
Parkinson Diseases in the 2020s and Beyond: Replacing Clinico-Pathologic Convergence With Systems Biology Divergence
Abstract
Parkinson disease has been considered for practical purposes a heterogeneous clinico-pathological entity. The operational definition requires clinical ascertainment of a levodopa-responsive parkinsonism with no “atypical” features, and pathological criteria based on the finding, usually at postmortem, of aggregates of α-synuclein in Lewy bodies and Lewy neurites. The underlying assumption has been that a molecular-biological disorder, targetable for disease modification as a whole, underlies this clinico-pathologic, convergent model of disease. The 2020s will be expected to mark the beginning of the end for this model, especially if therapeutic success in a specific molecular subtype, such as PD-GBA, is not translated to “sporadic PD”. The complex and dynamic biological abnormalities of aging, which have informed the evolution of other fields in medicine into divergent, systems-biology models, will also provide the template for the development of disease modifying therapies for neurodegenerative disorders. In the 2020s and 2030s we will no longer ask whether any given molecule may be neuroprotective in early Parkinson disease but, rather, which subtype (which endophenotype) among the Parkinson diseases would be the best mechanistic recipient for such molecule and which would not. The next breakthrough in Parkinson’s research will be conceptual: the recognition that discoveries in a subtype of PD will apply only or largely to that subtype and not construed to represent “a piece” that seamlessly inserts into, and helps explains, a unifying “Parkinson’s puzzle”. Successful neuroprotection for each PD subtype will likely require pharmacotherapeutic combinations (“drug cocktails”) to harness synergistic potential benefits when more than the dominant pathogenic mechanism is targeted, as identified from forthcoming population-based unbiased biomarker discovery programs.
INTRODUCTION
Dr. JB declared to a patient after his physical exam is complete: “based on my experience and available criteria, you have Disease X. We can support this diagnosis by Test Y. While Disease X may vary from patient to patient, it is invariably progressive although the speed and nature of such progression cannot be accurately predicted. I suggest we start Treatment Z soon”.
JB was an oncologist delivering the diagnosis of breast cancer, circa 1970. The treatment he proposed was radical mastectomy. JB did not anticipate that in 1981 a clinical trial would show that radical mastectomy and breast-preserving surgery yielded no differences in disease progression [1]. A similar proportion of patients survived in both arms.
In defense of an “only-radical-mastectomy-is-sensible” worldview, the following arguments could have been raised to explain how the results of a negative study were “artifactual”: the study design was not optimal, the radical surgery arm was not radical enough while the conservative surgery arm was too radical, and the wrong patients were chosen: their disease was far too advanced for the radical treatment to prevail over the conservative one.
JB might as well have been a neurologist delivering the diagnosis of Parkinson disease (PD), circa 2005. Test Y was DATscan and Treatment Z selegiline. Unlike the recalibration that began in oncology in the 1980s, when breast (and other) cancers began to be subdivided based on molecular biological markers, and no treatment was deemed sufficient to address all types of breast cancer, negative anti-amyloid trials have yet to force a similar recalibration in Alzheimer’s therapeutic development. Despite several negative Phase 3 trials [2–5], we have defended the anti-amyloid enterprise over the past 18 years and are prepared to do the same for any future negative anti-synuclein trial as the next big frontier in the treatment of sporadic PD.
In oncology, negative trials became opportunities to reconsider the approach to treatment. In neurodegenerative diseases, negative trials have been considered artifacts of trial design and execution. A positive outcome, we have insisted, would have been within reach if only these studies were better designed and had recruited patients at an earlier stage.
