
One Step Into the Future: New iPSC Tools to Advance Research in Parkinson’s Disease and Neurological Disorders
Abstract
Studying Parkinson’s disease (PD) in the laboratory presents many challenges, the main one being the limited availability of human cells and tissue from affected individuals. As PD is characterized by a loss of dopaminergic (DA) neurons in the brain, it is nearly impossible for researchers to access and extract these cells from living patients. Thus, in the past PD research has focused on the use of patients’ post-mortem tissues, animal models, or immortalized cell lines to dissect cellular pathways of interest. While these strategies deepened our knowledge of pathological mechanisms in PD, they failed to faithfully capture key mechanisms at play in the human brain. The emergence of induced pluripotent stem cell (iPSC) technology is revolutionizing PD research, as it allows for the differentiation and growth of human DA neurons in vitro, holding immense potential not only for modelling PD, but also for identifying novel therapies. However, to reproduce the complexity of the brain’s environment, researchers are recognizing the need to further develop and refine iPSC-based tools. In this review, we provide an overview of different systems now available for the study of PD, with a particular emphasis on the potential and limitations of iPSC as research tools to generate more relevant models of PD pathophysiology and advance the drug discovery process.
INTRODUCTION
Parkinson’s disease (PD) is a devastating neurodegenerative disorder, affecting more than 1% of the population over 65 years of age. It is characterized by debilitating motor symptoms, including bradykinesia, tremors and muscle rigidity, as well as non-motor symptoms that include intestinal dysfunction, depression, cognitive decline and sleep disturbances [1]. While the majority of PD cases are idiopathic, about 10% have been linked to genetic mutations with Mendelian inheritance within families. Specifically, such mutations have been found to occur in genes including α-synuclein (SNCA), Leucine-rich repeat kinase 2 (LRRK2), glucocerebrosidase (GBA), Parkin (PARK2 gene) and PTEN-induced putative kinase (PINK1). Independent of familial history, Gene wide association studies (GWAS) have also unravelled several gene variants associated with an increased risk of PD development [2–4]. These genetic data have helped scientists create models explaining the pathogenic causes of this disorder. Classical hallmarks of PD are the selective loss of dopaminergic (DA) neurons in the substantia nigra pars compacta, accompanied by the presence of inclusions within neurons called “Lewy bodies”. These inclusions are primarily comprised of the α-synuclein protein [5], which forms cellular fibrillar aggregates within the cells that may be able to spread from cell to cell in a prion-like manner [6–8]. After two decades of research on PD genes, several cellular pathways have been implicated in PD pathogenesis, including mitochondrial dysfunction [9–11], perturbed electrical activity of neurons [12, 13], and dysregulated protein homeostasis due to autophagy, lysosomal and proteasomal defects [14, 15]. However, to date, there are no therapies to effectively halt the progression of the disease, and treatment of PD is limited to symptom management. It is therefore of utmost importance to refine the models we use in fundamental research, to better understand the pathophysiology of PD for developing effective therapeutic strategies. Here we present existing PD models, and discuss the future of PD modelling by outlining the next generation of more relevant models of PD pathophysiology, made possible through the advent of human induced pluripotent stem cell (iPSC) technology and genome editing. We will also discuss the potential of using iPSCs in drug discovery assays.
FROM ANIMAL TO HUMAN MODELS
PD animal models
When investigating the pathological mechanisms contributing to PD, the availability of human post-mortem brain tissue is limited. Nonetheless, our understanding of the composition of Lewy bodies and their distribution within the brain has increased significantly through studies using post-mortem human brain tissue from PD patients. Over the past few decades, a combination of animal and cell models has also helped elucidate some of the potential causes underlying PD. However, many of these models have limitations in truly modelling PD. Mammalian and non-mammalian animal models of PD can be divided into toxin-induced and gene-related PD models. 6-OHDA (2,4,5-trihydroxyphenethylamine) injection within animal brains, tested on dogs, cats, primates and rodent models, is toxic for DA neurons and leads to PD-like behaviour. However, neurodegeneration is restricted to DA neurons and 6-OHDA does not affect other brain areas implicated in PD. MPTP (1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine) administration has long been used as a toxin-induced PD model. An MPTP metabolite, MPP+, inhibits complex I of the mitochondrial electron transport chain and causes neuronal death through oxidative stress. MPP + induces damage to the nigrostriatal DA connections, loss of DA cells, inflammation, and generation of reactive oxygen species. However, MPTP treatment doesn’t induce Lewy bodies or synuclein pathology in the majority of studies [16–20]. Only one group reported inclusions positive for synuclein and ubiquitin after continuous MPTP infusion [21]. Rotenone and paraquat, in animals, induces Lewy body formation but their effects still remain ambiguous due to numerous contradictory results in studies [16]. Overall, these toxin-induced PD models commonly induce oxidative stress and death of DA neurons, as observed in PD, but they do not completely model the pathological mechanisms occurring in PD patients [16–19, 21, 22]. Murine models have also been engineered to recapitulate genetic alterations, in PARK2, PINK1, DJ-1 (parkinsonism associated deglycase), SNCA, LRRK2 and GBA genes, observed in PD patients. Duplication or triplication of SNCA and misense mutations of α-synuclein (A30P, A53T and E46K) have been identified in familial PD. Thus, transgenic mice (knock-out and overexpression), grafting models, intracerebral injections, and virally induced expression of α-synuclein have been developed to replicate α-synuclein overexpression and mutations [23, 24]. Mutations in LRRK2, such as G2019S and R1441C, cause late-onset autosomal dominant forms of PD [25–27]. Similarly to SNCA models, LRRK2 mice models present motor impairments and reduction of striatal DA neurons, but do not display disruption of the nigrostriatal DA neurons [28–30]. Only viral vector-based model has been reported to induce degeneration of nigral DA neurons in rat [31]. Homozygous mutations in PARK2 and PINK1 genes are linked to autosomal recessive PD. Many knock-out mice have been generated for these genes and presented a moderate reduction in striatal DA neuron levels accompanied by low locomotor activity, without Lewy body formation [32–34]. Long-term overexpression of human wild-type and T240R mutant parkin in rat substantia nigra induces progressive DA neurodegeneration [35]. Bacterial artificial chromosome transgenic mice expressing a truncated mutant parkin exhibit age-dependent motor deficits and DA neurodegeneration [36]. DJ-1 mutations are linked to autosomal recessive and early-onset PD and different mice models recapitulate nigrostriatal DA deficits and progressive DA cell loss [37, 38]. Finally, GBA mutations are the greatest risk factor for PD discovered to date and are associated with an enhanced risk of developing synucleinopathies. Thus, different groups have investigated the link between GBA mutations and synuclein levels. They showed a correlation between β-glucosidase (GCase) levels and activity and synuclein degradation [39–42].
In these murine models, many of the hallmarks for PD, including impairment in mitochondrial and lysosomal function, synuclein propagation and synaptic dysfunction, have been observed but they do not completely model PD [16–19, 22]. Recently, small vertebrate fish, such as zebrafish and medakas, have been used to model PD pathology. DA neurons in these models are more sensitive to 6-OHDA or MPTP exposure or genetic impairment, but do not present with Lewy body-like inclusions. Interestingly, these fish show abnormal swimming behavior [43]. The invertebrate models, C. elegans and Drosophila melanogaster, show similar phenotypes as vertebrate models after exposure to rotenone, paraquat, MPTP or 6-OHDA. Many genetic models have been generated in these species. However, in C. elegans and Drosophila, there is no orthologous gene for α-synuclein, and studies were performed on animals overexpressing human α-synuclein [44–46].
While using animal models presents physiological advantages for studying PD compared to using cell lines, they incompletely recapitulate PD phenotypes. Many drug trials have been based on results from experimental animal models. Unfortunately, these results failed to translate into clinical therapies in the neurodegenerative field. This failure could be explained by the intrinsic differences between animals and human. Often, animals that appear to recapitulate the disease phenotype exhibit intrinsic differences with respect to the human disease. The recent development of techniques to generate stem cells from somatic human cells represents a major technological advance, providing a unique and novel approach to grow human neurons on a routine basis.
Stem cell technology to create human models of PD
Over the past decade, the emergence of iPSCs has opened up new avenues in PD research. With their self-renewal ability and potential to differentiate into many lineages, iPSC represent an opportunity to study the pathogenesis of PD at the cellular level in a human model, without raising the ethical concerns previously triggered by the use of embryonic stem cells [47]. iPSCs can be directly generated from somatic cells, obtained from the skin, blood or urine of an individual, by expressing the reprogramming factors OCT4, SOX2, KLF4, and c-MYC. This technology was first described in 2006 in a seminal study by Kazutoshi Takahashi and Shinya Yamanaka as a technique to create mouse iPSCs [48], and followed up with two studies in 2007 describing the generation of human iPSCs samples in follow-up studies in 2007 by Yamanaka and Thomson, respectively [49, 50]. In 2009, Soldner and colleagues were the first to describe generation of an iPSC cell-line from a patient with sporadic PD, and the subsequent differentiation of these cells into DA neurons [51]. Nguyen and others, in 2011, described for the first time a pathological phenotype manifesting in iPSC-derived DA neurons from a PD patient bearing a mutation in the LRRK2 gene [14]. Since then an increasing number of iPSC lines have been generated, including iPSCs from patients with mutations in familial PD genes, as well as from patients with sporadic PD without an identified causative genetic mutation. Currently, several organizations, such as the European Bank for induced pluripotent Stem Cells (EBiSC) and the New York Stem Cell Foundation (NYSCF), are centralizing and making iPSC lines derived from patients with PD available to researchers and industries. Taking advantage of such lines, many studies have started to focus on dissecting the pathological mechanisms of PD in iPSC-derived DA neurons from patients compared to neurons from healthy individuals (Table 1) [52–76].
