
Modeling Parkinson’s Disease Using Patient-specific Induced Pluripotent Stem Cells
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disorder. It is characterized by the degeneration of nigral dopaminergic (DA) neurons. While over 90% of cases are idiopathic, without a clear etiology, mutations in many genes have been linked to rare, familial forms of PD. It has been quite challenging to develop effective animal models of PD that capture salient features of PD. The discovery of induced pluripotent stem cells (iPSCs) makes it possible to generate patient-specific DA neurons to study PD. Here, we review the methods for the generation of iPSCs and discuss previous studies using iPSC-derived neurons from monogenic forms of PD. These investigations have revealed several converging pathways that intersect with the unique vulnerabilities of human nigral DA neurons. With the rapid development in stem cell biology, it is possible to generate patient-specific neurons that will be increasingly similar to those in the brain of the patient. Combined with the ability to edit the genome to generate isogenic iPSCs, the generation and analysis of patient-specific midbrain DA neurons will transform PD research by providing a valuable tool for mechanistic study and drug discovery.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after Alzheimer’s disease. PD incidence increases with age; its incidence is 1.5% among population over the age of 55, 1.8% among people over the age of 65 and 2.5% among population over the age of 70 [1]. With increasing longevity worldwide, the societal impact of PD will become more severe. PD patients typically suffer from debilitating motor deficits such as tremor, limb rigidity and slowness of movements (bradykinesia) [2]. They often also have non-motor symptoms such as dementia, depression, sleep disorders, constipation, loss of smell, etc. [3]. Neuropathologically, PD is defined by the progressive loss of dopaminergic (DA) neurons in substantia nigra pars compacta. Approximately 90% of PD cases are idiopathic, without a clear family history. By studying patients with rare, familial forms of PD, mutations in a number of genes have been linked to PD [4]. Among these genes, mutations of leucine-rich repeat kinase 2 (LRRK2), SNCA (encoding α-synuclein), Vacuolar Protein Sorting 35 (VPS35) have been linked to autosomal dominant PD, whereas mutations of PARK2 (encoding Parkin), PTEN-induced putative kinase (PINK1) and PARK7 (encoding DJ-1) cause autosomal recessive PD [5]. Despite the identification of these genetic mutations, the lack of a clear understanding on how these mutations cause PD hampers the development of disease-modifying therapies.
One major barrier to PD research is the lack of live brain tissue for invasive studies, such as experiments that require the destruction of live cells to analyze cellular components. As a substitute, the field has been relying on animal models of PD. Pharmacological ablation of nigral DA neurons in animal models of PD has been very useful in studying the degeneration of nigral DA neurons, but they do not reproduce the complex phenotypes seen in PD [6, 7]. Genetic mouse models of PD have significant challenges in recapitulating the selective degeneration of nigral DA neurons in PD patients [8]. Patient-specific induced pluripotent stem cells (iPSCs) represent a very useful and complementary approach to studying PD [9]. Neurons derived from iPSCs of patients with monogenic forms of PD can be used to study how mutations of a single gene generate PD-relevant phenotypes. Such information, taken together with those generated from other approaches, such as animal models and immortalized cell lines, has revealed important insights on mechanisms of PD pathogenesis. Based on mechanistic understanding of PD, a drug discovery program can be implemented on patient-specific neurons to identify agents that are protective against PD phenotypes. The discussions below explore some of these possibilities that are now available to PD research and drug discovery.
Generation of iPSCs
The discovery of induced pluripotency is a landmark breakthrough [10] that enables the conversion of easily accessible types of human somatic cells, such as skin or blood cells, into iPSCs. Shinya Yamanaka and colleagues make the initial discovery in 2006 by using retroviral transduction of Oct4, Sox2, Klf4 and c-myc (known as Yamanaka factors) into mouse embryonic fibroblasts or tail fibroblasts. Overexpression of Yamanaka factors reprograms the epigenome of the cells from that of a fibroblast to a pluripotent state and generates iPSCs [11]. Subsequently, adult human dermal fibroblasts are reprogrammed to iPSCs with the same method in 2007 [12]. These experiments have galvanized stem cell research by making it possible to use patient-specific iPSC-derived cells to study human diseases. Gene expression studies and functional studies show that variations between different lines of human embryonic stem cells (hESCs) and variations between different lines of iPSCs have significant overlap. For virtually all practical purposes, iPSCs and hESCs are essentially equivalent [13]. Two main advantages of iPSCs are that they avoid the destruction of human embryos and can be generated from readily available cells from any donors.
Generation of footprint-free iPSCs
Reprogramming methods that utilize retrovirus, lentivirus, or transposon introduce transgenes into the genome of the cells being reprogrammed. It carries the risk of insertional mutations and oncogenic transformation, as both c-Myc and Klf4 are oncogenes. Several methods have been developed to reprogram somatic cells to iPSCs without genomic integration, generating footprint-free iPSCs that have the same genome as the original source cells. Using non-integrating episomal plasmids that support prolonged expression of foreign genes, Yamanaka’s group has developed a simple and robust method to reprogram human somatic cells (e.g., human fibroblasts) to iPSCs [14]. Similarly, a plasmid of “minicircle” DNA that contains a single cassette of four reprogramming factors (Oct4, Sox2, Lin28, Nanog) plus a green fluorescent protein (GFP) reporter gene is used to reprogram human somatic cells to iPSCs [15]. Modified RNAs encoding the four Yamanaka factors have been used to reprogram multiple human cell types to pluripotency [16]. Another strategy is to use recombinant proteins. For example, iPSCs are generated from human fibroblasts by directly delivering four reprogramming proteins (Oct4, Sox2, Klf4, and c-Myc) fused with a cell penetrating peptide [17]. Similarly, purified recombinant Oct4 and Sox2 fused with the cell-penetrant TAT peptide are used to generate iPSCs for clinical applications [18, 19], as the reprogramming factors do not include any oncogenes and only transiently modify the epigenome of the source cells. Some integration-free viruses, such as adeno-associated viruses (AAV) [20] and Sendai Virus (SeV) [21], are used in iPSC derivation. Both these viruses will be gradually diluted and degraded with each cell division. These footprint-free reprogramming methods provide many options for a variety of applications, depending on the circumstances.
Xeno-free iPSCs
Human iPSCs and hESCs are routinely cultured on mouse embryonic fibroblast feeders. The presence of animal-derived components, which may contain pathogens found in animals, pose unacceptable risks for clinical applications such as transplantation of iPSC-derived cells. Much effort has been directed at establishing xeno-free conditions for the derivation and culture of human iPSCs [22, 23]. Cell culture plates can be coated with a xeno-free medium containing high concentrations of basic fibroblast growth factor and fibronectin [24]. Xeno-free synthetic culture plates are used in the production of clinical grade iPSCs and their subsequent differentiation to retina cells [25]. Combined with vitronectin as an extracellular matrix, a formulated medium with inhibitors of protein kinase C and histone deacetylases maintains long-term culture of xeno-free iPSCs [26]. Okano’s group establishes a procedure to obtain xeno-free and self-renewing neuroepithelial-like stem cells from feeder-free human iPSCs using a xeno-free medium. The neuroepithelial-like stem cells are cultured for long periods in the medium and can be cryopreserved using a defined serum-free freezing reagent for massive cell banking [27]. The dual use of E8 medium and vitronectin-coated culture surfaces can also be employed to derive and culture xeno-free human iPSCs [28]. A detailed protocol has been published on how to produce a consistent E8 medium for the derivation, maintenance, expansion and cryopreservation of iPSCs [29].
MODELING PD USING iPSCs
The past 20 years witness remarkable discoveries of the genetic influences on PD. Inherited mutations that cause familial PD have been identified in over 18 genes, collectively accounting for about 10% of all PD patients [30, 31]. The rest of PD cases are idiopathic, with unclear etiology. Somatic cells (usually skin fibroblasts) from PD patients are reprogrammed to iPSCs, which are differentiated to midbrain DA neurons. Using iPSC-derived DA neurons from PD patients with mutations in a certain gene, many investigators are uncovering mechanistic insights on how mutations of these genes are linked to PD. Here, we review the generation and analysis of patient-specific midbrain DA neurons as models of PD (see Table 1 for summary), with a brief discussion on the use of patient-specific glial cells.
Table 1
Phenotypes in iPSCs-derived neurons from monogenic forms of PD
| Protein/Gene | Mutation | Phenotype |
|---|---|---|
| α-synuclein/SNCA | SNCA triplication [54, 55] | • Elevated α-synuclein levels |
| SNCA triplication [56] |
| |
| A53T [59] | • Nitrosative stress and endoplasmic reticulum stress | |
| A53T [60] |
| |
| LRRK2/LRRK2 | G2019S [73] |
|
| G2019S [74] | • Increased phosphorylation and activation of ERK1/2 | |
| G2019S [75, 76] | • Reduced amount of neurites and neurite aborization | |
| G2019S [77, 78] | • Increased mitochondrial DNA damage and delayed initiation of mitophagy | |
| Parkin/PARK2 | Deletions of exon 3 and/or exon 5 [91, 94, 96] |
|
| Deletion of exons 2–4 or exons 6-7 [92] | • Elevated α-synuclein levels | |
| V324A [93] | • Abnormal mitochondrial morphology and/or function | |
| Deletion of exon 3 or exons 3-4 [99] | ||
| R275W or R42P [99, 121] | ||
| PINK1/PINK1 | Q456Stop [93] |
|
| V170G [106, 108] | • Impaired stress-induced recruitment of parkin to mitochondria | |
| Q456Stop [108] | ||
| Q456Stop [109] | • Decreased mitochondrial membrane potential and ATP content | |
| DJ-1/PARK7 | E64D [114] |
|
Common phenotypes caused by mutations in different genes are grouped in colors with the same color scheme in Fig. 1. References are cited in square brackets.
