
The Search for Environmental Causes of Parkinson’s Disease: Moving Forward
Abstract
It is widely believed that environmental exposures contribute to the vast majority of late-onset sporadic Parkinson’s disease (PD), alone or via interactions with genetic factors. The search for environmental causes of PD has however been hampered by lack of understanding the prodromal phase of PD development and the difficulties in exposure assessment during this prolonged period. On the other hand, the existence of this prodromal period, along with an increasingly better understanding of PD prodromal symptoms, provides an exciting opportunity to identify environmental factors that initiate PD pathogenesis and/or modify its progression. For prevention efforts, this prodromal stage is of a major interest. Targeting factors that enter the body via the nose or gut has become even more important since the discovery of α-synuclein aggregates in the enteric and olfactory nervous systems. In this paper, we speculate about novel research hypotheses and approaches that may help us better define the role of environment in PD etiology, especially during its extended and complex prodromal phase.
INTRODUCTION
The causes for late-onset sporadic Parkinson’s disease (PD) remain elusive, and PD is likely the cumulative result of numerous genetic and environmental insults and their interactions in the context of brain aging. Research on the environmental triggers and modifiers for PD development is incredibly important for a number of reasons. First, late-onset sporadic PD takes decades to develop, and by the time of diagnosis, neurodegenerative changes are too advanced to decelerate, stop, or reverse. Therefore, our battle against PD critically depends on disease early identification and intervention which in turn rely on a good understanding of disease etiology and actions upon modifiable risk factors. Second, despite recent great success in unveiling the genetic basis of late-onset sporadic PD, genetic findings may only explain a small portion of the cases and cannot be easily extended to disease prevention. On the other hand, during the decades of prodromal stage PD, many environmental factors may come into play at various time points that may trigger PD pathogenesis and modify its progression [1]. It is reasonable to assume that in a majority, if not all, late-onset PD cases, there are environmental contributions that determine or modify the risk and age of PD clinical onset. Unfortunately, we are far away from identifying these factors, defining their roles, and quantifying their contributions. Third, unlike genetic factors, environmental factors are potentially modifiable, and will therefore have profound implications for PD prevention and treatment. Finally, in the face of the rapid growth of aging populations across the globe, PD emerges as the fastest growing neurological disease in terms of both prevalence and death [2, 3]. Adding to this potential public health crisis, recent evidence, albeit preliminary and inconsistent, suggests that there is an increasing trend in PD incidence over the past a few decades [4], possibly indicating a role for environmental factors. Taken together, there is an urgent need for actions to be taken by funding agencies and PD researchers to identify environmental contributions to PD development.
WHAT DO WE KNOW NOW ABOUT ENVIRONMENTAL FACTORS AND PD?
In the past two decades, scientists have identified over a dozen environmental factors associated with the risk of developing PD, and for a majority, findings are reasonably consistent across studies [1, 5]. Examples include inverse associations with smoking [6, 7], coffee drinking [8, 9], vigorous exercise [10, 11], ibuprofen use [12, 13], and plasma urate [14, 15], as well as positive associations with overall pesticide exposure [16, 17], use of specific pesticides [18, 19], and traumatic brain injury [20, 21]. For most of these associations, plausible biological hypotheses have been proposed. However, causal inference for these epidemiological findings has been very difficult. Apart from limited and often inconsistent experimental data, for most of these epidemiological observations, reverse causation is a viable potential explanation - that PD development prior to clinical diagnosis changes lifestyle and behavior rather than the other way around. Possible exceptions are the use of certain pesticides. For example, epidemiological findings on rotenone and paraquat [18, 19] are supported by strong experimental evidence, so much so that these chemicals are being used to generate rodent models for PD therapeutic research [22]. Even for pesticides, there are many important questions unanswered. Therefore, despite its importance and a reasonable accumulation of literature, our understanding of environmental contributions to PD is still in its infancy.
WHAT ARE THE MAJOR CHALLENGES IN SEARCHING FOR ENVIRONMENTAL CAUSES OF PD?
The fact that late-onset sporadic PD takes decades to develop and the lack of understanding of this prolonged prodromal phase present a major challenge to understand environmental contributions to PD. In this disease development paradigm, causative exposures that initiate PD pathological process may have to occur and be documented decades before disease clinical diagnosis, which is often infeasible in epidemiological studies. Further, once neurodegeneration is initiated, many environmental and genetic factors may come into play to modify PD progression during the decades of prodromal disease development. As a consequence, even the most robust epidemiological findings, including those from longitudinal cohorts, are subject to alternative explanations. For instance, smokers have a robust and substantially lower risk for PD than non-smokers in all types of epidemiologic studies [23]. While a causal interpretation that cigarette smoking reduces PD risk is appealing, alternative hypotheses such as reverse causation and confounding by personality and other unknown risk factors are equally possible [24]. Similar analogies can be easily extended to most, if not all, of the presumed “protective” modifiable risk factors afore-mentioned.
Another major challenge is the lack of a good understanding of “environment” which encompasses a broad range of exposures that humans are in contact with, ranging from chemicals (e.g., pesticides), physical agents, microbes and viruses, to climate, lifestyle, socioeconomic conditions, and host-environment interactions. Unlike for the human genome, we do not have a “blueprint” of our environment; adding to the complexity, environmental exposures change over time, and their health consequences are likely to be complex, cumulative, interactive, and dynamic. While these are common challenges in environmental research for almost all chronic diseases, they are particularly challenging for neurodegenerative diseases such as PD. Neurons are long-lived, and thus the same aging neurons are subject to potential environmental influences over our entire lifespan. Further, PD is no longer considered a disease of the brain, it involves multiple systems and organs; when it comes to study environmental contributions to PD, we have to be mindful about environmental exposures through multiple routes of entry and at multiple susceptibility windows, which are yet to be better defined. All this, coupled with a decades-long disease initiation and prodromal development period, makes reliable and valid exposure assessment in epidemiologic studies that captures the most relevant etiological periods very difficult to achieve.
