
The Role of TMEM230 Gene in Parkinson’s Disease
Abstract
Parkinson’s disease (PD) is a common neurodegenerative disease whose pathogenesis remains unknown. TMEM230 gene, encoding a transmembrane protein in secretory and recycling vesicle, has been recently identified as a novel disease-causing gene of autosomal dominant PD with Lewy pathology and typical clinical symptoms. Although its mutation and variants seem to be rare in PD patients, functional studies have indicated that TMEM230 protein probably plays an important role in secretory and recycling pathway and may be involved in Lewy pathological mechanism. Here we summarize current genetic and functional reports about TMEM230 and focus on its relation with PD.
INTRODUCTION
Parkinson’s disease (PD) (OMIM 168600) is the second most common neurodegenerative disease after Alzheimer’s disease (AD), with incidence rate of 0.014% per year in total population and 0.16% per year in people over the age of 65 in high-income countries [1]. The typical clinical symptoms of PD include progressive bradykinesia in combination with rest tremor, rigidity, postural instability, and numerous non-motor symptoms [2, 3]. The loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and the Lewy pathology including Lewy bodies and Lewy neurites have been identified as pathological features of PD [2]. PD was once thought to be a nongenetic disease and mainly caused by environmental factors, but as a result of advances in genetic research, this notion has gradually changed and there is growing recognition of genetic mechanisms in the pathogenesis of the disease [4]. To date, at least 23 disease-causing loci and 19 genes have been reported in monogenic PD pedigree, though the list of risk-associated genes is steadily growing [3, 5]. PD is now viewed as a complex neurodegenerative disorder resulting from genetic, environmental and other, yet unknown factors [4].
Unlike sporadic PD which accounts for about 90% of total PD cases, a considerable number of patients with monogenic form of PD have been found to have young-onset of symptoms, atypical clinical features, and lack Lewy pathology [1, 6, 7]. Only a few of PD causing mutations had been reported to be related to clinically typical PD with Lewy pathology [6]. Recently, Deng et al. reported that transmembrane protein 230 gene (TMEM230), may play a pathogenic role in a rare autosomal dominant PD (ADPD) with typical motor features and Lewy body pathology [8]. This finding provides new insights into the pathogenesis of PD-related neurodegeneration. The primary aim of this article is to review current genetic and functional data about TMEM230 and suggest how the discovery of this disease-causing gene can lead to pathogenesis-targeted therapy for PD.
PARK21 AND THE TMEM230 GENE
In 2014, a large Canadian Mennonite family with a PD phenotype was reported with c.2564A > G (p.N855S) variant in the DNAJC13 gene, located on chromosome 3q22 [9]. But this variant did not fully cosegregate with the disease as demonstrated by its presence in one unaffected individual died at the age of 87 years and absence in two PD cases and one parkinsonism case with progressive supranuclear palsy pathology, who belong to different branches and couldn’t be explained as sporadic cases because extremely low possibility (<10- 3) [8]. The causal gene locus was termed as PARK21 (OMIM 616361) in Online Mendelian Inheritance in Man (OMIM) according to the chronology of identification of the disease-causing gene loci. However, in 2016, Deng and colleagues proposed that TMEM230 c.422G > T (p.R141L) mutation, mapped to chromosome 20p13-p12.3, was the pathogenic mutation of PD in the same family with 13 available patients [8]. Though a TMEM230 mutation–freed patient with atypical parkinsonism phenotype which was still mild after 23 years of disease progression was evidenced, the TMEM230 mutation was the best genetic explanation for PD in this family under current known data, which may be explained by other conditions such as environmental or inconsistent genetic factors [10, 11]. The Canadian Mennonite family of mixed European ancestry included 14 enrolled family members with ADPD, mean age at onset of 67.0 years, and typical presentation of late-onset, levodopa-responsive PD [8, 12]. These patients had rigidity, bradykinesia, and rest tremor (in 57% patients), and dementia was present in 21% of cases [13]. Additionally, neuronal loss in substantia nigra and nucleus basalis, and Lewy bodies were found in brainstem at autopsies including α-synuclein stains, performed in three of cases [10, 13].
The TMEM230 gene, also called as chromosome 20 open reading frame 30 (C20orf30), covers a genomic region of about 13.2 kb with five exons [14]. TMEM230 mRNA expression is high in many tissues, including several regions of nervous system, such as midbrain, cerebellum, neocortex and spinal cord [8, 15]. Its four mRNA transcriptional variants encode two protein isoforms: the isoform-1 of 183 amino acids and the isoform-2 of 120 amino acids [8]. The isoform-2 accounts for more than 95% of total protein isoforms in humans, and presents alone in species spanning zebrafish to most mammals [8]. The highly conserved amino acid sequence of isoform-2 contains two transmembrane segments, with N-terminal and C-terminal regions exposing to the cytosol [8]. There is no other known protein with sequence identical or similar to TMEM230.
TMEM230 VARIANTS IDENTIFIED IN PD
Three other PD-associated TMEM230 variants, including two variants (p.Y92C and p.*184Wext*5) which were found through analyzing 832 North American PD cases and one variant (p.*184PGext*5) which were detected by 9 PD cases of 7 families from China were reported in original Deng et al.’s study [8]. The asymptomatic carriers with p.Y92C and p.*184PGext*5 variants suggested incomplete penetrance, similar to LRRK2 p.G2019S variant [8, 16].
