
Gene Therapy for Parkinson’s Disease, An Update
Abstract
The current mainstay treatment of Parkinson’s disease (PD) consists of dopamine replacement therapy which, in addition to causing several side effects, does not delay disease progression. The field of gene therapy offers a potential means to improve current therapy. The present review gives an update of the present status of gene therapy for PD. Both non-disease and disease modifying transgenes have been tested for PD gene therapy in animal and human studies. Non-disease modifying treatments targeting dopamine or GABA synthesis have been successful and promising at improving PD symptomatology in randomized clinical studies, but substantial testing remains before these can be implemented in the standard clinical treatment repertoire. As for disease modifying targets that theoretically offer the possibility of slowing the progression of disease, several neurotrophic factors show encouraging results in preclinical models (e.g., neurturin, GDNF, BDNF, CDNF, VEGF-A). However, so far, clinical trials have only tested neurturin, and, unfortunately, no trial has been able to meet its primary endpoint. Future clinical trials with neurotrophic factors clearly deserve to be conducted, considering the still enticing goal of actually slowing the disease process of PD. As alternative types of gene therapy, opto- and chemogenetics might also find future use in PD treatment and novel genome-editing technology could also potentially be applied as individualized gene therapy for genetic types of PD.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disease and is estimated to afflict up to 5.8 million people worldwide [1], with the increased mean age of the population in the Western world, the prevalence of the disease is projected to rise. PD is characterized by a slow asymmetric debut of motor symptoms in the form of muscle rigidity, impaired gait, hypokinesia and tremor as well as several non-motor symptoms, including disturbed emotions, cognition, sleep pattern, and autonomous dysfunction [2].
The underlying pathophysiology consists of the cellular accumulation of synuclein-α (SNCA), aggregating in insoluble constructs leading to cellular dysfunction and toxicity [3]. This is especially apparent with the loss of dopaminergic neurons (DA) in the substantia nigra pars compacta (SNc), disrupting the many pathways relying on the dopaminergic tonus arising from this neuron population. The imbalance leads to aberrant signaling in the circuits of the striatal projections, giving rise to the parkinsonian phenotype [2, 4]. Aggregation of SNCA is also a pathologic hallmark of a group of other brain diseases such as lewy body dementia [5] and multiple system atrophy [6]. This review will focus on gene therapy for PD, but as many of the proposed therapies do not necessarily specifically target PD, it is likely that they could also prove feasible for treating other α-synucleinopathies.
Current treatment regimens for PD mainly involve administration of levo-dopa (L-DOPA), dopamine agonists or MAO-B inhibitors or surgery in the form of deep brain stimulation (DBS) or neuroablative surgery [7, 8]. However, these treatments are symptomatic and do not prevent PD progression and may be associated with significant side effects. Thus, the exploration of novel treatment avenues is clearly warranted [9].
Gene therapy was first described in 1972 as a means to “replace bad DNA with good DNA” [10]. The basic principle still stands but has evolved to become somewhat more sophisticated. Gene therapy can be used to treat diseases by the introduction of therapeutic genes or by replacing, silencing, or correcting faulty genes. Many different approaches exist, but the primary strategy is the use of engineered non-replicating viral vectors; predominantly various serotypes of recombinant adeno-associated virus (AAV) or lentivirus [11]. Several studies using viral vectors have provided evidence for both safety and high fidelity of gene expression [12–15]. Novel methods of gene delivery are currently being developed and have been successfully utilized in murine models of PD [16, 17]. In more recent years, gene therapy has also been tested in several human clinical trials as reviewed below. With the approval of AAV vectors for gene therapy in Europe and recently in the US [18, 19], it is expected that the gene therapeutic approach will gain further ground in the coming years. This review will present an up-to-date overview of in vivo gene therapy for PD.
So far, several possible targets for genetic treatment of PD have been identified. These targets can be classified as either disease modifying or non-disease modifying. The non-disease modifying treatments are aimed at ailing parkinsonistic symptoms by attempting to normalize aberrant firing in the basal ganglia by expression of either dopaminergic or GABAgenic enzymes. These treatments are symptomatic and do not alter the underlying pathophysiological process.
Disease modifying strategies revolve around stopping PD-mediated cell death and/or regenerating lost neurons. The most investigated approach has been nigral overexpression of growth factors found to have neuroprotective properties (see Tables 4 and 5) [20]. Other possible targets encompass genomic repair of faulty genes [21], giving rise to toxic SNCA constructs.
NON-DISEASE MODIFYING TARGETS
The enzyme aromatic acid decarboxylase (AADC) is part of the dopamine synthetic apparatus (Fig. 1) and is responsible for the conversion of L-DOPA to dopamine [22]. The primary clinical approach towards treatment of PD is the use of L-DOPA, a precursor of dopamine able to cross the blood-brain barrier, thus enabling oral treatment. L-DOPA is a symptomatic treatment unable to halt disease progression and is associated with several side effects, including so-called OFF-states. OFF-states are periods of immobility and incapacity often accompanied by depression or hypomania, due to erratic function of the drug. These periods have been attributed to unpredictable gastric emptying, overload of the blood-brain barrier carrier system responsible for transporting L-DOPA as well as insufficient aromatic L-amino acid decarboxylase (AADC) levels [23–26]. The absorption issues have largely been overcome using new formulations of L-DOPA and various pump systems.
Fig.1
L-tyrosine is converted to L-dihydroxyphenylalanine (L-DOPA) by tyrosine hydroxylase (TH) using the co-factor tetrahydrobiopterin, under the rate limit of GTP cyclohydroxylase 1 (GCH1). L-DOPA is further converted into dopamine by alpha-amino acid decarboxylase (AADC). The three enzymes in red are encoded by Prosavin gene therapy. PTPS: pyruvoyltetrahydropterin synthase; SPR: sepiapterin reductase.
