
Repairing the Brain: Gene Therapy
Abstract
In vivo gene therapy for neurodegenerative disorders has turned out to be a formidable challenge. It is a field not much older than twenty years, but we were many who would have predicted a much easier path towards the clinic using this treatment modality. For Parkinson’s disease patients, this has meant a frustrating wait, seeing many promising therapies being forgotten after a few pre-clinical proof-of-concept studies. The reasons for this are both scientific and economical. However, this is slowly but surely changing and over the next two decades we will see a very exciting development in this field. In a foreseeable future, gene therapy will be a very natural component of many clinical therapies, not least in Parkinson’s disease.
THE RISE AND FALL OF THE FIRST GENERATION OF GENE THERAPY IN PARKINSON’S DISEASE
Since the dawn of in vivo gene transfer, the treatment of Parkinson’s disease (PD) has always been listed as one of its prime targets [1, 2]. Many attempts have been made, each one with its own twist. However, in retrospect, with very few exemptions, they can be clustered into two groups; neuroprotective/restorative therapies [3–5] and symptomatic therapies modulating the dopamine system/basal ganglia [6–10]. It should be noted that all implemented therapies have had symptomatic relief as the primary endpoint in Phase II and not neuroprotection even when that is theoretically possible. There are many good reasons for this, not least that the disease heterogeneity and slow progression would require clinical trials an order of magnitude larger than what is feasible with gene therapy to prove disease modification.
The scientific rational for the implemented approaches is outside the scope of this opinion piece but is very well covered in previous reviews [2, 11–13]. In the enthusiasm around early gene therapy, multiple companies were formed including Neurologix [7], Ceregene [4, 5], Oxford Biomedica [14], and Voyager Therapeutics [6], all with the focus on gene therapy in PD. Now a decade later, the results are less than encouraging. The two first companies failed to show convincing benefits over current therapeutic options to remain viable and have disappeared. Oxford Biomedica have had signs of efficacy intermingled with aggravated L-DOPA induced dyskinesias (LIDs) [14, 15] and have gone back to the drawing board with the aim to refine the vector [16]. The latest news is that this approach is licensed out to Axovant and thus its future remains unpredictable. Voyager therapeutics has been very successful in attracting funding and has expanded its portfolio significantly. However, the AADC pro-drug approach remains in their active pipeline and is planned to enter a Phase 2 double-blind placebo-controlled trial. It is with this trial we will see if there is adequate therapeutic potential of this approach to warrant further development. The published results from the phase 1 trials have been puzzling in that they show greater therapeutic efficacy OFF compared to ON L-DOPA [6, 17] which does not fit well with the current understanding of the proposed action of this therapy which is aimed to potentiate the conversion of peripherally delivered L-DOPA into DA.
The Ceregene sponsored Neurturin trials failed to meet the primary endpoint of symptomatic relief in Phase II. It has been broadly debated why this happened [12, 18–20], was the degeneration of the DA system too severe in the selected patient population? Was the dosage too low or the AAV serotype suboptimal? Is it so that we do not fully understand the mechanism of action of neurotrophic factors in PD and thus targeted a neuroprotective strategy more suitable for disease models of PD than the real thing? Most likely it is a combination of all factors. The ongoing AAV-mediated GDNF trial conducted at the NINDS [21] will hopefully shine some light on the feasibility of the approach.
Prediction
The current generation of clinical gene therapy will not be the prevailing therapy for PD in 20 years. They will be replaced by refined alternatives described below. It is possible that the heterogeneity of PD will mean that the cause will not be identified in every case and that preventive measures cannot be taken for some patients. Those patients will hopefully be served well by restorative measures described below. However, the foundation which these pioneering gene therapy trials in PD have provided with regards to safety data, viral vector production and clinical trial design should not be underestimated. It is on this foundation all approaches described below will stand.
THE MERGER OF CELL AND GENE THERAPY
In 20 years, the front-line pre-clinical research for PD will have moved beyond symptomatic relief and circuit reconstruction and will focus on curative prevention of disease. This will however not mean that symptomatic therapies and circuit repair will become obsolete. On the contrary, it is expected that such clinical therapies will peak in complexity and potential about that time. Cell repair will be covered in detail in elsewhere in this issue, but in essence it aims to reconstruct the dopamine neurons in the Parkinsonian brain, originally residing in the substantia nigra. Cell sources differ significantly between the approaches from fetal ventral mesencephalon to replenishable sources such as embryonic stem cells and induced pluripotent stem (IPS) cells.
If nothing exceptional has occurred between the time of writing of this review and publication, the first PD patient will have received an IPS-based DA neuroblast transplant in September 2018 [22]. This is the result of a national fast track approach in Japan and everyone in the field does hope that the outcome will be positive. Otherwise this trial could again put an unfortunate delay on the development of brain repair therapies [23].