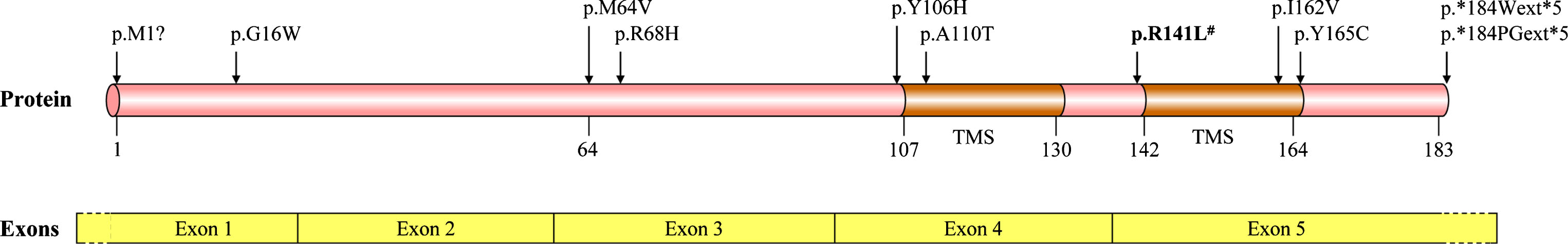
Subsequently, two novel variants p.G16W and p.M64V, and six known missense variants including p.Y106H (rs746223968), p.I162V (rs368707598), p.R68H (rs780460399), p.Y165C (rs758033952), p.M1? (rs768390203) and p.A110T were detected only in PD patients [17–22], though many other studies failed to find PD-related pathogenic variant in TMEM230 gene (Table 1) [23–31]. Because of these variants only observed in PD patients, and the incompletion of population genetic databases (e.g., ExAC and gnomAD) caused by the age-dependent and incomplete penetrance of the disorder, these variants probably exert a disease-causing or susceptibility role of PD. In 15 studies published, approximately 0.28% PD patients were found to harbor potential PD-related variants with full detection information of coding regions of the TMEM230 gene (Table 1) [8, 17–30]. Interestingly, most of the PD-related mutations and variants of TMEM230 gene detected to date were in the highly conserved sequence of isoform-2, and about half of these were in the regions around two transmembrane segments of this protein. This suggests that abnormal function of transmembrane segments in TMEM230 protein plays an important role in neurodegeneration (Fig. 1). Additionally, non-coding variants c.*746G > A (rs45610034), c.68 + 182G > A (rs149865687) and c.*161C > T were reported to be associated with PD risk or age at onset, though none of them passed the Bonferroni correction test and a large sample size will be needed to confirm the association [27, 31].
Table 1
Mutation/variants associated with PD detected in coding region of the TMEM230 gene
| Report | Geographic distribution/Ethnic background | Number of patients with PD [controls] | Detection region | Nucleotide change detected | Location | Amino acid change | Zygosity | Frequency in cases | MAF (ExAC) | MAF (gnomAD) |
|---|---|---|---|---|---|---|---|---|---|---|
| Deng et al. 2016 [8] | Canada | 1 PD family | Exome | c.422G > T | Exon 5 | p.R141L | Het | 12/13 | − | 8.963×10−6 (European Non-Finnish) |
| Northern America | 433 FPD and 399 SPD [1238] | Coding regions | c.551A > G | Exon 5 | p.*184Wext*5 | Het | 1/433 in FPD | − | − | |
| China | 225 FPD and 349 SPD [528] | Coding regions | c.550_552del TAGins CCCGGG | Exon 5 | p.*184PGext*5 | 5 Hom and 4 Het | 9/225 in FPD | − | − | |
| Giri et al. 2017 [17] | Caucasian | 1450 PD [2267] | Exome | c.316T > C | Exon 4 | p.Y106H | Het | 1/1450 | 1.501×10−5 (European Non-Finnish) | 1.798×10−5 (European Non-Finnish) |
| c.484A > G | Exon 5 | p.I162V | Het | 1/1450 | − | 1.578×10−5 (European Non-Finnish) | ||||
| Caucasian | 86 FPD [10] | Exome | − | − | − | − | − | − | − | |
| Baumann et al. 2017 [18] | Europe | 53 PD cases | Exome | c.203G > A | Exon 3 | p.R68H | Het | 1/53 | 4.548×10−5 (European Non-Finnish) | 5.541×10−5 (European Non-Finnish) |
| Quadri et al. 2017 [19] | Taiwan | 98 FPD and 717 SPD [417] | Exon 5 | c.494A > G | Exon 5 | p.Y165C | Het | 1/717 in SPD | 1.156×10−4 (East Asian) | 2.319×10−4 (East Asian) |
| Dutch | 31 FPD and 59 SPD | Exon 5 | − | − | − | − | − | − | − | |
| Caucasian | 266 PD probands | Coding regions | c.1A > G | Exon 1 | p.M1? | Het | 1/226 | 7.06×10−5 (Total) | 1.056×10−4 (European Non-Finnish) | |
| Yang et al. 2017 [20] | Southwestern China | 11 FPD and 355 SPD | Exons and exon-intron boundaries | c.46G > T | Exon 1 | p.G16W | Het | 1/355 in SPD | − | − |
| c.328G > A | Exon 4 | p.A110T | Het | 2/355 in SPD | − | 1.444×10−5 (Total) | ||||