Table 1
iPSC models of Parkinson’s disease
| PD model | Authors | Date | Cells used to derive iPSC | Control | Highlight | |
| Sporadic | Soldner et al. | 2009 | Skin fibroblasts | NA | Generation of iPSC line | |
| Cooper et al. | 2010 | Skin fibroblasts | NA | Improved small molecule differentiation protocol for generation of midbrain DA neurons | ||
| Sánchez-Danés et al. | 2012 | Epidermal keratinocutes and fibroblasts | Unrelated healthy individual | Dysfunctional autographic system described in sporadic and LRRK2 lines | ||
| Zhang et al. | 2017 | PBMCs | NA | Generation of iPSC line | ||
| LRRK2 | p.Gly2019Ser | Nguyen et al. | 2011 | Skin fibroblasts | Unrelated healthy individual | First study showing a pathological phenotype in PD iPSC-derived neurons |
| p.Gly2019Ser | Sánchez-Danés et al. | 2012 | Skin fibroblasts | Unrelated healthy individual | Dysfunctional autographic system described in sporadic and LRRK2 lines | |
| p.Gly2019Ser/ p.Arg1441Cys | Cooper et al. | 2012 | Skin fibroblasts | Unrelated healthy individual | Mitochondrial dysfunction in patient lines | |
| p.Gly2019Ser | Orenstein et al. | 2013 | Skin fibroblasts | Unrelated healthy individual | Effect of LRRK2 mutation on chaperone-mediated autophagy | |
| p.Gly2019Ser/ p.Arg1441Cys | Lopez-de Maturana et al. | 2016 | Skin fibroblasts | Unrelated healthy individual | Effect of mutations on NFkB pathway | |
| pAsn551Lys/ p.Ser1647Thr/ p.Arg1398His/ p.Arg1628Pro | Ma et al. | 2017 | PBMCs | NA | Generation of iPSC lines | |
| PINK1 | p.Gln456Xaa / p.Val170Gly | Seiber et al. | 2011 | Skin fibroblasts | Healthy family member | Mitochondrial defects in mutant lines |
| p.Gln456Xaa | Chung et al. | 2016 | Skin fibroblasts | Unrelated healthy individual | Compared floor-plate vs neural-rosette differentiation protocols | |
| p.Gln456Xaa | Cooper et al. | 2012 | Skin fibroblasts | Unrelated healthy individual | Mitochondrial dysfunction in patient lines | |
| SNCA (α-synuclein) | p.Ala53Thr/ p.Glu46Lys | Soldner et al. | 2011 | Skin fibroblasts | ||
| Triplication | Byers et al. | 2011 | Skin fibroblasts | Healthy family member | Confirmed synuclein over expression in the triplication line | |
| Triplication | Devine et al. | 2011 | Skin fibroblasts | Healthy family member | Confirmed synuclein over expression in the triplication line | |
| Triplication | Oliveira et al. | 2015 | Skin fibroblasts | Unrelated healthy individual and healthy family member | Impact of synuclein over expression on DA neuron differentiation | |
| GBA-1 (GCase) | p.Asn370Ser/ c.84dupG | Mazulli et al. | 2011 | Skin fibroblasts | Unrelated healthy individual | Relation between synuclein and GCase mutation |
| PARK2 (Parkin) | Deletion | Imaizumi et al. | 2012 | Skin fibroblasts | Unrelated healthy individual | Oxidative stress and mitochondrial dysfunction in PARK2 lines |
| Deletion | Jiang et al. | 2012 | Skin fibroblasts | Unrelated healthy individual | Link between Parkin and dopamine use in DA neurons | |
| Deletion | Ren et al. | 2015 | Skin fibroblasts | Unrelated healthy individual | Effect of deletions on neuronal arborization | |
| Deletion | Chang et al. | 2015 | Skin fibroblasts | Unrelated healthy individual | Proteasomal dysfunction and oxidative stress in mutant neurons | |
| p.Val324Ala | Chung et al. | 2016 | Skin fibroblasts | Unrelated healthy individual | Compared floor-plate vs neural-rosette differentiation protocols | |
| Deletion | Suzuki et al. | 2017 | Skin fibroblasts | Unrelated healthy individual | Monitored mitophag in iPSC using mtKeima | |
| Familial (LRRK2, SNCA, PARK2, GBA-1, PINK1) | Refer to paper for specific mutations | Holmqvist et al. | 2016 | Skin fibroblasts | NA | Generation of iPSC lines from 43 PD patients |
| Refer to paper for specific mutations | Momcilovic et al. | 2016 | Skin fibroblasts | NA | Generation of iPSC lines from 10 PD patients | |
Although this strategy represents a significant advance, there are limitations to the use of iPSCs for modelling PD. Neurons derived from iPSCs are often considered embryonic, unlike the aged neurons found in adult brains [77, 78]. In addition, they are grown outside the native environment of the brain, in artificial dishes, and maintained in culture media made of a combination of small molecules that does not recapitulate the complexity of factors received by cells in vivo. Thus, many factors required for their normal growth and differentiation may be lacking. Since PD is a late-onset and multi-factorial disorder, neurons obtained from the iPSCs of PD patients are expected to present very subtle pathological phenotypes. As there is intrinsic line-to-line variability amongst iPSC lines, partly due to differences in genetic backgrounds, it is very challenging to identify these subtle phenotypical differences by comparing patient lines to lines derived from unrelated healthy controls. In addition, when differentiated in a dish, cells are grown as almost pure neuronal populations. Obtaining neurons that behave in the same way as they would in the body requires the presence of additional cells in the culture. These could be astrocytes, oligodendrocytes, microglia, or different neuronal subtypes whose presence may elicit the formation of synapses between neurons, promote myelination of the neurons and elicit electrical activity and firing between neurons. To address these issues, scientists have designed new ways to generate more elaborate iPSC models.
TOWARDS MORE RELEVANT MODELS OF PD PATHOPHYSIOLOGY USING IPSCs
IPSC genome editing
Inherent line-to-line variability makes it challenging to identify phenotypical traits of diseased versus healthy iPSC lines. Differences in genetic background between lines can explain part of this phenomenon. Thus, when studying genetic forms of PD, it is worth attempting to control these known sources of variation. Using genome editing techniques, scientists can either introduce a pathogenic mutation in a control line or correct a mutation in a patient line back to its control counterpart (Fig. 1). This allows for comparison of two lines that have the same genetic background, differing only in the mutation of interest. Such lines are called isogenic. The standardization of genome editing techniques and development of CRISPR technology over the past decade has greatly facilitated the generation of isogenic iPSC lines for the study of PD.
Fig.1
The iPSC toolbox. Representation of the multiple tools available for creating in vitro models using iPSC. The upper left panel represents genome-editing techniques that allowing for the creation of isogenic pairs of stem cell lines. Clockwise, the other panels depict models of increasing complexity such as co-cultures, 3D organoids and assembly of multiple organoids on a chip. The main strengths and limitations of the different models are highlighted in the adjacent squares.
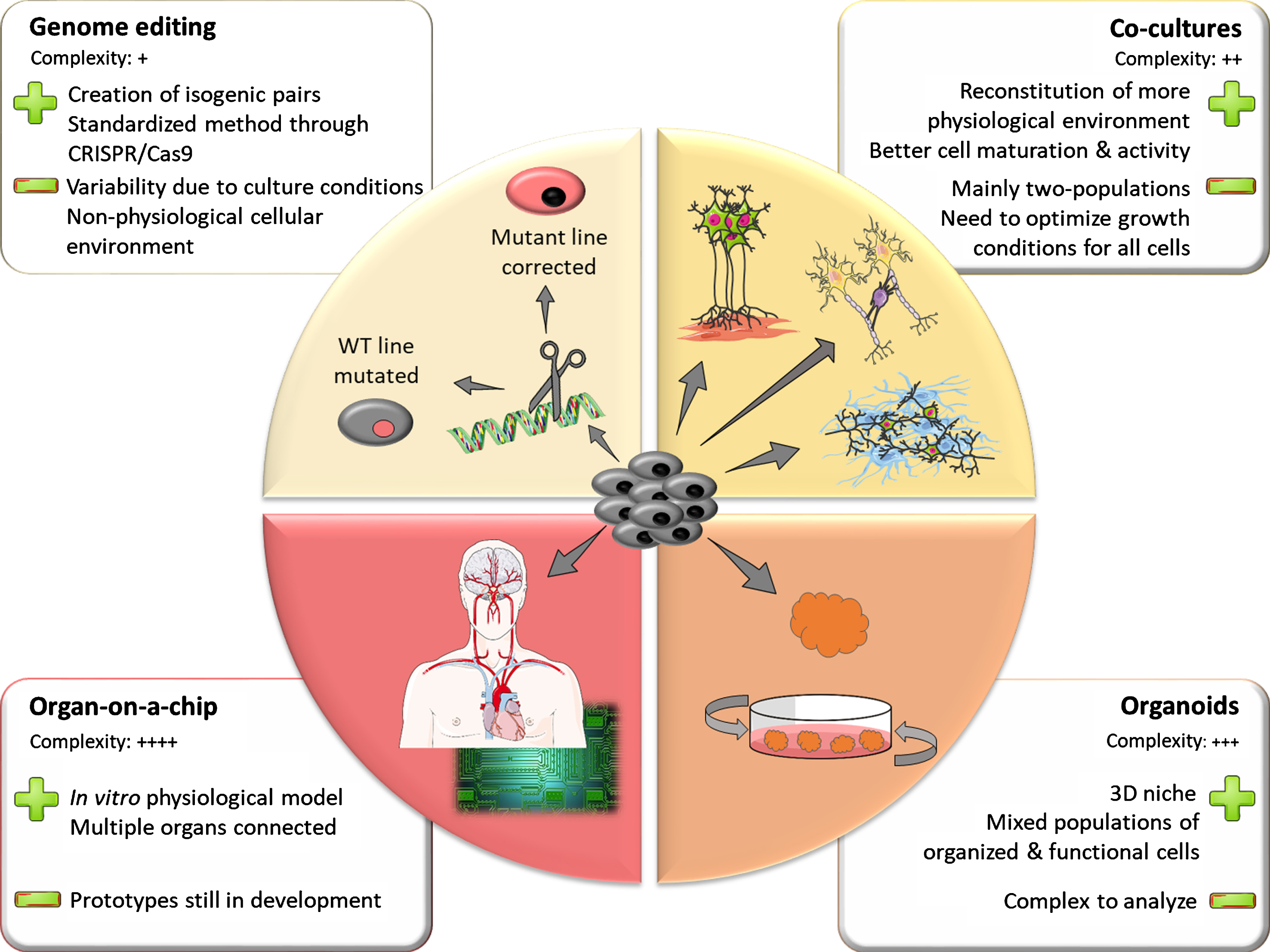
However, technical challenges remain in making the generation of isogenic pairs a standard in the iPSC field. Genome editing using homologous recombination (HR) [79, 80], while efficient in mouse cell lines, is quite inefficient in human stem cell lines [81]. This could be due to a lower rate of spontaneous HR in human cells. To overcome this problem, more efficient delivery strategies for the HR construct were designed. The one with the most success in human iPSCs was the helper-dependent adenoviral vectors (HDAdVs) [82]. HDAdVs are poorly immunogenic non-integrative viral vectors, which can efficiently transduce a wide range of cell types. In 2012, Liu and colleagues successfully used this technique, becoming the first team to model PD through generation of isogenic human iPSC lines [83]. In this study, a patient iPSC line bearing the LRRK2 c.6055G > A (p.Gly2019Ser, G2019S) mutation was corrected back to its WT counterpart, then both lines were differentiated into neural stem cells. The authors identified an aberrant nuclear phenotype in mutant cells that was not present in corrected cells. They also confirmed this previously undescribed phenotype in patients’ post-mortem tissues, illustrating the strength of modelling a disease with isogenic human lines.