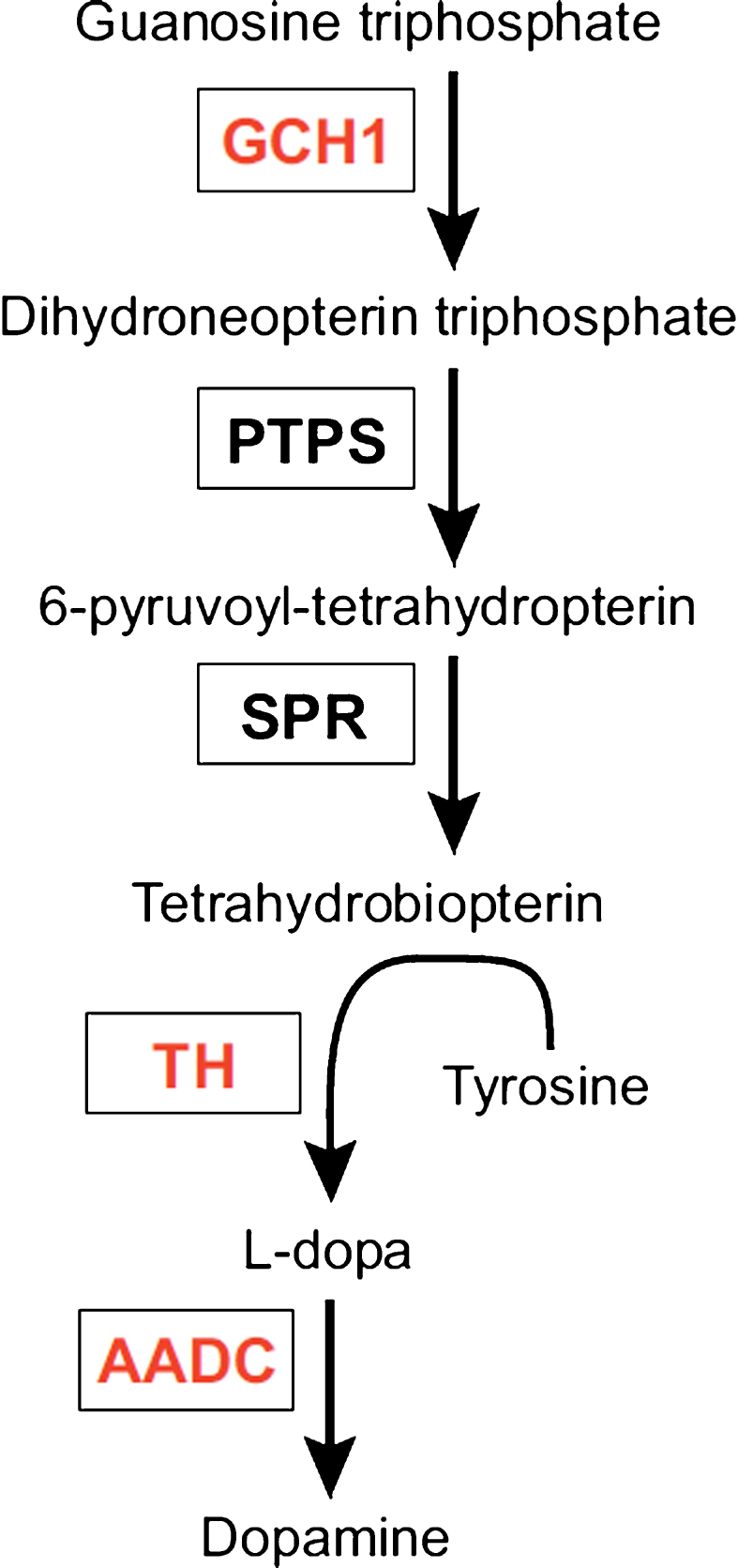
Several rodent studies found striatal vector-mediated AADC overexpression to be well tolerated and able to improve parkinsonian symptoms [27–29]. Further studies in non-human primates (NHP) [30–33] described a decreased need for medication, as well as improved motor function off medication, once a certain level of transfection was achieved (see Table 1) [31]. The promising results in NHPs led to the first phase 1 clinical trial with bilateral AAV2-induced AADC expression in the putamen of severely affected PD patients published in 2008 [14]. The authors found a modest improvement in the unified Parkinson’s disease rating scale (UPDRS) and, importantly, found no adverse effects of AAV-mediated AADC overexpression in humans. Furthermore, phase 1 trials (see Table 1) reached the same conclusions regarding improved symptomatology in the patients while in OFF-state as well as a general reduction in UPDRS and a trend towards lower need for L-DOPA [12, 25]. A follow-up study on patients from a phase 1 study by Christine et al. [12] showed a discrete increase in UPDRS scores as well as a minor reduction in activity of the AADC ligand 18Fluoro-L-m-tyrosine. Both were, however, still well above baseline. The authors attribute the decline to ongoing degeneration of non-transfected still-functioning nigral neurons, implying AAV-therapy to be permanent with no decline in expression of transfected cells. There are currently two ongoing and one planned open label study aimed at examining the safety profile of AADC therapy, optimizing dosage and delivery method [34, 35]. These studies will hopefully pave the way for future randomized placebo-controlled trials.
Table 1
NHP trials and clinical trials utilizing AADC
| Author | Method | Subjects | Results | Reference |
| Bankiewicz et al. (2000) | Bilateral convection enhanced delivery AAV induced AADC expression in nu. caudatus, putamen and globus pallidus. | MPTP-treated rhesus monkeys (n = 4). | Higher increase in gene expression using CED than conventional injection. Strong AADC expression in striatum. | [33] |
| Daadi et al. (2006) | Bilateral convection enhanced delivery AAV induced AADC expression in nu. caudatus, putamen and globus pallidus. | MPTP-treated macaque monkeys (n = 4). | Verification of strong striatal AADC expression after 3 years. | [32] |
| Forsayeth et al. (2006) | Dose ranging study with bilateral putaminal injection of AAV2-AADC in escalating doses. | MPTP-treated macaque monkeys (n = 12). | A minimum dose is needed to increase L-DOPA response and FMT-activity in striatum. | [31] |
| Bankiewicz et al. (2006) | Unilateral AAV-2 induced AADC expression in striatum. | MPTP-lesioned macaque monkeys (n = 8). | PET-verified increase in AADC activity after 2 years. Consistently higher L-DOPA sensitivity in treatment group. | [30] |
| Eberling et al. (2008) | Open label study. Bilateral AAV2-induced AADC expression in putamen. | Severely affected PD patients (n = 5). | Modest improvement of OFF-state UPDRS-score. 30% increase in putaminal FMT activity at 6-month follow-up. | [14] |
| Christine et al. (2009) | Open label study. Bilateral AAV2-induced AADC expression in putamen. High and low dosage. | Moderately affected PD patients (n = 10). | Improvement of UPDRS scores. Longer time in ON-state. Therapy well tolerated. High dose with higher FMT-activity than low dose at 6-month follow-up. | [12] |
| Hadaczek et al. (2010) | 8-year follow up on the study by Bankiewicz et al. (2006). | MPTP-lesioned macaque monkeys (n = 2). | No decrease in FMT signal and continued robust AADC expression. | [183] |
| Muramatsu et al. (2010) | Open label study. Bilateral AAV2-induced AADC expression in putamen. | Moderately to severe affected PD patients (n = 6). | Decrease in UPDRS scores by 46% in OFF-state. At 6-month follow-up. Persistent FMT activity at 24 weeks. | [25] |
| Mittermeyer et al. (2012) | 4-year follow-up on Christine et al. (2009). | Moderately affected PD patients. | Discrete decline in AADC activity at 4-year follow-up. Slight increase in UPDRS-score. | [24] |
Clinical and preclinical studies with AADC-gene therapy. FMT, 18Fluoro-L-m-tyrosine, a tracer specific for AADC.
Should the AADC therapy appear efficient, it is not suited as a stand-alone treatment due to the continued need for exogenous L-DOPA. However, as the patients in all the clinical trials have reported improved L-DOPA response as well as a general improvement of UPDRS scores, it is possible that AADC gene therapy can be included in future treatment aimed at ameliorating otherwise incontrollable motor fluctuations and be used on par with DBS in PD therapy.
AADC, TH, and GCH
An enticing approach to symptomatic treatment of PD, is to completely reconstruct the dopamine synthesis apparatus by introducing all the genes responsible for the conversion of tyrosine to dopamine (Fig. 1) in cells of the target area. This requires the enzyme tyrosine hydroxylase (TH) along with the co-factor tetrahydrobiopterin (BH4) for converting the amino acid L-tyrosine into L-DOPA which in turn is converted into dopamine by AADC. The rate limiting step for the synthesis of BH4 is the enzyme GTP cyclohydroxylase 1 (GCH) [36]. By expressing the three enzymes (TH, GCH and AADC) in the striatum, it should be theoretically possible to reduce, if not eliminate, the need for exogenous L-DOPA, which could ensure a stable dopamine tonus eliminating wearing off effects and off-target effects.
