
Non-Dopaminergic Therapies
Abstract
Currently all aspects of Parkinson’s disease (PD) treatment are less than ideal and would benefit from new interventions. We focus on problems associated with brain neurotransmitters amenable to more specific subreceptor and temporal manipulation. In addition, we consider potentially treatable CNS non-PD co-pathologies or co-morbidities that may exacerbate progression of various aspects of PD.
INTRODUCTION
The focus of this article is anticipated and hypothesized non-dopaminergic pharmacotherapies for patients with Parkinson’s disease (PD) who are in the motor symptomatic stage of the disease. Although depletion of dopamine is the most notable neurotransmitter change in PD, other neurochemical changes affecting cholinergic, serotoninergic, noradrenergic and other neurotransmitter systems occur and contribute to PD symptomatology. Current unmet critical needs in the management of symptomatic patients with PD include dopamine-unresponsive axial motor impairments and non-motor symptoms, such as dementia, depression, anxiety, psychosis and pain. Apart from PD-specific brain changes, the presences of comorbid systemic or central nervous system (CNS) co-pathologies have important implications for patients suffering from PD as they can aggravate and complicate the clinical course of PD. The synergistic detrimental interactions between CNS co-pathologies, medical co-morbidities, and PD will allow new therapeutic approaches. Major systemic medical comorbidities include cardiovascular, particularly cerebral small vessel disease, and metabolic disorders, notably diabetes mellitus. Important CNS co-pathologies include glucocerebrosidase (GBA) mutations, Alzheimer amyloid and tau proteinopaties. Finally, it is important to note, that even with the expected development of disease modifying treatments targeting α-synuclein, symptomatic treatment of the disease will remain important for many decades until disease-modifying treatments can completely prevent the emergence of symptomatic PD.
GENERAL TRENDS IN PHARMACOTHERAPEUTICS FOR PD
Anticipated advances in PD therapeutics will involve more targeted, receptor-specific, agents to better sculpt the response to drugs as well as in novel delivery systems that promote delivery of drugs to specific sites and/or on demand to mimic natural stimulation. A limitation of neurotransmitter precursors is that they stimulate all pre- and post-synaptic receptor subtypes of the neurotransmitter. This may be true for serotonin and norepinephrine precursors. Levodopa may be the exception, at least for the motor system. The same problem of generalized increase of a neurotransmitter is true of drugs that raise the concentration of a neurotransmitter by reducing its re-uptake or breakdown as occurs with cholinesterase inhibitors. Likewise the continuous and constant delivery of drugs, a goal for drugs like levodopa (see previous chapter) is not physiological and delivery that was on demand would more closely mimic the normal functioning of the brain. The best examples of exquisite control of locus and time specific delivery of drugs may require additional genetic manipulation using implanted light sources (phototherapy) or oral administration of another normally inert substance to stimulate designer receptors for G-protein coupled receptors (designer receptor exclusively activated by designer drugs or DREADDs). Although we are emphasizing specific receptor target engagement, looking at the effects of drugs on specific neural circuit disturbances may be equally important [1]. It is anticipated that with the expansion of imaging techniques and development of neural circuit biomarkers underlying specific symptoms, such as anxiety or depression, will allow testing of more precise and targeted drugs in more homogenous patient groups [2].
MOTOR AND COGNITIVE SYMPTOMS POORLY RESPONSIVE TO DOPAMINERGIC STIMULATION
Impairments in mobility, particularly freezing of gait and falls, and cognitive impairment impair quality of life in patients with PD [3]. The increasing refractoriness of these disabilities to dopaminergic therapy implicates non-dopaminergic mechanisms. The co-occurrence of axial motor impairments and dementia suggests a shared or overlapping pathophysiological role of the cholinergic system contributing to these symptoms [4, 5]. Similarly as shown by the successful dopaminergic replacement therapy for PD, more centrally active and effective cholinergic augmentation therapy could potentially make a substantial clinical impact in the management of these dopaminergic medication-refractory symptoms [6].