Fig. 1
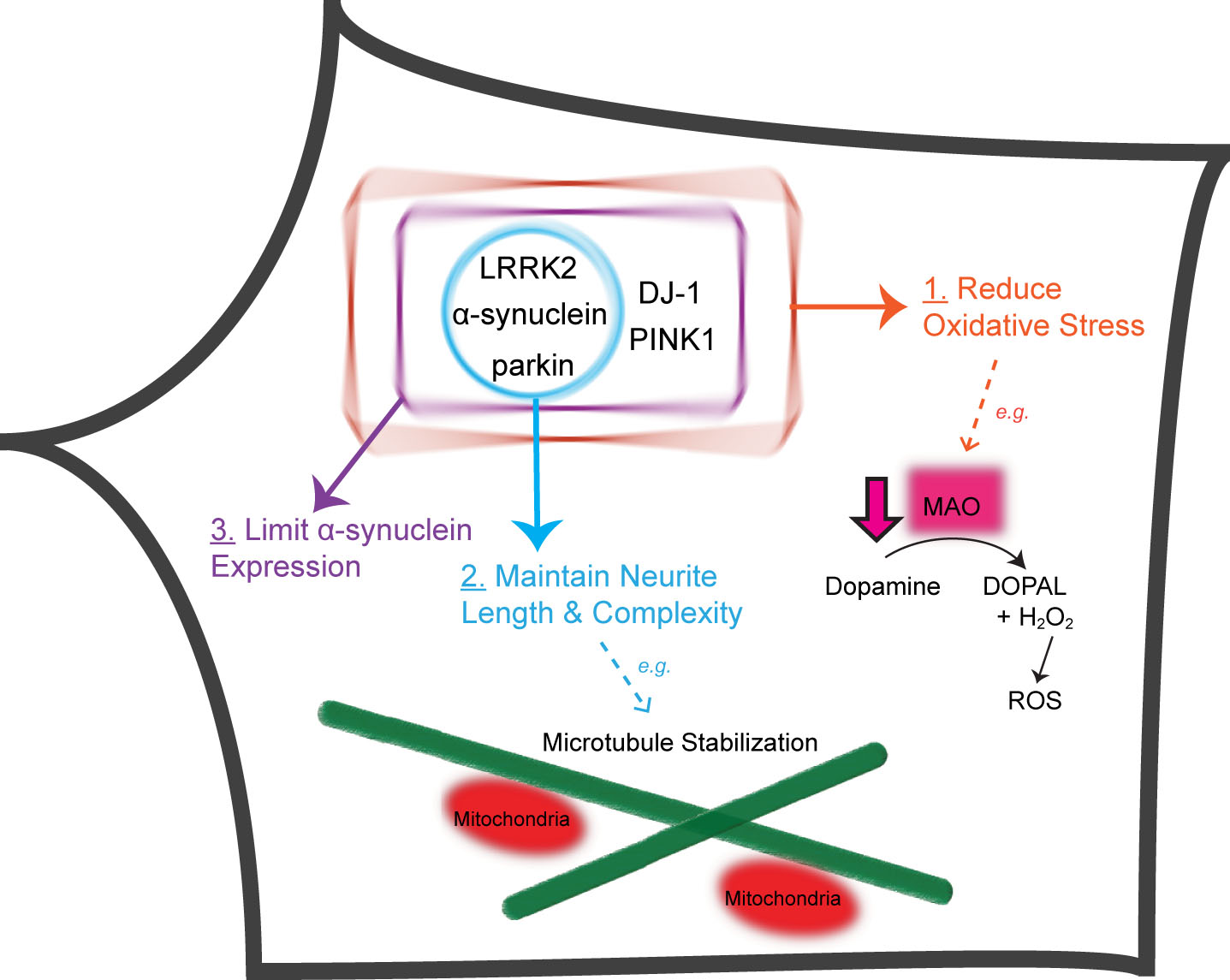
Five PD-linked genes converge on three pathways to protect against vulnerabilities of nigral DA neurons. Proteins encoded by five PD-linked genes (α-synuclein, LRRK2, parkin, PINK1 and DJ-1) limit oxidative stress stemming from dopamine metabolism and mitochondrial dysfunction (Pathway 1). α-synuclein, LRRK2 and parkin maintain the length and complexity of neuronal processes, e.g., through microtubule stabilization (Pathway 2). DJ-1, PINK1, α-synuclein, LRRK2 and parkin normally function to prevent the accumulation of α-synuclein (Pathway 3). DOPAL, 3,4-Dihydroxyphenylacetaldehyde; H2O2, hydrogen peroxide.
α-synuclein
α-synuclein, encoded by SNCA gene, is a synaptic vesicle protein that has been shown to negatively regulate dopamine and norepinephrine release. Neurons from mice lacking α-synuclein are able to recover quicker (i.e., release more catecholamines) from repetitive stimulation compared to wild-type neurons [32–34]. This could be related to α-synuclein’s role as a potent inhibitor of phospholipase D2, which converts phosphatidylcholine to phosphatidic acid and diacyglycerol, both of which positively regulate neurotransmitter release [32, 35]. While α-synuclein has been shown to be a chaperone in vitro; paradoxically, it is a main component of Lewy bodies—intracellular protein aggregates found in brains of patients with PD and other neurodegenerative disorders [37]. More in-depth discussions of the function of α-synuclein can be found in many excellent reviews [38, 39].
Missense mutations (A30P [40–42], A53E [43, 44], A53T [45], A53V [44], E46K [46], G51D [47–49], H50Q [50]) and copy number variations (duplication or triplication) of SNCA have been reported in PD patients [51, 52]. Genome-wide association studies identify SNCA as one of the genes associated with idiopathic PD, suggesting that changes in the expression level of SNCA may contribute to idiopathic PD [53]. Missense mutations of α-synuclein or its overexpression by copy number variations cause misfolding of the protein and its aggregation in Lewy bodies [37].
In DA neurons differentiated from iPSCs of PD patients with SNCA triplication mutation, the level of α-synuclein protein is twice the amount as that in neurons derived from normal iPSCs [54]. An independent study using iPSCs with SNCA triplication confirms the increased expression level of α-synuclein and demonstrates increased oxidative stress in these DA neurons [55]. Neurons with SNCA triplication display normal cellular and mitochondrial morphology but substantial changes in growth, viability, cellular energy metabolism and stress resistance, especially when challenged with starvation or toxins [56]. Another study finds that SNCA triplication in iPSC-derived neural precursor cells leads to a reduced capacity to differentiate into DA neurons, decreased neurite outgrowth and lower neuronal activity compared to normal neurons. The delayed maturation phenotype is accompanied by a significant reduction in the mRNA level of nuclear receptor related 1 protein (NURR1), G-protein-regulated inward-rectifier potassium channel 2 (GIRK-2) and tyrosine hydroxylase (TH) [57].
To remove potentially confounding influence of different genetic backgrounds when comparing PD patients and normal subjects, isogenic pairs of iPSCs with or without the A53T point mutation of α-synuclein are generated using zinc-finger nuclease-mediated genomic editing to introduce the mutation to normal iPSCs or to correct the mutation in patient iPSCs [58]. The correction of A53T mutation reverses nitrosative stress and endoplasmic reticulum stress in iPSC-derived neurons [59]. In the presence of A53T mutation, increased S-nitrosylation of the transcription factor MEF2C affects the transcriptional regulation of mitochondrial functions by PGC1α, a master regulator of mitochondrial biogenesis. Activation of this pathway by A53T mutation increases apoptotic cell death in iPSC-derived midbrain DA neurons [60]. Blocking the pathway by isoxazole, a small-molecule compound identified in a high-throughput screen using these isogenic iPSC-derived neurons, protects against the toxicity of environmental PD toxins that target mitochondria [60].
In addition to mutations of SNCA, isogenic iPSCs have been generated to study the impact of various single nucleotide polymorphisms identified in genome-wide association studies of idiopathic PD patients. Using CRISPR/Cas9-mediated genomic editing, isogenic pairs of iPSCs with different SNPs are generated. This strategy enables the identification of a distal enhancer in intron 4 of the human SNCA gene. A G allele in an SNP in this enhancer decreases the recruitment of brain-specific transcription factors EMX2 and NKX6-1. This increases the interaction of the enhancer with the promoter of SNCA and thus elevates the expression level of SNCA [61].
Leucine-rich repeat kinase 2 (LRRK2)
The LRRK2 gene encodes a protein with the same name that possesses both kinase and GTPase activities surrounded by protein-interacting domains. Being a multi-domain protein, it is perhaps unsurprising for LRRK2 to have pleiotropic roles in nature. For instance, LRRK2 has been identified to influence outgrowth of neurons [62] possibly through its phosphorylation of ERM proteins [63] and tubulin [64]. Neurite growth may be associated with the role of LRRK2 in macroautophagy [65]. Furthermore, through its interaction with Rab5b and Rab7, LRRK2 has been shown to modulate synaptic vesicle endocytosis and endocytic membrane trafficking, respectively [66, 67]. The functions of LRRK2 are covered in many excellent reviews [68, 69].
Missense mutations in the LRRK2 gene are the most common genetic cause of PD. The LRRK2 G2019S mutation (c.G6055>A) is found in 4% of familial cases and 1% of sporadic cases worldwide. Penetrance of LRRK2 mutations is age-dependent and estimated to be 24–26% in two large scale studies [70, 71]. The most frequent LRRK2 mutation G2019S has a very strong founder effect [72]. In iPSC-derived DA neurons from PD patients with LRRK2 G2019S mutation, there is increased expression of key oxidative stress response genes and α-synuclein. Consequently, DA neurons are more sensitive to caspase-3 activation [73]. The phenotype on increased oxidative stress is confirmed in an independent study [74]. G2019S mutation significantly increases the phosphorylation and activation of extracellular signal regulated kinase 1/2 (ERK1/2) in iPSC-derived DA neurons. Application of the MEK inhibitor PD0325901, which blocks ERK activation, rescues iPSC-derived neurons from degeneration and reduces cell death under oxidative stress [74]. The phenotype on increased expression of α-synuclein [73] is also confirmed in an independent study, which shows that knockdown of α-synuclein reduces the toxicity of LRRK2 G2019S mutation in iPSC-derived DA neurons [75]. Two studies independently show that LRRK2 G2019S mutation significantly reduces the numbers of neurites and neurite arborization [76], as well as progressive degeneration of neurites when iPSC-derived neurons mature [75]. LRRK2 G2019S mutation increases mitochondrial DNA damage [77] and delays the initiation of mitophagy by affecting the pausing of damaged mitochondria being transported on microtubules [78].