An early study by Kaplitt et al. (1994) reported behavioral recovery in 6-OHDA treated rats subjected to AAV-TH in the striatum [37]. This approach was investigated in an NHP study by During et al. (1998), who injected a mix of AAV-TH and AAV-AADC in the striatum of green monkeys [38]. They were, however, unable to detect any behavioral effect of the treatment compared to controls injected with empty vector. Nevertheless, they could detect an increased dopamine production in the striatum. The lack of clinical effect in their study could possibly be due to a subclinical restoration of dopamine levels because of the low amounts of vector injected [38] or lack of BH4.
Later, most research on reconstitution of the dopaminergic apparatus has focused on expressing all three dopaminergic enzymes. However, the delivery of all three genes represents a hurdle, as the three enzymes combined are too big to fit into a single AAV genome. This challenge has been met in two ways. Either by injecting a mixture of three different AAV-vectors, each containing one of the desired transgenes, or by using LV, which has a higher transgene expression capacity. Rodent studies indicate that both methods are well tolerated and efficient at reducing the parkinsonian phenotype in PD models [39–47].
The success prompted further investigation in NHPs (see Table 2) with both the tricistronic and lentiviral approach. An early study utilizing the tricistronic vectors showed overexpression of TH, AADC and GCH [48] to be both possible and well tolerated. Bearing in mind the lack of clinical effect seen in the study by During et al. (1998), Muramatsu and colleagues (2010) injected an increased titer and added AAV2-GCH to the injections and presented a significant increase in dopamine production as well as a robust rescue of the parkinsonian phenotype in MPTP-treated marmosets [48], with the effect persisting at 15 year follow-up in one monkey [49].
Table 2
NHP trials and clinical trials combining TH and AADC
| Author | Method | Subjects | Results | Reference |
| During et al. (1998) | Treatment with mix of AAV-TH and AAV-AADC in the striatum. | MPTP-treated green monkeys (n = 12). | Biochemically measured increase of dopamine production. No behavioral difference from non-transfected individuals. | [38] |
| Muramatsu et al. (2002) | Unilateral AAV induced expression of TH, GCH and AADC in the striatum. | MPTP-treated marmosets (n = 4). | Bilateral improvement of motor skills; improved fine motor skills. | [48] |
| Sehara et al. (2017) | Follow-up on Muramatsu et al. (2002). | MPTP-treated marmoset (n = 1). | Continuous effect on motor skills. Persistent expression of transfected genes. | [49] |
| Jarraya et al. (2009) | LV-induced bilateral putaminal expression of TH, GCH and AADC. | MPTP-treated macaques (n = 26). | Correction of motor deficits at 44 months. No observed dyskinesias. | [50] |
| Palfi et al. (2014) | Open label study. LV-induced putaminal expression of TH, GCH and AADC. | Patients with advanced bilateral PD (n = 15). | Therapy is deemed safe and tolerable. Improved UDPRS scores and lower need for medication. | [51] |
Clinical and preclinical studies with combined TH-, AADC- and GCH-gene therapy.
It has been proposed that the packaging of all genes within one vector ensures joint expression of the proteins and thus excludes the hassle of a tricistronic system. The LV approach was shown to be efficient in rats [39]. Prompting a study on the effects of a LV vector expressing AADC, TH and GCH in the putamen of MPTP-treated macaque monkeys. The therapy was well tolerated, and the subjects showed marked improvement of parkinsonian symptoms with no signs of dyskinesias. Microdialysis showed an approximately 50% restoration of normal dopamine tonus [50]. A subsequent phase 1 trial examining the effect of injection of LV-TH-GCH-AADC under the name of Prosavin to the putamen was sponsored by Oxford Biomedica [51]. The authors of the study found the drug to be well tolerated and able to reduce UPDRS scores in patients off medication by 12 points as well as decrease the general need for medication. The study is ongoing with several planned follow-up visits to further evaluate long-term safety and efficacy [52]. The mechanism behind the apparent effect of Prosavin is not yet fully understood, but it is theorized, that increased dopamine levels in the extracellular environment achieved by transfected cells can normalize firing rates of afferent projection axons in the same manner as DBS [38, 51, 53]. The only clinical trial on Prosavin, thus advocates further investigations regarding the vector dosage before it is deemed feasible to progress to a phase 2 study [51].
Vesicular monoamine transporter 2 (VMAT2) is a macromolecule which transports dopamine into intracellular synaptic vesicles of dopaminergic neurons for release into the synaptic cleft [26]. A study by Sun et al. (2004) compared the effect of injecting a herpes simplex (HSV) vector containing either TH, GCH and AADC or TH, GCH, AADC and VMAT2 to striatal GABAergic neurons in a 6-OHDA-rat model. The authors hypothesized that adding VMAT2 to the vector would further stabilize the dopaminergic tonus by a more stable release of dopamine. They found the 4-gene vector to be superior to the 3-gene vector in controlling amphetamine-induced rotations as well as K+-induced dopamine release and increased levels of striatal dopamine in general [45].
Glutamic acid decarboxylase (GAD)
A significant consequence of decreased striatal dopaminergic tonus is an increase in excitatory activity of the circuits leading from the predominantly glutamatergic subthalamic nucleus (STN). The depolarization pattern of the STN is affected in the form of increased firing frequency (∼30 Hz) along with spike bursts of activity with subsequent afterhyperpolarization [54]. The excitatory signal affects the internal globus pallidus (GPi) and substantia nigra pars reticulata (SNr). Increased activation of GPi and SNr exerts inhibitory effect on the thalamocortical circuits resulting in the classical parkinsonian motor symptoms [54]. Baron et al. (2002) showed that infusion of the GABAA agonist muscimol into the STN and GPi in MPTP-treated monkeys resulted in mitigation of subthalamic hyperactivity and amelioration of PD symptomatology [55]. The use of gene therapy in this approach is attractive since the use of permanently implanted pumps for CNS stimulation is undesirable due to the risk of infection, catheter-related issues, etc.
An early study investigating subthalamic GAD overexpression in a rat model of PD found the therapy to be both well tolerated and able to diminish parkinsonian motor symptoms [56]. The authors even found the therapy to exert neuroprotective effect on nigral neurons. A subsequent rodent study was unfortunately unable to replicate the neuroprotective effect, but nevertheless reproduced the improved motor effects [57].
In a subsequent NHP trial by Emborg et al. [58] (see Table 3) the effect of unilateral GAD expression was pronounced and prompted the advance to human trials. Phase 1 clinical trials funded by the company Neurologix found no adverse effects related to the treatment and reported a significant sustained improvement of UPDRS scores in both ON- and OFF-state [59, 60]. The latter data should, however, be interpreted with caution as they originate from an open label study. The safety profile gave rise to a phase 2 study by LeWitt et al. (2011) that confirmed AAV-GAD to be an efficient treatment for PD with a significant decrease in UPDRS-score 6 months after transfection as compared to patients subjected to sham surgery [13]. A recently published follow-up study on this work shows the effect to be persistent for 12 months regarding both attenuation of metabolic hyperactivity in the thalamus, striatum, prefrontal anterior cingulate gyrus and orbitofrontal cortices as wells as an improved UPDRS-score [61]. Further studies have yet to be undertaken as the funding company Neurologix no longer exists and, as such, a scheduled long-term follow-up study and an additional phase 2 trial have been cancelled. Thus, the development of GAD as a possible therapy for PD has, so far, come to a halt.
