
Updating Our Definitions of Parkinson’s Disease for a Molecular Age
Abstract
Clinical definitions of Parkinson’s disease (PD) are over 200 years old, while neuropathological definitions— which are still the basis of how we define the disease now— are over 100 years old. We argue that for both clinical care and therapeutic development, these definitions need updating for the molecular age in which we live. We highlight specific instances in which genetic or biochemical biomarkers are increasingly used for clinical trial enrollment in the neurodegenerative diseases, suggesting that molecular definition(s) of PD are already emerging. We review candidate biomarkers for PD-related pathologies and highlight the need for further validation.
James Parkinson provided the first clinical definition of the “shaking palsy” that now bears his name in 1817. A little less than 100 years later, Frederick Lewy reported the inclusion bodies that are still used today in the neuropathological diagnosis of Parkinson’s disease (PD) [1]. While both the clinical and neuropathological definitions of PD have been updated many times since, two aspects stand out. First, our fundamental definition of PD is over 100 years old. Second, what has been notably missing in these updates is integration of the vast amounts of molecular data that now characterize much of our understanding of many other areas of medicine, including the neurodegenerative diseases. Indeed, although both Mendelian genetic approaches and genome-wide association studies have identified 40+ genetic loci that confer varying degrees of risk for PD [2], genetic data have yet to influence how we diagnose PD, how we prognosticate for patients, or how we make treatment choices. Similarly, despite large-scale efforts at the international scale to develop biochemical biomarkers for PD, none of this information has crossed over into the clinic. The lack of molecular integration in our approach to PD comes into focus when we consider other areas of medicine where, for example, it would be unimaginable to diagnose, prognosticate, or determine a therapeutic plan for most types of cancer at this point without molecular guidance, or where biochemical tests (e.g., troponin and lipid measurements) routinely affect decision-making about the care of cardiology patients. It should also be noted that revised clinical criteria to the other major neurodegenerative disease, Alzheimer’s disease (AD), include biomarker evidence of amyloid-β (Aβ) and tangle pathology, as well as neurodegeneration, (i.e., the core pathological features of the disease [3]).
Here, we pose three related questions as we consider the future of PD research and clinical care. First: Do we currently use molecular data in any of our various working definitions of PD? Second: What are the likely benefits of incorporating molecular data into our definitions of PD? Third: What are the likely barriers to a molecular view of PD? While we do not have complete answers to all of these questions and while we need to acknowledge the paucity of clinically validated molecular markers of PD, we believe that the partial answers that are emerging suggest that, for therapeutic development, clinical trial, and clinical management purposes, we should begin the process of updating our definitions of PD for the molecular age in which we live.
DO WE CURRENTLY USE MOLECULAR DATA IN ANY OF OUR VARIOUS WORKING DEFINITIONS OF PD?
We believe the answer is, increasingly, yes. That is, while clinical definitions of PD are still based on symptomatology, confirmed by the “gold standard” of neuronal α-synuclein-containing Lewy bodies at autopsy, our working definitions of PD for mechanistic research purposes as well as clinical trial entry are already molecular in many cases. In preclinical models, animals with mutations (or loss of function) in PD-associated genes such as GBA or LRRK2 are studied, and conclusions drawn from these models are not assumed to hold true for all of PD, but rather for the molecularly-defined subsets of PD patients who carry mutations in these specific genes. Beyond the preclinical research realm, Phase I and II trials of LRRK2 or GBA-targeting agents are not enrolling all-comers with PD, but rather those PD patients molecularly defined by mutations in these targets. Even the ongoing Phase III trial of inosine for urate elevation in PD, which does not target a genetically-defined subset of patients, does use biochemical measures of urate from the blood in its entry criteria, as well as for proof of target engagement [4]. Thus, many signs point to the entry of molecular information— in the form of genetic data and biochemical measures from patient-derived biofluids— into research and clinical trial criteria now. Moreover, they suggest that we need to be thinking of how this data might enter the PD clinic in the near future and building in some of the tools (e.g., genetic counseling) needed for such a transition.
2WHAT ARE THE LIKELY BENEFITS OF INCORPORATING MOLECULAR DATA INTO OUR DEFINITIONS OF PD?