THE CLINICO-PATHOLOGIC MODEL OF DISEASE: 1912-CIRCA 2020
The PD story line has been compellingly linear, following a logical cause-effect sequence: the disease begins in the peripheral nervous system, with the accumulation of α-synuclein in the Meissner plexus and olfactory bulb, spreading centrally via permissive templating or prion-like transmission through the vagus nerve into the brainstem, creating the third of five Braak stages of progression [6], a point when a clinical diagnosis becomes possible. Events prior to this stage are considered “prodromal” and the “risk of conversion” from prodrome to disease variably anchored on the appearance of motor features [7]. Any “exceptions” only prove the rule: PD is accepted to be clinically and biologically heterogenous but ultimately a single, relatively scripted disease entity. Biomarkers are being developed from cohorts of clinically diagnosed individuals (biological measures, as the dependent variables, are categorized based on clinical phenotypes, the independent variable) [8] and pathogenesis of PD formulated from studies on cell culture and experimental animals [9]. Research on extremes of clinical phenotype (e.g., young-onset PD) or genotype (e.g., PD-GBA) provide pieces to a disease puzzle whose face is ever increasing and will eventually come into full view as these pieces are seamlessly integrated [10].
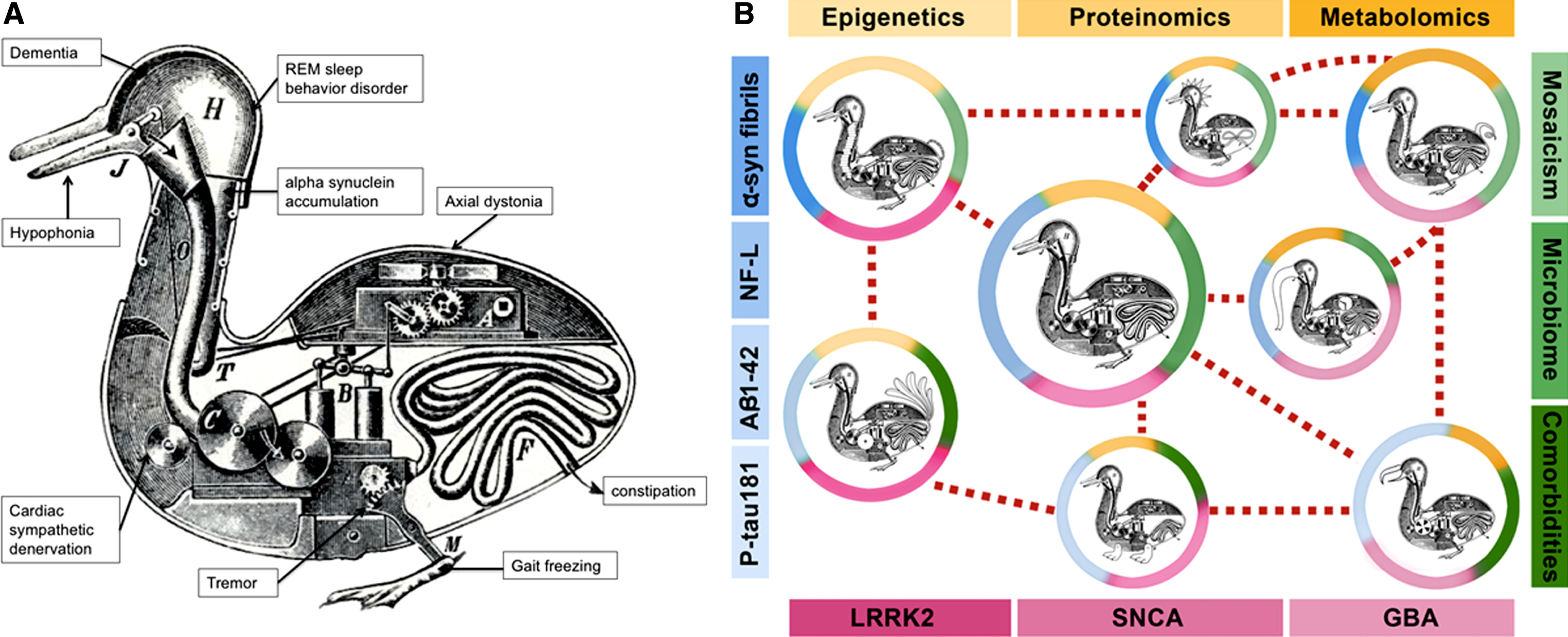
Characterization of the phenotypic and neurochemical range of the clinico-pathologic framework of PD has been very important to the development of symptomatic treatments. Dopamine deficiency is an eloquent neurochemical common denominator for which dopaminergic therapies have been an effective, indeed miraculous, answer for many of the clinical features. But enshrining common clinical and neurochemical features as the converging pathogenic basis of a disease and from which to validate genetic and molecular underpinnings represents a form of reductionism, similar to the idea of referring to all swimming birds with short necks and webbed feet as ducks (Fig. 1). We have had a long run giving primacy to the developmental sequence: phenotype to genotype to cellular biology (which can work in some monogenetic disorders). We have yet to consider analyses based on a more agnostic, nearly reversed order: genotype to cellular biology to phenotype. The amalgamation of a range of clinical features drawn into a phenotype by James Parkinson in 1817 and the identification of a common pathological lesion by Friedrich Heinrich Lewy in 1912 [11] created a unifying nosology that has proven sensitive to symptomatic therapies (at least in the early stages) but inclusive of too many biological abnormalities and, as a result, refractory to targeted molecular modification.