Table 3
NHP trials and clinical trials utilizing GAD
| Author | Method | Subjects | Results | Reference |
| Emborg et al. (2007) | Unilateral AAV-mediated GAD expression in STN after MPTP-lesion. | MPTP-treated rhesus monkeys (n = 13). | Well tolerated. No overall decrease in morbidity. Sub-analysis with decreased bradykinesia -16%, gross motor skills (–26%) and tremor (–36%). Unilaterally decreased FDG-metabolism. | [58] |
| Kaplitt et al. (2007) | Open-label study. Unilateral AAV-mediated GAD expression in STN. | PD patients. Hoehn Yahr stage ≤3 (n = 12) with substantial motor fluctuations and age <70. | 28% reduced UPDRS score. PET measured reduction in thalamic metabolism. Well tolerated therapy. | [59] |
| Feigin et al. (2007) | Open-label study. Unilateral AAV-mediated GAD expression in STN. | PD patients in Hoehn Yahr stage ≤3 (n = 12). | FDG and PET verified decline of glutamatergic hyperactivity and reduction of aberrant activity in basal ganglia. | [60] |
| Lewitt et al. (2011) | Phase 2 randomized controlled trial. Bilateral injection of AAV2-GAD. | PD patients scoring ≥25 on UPDRS motor score (n = 37). | UPDRS score decreased by 8.1 points in AAV2-GAD group as compared to 4.7 in sham group no serious adverse events related to surgery or treatment. | [13] |
| Niethammer et al. (2017) | 12-month follow up on LeWitt et al. (2011). | PD patients scoring ≥25 on UPDRS motor score (n = 37). | Effect from Lewitt et al. (2011) persisting | [61] |
Clinical and preclinical studies with GAD-gene therapy.
DISEASE MODIFYING TARGETS
A substantial amount of research has focused on growth factors as a treatment target for neurodegenerative diseases, including PD. Particular focus has been placed on the glial cell-line derived neurotrophic factor family of ligands (GFL) [62] that encompasses glial cell-line derived neurotrophic factor (GDNF), neurturin (NRTN), artemin (ARTN) and persephin (PSPN) [63]. The GFL signals by activating the RET receptor complex with the GDNF family receptor α (GFRα) as a co-factor, initiating the MAP kinase and PI3-kinase pathways promoting neuronal survival and neuritogenesis [64, 65]. Activation of GFRα also activates the transcriptional factors Nurr1 and Pitx3 which are crucial for the activation of genes encoding TH, VMAT2, DAT [42] and AADC [66, 67].
GDNF
Several studies investigating direct GDNF delivery to the SN and striatum have shown that GDNF induces neurite sprouting and reduces parkinsonian behavior in toxin models of PD [62, 68–70]. The latter was reproduced in phase 1 trials [66, 71], but not in a subsequent phase 2 trial [11]. The lack of effect of GDNF infusion has been attributed to insufficient spread of the drug, potentially due to pump malfunction, as well as an insufficient number of residual neurons for GDNF to affect [11]. For this reason, the gene therapeutic approach was explored. Several animal studies indicated that injection of AAV-GDNF in the putamen is both safe and efficient at treating parkinsonian behavior in murine PD models [72–75]. As alternative to viral vector delivery, a recent study by Espadas-Alvarez et al. (2017) achieved GDNF overexpression in dopaminergic cells using a novel transfection method called neurotensin-polyplex, utilizing the neurotensin receptor for introducing nanoparticles containing plasmids for expression of GDNF in dopaminergic cells under control of the tetracycline response element and reverse tetracycline-controlled transactivator. This method enables controlled expression of GDNF by supplying tetracycline, rendering otherwise constant expression controllable due to the need for exogenous stimulation [76]. Another new approach to the delivery of GDNF gene therapy was reported by Yurek et al. (2017) who delivered nanoparticles consisting of a 30-mer lysine polymer and a plasmid encoding GDNF to the midbrain of rats [77]. They showed that subsequent treatment with 6-OHDA increased the number of surviving TH+-cells and also improved rotational behavior in a cylinder test [77].
The anti-parkinsonistic effect of viral vector-mediated GDNF overexpression in dopaminergic midbrain neurons was also tested in aged and MPTP-treated monkeys with promising results [78–81] (see Table 4). Among the NHP studies, one study raised significant safety concern, as parkinsonian rhesus monkeys injected bilaterally in the SN with AAV-GDNF showed no motor improvement, but instead presented a significant weight loss [82]. However, based on convincing data from other studies delivering NRTN, the sibling of GDNF, to the SN [83–85], the FDA has approved an American phase 1 trial currently recruiting advanced PD patients for an open label safety, dosage elevation study, examining the effect of bilateral delivery of an AAV2-GDNF vector to the putamen with a 12-month follow-up [86].
Table 4
NHP trials and clinical trials utilizing GDNF
| Author | Method | Subjects | Results | Reference |
| Kordower et al. (2000a) | Unilateral injection of LV-GDNF into the caudate nucleus, putamen and SN. | Aged non-lesioned macaques approx. 25-years-old (n = 8). | Robust antero- and retrograde distribution of GDNF-vector. Strong trend towards unilateral improved 18 F-DOPA uptake. 85% increase in TH-positive neurons on treated side. | [184] |
| Kordower et al. (2000b) | Unilateral MRI-guided injection of LV-GDNF in either putamen, SN or caudate nucleus after MPTP lesion. | Unilaterally MPTP-treated young macaques (n = 12). | 44% increase in TH-immunoreactive cells in striatum compared to controls. >300% increase in FD-uptake in left striatum. Rescue of motor behavior in all groups. | [184] |
| Kozlowski et al. (2001) | Bilateral injection of AAV-GDNF vector in caudate nucleus. Euthanasia 1 week after operation. | St. Kitts green monkeys (n = 4). | First proof, that humane GDNF can be expressed in primates utilizing an AAV-vector. Increased GDNF amount in striatum. | [185] |
| Palfi et al. (2002a) | Unilateral injection of LV-GDNF into the caudate nucleus, putamen and SN. | Macaques aged between 24 to 27 years (n = 4). | Unilateral >800% increase in TH-positive cells in striatum. | [186] |
| Palfi et al. (2002b) | Unilateral injection of LV-GDNF into the caudate nucleus, putamen and SN 1 week after MPTP-lesion. | Unilaterally MPTP lesioned Rhesus monkeys (n = 8). | Overall GDNF-mediated increase in TH-positive cells in striatum by >700%. | [186] |
| Eslamboli et al. (2005) | Unilateral injection of AAV2-GDNF in the striatum and SN 4 weeks prior to 6-OHDA lesion. | Marmoset monkeys (n = 11) with unilateral 6-OHDA mediated nigral forebrain bundle lesion. | 19% survival of nigral dopaminergic cells. Uncertain mitigation of parkinsonian behavior. | [78] |
| Eberling et al. (2009) | Unilateral injection of AAV2-GDNF in putamen after MPTP-lesioning. | Unilateral MPTP-lesion Rhesus monkeys. | Bilaterally increased striatal metabolism. 60±6% decrease in CRS scores. | [81] |
| Su et al. (2009a) | Bilateral CED of AAV2-GDNF in either putamen with high- or low titer or to SN. | Macaques aged >20 years (n = 17). | No detectable immunological response to therapy after 6 months. SN group showed significant weight loss (–19.4%) not attributable to therapy. | [82] |
| Su et al. (2009b) | Bilateral CED of AAV2-GDNF in putamen 4 months after MPTP-lesion. | Unilaterally MPTP-treated macaques (n = 11). | No detectable immunological response. Bilaterally enlarged TH+ fibers in striatum. | [82] |
| Kells et al. (2010) | Bilateral AAV2-GDNF delivery to putamen after lesion. | Unilaterally MPTP-treated macaques (n = 15). | Robust improvement of motor behavior at 24 months. Effect proportional with severity of lesion. 3-fold increase in striatal dopamine. | [80] |
| Emborg et al. (2014) | Unilateral MRI-guided convection enhanced delivery of either low titer or high titer AAV5-GDNF vector to putamen. | Macaque monkeys (n = 4). | Higher GDNF expression with higher dose in both SN and striatum. | [187] |
Preclinical studies with GDNF-gene therapy. CRS: Clinical rating scale consisting of 14 parkinsonian features consisting of tremor, locomotion, freezing, fine motor skills, bradykinesia, hypokinesia, balance, posture, startle response, gross motor skills. Evaluated bilaterally where applicable and given a score from 0–3 with 0 being the best. Severe parkinsonism being viewed as a score >20. CED: Convection enhanced delivery.