NOW IS A MAJOR OPPORTUNITY TO MOVE FORWARD!
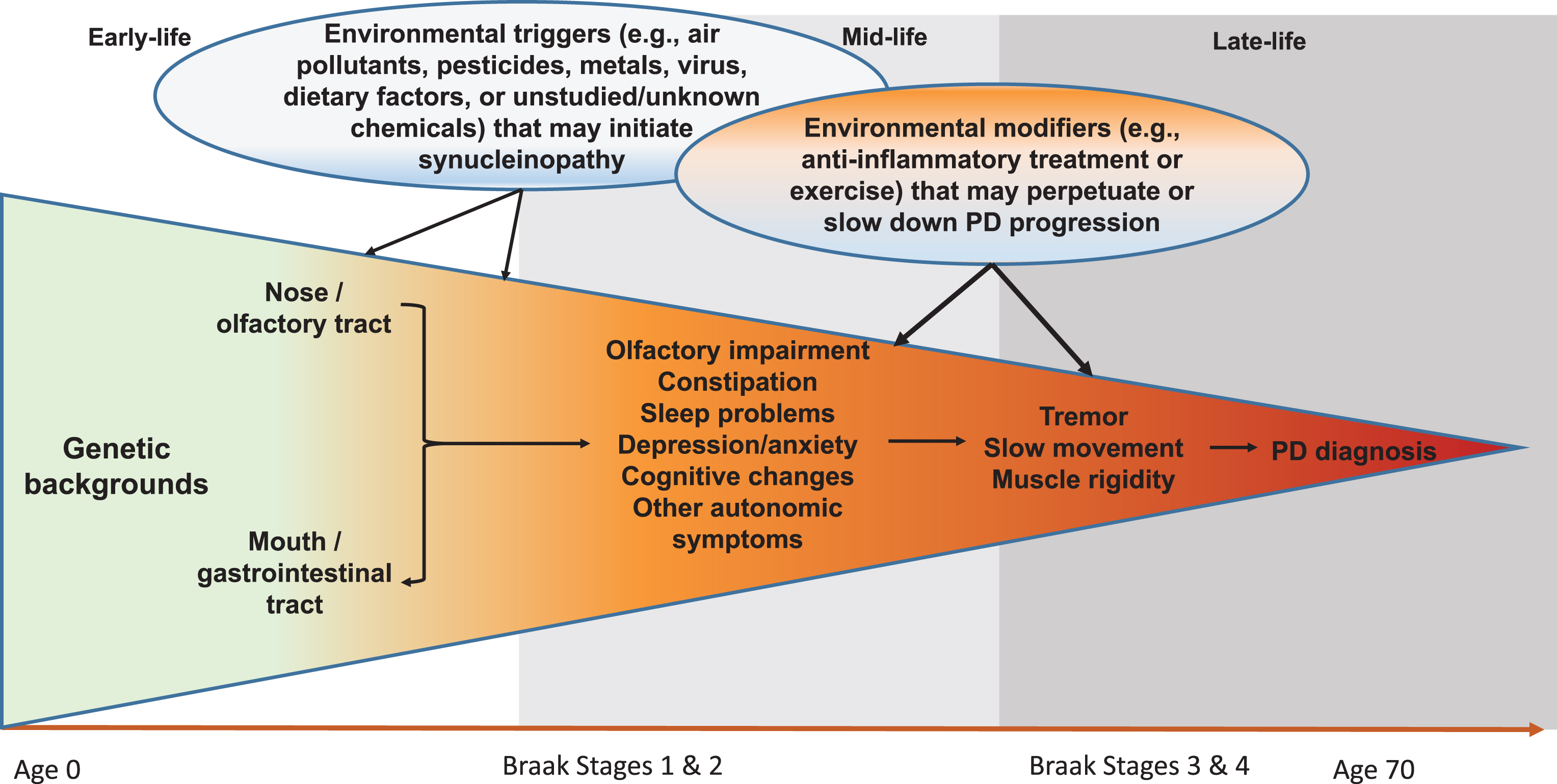
Although still somewhat controversial, the Braak hypothesis [25] presents a unique hypothetical framework of PD development that may allow us to better conceptualize steps of PD prodromal development and environmental contributions. According to this hypothesis, PD Lewy pathology develops in six sequential stages, first in the olfactory bulb or enteric nerves (stage 1), years if not decades, before spreading to the substantia nigra where dopaminergic neuron death occurs (stage 3). In support of this hypothesis, recent clinical and epidemiological studies have clearly documented a wide range of nonmotor symptoms in PD patients, and some symptoms such as olfactory impairment [26, 27], REM sleep behavior disorder (RBD) [28, 29], and constipation [30, 31] may have developed years, if not decades, prior to PD clinical diagnosis. While there are still substantial challenges to adequately define prodromal PD, by using these symptoms as noninvasive intermediate phenotypes, we may be able to bring new insights into this “black-box” of PD prodromal development by identifying factors that initiate PD pathogenesis, lead to these intermediate phenotypes, or modify progression to clinical PD (Fig. 1). This framework may fundamentally improve understanding of PD prodromal development and contributions from environmental factors.
Fig.1
Systematic and Life-long Approach to Study Environmental Triggers and Modifiers for PD Development. Late-onset sporadic PD takes decades to develop. In early- to mid-life, some environmental toxicants (e.g., pesticides, air pollutants, virus) may enter the body via the nose or the mouth, which may induce pathological synucleinopathy in susceptible individuals via mechanisms such as inflammation or microbiome dysbiosis; over time, the pathology may progress to the central olfactory structures and/or the lower brain stem and induce symptoms such as olfactory impairment and sleep disturbances. A portion of affected individuals may further develop motor deficits over years, which may eventually lead to a PD diagnosis. During this prolonged process, many environmental factors may come into play at various time windows to perpetuate or slow down the disease prodromal progression. Modified from Chen H (2018). J Parkinsons Dis. 8, 1–12.