For the clinic, in the near term, the benefits of incorporating molecular data into our definitions of PD are likely to revolve around the ability to offer experimental therapeutics for the patients most likely to respond. To put real-world numbers on this benefit, at the University of Pennsylvania (Penn), where one of us practices PD patient care, 23 carriers of GBA mutations have been identified over >10 years of research efforts with a robust research patient cohort; of these, fewer than 10 are eligible at this point for the GBA-targeted clinical trials that are currently enrolling. If molecular characterization were to encompass the entire clinical PD population at this site (>2000 patients), we estimate that >100 (and possibly >200) PD patients with GBA mutations would be identified, offering these patients the possibility of enrolling in a targeted clinical trial for their currently incurable disease. Indeed, September 2018 saw the launch of the Molecular Integration in Neurological Diagnosis (MIND) Initiative at Penn to do exactly this all-comers genetic characterization.
For the clinical research arena, the potential benefit is in the form of more efficient trials. As we have previously described [5, 6], within-PD heterogeneity is significant, with approximately 1/3 of PD patients in the international Parkinson’s Progression Markers Initiative research cohort (modeled on the type of cohort most likely to be used in a neuroprotective trial) showing no increase in their MDS-UPDRS-III score over 2 years of follow-up, which is the average length of a neuroprotective trial, while another 1/3 of PD patients in the same cohort show an average increase of >30 points. To discern a “signal” amidst the “noise” of this degree of within-disease heterogeneity, trials have to be quite large and, as a consequence, costly. Integration of molecular data in the selection of PD clinical trial participants may result in greater trial efficiency. Indeed, as mentioned in the preceding section, current clinical trials targeting GBA, LRRK2, or inosine already use molecular information in their enrollment criteria. To compare with AD, it has become clear that the incorporation of molecular data when recruiting patients for trials is crucial; around 30% of patients with clinical signs of AD do not have AD pathology, as determined by Aβ PET or CSF biomarker results [7], which is also supported by autopsy findings [8]. If unnoticed, such “AD mimics” would introduce considerable noise in a trial of disease-modifying drug candidates directed against Aβ.
For the preclinical research arena, we believe that loose definitions may have led to unnecessary dissension and/or red herrings in the past. For example, a model of neuronal toxicity based on rotenone treatment is unlikely to be the same biologically as a model based on brain injection of abnormal conformations of alpha-synuclein or one based on transgenic expression of a mutant form of LRRK2. Yet all three are considered PD models. Rather than focusing on the “PD” here, one might try to match the mechanisms being studied to the specific PD patients in which we have evidence of dysfunction in this exact mechanism.
3WHAT ARE THE LIKELY BARRIERS TO A MOLECULAR VIEW OF PD?
The most likely barrier to embracing a molecular view of PD follows directly from the points made previously: we may end up concluding that PD is not one disease, but many, and this will be difficult to accept. To some extent, fragmentation of the relatively monolithic view of what constitutes PD is already happening. As mentioned previously, trials of GBA substrate reduction are not enrolling PD patients who lack GBA mutations for the simple reason that PD patients who have GBA mutations are most likely to benefit from targeting this pathway. Other glimpses of this fragmentation are also apparent to the careful observer: the division between tremor-predominant PD and non-tremor-predominant PD can be found as early as Gowers’ statement in the 1880 s that “In the majority of cases (two-thirds) the tremor precedes the weakness; occasionally this relation is reversed,” which is remarkably similar to the ratio found by Jankovic in 800 patients (441:233) from the DATATOP cohort in 1990 [9]. What was lacking was a molecular “tag” for these subtypes, leading to debate in the literature of the significance of the tremor-predominant subtype if individuals might change their clinical presentation over time [10]. Indeed, this debate motivated our 2016 report that a single nucleotide polymorphism (SNP) near SNCA defines a subtype of PD characterized by clinical tremor-predominance, decreased cerebellar SNCA RNA expression, and slower motor progression [11]. Importantly, while the “tremor-predominant” clinical state may be demonstrably transient, a SNP genotype is not. Thus, while our report that the rs356182 SNCA SNP characterizes a clinically important subtype of PD may be disproved over time, the SNP status of an individual patient, unlike his or her tremor-predominant state, is not similarly debatable.