NRTN
NRTN has been the subject of a great deal of attention as it has been also been found to exert protective effect on dopaminergic midbrain neurons [62]. NRTN roughly use the same pathways as GDNF, both activating Nurr-1. While GDNF mainly acts through the co-receptor GFRα1, NRTN has been found to rely on the co-receptor GFRα2 [87]. NRTN has been shown to improve motor behavior in both rodents [88–90] and NHPs [83, 91, 92] (see Table 5). Like the animal experiments with GDNF, NRTN seemed to be good news for PD patients. Especially after a successful phase 1 trial, testing bilateral AAV2-NRTN infusion into the putamen, sponsored by the company Ceregene [93] (see Table 5). A subsequent phase 2 trial [9] with the same approach was sadly unable to meet its primary endpoint, as there was no significant difference in motor scores in the treatment group compared to controls at 12-month follow-up. Some secondary endpoints were, however, met as there was a significant improvement in self-reported activities of daily living (ADL) and a minor but significant improvement in motor scores at 18-month follow-up. These findings could point towards a delayed effect of AAV2-NRTN. However, the improvement in motor scores at 18-months was observed without blinding of the patients, and consequently the risk of observer bias means that the results should be interpreted with caution (see Table 5). Nonetheless, post-mortem studies of six subjects from clinical trials testing AAV2-NRTN delivery to putamen performed by Bartus et al. [94, 95] appeared to show that longer exposure to NRTN was associated with a small increased number of TH-positive cells.
Table 5
NHP trials and clinical trials utilizing NRTN
| Author | Method | Subjects | Results | Reference |
| Kordower et al. (2006) | Unilateral infusion of AAV2-NRTN into the striatum, caudate nucleus and SN, 4 days after MTPT lesion. | Unilaterally MPTP-treated macaques (n = 20). | Therapy well tolerated. 88% reduction in CRS score. Antero- and retrograde transport of vector found. Preservation of nigral neurons and striatal terminals after 10 months. | [91] |
| Herzog et al. (2007) | Unilateral injection of AAV2-NRTN into the caudate nucleus and putamen. | Macaques aged >20 years (n = 3). | Well tolerated therapy at 8 months. ∼20% Increased striatal metabolism. Increased number of TH+ cells in striatum. | [92] |
| Herzog et al. (2008) | Bilateral injection of AAV2-NRTN in the caudate nucleus and putamen in high, medium and low dose. | Naive cynomolgus monkeys (n = 12). | No observed adverse effects. 60–65% increase in striatal TH+ density. No difference between high and medium titer. No increase in nigral TH+ at 3 months. | [83] |
| Marks et al. (2008) | Open label study. Bilateral intraputaminal injections of either high or low dose AAV2-NRTN. | PD-patients in Hoehn-Yahr stage 3-4 aged between 35–70 years. | No serious adverse events at 1 year. 36% improvement of UPDRS OFF-state motor scores. No change in striatal 18F-L-DOPA uptake from baseline. | [93] |
| Marks et al. (2010) | Double blind sham-surgery controlled randomized trial. Bilateral putaminal injection of AAV2-NRTN. | PD-patients aged 35–75 years scoring >30 in the motor part of UPDRS in OFF-state averaging at 3.03 in Hoehn Yahr scale (n = 58). | Vector not superior to sham surgery at 12 months. 6 subjects developed antibodies against vector. Several secondary endpoints met. Subpopulation with improved motor score at 18-months. | [9] |
| Bartus et al. (2013) | Open label study. Bilateral injection of AAV2-NRTN to SN, and either low- or high dose injection in putamen. | PD-patients in Hoehn-Yahr stage 3-4 aged between 35–70 years (n = 12). | No serious adverse events at 2-year follow-up. Approximately 5-point decrease in UPDRS-motor-score in OFF-periods. No change in ADL. | [85] |
| Warren Olanow et al. (2015) | Randomized double blinded sham-surgery controlled study. Bilateral injection of AAV2-NRTN into SN and putamen. | PD-patients in Hoehn-Yahr stage 3-4 aged between 35–70 years (n = 51). | Therapy did not prove superior to sham surgery in improving motor score at 15-24-month follow-up. Therapy well tolerated. | [84] |
Clinical and preclinical studies with NRTN-gene therapy. UPDRS, United Parkinson Disease Rating Scale; ADL, Activities of daily living.
The failure of the AAV2-NRTN study to meet its primary endpoint might be due to poor transfection rate or potentially poor retrograde transport in nigral dopamine neurons after putamen administration of the vector. For this reason, Bartus et al. (2011) tested intranigral delivery of AAV2-NRTN in rodents and monkeys and found it to be both feasible and safe [96] (see Table 5), despite earlier findings that it might produce unwanted weight loss in rodents [82]. Subsequently, in a clinical trial, AAV2-NRTN delivery into the SN in addition to putamen and higher vector titers were found to be well tolerated and reduced UPDRS scores in the treated subjects [85]. The ensuing randomized phase 2 trial utilizing the same approach [84] (see Table 5), with a 15-24-month follow-up, sadly also failed to meet its primary endpoint, as AAV2-NRTN was not found to be superior at improving motor scores compared to sham surgery. The NRTN in the form used in previously mentioned studies has high binding affinity for extracellular heparin which may lead to decreased tissue diffusion [97]. Therefore Runeberg-Roos et al. (2016) has tested a new isoform of NRTN with lower affinity for heparin in rodent brains that should enable further diffusion distances and deserves further attention [97].