Promoting centrally active cholinergic pharmacotherapy for the management of falls, freezing of gait and dementia
Cholinergic losses have been associated with slow gait speed, sensory integration during postural control, falls, freezing of gait and dementia [4, 5]. These observations emphasize the clinical potential of cholinergic drugs to manage balance and gait disturbances in PD. Phase 2 clinical trials of acetylcholinesterase (AChE)-inhibitors, donepezil and rivastigmine, modestly reduced falls in patients with PD although phase 3 trials of these agents for cognitive problems failed to find a reduction of falls in the tallying of the adverse effects [7]. Importantly, the use of cholinesterase inhibitors did not result in a significant improvement of freezing of gait in these studies. Pharmacological support for a so-called ‘malignant’ hypocholinergic subtype of PD also comes from evidence of adverse effects of anti-cholinergic drugs in patients with PD, including impaired cognition, hallucinations and motor complications, such as freezing of gait [8]. Although currently available cholinesterase inhibitors are approved for the management of dementia in PD, the relative effectiveness of these drugs is modest at best due to a number of reasons. First, limited penetrance of the blood-brain barrier results in sub-optimal cholinesterase inhibition [9]. For example, our prior in vivo PET imaging study found that 3 months of donepezil (10 mg per day) therapy resulted in only a very modest mean cortical donepezil induced AChE inhibition of 19.1% in patients with Alzheimer disease [10]. Effectively, only about 40% of the treated patients had mildly improved cognitive outcome measures whereas the others did not. Second, dose escalation is limited by peripheral cholinergic side effects. Third, these drugs lack receptor specificity for selective nicotinic or muscarinic functions in the brain. An increase of synaptic acetylcholine levels in all brain regions by cholinesterase inhibitors may have unintended negative consequences. For example, augmentation of (extra-) synaptic cholinergic levels may inhibit presynaptic cholinergic signaling by stimulation of presynaptic muscarinic type 2 receptors and non-physiological and tonic stimulation of postsynaptic muscarinic and nicotinic receptors [11]. Thus, centrally active cholinergic drugs that are receptor sub-type specific may have the potential to produce more clinically meaningful effects in the future management of postural instability, falls, freezing of gait and dementia. Importantly, successful use of centrally active cholinergic drugs will also depend on selecting patients with cholinergic deficits. In this respect, a personalized medicine approach may be prudent to maximize enhancement and minimize impairments of cholinergic treatments. Selection could be achieved by biomarkers, such as neuroimaging, but clinical predictor variables, including cognitive impairment, REM sleep behavior disorder, falls and slow gait speed may be sufficient in routine clinical practice. Finally, invasive and non-invasive (e.g., transcranial direct current stimulation) neuromodulation stimulation approaches may selectively target cholinergic-dependent network functions to manage postural instability, gait difficulties and cognitive impairment in PD.
Targeting the main output structures of the basal ganglia: Glutamate and GABA for freezing of gait
Models of the basal ganglia postulate excessive inhibition of the main output structures of the basal ganglia. The striatal nuclei provide inhibitory control over the globus pallidum pars interna and substantia nigra pars reticulata [12]. The subthalamic nucleus sends glutaminergic excitatory efferents to the globus pallidum pars interna/substantia nigra pars reticulata stimulating their GABA-ergic inhibition of the thalamic and brainstem structures that control muscle tone and mobility functions [13]. A striking observation in decerebrate animals is that GABA agonists suppress locomotion and antagonists induce locomotion. The multiple GABA and glutamate receptors offer the potential of blocking excessive inhibition of the main output structure of the basal ganglia and thereby addressing a critical unmet need in the management of PD.
DEPRESSION AND ANXIETY
Raising serotoninergic and noradrenergic extracellular concentrations with re-uptake inhibitors is the central tenet of clinical pharmacotherapy of mood disorders in PD today. These agents are slow to improve symptoms and many people with PD do not respond. Recently, attention is shifting to glutaminergic systems that might have immediate antidepressant activity. For example, ketamine may be a prototype for quick antidepressant effects although ketamine itself would have unacceptable adverse effects with prolonged use. Nevertheless, the effects of ketamine are spurring evaluation of various glutaminergic agents targeting different glutamate subreceptors [14].