NOVEL ENVIRONMENTAL PERSPECTIVES
The Braak hypothesis implicates the olfactory pathway and digestive tract as potential origins of PD development [25]. Importantly, these are the two anatomic sites where the human body directly interacts with the environment, where inflammation commonly occurs, and where paths to the brain are well established. It is possible that environmental toxicants such as air pollutants, pesticides, dietary contaminants, or viruses may enter the human body via the nose and/or mouth, and may initiate PD pathogenesis at the olfactory bulb and/or the gut enteric nerves; over time, the pathogenesis may spread to the brain via the olfactory and/or the vagus nerve, and eventually lead to dopaminergic neuron deaths in the substantia nigra [32]. All these observations make investigations of environmental exposures that enter via the olfactory or digestive tracts relevant and attractive for PD etiology. A few common and relevant exposures of interest are discussed below.
Air pollutants
The olfactory pathway represents an entry point for airborne pollutants that bypasses the blood-brain barrier (BBB), and has been shown as a route for pathogenic α-synuclein transmission to the brain [33]. Therefore, it is plausible to hypothesize that airborne pollutants may contribute to PD pathogenesis by initiating α-synuclein neuropathology in the olfactory system, which may later progress to PD in the context of brain aging [33]. Epidemiological evidence on air pollutants and PD is provocative, but limited and somewhat inconsistent [34–37]. Interestingly, α-synuclein aggregates were observed in the olfactory bulb of toddlers, children, teens, and young adults who prematurely died in Mexico City where air pollution is high [38]. In the US where the air pollutant levels are substantially lower, air pollutants have recently been linked to olfactory impairment among older adults [39, 40]. This is accompanied by a growing literature on potential harmful effects of air pollutants on cognitive function and dementia [41], as well as on neuroinflammation as a potential pathway between air pollutants and neurodegenerative outcomes [42]. We expect future studies not only connect air pollutants, olfactory impairment, and PD development, but also ascertain underlying biological mechanisms.
The gut microbiome
The gut microbiome has quickly been recognized as a major contributor to human physiology as it influences the immune system, and it is responsible for the uptake of nutrients, medications, and environmental toxicants. Evidence is mounting that the microbiome can affect various aspects of neurological functions, brain activities, and behaviors in both animal models and human studies [43]. The Braak hypothesis further puts the microbiome to the forefront of PD etiological research [43, 44] as it implicates gut enteric nerves as an initiation site of PD pathology [25]. In support of this, constipation is one of the most prevalent prodromal symptoms of PD [45] which may have developed 1-2 decades prior to PD diagnosis [30, 31, 46]. Recent studies have compared the gut microbiomes of PD patients with those of controls [47–53]. Although results were not entirely consistent, the studies generally identified a pro-inflammatory microbiome in PD patients as compared with controls. Interestingly, one recent study further reported similarities in microbiome dysbiosis between PD and idiopathic RBD patients, suggesting that the microbiome may change in prodromal PD [53]. As the gut microbiome can be affected by many PD-relevant environmental factors (e.g., smoking, dietary factors, and pesticides), examinations of potential environmental influences on the gut microbiome in the context of PD development are warranted. Similar analogies could also be readily made to the nasal or oral microbiomes, which have been largely ignored thus far in PD research [53, 54].
New insights into pesticides and PD development
Pesticides are among the few well-documented harmful environmental contributors to PD [18, 19]. However, when, where, and how specific pesticides contribute to PD development remains largely unknown. Pesticides access the body via inhalation, eating/drinking, or skin contact [55]; therefore, the olfactory or digestive entry points can be readily extended to pesticide exposures. If pesticides enter the body via nose or the digestive tract, they may initiate synucleinopathy in the olfactory structure or gut, which may later spread to the brain over time via olfactory nerve or the gut-to-brain axis [56]. Research on pesticides in relation to constipation and olfactory impairment in the context of aging may provide information critically relevant to PD development. Although olfactory and gastrointestinal symptoms have been documented in pesticide-based animal models of PD [57, 58], human empirical evidence is sparse and indirect. One study in North Carolina found that farmworkers had higher olfactory thresholds than non-farming laborers [59], but the study could not directly attribute this observation to pesticide use. Interestingly, recently evidence suggests that some pesticides alter the gut microbiome. In murine models, the organophosphate insecticide diazinon perturbed the community structure, functional metagenome, and metabolic profile of the gut microbiome [60, 61] by modulation of quorum sensing, a key mechanism that regulates bacterial populations, composition, and importantly, their functional genes. We expect future detailed research to improve our understanding of the roles of specific pesticides in PD development by delineating potential routes of entry, effects at various stages of PD development, and potential biological or pathological mechanisms.
Other relevant environmental exposures
Although not the focus of this article, the Braak hypothesis also provides strong rationales to systematically examine several other environmental exposures that have not been well studied in the context of PD development such as organic solvents [62], high temperature cooked meats and heterocyclic amines [63], respiratory or GI infections and inflammation [64–66], and the use of antibiotics and antiviral therapies [67].
OTHER IMPORTANT CONSIDERATIONS IN DEFINING ENVIRONMENTAL CONTRIBUTIONS TO PD
Gene-environment interactions and epigenetics
Many biologic processes have evolved to be accomplished through multiple mechanisms that involve both genetic and environmental contributions, and both have to come together before a biologic system fails. Identification of genetic factors that modify the association of specific environmental stressors with PD will improve our understanding of etiological mechanisms. The success in understanding PD gene-environment interactions thus far has been largely limited to interactions of genetic factors with specific pesticides. For example, studies suggest that the potential adverse effects of paraquat on PD may be substantially augmented by genetic variants that are related to dopamine transporter function [68] or DNA base excision repair failure [69], possibly by rendering the system unable to compensate for the combined synergistic insults. While these individual studies provide crucial leads in understanding specific gene-environment interactions and pathways, we expect increasingly available and more statistically powerful pooled analyses to identify [70, 71], cross-validate, or refute [72] potential gene-environmental interactions in PD etiology.