A potential explanation why positive results from phase 1 studies have been hard to confirm in phase 2 trials could be that the apparent clinical benefit from GDNF and NRTN in open-label trials could be due to placebo effects in PD-patients [98]. Another point of criticism is that the clear effect of GFLs in 6-OHDA and MPTP animal models may not be valid at evaluating growth factors, as these animal models do not adequately mirror the underlying pathophysiology of PD [99]. Many of the animal studies cited in this manuscript rely on retrograde transport of gene therapeutic vectors expressed in SN after injection into the striatum. It has, however, been shown by Venda et al. (2010) that SNCA reduces intracellular transport through impairment of the Golgi apparatus [100]. The same transport impairment has been found in human post-mortem studies [94, 95]. This implies that it might be a good idea to test different injection sites when testing antiparkinsonian vectors. In a study by Decressac et al. (2011) investigating GDNF expression in a SNCA-expressing model of PD, there was no beneficial effect of GDNF on PD behavior in mice [101]. Consequently, Decressac et al. proposed that the reason for failure in the randomized human trials might be due to the presence of SNCA impairing expression of Nurr1, which blocks the pathway facilitating the neuroprotective function of the GFL. This appears to speak against the proposed idea of neuroprotection through expression of GFL alone. However, caution may be warranted since the SNCA-expressing mouse model used in this study appears to express SNCA at a far greater level than that in PD and other α-synucleinopathies [102].
Nurr1
Nurr1 is an intranuclear receptor found to play a significant role in the development of dopaminergic neurons and transcriptional regulation of AADC, TH, DAT and VMAT2 [103]. Increased levels of SNCA impairs Nurr1 function [104], thus hampering the effect of GDNF and other members of GFL. In a paper from 2015, Oh et al. examined the effect of AAV-mediated overexpression of Nurr1 and its co-transcription factor Foxa2 in the midbrain of a MPTP-mouse model [105]. They could rescue 69% of the neurons ipsilateral to the injection site, including a large amount of striatal fibers, as well as detect a decrease of the proinflammatory cytokines IL-1β and iNOS. The neuroprotective effect was also found in behavioral studies with bilateral overexpression of Nurr1 and Foxa2 [105]. Further studies overexpressing Nurr1 is warranted before any conclusions can be made, but the perspectives are intriguing, as overexpression alone is neuroprotective. Also, by expressing Nurr1 and Foxa2 in conjunction with GDNF or NRTN it is possible that the GFL may still prove to be efficient PD therapy [106].
Cerebral dopamine neurotrophic factor (CDNF) and mesencephalic astrocyte-derived neural factor (MANF)
CDNF and MANF constitute a family of neural growth factors not related to GFL. The CDNF/MANF family has been found to exhibit a neurotrophic effect on nigral dopaminergic neurons through a hitherto unknown mechanism [20]. The factors are found in both in the extracellular environment in response to ischemia and status epilepticus as well as in the endoplasmatic reticulum (ER) where they reduce ER stress by modulating the unfolded protein response [107–109].
The unfolded protein response is an intracellular response to accumulation of misfolded proteins and works through halting protein translation, increasing molecular chaperone production, and increasing protein degradation [107, 110]. The CDNF/MANF family’s mode of action could prove to be an interesting take on PD modulation, given the role of ER stress in neurodegenerative diseases [111]. The CDNF/MANF family is a relatively new discovery with MANF first described by Petrova et al. (2003) [112] and CDNF by Lindholm et al. (2007) [113]. MANF was shown by Petrova et al. [112] to promote survival of dopaminergic neurons in vitro and has later been shown to be on par with GDNF at protecting and restoring dopaminergic neurons and motor behavior in a 6-OHDA rat model with injection directly into the striatum [113]. To date, three studies have used gene therapy to overexpress CDNF in 6-OHDA based rodent models. One study made use of an AAV2-vector to deliver the gene encoding CDNF to the striatum in a rat model. Subsequent medial forebrain bundle lesion with 6-OHDA showed a significantly improved amphetamine-induced rotation test, but only partial protection of TH+ fibers and neurons [114]. Another study examined the effect of striatal AAV2-CDNF delivery in a rat model, 6 weeks after 6-OHDA treatment and found a significant improvement of spontaneous locomotion as well as decreased amphetamine-induced rotations [115]. It was also possible to detect a significant protection of TH-expressing cells measured by immunohistochemistry. In addition, a striatal increase in activity of the DAT-ligand [11C]-2β-carbomethoxy-3β-(4-fluorophenyl)-tropane points towards a functional recovery of the striatum [115]. A third study used a LV vector to compare the effect of LV-mediated nigral expression of either CDNF, MANF or a combination of the two in a 6-OHDA rat model [116]. The study was unable to completely reproduce the impressive results from the former studies. However, combined overexpression of MANF and CDNF resulted in decreased amphetamine-induced rotations as well as an increased number of TH+ cell bodies in the SN and increased TH+-fiber density in the striatum, suggesting a synergistic effect of the two factors [116]. A recent study investigated the effect of AAV9-MANF in the striatum of 6-OHDA-treated rats [117]. They found the protein to be widely expressed with signs of both antero- and retrograde transport with no significant inflammatory response as well as sustained MANF expression for 16 weeks. Injection of AAV9-MANF was able to halt the neurodegeneration caused by 6-OHDA and appeared to possibly regenerate lost nerve terminals [117]. Supported by the positive results using CDNF, a study in MPTP-treated monkeys is currently being conducted [20].
Brain-derived neurotrophic factor (BDNF)
BDNF belongs to the neurotrophin family of growth factors and primarily binds to the receptor tropomyosin-related kinase B (TrkB) [118]. It has been found to promote neuron survival and to protect dopaminergic neurons from toxic insults [119, 120]. BDNF can induce neurogenesis after intraventricular injection in rats and this could potentially compensate for cell loss in PD [121]. BDNF has received much of attention as a therapeutic option in several neurodegenerative diseases [122] and in relation to neurotraumatology [123]. Native BDNF has been found to be reduced in the SN of PD patients [124], and inhibition of nigral BDNF expression in rats induces parkinsonian symptoms [125]. Relatively few studies have examined gene therapy utilizing BDNF as a therapeutic option in PD. This is surprising given the apparent effect of continuous infusion of a BDNF ligand on parkinsonistic behavior and dopaminergic cell survival in an older monkey study [126].
A study by Klein et al. (1999) injected an AAV-vector encoding BDNF in the SNc of rats with a partial 6-OHDA lesion [127]. The animals treated with BDNF showed a general increase in locomotor activity and a reduction in amphetamine-induced rotations, but no signs of neuroprotection. Another study attempted to compare the effect of BDNF vs. GDNF in a 6-OHDA rat model and found GDNF to be superior in terms of both neural protection as well as at reducing parkinsonian symptoms [128]. In addition, it was impossible to detect a statistical difference in the behavior of rats treated solely with HSV1-GDNF and rats treated with a combination of HSV1-GDNF and HSV1-BDNF, thus suggesting that GDNF is a superior therapeutic option [128]. Despite the apparent earlier failure of BDNF as gene therapeutic treatment for PD symptoms, BDNF was reinvestigated in a fairly recent study by Hernandez-Chan et al. (2015) [129]. The authors utilized the novel gene delivery method mentioned above called neurotensin-polyplex [17] to overexpress BDNF in a 6-OHDA rat model of early parkinsonism. The neurotensin-polyplex is a non-viral delivery method that utilizes neurotensin receptors to induce receptor-mediated internalization of nano-coated genetic cargo specifically in dopaminergic neurons [17]. The authors proposed that the scarcity of dopaminergic nerve terminals in the striatum in earlier studies was not sufficient to facilitate proper retrograde transport of the vector and might thus not facilitate nigral expression of BDNF at therapeutic amounts. Using a less severe PD model, it was possible to detect massive neuritogenesis, but no outright neurogenesis in both SN and the striatum [129]. In the behavioral tests, it was possible to detect substantial improvement in parkinsonian behavior [129]. These results point towards a revival of BDNF as a potential candidate for PD therapy. There is, however, still a long way to go, as BDNF must be tested in other models of PD. Also, estimations of potential long-term effects of elevated BDNF levels should be considered before BDNF-therapy can advance to non-human primate studies, as an earlier study raised safety concerns regarding elevation of striatal BDNF levels [130]. Thus Lapchak and colleagues (1993) found that chronic delivery of BDNF to the SN in healthy rats produced a hypodopaminergic phenotype with increased rotatory behavior and decreased TH mRNA on the injected side [130].