Another key barrier is the lack of well-validated biomarkers for the PD-related pathologies that constitute our current “definitions” of disease. CSF total tau, phospho-tau and Aβ42 (preferably in a ratio with the less aggregation-prone Aβ40 that may be used to normalize for inter-individual differences in Aβ production and clearance) may be used to exclude individuals with AD-related pathologies, although this is certainly complicated by the co-occurrence of AD-related pathologies in the vast majority of PD patients at autopsy [12]. CSF or serum/plasma neurofilament light concentrations may be used to differentiate PD from atypical parkinsonian disorders, as neurofilament light levels are less elevated in PD [13]. However, when it comes to positive identification of Lewy body pathology, CSF α-synuclein concentration is not specific; currently available tests do not differentiate normal from pathological (inclusion-specific or -enriched) forms of α-synuclein and show only minor changes in PD [14]. Novel tests for oligomeric or phosphorylated forms of α-synuclein show promising results but are far from validated [15, 16]. The most promising fluid-based biomarker assay for Lewy body pathology at present detects the amplified biochemical signal of α-synuclein seeds that may be Lewy body-derived in the CSF from PD patients but not in healthy controls [17, 18]. In regards to positron emission tomography, intense research is ongoing to identify Lewy body-specific ligands but there is no available method yet. Our hope, however, is that the genetic and biochemical characterization of patient-derived materials, using both targeted and unbiased approaches [19], will make it possible to subgroup clinical PD patients regarding prognosis, risk of various co-morbidities, and extent and intensity of pathological processes. We note, additionally, that while genetic markers such as GBA and LRRK2 mutation status are most mature at this point, biochemical biomarkers are important to develop as well, since they may reflect both genetic and environmental influences. Patient-derived molecular signatures—reflecting both genetic and environmental influences— should be useful when developing and evaluating novel drug candidates.
What might it mean to have many types of PD, rather than our current conception of PD as one disease affecting a million people worldwide? Certainly, growing pains are expected. Indeed, we don’t yet know what to make of the fact that among the 55 LRRK2 mutation carriers with neuropathology reported to date, a substantial fraction appear not to have the Lewy body pathology that defines PD (reviewed in [20])! It does seem unsatisfactory to then conclude that these individuals do not have PD.
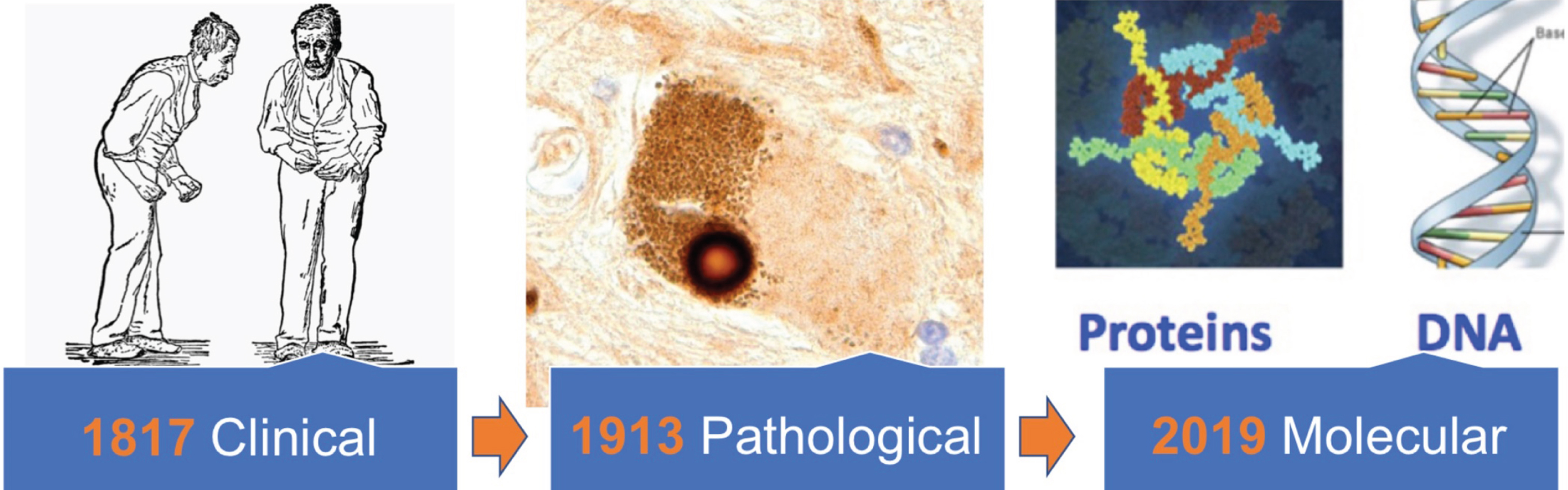
We suspect, however, that the potential benefits far outweigh these growing pains. Indeed, we predict that for PD, as for many forms of cancer over the previous decades, molecular definition (see Fig. 1) could be truly transformative.
Fig.1
Clinical definitions of Parkinson’s disease (represented here by the 1886 William Gowers illustration) are over 200 years old, while the Lewy body that still forms the basis of our pathological definitions was first described over 100 years ago. We argue that a molecular “update” to our disease definitions, based on characterization of DNA and proteins obtained from the biofluids of individual patients, is needed.