Until the day when we can direct and regulate the circuit integration of de novo neurons, extrinsic regulation of their activity will be an attractive option [24–26]. The Japanese trial as well as those planned elsewhere using human embryonic stem cells do all pursue an ectopic graft placement, i.e., in the putamen instead of the substantia nigra pars compacta where the somata of the nigral neurons normally reside. This is currently a necessity as the post-synaptic dopamine stimulation is required in the putamen for the recovery of motor impairment in PD [27]. Transplanted neuroblasts do not yet show efficient axonal outgrowth from the substantia nigra (SN) to the putamen [28], at least not efficient enough to cover the distance in the human brain. Due to this fact, the ectopic DA transplant does not readily have access to the afferent regulation of the SN and is subject to the highly inhibitory environment of the putamen. We have found that this results in a state where the transplanted cells don’t show their full therapeutic potential [24]. Fortunately, this can be remedied using gene therapy. Using chemogenetic receptors expressed only in the transplanted DA neurons, we were able to independently introduce an excitatory and an inhibitory regulation of the transplant. We and others have found that the behavioural recovery can be significantly increased with the excitatory stimulation of the DA neurons while it is diminished if the DA neurons is silenced [24–26]. This provides a powerful bidirectional paradigm for clinical regulation of cell transplants, regardless of cell source, which will not only provide an attractive safety mechanism for handling graft-induced dyskinesias [29] but one which could also provide significant benefits for the patient.
Prediction
Gene transfer will be a key component of clinical cell transplantation within the next 20 years. All transplanted neurons will contain genetic alterations in one way or the other. Chemogenetics will be one such alteration providing a significant potentiation of the transplanted neurons but will not be the only change. We will see genome editing repairing disease-causing mutation e.g., in LRRK2 enabling personalized approaches but we will also see genetically engineered cells evading an immune response and thus allowing broadly utilized cell sources.
TAILORED AAV CAPSIDS HOMING FROM THE PERIPHERY TO THE CNS
Gene therapy for PD has been pursued using almost all recombinant viral vehicles developed, including Retrovirus [30], Adenovirus [31], Herpex simplex virus [32], Lentivirus [14] and of course the Adeno-associated virus (AAV) [5–8]. The AAV vector has such distinct advantages over the other vectors with regards to efficacy and low immunogenicity in the CNS that it has become the de facto standard vehicle for CNS gene transfer despite its intrinsic limitation in the size of packaged genetic material [33]. One key property of the AAV which has been especially attractive for CNS delivery has been the broad diversity of naturally available AAV serotypes and their varied tropisms [34, 35]. Furthermore, the AAV capsid has turned out to be extremely malleable. Parts of the capsid can be exchanged between different serotypes to generate new variants with different functions [36] and random peptides can be displayed on the capsid surface to result in even bigger changes in vector function [37]. We can even utilize our knowledge of other virus types to engage in molecular mimicry where peptides from other viruses (or other proteins) are used to transfer desirable functions onto the clinically suitable AAV such as the retrograde infectivity of neurons from the terminal [38]. This development has been greatly facilitated by recent advances in parallel gene synthesis, deep sequencing and computational modelling [39] where novel capsid variants can now be systematically screened millions at the time also in humanized modelling systems or non-human primates. In the coming decade we will see an explosion in synthetic capsid variants with tailored properties for each individual therapy.
Prediction
Gene therapy in PD will in 20 years be conducted solely using tailored synthetic vectors. Intraparenchymal injections will be replaced with intrathecal or systemic injections. Each therapy will depend on its own tailored vector with optimized function such as transport over the brood brain barrier or homing to the targeted neuronal population based on either connectivity, gene expression profile or disease state.
BEYOND BRAIN REPAIR: PREVENTION OF DISEASE THROUGH GENE REPAIR
In 20 years, we will have moved away from the focus on monogenetic familiar forms of PD as the focus for genome editing and repair and all emphasis will be on the idiopathic forms [40]. This is not because of financial or technical constraints but it will be because all monogenetic forms will have seen novel therapies become available in the form of gene repair.
Gene repair has gone through multiple steps towards reaching that point, but many remain before we have suitable and safe therapies. Gene disruption techniques such as the CRISPR/Cas9-mediated NHEJ [41–43] are filling our journals with very interesting proof-of-concept approaches for therapy based on gene removal [44, 45]. At the time of writing, the first rouge application of CRISPR/Cas9 applied to In vitro fertilization appears to have resulted in the first birth of a genome edited child in China [46]. In this case the target was the CCR5 receptor for very weak scientific reasons and with even weaker regulatory oversight. While NHEJ will no doubt find suitable clinical applications over the next 10 years, the real revolution for in vivo genome editing comes when we can perform efficient and safe gene repair in situ.
This is a field that is seeing active development as well. In its simplest form, gene replacement can be conducted without the use of genome editing. An example can be seen in a recent paper on the development of AAV-mediated therapy for autosomal dominant retinitis pigmentosa. With this approach, the disease-causing gene (mutant rhodopsin) is silence using short hairpin (sh) RNA interference [47]. In the same AAV vector an intact rhodopsin gene (with silent mutations allowing escape from RNA interference) is expressed from a short opsin promoter. Such approach has shown promise in canine models and could become suitable for clinical translation. This approach may also be suitable for gene replacement in PD. However, the challenge there will be the regulation of gene dosage. While the jury is still out on how detrimental it is for the adult dopamine neurons to have α-Synuclein totally removed (e.g., using shRNA) [48, 49], it is very clear that an increase of any form of α-Synuclein is not very desirable. Thus, going forward, gene dosage for PD gene repair will be a necessity. With α-Synuclein, multiple approaches are being studied to reduce α-Synuclein load either by targeted methylation of the locus [50] or exchanging regulatory sequences using knock-in [51]. Knock-in approaches are very attractive for gene repair and gene dosage. Such techniques will allow the chromatin to control tightly the gene and thus ensure perfectly regulated gene expression in every cell regardless of viral titer and local concentration gradients.