However, editing the genome by relying on natural HR was quickly superseded by the use of site-specific nucleases (SSN) to induce double stranded breaks (DSB) in DNA. Indeed, it was shown that generating a DSB in DNA could greatly increase the rates of homology-directed repair at the break site [84, 85]. Applying this strategy, while providing an exogenous repair template bearing a mutation of interest, enhanced gene editing success rates by orders of magnitude compared to classical HR. In the years following this discovery, SSN were designed to target specific genomic sites. Zinc finger nucleases (ZFNs), engineered proteins consisting of a fusion of a zinc finger DNA-binding domain and the nuclease domain of the FokI restriction enzyme [86], were extensively used in modelling of PD. In 2013, Reinhardt and colleagues were the first to describe the generation of isogenic iPSC PD lines using ZFN-based genome editing [87]. In this study, the researchers corrected three patient iPSC lines bearing the LRRK2 G2019S mutation and additionally introduced this mutation in a line from a healthy individual. The analysis of these four isogenic pairs revealed mutation-dependent defects in axonal length and sensitivity to PD stressors. In the following years, scientists used ZFN genome editing to investigate the effects of mutations in other PD-related genes such as SNCA [88–90] or GBA-1 [91, 92].
ZFN design requires expertise and a considerable amount of time, making ZFN based genome editing challenging to implement for many teams. Transcription activator-like effector nucleases (TALENs) [93], a different type of SSNs, present the same issues. It was the advent of the CRISPR/Cas9 tools in 2013 [94] that democratized the use of genome engineering. This technique, inspired by a bacterial immune mechanism [95], allows for DNA targeting using a combination of a specific guide RNA construct (gRNA) and the Cas9 nuclease. The versatility of DNA editing through distinct gRNAs allowed many groups to implement genome engineering, in a faster and more standardized manner. In 2016, Soldner and others used CRISPR/Cas9 methodology to introduce single nucleotide polymorphisms (SNPs) in the PD-related SNCA gene in human embryonic stem cells [96]. They identified SNP variants associated with α-synuclein overexpression that correlated with findings from GWAS.
Targeted genetic manipulation of iPSCs, using the now standardized CRISPR/Cas9 system to create isogenic controls will undoubtedly improve the utility of iPSCs to model PD. However, in addition to genetic background, other apparently stochastic factors in the differentiation process, such as temperature, cell density, number of cell passages and concentrations of specific growth factors, also seem to contribute substantially to the line-to-line variability of iPSCs. Moreover, cellular phenotype is a result of the interaction between a cell’s genotype and its environment. Therefore, significant efforts are now being made to generate more authentic cellular environments to better model PD in vitro.
Reaching the next step: Modelling cellular interactions using co-culture systems
Every tissue and system in the human body is composed of various cell types interacting with each other. In most cases, cells depend on these interactions to establish and maintain their proper physiological function. Therefore, to generate more faithful in vitro models of cellular function and dysfunction, it is critical to recreate the natural interactions between cell populations. To this end, co-culture systems such as microfluidic platforms [97, 98], membrane filters [99], three-dimensional scaffolds [100] and microarrays [101] have been developed. These permit the growth of two or more different cellular populations, within partially separated or common chambers.
Recently, scientists have started developing iPSC-based co-cultures to create models of the nervous system (Fig. 1). For instance, as a tool to study neuromuscular disorders such as amyotrophic lateral sclerosis (ALS) or spinal muscular atrophy (SMA), several teams have worked on reconstituting neuromuscular junctions by co-culturing stem cell-derived motor neurons with muscle cells [102–106]. Similarly, co-culturing iPSC-derived neurons with glial cells was shown to accelerate their maturation process and improve their electrophysiological activity [107–109]. In a recent study, Du and colleagues co-cultured iPSC-derived astrocytes with iPSC-derived DA neurons using a transwell co-culture system [110]. They showed that the presence of astrocytes in the culture could rescue mitochondrial defects induced in DA neurons by PD-related toxins such as rotenone. This observation highlights a crucial role for astrocytes as neuromodulators in the pathogenesis of PD. Thus, not only can co-cultures improve the quality of our models by accelerating cell maturation and promoting activity, but they can also increase our understanding of some of the more complex aspects of these neurological disorders.
To date, most co-culture models have involved only two defined cell populations. This is due to the intrinsic complexity associated with connecting different cell populations, both in terms of the analysis and technical considerations. Indeed, one challenge when co-culturing distinct cell types is optimizing a common growth medium to sustain all cells [111]. This becomes even harder when working with iPSC-derived cells that require specific growth factor cocktails to differentiate properly. To overcome this challenge and model even more complex systems involving many cell types, scientists are now turning to three-dimensional (3D) systems to mimic organ development in vitro.
Human brain organoids: Promising new models for neurological disorders
In 1992, the first 3D neuronal models composed of neural stem cells or neuronal progenitors, called neurospheres, were described [112]. Neurospheres are aggregates of cells that are not stereotypically organized in space, which means they do not recapitulate the cytoarchitecture of the brain. Recent progress in 3D cultures has given rise to 3D neural cell aggregates that have the ability to recapitulate some neuropathological phenotypes, such as the amyloid-beta accumulation commonly observed in Alzheimer’s disease (AD) [113–116]. Even though neuronal aggregates can model some aspects of neurodegenerative diseases, they still lack the anatomical organization of the brain, where distinct areas contribute differentially to the development of AD or PD [117].
In 2013, Lancaster described a novel 3D model recapitulating different areas of the human brain: the cerebral organoid [118]. Kept in culture, an organoid forms a complex neuronal self-organized tissue composed of a mixed population of neurons, astrocytes and oligodendrocytes, with some architectural similarities to the real human brain. Compared to the neurospheres generated previously, the organoid’s cells are organized in layers that include ventricular-like zones at early stages, composed of progenitors. Cells are functional, have spontaneous electrical activity in networks, and are in a niche-like environment. Interestingly, brain organoids can be cultured for long periods to obtain mature cells, morphologically and functionally, in contrast to neurosphere cultures [118–121].
Since 2013, different types of brain organoids have been generated, based on adaptations of the initial protocol published by Lancaster. Initially, no growth factors were included in the medium, and cells were self-differentiating. Now however, different laboratories directly drive the stem cells towards specific cell lineages. The key for efficient brain organoid generation is the appropriate combination of inductive signals and physical factors that drive pluripotent stem cells to form 3D structures. The modulation of this combination gives rise to multiple types of minibrains with different advantages for the investigation of neurodevelopmental and neurodegenerative disorders. Protocols now exist for making human cerebral [118, 122, 123], forebrain-like (dorsal and ventral) [124, 125], cerebellar [126], cortical-like (dorsal and ventral) [127, 128], hippocampal and choroid plexus-like tissue [129], midbrain [119, 120, 125], hypothalamic [125], and pallium and subpallium [130] brain organoids. Over the past five years, laboratories have focused on differentiation of brain organoids towards a specific fate, with defined media and scaffolds (Matrigel or Cell-Mate3D), in order to study different neuropathologies [118, 122, 123]. Such methods limit the cellular diversity compared to undirected whole-brain organoid methods.
IPSC-derived human brain organoids can be used to study neurodevelopment. Indeed, brain organoid development recapitulates early to mid-fetal development, and the epigenomic signatures of the human fetal brain [125, 131, 132]. So far, cerebral organoids have been used to study pathologies including microcephaly [118], Zika virus infection [133–136], and autism spectrum disorders [130, 137]. As well, human brain organoid technology has been used to investigate aspects of neurodegenerative disorders. Two groups generated cerebral organoids from the iPSCs of AD patients carrying familial mutations for presenilin1 or an amyloid precursor protein duplication, and successfully recapitulated the aggregation of amyloid-beta protein and tau pathology (hyperphosphorylation and aggregation), two neuropathological markers of AD. Treatment of the 3D cultures with drugs targeting either amyloid-beta aggregation or tau phosphorylation decreased the pathological markers [114, 138]. These promising results demonstrated that human brain organoids represent a relevant model to accelerate drug discovery.
The development of different types of brain organoids represents a major technological advance in the stem cell field, and a novel bridge between traditional 2D cultures and in vivo animal models for personalized drug screening. In particular, the development of midbrain organoids represents a new drug discovery model for PD (Fig. 1). Two groups published similar protocols for generating human midbrain organoids based on specific inductive signals introduced at specific timepoints during 3D culture to drive the stem cells towards a midbrain fate [119, 120]. The midbrain organoids were shown to contain functional midbrain neurons producing neuromelanin granules, a by-product of dopamine synthesis. Thirty per cent of the neuronal population is myelinated due to the presence of oligodendrocytes. Interestingly, Monzel and colleagues showed that nodes of Ranvier and spontaneous saltatory transmission were observed [119, 120]. So far, studies with PD patient-derived midbrain organoids have not been published, thus the pathological mechanisms in the midbrain organoid model are still unexplored.
Despite the remarkable features of brain organoids, circuits connecting multiple brain regions have not been observed. The connection or fusion of different type of brain organoids, for instance the midbrain with the cortical-like organoid, would be informative to model interactions between brain regions. Compared to animal models, brain organoids also lack connections with other organs. For PD, recent studies suggest that gut microbia might affect brain activity and trigger development of pathology [139]. Connecting different organs together, such as the gut and the brain, can potentially be an informative model for modelling PD in a dish. The development of organs connected on a chip could provide new tools for more accurate disease modeling.
Organs-on-a chip: Connection of brain organoids to other body organoids
Recent advances in biomaterials, 3D tissue engineering and microscale technologies have led to the emergence of “organs-on-a-chip” (Fig. 1). In an in vitro microphysiological system, different types of organoids are co-cultured in a continuous liquid flow system to mimic the physiology of the body. This technology potentially represents a more physiological platform for drug discovery and toxicity testing, because it mimics cell-cell and cell-matrix interactions, and provides appropriate chemical and mechanical forces, such as shear stress, and electrical cues through microelectrodes arrays [119]. Moreover, the development of incorporated biosensors can allow monitoring of biomarkers, chemokines and analytes, to better understand disease mechanisms. A number of companies and laboratories have developed connected organs with multiple fluid flow circuits operated by pumps, thus incorporating blood and excretory flow circuit with organoids, to carry out brain neurosphere analyses such as neurotoxicity screening, high content imaging, kinetic and electrophysiological assays [140] (cf also Mimetas®, DRAPER®, StemoniX® websites). Brain-on-a-chip models have been developed to model neurodegenerative disorders. So far, only neurospheres, but not complex brain organoids, have been incorporated on chips to model AD [140] or PD [141]. Interestingly, for the AD model, fluid flow mimicked the cerebrospinal fluid flow, which has a major role in cell communication and metabolism. This flow promoted differentiation of neurons, decreased cell death, and promoted the response of neurons to amyloid-beta protein addition [140].