Fig.1
A) Reductionism: the Duck of Vaucanson. This engraving of the Canard Digérateur or “Digesting Duck” illustrated the famous mechanical duck made by Jacques de Vaucanson in the 18th century, which supposedly ate grain and excreted droppings in front of an audience unaware such stool pellets were not manufactured by the contraption but placed surreptitiously. The “Duck of Vaucanson” served to illustrate Descartes view (in De Homine, 1662) that all animals could be reductively explained as automata. We have reductively attempted to explain the large range of clinical, pathologic, genetic, and molecular findings into a single “Duck of Parkinson,” represented by each of the wheels, levers, and pipes of a single contraption (Copyright in public domain via Wikimedia Commons). B) Systems biology: Looks like a duck, walks like a duck, but there are differences. Modifications of the Duck of Vaucanson illustrate the systems biology model of Parkinson diseases. It acknowledges distinctive self-regulating “mechanical” systems within each of these “birds” (distinct pathophysiological processes), yet sharing enough external and internal duck-like features to belong to the same “family” but with a phenotype shaped by the “pond where they swim” (colored circles surrounding the ducks). Each “pond” includes a combination of genetic, molecular, and environmental traits that combine in systems biology networks (shades of yellow, blue, green, and red). These “Parkinson ducks” represent an oversimplification of more complicated interactions (e.g., mosaicism might have an effect on all genetic factors, mono- and polygenic; similarly, microbiota on metagenomics, etc.). LRRK2: leucine-rich repeat kinase 2 gene; SNCA: alpha-synuclein gene; GBA: glucocerebrosidase gene; P-tau181: tau phosphorylated at threonine 181; α-syn fibrils: oligomeric (presumably “toxic”) forms of α-synuclein; Aβ1-42: amyloid beta 1-42; NF-L: neurofilament light chain.
PARKINSON DISEASES: PREDICTIONS FOR THE NEXT CENTURY
Like other fields of medicine, neurodegenerative disorders will move from reductionism to systems biology, in which genotype and cellular biological signals prevail as anchors for how diseases are diagnosed and treated. With the recognition that phenotype does not predict biology or pathophysiology, we will no longer espouse the concept that greater granularity in the clinical characterization of patients, based on more sophisticated semiologic, neuroimaging, or wearable technologies-based activity sensors, will allows us to select patient subtypes for specific disease-modifying treatments. Even after the needed implementation of an agnostic, non-hypothesis driven biomarker development program (biomarker-driven subtyping), we may be disappointed to discover that relatively homogenous molecular clusters may still fall into clinically heterogenous populations, defying further the primacy of semiology as arbiter of biology. Big data will not be used to find order in complexity but to confirm or refine what we already know. Many concepts associated with the relief of convergence (explaining phenomena to make sense to us) will be replaced by the anxiety of divergence (no single explanation can sum the range of observations) (Table 1).