| Wei et al. 2018 [21] | Southwestern China | 120 FPD [650] | Exons and exon-intron boundaries | c.46G > T | Exon 1 | p.G16W | Het | 1/120 | − | − |
| Tejera-Parrado et al. 2018 [22] | Southern Spanish | 148 FPD and 555 SPD [695] | Exons and exon-intron boundaries | c.190A > G | Exon 3 | p.M64V | − | 1/703 | − | − |
| Yan et al. 2017 [23] | China | 192 FPD and 1043 SPD [1252] | Exons and exon-intron boundaries | − | − | − | − | 0/1235 | − | − |
| Wu et al. 2017 [24] | Eastern China | 122 PD probands | Coding regions | − | − | − | − | 0/122 | − | − |
| Fan et al. 2017 [25] | Taiwan | 180 FPD | Exons and exon-intron boundaries | − | − | − | − | 0/180 | − | − |
| 500 SPD [992] | c.68G>A, c.275A>G, c.422G>T and c.551A>G | − | − | − | − | 0/500 | − | − | ||
| He et al. 2017 [26] | China | 207 FPD and 207 SPD [400] | Stop codon region | − | − | − | − | 0/414 | − | − |
| Shi et al. 2017 [27] | China | 550 SPD [560] | Coding regions and exon-intron boundaries | − | − | − | − | 0/550 | − | − |
| Ma et al. 2017 [28] | Singapore | 99 PD [99] | Coding regions | − | − | − | − | 0/99 | − | − |
| Buongarzone et al. 2017 [29] | Italy | 86 FPD | Exons | − | − | − | − | 0/86 | − | − |
| Conedera et al. 2018 [30] | Japan | 182 PD | Coding regions and exon-intron boundaries | − | − | − | − | 0/182 | − | − |
| Combined | All world | 6904 PD [7299] | Coding regions | Missense mutation/variants only in PD | Coding regions | Missense mutation/variants only in PD | All | 19/6115 (2.752×10−3) | − | − |
FPD, familial PD; SPD, sporadic PD; Het, heterozygous; Hom, homozygous; MAF, minor allele frequency; ExAC, Exome Aggregation Consortium database; gnomAD, Genome Aggregation Database. Data in reference 31 was not extracted due to the lack of detailed information [31].
Fig. 1
Missense mutation and variants associated with PD detected in TMEM230 coding regions. # The initial mutation detected in a large family with autosomal dominant and Lewy pathology confirmed PD. TMS, transmembrane segment.
THE POTENTIAL PATHOLOGICAL MECHANISM OF TMEM230 IN PD
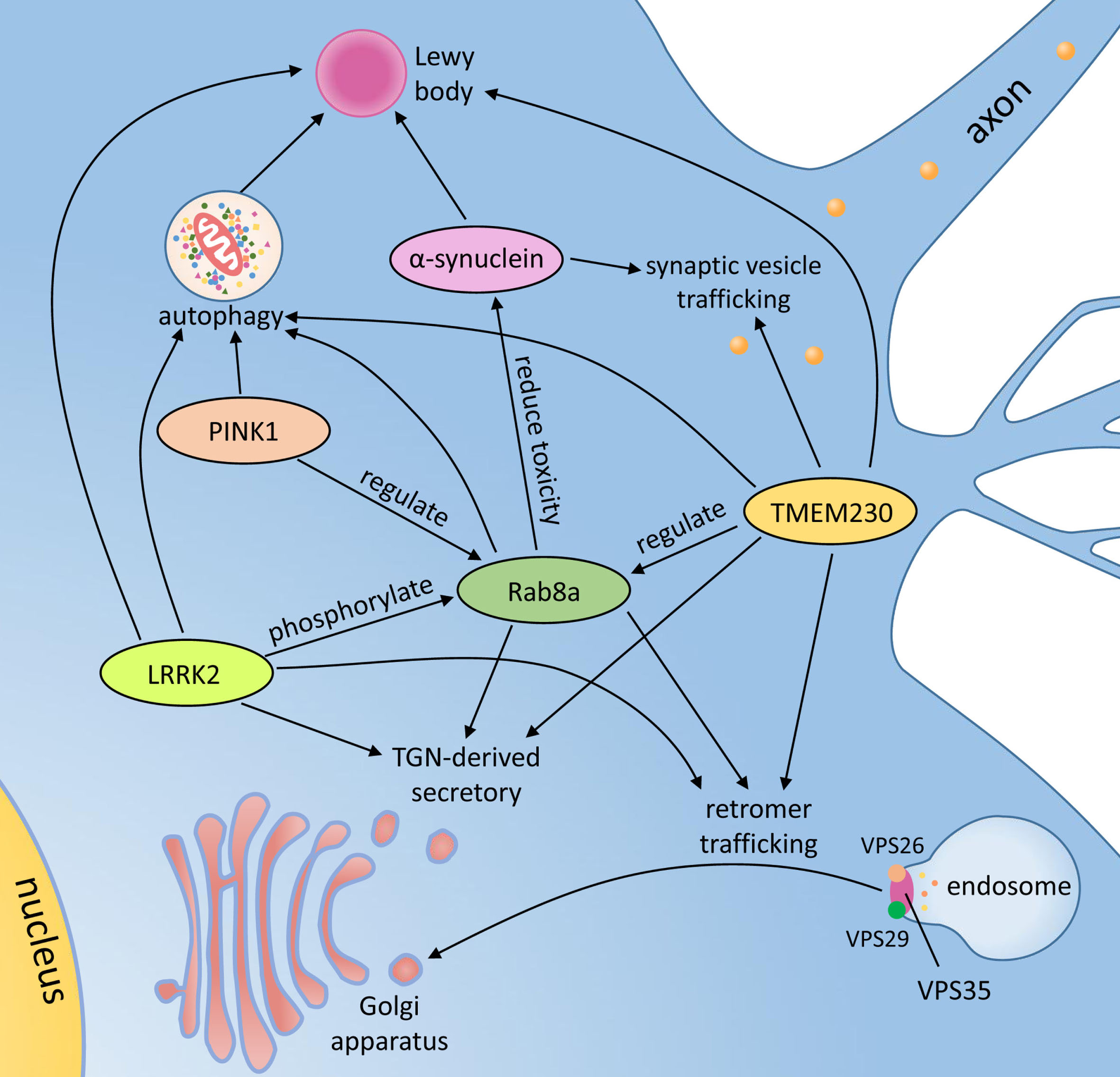
The TMEM230 protein is localized to vesicle structures in human SH-SY5Y cells, and the mouse ortholog distributes to the same subcelluar structures in brain neurons including dopaminergic neurons in the substantia nigra [8]. These vesicle structures predominantly co-localize with STX6, a protein mainly enriched in the trans-Golgi network (TGN), and with vesicular monoamine transporter type 2 (VMAT2), vacuolar protein sorting-35 (VPS35), Rab5a and Rab11a proteins, suggesting that TMEM230-positive vesicles are involved in the function of synaptic vesicles and recycling of endosomes [8]. Thus, TMEM230 appears to play an important role in many cellular functions, including synaptic vesicles trafficking, retromer trafficking, secretory autophagy and Golgi-derived vesicle secretion. As such, it shares in pathogenic pathways implicated other PD-causing genes, such as the SNCA, LRRK2, VPS35 and PINK1 [32].
Interaction with SNCA