Knock-in approaches in the CNS mediated by genome editing have now started to see proof-of concept with the HITI technique [52, 53]. It is using the fact that AAV vectors are very attractive for template delivery and that a double-strand break in the genome opens up for insertion of genetic material [54]. However, the “passive” nature of this insertion together with the requirement of a double-strand break (which will also introduce NHEJ) makes this approach intrinsically ineffective and not very suitable for clinical translation, at least not in PD. It is also not yet shown to be capable of repairing a gene but has only been used to insert a coding sequencing flanking the endogenous allele.
Potent editing approaches also require potent means of regulation. Gene editing ex vivo and even in some peripheral organs in vivo can be achieved using transient expression of nucleases using either mRNA or protein delivery [55, 56]. This is not yet possible for the post mitotic cells of the CNS and thus we still have to depend on constitutive transgene delivery for the foreseeable future. However, attempts have been made to make the Cas9 protein Self-inactivating after viral delivery. One example is the KamiCas9 approach presented by Deglon and collaborators [45]. This approach ensures that the Cas9 is itself inactivated as soon as the cell starts to produce all components required for the NHEJ. Through the use of PolIII promoters of different strength, the authors could drive the equilibrium so that most cells edit the target gene before the Cas9 is disrupted. The first application of this approach was gene editing of a virally derived huntingtin gene in the mouse striatum.
Base editing approaches show much greater therapeutic potential but do have other challenges. In base editing, no double strand break is made and there is no requirement for a template DNA strand to be used for insertion and repair. Instead, a cytidine or adenine deaminase enzyme chemically alters a nucleotide on one of the DNA strands in situ [56, 57]. Through a DNA repair process, the other strand will be modified to adhere to the Watson-Crick base pairing. The deaminase will have to be accurately and efficiently brought to the correct nucleotide to allow for the editing to occur but also to restrict the editing of other bases in the genome [58]. This is a formidable challenge, but fusion proteins with the Cas9 protein and tailored sgRNA have shown initial promise [57, 59, 60]. Further restrictions of its applicability relate to the available enzymes only permitting A to G or C to U mutation and the available Cas9 proteins restricting the possible positions of Cas9 binding in the targeted genes. However, the ongoing and historically successful mutation efforts will very likely result in a battery of base editing tools [61] which will allow for the editing of any base at any position of the gene and this will happen well within the next 20 years.
Prediction
The tool of the future for monogenetic familiar forms of PD will be a synthetically designed viral vector capsid with the capacity to transport the repair machinery from the blood into the CNS delivering the cargo throughout the body and the CNS. This will be performed in the early neonatal stage and will be seen as severe as a flu shot. The cargo of this viral vector will be a base editing enzyme which will accurately edit the erroneous base and will then self-delete when the mission is accomplished.
NEURONAL REJUVENATION
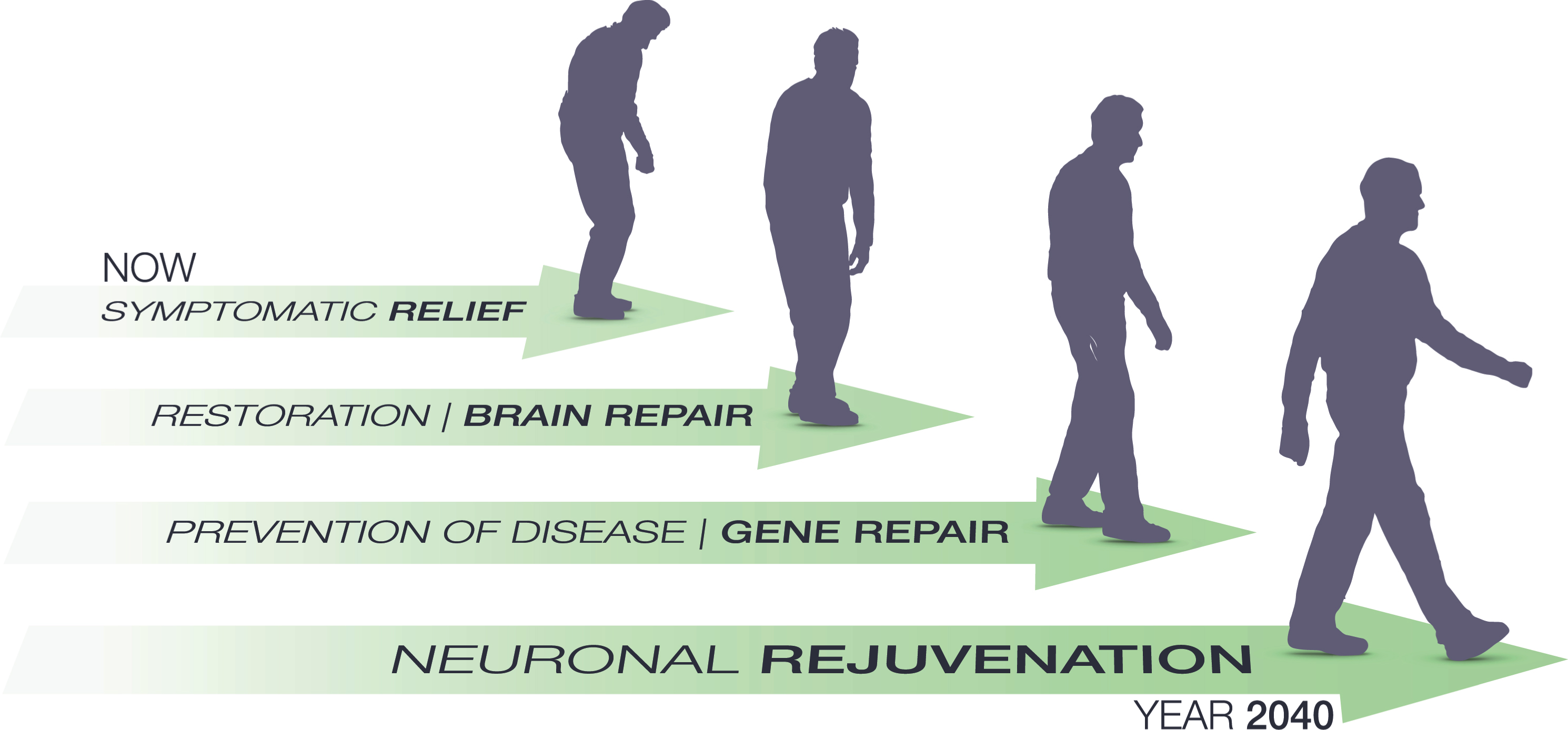
Fig.1