TREATING PAIN
Pain is a neglected, but common, feature of PD and is expected to receive more attention and a search for new targets for therapies. Several serotonin receptor subtypes modulate pain in animals and humans. For example, 5-HT1, 5-HT2, 5-HT3, and 5-HT7 receptors in the CNS modulate pain. However, inhibition, excitation, or maintenance of nociceptive responses depends on administration route, distribution of the receptors, dose of agonists or antagonists, pain type and duration of pain [15]. As may be the case for depression, ketamine and related drugs may also play an important role in future pain management in PD. This complicated field requires much more work but we expect better treatments for pain in PD.
MANAGING THE PATHOPHYSIOLOGICAL CONSEQUENCES OF GBA MUTATIONS IN PD
Mutations of GBA gene increase the risk of developing PD and produce a more rapid and malignant phenotype of parkinsonism. One approach to treating the cerebral consequences of GBA mutations is by substrate reduction using small molecules that cross the blood-brain barrier and reduce synthesis of glucocerebroside by inhibition of the ceramide-specific glucosyltransferase. Likewise small molecules capable of entering the brain that act as chaperones for GBA transfer to the lysozomes are currently being investigated [16]. Moreover, dysfunction of GBA has also been demonstrated in PD without mutations of GBA. Therefore, GBA enzyme function represents an interesting pharmacological target for PD. Cell and animal models suggest that increasing GBA enzyme activity can reduce α-synuclein levels. Clinical trials of ambroxol, a GBA chaperone, are currently ongoing in PD and PD dementia, as is a trial of substrate reduction therapy [16]. GBA mutation status may also inform outcome after deep brain stimulation (’surgicogenomics’) where mutation carriers were shown to have a more rapid post-operative cognitive and non-motor disease progression compared to non-carriers [17].
REDUCING THE ACCUMULATION OF ABNORMAL PROTEINS: α-SYNUCLEIN, β-AMYLOID AND TAU
Proteinopathies, such as α-syncleinopathy, β-amyloidopathy, TDP-43 or tauopathy are an important cause of neurodegeneration, including neuronal loss and axonal degeneration. Proteinopathy and other changes in subcortical projection systems may also result in neurotransmitter changes, including cholinergic and dopaminergic systems. Therefore, early intervention that may attenuate or prevent the build-up of these abnormal proteineous deposits may have an important disease-modifying effect in PD. There is emerging interest in the so-called ‘glymphatic’ system defined as a brain-wide paravascular pathway for cerebrospinal fluid and interstitial fluid exchange that facilitates efficient clearance of solutes and waste from the brain [18]. This brain garbage removal system may have important implications for proteinopathies, where potentially toxic soluble phase proteins may be cleared from the brain before these may transfect brain cells and/or become pathological protein deposits. Although much interest in this field has focused on Alzheimer disease, similar level of enthusiasm may be warranted for α-synucleinopathies, such as PD. Obviously, very early target engagement would be best served in the presymptomatic or prodromal proteinopathy stages of PD. However, there remains a potential important role for glymphatic clearance promoting treatments in the motor symptomatic phase of this disease. This is for two main reasons. First, early motor phase disease may have α-synucleinopathy limited to brainstem or olfactory tubercle regions without significant involvement of the limbic or neocortex. Second, Alzheimer and/or other proteinopathies may complicate advancing stage PD and increase the risk of dementia. Interestingly, natural sleep or anesthesia are associated with a 60% increase in the interstitial space, resulting in a striking increase in convective exchange of cerebrospinal fluid with interstitial fluid. In turn, convective fluxes of interstitial fluid increased the rate of β-amyloid clearance during sleep in mice [19]. Normal pressure hydrocephalus is unlikely to be a diagnostic entity on its own but a collection of clinical signs that emerge from a spectrum of neurodegenerative and vascular disorders. The partial overlap of postural instability and gait difficulty motor features between normal pressure hydrocephalus and PD may suggest a spectrum disorder involving abnormal glymphatic clearance [20]. Therefore, sleep promotion, cerebrospinal fluid (CSF) diversion by shunting, and other targeted glymphatic clearance therapies may modify the natural course of PD. Potential glymphatic clearance enhancing drugs may target tight junctions at the blood-brain barrier or astrocytic aquaporin-4 water channels. Zinc may also play a role in tight junction regulation of in blood-brain barrier permeability associated with efflux of macromolecules [21].