Epigenetic research is another exciting approach to understanding PD pathogenesis by studying mechanisms that regulate gene function through DNA methylation and histone modification without alternating DNA sequence itself. Most PD epigenetic studies to date have targeted methylation in candidate genes using blood or saliva samples [73], and interpretation of study findings are limited by the fact that epigenetic regulations are often tissue specific. Nevertheless, recent epigenome-wide association and network analyses showed that PD status was associated with DNA methylation changes in blood and saliva that implicated the immune system, with some of the differences not due to differential white blood cell composition that distinguished PD patients from controls [74, 75]. This observation converges nicely with the increasing genomic [76, 77], epidemiologic [66], and experimental [78] evidence for the importance of immunity and inflammation in PD development. Epigenetic approaches, as well as other ‘omics’ tools (proteinomics, transcriptomics, lipidomics, metabolomics), based on human tissue samples and induced human stem cells can also be employed for the “meet in the middle” approach, an informative strategy for the integration of such technologies into epidemiological research [79]. We expect such synergistic approaches to provide more insights into biological plausibility and to help establish causality of epidemiological findings.
A life-long and exposome approach to PD etiological research
Most epidemiological studies on PD have focused on a snapshot of a single environmental exposure often in mid to late adulthood, largely ignoring the multidimensional complexity of the disease that may require a life-course perspective. Some experimental studies showed that prenatal or early developmental exposures to certain pesticides (e.g., dieldrin [80], paraquat or maneb [81], atrazine [82]) can lead to dopaminergic neuron degeneration later in life. Others reported that such early-life exposures rendered animals more susceptible to a second toxic environmental insult later in life [81, 83]. These findings suggest that early-life exposures may be contributing to PD development by setting the stage for late-life susceptibility to PD, and point out the importance of preventing repeated exposures during vulnerable periods. Although a life-course approach to late-onset neurodegenerative diseases such as PD is methodologically formidable, we expect future studies to carefully consider such possibilities.
The latest genetic research embarked on generating a polygenic risk score that encompasses all known risk alleles and its use has been promoted to define individual’s overall genetic susceptibility to PD [84] or to predict disease progression [85]. In comparison, a “composite environmental index” is much more difficult to develop, and a vast majority of the epidemiological studies have focused on a single risk factor (e.g., smoking). Nevertheless, epidemiological studies have made first efforts to simultaneously consider multiple environmental exposures. For example, Lee et al. reported potential joint effects from traumatic brain injury and paraquat exposure on PD risk [86] and Kim et al. simultaneously considered smoking, caffeine intake, physical activity and several other factors [87]. These findings are provocative, suggesting stronger associations when composite exposures and their possible synergism are considered. In addition, biomarkers of long-term and lifelong environmental exposures are being developed, such as blood methylation profiles for long-term organophosphate exposures [88]. Further, some major scientific and technological advances that inform the external assessment of the exposome have been made [91, 92], including geographic information systems, remote sensing, global positioning system and geolocation technologies, portable and personal sensing, including smartphone-based sensors and self-reported questionnaire assessments relying on Internet-based platforms. However, all of these methodological and technological improvements in exposure assessment need to be accompanied by new data analysis and interpretation methodologies and also require developing protocols for ethical sharing of sensitive data.
When it comes to considering multiple exposures over time, the concepts of “exposome” and “neuroexposome” are particularly appealing. Chris Wild first proposed the concept of “exposome” more than a decade ago as a measure of the totality of human environmental exposures over one’s lifetime [89]. Heffernan and Hare [90] recently further adapted this concept for neurological diseases. They proposed to cultivate the strengths of “omics” technologies (e.g., genomics and metabolomics), traditional epidemiological surveys, and detailed clinical assessments across the lifespan while considering the unique features of the brain (e.g., BBB permeability and neuron longevity).
This exposome concept, while challenging to implement in the real world, together with the Braak hypothesis, provides a theoretical framework for scientists to design future studies to decipher the environmental causes of PD and develop early interventions to halt the progression to the characteristic motor dysfunction in PD.
CONFLICT OF INTEREST
The author has no conflict of interest to report.
ACKNOWLEDGMENTS
Dr. Chen is supported by a start-up fund from Michigan State University (GE100455), the Parkinson’s Foundation (Grant No. PF-IMP-1825), and the Office of the Assistant Secretary of Defense for Health Affairs, through the Parkinson’s Research Program (Award No. W81XWH-17-1-0536). Opinions, interpretations, conclusions and recommendations are those of the author and are not necessarily endorsed by the Department of Defense. Dr Ritz was supported by the NIEHS grants ES10544, P01ES016732, U54ES12078, R01ES013717, R21ES022391, R21ES024356, and the NINDS grant: P50NS038367 and by pilot funding from SCEHSC# 5P30 ES07048 and The American Parkinson Disease Association. We would also like to thank Mr. Frank Purdy for proof reading the manuscript.