One of the major advantages to combining iPSC-derived tissue and microfluidic chips is the possibility of deriving multiple organs from the same donor or patient. As mentioned above, connection of gut and brain organoids from a PD patient would be an interesting chip to develop, to investigate the impact of microbiota on the development of pathological markers, such as α-synuclein, which is suspected to spread from the enteric nervous system to the brain via the vagus nerve [142]. “Gut-on-a chip” systems have already been developed to test the inflammatory impact of microbial infection on intestinal epithelial cells [143]. Future technologies may connect, on a single chip, gut-organoids and brain-organoids derived from iPSCs [120, 144–147]. Such microphysiological systems, that mimic biologically relevant human conditions, could help model diseases and provide a better understanding of the pathological mechanisms at play. Moreover, adding a blood brain barrier to envelop the neural network system is a major potential breakthrough for the 3D neural network model [148]. Recently, one group generated human vascularized brain organoids from a patient, by co-culturing his iPSC-derived endothelial cells with the brain organoids [149]. This promising study demonstrated the feasibility of organoid vascularization. So far, neuronal organoids lack vascularization, leading to poor diffusion of oxygen and nutrients to the center of the tissue, which can often become necrotic with age [138, 150, 151]. This is one of the major limitations of organoid growth and one explanation for the heterogeneity observed within the tissue. Concurrently, another group showed vascularization of a human brain organoid transplanted in an adult mouse brain [152], suggesting a connectivity between the grafted organoid and the host. Recently, Berger and others developed millifluidic cultures to improve human midbrain organoid vitality and differentiation in bioreactors. They observed a decrease in the necrotic core size with continuous medium flow during culture, rather than shaking culture [153]. These pioneering studies present immense promise for truly generating more relevant models of PD through organoids.
USING iPSCs FOR DRUG DISCOVERY
No cure exists for PD despite huge pharmaceutical investments over recent decades. The lack of translation of promising results from animal models into human therapies, toxic side effects from many of these promising therapies and the requirement for drugs to pass the blood brain barrier accounts for many of the clinical failures so far. These issues need to be addressed, at the earliest stage of fundamental research, in order to develop next generation therapeutic compounds. The iPSCs models described here provide a different strategy that could open up new avenues for personalized medicine, by providing access to neurons from affected individuals, which would otherwise be inaccessible. To date, iPSCs represent a powerful and promising tool to identify new therapies for patients with PD.
IPSCs have a major clinical advantage over the classical cellular and animal models: they are generated from an individual’s own somatic cells through a non-invasive procedure, they carry the same genetic background as the person they were derived from and they can be grown in a physiological 3D environment, leading to complex neuronal models. Moreover, iPSCs represent an unlimited renewable resource that can be distributed across many researchers worldwide, ensuring that many labs can all work with the same patient cell-lines, developing assays that are both innovative, but also reproducible and that have the potential one day to be adapted for drug discovery purposes. These preclinical trials in a dish can speed up the discovery of new and effective compounds, while at the same time excluding potentially ineffective or toxic ones [154]. Recently, one group integrated radio frequency identification within a liver organoid to trace and track organoids after transplantation [155]. This proof of principle expands the use of chips to potentially record real-time data for drug assays in large quantities of organoids.
CONCLUDING REMARKS
Like all neurodegenerative diseases, PD remains a challenge for drug discovery. There are limited treatments and no cures available, despite huge investments. PD patients exhibit clinical, pathological and genetic heterogeneity that, combined with lack of efficacy or toxic side effects of drugs, may explain the failures of drug trials [156]. New tools are needed to better predict and target future therapies, especially at presymptomatic stages. The technologies described in this review represent promising platforms for human research. Worldwide, institutions and universities are creating biobanks, including open access biobanks [157], that could allow researchers to more readily generate such tools, to be used for finding personalized drug candidates or therapies to test in clinical trials [158, 159]. However, since the goal of models is to reproduce the complexity of the human body, they need to be highly elaborate. As a consequence, there are many challenges to face. First, the production of iPSCs, via fibroblast or blood cell reprogramming, induces a loss of epigenetic memory [160]. 2D or 3D cultures derived from iPSCs recapitulate fetal gene expression profiles and thus represent a hurdle for modeling late-onset disorders. Inducing or accelerating the aging process in iPSC-derived cultures, to improve late-onset age-related disease features, is a critical point to consider and improve in existing iPSC models for AD and PD. So far, one group has reported induction of aging in an iPSCs model using progerin [62]. However, even in such aged cells, the original potential epigenetics disease markers carried by the donor cannot be recovered, representing a limitation of this model. Secondly, it is important to keep in mind that genetic factors contribute to approximately 10% of PD cases, meaning that environmental factors have a major impact on the development of pathology. Recently, a group investigated the interaction between genome and environment and demonstrated an impact of local environment on the transcriptome and clinical endophenotypes, stronger than genetic ancestry. They demonstrated that local air pollution directly affected gene expression, and pathways affecting cardio-metabolic and respiratory traits, when controlling for genetic ancestry [161]. Thus, it is crucial to consider and include environmental contributors and complex polygenic inheritance in existing genetic iPSC models.
These iPSC-based tools and models will bring researchers closer to understanding the pathological mechanisms involved in PD. In addition, they are amenable for use in large scale compound and biologics screens to develop personalized treatments for patients. In parallel, many researchers are working on directly using iPSC in personalized cell therapies, by transplanting iPSC-derived DA neurons back into PD patients. Although this is beyond the scope of this review, other authors provide detailed overviews of this specific aspect of iPSC research [162, 163]. This technique, developed and recognized as promising in animal models [164, 165], is now being tested in several clinical trials. However, although cell therapies might significantly alleviate symptoms for a period of time, they may not be a definitive cure for the disease, given that PD is not limited to DA neuron loss [166]. Hence, generating faithful PD models is of utmost importance, to dissect the mechanisms of pathogenesis and design effective drugs to halt the progression of the disease.
Overall, although considerable work is still needed to optimize their use, iPSCs represent a promising tool for PD modeling and personalized drug discovery.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
NVM is supported by Jeanne Timmins Costello, Fonds de Recherche du Québéc-Santé and Parkinson Canada fellowships. FL is supported by a Parkinson Canada fellowship. EF is supported by a Foundation Grant from the Canadian Institutes of Health Research, the Consortium Québécois sur la Découverte du Médicament, Fonds de Recherche du Québéc-Santé Quebec Parkinson Network and the Michael J.Fox Foundation. TMD is supported by a Parkinson’s Canada New Investigator Award, the Consortium Québécois sur la Découverte du Médicament, the Michael J. Fox Foundation and through funding of the brain organoid program at the MNI by the Van Berkom-Saucier Foundation.
REFERENCES
[1] | Lees AJ , Hardy J , Revesz T ((2009) ) Parkinson’s disease. Lancet 373: , 2055–2066. |
[2] | Bekris LM , Mata IF , Zabetian CP ((2010) ) The genetics of Parkinson disease. J Geriatr Psychiatry Neurol 23: , 228–242. |
[3] | Chang D , Nalls MA , Hallgrimsdottir IB , Hunkapiller J , van der Brug M , Cai F , International Parkinson’s Disease Genomics Consortium; 23andMe Research Team, Kerch-ner GA , Ayalon G , Bingol B , Sheng M , Hinds D , Behrens TW , Singleton AB , Bhangale TR , Graham RR ((2017) ) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49: , 1511–1516. |
[4] | Foo JN , Tan LC , Irwan ID , Au WL , Low HQ , Prakash KM , Ahmad-Annuar A , Bei J , Chan AY , Chen CM , Chen YC , Chung SJ , Deng H , Lim SY , Mok V , Pang H , Pei Z , Peng R , Shang HF , Song K , Tan AH , Wu YR , Aung T , Cheng CY , Chew FT , Chew SH , Chong SA , Ebstein RP , Lee J , Saw SM , Seow A , Subramaniam M , Tai ES , Vithana EN , Wong TY , Heng KK , Meah WY , Khor CC , Liu H , Zhang F , Liu J , Tan EK ((2017) ) Genome-wide association study of Parkinson’s disease in East Asians. Hum Mol Genet 26: , 226–232. |