PARK2
The PARK2 gene encodes parkin [79], an E3 ligase [80] that ubiquitinates a variety of substrates [81]. Mutations of parkin are the most frequent cause of recessively inherited PD [82]. PD patients with parkin mutations have early onset of PD symptoms; they account for almost 50% of all cases before the age of 45 [83]. In contrast to the situation in PD patients with parkin mutations, parkin knockout mice [84, 85] or rats [86] do not exhibit any significant degeneration of nigral DA neurons or PD-like locomotor symptoms. Our earlier studies in cell lines and neuronal cultures show that parkin suppresses dopamine-induced oxidative stress [87] by limiting the transcription of monoamine oxidases (MAO) A and B [88] because parkin ubiquitinates and degrades estrogen-related receptors [89], a family of nuclear receptors that regulate the transcription of many nuclear-encoded mitochondrial proteins such as MAOs [90]. In iPSC-derived midbrain DA neurons that we generate from PD patients with parkin mutations, there is a marked increase of dopamine-induced oxidative stress, as the expression of MAOA and MAOB is significantly elevated [91]. Independent studies have confirmed increased oxidative stress in iPSC-derived neurons with parkin mutations [92, 93]. We also find that the precision of dopaminergic transmission is significantly disrupted by increased spontaneous DA release and decreased DA reuptake [91]. Consistent with this change in neural transmission, dopamine induces oscillatory neuronal activities in iPSC-derived neurons from PD patients with parkin mutations but not from normal subjects [94]. This phenotype mirrors widespread rhythmic bursting of neuronal activities in the basal ganglia of PD patients but not of normal subjects or of PD patients on dopaminergic drugs [95]. In iPSC-derived neurons, parkin mutations reduce the length and complexity of neuronal processes, particularly those of midbrain DA neurons [96]. It is caused by a marked decrease of microtubule stability in neurons with parkin mutations, as the phenotype can be rescued by taxol or overexpression of parkin and can be mimicked in normal neurons treated with colchicine [96]. An independent study confirms that parkin mutations reduce microtubule stability in iPSC-derived neurons [97].
With rare exceptions, most PD patients with parkin mutations do not have Lewy bodies [98]. In iPSC-derived neurons from two parkin patients, increased accumulation of α-synuclein is seen in one patient with Lewy bodies, but not in the other patient without Lewy bodies [92]. In our study, there is no significant difference in α-synuclein protein levels in iPSC-derived midbrain DA neurons from two PD patients with parkin mutations and two normal subjects [91]. Increased levels of α-synuclein protein are observed in two other studies using iPSC-derived neurons from PD patients with parkin mutations [93, 99].
Many studies using transfected parkin in cell lines have shown that parkin is recruited to mitochondria to ubiquitinate a variety of substrates for the induction of mitophagy [100, 101]. The recruitment of endogenous parkin to mitochondria is not robustly seen in cell lines [102, 103], mice [104, 105] or iPSC-derived human neurons [91, 106]. Although these issues have been extensively reviewed [101–103], the large body of literature on the role of parkin in mitophagy and the significant interest in the field have led to several independent studies of this issue in iPSC-derived neurons with parkin mutations. Significantly increased percentages of iPSC-derived neurons with abnormal mitochondria (usually with elongated shape and larger volume) are observed in three separate studies [92, 93, 99]. However, two different studies using qPCR show that there is no significant change in mitochondria DNA copy number [91, 92]. It seems that parkin mutations in iPSC-derived neurons may alter mitochondrial morphology in a portion of the cells, particularly TH+ neurons [99], but the modest changes may be balanced by robust biogenesis of mitochondria.
PINK1
Phosphatase and tensin homolog (PTEN)-induced Putative Kinase 1 (PINK1) is a mitochondria-targeted Ser/Thr protein kinase. Under normal condition, PINK1 is quickly cleaved by proteases on mitochondrial outer membrane and is thus released to cytosol for degradation, which limits the steady state level of PINK1 [107]. Insults that depolarize mitochondrial inner membrane arrest the mitochondrial transport of PINK1, which triggers the phosphorylation of parkin on the ubiquitin-like domain and activates the E3 ligase activity of parkin. Mitochondrially recruited parkin ubiquitinates many mitochondrial outer membrane proteins and induces autophagic clearance of depolarized mitochondria [101].
In two separate studies using the same line of iPSC-derived neurons from PD patients with PINK1 mutations, valinomycin-induced mitochondrial recruitment of exogenously expressed parkin is abrogated by PINK1 mutations [106, 108]. Loss of PINK1 abolishes the degradation of mitochondrial proteins only in fibroblasts, but not in isogenic iPSC-derived neurons, which do not exhibit significant mitophagy even with parkin overexpression and valinomycin treatment for 16 hours [106]. Midbrain DA neurons derived from iPSCs of PD patients with PINK1 mutations exhibit α-synuclein accumulation, abnormal mitochondrial morphology and increased cytosolic dopamine levels [93]. In iPSC-derived neurons, PINK1 mutations reduce complex I activity, which leads to a reduction in mitochondrial membrane potential. Phosphorylation of Ser250 in NdufA10 regulates the activity of ubiquinone reductase in mitochondrial complex I. A phosphomimetic mutant of NdufA10 reverses the deficiencies in complex I activity and ATP synthesis in iPSC-derived neurons with PINK1 mutations [109]. Consistent with this, coenzyme Q10 rescues the cellular vulnerability associated with mitochondrial dysfunction in iPSC-derived neurons from PD patients with PINK1 mutations [110]. In a separate study, increased expression of LRRK2 is observed in iPSC-derived neurons with PINK1 mutations [111].
DJ-1
Mutations of DJ-1 cause early-onset recessive PD [112]. Human DJ-1 is a small protein with 189 amino acid residues, three of which are cysteines. It normally forms a homodimer that exhibits anti-oxidant activities [113]. In iPSC-derived midbrain DA neurons from PD patients with homozygous DJ-1 mutations, there are increased dopamine oxidation and neuromelanin-like pigmented aggregates [114]. Increased oxidative stress stemming from dopamine metabolism triggers mitochondrial oxidative stress, both of which are significantly attenuated by blocking dopamine synthesis [114]. Dopamine-induced oxidative stress inactivates glucocerebrosidase, which inhibits lysosomal functions and leads to increased expression of α-synuclein [114]. The phenotypes are confirmed in an isogenic pair of iPSC-derived midbrain DA neurons with or without DJ-1 mutations. More interestingly, iPSC-derived normal human midbrain DA neurons have significantly higher level of dopamine than iPSC-derived wild-type mouse midbrain DA neurons. Increasing dopamine level by L-DOPA treatment dramatically elevates dopamine oxidation in iPSC-derived midbrain DA neurons from DJ-1 knockout mice but not neurons derived from wild-type mice [114]. Thus, DJ-1 mutations reveal the vulnerability of midbrain DA neurons in handling dopamine-induced oxidative stress, which is more severe in human than in mouse.
Risk alleles
The majority of PD cases do not have any family history and are likely influenced by both environmental and genetic susceptibility factors. With an odds ratio around 5, mutations in the gene encoding glucocerebrosidase (GBA) are currently recognized to be the greatest risk factor for PD [2, 115]. GBA mutations decrease the enzymatic activity of glucocerebrosidase, leading to the accumulation of glucosylceramide and glucosylsphingosine in lysosomes and the potential development of symptoms for Gaucher’s disease and PD [115]. In iPSC-derived DA neurons from PD patients carrying mutated GBA, protein levels and enzymatic activity of glucocerebrosidase are significantly lower, concomitant with higher levels of α-synuclein and glucosylceramide compared to isogenic controls [116]. These GBA mutant neurons also display abnormalities towards the autophagic/lysosomal pathway and calcium homeostasis [116]. The association between elevated a-synuclein and diminished glucocerebrosidase activity with GBA mutations has also been reported in a twin study through analyzing their iPSC-derived DA neurons [117]. The monozygotic twins are discordant for PD but both heterozygous for a mutated GBA. Intriguingly, neurons derived from the affected twin display lower dopamine level and upregulated MAOB expression, compared to the unaffected twin. The study highlights the interplay between genetic and environmental factors in shaping phenotypes.
With the rise of genome-wide association studies, variants in loci including SNCA and LRKK2 have been confirmed, while others—MAPT, VPS13C, BST1, DDGRK1 and many more—have been identified as novel risk factors for PD development [2]. To date, iPSC technology has been applied to only a few of these new targets. For example, iPSC-derived neurons from patients with MAPT mutations show irregularities in the expression of tau proteins, tau hyperphosphorylation and mitochondrial axonal transport compared to controls [118]. Axonal degeneration has also been found in iPSC-derived neurons carrying mutated MAPT. Isogenic repair of the mutation restores axonal morphology [119].
Utilizing iPSC-derived glial cells in modeling PD
Whether glial cells instigate or react to neuronal death remains to be settled. However, many lines of evidence have implicated the involvement of glia in PD. Neuroinflammation observed in PD patients has been mainly associated with activation of microglial cells, which releases inflammatory cytokines, such as IL-1β, IL-6 and TNF-α (tumor necrosis factor-α). In particular, TNF-α is known to trigger neuronal death [120]. Elevated levels of TNF-α have been found in the brain and cerebrospinal fluid of PD patients [121]. Activated microglial cells may also inflict neuronal damage by producing reactive oxygen and nitrogen species [122]. Paradoxically, astrocytes and microglial cells secrete growth factors, such as BDNF (brain-derived neurotrophic factor) and GDNF (glial cell line-derived neurotrophic factor), that are critical for neuronal survival [122, 123]. Supplementing GDNF to MPTP-treated primate models of PD significantly increases levels of DA neurons and improves PD motor symptoms [124, 125]; however, the therapeutic effect of BDNF for PD patients remains to be established [126].
As the complex relationship between glia and neurons is disrupted in PD, co-culturing glial cells and neurons both derived from iPSCs of PD patients should therefore be another platform to advance insights into the multifactorial pathogenesis of PD. For example, α-synuclein has been shown to not only impact neurons but also directly activate and induce TNF-α release from primary microglial cultures [127]. In transgenic mice overexpressing wild-type α-synuclein, significantly more activated microglial are observed in the substantia nigra compared to age-matched control mice [127]. Moreover, LRKK2 is known to be expressed in microglial. Activating microglial from transgenic mice overexpressing PD-linked LRRK2-R1441G mutation leads to more secretion of inflammatory cytokines compared to those from activated wild-type microglial [128].