Mutations in synuclein alpha (SNCA) gene, which encodes α-synuclein protein, the key component of Lewy body inclusions, have resulted in ADPD [6, 33]. As the first PD-related gene identified and labeled PARK1, the typical PD clinical phenotype of patients was also associated with characteristic Lewy body pathology [6, 33]. Although the physiological function of α-synuclein protein remains enigmatic, mounting evidence suggests a regulatory function in synapse, such as vesicle trafficking, synaptic vesicle pool maintenance and neurotransmitter release [33]. TMEM230 protein was detected in α-synuclein-positive Lewy bodies and Lewy neurites both in sporadic PD and dementia with Lewy bodies (DLB) cases [8]. Similar to α-synuclein, the TMEM230 protein was observed in the synaptic vesicle pool region in the rat brain neuron presynapse [8, 34]. Expression of PD-related TMEM230 variants resulted in significantly slower movement of synaptic vesicles and increased α-synuclein protein level compared to wild-type protein possibly due to impairment of autophagy-mediated clearance [8, 32]. In addition, the Rab8a protein whose function is connected with TMEM230, also interacts with α-synuclein, and its overexpression reduces α-synuclein-induced toxicity in vitro and improves α-synuclein-induced behavioral defects in fruit flies [32, 35]. Intriguingly, tmem230a, the zebrafish ortholog of human TMEM230, could affect angiogenic blood vessel growth though Delta/Notch signaling pathway [36], which may be involved in neurodegenerative disease and reduced by overexpressive or mutant α-synuclein protein [37, 38].
Interaction with LRRK2
Leucine-rich repeat kinase 2 (LRRK2) mutations represent the most common genetic cause of ADPD and nearly half of LRRK2-related PD cases had Lewy bodies [6, 39]. LRRK2 protein has been found to phosphorylate several members of Rab family which plays a key role in all forms of intracellular vesicular trafficking [40]. The Rab8a protein is one of substrates of LRRK2, and its phosphorylation may be increased 2-3 fold by LRRK2 p.G2019S mutation [40]. Thus LRRK2 kinase activity-dependent phosphorylation may lead to deficits in cell polarization, neurite outgrowth and directed migration [41]. The Rab8a-mediated secretory vesicle and retromer trafficking were impaired when TMEM230 lost function, similar to lack of LRRK2 protein [32]. This suggests that TMEM230 and LRRK2 may share Rab8a-mediated vesicle trafficking pathway in development of PD and Lewy pathology. Additionally, Notch signaling pathway which may be associated with TMEM230 was also regulated by LRRK2 through endosomal pathway [36, 42].
Interaction with VPS35
VPS35, a causal gene linked to ADPD, encodes a subunit of retromer complex [43]. The TMEM230 protein partially co-localizes with VPS35 protein and both regulate retromer trafficking function [8]. The expression of TMEM230-R141L mutant protein changed VPS35 and itself from perinuclear to punctate cytoplasmic distribution [32]. However, only one VPS35-PD autopsy report showed no immunostaining for α-synuclein and there was no neuronal loss or intraneuronal inclusions in the cortex and basal ganglia; the substantia nigra tissue was not available [44]. Further studies are warranted to clarify the similarities and differences between TMEM230 and VPS35 in pathogenesis of PD.
Interaction with PINK1
Many mutations of phosphatase and tensin homolog-induced putative kinase 1 (PINK1) gene have been identified in different families with autosomal recessive PD [45]. One early-onset PD patient with two compound heterozygous PINK1 mutations was reported to have neuronal loss and Lewy pathology in the SNpc [46]. This gene encodes PINK1 protein, a serine/threonine protein kinase whose activation caused phosphorylation of Rab8a at residue of serine 111 and significantly impaired Rab8a activation [47]. Further studies of Rab8a-involved pathway may help to elucidate the association between TMEM230 and PINK1 in pathogenesis of PD.