Vascular endothelial growth factor (VEGF)
The VEGF family of proteins consists of VEGF-A VEGF-B, VEGF-C, VEGF-D, and placental growth factor (PLGF), with VEGF-A, and to some extent VEGF-B, being the most physiologically active [131]. The VEGF family has been intensely studied due to its angiogenic role in tumor growth [132]. However, subsequent research also found the factors able to exert neurotrophic, potentially neuroprotective, effects in the CNS [16]. The VEGF family peptides mainly mediate their effects via the tyrosine kinase receptor and vascular endothelial growth factor receptor 2 (VEGFR-2) using the neuropillins NP1 and NP2 as co-receptors [133] with vascular endothelial growth factor receptor 1 (VEGFR-1) acting as a decoy receptor [134]. Neuroprotective/neuroptrophic effects have been observed by overexpressing VEGF in both the striatum [73, 135] and SNPc [16] in 6-OHDA-treated rodents. These effects are believed to be mediated by VEGFR-2 activation, leading to increased angiogenesis with improvement of microcirculation and through microglial proliferation causing increased extracellular levels of the antioxidant glutathione and increased GDNF secretion [134]. In addition to the neurotrophic effects, VEGF has also been found to prevent apoptosis by outcompeting the apoptosis-inducing factor Sema3A for neuropillin-binding sites [136]. Also, VEGF-A has been found to activate the anti-apoptotic PI3K/AKT pathway [137], lending additional credence to further investigation of the VEGF family as a future target for PD treatment. Due to the implication of VEGF in oncogenesis [132] it is a relevant concern that overexpression may increase the risk of CNS cancers. However, gene therapy with VEGF-A has already been utilized in patients suffering from coronary disease with good results [138], which speaks for the feasibility of VEGF gene therapy for vascular diseases. The usage of VEGF as a neuroprotective factor is complicated by the fact that the effect is dose-dependent with high levels of VEGF inducing brain edema [139]. As such the dosage of VEGF should be carefully studied in animal models before advancing to human trials.
OTHER TARGETS AND STRATEGIES
In addition to the above-mentioned peptides for gene therapeutic treatment of PD, several other possible genes deserve attention as potential targets in PD. One such target is the neuron restrictive silencing factor (NRSF), a zinc finger transcription factor found to be involved in restriction of neuronal factors in non-neural cells and regulation of neurogenesis [140]. Yu and associates (2013) reported that NRSF knockout mice were more susceptible to MPTP-induced dopaminergic cell death, as lack of NRSF leads to lower levels of BDNF and TH [140]. It is possible that by upregulating NRSF in conjunction with BDNF, it would be possible to enhance the neuroprotective effects of BDNF. However, sparse data are available on NRSF in PD, but as the role of long coding RNAs have been associated with several neurodegenerative diseases, epigenetic regulation provides a novel interesting avenue of research regarding several diseases [141].
Neuropeptide Y (NPY) is an abundantly expressed neuropeptide in the CNS that exerts neuroprotective effects by acting through the Y2 receptor (Y2R) [142, 143]. Cannizzaro et al. [144] detected increased levels of NPY mRNA in the basal ganglia of PD patients, which has been interpreted as a compensatory, yet unsuccessful neuroprotective mechanism of the basal ganglia to prevent tissue damage. So far, no studies have directly tested the effect of gene therapy with NPY in PD models, but promising results have been obtained in rodent PD model using direct infusion of NPY ligands [142] and in other neurodegenerative diseases [145] which encourage further research on this target.
The transcription factor EB (TFEB) has been found to be a key player in regulating the autophagy-lysosomal pathway [146] which is found to be impaired in PD [147]. By overexpressing TFEB, Decressac et al. (2013) reported neuroprotection as well as rescue of a parkinsonian phenotype in a rat model overexpressing SNCA in the SN and ventral tegmental area [148]. These results point to TFEB as another potential future target in the pursuit of therapies for PD.
Optogenetics
Optogenetics is a rapidly emerging technology using targeted expression of light-sensitive ion channels (opsins) or G-protein coupled receptors which enables precise control of neural activity of transfected specific cell populations. The opsins are activated using light pulses with a wavelength specific for the used opsins [149]. The technology has so far been used to decipher hitherto unknown neural mechanisms and may prove to be a viable therapeutic option in itself. The technology has been explored in a wide number of fields, such as management of pain [150], epilepsy [151], depression [152], cardiac arrhythmias [153], retinal dystrophy [154] and PD. The primary focus of optogenetic studies of PD has so far been aimed at deconstructing the parkinsonian circuitry and mapping the effects of DBS. Nevertheless, several recent publications show optogenetic stimulation to improve motor behavior in parkinsonian rodents. For instance, two studies show that high frequency optogenetic stimulation of the STN or the 5th layer of the neocortical M1 area improves locomotor behavior in 6-OHDA-treated mice [155, 156]. This suggests that optogenetic stimulation might be used as a more selective alternative to electrical DBS, stimulating selective STN populations as opposed to the unspecific effects of electrical DBS. The two studies also suggest the neocortex as a new possible target for DBS. Consistent with these latter studies, Yoon et al. found that optogenetic low frequency inhibition of glutamatergic cells in the STN using the light sensitive Cl--channel NpHR was both able to reduce parkinsonian behavior [157] and to reduce L-DOPA-induced dyskinesia in 6-OHDA treated rats [158].
Kravitz et al. (2010) [159] investigated the classic concept of parkinsonian motor deficits being driven by an imbalance between the “direct” and “indirect” basal ganglial pathways. The authors overexpressed channelrhodopsin-2 (ChR2) in either the D2- or D1-expressing medium spiny projection neurons in the striatum of mice. Bilateral excitation of the D2-expressing neurons of the indirect pathway induced a parkinsonian phenotype in the mice. On the other hand, they completely rescued PD behavior in 6-OHDA-treated mice by activating the D1-expressing neurons of the direct pathway. Further research on the subject by the same group showed that activation of the two pathways induced changes in the firing pattern of the SN pars reticulata (SNr) [160]. By activating the direct pathway, it was possible to detect a reduction in the firing rate of a subset of nigral neurons. By activating the indirect pathway, it was possible to detect increased activity in one subset of neurons as well as reduced activity in another subset. The authors explain this modulation of firing rate with the possibility of the inhibited nigral cells being silenced to avoid executing multiple motor actions simultaneously [160]. If this holds true in further studies, it would be a theoretical possibility to tailor optogenetic stimulation when needed, rather than the continuous stimulation protocols presently used.
In a recent study Moon et al. (2017) NpHR was expressed in the entopeduncular nucleus (EN) of an AAV2-α-synuclein-induced hemiparkinsonian rat model to monitor the effect of inhibition of the rat equivalent of GPi on thalamic activity and ultimately on motor behavior [161]. By inhibiting EN activity, improved contralateral forelimb use in amphetamine-induced rotations, cylinder test and stepping test was seen.