[5] | Spillantini MG , Schmidt ML , Lee VM , Trojanowski JQ , Jakes R , Goedert M ((1997) ) Alpha-synuclein in Lewy bodies. Nature 388: , 839–840. |
[6] | Shimozawa A , Ono M , Takahara D , Tarutani A , Imura S , Masuda-Suzukake M , Higuchi M , Yanai K , Hisanaga SI , Hasegawa M ((2017) ) Propagation of pathological alpha-synuclein in marmoset brain. Acta Neuropathol Commun 5: , 12. |
[7] | Bernis ME , Babila JT , Breid S , Wusten KA , Wullner U , Tamguney G ((2015) ) Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol Commun 3: , 75. |
[8] | Recasens A , Dehay B , Bove J , Carballo-Carbajal I , Dovero S , Perez-Villalba A , Fernagut PO , Blesa J , Parent A , Perier C , Farinas I , Obeso JA , Bezard E , Vila M ((2014) ) Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 75: , 351–362. |
[9] | Deng H , Dodson MW , Huang H , Guo M ((2008) ) The Parkinson’s disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc NatlAcadSci USA 105: , 14503–14508. |
[10] | Langston JW , Ballard PA, Jr. ((1983) ) Parkinson’s disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N Engl J Med 309: , 310. |
[11] | Zhu J , Chu CT ((2010) ) Mitochondrial dysfunction in Parkinson’s disease. J Alzheimers Dis 20: (Suppl 2), S325–334. |
[12] | Kurz A , Double KL , Lastres-Becker I , Tozzi A , Tantucci M , Bockhart V , Bonin M , Garcia-Arencibia M , Nuber S , Schlaudraff F , Liss B , Fernandez-Ruiz J , Gerlach M , Wullner U , Luddens H , Calabresi P , Auburger G , Gispert S ((2010) ) A53T-alpha-synuclein overexpression impairs dopamine signaling and striatal synaptic plasticity in old mice. PLoS One 5: , e11464. |
[13] | Good CH , Hoffman AF , Hoffer BJ , Chefer VI , Shippenberg TS , Backman CM , Larsson NG , Olson L , Gellhaar S , Galter D , Lupica CR ((2011) ) Impaired nigrostriatal function precedes behavioral deficits in a genetic mitochondrial model of Parkinson’s disease. FASEB J 25: , 1333–1344. |
[14] | Hara T , Nakamura K , Matsui M , Yamamoto A , Nakahara Y , Suzuki-Migishima R , Yokoyama M , Mishima K , Saito I , Okano H , Mizushima N ((2006) ) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: , 885–889. |
[15] | Anglade P , Vyas S , Javoy-Agid F , Herrero MT , Michel PP , Marquez J , Mouatt-Prigent A , Ruberg M , Hirsch EC , Agid Y ((1997) ) Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol Histopathol 12: , 25–31. |
[16] | Blesa J , Przedborski S ((2014) ) Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front Neuroanat 8: , Article 155. |
[17] | Blesa J , Trigo-Damas I , Quiroga-Varela A , LopezGonzalez del Rey N ((2016) ) Animal models of Parkinson’s disease. In Challenges in Parkinson’s Disease, DorszewskaJ, WojciechK, eds. IntechOpen, doi: 10.5772/63328. Available from: https://www.intechopen.com/books/challenges-in-parkinson-s-disease/animal-models-of-parkinson-s-disease |
[18] | Vingill S , Connor-Robson N , Wade-Martins R ((2017) ) Are rodent models of Parkinson’s disease behaving as they should? Behav Brain Res 352: , 133–141. |
[19] | Creed RB , Goldberg MS ((2018) ) New developments in genetic rat models of Parkinson’s disease. Mov Disord 33: , 717–729. |
[20] | Morissette M , Di Paolo T ((2018) ) Non-human primate models of PD to test novel therapies. J Neural Transm (Vienna) 125: , 291–324. |
[21] | Fornai F , Schluter OM , Lenzi P , Gesi M , Ruffoli R , Ferrucci M , Lazzeri G , Busceti CL , Pontarelli F , Battaglia G , Pellegrini A , Nicoletti F , Ruggieri S , Paparelli A , Sudhof TC ((2005) ) Parkinson-like syndrome induced by continuous MPTP infusion: Convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc NatlAcadSci USA 102: , 3413–3418. |
[22] | Morissette M , Di Paolo T ((2018) ) Non-human primate models of PD to test novel therapies. J Neural Transm 125: , 291–324. |
[23] | Singleton AB , Farrer M , Johnson J , Singleton A , Hague S , Kachergus J , Hulihan M , Peuralinna T , Dutra A , Nussbaum R , Lincoln S , Crawley A , Hanson M , Maraganore D , Adler C , Cookson MR , Muenter M , Baptista M , Miller D , Blancato J , Hardy J , Gwinn-Hardy K ((2003) ) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302: , 841. |
[24] | Vekrellis K , Xilouri M , Emmanouilidou E , Rideout HJ , Stefanis L ((2011) ) Pathological roles of alpha-synuclein in neurological disorders. Lancet Neurol 10: , 1015–1025. |
[25] | Hinkle KM , Yue M , Behrouz B , Dachsel JC , Lincoln SJ , Bowles EE , Beevers JE , Dugger B , Winner B , Prots I , Kent CB , Nishioka K , Lin WL , Dickson DW , Janus CJ , Farrer MJ , Melrose HL ((2012) ) LRRK2 knockout mice have an intact dopaminergic system but display alterations in exploratory and motor co-ordination behaviors. Mol Neurodegener 7: , 25. |
[26] | Tong Y , Pisani A , Martella G , Karouani M , Yamaguchi H , Pothos EN , Shen J ((2009) ) R1441C mutation in LRRK2impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci USA 106: , 14622–14627. |
[27] | Tong Y , Yamaguchi H , Giaime E , Boyle S , Kopan R , Kelleher RJ , 3rd, Shen J ((2010) ) Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc Natl Acad Sci USA 107: , 9879–9884. |
[28] | Ramonet D , Daher JP , Lin BM , Stafa K , Kim J , Banerjee R , Westerlund M , Pletnikova O , Glauser L , Yang L , Liu Y , Swing DA , Beal MF , Troncoso JC , McCaffery JM , Jenkins NA , Copeland NG , Galter D , Thomas B , Lee MK , Dawson TM , Dawson VL , Moore DJ ((2011) ) Dopaminergic neuronal loss, reduced neurite complexity and autophagic abnormalities in transgenic mice expressing G2019S mutant LRRK2. PLoS One 6: , e18568. |
[29] | Chen CY , Weng YH , Chien KY , Lin KJ , Yeh TH , Cheng YP , Lu CS , Wang HL ((2012) ) (G2019S) LRRK2 activates MKK4-JNK pathway and causes degeneration of SN dopaminergic neurons in a transgenic mouse model of PD. Cell Death Differ 19: , 1623–1633. |
[30] | Shaikh KT , Yang A , Youshin E , Schmid S ((2015) ) Transgenic LRRK2 (R1441G) rats-a model for Parkinson disease? PeerJ 3: , e945. |
[31] | Dusonchet J , Kochubey O , Stafa K , Young SM Jr , Zuf-ferey R , Moore DJ , Schneider BL , Aebischer P ((2011) ) A rat model of progressive nigral neurodegeneration induced by the Parkinson’s disease-associated G2019S mutation in LRRK2. J Neurosci 31: , 907–912. |
[32] | Goldberg MS , Fleming SM , Palacino JJ , Cepeda C , Lam HA , Bhatnagar A , Meloni EG , Wu N , Ackerson LC , Klapstein GJ , Gajendiran M , Roth BL , Chesselet MF , Maidment NT , Levine MS , Shen J ((2003) ) Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278: , 43628–43635. |
[33] | Gispert S , Ricciardi F , Kurz A , Azizov M , Hoepken HH , Becker D , Voos W , Leuner K , Muller WE , Kudin AP , Kunz WS , Zimmermann A , Roeper J , Wenzel D , Jendrach M , Garcia-Arencibia M , Fernandez-Ruiz J , Huber L , Rohrer H , Barrera M , Reichert AS , Rub U , Chen A , Nussbaum RL , Auburger G ((2009) ) Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PLoS One 4: , e5777. |
[34] | Perez FA , Palmiter RD ((2005) ) Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci U SA 102: , 2174–2179. |
[35] | Van Rompuy AS , Lobbestael E , Van der Perren A , Van den Haute C , Baekelandt V ((2014) ) Long-term overexpression of human wild-type and T240R mutant Parkin in rat substantia nigra induces progressive dopaminergic neurodegeneration. J Neuropathol Exp Neurol 73: , 159–174. |
[36] | Lu XH , Fleming SM , Meurers B , Ackerson LC , Mortazavi F , Lo V , Hernandez D , Sulzer D , Jackson GR , Maidment NT , Chesselet MF , Yang XW ((2009) ) Bacterial artificial chromosome transgenic mice expressing a truncated mutant parkin exhibit age-dependent hypokinetic motor deficits, dopaminergic neuron degeneration, and accumulation of proteinase K-resistant alpha-synuclein. J Neurosci 29: , 1962–1976. |
[37] | Rousseaux MW , Marcogliese PC , Qu D , Hewitt SJ , Seang S , Kim RH , Slack RS , Schlossmacher MG , Lagace DC , Mak TW , Park DS ((2012) ) Progressive dopaminergic cell loss with unilateral-to-bilateral progression in a genetic model of Parkinson disease. Proc Natl Acad Sci U S A 109: , 15918–15923. |
[38] | Goldberg MS , Pisani A , Haburcak M , Vortherms TA , Kitada T , Costa C , Tong Y , Martella G , Tscherter A , Martins A , Bernardi G , Roth BL , Pothos EN , Calabresi P , Shen J ((2005) ) Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial Parkinsonism-linked gene DJ-1. Neuron 45: , 489–496. |
[39] | O'Regan G , deSouza RM , Balestrino R , Schapira AH ((2017) ) Glucocerebrosidase mutations in Parkinson disease. J Parkinsons Dis 7: , 411–422. |
[40] | Fishbein I , Kuo YM , Giasson BI , Nussbaum RL ((2014) ) Augmentation of phenotype in a transgenic Parkinson mouse heterozygous for a Gaucher mutation. Brain 137: , 3235–3247. |
[41] | Rockenstein E , Clarke J , Viel C , Panarello N , Treleaven CM , Kim C , Spencer B , Adame A , Park H , Dodge JC , Cheng SH , Shihabuddin LS , Masliah E , Sardi SP ((2016) ) Glucocerebrosidase modulates cognitive and motor activities in murine models of Parkinson’s disease. Hum Mol Genet 25: , 2645–2660. |
[42] | Migdalska-Richards A , Daly L , Bezard E , Schapira AH ((2016) ) Ambroxol effects in glucocerebrosidase and alpha-synuclein transgenic mice. Ann Neurol 80: , 766–775. |
[43] | Matsui H , Takahashi R ((2018) ) Parkinson’s disease patho-genesis from the viewpoint of small fish models. J Neural Transm (Vienna) 125: , 25–33. |
[44] | Chakraborty S , Bornhorst J , Nguyen TT , Aschner M ((2013) ) Oxidative stress mechanisms underlying Parkinson’s disease-associated neurodegeneration in C. elegans. IJMS 14: , 23103–23128. |
[45] | Hewitt VL , Whitworth AJ ((2017) ) Mechanisms of Parkinson’s disease: Lessons from Drosophila. Curr Top Dev Biol 121: , 173–200. |
[46] | Ko WKD , Bezard E ((2017) ) Experimental animal models of Parkinson’s disease: A transition from assessing symptomatology to α-synuclein targeted disease modification. Exp Neurol 298: , 172–179. |