REFERENCES
[1] | Chen H ((2018) ) The changing landscape of Parkinson epidemiologic research. J Parkinsons Dis 8: , 1–12. |
[2] | GBD 2016 Parkinson’s Disease Collaborators ((2018) ) Global, regional, and national burden of Parkinson’s disease, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 17: , 939–953. |
[3] | Darweesh SKL , Raphael KG , Brundin P , Matthews H , Wyse RK , Chen H , Bloem BR ((2018) ) Parkinson Matters. J Parkinsons Dis 8: , 495–498. |
[4] | Savica R , Grossardt BR , Bower JH , Ahlskog JE , Rocca WA ((2017) ) Time trends in the incidence of Parkinson’s disease: A 30-year study. JAMA Neurol 32: , 227–234. |
[5] | Ascherio A , Schwarzschild MA ((2016) ) The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol 15: , 1257–1272. |
[6] | Chen H , Huang X , Guo X , Mailman RB , Park Y , Kamel F , Umbach DM , Xu Q , Hollenbeck A , Schatzkin A , Blair A ((2010) ) Smoking duration, intensity, and risk of Parkinson disease. Neurology 74: , 878–884. |
[7] | Ritz B , Lee PC , Lassen CF , Arah OA ((2014) ) Parkinson disease and smoking revisited: Ease of quitting is an early sign of the disease. Neurology 83: , 1396–1402. |
[8] | Ascherio A , Weisskopf MG , O’Reilly EJ , McCullough ML , Calle EE , Rodriguez C , Thun MJ ((2004) ) Coffee consumption, gender, and Parkinson’s disease mortality in the cancer prevention study II cohort: The modifying effects of estrogen. Am J Epidemiol 160: , 977–984. |
[9] | Liu R , Guo X , Park Y , Huang X , Sinha R , Freedman ND , Hollenbeck AR , Blair A , Chen H ((2012) ) Caffeine intake, smoking, and risk of Parkinson disease in men and women. Am J Epidemiol 175: , 1200–1207. |
[10] | Chen H , Zhang SM , Schwarzschild MA , Hernan MA , Ascherio A ((2005) ) Physical activity and the risk of Parkinson disease. Neurology 64: , 664–669. |
[11] | Xu Q , Park Y , Huang X , Hollenbeck A , Blair A , Schatzkin A , Chen H ((2010) ) Physical activities and future risk of Parkinson disease. Neurology 75: , 341–348. |
[12] | Chen H , Jacobs E , Schwarzschild MA , McCullough ML , Calle EE , Thun MJ , Ascherio A ((2005) ) Nonsteroidal antiinflammatory drug use and the risk for Parkinson’s disease. Ann Neurol 58: , 963–967. |
[13] | Gao X , Chen H , Schwarzschild MA , Ascherio A ((2011) ) Use of ibuprofen and risk of Parkinson disease. Neurology 76: , 863–869. |
[14] | Chen H , Mosley TH , Alonso A , Huang X ((2009) ) Plasma urate and Parkinson’s disease in the Atherosclerosis Risk in Communities (ARIC) study. Am J Epidemiol 169: , 1064–1069. |
[15] | Gao X , O’Reilly EJ , Schwarzschild MA , Ascherio A ((2016) ) Prospective study of plasma urate and risk of Parkinson disease in men and women. Neurology 86: , 520–526. |
[16] | Ascherio A , Chen H , Weisskopf MG , O’Reilly E , McCullough ML , Calle EE , Schwarzschild MA , Thun MJ ((2006) ) Pesticide exposure and risk for Parkinson’s disease. Ann Neurol 60: , 197–203. |
[17] | Liew Z , Wang A , Bronstein J , Ritz B ((2014) ) Job exposure matrix (JEM)-derived estimates of lifetime occupational pesticide exposure and the risk of Parkinson’s disease. Arch Environ Occup Health 69: , 241–251. |
[18] | Tanner CM , Kamel F , Ross GW , Hoppin JA , Goldman SM , Korell M , Marras C , Bhudhikanok GS , Kasten M , Chade AR , Comyns K , Richards MB , Meng C , Priestley B , Fernandez HH , Cambi F , Umbach DM , Blair A , Sandler DP , Langston JW ((2011) ) Rotenone, paraquat, and Parkinson’s disease. Environ Health Perspect 119: , 866–872. |
[19] | Costello S , Cockburn M , Bronstein J , Zhang X , Ritz B ((2009) ) Parkinson’s disease and residential exposure to maneb and paraquat from agricultural applications in the central valley of California. Am J Epidemiol 169: , 919–926. |
[20] | Fang F , Chen H , Feldman AL , Kamel F , Ye W , Wirdefeldt K ((2012) ) Head injury and Parkinson’s disease: A population-based study. Mov Disord 27: , 1632–1635. |
[21] | Gao J , Liu R , Zhao E , Huang X , Nalls MA , Singleton AB , Chen H ((2015) ) Head injury, potential interaction with genes, and risk for Parkinson’s disease. Parkinsonism Relat Disord 21: , 292–296. |
[22] | Le W , Sayana P , Jankovic J ((2014) ) Animal models of Parkinson’s disease: A gateway to therapeutics? Neurotherapeutics 11: , 92–110. |
[23] | Breckenridge CB , Berry C , Chang ET , Sielken RL Jr , Mandel JS ((2016) ) Association between Parkinson’s disease and cigarette smoking, rural living, well-water consumption, farming and pesticide use: Systematic review and meta-analysis. PLoS One 11: , e0151841. |
[24] | Ritz B , Rhodes SL ((2010) ) After half a century of research on smoking and PD, where do we go now? Neurology 74: , 870–871. |
[25] | Braak H , Bohl JR , Müller CM , Rüb U , de Vos RAI , Tredici KD ((2006) ) Stanley Fahn Lecture 2005: The staging procedure for the inclusion body pathology associated with sporadic Parkinson’s disease reconsidered. Mov Disord 21: , 2042–2051. |
[26] | Chen H , Shrestha S , Huang X , Jain S , Guo X , Tranah GJ , Garcia ME , Satterfield S , Phillips C , Harris TB , Health ABCS ((2017) ) Olfaction and incident Parkinson disease in US white and black older adults. Neurology 89: , 1441–1447. |