Path and milestones predicted for gene therapy in Parkinson’s disease over the next 20 years. Clinical gene therapy is currently aiming for symptomatic relief as its primary end point. This will gradually shift to brain repair when it is merged will cell therapy or in vivo re-programming for circuit repair. Over the next decade we hope for a significant increase in mechanistic insights into the idiopathic Parkinson’s disease which will then pave the way for gene therapy to move into prevention of disease. As a final step we will see these measures being refined and validated to warrant their applications in normal healthy aging. Through prevention and reversal of detrimental processes in the brain such as DNA damage, protein accumulation and oxidative stress we may see the first attempts towards brain rejuvenation using gene therapy before the year 2040. It should be noted that most patients will receive a combination of these therapies and the most successful therapies will most likely target two or more of the modalities, e.g., both symptomatic and restorative.
Aging is the principal risk factor for PD [62]. It is also a major contributor to most other neurodegenerative disorders. In addition, even for those of us fortunate enough to evade a clinical diagnosis, aging will with time endow us with subclinical manifestations of many of the hallmarks of PD such as rigidity, bradykinesia, postural instability and tremor. To add insult to injury we will also see our cognitive performance gradually decline. If we could choose, most of us would like to avoid these changes altogether and remain neurologically top performing until the inevitable end. In this endeavour gene therapy will play an important role. Neuronal aging and its prevention or reversal is still a largely unexplored field [63]. However, we can learn a lot from the “accelerated aging” seen in PD but also from the healthy aging brain [62]. The genome of our neurons is continuously attacked through our lifespan inducing random mutation throughout [64]. Most mutations are corrected in the young nerve cells using the base excision repair (BER) pathway but with time the uncorrected mutations accumulate and the BER pathway becomes less and less active. These changes are thought to contribute negatively to the aging, not only of the brain. Another cellular event which has attracted significant interest in the neuronal aging field is the Nicotinamide adenine dinucleotide (NAD+) biosynthesis [65]. In the pathophysiology of aging, NAD+ is both linked to the afore mentioned DNA repair through its requirement in the poly-ADP-ribose polymerase (PARP) function and in histone methylation through Sirtuins. Reduction in NAD+ levels are observed with age in most tissues and thus has detrimental effects on both DNA repair and Sirtuin function. Interestingly overexpression of the brain-specific SIRT 1 reverts many age-related changes, prolongs lifespan [66] and protects against neurodegeneration in AD [67]. Thus, the NAD+ biosynthesis, e.g., through the Namnpt gene is an interesting target for neuronal rejuvenation.
Neurons have to survive many challenges throughout a lifetime. Due to the lack of self-renewal, every dysfunctional protein or organelle may become a treat to survival of the neuron and gradually they accumulate [68]. Therefore, the self-eating or macroautophagy is a crucial housekeeping event, not least in dopamineric neurons [69, 70]. Macroautophagy is impaired with aging and with its decrease comes protein aggregation and neurodegeneration [70, 71]. Controlling macroautophagy may thus also be a potent component in neuronal rejuvenation. However, it is also so that more is not always better in this case [72] so much more knowledge is required.