Protocols to differentiate hPSCs (human pluripotent stem cells) to astrocytes have been published since the first neural differentiation method in 2001 using FGF-2 (fibroblast growth factor 2) [129]. For the majority, embryoid bodies are utilized as starting point. Through various growth factor cocktails, embryoid bodies are differentiated to neuroectodermal derivatives, neural progenitor cells, and finally astrocytes [130]. Since microglial are of mesodermal lineage and distinct from astrocytes, IL-34 and CSF1 (colony-stimulating factor 1) have been used to differentiate embryoid bodies into cell lawns resembling endothelial cells (i.e., cystic embryoid bodies) to later become mature microglia-like cells [131]. Applications of glial cell differentiation protocols have shed light into the pathogenesis of Alzheimer’s disease [132] and Huntington’s disease [133]. Similar studies on PD will undoubtedly improve our understanding of the complex interactions between glia and neurons in PD pathogenesis.
CONCLUSIONS
The discovery of induced pluripotent stem cells has transformed biomedical research by enabling researchers to use the human model system to study diseases. It has been particularly fruitful for PD, as genetic animal models of PD have significant challenges in recapitulating the critical features of PD—loss of nigral DA neurons and the ensuing locomotor phenotypes [8]. Rapid development in stem cell technology will make the system more useful. One current issue in using iPSC-derived neurons is its immaturity [134, 135], which may not adequately reflect age-related disorders such as PD. The use of progerin [136] and astrocytes [137] allows improved modeling of age-related changes in iPSC-derived neurons. Another approach is to transplant human iPSC-derived neurons in rat brains [138] or even brains of non-human primates [139], which would enable more effective modeling of age-dependent pathologies. The long-term engraftment of hPSC-derived brain organoids in mouse brains [140] offers another novel solution that provides physiological environment in vivo to promote the maturation and function of transplanted human neurons.
From analyzing phenotypes found in iPSC-derived neurons from monogenic forms of PD, several converging pathways appear to intersect with the unique vulnerabilities of human nigral DA neurons (Fig. 1). First, increased oxidative stress is found in iPSC-derived DA neurons from PD patients with α-synuclein [55, 59], LRRK2 [73, 74], parkin [91–93], PINK1 [93] or DJ-1 mutations [114]. Mitochondria are the most significant source of oxidative stress, as they handle the catabolic breakdown of dopamine. Monoamine oxidases, which are anchored on mitochondrial outer membrane [141], catalyze the oxidative deamination of cytosolic dopamine, a reaction that produces H2O2 and its derivative free radicals. Dopamine-induced oxidative stress is increased by elevated expression of monoamine oxidases when parkin is mutated [91]. Mutations of DJ-1 in human midbrain DA neurons increase dopamine-induced oxidative stress, which triggers mitochondrial dysfunction downstream [114]. Depolarization of mitochondrial inner membrane potential increases oxidative stress as electrons normally flowing through the mitochondrial respiratory chain are shunted to O2 in the cell. Ca2 +-dependent autonomous pacemaking [142] and lack of Ca2 + buffer proteins [143] exacerbate the stress on mitochondria because they serve as a major store of intracellular Ca2 + [144]. A significant decrease of mitochondrial inner membrane potential triggers the stabilization of PINK1, which phosphorylates parkin to activate and recruit it to mitochondria, where parkin ubiquitinates many mitochondrial proteins to trigger autophagy [101]. Relatively modest morphological or functional changes in mitochondria have been observed in iPSC-derived neurons with parkin mutations [92, 93, 99] or PINK1 mutations [109]. Several studies show that parkin mutations [91, 92] or PINK1 mutations [106, 108] do not grossly affect mitochondria copy number, or even mitophagy [106]. The different results between iPSC-derived human midbrain DA neurons and immortalized cell lines are not easy to reconcile. One possibility could be that human nigral DA neurons normally increase mitochondrial DNA (mtDNA) copy numbers with age to maintain the amount of wild-type mtDNA despite age-dependent increase of deletion mutations in mtDNA [145]. Thus, strong mitochondrial biogenesis in human DA neurons may balance out the modest defects in mitophagy. Further studies are needed to address the role of mitochondria in the selective vulnerability of nigral DA neurons, which paradoxically have significantly lower mitochondrial mass than VTA DA neurons and other non-DA neurons [146] and are quite resistant to mutations of mtDNA [147].
Second, reduced neurite length and complexity are found in iPSC-derived neurons from PD patients with mutations in a-synuclein [57], LRRK2 [75, 76], or parkin [96, 97]. This phenotype critically reflects a key vulnerability of nigral DA neurons, which have massive axon arbors. The total length of axon arbors of a single rat nigral DA neuron averages 45 cm [148]. It is estimated that a single human nigral DA neuron may have axon arbors with a total length of 4.6 m [149], which may make these neurons much more vulnerable than other types of neurons to microtubule disruption [150], particularly in human [151]. Microtubules not only maintain the morphology and structural integrity of neuronal processes but also serve an obligatory function in the transportation of vesicles, including mitochondria. Abnormal mitochondrial transport has been reported in iPSC-derived neurons from PD patients with LRRK2 G2019S mutation [78].
Third, increased expression of α-synuclein has been observed in iPSC-derived neurons from PD patients with mutations in α-synuclein [54, 55], LRRK2 [73, 75], parkin [93, 99], PINK1 [93] or DJ-1 [114]. The significant role of α-synuclein in PD has been well documented [152]. Debates on whether α-synuclein is transmitted between neurons and whether such transmission causes the selective degeneration of nigral DA neurons in PD [153, 154] make it very important to generate patient-specific midbrain DA neurons to address these issues. It remains unclear how α-synuclein accumulation causes the selective loss of nigral DA neurons. One possibility is that α-synuclein increases cytosolic dopamine concentration more significantly in nigral DA neurons than in VTA DA neurons [155]. Dopamine-induced oxidative stress, in conjunction with mitochondrial oxidative stress downstream of α-synuclein overexpression, may impact nigral DA neurons much more than other types of cells [156].
These three pathways intersect with the unique vulnerabilities of human nigral DA neurons, which produce a large amount of dopamine for controlled release in their massive axon arbors. The need for nigral DA neurons to release an oxidation-prone neurotransmitter in a continuous, autonomous pacemaking manner underlies the vulnerabilities that are revealed by mutations of PD-linked genes. Mechanism-based drug development programs can be implemented to mimic the normal functions of these genes in suppressing oxidative stress, maintaining neuronal morphology and limiting α-synuclein expression. Patient-specific iPSC-derived neurons would provide an excellent platform for screening compounds that block the deleterious effects of these mutations. The observation that five different monogenic causes of PD converge on three common pathways suggests that it is possible to use iPSC-derived neurons with different monogenic etiologies to cross-validate hits identified from screens. Isogenic pairs of iPSC-derived neurons can be generated by introducing PD-linked mutations to normal iPSCs or by repairing mutations in patient iPSCs. Such isogenic pairs of iPSC-derived midbrain DA neurons would be an even better platform for screening compounds, as they remove the potentially confounding influence of different genetic backgrounds between normal subjects and PD patients. Furthermore, iPSC-derived midbrain DA neurons from idiopathic PD patients can be used to validate the hits and generate valuable information on how well the findings can be extrapolated to idiopathic PD. This is highly useful and unique to the human model system, where efficacy can be established in patient-specific neurons in a dish to estimate their chance of success in patients. In addition, safety studies can be done on other types of cells (e.g., liver cells or kidney cells) differentiated from the same set of iPSCs. There is an increasing trend for pharmaceutical companies to transition their drug discovery programs to the human model system as embodied in iPSCs. The stage is set for more mechanistic study and drug discovery research on PD based on patient-specific iPSC-derived cells. The future is indeed much brighter with the advent of this transformative technology.
CONFLICT OF INTEREST
The authors declare no competing financial interests.
ACKNOWLEDGMENTS
This work was supported by National Institute of Neurological Diseases and Stroke grant R56NS102148, Department of Veterans Affairs Merit Awards I01BX002452 and I01BX003831 and New York State Department of Health NYSTEM Contract C029556.