Table 1
Reductionism and related ideas that will die
| Idea | Why it makes sense | Why it will die |
| Unification | Few principles must explain many natural phenomena. Mathematics can explain natural patterns. | Mathematics isn’t physics. We can only construct approximate models.* |
| Essentialism | People and events must belong to discrete categories. | There exists a continuous spectrum of intermediates. |
| Cause-Effect | Events must be organized into chains or causes and effects. A gene seems to cause a trait like height or a disease such as cancer. | Complex dynamical systems of living organisms have patterns of information flow that defy our tools for storytelling. |
| Linnaean Classification | The vast biological diversity can be ordered based on the description of their similarities and differences. | Taxonomies do not equate with basic biological processes, impeding discovery of treatments. |
| One genome per individual | Single-cell sequencing technology works because all 37 trillion cells have the same copy of one’s genome. | A high proportion of brain cells have structural DNA variants (mosaicism). |
| Race | Skin color, hair form, cranial shape cluster into some diseases. Racial groups may give order to biology. | Racial patterns are complex genetic mixtures created by the sharing of similar exposures. |
| Nature versus Nurture | You can separate one from the other like Newtonian space and time: heritability is immutable. | As Einsteinian spacetime, they are intertwined. Heritability is affected by the environment. |
| Big Data | Larger n is better because we can detect small effects. More events and effects become salient. | Significant effects on low n means effect is bigger. Big data may be 99% irrelevant. |
| Underlying constructs | ||
| Reductionism: PD as a clinico-pathologic entity | Systems Biology: PD as a collection of biological entities | |
| A complex system is nothing but the sum of its parts and can be reduced to its individual constituents. Exceptions to this model are physiological “noise” obscuring the “true” signal. | “Noise” turn into profiles of unique biological systems or subsystems evolving in humans into intricate phenotypes that cannot be reduced. | |
Inspired from “This Idea Must Die: Scientific Theories That Are Blocking Progress” [15]. *Even the most sacred unifications are approximations: equations describing electricity and magnetism are perfectly symmetric only in an empty space. Unification from Marcelo Gleiser (Theoretical physicist); Essentialism from Richard Dawkins (Evolutionary biologist); Cause and Effect from W. Daniel Hillis (Physicist); Linnaean Classification (“Numbering Nature”) from Kurt Gray (Social psychologist); One genome per individual from Eric J. Topol (Professor of genomics); Race from Nina Jablonski (Biological anthropologist); Nature versus Nurture from Timo Hannay (Director of Digital Science); and Big Data from Melanie Swan (Applied genomics expert).
Prediction 1. The death of Parkinson’s as a puzzle of many interdigitating cellular derangements
In 2022, following earlier therapeutic success in a cohort of PD-GBA patients, the first anti-GBA therapies will suffer a setback when tested in a cohort of “sporadic PD” (individuals with clinically-defined diagnosis, possibly selected on the basis of low glucocerebrosidase levels, but without harboring GBA mutations). Clinician scientists will consider the negative outcome evidence that PD-GBA is a distinct disease and not a piece in the puzzle to “understand PD”, although some will advocate for a repeat trial with “prodromal” patients. By 2025, we will no longer expect that therapies that might work for any disease subtype, such as for PD-GBA, might work for everyone with “idiopathic” PD. Extensive efforts will be undertaken to construct separate diagnostic criteria that incorporate clinical as well as genetic, molecular, and pathologic biomarkers for each PD subtype/endophenotype.
Prediction 2. Animal models to confirm human disease-based hypothesis; not to create new hypothesis
Animal models will not be relied upon to “recapitulate the complexity of a human disease” but to understand specific mechanisms suggested by studies on biospecimens and neuroimaging techniques in humans. Potential therapies demonstrated to affect a presumed pathogenic pathway in an animal model will require a demonstration that the same mechanism (biomarker of pathogenesis) is active and pathogenic in the intended recipients. NIH research applications will no longer require that prospective trials in humans should be first reproduced in mice or insist on review policies that enforce animal models as gold standard for predicting the safety and particularly the efficacy of drugs. High-risk/high-gain non-hypothesis-based exploratory proposals will begin to replace hypothesis-based proposals.
Prediction 3. Smaller, smarter clinical trials
Future trials will select and stratify study participants by mechanism and not phenotype, composed of tens of biologically homogenous PD subjects (with “homogeneity” defined by the upper or lower quartiles or quintiles of any biological/molecular abnormality suitable for a targeted biological/molecular intervention) rather than hundreds of clinically probable early or even “prodromal” PD subjects lacking such biomarker(s). Treatments with putative disease modifying properties, considered ineffective in prior large clinical trials, will be reexamined as potentially useful for a smaller subset of biologically-defined subsets. Traditional phases of clinical trial development (preclinical and clinical phases 1 through 3) will be replaced with nimbler learning and confirming phases [12]. Basket and other adaptive trial designs will become standard. Trials of combined drugs or “cocktail” treatments will harness additional potential benefits by targeting more than one dominant pathogenic mechanism in any given disease subtype [13].