Early use of serotoninergic drugs may provide an alternative way to decrease β-amyloidopathy in PD [22]. Serotoninergic medications are suggested to stimulate a serotonin G-protein-coupled receptor-linked intracellular cascade that favors the cleavage of amyloid precursor protein by α-secretase, leading to reductions in toxic β-amyloid peptide generation and reduced plaque formation [23]. Serotonin medications may also promote efflux of toxic β-amyloid peptide species from the brain into the blood [24].
DIABETES MELLITUS AND PD
There is increasing interest in detrimental interactions between cardiometabolic syndromes and neurodegeneration. Diabetes mellitus is emerging not only as a risk factor but also as an important negative prognostic factor for progression of PD [25]. Furthermore, patients with diabetes mellitus but without PD have lower striatal dopamine nerve terminal binding and abnormal CSF α-synuclein or tau levels compared to non-diabetic healthy control subjects [25]. Therefore, more effective treatment of risk factors for the metabolic syndrome may have the potential to improve long-term outcome in PD. This should be done earlier rather than later into the course of PD. Furthermore, more targeted CNS mechanistic treatment approaches may be needed, such as intranasal insulin. Restoration of brain insulin signaling could have neuroprotective effects and anti-diabetic drugs are currently being repurposed as potential PD treatments [26]. As a prime example of so-called ‘drug repurposing’, the anti-diabetic drug exenatide, a glucagon-like peptide-1 receptor agonist, improves motor ratings in patients with PD, which may represent either a neuroprotective or symptomatic effects [27]. Although these preliminary observations are promising, it is important to await the results of further clinical trials before implementing this treatment in routine clinical practice.
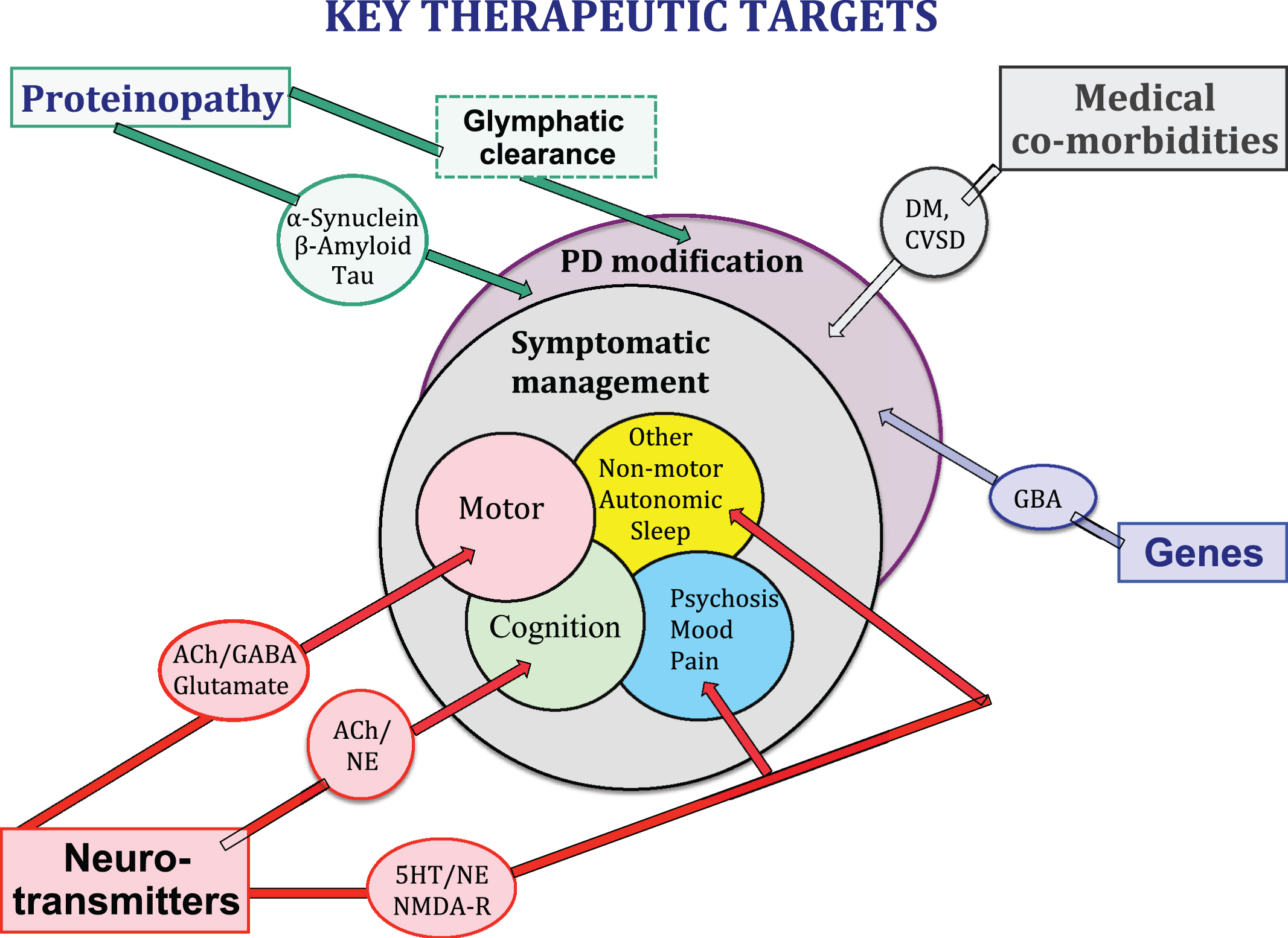
Fig.1