In summary, there is a growing body of evidence that TMEM230 protein and its interaction with Rab8a, SNCA, LRRK2 and PINK1 may lead to PD-related neurodegeneration (Fig. 2).
Fig. 2
The potential pathological mechanisms and associated proteins of TMEM230. TGN, trans-Golgi network; TMEM230, transmembrane protein 230; PINK1, phosphatase and tensin homolog-induced putative kinase 1; LRRK2, leucine-rich repeat kinase 2; VPS35, vacuolar protein sorting-35.
THE POTENTIAL ROLE OF TMEM230 IN OTHER DISEASES
The TMEM230 gene may be potentially related with other neurodegenerative diseases with Lewy pathology such as DLB and AD, multiple system atrophy (MSA) [48, 49]. In AD patients, the TMEM230 protein was increased in hippocampal neurons and aggregated in granulovacuolar and dystrophic neurites, two prominent pathological features of AD [50]. No MSA-risk variants have been found in the TMEM230 gene in 110 cases of MSA [51]. Furthermore, He et al. did not find stop codon variants in the TMEM230 gene in 200 Chinese patients with essential tremor [26].
CONCLUSION
Even after exciting acceleration of PD research during the past 50 years since the discovery of levodopa, the pathogenesis of this complex disorder remains enigmatic [2]. Notable discoveries, especially the advances in genetics of PD in recent 20 years have greatly changed our understanding on etiology and pathogenesis of PD [1]. It seems increasingly clear that PD is a highly complex neurological disease with heterogeneous clinical presentation, variable pathological features, and multifactorial causes [52]. Reveal of the association of phenotype-genotype, especially analysis of protein interaction network involving in Lewy body-confirmed PD-related genes, which highly mimics idiopathic PD, will help to understand the main underlying pathogenic mechanism of this complex disorder [52]. But only a few of PD-related mutations have been associated to PD with Lewy body pathology which remains a core feature of most PD cases, without precise mechanism known [1, 6].
Only a few PD-related TMEM230 variants could not cover up its significance in discovering pathogenesis of PD. Copy number variations including duplication and triplication in the TMEM230 gene, perhaps share a similar mechanism resulting in PD with dementia as the SNCA gene [53], as well as its epigenetic or non-coding regulatory factors, cannot be ignored in future studies. The application of quantitative PCR, digital PCR, whole genome sequencing and epigenetic strategies may help to identify more pathogenic mechanisms involving TMEM230 in PD, especially phenotype with dementia and other neurodegenerative disorders. Future research should focus on development of TMEM230 genetic animal models to better understand the role of TMEM230 in pathogenesis of neurodegeneration. These studies may also provide insight into potential treatment and prevention of PD and related disorders.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
H.D.’s research was supported by the National Key Research and Development Program of China [grant number 2016YFC1306604], the National Natural Science Foundation of China [grant number 81670216], the Natural Science Foundation of Hunan Province [grant numbers 2015JJ4088, 2016JJ2166 and 2017JJ3469], Grant for the Foster Key Subject of the Third Xiangya Hospital of Central South University [Clinical Laboratory Diagnostics], and the New Xiangya Talent Project of the Third Xiangya Hospital of Central South University [grant number 20150301], China. J.J. was supported by the Parkinson Foundation and Michael J. Fox Foundation for Parkinson Research.
REFERENCES
[1] | Ascherio A , Schwarzschild MA ((2016) ) The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol 15: , 1257–1272. |
[2] | Kalia LV , Lang AE ((2015) ) Parkinson’s disease. Lancet 386: , 896–912. |
[3] | Obeso JA , Stamelou M , Goetz CG , Poewe W , Lang AE , Weintraub D , Burn D , Halliday GM , Bezard E , Przedborski S , Lehericy S , Brooks DJ , Rothwell JC , Hallett M , DeLong MR , Marras C , Tanner CM , Ross GW , Langston JW , Klein C , Bonifati V , Jankovic J , Lozano AM , Deuschl G , Bergman H , Tolosa E , Rodriguez-Violante M , Fahn S , Postuma RB , Berg D , Marek K , Standaert DG , Surmeier DJ , Olanow CW , Kordower JH , Calabresi P , Schapira AHV , Stoessl AJ ((2017) ) Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov Disord 32: , 1264–1310. |
