang V , Adey M , Leirer VO Development and validation of a geriatric depression screening scale: A preliminary report. J Psychiatr Res 17: , 37–49. |
[17] | Folstein MF , Folstein SE , McHugh PR ((1975) ) “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 12: ,189–198. |
[18] | Dalrymple-Alford JC , MacAskill MR , Nakas CT , Livingston L , Graham C , Crucian GP , Melzer TR , Kirwan J , Keenan R , Wells S , Porter RJ , Watts R , Anderson TJ ((2010) ) The MoCA: Well-suited screen for cognitive impairment in Parkinson disease. Neurology 75: , 1717–1725. |
[19] | ((2013) ) R: A language and environment for statistical computing. R Foundation for Statistical Computing [program]. |
[20] | Bates D , Mächler M , M Bolker B , Walker S ((2014) )Package Lme4: Linear Mixed-Effects Models Using Eigen and S4. |
[21] | Ascherio A , LeWitt PA , Xu K , Eberly S , Watts A , Matson WR , Marras C , Kieburtz K , Rudolph A , Bogdanov MB , Schwid SR , Tennis M , Tanner CM , Beal MF , Lang AE , Oakes D , Fahn S , Shoulson I , Schwarzschild MA ((2009) ) Urate as a predictor of the rate of clinical decline in Parkinson disease. Arch Neurol 66: , 1460–1468. |
[22] | Chen X , Burdett TC , Desjardins CA , Logan R , Cipriani S , Xu Y , Schwarzschild MA ((2013) ) Disrupted and transgenic urate oxidase alter urate and dopaminergic neurodegenera-tion. Proc Natl Acad Sci USA 110: , 300–305. |
[23] | Bhattacharyya S , Bakshi R , Logan R , Ascherio A , Mack-lin EA , Schwarzschild MA ((2016) ) Oral inosine persistently elevates plasma antioxidant capacity in Parkinson’s disease. Mov Disord 31: , 417–421. |
[24] | Rodriguez-Oroz MC , Lage PM , Sanchez-Mut J , Lamet I , Pagonabarraga J , Toledo JB , Garcia-Garcia D , Clavero P , Samaranch L , Irurzun C , Matsubara JM , Irigoien J , Bescos E , Kulisevsky J , Perez-Tur J , Obeso JA ((2009) ) Homocys-teine and cognitive impairment in Parkinson’s disease: A biochemical, neuroimaging, and genetic study. Mov Disord 24: , 1437–1444. |
[25] | O'Suilleabhain PE , Sung V , Hernandez C , Lacritz L , Dewey RBJ , Bottiglieri T , Diaz-Arrastia R ((2004) ) Elevated plasma homocysteine level in patients with Parkinson disease: Motor, affective, and cognitive associations. Arch Neurol 61: , 865–868. |
[26] | Lashley T , Holton JL , Gray E , Kirkham K , O'Sullivan SS , Hilbig A , Wood NW , Lees AJ , Revesz T ((2008) ) Cortical alpha-synuclein load is associated with amyloid-beta plaque burden in a subset of Parkinson’s disease patients. Acta Neuropathol 115: , 417–425. |
[27] | Barone P , Burn DJ , van Laar T , Hsu C , Poewe W , Lane RM ((2008) ) Rivastigmine versus placebo in hyperhomocysteine-mic Parkinson’s disease dementia patients. Mov Disord 23: , 1532–1540. |
[28] | Bostom AG , Silbershatz H , Rosenberg IH , Selhub J , D'Agostino RB , Wolf PA , Jacques PF , Wilson PW ((1999) ) Nonfasting plasma total homocysteine levels and all-cause and cardiovascular disease mortality in elderly Framingham men and women. Arch Intern Med 159: , 1077–1080. |
[29] | Poddar R , Paul S ((2009) ) Homocysteine-NMDA receptor-mediated activation of extracellular signal-regulated kinase leads to neuronal cell death. J Neurochem 110: , 1095–1106. |
[30] | Lee E-SY , Chen H , Soliman KFA , Charlton CG (2005) Effects of homocysteine on the dopaminergic system and behavior in rodents. Neurotoxicology 26: , 361–371. |
[31] | Bhattacharjee N , Paul R , Giri A , Borah A ((2016) ) Chronic exposure of homocysteine in mice contributes to dopamine loss by enhancing oxidative stress in nigrostriatum and produces behavioral phenotypes of Parkinson’s disease. Biochem Biophys Rep 6: , 47–53. |
[32] | Clarke R , Lewington S , Donald A , Johnston C , Refsum H , Stratton I , Jacques P , Breteler MM , Holman R ((2001) ) Underestimation of the importance of homocysteine as a risk factor for cardiovascular disease in epidemiological studies J Cardiovasc Risk 8: , 363–369. |
[33] | Wei Z , Tiandong W , Yang L , Huaxing M , Guowen M , Yalan F , Xiaoyuan N ((2016) ) Parkinson’s disease and homocysteine: A community-based study in a folate and vitamin B12 deficient population. Parkinsons Dis (2016) , 9539836. |
[34] | Muller T , Muhlack S ((2010) ) Acute homocysteine rise after repeated levodopa application in patients with Parkinson’s disease. Parkinsonism Relat Disord 16: , 688–689. |