[47] | Welin S ((2002) ) Ethical issues in human embryonic stem cell research. Acta Obstet Gynecol Scand 81: , 377–382. |
[48] | Takahashi K , Yamanaka S ((2006) ) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: , 663–676. |
[49] | Yu J , Vodyanik MA , Smuga-Otto K , Antosiewicz-Bourget J , Frane JL , Tian S , Nie J , Jonsdottir GA , Ruotti V , Stewart R , Slukvin, II , Thomson JA ((2007) ) Induced pluripotent stem cell lines derived from human somatic cells. Science 318: , 1917–1920. |
[50] | Takahashi K , Tanabe K , Ohnuki M , Narita M , Ichisaka T , Tomoda K , Yamanaka S ((2007) ) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: , 861–872. |
[51] | Soldner F , Hockemeyer D , Beard C , Gao Q , Bell GW , Cook EG , Hargus G , Blak A , Cooper O , Mitalipova M , Isacson O , Jaenisch R ((2009) ) Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136: , 964–977. |
[52] | Cooper O , Hargus G , Deleidi M , Blak A , Osborn T , Marlow E , Lee K , Levy A , Perez-Torres E , Yow A , Isacson O ((2010) ) Differentiation of human ES and Parkinson’s disease iPS cells into ventral midbrain dopaminergic neurons requires a high activity form of SHH, FGF8a and specific regionalization by retinoic acid. Mol Cell Neurosci 45: , 258–266. |
[53] | Byers B , Cord B , Nguyen HN , Schule B , Fenno L , Lee PC , Deisseroth K , Langston JW , Pera RR , Palmer TD ((2011) ) SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PLoS One 6: , e26159. |
[54] | Devine MJ , Ryten M , Vodicka P , Thomson AJ , Burdon T , Houlden H , Cavaleri F , Nagano M , Drummond NJ , Taanman JW , Schapira AH , Gwinn K , Hardy J , Lewis PA , Kunath T ((2011) ) Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat Commun 2: , 440. |
[55] | Mazzulli JR , Xu YH , Sun Y , Knight AL , McLean PJ , Caldwell GA , Sidransky E , Grabowski GA , Krainc D ((2011) ) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146: , 37–52. |
[56] | Seibler P , Graziotto J , Jeong H , Simunovic F , Klein C , Krainc D ((2011) ) Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci 31: , 5970–5976. |
[57] | Cooper O , Seo H , Andrabi S , Guardia-Laguarta C , Graziotto J , Sundberg M , McLean JR , Carrillo-Reid L , Xie Z , Osborn T , Hargus G , Deleidi M , Lawson T , Bogetofte H , Perez-Torres E , Clark L , Moskowitz C , Mazzulli J , Chen L , Volpicelli-Daley L , Romero N , Jiang H , Uitti RJ , Huang Z , Opala G , Scarffe LA , Dawson VL , Klein C , Feng J , Ross OA , Trojanowski JQ , Lee VM , Marder K , Surmeier DJ , Wszolek ZK , Przedborski S , Krainc D , Dawson TM , Isacson O ((2012) ) Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med 4: , 141ra190. |
[58] | Imaizumi Y , Okada Y , Akamatsu W , Koike M , Kuzumaki N , Hayakawa H , Nihira T , Kobayashi T , Ohyama M , Sato S , Takanashi M , Funayama M , Hirayama A , Soga T , Hishiki T , Suematsu M , Yagi T , Ito D , Kosakai A , Hayashi K , Shouji M , Nakanishi A , Suzuki N , Mizuno Y , Mizushima N , Amagai M , Uchiyama Y , Mochizuki H , Hattori N , Okano H ((2012) ) Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol Brain 5: , 35. |
[59] | Jiang H , Ren Y , Yuen EY , Zhong P , Ghaedi M , Hu Z , Azabdaftari G , Nakaso K , Yan Z , Feng J ((2012) ) Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat Commun 3: , 668. |
[60] | Sanchez-Danes A , Richaud-Patin Y , Carballo-Carbajal I , Jimenez-Delgado S , Caig C , Mora S , Di Guglielmo C , Ezquerra M , Patel B , Giralt A , Canals JM , Memo M , Alberch J , Lopez-Barneo J , Vila M , Cuervo AM , Tolosa E , Consiglio A , Raya A ((2012) ) Disease-specific pheno-types in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med 4: , 380–395. |
[61] | Chung SY , Kishinevsky S , Mazzulli JR , Graziotto J , Mre-jeru A , Mosharov EV , Puspita L , Valiulahi P , Sulzer D , Milner TA , Taldone T , Krainc D , Studer L , Shim JW ((2016) ) Parkin and PINK1 patient iPSC-derived midbrain dopamine neurons exhibit mitochondrial dysfunction and alpha-synuclein accumulation. Stem Cell Reports 7: , 664–677. |
[62] | Miller JD , Ganat YM , Kishinevsky S , Bowman RL , Liu B , Tu EY , Mandal PK , Vera E , Shim JW , Kriks S , Taldone T , Fusaki N , Tomishima MJ , Krainc D , Milner TA , Rossi DJ , Studer L ((2013) ) Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13: , 691–705. |
[63] | Orenstein SJ , Kuo SH , Tasset I , Arias E , Koga H , Fernandez-Carasa I , Cortes E , Honig LS , Dauer W , Consiglio A , Raya A , Sulzer D , Cuervo AM ((2013) ) Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci 16: , 394–406. |
[64] | Fernandez-Santiago R , Carballo-Carbajal I , Castellano G , Torrent R , Richaud Y , Sanchez-Danes A , Vilarrasa-Blasi R , Sanchez-Pla A , Mosquera JL , Soriano J , Lopez-Barneo J , Canals JM , Alberch J , Raya A , Vila M , Consiglio A , Martin-Subero JI , Ezquerra M , Tolosa E ((2015) ) Aberrant epigenome in iPSC-derived dopaminergic neurons from Parkinson’s disease patients. EMBO Mol Med 7: , 1529–1546. |
[65] | Oliveira LM , Falomir-Lockhart LJ , Botelho MG , Lin KH , Wales P , Koch JC , Gerhardt E , Taschenberger H , Outeiro TF , Lingor P , Schule B , Arndt-Jovin DJ , Jovin TM ((2015) ) Elevated alpha-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell Death Dis 6: , e1994. |
[66] | Ren Y , Jiang H , Hu Z , Fan K , Wang J , Janoschka S , Wang X , Ge S , Feng J ((2015) ) Parkin mutations reduce the complexity of neuronal processes in iPSC-derived human neurons. Stem Cells 33: , 68–78. |
[67] | Chang KH , Lee-Chen GJ , Wu YR , Chen YJ , Lin JL , Li M , Chen IC , Lo YS , Wu HC , Chen CM ((2016) ) Impairment of proteasome and anti-oxidative pathways in the induced pluripotent stem cell model for sporadic Parkinson’s disease. Parkinsonism Relat Disord 24: , 81–88. |
[68] | Holmqvist S , Lehtonen S , Chumarina M , Puttonen KA , Azevedo C , Lebedeva O , Ruponen M , Oksanen M , Djelloul M , Collin A , Goldwurm S , Meyer M , Lagarkova M , Kiselev S , Koistinaho J , Roybon L ((2016) ) Creation of a library of induced pluripotent stem cells from Parkinso-nian patients. NPJ Parkinsons Dis 2: , 16009. |
[69] | Lopez de Maturana R , Lang V , Zubiarrain A , Sousa A , Vazquez N , Gorostidi A , Aguila J , Lopez de Munain A , Rodriguez M , Sanchez-Pernaute R ((2016) ) Mutations in LRRK2 impair NF-kappaB pathway in iPSC-derived neurons. J Neuroinflammation 13: , 295. |
[70] | Momcilovic O , Sivapatham R , Oron TR , Meyer M , Mooney S , Rao MS , Zeng X ((2016) ) Derivation, characterization, and neural differentiation of integration-free induced pluripotent stem cell lines from Parkinson’s disease patients carrying SNCA, LRRK2, PARK2, and GBA mutations. PLoS One 11: , e0154890. |
[71] | Ma D , Ng EY , Zeng L , Lim CY , Zhao Y , Tan EK ((2017) ) Development of a human induced pluripotent stem cell (iPSC) line from a Parkinson’s disease patient carrying the N551K variant in LRRK2 gene. Stem Cell Res 18: , 51–53. |
[72] | Ma D , Ng SH , Zeng L , Zhao Y , Tan EK ((2017) ) Generation of a human induced pluripotent stem cell (iPSC) line carrying the Parkinson’s disease linked LRRK2 variant S1647T. Stem Cell Res 18: , 54–56. |
[73] | Ma D , Tio M , Ng SH , Li Z , Lim CY , Zhao Y , Tan EK ((2017) ) Derivation of human induced pluripotent stem cell (iPSC) line with LRRK2 gene R1398H variant in Parkinson’s disease. Stem Cell Res 18: , 48–50. |
[74] | Ma D , Zhou W , Ng EY , Zeng L , Zhao Y , Tan EK ((2017) ) Reprogramming of a human induced pluripotent stem cell (iPSC) line from a Parkinson’s disease patient with a R1628P variant in the LRRK2 gene. Stem Cell Res 18: , 45–47. |
[75] | Suzuki S , Akamatsu W , Kisa F , Sone T , Ishikawa KI , Kuzumaki N , Katayama H , Miyawaki A , Hattori N , Okano H ((2017) ) Efficient induction of dopaminergic neuron differentiation from induced pluripotent stem cells reveals impaired mitophagy in PARK2 neurons. Biochem Biophys Res Commun 483: , 88–93. |
[76] | Zhang S , Liu L , Hu Y , Lv Z , Li Q , Gong W , Sha H , Wu H ((2017) ) Derivation of human induced pluripotent stem cell (iPSC) line from a 79year old sporadic male Parkinson’s disease patient. Stem Cell Res 19: , 43–45. |
[77] | Hrvatin S , O'Donnell CW , Deng F , Millman JR , Pagliuca FW , DiIorio P , Rezania A , Gifford DK , Melton DA ((2014) ) Differentiated human stem cells resemble fetal, not adult, beta cells. Proc Natl Acad Sci USA 111: , 3038–3043. |
[78] | Spence JR , Mayhew CN , Rankin SA , Kuhar MF , Vallance JE , Tolle K , Hoskins EE , Kalinichenko VV , Wells SI , Zorn AM , Shroyer NF , Wells JM ((2011) ) Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature 470: , 105–109. |
[79] | Thomas KR , Capecchi MR ((1987) ) Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell 51: , 503–512. |
[80] | Smithies O , Gregg RG , Boggs SS , Koralewski MA , Kucherlapati RS ((1985) ) Insertion of DNA sequences into the human chromosomal beta-globin locus by homologous recombination. Nature 317: , 230–234. |
[81] | Zwaka TP , Thomson JA ((2003) ) Homologous recombination in human embryonic stem cells. Nat Biotechnol 21: , 319–321. |
[82] | Khan IF , Hirata RK , Wang PR , Li Y , Kho J , Nelson A , Huo Y , Zavaljevski M , Ware C , Russell DW ((2010) ) Engineering of human pluripotent stem cells by AAV-mediated gene targeting. Mol Ther 18: , 1192–1199. |
[83] | Liu GH , Qu J , Suzuki K , Nivet E , Li M , Montserrat N , Yi F , Xu X , Ruiz S , Zhang W , Wagner U , Kim A , Ren B , Li Y , Goebl A , Kim J , Soligalla RD , Dubova I , Thompson J , Yates J , 3rd, Esteban CR , Sancho-Martinez I , Izpisua Belmonte JC ((2012) ) Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 491: , 603–607. |
[84] | Rouet P , Smih F , Jasin M ((1994) ) Introduction of doublestrand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol 14: , 8096–8106. |