Neurotransmitter systems, proteonopathies, genes, CNS co-pathologies and medical co-morbidities all contribute to the clinical expression of the PD syndrome and may provide key therapeutic targets for symptomatic control of clinical symptoms and/or clinical disease modification.
CEREBRAL SMALL VESSEL DISEASE
There is a blurring of boundaries between cerebrovascular disease and PD. Although pure vascular parkinsonism remains a poorly understood entity, co-existing cerebrovascular disease and PD is common [28]. This has clinical relevance as co-existing cerebrovascular disease may aggravate motor parkinsonism, in particular postural instability and gait difficulties. The risk factors for cerebral small vessel disease (CSVD) are not identical to those for large vessel disease. A variety of pathophysiological mechanisms and pharmacological interventions targeting CSVD have been proposed [29]. Further research is needed before making specific recommendations how to best manage this entity in clinical practice apart from standard cardiovascular risk factor reductions. CSVD is now the focus of several large collaborative groups and targeted treatments should ensue in the coming decades.
CONCLUSION
Management of many of the motor and non-motor aspects of PD needs better therapies. This chapter discusses expected improvements to emerge in coming decades. Suggested improvements include further development of more targeted non-dopaminergic neurotransmission enhancing treatments, novel locally activity and on-demand treatment delivery systems, proteinopathy clearing and genetic targeted treatments. The chapter emphasizes that PD is not just α-synucleinopathy that exclusively dictates the progression of the disease but important disease-modifying effects are expected to emerge from treatments of non-PD CNS co-pathologies and systemic medical co-morbidities.
CONFLICT OF INTEREST
The authors declare no relevant conflict of interests.
ACKNOWLEDGMENTS
Nicolaas Bohnen has received research support from the NIH, Michael J. Fox Foundation, the Department of Veteran Affairs, Chase Pharmaceuticals and Axovant Sciences.