Prediction 4. Larger, phenotype-agnostic biomarker studies of aging
Deeply-phenotyped large population-based aging cohorts will facilitate analyses anchored on outlier biological signals, especially those reflective of pathogenic mechanisms for which putative therapies already exist. Biomarkers of interest may not segregate into homogenous phenotypic clusters. Discoveries made on biomarker-driven phenotype-agnostic studies of aging will reinvigorate the efforts on repurposing medications for disease modification. Proof-of-concept clinical trials of putative disease-modifying drugs, targeting specific pathogenic mechanisms, will be embedded within biomarker-development cohorts of subjects with evidence of the pathogenic dysfunction of interest, and accounting for the confounding or attenuating role of aging and concomitant pathologies (e.g., vascular, amyloid).
CONCLUSIONS AND FINAL PREDICTIONS
The decades following 2020 will witness a seismic change in the approach to ushering precision medicine for PD. The “sum of parts” clinico-pathologic reductionism that has defined Parkinson as a single but heterogenous disease will be fully replaced with biomarker development and disease modification efforts responsive to intra-individual combinations of clinical, pathologic, genetic, and molecular/biologic features. The idea of PD representing many diseases has received ceremonious lip service for most of the past 15 years [14] but walking the talk will soon begin in earnest. An agnostic, non-hypothesis-driven approach to biomarker development, anticipated to be far more expensive than the ongoing phenotype-based biomarker programs because of its inclusivity, will be needed in order to distinguishing biomarkers of causative mechanisms from non-specific or late mechanisms (reflecting reactive plasticity or self-perpetuating neurodegenerative cycles or biomarkers of response to injury), which are inappropriate or inadequate as therapeutic targets. Such diagnostic shift will also engender a therapeutic testing paradigm for neuroprotection, emulating the approach in oncology, using a tailored biology-specific but multiple-mechanism strategy.
CONFLICTS OF INTEREST
Dr. Espay has received grant support from the NIH, Great Lakes Neurotechnologies and the Michael J Fox Foundation; personal compensation as a consultant/scientific advisory board member for Abbvie, Adamas, Acadia, Acorda, Neuroderm, TEVA, Impax, Sunovion, Lundbeck, Osmotica Pharmaceutical, and US World Meds; publishing royalties from Lippincott Williams & Wilkins, Cambridge University Press, and Springer; and honoraria from Abbvie, UCB, USWorldMeds, Lundbeck, Acadia, Sunovion, the American Academy of Neurology, and the Movement Disorders Society.
Dr. Lang has served as an advisor for Abbvie, Allon Therapeutics, Avanir Pharmaceuticals, Biogen Idec, Boerhinger-Ingelheim, Ceregene, Lilly, Medtronic, Merck, Novartis, NeuroPhage Pharmaceuticals, Teva and UCB; received honoraria from Medtronic, Teva, UCB, AbbVie; received grants from Brain Canada, Canadian Institutes of Health Research, Edmond J Safra Philanthropic Foundation, Michael J. Fox Foundation, the Ontario Brain Institute, National Parkinson Foundation, Parkinson Society Canada, Tourette Syndrome Association, W. Garfield Weston Foundation; received publishing royalties from Saunders, Wiley-Blackwell, Johns Hopkins Press, and Cambridge University Press; and has served as an expert witness in cases related to the welding industry.
ACKNOWLEDGMENTS
This work was not supported or sponsored by any agency or individual. The text was fully created without the influence of companies or foundations with which the authors have had financial relationships. There was no ghost writing.