REFERENCES
[1] | Latourelle JC , Pankratz N , Dumitriu A , Wilk JB , Goldwurm S , Pezzoli G , Mariani CB , DeStefano AL , Halter C , Gusella JF , Nichols WC , Myers RH , Foroud T , Progeni and GenePd Investigators ((2009) ) Genomewide association study for onset age in Parkinson disease. BMC Med Genet 10: , 98. |
[2] | Kalia LV , Lang AE ((2015) ) Parkinson’s disease. Lancet 386: , 896–912. |
[3] | Chaudhuri KR , Healy DG , Schapira AH ((2006) ) Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol 5: , 235–245. |
[4] | Gasser T , Hardy J , Mizuno Y ((2011) ) Milestones in PD genetics. Mov Disord 26: , 1042–1048. |
[5] | Singleton A , Hardy J ((2016) ) The evolution of genetics: Alzheimer’s and Parkinson’s diseases. Neuron 90: , 1154–1163. |
[6] | Beal MF ((2001) ) Experimental models of Parkinson’s disease. Nat Rev Neurosci 2: , 325–334. |
[7] | Duty S , Jenner P ((2011) ) Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br J Pharmacol 164: , 1357–1391. |
[8] | Dawson TM , Ko HS , Dawson VL ((2010) ) Genetic animal models of Parkinson’s disease. Neuron 66: , 646–661. |
[9] | Pu J , Jiang H , Zhang B , Feng J ((2012) ) Redefining Parkinson’s disease research using induced pluripotent stem cells. Curr Neurol Neurosci Rep 12: , 392–398. |
[10] | Yamanaka S ((2013) ) The winding road to pluripotency (Nobel Lecture). Angew Chem Int Ed Engl 52: , 13900–13909. |
[11] | Takahashi K , Yamanaka S ((2006) ) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126: , 663–676. |
[12] | Takahashi K , Tanabe K , Ohnuki M , Narita M , Ichisaka T , Tomoda K , Yamanaka S ((2007) ) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131: , 861–872. |
[13] | Choi J , Lee S , Mallard W , Clement K , Tagliazucchi GM , Lim H , Choi IY , Ferrari F , Tsankov AM , Pop R , Lee G , Rinn JL , Meissner A , Park PJ , Hochedlinger K ((2015) ) A comon of genetically matched cell lines reveals the equivalence of human iPSCs and ESCs. Nat Biotechnol 33: , 1173–1181. |
[14] | Okita K , Matsumura Y , Sato Y , Okada A , Morizane A , Okamoto S , Hong H , Nakagawa M , Tanabe K , Tezuka K , Shibata T , Kunisada T , Takahashi M , Takahashi J , Saji H , Yamanaka S ((2011) ) A more efficient method to generate integration-free human iPS cells. Nat Methods 8: , 409–412. |
[15] | Jia F , Wilson KD , Sun N , Gupta DM , Huang M , Li Z , Panetta NJ , Chen ZY , Robbins RC , Kay MA , Longaker MT , Wu JC ((2010) ) A nonviral minicircle vector for deriving human iPS cells. Nat Methods 7: , 197–199. |
[16] | Warren L , Manos PD , Ahfeldt T , Loh YH , Li H , Lau F , Ebina W , Mandal PK , Smith ZD , Meissner A , Daley GQ , Brack AS , Collins JJ , Cowan C , Schlaeger TM , Rossi DJ ((2010) ) Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7: , 618–630. |
[17] | Nordin F , Ahmad RNR , Farzaneh F ((2017) ) Transactivator protein: An alternative for delivery of recombinantroteins for safer reprogramming of induced pluripotent stem cell. Virus Res 235: , 106–114. |
[18] | Thier M , Munst B , Edenhofer F ((2010) ) Exploring refined conditions for reprogramming cells by recombinant Oct4 protein. Int J Dev Biol 54: , 1713–1721. |
[19] | Thier M , Munst B , Mielke S , Edenhofer F ((2012) ) Cellular reprogramming employing recombinant sox2 protein. Stem Cells Int 2012: , 549846. |
[20] | Zhou W , Freed CR ((2009) ) Adenoviral gene delivery can reprogram human fibroblasts to induced pluripotent stem cells. Stem Cells 27: , 2667–2674. |
[21] | Fusaki N , Ban H , Nishiyama A , Saeki K , Hasegawa M ((2009) ) Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci 85: , 348–362. |
[22] | Villa-Diaz LG , Ross AM , Lahann J , Krebsbach PH ((2013) ) Concise review: The evolution of human pluripotent stem cell culture: From feeder cells to synthetic coatings. Stem Cells 31: , 1–7. |
[23] | Fan Y , Wu J , Ashok P , Hsiung M , Tzanakakis ES ((2015) ) Production of human pluripotent stem cell therapeutics under defined xeno-free conditions: Progress and challenges. Stem Cell Rev 11: , 96–109. |
[24] | Ding Y , Yang H , Yu L , Xu CL , Zeng Y , Qiu Y , Li DS ((2015) ) Feeder-free and xeno-free culture of human pluripotent stem cells using UCBS matrix.. Cell Biol Int 39: , 1111–1119. |
[25] | Tucker BA , Anfinson KR , Mullins RF , Stone EM , Young MJ ((2013) ) Use of a synthetic xeno-free culture substrate for induced pluripotent stem cell induction and retinal differentiation. Stem Cells Transl Med 2: , 16–24. |
[26] | Kim HT , Lee KI , Kim DW , Hwang DY ((2013) ) An ECM-based culture system for the generation and maintenance of xeno-free human iPS cells. Biomaterials 34: , 1041–1050. |
[27] | Isoda M , Kohyama J , Iwanami A , Sanosaka T , Sugai K , Yamaguchi R , Matsumoto T , Nakamura M , Okano H ((2016) ) Robust production of human neural cells by establishing neuroepithelial-like stem cells from peripheral blood mononuclear cell-derived feeder-free iPSCs under xeno-free conditions. Neurosci Res 110: , 18–28. |
[28] | Chen G , Gulbranson DR , Hou Z , Bolin JM , Ruotti V , Probasco MD , Smuga-Otto K , Howden SE , Diol NR , Propson NE , Wagner R , Lee GO , Antosiewicz-Bourget J , Teng JM , Thomson JA ((2011) ) Chemically defined conditions for human iPSC derivation and culture. Nat Methods 8: , 424–429. |
[29] | Beers J , Gulbranson DR , George N , Siniscalchi LI , Jones J , Thomson JA , Chen G ((2012) ) Passaging and colony expansion of human pluripotent stem cells by enzyme-free dissociation in chemically defined culture conditions. Nat Protoc 7: , 2029–2040. |
[30] | Hernandez DG , Reed X , Singleton AB ((2016) ) Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J Neurochem 139: (Suppl 1), 59–74. |
[31] | Ferreira M , Massano J ((2017) ) An updated review of Parkinson’s disease genetics and clinicopathological correlations. Acta Neurol Scand 135: , 273–284. |
[32] | Abeliovich A , Schmitz Y , Farinas I , Choi-Lundberg D , Ho WH , Castillo PE , Shinsky N , Verdugo JM , Armanini M , Ryan A , Hynes M , Phillips H , Sulzer D , Rosenthal A ((2000) ) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25: , 239–252. |
[33] | Yavich L , Tanila H , Vepsalainen S , Jakala P ((2004) ) Role of alpha-synuclein in presynaptic dopamine recruitment. J Neurosci 24: , 11165–11170. |
[34] | Yavich L , Jakala P , Tanila H ((2006) ) Abnormal compartmentalization of norepinephrine in mouse dentate gyrus in alpha-synuclein knockout and A30P transgenic mice. J Neurochem 99: , 724–732. |
[35] | Frohman MA , Morris AJ ((1996) ) Rho is only ARF the story. Phospholipid signalling. Curr Biol 6: , 945–947. |
[36] | Kim TD , Paik SR , Yang CH ((2002) ) Structural and functional implications of C-terminal regions of alpha-synuclein. Biochemistry 41: , 13782–13790. |
[37] | Spillantini MG , Schmidt ML , Lee VM , Trojanowski JQ , Jakes R , Goedert M ((1997) ) Alpha-synuclein in Lewy bodies. Nature 388: , 839–840. |
[38] | Koprich JB , Kalia LV , Brotchie JM ((2017) ) Animal models of alpha-synucleinopathy for Parkinson disease drug development. Nat Rev Neurosci 18: , 515–529. |
[39] | Goedert M , Jakes R , Spillantini MG ((2017) ) The synucleinopathies: Twenty years on.S53-S. J Parkinsons Dis 7: , 71. |
[40] | Kruger R , Kuhn W , Muller T , Woitalla D , Graeber M , Kosel S , Przuntek H , Epplen JT , Schols L , Riess O ((1998) ) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 18: , 106–108. |
[41] | Kruger R , Kuhn W , Leenders KL , Sprengelmeyer R , Muller T , Woitalla D , Portman AT , Maguire RP , Veenma L , Schroder U , Schols L , Epplen JT , Riess O , Przuntek H ((2001) ) Familial parkinsonism with synuclein pathology: Clinical and PET studies of A30P mutation carriers. Neurology 56: , 1355–1362. |
[42] | Seidel K , Schols L , Nuber S , Petrasch-Parwez E , Gierga K , Wszolek Z , Dickson D , Gai WP , Bornemann A , Riess O , Rami A , Den Dunnen WF , Deller T , Rub U , Kruger R ((2010) ) First appraisal of brain pathology owing to A30P mutant alpha-synuclein. Ann Neurol 67: , 684–689. |
[43] | Martikainen MH , Paivarinta M , Hietala M , Kaasinen V ((2015) ) Clinical and imaging findings in Parkinson disease associated with the A53E SNCA mutation. Neurol Genet 1: , e27. |
[44] | Pasanen P , Myllykangas L , Siitonen M , Raunio A , Kaakkola S , Lyytinen J , Tienari PJ , Poyhonen M , Paetau A ((2014) ) Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging 35: , 2180 e2181–2185. |
[45] | Polymeropoulos MH , Lavedan C , Leroy E , Ide SE , Dehejia A , Dutra A , Pike B , Root H , Rubenstein J , Boyer R , Stenroos ES , Chandrasekharappa S , Athanassiadou A , Papapetropoulos T , Johnson WG , Lazzarini AM , Duvoisin RC , DiIorio G , Golbe LI , Nussbaum RL ((1997) ) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276: , 2045–2047. |
[46] | ((2015) ) Correction: Behoral phenotyping of Parkin-deficient mice: Looking for early preclinical features of Parkinson’s disease. PLoS One 10: , e0118526. |