[27] | Ross GW , Petrovitch H , Abbott RD , Tanner CM , Popper J , Masaki K , Launer L , White LR ((2008) ) Association of olfactory dysfunction with risk for future Parkinson’s disease. Ann Neurol 63: , 167–173. |
[28] | Postuma RB , Gagnon JF , Vendette M , Fantini ML , Massicotte-Marquez J , Montplaisir J ((2009) ) Quantifying the risk of neurodegenerative disease in idiopathic REM sleep behavior disorder. Neurology 72: , 1296–1300. |
[29] | Postuma RB , Iranzo A , Hogl B , Arnulf I , Ferini-Strambi L , Manni R , Miyamoto T , Oertel W , Dauvilliers Y , Ju YE , Puligheddu M , Sonka K , Pelletier A , Santamaria J , Frauscher B , Leu-Semenescu S , Zucconi M , Terzaghi M , Miyamoto M , Unger MM , Carlander B , Fantini ML , Montplaisir JY ((2015) ) Risk factors for neurodegeneration in idiopathic rapid eye movement sleep behavior disorder: A multicenter study. Ann Neurol 77: , 830–839. |
[30] | Abbott RD , Petrovitch H , White LR , Masaki KH , Tanner CM , Curb JD , Grandinetti A , Blanchette PL , Popper JS , Ross GW ((2001) ) Frequency of bowel movements and the future risk of Parkinson’s disease. Neurology 57: , 456–462. |
[31] | Gao X , Chen H , Schwarzschild MA , Ascherio A ((2011) ) A prospective study of bowel movement frequency and risk of Parkinson’s disease. Am J Epidemiol 174: , 546–551. |
[32] | Reichmann H ((2011) ) View point: Etiology in Parkinson’s disease. Dual hit or spreading intoxication. J Neurol Sci 310: , 9–11. |
[33] | Rey NL , Steiner JA , Maroof N , Luk KC , Madaj Z , Trojanowski JQ , Lee VM , Brundin P ((2016) ) Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med 213: , 1759–1778. |
[34] | Ritz B , Lee PC , Hansen J , Lassen CF , Ketzel M , Sorensen M , Raaschou-Nielsen O ((2016) ) Traffic-related air pollution and Parkinson’s disease in Denmark: A case-control study. Environ Health Perspect 124: , 351–356. |
[35] | Liu R , Young MT , Chen JC , Kaufman JD , Chen H ((2016) ) Ambient air pollution exposures and risk of Parkinson disease. Environ Health Perspect 124: , 1759–1765. |
[36] | Lee PC , Liu LL , Sun Y , Chen YA , Liu CC , Li CY , Yu HL , Ritz B ((2016) ) Traffic-related air pollution increased the risk of Parkinson’s disease in Taiwan: A nationwide study. Environ Int 96: , 75–81. |
[37] | Palacios N , Fitzgerald KC , Hart JE , Weisskopf M , Schwarzschild MA , Ascherio A , Laden F ((2017) ) Air pollution and risk of Parkinson’s disease in a large prospective study of men. Environ Health Perspect 125: , 087011. |
[38] | Calderon-Garciduenas L , Gonzalez-Maciel A , Reynoso-Robles R , Kulesza RJ , Mukherjee PS , Torres-Jardon R , Ronkko T , Doty RL ((2018) ) Alzheimer’s disease and alpha-synuclein pathology in the olfactory bulbs of infants, children, teens and adults </=40 years in Metropolitan Mexico City. APOE4 carriers at higher risk of suicide accelerate their olfactory bulb pathology. Environ Res 166: , 348–362. |
[39] | Ajmani GS , Suh HH , Wroblewski KE , Kern DW , Schumm LP , McClintock MK , Yanosky JD , Pinto JM ((2016) ) Fine particulate matter exposure and olfactory dysfunction among urban-dwelling older US adults. Environ Res 151: , 797–803. |
[40] | Adams DR , Ajmani GS , Pun VC , Wroblewski KE , Kern DW , Schumm LP , McClintock MK , Suh HH , Pinto JM ((2016) ) Nitrogen dioxide pollution exposure is associated with olfactory dysfunction in older U.S. adults. Int Forum Allergy Rhinol 6: , 1245–1252. |
[41] | Power MC , Adar SD , Yanosky JD , Weuve J ((2016) ) Exposure to air pollution as a potential contributor to cognitive function, cognitive decline, brain imaging, and dementia: A systematic review of epidemiologic research. Neurotoxicology 56: , 235–253. |
[42] | Jayaraj RL , Rodriguez EA , Wang Y , Block ML ((2017) ) Outdoor ambient air pollution and neurodegenerative diseases: The neuroinflammation hypothesis. Curr Environ Health Rep 4: , 166–179. |
[43] | Tremlett H , Bauer KC , Appel-Cresswell S , Finlay BB , Waubant E ((2017) ) The gut microbiome in human neurological disease: A review. Ann Neurol 81: , 369–382. |
[44] | Parashar A , Udayabanu M ((2017) ) Gut microbiota: Implications in Parkinson’s disease. Parkinsonism Relat Disord 38: , 1–7. |
[45] | Chen H , Zhao EJ , Zhang W , Lu Y , Liu R , Huang X , Ciesielski-Jones AJ , Justice MA , Cousins DS , Peddada S ((2015) ) Meta-analyses on prevalence of selected Parkinson’s nonmotor symptoms before and after diagnosis. Transl Neurodegener 4: , 1. |
[46] | Savica R , Carlin JM , Grossardt BR , Bower JH , Ahlskog JE , Maraganore DM , Bharucha AE , Rocca WA ((2009) ) Medical records documentation of constipation preceding Parkinson disease: A case-control study. Neurology 73: , 1752–1758. |
[47] | Keshavarzian A , Green SJ , Engen PA , Voigt RM , Naqib A , Forsyth CB , Mutlu E , Shannon KM ((2015) ) Colonic bacterial composition in Parkinson’s disease. Mov Disord 30: , 1351–1360. |
[48] | Scheperjans F , Aho V , Pereira PA , Koskinen K , Paulin L , Pekkonen E , Haapaniemi E , Kaakkola S , Eerola-Rautio J , Pohja M , Kinnunen E , Murros K , Auvinen P ((2015) ) Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord 30: , 350–358. |