[4] | Rousseaux MWC , Shulman JM , Jankovic J ((2017) ) Progress toward an integrated understanding of Parkinson’s disease. F1000Res 6: , 1121. |
[5] | Deng H , Wang P , Jankovic J ((2017) ) The genetics of Parkinson disease. Ageing Res Rev 42: , 72–85. |
[6] | Langston JW , Schule B , Rees L , Nichols RJ , Barlow C ((2015) ) Multisystem Lewy body disease and the other parkinsonian disorders. Nat Genet 47: , 1378–1384. |
[7] | Alcalay RN , Caccappolo E , Mejia-Santana H , Tang MX , Rosado L , Orbe Reilly M , Ruiz D , Louis ED , Comella CL , Nance MA , Bressman SB , Scott WK , Tanner CM , Mickel SF , Waters CH , Fahn S , Cote LJ , Frucht SJ , Ford B , Rezak M , Novak KE , Friedman JH , Pfeiffer RF , Marsh L , Hiner B , Payami H , Molho E , Factor SA , Nutt JG , Serrano C , Arroyo M , Ottman R , Pauciulo MW , Nichols WC , Clark LN , Marder KS ((2014) ) Cognitive and motor function in long-duration PARKIN-associated Parkinson disease. JAMA Neurol 71: , 62–67. |
[8] | Deng HX , Shi Y , Yang Y , Ahmeti KB , Miller N , Huang C , Cheng L , Zhai H , Deng S , Nuytemans K , Corbett NJ , Kim MJ , Deng H , Tang B , Yang Z , Xu Y , Chan P , Huang B , Gao XP , Song Z , Liu Z , Fecto F , Siddique N , Foroud T , Jankovic J , Ghetti B , Nicholson DA , Krainc D , Melen O , Vance JM , Pericak-Vance MA , Ma YC , Rajput AH , Siddique T ((2016) ) Identification of TMEM230 mutations in familial Parkinson’s disease. Nat Genet 48: , 733–739. |
[9] | Vilarino-Guell C , Rajput A , Milnerwood AJ , Shah B , Szu-Tu C , Trinh J , Yu I , Encarnacion M , Munsie LN , Tapia L , Gustavsson EK , Chou P , Tatarnikov I , Evans DM , Pishotta FT , Volta M , Beccano-Kelly D , Thompson C , Lin MK , Sherman HE , Han HJ , Guenther BL , Wasserman WW , Bernard V , Ross CJ , Appel-Cresswell S , Stoessl AJ , Robinson CA , Dickson DW , Ross OA , Wszolek ZK , Aasly JO , Wu RM , Hentati F , Gibson RA , McPherson PS , Girard M , Rajput M , Rajput AH , Farrer MJ ((2014) ) DNAJC13 mutations in Parkinson disease. Hum Mol Genet 23: , 1794–1801. |
[10] | Deng HX , Siddique T ((2017) ) Identification of TMEM230 mutations in familial Parkinson’s disease (response to comments). bioRxiv doi: 10.1101/170852 |
[11] | Farrer MJ , Milnerwood AJ , Follett J , Guella I ((2017) ) TMEM230 is not a gene for Parkinson disease. bioRxiv doi: 10.1101/097030 |
[12] | Pagano G , Ferrara N , Brooks DJ , Pavese N ((2016) ) Age at onset and Parkinson disease phenotype. Neurology 86: , 1400–1407. |
[13] | Appel-Cresswell S , Rajput AH , Sossi V , Thompson C , Silva V , McKenzie J , Dinelle K , McCormick SE , Vilarino-Guell C , Stoessl AJ , Dickson DW , Robinson CA , Farrer MJ , Rajput A ((2014) ) Clinical, positron emission tomography, and pathological studies of DNAJC13 p.N855S Parkinsonism. Mov Disord 29: , 1684–1687. |
[14] | Mandemakers W , Quadri M , Stamelou M , Bonifati V ((2017) ) TMEM230: How does it fit in the etiology and pathogenesis of Parkinson’s disease? Mov Disord 32: , 1159–1162. |
[15] | Fagerberg L , Hallstrom BM , Oksvold P , Kampf C , Djureinovic D , Odeberg J , Habuka M , Tahmasebpoor S , Danielsson A , Edlund K , Asplund A , Sjostedt E , Lundberg E , Szigyarto CA , Skogs M , Takanen JO , Berling H , Tegel H , Mulder J , Nilsson P , Schwenk JM , Lindskog C , Danielsson F , Mardinoglu A , Sivertsson A , von Feilitzen K , Forsberg M , Zwahlen M , Olsson I , Navani S , Huss M , Nielsen J , Ponten F , Uhlen M ((2014) ) Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteomics 13: , 397–406. |
[16] | Healy DG , Falchi M , O’Sullivan SS , Bonifati V , Durr A , Bressman S , Brice A , Aasly J , Zabetian CP , Goldwurm S , Ferreira JJ , Tolosa E , Kay DM , Klein C , Williams DR , Marras C , Lang AE , Wszolek ZK , Berciano J , Schapira AH , Lynch T , Bhatia KP , Gasser T , Lees AJ , Wood NW ((2008) ) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol 7: , 583–590. |
[17] | Giri A , Mok KY , Jansen I , Sharma M , Tesson C , Mangone G , Lesage S , Bras JM , Shulman JM , Sheerin UM , International Parkinson’s Disease Consortium , Diez-Fairen M , Pastor P , Marti MJ , Ezquerra M , Tolosa E , Correia-Guedes L , Ferreira J , Amin N , van Duijn CM , van Rooij J , Uitterlinden AG , Kraaij R , Nalls M , Simon-Sanchez J ((2017) ) Lack of evidence for a role of genetic variation in TMEM230 in the risk for Parkinson’s disease in the Caucasian population. Neurobiol Aging 50: , 167.e11–167.e13. |