[85] | Rouet P , Smih F , Jasin M ((1994) ) Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci U S A 91: , 6064–6068. |
[86] | Urnov FD , Rebar EJ , Holmes MC , Zhang HS , Gregory PD ((2010) ) Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11: , 636–646. |
[87] | Reinhardt P , Schmid B , Burbulla LF , Schondorf DC , Wagner L , Glatza M , Hoing S , Hargus G , Heck SA , Dhingra A , Wu G , Muller S , Brockmann K , Kluba T , Maisel M , Kruger R , Berg D , Tsytsyura Y , Thiel CS , Psathaki OE , Klingauf J , Kuhlmann T , Klewin M , Muller H , Gasser T , Scholer HR , Sterneckert J ((2013) ) Genetic correction of aLRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12: , 354–367. |
[88] | Chung CY , Khurana V , Auluck PK , Tardiff DF , Mazzulli JR , Soldner F , Baru V , Lou Y , Freyzon Y , Cho S , Mungenast AE , Muffat J , Mitalipova M , Pluth MD , Jui NT , Schule B , Lippard SJ , Tsai LH , Krainc D , Buchwald SL , Jaenisch R , Lindquist S ((2013) ) Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science 342: , 983–987. |
[89] | Ryan SD , Dolatabadi N , Chan SF , Zhang X , Akhtar MW , Parker J , Soldner F , Sunico CR , Nagar S , Talantova M , Lee B , Lopez K , Nutter A , Shan B , Molokanova E , Zhang Y , Han X , Nakamura T , Masliah E , Yates JR , 3rd, Nakanishi N , Andreyev AY , Okamoto S , Jaenisch R , Ambasudhan R , Lipton SA ((2013) ) Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell 155: , 1351–1364. |
[90] | Soldner F , Laganiere J , Cheng AW , Hockemeyer D , Gao Q , Alagappan R , Khurana V , Golbe LI , Myers RH , Lindquist S , Zhang L , Guschin D , Fong LK , Vu BJ , Meng X , Urnov FD , Rebar EJ , Gregory PD , Zhang HS , Jaenisch R ((2011) ) Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146: , 318–331. |
[91] | Schondorf DC , Aureli M , McAllister FE , Hindley CJ , Mayer F , Schmid B , Sardi SP , Valsecchi M , Hoffmann S , Schwarz LK , Hedrich U , Berg D , Shihabuddin LS , Hu J , Pruszak J , Gygi SP , Sonnino S , Gasser T , Deleidi M ((2014) ) iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun 5: , 4028. |
[92] | Shaltouki A , Sivapatham R , Pei Y , Gerencser AA , Momcilovic O , Rao MS , Zeng X ((2015) ) Mitochondrial alterations by PARKIN in dopaminergic neurons using PARK2 patient-specific and PARK2 knockout isogenic iPSC lines. Stem Cell Reports 4: , 847–859. |
[93] | Bogdanove AJ , Voytas DF ((2011) ) TAL effectors: Customizable proteins for DNA targeting. Science 333: , 1843–1846. |
[94] | Jinek M , East A , Cheng A , Lin S , Ma E , Doudna J ((2013) ) RNA-programmed genome editing in human cells. Elife 2: , e00471. |
[95] | Jinek M , Chylinski K , Fonfara I , Hauer M , Doudna JA , Charpentier E ((2012) ) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: , 816–821. |
[96] | Soldner F , Stelzer Y , Shivalila CS , Abraham BJ , Latourelle JC , Barrasa MI , Goldmann J , Myers RH , Young RA , Jaenisch R ((2016) ) Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature 533: , 95–99. |
[97] | Wu MH , Huang SB , Lee GB ((2010) ) Microfluidic cell culture systems for drug research. Lab Chip 10: , 939–956. |
[98] | El-Ali J , Sorger PK , Jensen KF ((2006) ) Cells on chips. Nature 442: , 403–411. |
[99] | Grobstein C ((1953) ) Morphogenetic interaction between embryonic mouse tissues separated by a membrane filter. Nature 172: , 869–870. |
[100] | Campbell JJ , Davidenko N , Caffarel MM , Cameron RE , Watson CJ ((2011) ) A multifunctional 3D co-culture system for studies of mammary tissue morphogenesis and stem cell biology. PLoS One 6: , e25661. |
[101] | Felton EJ , Copeland CR , Chen CS , Reich DH ((2012) ) Heterotypic cell pair co-culturing on patterned microarrays. Lab Chip 12: , 3117–3126. |
[102] | Toma JS , Shettar BC , Chipman PH , Pinto DM , Borowska JP , Ichida JK , Fawcett JP , Zhang Y , Eggan K , Rafuse VF ((2015) ) Motoneurons derived from induced pluripotent stem cells develop mature phenotypes typical of endogenous spinal motoneurons. J Neurosci 35: , 1291–1306. |
[103] | Puttonen KA , Ruponen M , Naumenko N , Hovatta OH , Tavi P , Koistinaho J ((2015) ) Generation of functional neuromuscular junctions from human pluripotent stem cell lines. Front CellNeurosci 9: , 473. |
[104] | Demestre M , Orth M , Fohr KJ , Achberger K , Ludolph AC , Liebau S , Boeckers TM ((2015) ) Formation and characterisation of neuromuscular junctions between hiPSC derived motoneurons and myotubes. Stem Cell Res 15: , 328–336. |
[105] | Umbach JA , Adams KL , Gundersen CB , Novitch BG ((2012) ) Functional neuromuscular junctions formed by embryonic stem cell-derived motor neurons. PLoS One 7: , e36049. |
[106] | Guo XF , Gonzalez M , Stancescu M , Vandenburgh HH , Hickman JJ ((2011) ) Neuromuscular junction formation between human stem cell-derived motoneurons and human skeletal muscle in a defined system. Biomaterials 32: , 9602–9611. |
[107] | Haenseler W , Sansom SN , Buchrieser J , Newey SE , Moore CS , Nicholls FJ , Chintawar S , Schnell C , Antel JP , Allen ND , Cader MZ , Wade-Martins R , James WS , Cowley SA ((2017) ) A highly efficient human pluripotent stem cell microglia model displays a neuronal-co-culture-specific expression profile and inflammatory response. Stem Cell Reports 8: , 1727–1742. |
[108] | Clark AJ , Kaller MS , Galino J , Willison HJ , Rinaldi S , Bennett DLH ((2017) ) Co-cultures with stem cell-derived human sensory neurons reveal regulators of peripheral myelination. Brain 140: , 898–913. |
[109] | Kuijlaars J , Oyelami T , Diels A , Rohrbacher J , Ver-sweyveld S , Meneghello G , Tuefferd M , Verstraelen P , Detrez JR , Verschuuren M , De Vos WH , Meert T , Peeters PJ , Cik M , Nuydens R , Brone B , Verheyen A ((2016) ) Sustained synchronized neuronal network activity in a human astrocyte co-culture system. Sci Rep 6: , 36529. |
[110] | Du F , Yu Q , Chen A , Chen D , Yan SS ((2018) ) Astrocytes attenuate mitochondrial dysfunctions in human dopamin-ergic neurons derived from iPSC. Stem Cell Reports 10: , 366–374. |
[111] | Goers L , Freemont P , Polizzi KM ((2014) ) Co-culture systems and technologies: Taking synthetic biology to the next level. J R Soc Interface 11: . |
[112] | Reynolds BA , Weiss S ((1992) ) Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 255: , 1707–1710. |
[113] | Hogberg HT , Bressler J , Christian KM , Harris G , Makri G , O'Driscoll C , Pamies D , Smirnova L , Wen Z , Hartung T ((2013) ) Toward a 3D model of human brain development for studying gene/environment interactions. Stem Cell Res Ther 4: Suppl 1, S4. |
[114] | Choi SH , Kim YH , Hebisch M , Sliwinski C , Lee S , D'Avanzo C , Chen H , Hooli B , Asselin C , Muffat J , Klee JB , Zhang C , Wainger BJ , Peitz M , Kovacs DM , Woolf CJ , Wagner SL , Tanzi RE , Kim DY ((2014) ) A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 515: , 274–278. |
[115] | Kim YH , Choi SH , D'Avanzo C , Hebisch M , Sliwinski C , Bylykbashi E , Washicosky KJ , Klee JB , Brustle O , Tanzi RE , Kim DY ((2015) ) A 3D human neural cell culture system for modeling Alzheimer’s disease. Nat Protoc 10: , 985–1006. |
[116] | Lee CT , Bendriem RM , Wu WW , Shen RF ((2017) ) 3D brain Organoids derived from pluripotent stem cells: Promising experimental models for brain development and neurodegenerative disorders. J Biomed Sci 24: , 59. |
[117] | Goedert M ((2015) ) Neurodegeneration. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 349: , 1255555. |
[118] | Lancaster MA , Renner M , Martin C-A , Wenzel D , Bicknell LS , Hurles ME , Homfray T , Penninger JM , Jackson AP , Knoblich JA ((2013) ) Cerebral organoids model human brain development and microcephaly. Nature 501: , 373–379. |
[119] | Monzel AS , Smits LM , Hemmer K , Hachi S , Moreno EL , van Wuellen T , Jarazo J , Walter J , Brüggemann I , Boussaad I , Berger E , Fleming RMT , Bolognin S , Schwamborn JC ((2017) ) Derivation of human midbrain-specific organoids from neuroepithelial stem cells. Stem Cell Reports 8: , 1144–1154. |
[120] | Jo J , Xiao Y , Sun AX , Cukuroglu E , Tran HD , Goke J , Tan ZY , Saw TY , Tan CP , Lokman H , Lee Y , Kim D , Ko HS , Kim SO , Park JH , Cho NJ , Hyde TM , Kleinman JE , Shin JH , Weinberger DR , Tan EK , Je HS , Ng HH ((2016) ) Midbrain-like organoids from human pluripotent stem cells contain functional dopaminergic and neuromelanin-producing neurons. Cell Stem Cell 19: , 248–257. |
[121] | Sloan SA , Darmanis S , Huber N , Khan TA , Birey F , Caneda C , Reimer R , Quake SR , Barres BA , Pasca SP ((2017) ) Human astrocyte maturation captured in 3D cerebral cortical spheroids derived from pluripotent stem cells. Neuron 95: , 779–790 e776. |
[122] | Lindborg BA , Brekke JH , Vegoe AL , Ulrich CB , Haider KT , Subramaniam S , Venhuizen SL , Eide CR , Orchard PJ , Chen W , Wang Q , Pelaez F , Scott CM , Kokkoli E , Keirstead SA , Dutton JR , Tolar J , O'Brien TD ((2016) ) Rapid induction of cerebral organoids from human induced pluripotent stem cells using a chemically defined hydrogel and defined cell culture medium. Stem Cells Transl Med. 5: , 970–979. |
[123] | Quadrato G , Nguyen T , Macosko EZ , Sherwood JL , Yang SM , Berger D , Maria N , Scholvin J , Goldman M , Kinney J , Boyden ES , Lichtman J , Williams ZM , McCarroll SA , Arlotta P ((2017) ) Cell diversity and network dynamics in photosensitive human brain organoids. Nature 545: , 48–53. |
[124] | Kadoshima T , Sakaguchi H , Nakano T , Soen M , Ando S , Eiraku M , Sasai Y ((2013) ) Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc Natl Acad Sci USA 110: , 20284–20289. |