[49] | Unger MM , Spiegel J , Dillmann KU , Grundmann D , Philippeit H , Burmann J , Fassbender K , Schwiertz A , Schafer KH ((2016) ) Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord 32: , 66–72. |
[50] | Hasegawa S , Goto S , Tsuji H , Okuno T , Asahara T , Nomoto K , Shibata A , Fujisawa Y , Minato T , Okamoto A , Ohno K , Hirayama M ((2015) ) Intestinal dysbiosis and lowered serum lipopolysaccharide-binding protein in Parkinson’s disease. PLoS One 10: , e0142164. |
[51] | Petrov VA , Saltykova IV , Zhukova IA , Alifirova VM , Zhukova NG , Dorofeeva YB , Tyakht AV , Kovarsky BA , Alekseev DG , Kostryukova ES , Mironova YS , Izhboldina OP , Nikitina MA , Perevozchikova TV , Fait EA , Babenko VV , Vakhitova MT , Govorun VM , Sazonov AE ((2017) ) Analysis of gut microbiota in patients with Parkinson’s disease. Bull Exp Biol Med 162: , 734–737. |
[52] | Hopfner F , Kunstner A , Muller SH , Kunzel S , Zeuner KE , Margraf NG , Deuschl G , Baines JF , Kuhlenbaumer G ((2017) ) Gut microbiota in Parkinson disease in a northern German cohort. Brain Res 1667: , 41–45. |
[53] | Heintz-Buschart A , Pandey U , Wicke T , Sixel-Doring F , Janzen A , Sittig-Wiegand E , Trenkwalder C , Oertel WH , Mollenhauer B , Wilmes P ((2018) ) The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eye movement sleep behavior disorder. Mov Disord 33: , 88–98. |
[54] | Pereira PAB , Aho VTE , Paulin L , Pekkonen E , Auvinen P , Scheperjans F ((2017) ) Oral and nasal microbiota in Parkinson’s disease. Parkinsonism Relat Disord 38: , 61–67. |
[55] | Mostafalou S , Abdollahi M ((2017) ) Pesticides: An update of human exposure and toxicity. Arch Toxicol 91: , 549–599. |
[56] | Pan-Montojo F , Anichtchik O , Dening Y , Knels L , Pursche S , Jung R , Jackson S , Gille G , Spillantini MG , Reichmann H , Funk RH ((2010) ) Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One 5: , e8762. |
[57] | Sasajima H , Miyazono S , Noguchi T , Kashiwayanagi M ((2015) ) Intranasal administration of rotenone in mice attenuated olfactory functions through the lesion of dopaminergic neurons in the olfactory bulb. Neurotoxicology 51: , 106–115. |
[58] | Johnson ME , Stringer A , Bobrovskaya L ((2018) ) Rotenone induces gastrointestinal pathology and microbiota alterations in a rat model of Parkinson’s disease. Neurotoxicology 65: , 174–185. |
[59] | Quandt SA , Walker FO , Talton JW , Summers P , Chen H , McLeod DK , Arcury TA ((2016) ) Olfactory function in Latino farmworkers: Subclinical neurological effects of pesticide exposure in a vulnerable population. J Occup Environ Med 58: , 248–253. |
[60] | Gao B , Bian X , Chi L , Tu P , Ru H , Lu K ((2017) ) Editor’s Highlight: Organophosphate diazinon altered quorum sensing, cell motility, stress response, and carbohydrate metabolism of gut microbiome. Toxicol Sci 157: , 354–364. |
[61] | Gao B , Bian X , Mahbub R , Lu K ((2017) ) Sex-specific effects of organophosphate diazinon on the gut microbiome and its metabolic functions. Environ Health Perspect 125: , 198–206. |
[62] | Goldman SM , Quinlan PJ , Ross GW , Marras C , Meng C , Bhudhikanok GS , Comyns K , Korell M , Chade AR , Kasten M , Priestley B , Chou KL , Fernandez HH , Cambi F , Langston JW , Tanner CM ((2012) ) Solvent exposures and Parkinson disease risk in twins. Ann Neurol 71: , 776–784. |
[63] | Agim ZS , Cannon JR ((2018) ) Alterations in the nigrostriatal dopamine system after acute systemic PhIP exposure. Toxicol Lett 287: , 31–41. |
[64] | Huang HK , Wang JH , Lei WY , Chen CL , Chang CY , Liou LS ((2018) ) Helicobacter pylori infection is associated with an increased risk of Parkinson’s disease: A population-based retrospective cohort study. Parkinsonism Relat Disord 47: , 26–31. |
[65] | Macerollo A , Lu MK , Huang HC , Chen HJ , Lin CC , Kao CH , Tsai CH , Chen JC ((2017) ) Colonic diverticular disease: A new risk factor for Parkinson’s disease? Parkinsonism Relat Disord 42: , 61–65. |
[66] | Villumsen M , Aznar S , Pakkenberg B , Jess T , Brudek T ((2018) ) Inflammatory bowel disease increases the risk of Parkinson’s disease: A Danish nationwide cohort study 1977-2014. Gut. 10.1136/gutjnl-2017-315666. |
[67] | Racette BA , Gross A , Vouri SM , Camacho-Soto A , Willis AW , Searles Nielsen S ((2018) ) Immunosuppressants and risk of Parkinson disease. Ann Clin Transl Neurol 5: , 870–875. |
[68] | Ritz BR , Manthripragada AD , Costello S , Lincoln SJ , Farrer MJ , Cockburn M , Bronstein J ((2009) ) Dopamine transporter genetic variants and pesticides in Parkinson’s disease. Environ Health Perspect 117: , 964–969. |
[69] | Sanders LH , Paul KC , Howlett EH , Lawal H , Boppana S , Bronstein JM , Ritz B , Greenamyre JT ((2017) ) Editor’s Highlight: Base excision repair variants and pesticide exposure increase Parkinson’s disease risk. Toxicol Sci 158: , 188–198. |
[70] | Chuang YH , Lee PC , Vlaar T , Mulot C , Loriot MA , Hansen J , Lill CM , Ritz B , Elbaz A ((2017) ) Pooled analysis of the HLA-DRB1 by smoking interaction in Parkinson disease. Ann Neurol 82: , 655–664. |