[18] | Baumann H , Wolff S , Munchau A , Hagenah JM , Lohmann K , Klein C ((2017) ) Evaluating the role of TMEM230 variants in Parkinson’s disease. Parkinsonism Relat Disord 35: , 100–101. |
[19] | Quadri M , Breedveld GJ , Chang HC , Yeh TH , Guedes LC , Toni V , Fabrizio E , De Mari M , Thomas A , Tassorelli C , Rood JP , Saddi V , Chien HF , Kievit AJ , Boon AJ , Stocchi F , Lopiano L , Abbruzzese G , Cortelli P , Meco G , Cossu G , Barbosa ER , Ferreira JJ , International Parkinsonism Genetics Network , Lu CS , Bonifati V ((2017) ) Mutations in TMEM230 are not a common cause of Parkinson’s disease. Mov Disord 32: , 302–304. |
[20] | Yang X , An R , Xi J , Zheng J , Chen Y , Huang H , Tian S , Zhao Q , Ning P , Xu Y ((2017) ) Sequencing TMEM230 in Chinese patients with sporadic or familial Parkinson’s disease. Mov Disord 32: , 800–802. |
[21] | Wei Q , Ou R , Zhou Q , Chen Y , Cao B , Gu X , Zhao B , Wu Y , Song W , Shang HF ((2018) ) TMEM230 mutations are rare in Han Chinese patients with autosomal dominant Parkinson’s disease. Mol Neurobiol 55: , 2851–2855. |
[22] | Tejera-Parrado C , Jesus S , Lopez-Ruiz A , Buiza-Rueda D , Bonilla-Toribio M , Bernal-Bernal I , Perinan MT , Vargas-Gonzalez L , Gomez-Garre P , Mir P ((2018) ) TMEM230 in Parkinson’s disease in a southern Spanish population. PLoS One 13: , e0197271. |
[23] | Yan W , Tang B , Zhou X , Lei L , Li K , Sun Q , Xu Q , Yan X , Guo J , Liu Z ((2017) ) TMEM230 mutation analysis in Parkinson’s disease in a Chinese population. Neurobiol Aging 49: , 219.e1–219.e3. |
[24] | Wu H , Zheng X , Cen Z , Xie F , Chen Y , Lu X , Luo W ((2017) ) Genetic analysis of the TMEM230 gene in Chinese patients with familial Parkinson disease. Parkinsonism Relat Disord 36: , 105–106. |
[25] | Fan TS , Lin CH , Lin HI , Chen ML , Wu RM ((2017) ) Lack of TMEM230 mutations in patients with familial and sporadic Parkinson’s disease in a Taiwanese population. Am J Med Genet B Neuropsychiatr Genet 174: , 751–756. |
[26] | He YC , Huang P , Li QQ , Sun Q , Li DH , Wang T , Shen JY , Chen SD ((2017) ) TMEM230 stop codon mutation is rare in Parkinson’s disease and essential tremor in eastern China. Mov Disord 32: , 301–302. |
[27] | Shi CH , Li F , Shi MM , Yang ZH , Mao CY , Zhang SY , Wang H , Cheng Y , Yang J , Wu J , Xu YM ((2017) ) Genetic analysis of the TMEM230 gene in Chinese Han patients with Parkinson’s disease. Sci Rep 7: , 1190. |
[28] | Ma D , Foo JN , Yulin Ng E , Zhao Y , Liu JJ , Tan EK ((2017) ) Screening for TMEM230 mutations in young-onset Parkinson’s disease. Neurobiol Aging 58: , 239.e9–239.e10. |
[29] | Buongarzone G , Monfrini E , Franco G , Trezzi I , Borellini L , Frattini E , Melzi V , Di Caprio AC , Ronchi D , Monzio Compagnoni G , Cogiamanian F , Ardolino G , Bresolin N , Comi GP , Corti S , Di Fonzo A ((2017) ) Mutations in TMEM230 are rare in autosomal dominant Parkinson’s disease. Parkinsonism Relat Disord 39: , 87–88. |
[30] | Conedera SA , Li Y , Funayama M , Yoshino H , Nishioka K , Hattori N ((2018) ) Genetic analysis of TMEM230 in Japanese patients with familial Parkinson’s disease. Parkinsonism Relat Disord 48: , 107–108. |
[31] | Ibanez L , Dube U , Budde J , Black K , Medvedeva A , Davis AA , Perlmutter JS , Benitez BA , Cruchaga C ((2017) ) TMEM230 in Parkinson’s disease. Neurobiol Aging 56: , 212.e1–212.e3. |
[32] | Kim MJ , Deng HX , Wong YC , Siddique T , Krainc D ((2017) ) The Parkinson’s disease-linked protein TMEM230 is required for Rab8a-mediated secretory vesicle trafficking and retromer trafficking. Hum Mol Genet 26: , 729–741. |
[33] | Deng H , Yuan L ((2014) ) Genetic variants and animal models in SNCA and Parkinson disease. Ageing Res Rev 15: , 161–176. |
[34] | Burre J ((2015) ) The synaptic function of alpha-synuclein. J Parkinsons Dis 5: , 699–713. |
[35] | Yin G , Lopes da Fonseca T , Eisbach SE , Anduaga AM , Breda C , Orcellet ML , Szego EM , Guerreiro P , Lazaro DF , Braus GH , Fernandez CO , Griesinger C , Becker S , Goody RS , Itzen A , Giorgini F , Outeiro TF , Zweckstetter M ((2014) ) alpha-Synuclein interacts with the switch region of Rab8a in a Ser129 phosphorylation-dependent manner. Neurobiol Dis 70: , 149–161. |
[36] | Carra S , Sangiorgio L , Pelucchi P , Cermenati S , Mezzelani A , Martino V , Palizban M , Albertini A , Gotte M , Kehler J , Deflorian G , Beltrame M , Giordano A , Reinbold R , Cotelli F , Bellipanni G , Zucchi I ((2018) ) Zebrafish Tmem230a cooperates with the Delta/Notch signaling pathway to modulate endothelial cell number in angiogenic vessels. J Cell Physiol 233: , 1455–1467. |