Prediction
In 20 years, gene therapy will have become so mainstream and safe that it will be used to also counteract the detrimental effects on the dopamine system of normal aging. Through potentiation of DNA repair, careful chromatin remodeling, stimulation of neurogenesis, plasticity and macroautophagy, we will be able to maintain a much better brain health throughout the lifespan. This will not only protect again clinically manifested neurodegenerative disorders such as PD and AD but will allow all of us remain neurologically young.
FINAL WORDS
Gene therapy will no doubt see many challenges in the coming decades. With the increasing complexity of the approaches comes significant safety and ethical concerns. It is my belief however that these hurdles are surmountable. Recent unauthorized shortcuts to the clinic with genome editing in China must not become the norm. We all need to decide on an ethical development route which protects and benefits the patients without stifling innovation. If this balancing act is executed successfully, the future for clinical gene therapy in PD will be very bright even within as short time frame as 20 years
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Bjorklund A , Bjorklund T , Kirik D ((2009) ) Gene therapy for dopamine replacement in Parkinson’s disease. Sci Transl Med 1: , 2ps2. |
[2] | Bjorklund T , Kirik D ((2009) ) Scientific rationale for the development of gene therapy strategies for Parkinson’s disease. Biochim Biophys Acta 1792: , 703–713. |
[3] | Kordower JH , Emborg ME , Bloch J , Ma SY , Chu Y , Leventhal L , McBride J , Chen EY , Palfi S , Roitberg BZ , Brown WD , Holden JE , Pyzalski R , Taylor MD , Carvey P , Ling Z , Trono D , Hantraye P , Deglon N , Aebischer P ((2000) ) Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson’s disease. Science 290: , 767–773. |
[4] | Marks WJ, Jr , Baumann TL , Bartus RT ((2016) ) Longterm safety of patients with Parkinson’s disease receiving rAAV2-neurturin (CERE-120) gene transfer. Hum Gene Ther 27: , 522–527. |
[5] | Marks WJ Jr , Ostrem JL , Verhagen L , Starr PA , Larson PS , Bakay RA , Taylor R , Cahn-Weiner DA , Stoessl AJ , Olanow CW , Bartus RT ((2008) ) Safety and tolerability of intra- putaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: An open-label, phase I trial. Lancet Neurol 7: , 400–408. |
[6] | Christine CW , Starr PA , Larson PS , Eberling JL , Jagust WJ , Hawkins RA , VanBrocklin HF , Wright JF , Bankiewicz KS , Aminoff MJ ((2009) ) Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 73: , 1662–1669. |
[7] | Kaplitt MG , Feigin A , Tang C , Fitzsimons HL , Mattis P , Lawlor PA , Bland RJ , Young D , Strybing K , Eidelberg D , During MJ ((2007) ) Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: An open label, phase I trial. Lancet 369: , 2097–2105. |
[8] | Bjorklund T , Carlsson T , Cederfjall EA , Carta M , Kirik D ((2010) ) Optimized adeno-associated viral vector-mediated striatal DOPA delivery restores sensorimotor function and prevents dyskinesias in a model of advanced Parkinson’s disease. Brain 133: , 496–511. |
[9] | Cederfjall E , Sahin G , Kirik D , Bjorklund T ((2012) ) Design of a single AAV vector for coexpression of TH and GCH1 to establish continuous DOPA synthesis in a rat model of Parkinson’s disease. Mol Ther 20: , 1315–1326. |
[10] | Shen Y , Muramatsu SI , Ikeguchi K , Fujimoto KI , Fan DS , Ogawa M , Mizukami H , Urabe M , Kume A , Nagatsu I , Urano F , Suzuki T , Ichinose H , Nagatsu T , Monahan J , Nakano I , Ozawa K ((2000) ) Triple transduction with adeno-associated virus vectors expressing tyrosine hydroxylase, aromatic-L-amino-acid decarboxylase, and GTP cyclohy- drolase I for gene therapy of Parkinson’s disease. Hum Gene Ther 11: , 1509–1519. |
[11] | Fiandaca MS , Bankiewicz KS ((2010) ) Gene therapy for Parkinson’s disease: From non-human primates to humans. Curr Opin Mol Ther 12: , 519–529. |
[12] | Olanow CW , Bartus RT , Volpicelli-Daley LA , Kordower JH ((2015) ) Trophic factors for Parkinson’s disease: To live or let die. Mov Disord 30: , 1715–1724. |
[13] | Bjorklund T , Kordower JH ((2010) ) Gene therapy for Parkinson’s disease. Mov Disord 25: (Suppl 1), S161–S173. |