REFERENCES
[1] | Farb DH , Ratner MH ((2014) ) Targeting the modulation of neural circuitry for the treatment of anxiety disorders. Pharmacol Rev 66: , 1002–1032. |
[2] | Schindlbeck KA , Eidelberg D ((2018) ) Network imaging biomarkers: Insights and clinical applications in Parkinson’s disease. Lancet Neurol 17: , 629–640. |
[3] | Lopez IC , Ruiz PJ , Del Pozo SV , Bernardos VS ((2010) ) Motor complications in Parkinson’s disease: Ten year follow-up study. Mov Disord 25: , 2735–2739. |
[4] | Bohnen NI , Frey KA , Studenski S , Kotagal V , Koeppe RA , Constantine GM , Scott PJ , Albin RL , Muller ML ((2014) ) Extra-nigral pathological conditions are common in Parkinson’s disease with freezing of gait: An in vivo positron emission tomography study. Mov Disord 29: , 1118–1124. |
[5] | Bohnen NI , Albin RL , Muller ML , Petrou M , Kotagal V , Koeppe RA , Scott PJ , Frey KA ((2015) ) Frequency of cholinergic and caudate nucleus dopaminergic deficits across the predemented cognitive spectrum of Parkinson disease and evidence of interaction effects. JAMA Neurol 72: , 194–200. |
[6] | Bohnen NI , Grothe MJ , Ray NJ , Muller MLTM , Teipel SJ ((2018) ) Recent advances in cholinergic imaging and cognitive decline-Revisiting the cholinergic hypothesis of dementia. Curr Geriatr Rep 7: , 1–11. |
[7] | Perez-Lloret S , Barrantes FJ ((2016) ) Deficits in cholinergic neurotransmission and their clinical correlates in Parkinson’s disease. NPJ Parkinsons Dis 2: , 16001. |
[8] | Perez-Lloret S , Negre-Pages L , Damier P , Delval A , Derkinderen P , Destee A , Meissner WG , Schelosky L , Tison F , Rascol O ((2014) ) Prevalence, determinants, and effect on quality of life of freezing of gait in Parkinson disease. JAMA Neurol 71: , 884–890. |
[9] | Zemek F , Drtinova L , Nepovimova E , Sepsova V , Korabecny J , Klimes J , Kuca K ((2014) ) Outcomes of Alzheimer’s disease therapy with acetylcholinesterase inhibitors and memantine. Expert Opin Drug Saf 13: , 759–774. |
[10] | Bohnen NI , Kaufer DI , Hendrickson R , Ivanco LS , Lopresti BJ , Koeppe RA , Meltzer CC , Constantine G , Davis JG , Mathis CA , Dekosky ST , Moore RY ((2005) ) Degree of inhibition of cortical acetylcholinesterase activity and cognitive effects by donepezil treatment in Alzheimer’s disease. J Neurol Neurosurg Psychiatry 76: , 315–319. |
[11] | Hasselmo ME , Sarter M ((2011) ) Modes and models of forebrain cholinergic neuromodulation of cognition. Neuropsychopharmacology 36: , 52–73. |
[12] | Nambu A ((2004) ) A new dynamic model of the cortico-basal ganglia loop. Prog Brain Res 143: , 461–466. |
[13] | Lewis SJ , Shine JM ((2016) ) The next step: A common neural mechanism for freezing of gait. Neuroscientist 22: , 72–82. |
[14] | Zanos P , Thompson SM , Duman RS , Zarate CA Jr , Gould TD ((2018) ) Convergent mechanisms underlying rapid antidepressant action. CNS Drugs 32: , 197–227. |
[15] | Cortes-Altamirano JL , Olmos-Hernandez A , Jaime HB , Carrillo-Mora P , Bandala C , Reyes-Long S , Alfaro-Rodriguez A ((2018) ) Review: 5-HT1, 5-HT2, 5-HT3 and 5-HT7 receptors and their role in the modulation of pain response in the central nervous system. Curr Neuropharmacol 16: , 210–221. |