[47] | Kiely AP , Asi YT , Kara E , Limousin P , Ling H , Lewis P , Proukakis C , Quinn N , Lees AJ , Hardy J , Revesz T , Houlden H , Holton JL ((2013) ) alpha-Synucleinopathy associated with G51D SNCA mutation: A link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol 125: , 753–769. |
[48] | Kiely AP , Ling H , Asi YT , Kara E , Proukakis C , Schapira AH , Morris HR , Roberts HC , Lubbe S , Limousin P , Lewis PA , Lees AJ , Quinn N , Hardy J , Love S , Revesz T , Houlden H , Holton JL ((2015) ) Distinct clinical and neuropathological features of G51D SNCA mutation cases compared with SNCA duplication and H50Q mutation. Mol Neurodegener 10: , 41. |
[49] | Lesage S , Anheim M , Letournel F , Bousset L , Honore A , Rozas N , Pieri L , Madiona K , Durr A , Melki R , Verny C , Brice A , French Parkinson’s Disease Genetics Study Group ((2013) ) G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol 73: , 459–471. |
[50] | Appel-Cresswell S , Vilarino-Guell C , Encarnacion M , Sherman H , Yu I , Shah B , Weir D , Thompson C , Szu-Tu C , Trinh J , Aasly JO , Rajput A , Rajput AH , Jon Stoessl A , Farrer MJ ((2013) ) Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord 28: , 811–813. |
[51] | Polymeropoulos MH , Lavedan C , Leroy E , Ide SE , Dehejia A , Dutra A , Pike B , Root H , Rubenstein J , Boyer R , Stenroos ES , Chandrasekharappa S , Athanassiadou A , Papapetropoulos T , Johnson WG , Lazzarini AM , Duvoisin RC , Di Iorio G , Golbe LI , Nussbaum RL ((1997) ) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276: , 2045–2047. |
[52] | Singleton AB , Farrer M , Johnson J , Singleton A , Hague S , Kachergus J , Hulihan M , Peuralinna T , Dutra A , Nussbaum R , Lincoln S , Crawley A , Hanson M , Maraganore D , Adler C , Cookson MR , Muenter M , Baptista M , Miller D , Blancato J , Hardy J , Gwinn-Hardy K ((2003) ) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302: , 841. |
[53] | Devine MJ , Gwinn K , Singleton A , Hardy J ((2011) ) Parkinson’s disease and alpha-synuclein expression. Mov Disord 26: , 2160–2168. |
[54] | Devine MJ , Ryten M , Vodicka P , Thomson AJ , Burdon T , Houlden H , Cavaleri F , Nagano M , Drummond NJ , Taanman JW , Schapira AH , Gwinn K , Hardy J , Lewis PA , Kunath T ((2011) ) Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat Commun 2: , 440. |
[55] | Byers B , Cord B , Nguyen HN , Schule B , Fenno L , Lee PC , Deisseroth K , Langston JW , Pera RR , Palmer TD ((2011) ) SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PLoS One 6: , e26159. |
[56] | Flierl A , Oliveira LM , Falomir-Lockhart LJ , Mak SK , Hesley J , Soldner F , Arndt-Jovin DJ , Jaenisch R , Langston JW , Jovin TM , Schule B ((2014) ) Higher vulnerability and stress sensitivity of neuronal precursor cells carrying an alpha-synuclein gene triplication. PLoS One 9: , e112413. |
[57] | Oliveira LM , Falomir-Lockhart LJ , Botelho MG , Lin KH , Wales P , Koch JC , Gerhardt E , Taschenberger H , Outeiro TF , Lingor P , Schule B , Arndt-Jovin DJ , Jovin TM ((2015) ) Elevated alpha-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell Death Dis 6: , e1994. |
[58] | Soldner F , Laganiere J , Cheng AW , Hockemeyer D , Gao Q , Alagappan R , Khurana V , Golbe LI , Myers RH , Lindquist S , Zhang L , Guschin D , Fong LK , Vu BJ , Meng X , Urnov FD , Rebar EJ , Gregory PD , Zhang HS , Jaenisch R ((2011) ) Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146: , 318–331. |
[59] | Chung CY , Khurana V , Auluck PK , Tardiff DF , Mazzulli JR , Soldner F , Baru V , Lou Y , Freyzon Y , Cho S , Mungenast AE , Muffat J , Mitalipova M , Pluth MD , Jui NT , Schule B , Lippard SJ , Tsai LH , Krainc D , Buchwald SL , Jaenisch R , Lindquist S ((2013) ) Identification and rescue of alpha-synuclein toxicity in Parkinson patient-derived neurons. Science 342: , 983–987. |
[60] | Ryan SD , Dolatabadi N , Chan SF , Zhang X , Akhtar MW , Parker J , Soldner F , Sunico CR , Nagar S , Talantova M , Lee B , Lopez K , Nutter A , Shan B , Molokanova E , Zhang Y , Han X , Nakamura T , Masliah E , Yates JR 3rd , Nakanishi N , Andreyev AY , Okamoto S , Jaenisch R , Ambasudhan R , Lipton SA ((2013) ) Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1alpha transcription. Cell 155: , 1351–1364. |
[61] | Soldner F , Stelzer Y , Shivalila CS , Abraham BJ , Latourelle JC , Barrasa MI , Goldmann J , Myers RH , Young RA , Jaenisch R ((2016) ) Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature 533: , 95–99. |
[62] | MacLeod D , Dowman J , Hammond R , Leete T , Inoue K , Abeliovich A ((2006) ) The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron 52: , 587–593. |
[63] | Parisiadou L , Xie C , Cho HJ , Lin X , Gu XL , Long CX , Lobbestael E , Baekelandt V , Taymans JM , Sun L , Cai H ((2009) ) Phosphorylation of ezrin/radixin/moesin proteins by LRRK2 promotes the rearrangement of actin cytoskeleton in neuronal morphogenesis. J Neurosci 29: , 13971–13980. |
[64] | Gillardon F ((2009) ) Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability–a point of convergence in parkinsonian neurodegeneration? J Neurochem 110: , 1514–1522. |
[65] | Plowey ED , Cherra SJ 3rd , Liu YJ , Chu CT ((2008) ) Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem 105: , 1048–1056. |
[66] | Shin N , Jeong H , Kwon J , Heo HY , Kwon JJ , Yun HJ , Kim CH , Han BS , Tong Y , Shen J , Hatano T , Hattori N , Kim KS , Chang S , Seol W ((2008) ) LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res 314: , 2055–2065. |
[67] | Gomez-Suaga P , Rivero-Rios P , Fdez E , Blanca Ramirez M , Ferrer I , Aiastui A , Lopez De Munain A , Hilfiker S ((2014) ) LRRK2 delays degradative receptor trafficking by impeding late endosomal budding through decreasing Rab7 activity. Hum Mol Genet 23: , 6779–6796. |
[68] | Roosen DA , Cookson MR ((2016) ) LRRK2 at the interface of autophagosomes, endosomes and lysosomes. Mol Neurodegener 11: , 73. |
[69] | Esteves AR , Cardoso SM ((2017) ) LRRK2 at the crossroad between autophagy and microtubule trafficking: Insights into Parkinson’s disease. Neuroscientist 23: , 16–26. |
[70] | Clark LN , Wang Y , Karlins E , Saito L , Mejia-Santana H , Harris J , Louis ED , Cote LJ , Andrews H , Fahn S , Waters C , Ford B , Frucht S , Ottman R , Marder K ((2006) ) Frequency of LRRK2 mutations in early- and late-onset Parkinson disease. Neurology 67: , 1786–1791. |
[71] | Marder K , Wang Y , Alcalay RN , Mejia-Santana H , Tang MX , Lee A , Raymond D , Mirelman A , Saunders-Pullman R , Clark L , Ozelius L , Orr-Urtreger A , Giladi N , Bressman S , Consortium LAJ ((2015) ) Age-specific penetrance of LRRK2 G2019S in the Michael J. Fox Ashkenazi Jewish LRRK2 Consortium. Neurology 85: , 89–95. |
[72] | Bardien S , Lesage S , Brice A , Carr J ((2011) ) Genetic characteristics of leucine-rich repeat kinase 2 (LRRK2) associated Parkinson’s disease. Parkinsonism Relat Disord 17: , 501–508. |
[73] | Nguyen HN , Byers B , Cord B , Shcheglovitov A , Byrne J , Gujar P , Kee K , Schule B , Dolmetsch RE , Langston W , Palmer TD , Pera RR ((2011) ) LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8: , 267–280. |
[74] | Reinhardt P , Schmid B , Burbulla LF , Schondorf DC , Wagner L , Glatza M , Hoing S , Hargus G , Heck SA , Dhingra A , Wu G , Muller S , Brockmann K , Kluba T , Maisel M , Kruger R , Berg D , Tsytsyura Y , Thiel CS , Psathaki OE , Klingauf J , Kuhlmann T , Klewin M , Muller H , Gasser T , Scholer HR , Sterneckert J ((2013) ) Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell 12: , 354–367. |
[75] | Skibinski G , Nakamura K , Cookson MR , Finkbeiner S ((2014) ) Mutant LRRK2 toxicity in neurons depends on LRRK2 levels and synuclein but not kinase activity or inclusion bodies. J Neurosci 34: , 418–433. |
[76] | Sanchez-Danes A , Richaud-Patin Y , Carballo-Carbajal I , Jimenez-Delgado S , Caig C , Mora S , Di Guglielmo C , Ezquerra M , Patel B , Giralt A , Canals JM , Memo M , Alberch J , Lopez-Barneo J , Vila M , Cuervo AM , Tolosa E , Consiglio A , Raya A ((2012) ) Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med 4: , 380–395. |
[77] | Sanders LH , Laganiere J , Cooper O , Mak SK , Vu BJ , Huang YA , Paschon DE , Vangipuram M , Sundararajan R , Urnov FD , Langston JW , Gregory PD , Zhang HS , Greenamyre JT , Isacson O , Schule B ((2014) ) LRRK2 mutations cause mitochondrial DNA damage in iPSC-derived neural cells from Parkinson’s disease patients: Reversal by gene correction. Neurobiol Dis 62: , 381–386. |
[78] | Hsieh CH , Shaltouki A , Gonzalez AE , Bettencourt da Cruz A , Burbulla LF , St Lawrence E , Schule B , Krainc D , Palmer TD , Wang X ((2016) ) Functional impairment in miro degradation and mitophagy is a shared feature in familial and sporadic Parkinson’s disease. Cell Stem Cell 19: , 709–724. |
[79] | Kitada T , Asakawa S , Hattori N , Matsumine H , Yamamura Y , Minoshima S , Yokochi M , Mizuno Y , Shimizu N ((1998) ) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: , 605–608. |