CONFLICTS OF INTEREST
HZ has served at scientific advisory boards for Eli Lilly, Roche Diagnostics, Wave, CogRx and Samumed, has received travel support from Teva and is one of the founders of Brain Biomarker Solutions in Gothenburg AB, a GU Ventures-based platform company at the University of Gothenburg.
ACP serves on the Executive Scientific Advisory Board of the Michael J. Fox Foundation. She has a scientific collaboration with 23andMe.
ACKNOWLEDGMENTS
Work in the authors’ laboratories is supported by grants from the NIH (RO1 NS082265, P30 AG010124, UO1 NS097056 to ACP), the Benaroya Fund (to ACP), the Penn Center for Precision Medicine (to ACP), a Biomarkers Across Neurodegenerative Disease Award (ACP), the Swedish Research Council (HZ), the European Research Council (HZ), the Knut and Alice Wallenberg Foundation (HZ) and Swedish State Support for Clinical Research. HZ is a Wallenberg Academy Fellow. ACP is the Parker Family Associate Professor of Neurology.
REFERENCES
[1] | Przedborski S ((2017) ) The two-century journey of Parkinson disease research. Nat Rev Neurosci 18: , 251–259. |
[2] | Chang D , Nalls MA , Hallgrímsdóttir IB , Hunkapiller J , van der Brug M , Cai F , Kerchner GA , Ayalon G , Bingol B , Sheng M , Hinds D , Behrens TW , Singleton AB , Bhangale TR , Graham RR ((2017) ) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49: , 1511–1516. |
[3] | Dubois B , Feldman HH , Jacova C , Hampel H , Molinuevo JL , Blennow K , DeKosky ST , Gauthier S , Selkoe D , Bateman R , Cappa S , Crutch S , Engelborghs S , Frisoni GB , Fox NC , Galasko D , Habert M-OO , Jicha GA , Nordberg A , Pasquier F , Rabinovici G , Robert P , Rowe C , Salloway S , Sarazin M , Epelbaum S , de Souza LC , Vellas B , Visser PJ , Schneider L , Stern Y , Scheltens P , Cummings JL ((2014) ) Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol 13: , 614–629. |
[4] | Schwarzschild MA , Ascherio A , Beal MF , Cudkowicz ME , Curhan GC , Hare JM , Hooper DC , Kieburtz KD , Macklin EA , Oakes D , Rudolph A , Shoulson I , Tennis MK , Espay AJ , Gartner M , Hung A , Bwala G , Lenehan R , Encarnacion E , Ainslie M , Castillo R , Togasaki D , Barles G , Friedman JH , Niles L , Carter JH , Murray M , Goetz CG , Jaglin J , Ahmed A , Russell DS , Cotto C , Goudreau JL , Russell D , Parashos SA , Ede P , Saint-Hilaire MH , Thomas C-A , James R , Stacy MA , Johnson J , Gauger L , Antonelle de Marcaida J , Thurlow S , Isaacson SH , Carvajal L , Rao J , Cook M , Hope-Porche C , McClurg L , Grasso DL , Logan R , Orme C , Ross T , Brocht AFD , Constantinescu R , Sharma S , Venuto C , Weber J , Eaton K ((2014) ) Inosine to increase serum and cerebrospinal fluid urate in Parkinson disease: A randomized clinical trial. JAMA Neurol 71: , 141–150. |
[5] | Tropea TF , Chen-Plotkin AS ((2017) ) Unlocking the mystery of biomarkers: A brief introduction, challenges and opportunities in Parkinson disease. Parkinsonism Relat Disord 46: , S15–S18. |
[6] | Chen-Plotkin AS , Albin R , Alcalay R , Babcock D , Bajaj V , Bowman D , Buko A , Cedarbaum J , Chelsky D , Cookson MR , Dawson TM , Dewey R , Foroud T , Frasier M , German D , Gwinn K , Huang X , Kopil C , Kremer T , Lasch S , Marek K , Marto JA , Merchant K , Mollenhauer B , Naito A , Potashkin J , Reimer A , Rosenthal LS , Saunders-Pullman R , Scherzer CR , Sherer T , Singleton A , Sutherland M , Thiele I , van der Brug M , Van Keuren-Jensen K , Vaillancourt D , Walt D , West A , Zhang J ((2018) ) Finding useful biomarkers for Parkinson’s disease. Sci Transl Med 10: , pii:eaam6003. |