[16] | Balestrino R , Schapira AHV ((2018) ) Glucocerebrosidase and Parkinson disease: Molecular, clinical, and therapeutic implications. Neuroscientist 24: , 540–559. |
[17] | Lythe V , Athauda D , Foley J , Mencacci NE , Jahanshahi M , Cipolotti L , Hyam J , Zrinzo L , Hariz M , Hardy J , Limousin P , Foltynie T ((2017) ) GBA-associated Parkinson’s disease: Progression in a deep brain stimulation cohort. J Parkinsons Dis 7: , 635–644. |
[18] | Iliff JJ , Wang M , Liao Y , Plogg BA , Peng W , Gundersen GA , Benveniste H , Vates GE , Deane R , Goldman SA , Nagelhus EA , Nedergaard M ((2012) ) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci Transl Med 4: , 147ra111. |
[19] | Xie L , Kang H , Xu Q , Chen MJ , Liao Y , Thiyagarajan M , O’Donnell J , Christensen DJ , Nicholson C , Iliff JJ , Takano T , Deane R , Nedergaard M ((2013) ) Sleep drives metabolite clearance from the adult brain. Science 342: , 373–377. |
[20] | Ringstad G , Valnes LM , Dale AM , Pripp AH , Vatnehol SS , Emblem KE , Mardal KA , Eide PK ((2018) ) Brain-wide glymphatic enhancement and clearance in humans assessed with MRI. JCI Insight 3: , 121537. |
[21] | Xiao R , Yuan L , He W , Yang X ((2018) ) Zinc ions regulate opening of tight junction favouring efflux of macromolecules via the GSK3beta/snail-mediated pathway. Metallomics 10: , 169–179. |
[22] | Kotagal V , Spino C , Bohnen NI , Koeppe R , Albin RL ((2018) ) Serotonin, beta-amyloid, and cognition in Parkinson disease. Ann Neurol 83: , 994–1002. |
[23] | Cirrito JR , Disabato BM , Restivo JL , Verges DK , Goebel WD , Sathyan A , Hayreh D , D’Angelo G , Benzinger T , Yoon H , Kim J , Morris JC , Mintun MA , Sheline YI ((2011) ) Serotonin signaling is associated with lower amyloid-beta levels and plaques in transgenic mice and humans. Proc Natl Acad Sci U S A 108: , 14968–14973. |
[24] | Brenn A , Grube M , Jedlitschky G , Fischer A , Strohmeier B , Eiden M , Keller M , Groschup MH , Vogelgesang S ((2014) ) St. John’s Wort reduces beta-amyloid accumulation in a double transgenic Alzheimer’s disease mouse model-role of P-glycoprotein. Brain Pathol 24: , 18–24. |
[25] | Pagano G , Polychronis S , Wilson H , Giordano B , Ferrara N , Niccolini F , Politis M ((2018) ) Diabetes mellitus and Parkinson disease. Neurology 90: , e1654–e1662. |
[26] | De Pablo-Fernandez E , Goldacre R , Pakpoor J , Noyce AJ , Warner TT ((2018) ) Association between diabetes and subsequent Parkinson disease: A record-linkage cohort study. Neurology 91: , e139–e142. |
[27] | Athauda D , Maclagan K , Budnik N , Zampedri L , Hibbert S , Aviles-Olmos I , Chowdhury K , Skene SS , Limousin P , Foltynie T ((2018) ) Post hoc analysis of the Exenatide-PD trial-Factors that predict response. Eur J Neurosci, doi: 10.1111/ejn.14096. |
[28] | Rektor I , Bohnen NI , Korczyn AD , Gryb V , Kumar H , Kramberger MG , de Leeuw FE , Pirtosek Z , Rektorova I , Schlesinger I , Slawek J , Valkovic P , Vesely B ((2018) ) An updated diagnostic approach to subtype definition of vascular parkinsonism - Recommendations from an expert working group. Parkinsonism Relat Disord 49: , 9–16. |
[29] | Bath PM , Wardlaw JM ((2015) ) Pharmacological treatment and prevention of cerebral small vessel disease: A review of potential interventions. Int J Stroke 10: , 469–478. |