[125] | Qian X , Nguyen HN , Song MM , Hadiono C , Ogden SC , Hammack C , Yao B , Hamersky G , Jacob F , Zhong C , Yoon K-J , Jeang W , Lin L , Li Y , Thakor J , Berg D , Zhang C , Kang E , Chickering M , Naeun D , Ho C-Y , Wen Z , Christian KM , Shi P-Y , Maher BJ , Wu H , Jin P , Tang H , Song H , Ming G-l ((2016) ) Brain region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165: , 1238–1254. |
[126] | Muguruma K , Nishiyama A , Kawakami H , Hashimoto K , Sasai Y ((2015) ) Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Reports 10: , 537–550. |
[127] | Bagley JA , Reumann D , Bian S , Levi-Strauss J , Knoblich JA ((2017) ) Fused cerebral organoids model interactions between brain regions. Nat Methods 14: , 743–751. |
[128] | Paşca AM , Sloan SA , Clarke LE , Tian Y , Makinson CD , Huber N , Kim CH , Park J-Y , O'Rourke NA , Nguyen KD , Smith SJ , Huguenard JR , Geschwind DH , Barres BA , Paşca SP ((2015) ) Functional cortical neurons and astro-cytes from human pluripotent stem cells in 3D culture. Nat Methods 12: , 671–678. |
[129] | Sakaguchi H , Kadoshima T , Soen M , Narii N , Ishida Y , Ohgushi M , Takahashi J , Eiraku M , Sasai Y ((2015) ) Generation of functional hippocampal neurons from self-organizing human embryonic stem cell-derived dor-somedial telencephalic tissue. Nat Commun 6: , 8896. |
[130] | Birey F , Andersen J , Makinson CD , Islam S , Wei W , Huber N , Fan HC , Cordes Metzler KR , Panagiotakos G , Thom N , O'Rourke NA , Steinmetz LM , Bernstein JA , Hallmayer J , Huguenard JR , Paşca SP ((2017) ) Assembly of functional forebrain spheroids from human pluripotent cells. Nature 545: , 54–59. |
[131] | Camp JG , Badsha F , Florio M , Kanton S , Gerber T , Wilsch-Bräuninger M , Lewitus E , Sykes A , Hevers W , Lancaster M , Knoblich JA , Lachmann R , Pääbo S , Huttner WB , Treutlein B ((2015) ) Human cerebral organoids recapitulate gene expression programs of fetal neocortex development. Proc Natl Acad Sci USA 112: , 15672–15677. |
[132] | Luo C , Lancaster MA , Castanon R , Nery JR , Knoblich JA , Ecker JR ((2016) ) Cerebral organoids recapitulate epige-nomic signatures of the human fetal brain. Cell Rep 17: , 3369–3384. |
[133] | Cugola FR , Fernandes IR , Russo FB , Freitas BC , Dias JLM , Guimarães KP , Benazzato C , Almeida N , Pignatari GC , Romero S , Polonio CM , Cunha I , Freitas CL , Brandão WN , Rossato C , Andrade DG , Faria DdP , Garcez AT , Buchpigel CA , Braconi CT , Mendes E , Sall AA , Zanotto PMdA , Peron JPS , Muotri AR , Beltrão-Braga PCB ((2016) ) The Brazilian Zika virus strain causes birth defects in experimental models. Nature 534: , 267–271. |
[134] | Dang J , Tiwari SK , Lichinchi G , Qin Y , Patil VS , Eroshkin AM , Rana TM ((2016) ) Zika virus depletes neural progenitors in human cerebral organoids through activation of the innate immune receptor TLR3. Cell Stem Cell 19: , 258–265. |
[135] | Garcez PP , Loiola EC , Madeiro da Costa R , Higa LM , Trindade P , Delvecchio R , Nascimento JM , Brindeiro R , Tanuri A , Rehen SK ((2016) ) Zika virus impairs growth in human neurospheres and brain organoids. Science 352: , 816–818. |
[136] | Wells MF , Salick MR , Wiskow O , Ho DJ , Worringer KA , Ihry RJ , Kommineni S , Bilican B , Klim JR , Hill EJ , Kane LT , Ye C , Kaykas A , Eggan K ((2016) ) Genetic ablation of AXL does not protect human neural progenitor cells and cerebral organoids from Zika virus infection. Cell Stem Cell 19: , 703–708. |
[137] | Mariani J , Coppola G , Zhang P , Abyzov A , Provini L , Tomasini L , Amenduni M , Szekely A , Palejev D , Wilson M , Gerstein M , Grigorenko E , Chawarska K , Pelphrey K , Howe J , Vaccarino FM ((2015) ) FOXG1-dependent dys-regulation of GABA/glutamate neuron differentiation in autism spectrum disorders. Cell 162: , 375–390. |
[138] | Raja WK , Mungenast AE , Lin Y-T , Ko T , Abdurrob F , Seo J , Tsai L-H ((2016) ) Self-organizing 3D human neural tissue derived from induced pluripotent stem cells recapitulate Alzheimer’s disease phenotypes. PLoS One 11: , e0161969. |
[139] | Nair AT , Ramachandran V , Joghee NM , Antony S , Ramalingam G ((2018) ) Gut microbiota dysfunction as reliable non-invasive early diagnostic biomarkers in the patho-physiology of Parkinson’s disease: A critical review. J Neurogastroenterol Motil 24: , 30–42. |
[140] | Park J , Lee BK , Jeong GS , Hyun JK , Lee CJ , Lee S-H ((2015) ) Three-dimensional brain-on-a-chip with an interstitial level of flow and its application as an in vitro model of Alzheimer’s disease. Lab Chip 15: , 141–150. |
[141] | Moreno EL , Hachi S , Hemmer K , Trietsch SJ , Baumuratov AS , Hankemeier T , Vulto P , Schwamborn JC , Fleming RMT ((2015) ) Differentiation of neuroepithelial stem cells into functional dopaminergic neurons in 3D microfluidic cell culture. Lab Chip 15: , 2419–2428. |
[142] | Gershanik OS ((2018) ) Does Parkinson’s disease start in the gut? Arq Neuropsiquiatr 76: , 67–70. |
[143] | Kim HJ , Li H , Collins JJ , Ingber DE ((2016) ) Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc Natl Acad Sci USA 113: , E7–15. |
[144] | Lancaster MA , Knoblich JA ((2014) ) Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc 9: , 2329–2340. |
[145] | McCracken KW , Catá EM , Crawford CM , Sinagoga KL , Schumacher M , Rockich BE , Tsai Y-H , Mayhew CN , Spence JR , Zavros Y , Wells JM ((2014) ) Modelling human development and disease in pluripotent stem-cell-derived gastric organoids. Nature 516: , 400–404. |
[146] | McCracken KW , Howell JC , Wells JM , Spence JR ((2011) ) Generating human intestinal tissue from pluripotent stem cells in vitro. Nat Protoc 6: , 1920–1928. |
[147] | Sato T , Clevers H ((2013) ) Growing self-organizing miniguts from a single intestinal stem cell: Mechanism and applications. Science 340: , 1190–1194. |
[148] | Cho H , Seo JH , Wong KHK , Terasaki Y , Park J , Bong K , Arai K , Lo EH , Irimia D ((2015) ) Three-dimensional blood-brain barrier model for in vitro studies of neurovascular pathology. Sci Rep 5: , 15222. |
[149] | Pham MT , Pollock KM , Rose MD , Cary WA , Stewart HR , Zhou P , Nolta JA , Waldau B ((2018) ) Generation of human vascularized brain organoids. Neuroreport 29: , 588–593. |
[150] | Watanabe M , Buth JE , Vishlaghi N , de la Torre-Ubieta L , Taxidis J , Khakh BS , Coppola G , Pearson CA , Yamauchi K , Gong D , Dai X , Damoiseaux R , Aliyari R , Liebscher S , Schenke-Layland K , Caneda C , Huang EJ , Zhang Y , Cheng G , Geschwind DH , Golshani P , Sun R , Novitch BG ((2017) ) Self-organized cerebral organoids with human-specific features predict effective drugs to combat Zika virus infection. Cell Rep 21: , 517–532. |
[151] | Lancaster MA ((2018) ) Brain organoids get vascularized. Nat Biotechnol 36: , 407–408. |
[152] | Mansour AA , Goncalves JT , Bloyd CW , Li H , Fernandes S , Quang D , Johnston S , Parylak SL , Jin X , Gage FH ((2018) ) An in vivo model of functional and vascularized human brain organoids. Nat Biotechnol 36: , 432–441. |
[153] | Berger E , Magliaro C , Paczia N , Monzel AS , Antony P , Linster CL , Bolognin S , Ahluwalia A , Schwamborn JC ((2018) ) Millifluidic culture improves human midbrain organoid vitality and differentiation. Lab Chip 18: , 3172–3183. |
[154] | Fermini B , Coyne ST , Coyne KP ((2018) ) Clinical trials in a dish: A perspective on the coming revolution in drug development. SLAS Discov 23: , 765–776. |
[155] | Takebe T , Wells JM , Helmrath MA , Zorn AM ((2018) ) Organoid center strategies for accelerating clinical translation. Cell Stem Cell 22: , 806–809. |
[156] | Munos BH , Chin WW ((2009) ) A call for sharing: Adapting pharmaceutical research to new realities. Sci Transl Med 1: , 9cm8. |
[157] | Poupon V , Seyller A , Rouleau GA ((2017) ) The Tanenbaum Open Science Institute: Leading a paradigm shift at the Montreal Neurological Institute. Neuron 95: , 1002–1006. |
[158] | Han C , Chaineau M , Chen CX , Beitel LK , Durcan TM ((2018) ) Open science meets stem cells: A new drug discovery approach for neurodegenerative disorders. Front Neurosci 12: , 47. |
[159] | Beaudin M , Klein CJ , Rouleau GA , Dupre N ((2017) ) Systematic review of autosomal recessive ataxias and proposal for a classification. Cerebellum Ataxias 4: , 3. |
[160] | Berry BJ , Smith AST , Young JE , Mack DL ((2018) ) Advances and current challenges associated with the use of human induced pluripotent stem cells in modeling neurodegenerative disease. Cells Tissues Organs, doi: 10.1159/000493018 |
[161] | Favé M-J , Lamaze FC , Soave D , Hodgkinson A , Gauvin H , Bruat V , Grenier J-C , Gbeha E , Skead K , Smargiassi A , Johnson M , Idaghdour Y , Awadalla P ((2018) ) Gene-by-environment interactions in urban populations modulate risk phenotypes. Nat Commun 9: , 827. |
[162] | Zhang Q , Chen W , Tan S , Lin T ((2017) ) Stem cells for modeling and therapy of Parkinson’s disease. Hum Gene Ther 28: , 85–98. |
[163] | Xu X , Huang J , Li J , Liu L , Han C , Shen Y , Zhang G , Jiang H , Lin Z , Xiong N , Wang T ((2016) ) Induced pluripotent stem cells and Parkinson’s disease: Modelling and treatment. Cell Prolif 49: , 14–26. |
[164] | Wang S , Zou C , Fu L , Wang B , An J , Song G , Wu J , Tang X , Li M , Zhang J , Yue F , Zheng C , Chan P , Zhang YA , Chen Z ((2015) ) Autologous iPSC-derived dopamine neuron transplantation in a nonhuman primate Parkinson’s disease model. Cell Discov 1: , 15012. |
[165] | Hallett PJ , Deleidi M , Astradsson A , Smith GA , Cooper O , Osborn TM , Sundberg M , Moore MA , Perez-Torres E , Brownell AL , Schumacher JM , Spealman RD , Isacson O ((2015) ) Successful function of autologous iPSC-derived dopamine neurons following transplantation in a nonhuman primate model of Parkinson’s disease. Cell Stem Cell 16: , 269–274. |
[166] | Lindvall O ((2016) ) Clinical translation of stem cell transplantation in Parkinson’s disease. J Intern Med 279: , 30–40. |