[71] | Lee PC , Ahmed I , Loriot MA , Mulot C , Paul KC , Bronstein JM , Ritz B , Elbaz A ((2018) ) Smoking and Parkinson disease: Evidence for gene-by-smoking interactions. Neurology 90: , e583–e592. |
[72] | Ahmed I , Lee PC , Lill CM , Searles Nielsen S , Artaud F , Gallagher LG , Loriot MA , Mulot C , Nacfer M , Liu T , Biernacka JM , Armasu S , Anderson K , Farin FM , Lassen CF , Hansen J , Olsen JH , Bertram L , Maraganore DM , Checkoway H , Ritz B , Elbaz A ((2014) ) Lack of replication of the GRIN2A-by-coffee interaction in Parkinson disease. PLoS Genet 10: , e1004788. |
[73] | Miranda-Morales E , Meier K , Sandoval-Carrillo A , Salas-Pacheco J , Vazquez-Cardenas P , Arias-Carrion O ((2017) ) Implications of DNA methylation in Parkinson’s disease. Front Mol Neurosci 10: , 225. |
[74] | Horvath S , Ritz BR ((2015) ) Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging (Albany NY) 7: , 1130–1142. |
[75] | Chuang YH , Paul KC , Bronstein JM , Bordelon Y , Horvath S , Ritz B ((2017) ) Parkinson’s disease is associated with DNA methylation levels in human blood and saliva. Genome Med 9: , 76. |
[76] | Witoelar A , Jansen IE , Wang Y , Desikan RS , Gibbs JR , Blauwendraat C , Thompson WK , Hernandez DG , Djurovic S , Schork AJ , Bettella F , Ellinghaus D , Franke A , Lie BA , McEvoy LK , Karlsen TH , Lesage S , Morris HR , Brice A , Wood NW , Heutink P , Hardy J , Singleton AB , Dale AM , Gasser T , Andreassen OA , Sharma M , International Parkinson’s Disease Genomics Consortium (IPDGC), North American Brain Expression Consortium (NABEC), and United Kingdom Brain Expression Consortium (UKBEC) Investigators ((2017) ) Genome-wide pleiotropy between Parkinson disease and autoimmune diseases. JAMA Neurol 74: , 780–792. |
[77] | Gagliano SA , Pouget JG , Hardy J , Knight J , Barnes MR , Ryten M , Weale ME ((2016) ) Genomics implicates adaptive and innate immunity in Alzheimer’s and Parkinson’s diseases. Ann Clin Transl Neurol 3: , 924–933. |
[78] | Kannarkat GT , Boss JM , Tansey MG ((2013) ) The role of innate and adaptive immunity in Parkinson’s disease. J Parkinsons Dis 3: , 493–514. |
[79] | Vineis P , van Veldhoven K , Chadeau-Hyam M , Athersuch TJ ((2013) ) Advancing the application of omics-based biomarkers in environmental epidemiology. Environ Mol Mutagen 54: , 461–467. |
[80] | Richardson JR , Caudle WM , Wang M , Dean ED , Pennell KD , Miller GW ((2006) ) Developmental exposure to the pesticide dieldrin alters the dopamine system and increases neurotoxicity in an animal model of Parkinson’s disease. FASEB J 20: , 1695–1697. |
[81] | Cory-Slechta DA , Thiruchelvam M , Barlow BK , Richfield EK ((2005) ) Developmental pesticide models of the Parkinson disease phenotype. Environ Health Perspect 113: , 1263–1270. |
[82] | Li Y , Sun Y , Yang J , Wu Y , Yu J , Li B ((2014) ) Age-dependent dopaminergic dysfunction following fetal exposure to atrazine in SD rats. Environ Toxicol Pharmacol 37: , 1275–1282. |
[83] | Thiruchelvam M , Richfield EK , Goodman BM , Baggs RB , Cory-Slechta DA ((2002) ) Developmental exposure to the pesticides paraquat and maneb and the Parkinson’s disease phenotype. Neurotoxicology 23: , 621–633. |
[84] | Nalls MA , Escott-Price V , Williams NM , Lubbe S , Keller MF , Morris HR , Singleton AB , International Parkinson’s Disease Genomics C ((2015) ) Genetic risk and age in Parkinson’s disease: Continuum not stratum. Mov Disord 30: , 850–854. |
[85] | Paul KC , Schulz J , Bronstein JM , Lill CM , Ritz BR ((2018) ) Association of polygenic risk score with cognitive decline and motor progression in Parkinson disease. JAMA Neurol 75: , 360–366. |
[86] | Lee PC , Bordelon Y , Bronstein J , Ritz B ((2012) ) Traumatic brain injury, paraquat exposure, and their relationship to Parkinson disease. Neurology 79: , 2061–2066. |
[87] | Kim IY , O’Reilly EJ , Hughes KC , Gao X , Schwarzschild MA , Hannan MT , Betensky RA , Ascherio A ((2018) ) Integration of risk factors for Parkinson disease in 2 large longitudinal cohorts. Neurology 90: , e1646–e1653. |
[88] | Paul K , Chuang Y , Cockburn M , Bronstein J , Horvath S , Ritz B ((2018) ) Organophosphate pesticide exposure and differential genome-wide DNA methylation. Sci Total Environ 645: , 1135–1143. |
[89] | Wild CP ((2005) ) Complementing the genome with an “exposome”: The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol Biomarkers Prev 14: , 1847–1850. |
[90] | Heffernan AL , Hare DJ ((2018) ) Tracing Environmental exposure from neurodevelopment to neurodegeneration. Trends Neurosci 41: , 496–501. |
[91] | Turner MC , Nieuwenhuijsen M , Anderson K , Balshaw D , Cui Y , Dunton G , Hoppin JA , Koutrakis P , Jerrett M ((2017) ) Assessing the exposome with external measures: Commentary on the state of the science and research recommendations. Annu Rev Public Health 38: , 215–239. |
[92] | Loh M , Sarigiannis D , Gotti A , Karakitsios S , Pronk A , Kuijpers E , Annesi-Maesano I , Baiz N , Madureira J , Oliveira Fernandes E , Jerrett M , Cherrie JW ((2017) ) How sensors might help define the external exposome. Int J Environ Res Public Health 14: . |