[14] | Palfi S , Gurruchaga JM , Ralph GS , Lepetit H , Lavisse S , Buttery PC , Watts C , Miskin J , Kelleher M , Deeley S , Iwamuro H , Lefaucheur JP , Thiriez C , Fenelon G , Lucas C , Brugieres P , Gabriel I , Abhay K , Drouot X , Tani N , Kas A , Ghaleh B , Le Corvoisier P , Dolphin P , Breen DP , Mason S , Guzman NV , Mazarakis ND , Radcliffe PA , Harrop R , Kingsman SM , Rascol O , Naylor S , Barker RA , Hantraye P , Remy P , Cesaro P , Mitrophanous KA ((2014) ) Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: A dose escalation, open-label, phase 1/2 trial. Lancet 383: , 1138–1146. |
[15] | Palfi S , Gurruchaga JM , Lepetit H , Howard K , Ralph GS , Mason S , Gouello G , Domenech P , Buttery PC , Hantraye P , Tuckwell NJ , Barker RA , Mitrophanous KA ((2018) ) Long- term follow-up of a Phase I/II study of ProSavin, a lentiviral vector gene therapy for Parkinson’s disease. Hum Gene Ther ClinDev 29: , 148–155. |
[16] | Stewart HJ , Ralph GS , Fong-Wong L , Strickland I , McCloskey L , Barnes L , Blount I , Wells O , Truran CJ , Kingsman AJ , Palfi S , Mitrophanous KA ((2016) ) Optimizing transgene configuration and protein fusions to maximize dopamine production for the gene therapy of Parkinson’s disease. Hum Gene Ther Clin Dev 27: , 100–110. |
[17] | Mittermeyer G , Christine CW , Rosenbluth KH , Baker SL , Starr P , Larson P , Kaplan PL , Forsayeth J , Aminoff MJ , Bankiewicz KS ((2012) ) Long-term evaluation of a phase 1 study of AADC gene therapy for Parkinson’s disease. Hum Gene Ther 23: ,377–381. |
[18] | Barker RA ((2015) ) Neurturin and Parkinson’s disease - the latest chapter. ACNR 15: , 20. |
[19] | Sullivan AM , O'Keeffe GW ((2016) ) Neurotrophic factor therapy for Parkinson’s disease: Past, present and future. Neural Regen Res 11: , 205–207. |
[20] | Kordower JH ((2015) ) Gene therapy for Parkinson’s disease: Still a hot topic? Neuropsychopharmacology 40: , 255–256. |
[21] | https://clinicaltrials.gov/ct2/show/study/NCT01621581 AAV2-GDNF for Advanced Parkinson’s Disease. |
[22] | Kikuchi T , Morizane A , Doi D , Magotani H , Onoe H , Hayashi T , Mizuma H , Takara S , Takahashi R , Inoue H , Morita S , Yamamoto M , Okita K , Nakagawa M , Parmar M , Takahashi J ((2017) ) Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 548: , 592–596. |
[23] | Barker RA , Barrett J , Mason SL , Bjorklund A ((2013) ) Fetal dopaminergic transplantation trials and the future of neural grafting in Parkinson’s disease. Lancet Neurol 12: , 84–91. |
[24] | Aldrin-Kirk P , Heuer A , Wang G , Mattsson B , Lundblad M , Parmar M , Bjorklund T ((2016) ) DREADD modulation of transplanted DA neurons reveals a novel parkinsonian dyskinesia mechanism mediated by the serotonin 5-HT6 receptor. Neuron 90: , 955–968. |
[25] | Steinbeck JA , Choi SJ , Mrejeru A , Ganat Y , Deisseroth K , Sulzer D , Mosharov EV , Studer L ((2015) ) Optogenetics enables functional analysis of human embryonic stem cell-derived grafts in a Parkinson’s disease model. Nat Biotechnol 33: , 204–209. |
[26] | Chen Y , Xiong M , Dong Y , Haberman A , Cao J , Liu H , Zhou W , Zhang SC ((2016) ) Chemical control of grafted human PSC-derived neurons in a mouse model of Parkinson’s disease. Cell Stem Cell 18: , 817–826. |
[27] | Winkler C , Kirik D , Bjorklund A , Dunnett SB ((2000) ) Transplantation in the rat model of Parkinson’s disease: Ectopic versus homotopic graft placement. Prog Brain Res 127: , 233–265. |
[28] | Grealish S , Diguet E , Kirkeby A , Mattsson B , Heuer A , Bramoulle Y , Van Camp N , Perrier AL , Hantraye P , Bjorklund A , Parmar M ((2014) ) Human ESC-derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson’s disease. Cell Stem Cell 15: , 653–665. |
[29] | Politis M , Wu K , Loane C , Quinn NP , Brooks DJ , Rehncrona S , Bjorklund A , Lindvall O , Piccini P ((2010) ) Serotonergic neurons mediate dyskinesia side effects in Parkinson’s patients with neural transplants. Sci Transl Med 2: , 38ra46. |
[30] | Bencsics C , Wachtel SR , Milstien S , Hatakeyama K , Becker JB , Kang UJ ((1996) ) Double transduction with GTP cyclohydrolase I and tyrosine hydroxylase is necessary for spontaneous synthesis of L-DOPA by primary fibroblasts. J Neurosci 16: , 4449–4456. |