[80] | Shimura H , Hattori N , Kubo S , Mizuno Y , Asakawa S , Minoshima S , Shimizu N , Iwai K , Chiba T , Tanaka K , Suzuki T ((2000) ) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase.302-305.. Nat Genet 25: . |
[81] | Panicker N , Dawson VL , Dawson TM ((2017) ) Activation mechanisms of the E3 ubiquitin ligase parkin. Biochem J 474: , 3075–3086. |
[82] | Hardy J ((2010) ) Genetic analysis of pathways to Parkinson disease. Neuron 68: , 201–206. |
[83] | Lücking CB , Durr A , Bonifati V , Vaughan J , De Michele G , Gasser T , Harhangi BS , Meco G , Denefle P , Wood NW , Agid Y , Brice A , French Parkinson’s Disease Genetics Study G, European Consortium on Genetic Susceptibility in Parkinson’s Disease ((2000) ) Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med 342: , 1560–1567. |
[84] | Goldberg MS , Fleming SM , Palacino JJ , Cepeda C , Lam HA , Bhatnagar A , Meloni EG , Wu N , Ackerson LC , Klapstein GJ , Gajendiran M , Roth BL , Chesselet MF , Maidment NT , Levine MS , Shen J ((2003) ) Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278: , 43628–43635. |
[85] | Perez FA , Palmiter RD ((2005) ) Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci U S A 102: , 2174–2179. |
[86] | Dave KD , De Silva S , Sheth NP , Ramboz S , Beck MJ , Quang C , Switzer RC 3rd , Ahmad SO , Sunkin SM , Walker D , Cui X , Fisher DA , McCoy AM , Gamber K , Ding X , Goldberg MS , Benkovic SA , Haupt M , Baptista MA , Fiske BK , Sherer TB , Frasier MA ((2014) ) Phenotypic characterization of recessive gene knockout rat models of Parkinson’s disease. Neurobiol Dis 70: , 190–203. |
[87] | Jiang H , Ren Y , Zhao J , Feng J ((2004) ) Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum Mol Genet 13: , 1745–1754. |
[88] | Jiang H , Jiang Q , Liu W , Feng J ((2006) ) Parkin suppresses the expression of monoamine oxidases. J Biol Chem 281: , 8591–8599. |
[89] | Ren Y , Jiang H , Ma D , Nakaso K , Feng J ((2011) ) Parkin degrades estrogen-related receptors to limit the expression of monoamine oxidases. Hum Mol Genet 20: , 1074–1083. |
[90] | Schreiber SN , Emter R , Hock MB , Knutti D , Cardenas J , Podvinec M , Oakeley EJ , Kralli A ((2004) ) The estrogen-related receptor alpha (ERRalpha) functions in PPARgamma coactivator 1alpha (PGC-1alpha)-induced mitochondrial biogenesis. Proc Natl Acad Sci U S A 101: , 6472–6477. |
[91] | Jiang H , Ren Y , Yuen EY , Zhong P , Ghaedi M , Hu Z , Azabdaftari G , Nakaso K , Yan Z , Feng J ((2012) ) Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat Commun 3: , 668. |
[92] | Imaizumi Y , Okada Y , Akamatsu W , Koike M , Kuzumaki N , Hayakawa H , Nihira T , Kobayashi T , Ohyama M , Sato S , Takanashi M , Funayama M , Hirayama A , Soga T , Hishiki T , Suematsu M , Yagi T , Ito D , Kosakai A , Hayashi K , Shouji M , Nakanishi A , Suzuki N , Mizuno Y , Mizushima N , Amagai M , Uchiyama Y , Mochizuki H , Hattori N , Okano H ((2012) ) Mitochondrial dysfunction associated with increased oxidative stress and alpha-synuclein accumulation in PARK2 iPSC-derived neurons and postmortem brain tissue. Mol Brain 5: , 35. |
[93] | Chung SY , Kishinevsky S , Mazzulli JR , Graziotto J , Mrejeru A , Mosharov EV , Puspita L , Valiulahi P , Sulzer D , Milner TA , Taldone T , Krainc D , Studer L , Shim JW ((2016) ) Parkin and PINK1 patient iPSC-derived midbrain dopamine neurons exhibit mitochondrial dysfunction and alpha-synuclein accumulation. Stem Cell Reports 7: , 664–677. |
[94] | Zhong P , Hu Z , Jiang H , Yan Z , Feng J ((2017) ) Dopamine induces oscillatory activities in human midbrain neurons with parkin mutations. Cell Rep 19: , 1033–1044. |
[95] | Brittain JS , Brown P ((2014) ) Oscillations and the basal ganglia: Motor control and beyond. Neuroimage 85: (Pt 2), 637–647. |
[96] | Ren Y , Jiang H , Hu Z , Fan K , Wang J , Janoschka S , Wang X , Ge S , Feng J ((2015) ) Parkin mutations reduce the complexity of neuronal processes in iPSC-derived human neurons. Stem Cells 33: , 68–78. |
[97] | Cartelli D , Amadeo A , Calogero AM , Casagrande FVM , De Gregorio C , Gioria M , Kuzumaki N , Costa I , Sassone J , Ciammola A , Hattori N , Okano H , Goldwurm S , Roybon L , Pezzoli G , Cappelletti G ((2018) ) Parkin absence accelerates microtubule aging in dopaminergic neurons. Neurobiol Aging 61: , 66–74. |
[98] | Farrer M , Chan P , Chen R , Tan L , Lincoln S , Hernandez D , Forno L , Gwinn-Hardy K , Petrucelli L , Hussey J , Singleton A , Tanner C , Hardy J , Langston JW ((2001) ) Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol 50: , 293–300. |
[99] | Shaltouki A , Sivapatham R , Pei Y , Gerencser AA , Momcilovic O , Rao MS , Zeng X ((2015) ) Mitochondrial alterations by PARKIN in dopaminergic neurons using PARK2 patient-specific and PARK2 knockout isogenic iPSC lines. Stem Cell Reports 4: , 847–859. |
[100] | Narendra D , Tanaka A , Suen DF , Youle RJ ((2008) ) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: , 795–803. |
[101] | Pickrell AM , Youle RJ ((2015) ) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85: , 257–273. |
[102] | Scarffe LA , Stevens DA , Dawson VL , Dawson TM ((2014) ) Parkin and PINK1: Much more than mitophagy. Trends Neurosci 37: , 315–324. |
[103] | Grenier K , McLelland GL , Fon EA ((2013) ) Parkin- and PINK1-dependent mitophagy in neurons: Will the real pathway please stand up? Front Neurol 4: , 100. |
[104] | Sterky FH , Lee S , Wibom R , Olson L , Larsson NG ((2011) ) Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc Natl Acad Sci U S A 108: , 12937–12942. |
[105] | Kageyama Y , Zhang Z , Roda R , Fukaya M , Wakabayashi J , Wakabayashi N , Kensler TW , Reddy PH , Iijima M , Sesaki H ((2012) ) Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J Cell Biol 197: , 535–551. |
[106] | Rakovic A , Shurkewitsch K , Seibler P , Grunewald A , Zanon A , Hagenah J , Krainc D , Klein C ((2013) ) Phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1)-dependent ubiquitination of endogenous Parkin attenuates mitophagy: Study in human primary fibroblasts and induced pluripotent stem cell-derived neurons. J Biol Chem 288: , 2223–2237. |
[107] | Greene AW , Grenier K , Aguileta MA , Muise S , Farazifard R , Haque ME , McBride HM , Park DS , Fon EA ((2012) ) Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 13: , 378–385. |
[108] | Seibler P , Graziotto J , Jeong H , Simunovic F , Klein C , Krainc D ((2011) ) Mitochondrial Parkin recruitment is impaired in neurons derived from mutant PINK1 induced pluripotent stem cells. J Neurosci 31: , 5970–5976. |
[109] | Morais VA , Haddad D , Craessaerts K , De Bock PJ , Swerts J , Vilain S , Aerts L , Overbergh L , Grunewald A , Seibler P , Klein C , Gevaert K , Verstreken P , De Strooper B ((2014) ) PINK1 loss-of-function mutations affect mitochondrial complex I activity via NdufA10 ubiquinone uncoupling. Science 344: , 203–207. |
[110] | Cooper O , Seo H , Andrabi S , Guardia-Laguarta C , Graziotto J , Sundberg M , McLean JR , Carrillo-Reid L , Xie Z , Osborn T , Hargus G , Deleidi M , Lawson T , Bogetofte H , Perez-Torres E , Clark L , Moskowitz C , Mazzulli J , Chen L , Volpicelli-Daley L , Romero N , Jiang H , Uitti RJ , Huang Z , Opala G , Scarffe LA , Dawson VL , Klein C , Feng J , Ross OA , Trojanowski JQ , Lee VM , Marder K , Surmeier DJ , Wszolek ZK , Przedborski S , Krainc D , Dawson TM , Isacson O ((2012) ) Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med 4: , 141ra190. |
[111] | Azkona G , Lopez de Maturana R , Del Rio P , Sousa A , Vazquez N , Zubiarrain A , Jimenez-Blasco D , Bolanos JP , Morales B , Auburger G , Arbelo JM , Sanchez-Pernaute R ((2018) ) LRRK2 expression is deregulated in fibroblasts and neurons from Parkinson patients with mutations in PINK1. Mol Neurobiol 55: , 506–516. |
[112] | Bonifati V , Rizzu P , van Baren MJ , Schaap O , Breedveld GJ , Krieger E , Dekker MC , Squitieri F , Ibanez P , Joosse M , van Dongen JW , Vanacore N , van Swieten JC , Brice A , Meco G , van Duijn CM , Oostra BA , Heutink P ((2003) ) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299: , 256–259. |
[113] | Wilson MA ((2011) ) The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxid Redox Signal 15: , 111–122. |
[114] | Burbulla LF , Song P , Mazzulli JR , Zampese E , Wong YC , Jeon S , Santos DP , Blanz J , Obermaier CD , Strojny C , Savas JN , Kiskinis E , Zhuang X , Kruger R , Surmeier DJ , Krainc D ((2017) ) Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 357: , 1255–1261. |
[115] | Sidransky E , Lopez G ((2012) ) The link between the GBA gene and parkinsonism. Lancet Neurol 11: , 986–998. |
[116] | Schondorf DC , Aureli M , McAllister FE , Hindley CJ , Mayer F , Schmid B , Sardi SP , Valsecchi M , Hoffmann S , Schwarz LK , Hedrich U , Berg D , Shihabuddin LS , Hu J , Pruszak J , Gygi SP , Sonnino S , Gasser T , Deleidi M ((2014) ) iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun 5: , 4028. |