REFERENCES
[1] | Veronesi U , Saccozzi R , Del Vecchio M , Banfi A , Clemente C , De Lena M , Gallus G , Greco M , Luini A , Marubini E , Muscolino G , Rilke F , Salvadori B , Zecchini A , Zucali R ((1981) ) Comparing radical mastectomy with quadrantectomy, axillary dissection, and radiotherapy in patients with small cancers of the breast. N Engl J Med 305: , 6–11. |
[2] | Ostrowitzki S , Lasser RA , Dorflinger E , Scheltens P , Barkhof F , Nikolcheva T , Ashford E , Retout S , Hofmann C , Delmar P , Klein G , Andjelkovic M , Dubois B , Boada M , Blennow K , Santarelli L , Fontoura P ((2017) ) A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther 9: , 95. |
[3] | Salloway S , Sperling R , Fox NC , Blennow K , Klunk W , Raskind M , Sabbagh M , Honig LS , Porsteinsson AP , Ferris S , Reichert M , Ketter N , Nejadnik B , Guenzler V , Miloslavsky M , Wang D , Lu Y , Lull J , Tudor IC , Liu E , Grundman M , Yuen E , Black R , Brashear HR ((2014) ) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370: , 322–333. |
[4] | Doody RS , Raman R , Farlow M , Iwatsubo T , Vellas B , Joffe S , Kieburtz K , He F , Sun X , Thomas RG , Aisen PS , Siemers E , Sethuraman G , Mohs R ((2013) ) A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369: , 341–350. |
[5] | Doody RS , Thomas RG , Farlow M , Iwatsubo T , Vellas B , Joffe S , Kieburtz K , Raman R , Sun X , Aisen PS , Siemers E , Liu-Seifert H , Mohs R ((2014) ) Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 370: , 311–321. |
[6] | Braak H , Del Tredici K , Rub U , de Vos RA , Jansen Steur EN , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: , 197–211. |
[7] | Berg D , Postuma RB , Adler CH , Bloem BR , Chan P , Dubois B , Gasser T , Goetz CG , Halliday G , Joseph L , Lang AE , Liepelt-Scarfone I , Litvan I , Marek K , Obeso J , Oertel W , Olanow CW , Poewe W , Stern M , Deuschl G ((2015) ) MDS research criteria for prodromal Parkinson’s disease. Mov Disord 30: , 1600–1611. |
[8] | Espay AJ , Schwarzschild MA , Tanner CM , Fernandez HH , Simon DK , Leverenz JB , Merola A , Chen-Plotkin A , Brundin P , Kauffman MA , Erro R , Kieburtz K , Woo D , Macklin EA , Standaert DG , Lang AE ((2017) ) Biomarker-driven phenotyping in Parkinson’s disease: A translational missing link in disease-modifying clinical trials. Mov Disord 32: , 319–324. |
[9] | Steiner JA , Quansah E , Brundin P ((2018) ) The concept of alpha-synuclein as a prion-like protein: Ten years after. Cell Tissue Res 373: , 161–173. |
[10] | Fujita KA , Ostaszewski M , Matsuoka Y , Ghosh S , Glaab E , Trefois C , Crespo I , Perumal TM , Jurkowski W , Antony PM , Diederich N , Buttini M , Kodama A , Satagopam VP , Eifes S , Del Sol A , Schneider R , Kitano H , Balling R ((2014) ) Integrating pathways of Parkinson’s disease in a molecular interaction map. Mol Neurobiol 49: , 88–102. |
[11] | Lees AJ ((2007) ) Unresolved issues relating to the shaking palsy on the celebration of James Parkinson’s 250th birthday. Mov Disord 22: (Suppl 17), S327–S334. |
[12] | Cedarbaum JM ((2018) ) Elephants, Parkinson’s disease, and proof-of-concept clinical trials. Mov Disord 33: , 697–700. |
[13] | Lang AE , Espay AJ ((2018) ) Disease modification in Parkinson’s disease: Current approaches, challenges, and future considerations. Mov Disord 33: , 660–677. |
[14] | Calne DB , Mizuno Y ((2004) ) The neuromythology of Parkinson’s disease. Parkinsonism Relat Disord 10: , 319–322. |
[15] | Brockman JE ((2015) ) This Idea Must Die: Scientific Theories That Are Blocking Progress, HarperCollins Publishers, New York. |