[31] | Choi-Lundberg DL , Lin Q , Chang YN , Chiang YL , Hay CM , Mohajeri H , Davidson BL , Bohn MC ((1997) ) Dopaminergic neurons protected from degeneration by GDNF gene therapy. Science 275: , 838–841. |
[32] | During MJ , Naegele JR , O'Malley KL , Geller AI ((1994) ) Long-term behavioral recovery in parkinsonian rats by an HSV vector expressing tyrosine hydroxylase. Science 266: , 1399–1403. |
[33] | Wu Z , Yang H , Colosi P ((2010) ) Effect of genome size on AAV vector packaging. Mol Ther 18: , 80–86. |
[34] | Dodiya HB , Bjorklund T , Stansell J , 3rd, Mandel RJ , Kirik D , Kordower JH ((2010) ) Differential transduction following basal ganglia administration of distinct pseudotyped AAV capsid serotypes in nonhuman primates. Mol Ther 18: , 579–587. |
[35] | Taymans JM , Vandenberghe LH , Haute CV , Thiry I , Deroose CM , Mortelmans L , Wilson JM , Debyser Z , Baekelandt V ((2007) ) Comparative analysis of adeno-associated viral vector serotypes 1, 2, 5, 7, and 8 in mouse brain. Hum Gene Ther 18: , 195–206. |
[36] | Grimm D , Lee JS , Wang L , Desai T , Akache B , Storm TA , Kay MA ((2008) ) In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol 82: , 5887–5911. |
[37] | Deverman BE , Pravdo PL , Simpson BP , Kumar SR , Chan KY , Banerjee A , Wu WL , Yang B , Huber N , Pasca SP , Gradinaru V ((2016) ) Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol 34: , 204–209. |
[38] | Davidsson M , Wang G , Aldrin-Kirk P , Cardoso T , Nolbrant S , Hartnor M , Parmar M , Bjorklund T ((2018) ) Barcoded Rational AAV Vector Evolution enables systematic in vivo mapping of peptide binding motifs. bioRxiv. doi: 10.1101/335372 |
[39] | Davidsson M , Diaz-Fernandez P , Schwich OD , Torroba M , Wang G , Bjorklund T ((2016) ) A novel process of viral vector barcoding and library preparation enables high-diversity library generation and recombination-free paired-end sequencing. Sci Rep 6: , 37563. |
[40] | Di Maio R , Hoffman EK , Rocha EM , Keeney MT , Sanders LH , De Miranda BR , Zharikov A , Van Laar A , Stepan AF , Lanz TA , Kofler JK , Burton EA , Alessi DR , Hastings TG , Greenamyre JT ((2018) ) LRRK2 activation in idiopathic Parkinson’s disease. Sci Transl Med 10: , eaar542. |
[41] | Cong L , Ran FA , Cox D , Lin S , Barretto R , Habib N , Hsu PD , Wu X , Jiang W , Marraffini LA , Zhang F ((2013) ) Multiplex genome engineering using CRISPR/Cas systems. Science 339: , 819–823. |
[42] | Mali P , Yang L , Esvelt KM , Aach J , Guell M , DiCarlo JE , Norville JE , Church GM ((2013) ) RNA-guided human genome engineering via Cas9. Science 339: , 823–826. |
[43] | Shalem O , Sanjana NE , Hartenian E , Shi X , Scott DA , Mikkelson T , Heckl D , Ebert BL , Root DE , Doench JG , Zhang F ((2014) ) Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343: , 84–87. |
[44] | Swiech L , Heidenreich M , Banerjee A , Habib N , Li Y , Trombetta J , Sur M , Zhang F ((2015) ) in vivo interrogation of gene function in the mammalian brain using CRISPR-Cas9. Nat Biotechnol 33: , 102–106. |
[45] | Merienne N , Vachey G , de Longprez L , Meunier C , Zimmer V , Perriard G , Canales M , Mathias A , Herrgott L , Beltraminelli T , Maulet A , Dequesne T , Pythoud C , Rey M , Pellerin L , Brouillet E , Perrier AL , du Pasquier R , Deglon N ((2017) ) The self-inactivating KamiCas9 system for the editing of CNS disease genes. Cell Rep 20: , 2980–2991. |
[46] | Cyranoski D , Ledford H ((2018) ) Genome-edited baby claim provokes international outcry. Nature 563: , 607–608. |
[47] | Cideciyan AV , Sudharsan R , Dufour VL , Massengill MT , Iwabe S , Swider M , Lisi B , Sumaroka A , Marinho LF , Appelbaum T , Rossmiller B , Hauswirth WW , Jacobson SG , Lewin AS , Aguirre GD , Beltran WA ((2018) ) Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc Natl Acad Sci USA 115: , E8547–E8556. |
[48] | Benskey MJ , Sellnow RC , Sandoval IM , Sortwell CE , Lipton JW , Manfredsson FP ((2018) ) Silencing alpha synuclein in mature nigral neurons results in rapid neuroinflammation and subsequent toxicity. Front Mol Neurosci 11: , 36. |
[49] | Zharikov AD , Cannon JR , Tapias V , Bai Q , Horowitz MP , Shah V , El Ayadi A , Hastings TG , Greenamyre JT , Burton EA ((2015) ) shRNA targeting alpha-synuclein prevents neurodegeneration in a Parkinson’s disease model. J Clin Invest 125: , 2721–2735. |