[117] | Woodard CM , Campos BA , Kuo SH , Nirenberg MJ , Nestor MW , Zimmer M , Mosharov EV , Sulzer D , Zhou H , Paull D , Clark L , Schadt EE , Sardi SP , Rubin L , Eggan K , Brock M , Lipnick S , Rao M , Chang S , Li A , Noggle SA ((2014) ) iPSC-derived dopamine neurons reveal differences between monozygotic twins discordant for Parkinson’s disease. Cell Rep 9: , 1173–1182. |
[118] | Iovino M , Agathou S , Gonzalez-Rueda A , Del Castillo Velasco-Herrera M , Borroni B , Alberici A , Lynch T , O’Dowd S , Geti I , Gaffney D , Vallier L , Paulsen O , Karadottir RT , Spillantini MG ((2015) ) Early maturation and distinct tau pathology in induced pluripotent stem cell-derived neurons from patients with MAPT mutations. Brain 138: , 3345–3359. |
[119] | Fong H , Wang C , Knoferle J , Walker D , Balestra ME , Tong LM , Leung L , Ring KL , Seeley WW , Karydas A , Kshirsagar MA , Boxer AL , Kosik KS , Miller BL , Huang Y ((2013) ) Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem Cell Reports 1: , 226–234. |
[120] | Wang Q , Liu Y , Zhou J ((2015) ) Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl Neurodegener 4: , 19. |
[121] | Mogi M , Harada M , Riederer P , Narabayashi H , Fujita K , Nagatsu T ((1994) ) Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett 165: , 208–210. |
[122] | Vila M , Jackson-Lewis V , Guegan C , Wu DC , Teismann P , Choi DK , Tieu K , Przedborski S ((2001) ) The role of glial cells in Parkinson’s disease. Curr Opin Neurol 14: , 483–489. |
[123] | Mena MA , Garcia de Yebenes J ((2008) ) Glial cells as players in parkinsonism: The “good,” the “bad,” and the “mysterious” glia. Neuroscientist 14: , 544–560. |
[124] | Kordower JH , Emborg ME , Bloch J , Ma SY , Chu Y , Leventhal L , McBride J , Chen EY , Palfi S , Roitberg BZ , Brown WD , Holden JE , Pyzalski R , Taylor MD , Carvey P , Ling Z , Trono D , Hantraye P , Deglon N , Aebischer P ((2000) ) Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson’s disease. Science 290: , 767–773. |
[125] | Gash DM , Zhang Z , Ovadia A , Cass WA , Yi A , Simmerman L , Russell D , Martin D , Lapchak PA , Collins F , Hoffer BJ , Gerhardt GA ((1996) ) Functional recovery in parkinsonian monkeys treated with GDNF. Nature 380: , 252–255. |
[126] | d’Anglemont de Tassigny X , Pascual A , Lopez-Barneo J ((2015) ) GDNF-based therapies, GDNF-producing interneurons, and trophic support of the dopaminergic nigrostriatal pathway. Implications for Parkinson’s disease. Front Neuroanat 9: , 10. |
[127] | Su X , Maguire-Zeiss KA , Giuliano R , Prifti L , Venkatesh K , Federoff HJ ((2008) ) Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol Aging 29: , 1690–1701. |
[128] | Gillardon F , Schmid R , Draheim H ((2012) ) Parkinson’s disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience 208: , 41–48. |
[129] | Zhang SC , Wernig M , Duncan ID , Brustle O , Thomson JA ((2001) ) In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat Biotechnol 19: , 1129–1133. |
[130] | Chandrasekaran A , Avci HX , Leist M , Kobolak J , Dinnyes A ((2016) ) Astrocyte differentiation of human pluripotent stem cells: New tools for neurological disorder research. Front Cell Neurosci 10: , 215. |
[131] | Muffat J , Li Y , Yuan B , Mitalipova M , Omer A , Corcoran S , Bakiasi G , Tsai LH , Aubourg P , Ransohoff RM , Jaenisch R ((2016) ) Efficient derivation of microglia-like cells from human pluripotent stem cells. Nat Med 22: , 1358–1367. |
[132] | Abud EM , Ramirez RN , Martinez ES , Healy LM , Nguyen CHH , Newman SA , Yeromin AV , Scarfone VM , Marsh SE , Fimbres C , Caraway CA , Fote GM , Madany AM , Agrawal A , Kayed R , Gylys KH , Cahalan MD , Cummings BJ , Antel JP , Mortazavi A , Carson MJ , Poon WW , Blurton-Jones M ((2017) ) iPSC-derived human microglia-like cells to study neurological diseases. Neuron 94: , 278–293 e279. |
[133] | Juopperi TA , Kim WR , Chiang CH , Yu H , Margolis RL , Ross CA , Ming GL , Song H ((2012) ) Astrocytes generated from patient induced pluripotent stem cells recapitulate features of Huntington’s disease patient cells. Mol Brain 5: , 17. |
[134] | Brennand K , Savas JN , Kim Y , Tran N , Simone A , Hashimoto-Torii K , Beaumont KG , Kim HJ , Topol A , Ladran I , Abdelrahim M , Matikainen-Ankney B , Chao SH , Mrksich M , Rakic P , Fang G , Zhang B , Yates JR 3rd , Gage FH ((2015) ) Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry 20: , 361–368. |
[135] | Mariani J , Simonini MV , Palejev D , Tomasini L , Coppola G , Szekely AM , Horvath TL , Vaccarino FM ((2012) ) Modeling human cortical developmentusing induced pluripotent stem cells. Proc Natl Acad Sci U S A 109: , 12770–12775. |
[136] | Miller JD , Ganat YM , Kishinevsky S , Bowman RL , Liu B , Tu EY , Mandal PK , Vera E , Shim JW , Kriks S , Taldone T , Fusaki N , Tomishima MJ , Krainc D , Milner TA , Rossi DJ , Studer L ((2013) ) Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13: , 691–705. |
[137] | Gunhanlar N , Shpak G , van der Kroeg M , Gouty-Colomer LA , Munshi ST , Lendemeijer B , Ghazvini M , Dupont C , Hoogendijk WJG , Gribnau J , de Vrij FMS , Kushner SA ((2018) ) A simplified protocol for differentiation of electrophysiologically mature neuronal networks from human induced pluripotent stem cells. Mol Psychiatry 23: , 1336–1344. |
[138] | Grealish S , Diguet E , Kirkeby A , Mattsson B , Heuer A , Bramoulle Y , Van Camp N , Perrier AL , Hantraye P , Bjorklund A , Parmar M ((2014) ) Human ESC-derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson’s disease. Cell Stem Cell 15: , 653–665. |
[139] | Kikuchi T , Morizane AD , Magotani H , Onoe H , Hayashi T , Mizuma H , Takara S , Takahashi R , Inoue H , Morita S , Yamamoto M , Okita K , Nakagawa M , Parmar M , Takahashi J ((2017) ) Human iPS cell-derived dopaminergic neurons function in a primate Parkinson’s disease model. Nature 548: , 592–596. |
[140] | Mansour AA , Goncalves JT , Bloyd CW , Li H , Fernandes S , Quang D , Johnston S , Parylak SL , Jin X , Gage FH ((2018) ) An in vivo model of functional and vascularized human brain organoids. Nat Biotechnol 36: , 432–441. |
[141] | Shih JC , Chen K , Ridd MJ ((1999) ) Monoamine oxidase: From genes to behor. Annu Rev Neurosci 22: , 197–217. |
[142] | Guzman JN , Sanchez-Padilla J , Chan CS , Surmeier DJ ((2009) ) Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci 29: , 11011–11019. |
[143] | Thompson L , Barraud P , Andersson E , Kirik D , Bjorklund A ((2005) ) Identification of dopaminergic neurons of nigral and ventral tegmental area subtypes in grafts of fetal ventral mesencephalon based on cell morphology, protein expression, and efferent projections. J Neurosci 25: , 6467–6477. |
[144] | Sulzer D , Surmeier DJ ((2013) ) Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov Disord 28: , 41–50. |
[145] | Dolle C , Flones I , Nido GS , Miletic H , Osuagwu N , Kristoffersen S , Lilleng PK , Larsen JP , Tysnes OB , Haugarvoll K , Bindoff LA , Tzoulis C ((2016) ) Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat Commun 7: , 13548. |
[146] | Liang CL , Wang TT , Luby-Phelps K , German DC ((2007) ) Mitochondria mass is low in mouse substantia nigra dopamine neurons: Implications for Parkinson’s disease. Exp Neurol 203: , 370–380. |
[147] | Perier C , Bender A , Garcia-Arumi E , Melia MJ , Bove J , Laub C , Klopstock T , Elstner M , Mounsey RB , Teismann P , Prolla T , Andreu AL , Vila M ((2013) ) Accumulation of mitochondrial DNA deletions within dopaminergic neurons triggers neuroprotective mechanisms. Brain 136: , 2369–2378. |
[148] | Matsuda W , Furuta T , Nakamura KC , Hioki H , Fujiyama F , Arai R , Kaneko T ((2009) ) Single nigrostriatal dopaminergic neurons form widely spread and highly dense al arborizations in the neostriatum. J Neurosci 29: , 444–453. |
[149] | Pissadaki EK , Bolam JP ((2013) ) The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front Comput Neurosci 7: , 13. |
[150] | Feng J ((2006) ) Microtubule: A common target for parkin and Parkinson’s disease toxins. Neuroscientist 12: , 469–476. |
[151] | Bolam JP , Pissadaki EK ((2012) ) Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov Disord 27: , 1478–1483. |
[152] | Lee VM , Trojanowski JQ ((2006) )Neuron 52: , 33–38 Mechanisms of Parkinson’s disease linked to pathological alpha-synucle. New targets for drug discovery. |
[153] | Brundin P , Melki R ((2017) ) Prying into the prion hypothesis for Parkinson’s disease. J Neurosci 37: , 9808–9818. |
[154] | Surmeier DJ , Obeso JA , Halliday GM ((2017) ) Parkinson’s disease is not simply a prion disorder. J Neurosci 37: , 9799–9807. |
[155] | Lieberman OJ , Choi SJ , Kanter E , Saverchenko A , Frier MD , Fiore GM , Wu M , Kondapalli J , Zampese E , Surmeier DJ , Sulzer D , Mosharov EV ((2017) ) alpha-synuclein-dependent calcium entry underlies differential sensitivity of cultured SN and VTA dopaminergic neurons to a Parkinsonian neurotoxin. eNeuro 4: , ENEURO.0167-17. 2017. |
[156] | Mosharov EV , Larsen KE , Kanter E , Phillips KA , Wilson K , Schmitz Y , Krantz DE , Kobayashi K , Edwards RH , Sulzer D ((2009) ) Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 62: , 218–229. |