[37] | Ables JL , Breunig JJ , Eisch AJ , Rakic P ((2011) ) Not(ch) just development: Notch signalling in the adult brain. Nat Rev Neurosci 12: , 269–283. |
[38] | Crews L , Mizuno H , Desplats P , Rockenstein E , Adame A , Patrick C , Winner B , Winkler J , Masliah E ((2008) ) Alpha-synuclein alters Notch-1 expression and neurogenesis in mouse embryonic stem cells and in the hippocampus of transgenic mice. J Neurosci 28: , 4250–4260. |
[39] | Kalia LV, Lang AE, Hazrati LN, Fujioka S, Wszolek ZK, Dickson DW, Ross OA, Van Deerlin VM, Trojanowski JQ, Hurtig HI, Alcalay RN, Marder KS, Clark LN, Gaig C, Tolosa E, Ruiz-Martinez J, Marti-Masso JF, Ferrer I, Lopez de Munain A, Goldman SM, Schule B, Langston JW, Aasly JO, Giordana MT, Bonifati V, Puschmann A, Canesi M, Pezzoli G, Maues De Paula A, Hasegawa K, Duyckaerts C, Brice A, Stoessl AJ, Marras C ((2015) ) Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol 72: , 100–105. |
[40] | Steger M , Tonelli F , Ito G , Davies P , Trost M , Vetter M , Wachter S , Lorentzen E , Duddy G , Wilson S , Baptista MA , Fiske BK , Fell MJ , Morrow JA , Reith AD , Alessi DR , Mann M ((2016) ) Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 5: , e12813. |
[41] | Madero-Perez J , Fdez E , Fernandez B , Lara Ordonez AJ , Blanca Ramirez M , Gomez-Suaga P , Waschbusch D , Lobbestael E , Baekelandt V , Nairn AC , Ruiz-Martinez J , Aiastui A , Lopez de Munain A , Lis P , Comptdaer T , Taymans JM , Chartier-Harlin MC , Beilina A , Gonnelli A , Cookson MR , Greggio E , Hilfiker S ((2018) ) Parkinson disease-associated mutations in LRRK2 cause centrosomal defects via Rab8a phosphorylation. Mol Neurodegener 13: , 3. |
[42] | Imai Y , Kobayashi Y , Inoshita T , Meng H , Arano T , Uemura K , Asano T , Yoshimi K , Zhang CL , Matsumoto G , Ohtsuka T , Kageyama R , Kiyonari H , Shioi G , Nukina N , Hattori N , Takahashi R ((2015) ) The Parkinson’s disease-associated protein kinase LRRK2 modulates notch signaling through the endosomal pathway. PLoS Genet 11: , e1005503. |
[43] | Deng H , Gao K , Jankovic J ((2013) ) The VPS35 gene and Parkinson’s disease. Mov Disord 28: , 569–575. |
[44] | Wider C , Skipper L , Solida A , Brown L , Farrer M , Dickson D , Wszolek ZK , Vingerhoets FJ ((2008) ) Autosomal dominant dopa-responsive parkinsonism in a multigenerational Swiss family. Parkinsonism Relat Disord 14: , 465–470. |
[45] | Kawajiri S , Saiki S , Sato S , Hattori N ((2011) ) Genetic mutations and functions of PINK1. Trends Pharmacol Sci 32: , 573–580. |
[46] | Samaranch L , Lorenzo-Betancor O , Arbelo JM , Ferrer I , Lorenzo E , Irigoyen J , Pastor MA , Marrero C , Isla C , Herrera-Henriquez J , Pastor P ((2010) ) PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133: , 1128–1142. |
[47] | Lai YC , Kondapalli C , Lehneck R , Procter JB , Dill BD , Woodroof HI , Gourlay R , Peggie M , Macartney TJ , Corti O , Corvol JC , Campbell DG , Itzen A , Trost M , Muqit MM ((2015) ) Phosphoproteomic screening identifies Rab GTPases as novel downstream targets of PINK1. EMBO J 34: , 2840–2861. |
[48] | Hamilton RL ((2000) ) Lewy bodies in Alzheimer’s disease: A neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol 10: , 378–384. |
[49] | Barker RA , Williams-Gray CH ((2016) ) Review: The of clinical features seen with alpha synuclein pathology. Neuropathol Appl Neurobiol 42: , 6–19. |
[50] | Siedlak SL , Jiang Y , Huntley ML , Wang L , Gao J , Xie F , Liu J , Su B , Perry G , Wang X ((2017) ) TMEM230 accumulation in granulovacuolar degeneration bodies and dystrophic neurites of Alzheimer’s disease. J Alzheimers Dis 58: , 1027–1033. |
[51] | Yang X , An R , Xi J , Zhen J , Chen Y , Huang H , Tian S , Zhao Q , Ning P , Xu Y ((2017) ) Sequence TMEM230 gene in patients with multiple system atrophy in a southwest Chinese population: A pilot study. J Neurol Sci 375: , 264–265. |
[52] | Thenganatt MA , Jankovic J ((2014) ) Parkinson disease subtypes. JAMA Neurol 71: , 499–504. |
[53] | Ross OA , Braithwaite AT , Skipper LM , Kachergus J , Hulihan MM , Middleton FA , Nishioka K , Fuchs J , Gasser T , Maraganore DM , Adler CH , Larvor L , Chartier-Harlin MC , Nilsson C , Langston JW , Gwinn K , Hattori N , Farrer MJ ((2008) ) Genomic investigation of alpha-synuclein multiplication and parkinsonism. Ann Neurol 63: , 743–750. |