[50] | Kantor B , Tagliafierro L , Gu J , Zamora ME , Ilich E , Grenier C , Huang ZY , Murphy S , Chiba-Falek O ((2018) ) Downregulation of SNCA expression by targeted editing of DNA methylation: A potential strategy for precision therapy in PD. Mol Ther 26: , 2638–2649. |
[51] | Soldner F , Stelzer Y , Shivalila CS , Abraham BJ , Latourelle JC , Barrasa MI , Goldmann J , Myers RH , Young RA , Jaenisch R ((2016) ) Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature 533: , 95–99. |
[52] | Suzuki K , Izpisua Belmonte JC ((2018) ) in vivo genome editing via the HITI method as a tool for gene therapy. J Hum Genet 63: , 157–164. |
[53] | Suzuki K , Tsunekawa Y , Hernandez-Benitez R , Wu J , Zhu J , Kim EJ , Hatanaka F , Yamamoto M , Araoka T , Li Z , Kurita M , Hishida T , Li M , Aizawa E , Guo S , Chen S , Goebl A , Soligalla RD , Qu J , Jiang T , Fu X , Jafari M , Esteban CR , Berggren WT , Lajara J , Nunez-Delicado E , Guillen P , Campistol JM , Matsuzaki F , Liu GH , Magistretti P , Zhang K , Callaway EM , Zhang K , Belmonte JC ((2016) ) in vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540: , 144–149. |
[54] | Bak RO , Porteus MH ((2017) ) CRISPR-mediated integration of large gene cassettes using AAV donor vectors. Cell Rep 20: , 750–756. |
[55] | Yu X , Liang X , Xie H , Kumar S , Ravinder N , Potter J , de Mollerat du Jeu X , Chesnut JD ((2016) ) Improved delivery of Cas9 protein/gRNA complexes using lipofectamine CRISPRMAX. Biotechnol Lett 38: , 919–929. |
[56] | Yeh WH , Chiang H , Rees HA , Edge ASB , Liu DR ((2018) ) in vivo base editing of post-mitotic sensory cells. Nat Commun 9: , 2184. |
[57] | Gaudelli NM , Komor AC , Rees HA , Packer MS , Badran AH , Bryson DI , Liu DR ((2017) ) Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551: , 464–471. |
[58] | Eid A , Alshareef S , Mahfouz MM ((2018) ) CRISPR base editors: Genome editing without double-stranded breaks. Biochem J 475: , 1955–1964. |
[59] | Kim YB , Komor AC , Levy JM , Packer MS , Zhao KT , Liu DR ((2017) ) Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat Biotechnol 35: , 371–376. |
[60] | Komor AC , Kim YB , Packer MS , Zuris JA , Liu DR ((2016) ) Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533: , 420–424. |
[61] | Li X , Wang Y , Liu Y , Yang B , Wang X , Wei J , Lu Z , Zhang Y , Wu J , Huang X , Yang L , Chen J ((2018) ) Base editing with a Cpfl-cytidine deaminase fusion. Nat Biotechnol 36: , 324–327. |
[62] | Reeve A , Simcox E , Turnbull D ((2014) ) Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res Rev 14: , 19–30. |
[63] | Wyss-Coray T ((2016) ) Ageing, neurodegeneration and brain rejuvenation. Nature 539: , 180–186. |
[64] | Hegde ML , Bohr VA , Mitra S ((2017) ) DNA damage responses in central nervous system and age-associated neurodegeneration. Mech Ageing Dev 161: , 1–3. |
[65] | Johnson S , Imai SI ((2018) ) NAD (+) biosynthesis, aging, and disease. F1000Res 7: , 132. |
[66] | Satoh A , Brace CS , Rensing N , Cliften P , Wozniak DF , Herzog ED , Yamada KA , Imai S ((2013) ) Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab 18: , 416–430. |
[67] | Rizzi L , Roriz-Cruz M ((2018) ) Sirtuin 1 and Alzheimer’s disease: An up-to-date review. Neuropeptides 71: , 54–60. |
[68] | Benito-Cuesta I , Diez H , Ordonez L , Wandosell F ((2017) ) Assessment of autophagy in neurons and brain tissue. Cells 6: , E25. |
[69] | Hara T , Nakamura K , Matsui M , Yamamoto A , Nakahara Y , Suzuki-Migishima R , Yokoyama M , Mishima K , Saito I , Okano H , Mizushima N ((2006) ) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: , 885–889. |
[70] | Sato S , Uchihara T , Fukuda T , Noda S , Kondo H , Saiki S , Komatsu M , Uchiyama Y , Tanaka K , Hattori N ((2018) ) Loss of autophagy in dopaminergic neurons causes Lewy pathology and motor dysfunction in aged mice. Sci Rep 8: , 2813. |
[71] | Rubinsztein DC , Marino G , Kroemer G ((2011) ) Autophagy and aging. Cell 146: , 682–695. |
[72] | Thorburn A ((2018) ) Autophagy and disease. J Biol Chem 293: , 5425–5430. |
[73] | Thorburn A ((2018) ) Autophagy and disease. J Biol Chem 293: , 5425–5430. |