[7] | Ashton NJ , Schöll M , Heurling K , Gkanatsiou E , Portelius E , Höglund K , Brinkmalm G , Hye A , Blennow K , Zetterberg H ((2018) ) Update on biomarkers for amyloid pathology in Alzheimer’s disease. Biomark Med 12: , 799–812. |
[8] | Jellinger KA , Attems J ((2015) ) Challenges of multimorbidity of the aging brain A critical udate. J Neural Transm 122: , 505–21. |
[9] | Jankovic J , McDermott M , Carter J , Gauthier S , Goetz C , Golbe L , Huber S , Koller W , Olanow C , Shoulson I , Stern M , Tanner C , Weiner W ((1990) ) Variable expression of Parkinson’s disease: A base-line analysis of the DAT ATOP cohort. Neurology 40: , 1529–1534. |
[10] | Alves G , Larsen JP , Emre M , Wentzel-Larsen T , Aarsland D ((2006) ) Changes in motor subtype and risk for incident dementia in Parkinson’s disease. Mov Disord 21: , 1123–1130. |
[11] | Cooper CA , Jain N , Gallagher MD , Weintraub D , Xie SX , Berlyand Y , Espay AJ , Quinn J , Edwards KL , Montine T , Van Deerlin VM V , Trojanowski J , Zabetian CP , Chen-Plotkin AS ((2017) ) Common variant rs356182 near SNCA defines a Parkinson’s disease endophenotype. Ann Clin Transl Neurol 4: , 15–25. |
[12] | Robinson JL , Lee EB , Xie SX , Rennert L , Suh E , Bredenberg C , Caswell C , Van Deerlin VM , Yan N , Yousef A , Hurtig HI , Siderowf A , Grossman M , McMillan CT , Miller B , Duda JE , Irwin DJ , Wolk D , Elman L , McCluskey L , Chen-Plotkin A , Weintraub D , Arnold SE , Brettschneider J , Lee VM-Y , Trojanowski JQ ((2018) ) Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain 141: , 2181–2193. |
[13] | Zetterberg H ((2016) ) Neurofilament light: A dynamic cross-disease fluid biomarker for neurodegeneration. Neuron 91: , 1–3. |
[14] | Mollenhauer B , DuBois BF , Drake D , Duong J , Blennow K , El-Agnaf O , Shaw LM , Masucci J , Taylor P , Umek RM , Dunty IM , Smith CL , Stoops E , Vanderstichele H , Schmid AW , Moniatte M , Zhang J , Kruse N , Lashuel HA , Teunissen C , Schubert T , Dave KD , Hutten SJ , Zetterberg H ((2018) ) Antibody-based methods for the measurement of alpha-synuclein concentration in human cerebrospinal fluid - method comparison and round robin study. J Neurochem. doi: 10.1111/jnc.14569. |
[15] | Kang JH , Mollenhauer B , Coffey CS , Toledo JB , Weintraub D , Galasko DR , Irwin DJ , Van Deerlin V , Chen-Plotkin AS , Caspell-Garcia C , Waligórska T , Taylor P , Shah N , Pan S , Zero P , Frasier M , Marek K , Kieburtz K , Jennings D , Tanner CM , Simuni T , Singleton A , Toga AW , Chowdhury S , Trojanowski JQ , Shaw LM ((2016) ) CSF biomarkers associated with disease heterogeneity in early Parkinson’s disease: The Parkinson’s Progression Markers Initiative study. Acta Neuropathol 131: , 935–949. |
[16] | Parnetti L , Cicognola C , Eusebi P , Chiasserini D ((2016) ) Value of cerebrospinal fluid α-synuclein species as biomarker in Parkinson’s diagnosis and prognosis. Biomark Med 10: , 35–49. |
[17] | Fairfoul G , McGuire LI , Pal S , Ironside JW , Neumann J , Christie S , Joachim C , Esiri M , Evetts SG , Rolinski M , Baig F , Ruffmann C , Wade-Martins R , Hu MTM , Parkkinen L , Green AJE ((2016) ) Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol 3: , 812–818. |
[18] | Shahnawaz M , Tokuda T , Waraga M , Mendez N , Ishii R , Trenkwalder C , Mollenhauer B , Soto C ((2017) ) Development of a biochemical diagnosis of Parkinson disease by detection of α-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol 74: , 163–172. |
[19] | Chen-Plotkin AS ((2014) ) Unbiased approaches to biomarker discovery in neurodegenerative diseases. Neuron 84: , 594–607. |
[20] | Schneider SA , Alcalay RN ((2017) ) Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov Disord 32: , 1504–1523. |