Optogenetics is an evolving field and provides crucial insights into neuronal circuitry and may in time provide viable treatment options for PD. This progress may be boosted by the recent development of an inhibitory opsin capable of activation by transcranial light [162]. Thus, optogenetic inhibition of the STN or stimulation of the M1 motor cortex could become a feasible alternative to DBS as this potentially avoids the need for implantation of electrodes or optic cables.
Chemogenetics
G-protein coupled receptors (GPCR) are found in abundance all over the body and contributes to a great number of physiological processes. Researchers have succeeded in engineering GPCRs only responsive to otherwise inert substances such as the clozapine metabolite clozapine-N-oxide (CNO) [163–165]. These receptors are known as designer receptors exclusively activated by designer drugs (DREADDs) and have been developed to initiate the activation of several different G-proteins, including Gi, Gs, and Gq [166]. By expressing DREADDs under a promotor specific for the desired cell population, it is possible to achieve pharmacological control over the cells without inducing off-target effects. Chemogenetics, similarly to optogenetics, has been widely used to probe neural circuits, but also carries the potential to work as a therapy by itself. So far, DREADDs have mainly seen use in other fields. In one in vivo study, though, DREADDs were utilized in a PD model. Pienaar et al. used an AAV-vector to express an excitatory DREADD in cholinergic cells in the rat pedunculopontine nucleus (PPN) [167]. The authors of this study utilized a novel rat model treated with the irreversible ubiquitin-proteasome inhibitor lactacystin, leading to disrupted breakdown of SNCA resulting in a parkinsonian phenotype [168]. After treatment, the transgenetic lactacystin-treated rats [168] showed a marked improvement in motor behavior, when treated with the otherwise pharmacologically inert CNO, potentially suggesting PPN as a future target for PD therapy using chemogenetics [167]. A standing concern regarding gene therapy is the fact that the therapy is irreversible and, in most cases, uncontrollable. Opto- and chemogenetics, in contrast, can be applied when necessary and side effects may therefore be few.
Genome editing
Mutations in several genes have been associated with both familial- and sporadic PD, including parkin, LRRK2, SNCA, PINK1, DJ-1, VPS35, DNAJC13, CHCHD2 [169]. Several of these mutations potentially influence neuroplasticity, immunomodulation, endosomal sorting, autophagy and mitochondrial function linked to the development of PD [170–177]. The recently developed “clustered regularly interspaced short palindromic repeats” technology (CRISPR) in conjunction with the endonuclease CAS9 [178] are able to induce double-stranded DNA-breaks and with that, the possibility of editing genes responsible for familial parkinsonism is now a theoretical possibility [179]. Thus the technology could be employed in adults with brain region-specific injections of vectors encoding CRISPR-CAS9 constructs to correct gene mutations responsible for focal disease activity [180]. It remains to be seen what will be the time window in the lifespan of individual patients for editing disease genes in this manner. With CRISPR-CAS9 comes also the possibility of altering the germ cell DNA and thus sparing future generations from familial parkinsonism [181]. However, this approach is, as of yet, not approved by any ethics committee.
CONCLUSION
Gene therapy offers a promising potential treatment avenue for PD with the theoretical possibility of targeting both non-disease and disease modifying targets. While encouraging results have been obtained in clinical trials using non-disease modifying treatment, a disease modifying gene therapeutic treatment remains to be identified as effective in slowing or reversing PD disease progression. Prosavin and AADC dopaminogenic therapies have been shown to be both safe and successful at reducing the need for medication in advanced PD patients. The same also appears to be the case for overexpression of GAD in the STN. These therapies should be further validated in clinical studies and could potentially be applied in conjunction with electrode implantation or as an alternative to it in order to avoid DBS-induced adverse effects such as speech impediments, paresthesia, cognitive impairment, and mood disturbances [182].
As for disease modifying targets, research targeting GFL family members (GDNF, NRTN, ARTN, PSPN) is still ongoing, with a planned clinical study examining putaminal overexpression of GDNF. The CDNF/MANF family might also be a viable alternative to the GFL family, as this family of growth factors exerts its neuroprotective effect via a different mechanism than the GFL family. The ongoing NHP trial with CDNF vs GDNF will be most interesting. The possibility of combining Nurr1 therapy with a member of the GFL family might also be an interesting approach. In the investigation of growth factors for PD, researchers are challenged by the validity of the PD animal models used. For instance, 6-OHDA and MPTP-treated animals phenotypically display parkinsonistic symptoms, but the pathophysiology does not adequately mirror PD. This warrants more accurate PD models and their implementation in PD research such as the lactacystine model for PPN and a more finely tuned SNCA model. By utilizing the novel CRISPR-CAS9 technology it should be possible to engineer animals suffering mutations that result in true PD rather than an artificially induced one. Another hurdle faced in exploring the use of growth factors, is the high PD disease load of the subjects in the trials. Thus, there must be dopaminergic neurons left to save if the growth factors are to have any effect. Future trials should attempt to investigate the effect of GFL gene therapy in patients at earlier disease stages before abandoning the target for potential therapy. At a longer perspective, gene therapy with growth factors could be relevant as an aggressive treatment regime in patients with early verified PD, rather than as a last-line of treatment.
Gene therapy with opto- and chemogenetics could prove a viable alternative option for treating symptoms of PD as they provide a more specific intervention as compared to DBS or ablation. Genome editing with the CRISPR-CAS9 technology might be another future form of personalized gene therapy for known mutations leading to PD. In conclusion, while gene therapy has yet to deliver the true cure for PD, there is increasing data supporting that this treatment modality could become an important avenue for future PD treatment.
CONFLICT OF INTERESTS
D Woldbye is cofounder and shareholder of two stock-listed gene therapy companies in Sweden. Dr. T. M. Axelsen has no conflict of interests to declare.
REFERENCES
[1] | Global Study of Disease Study 2013, Collaborators ((2015) ) Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 386: , 743–800. |
[2] | Obeso JA , Rodriguez-Oroz MC , Benitez-Temino B , Blesa FJ , Guridi J , Marin C , Rodriguez M ((2008) ) Functional organization of the basal ganglia: Therapeutic implications for Parkinson’s disease. Mov Disord 23: (Suppl 3), 548–559. |
[3] | Spillantini MG , Schmidt ML , Lee VM , Trojanowski JQ , Jakes R , Goedert M ((1997) ) Alpha-synuclein in Lewy bodies. Nature 388: , 839–840. |
[4] | Berglund MM , Hipskind PA , Gehlert DR ((2003) ) Recent developments in our understanding of the physiological role of PP-fold peptide receptor subtypes. Exp Biol Med 228: , 217–244. |
[5] | Walker Z , Possin KL , Boeve BF , Aarsland D ((2015) ) Lewy body dementias. Lancet 386: , 1683–1697. |
[6] | Wenning GK , Ben Shlomo Y , Magalhaes M , Daniel SE , Quinn NP ((1994) ) Clinical features and natural history of multiple system atrophy. An analysis of 100 cases. Brain 117: , 835–845. |
[7] | Fahn S ((2015) ) The medical treatment of Parkinson disease from James Parkinson to George Cotzias. Mov Disord 30: , 4–18. |
[8] | Verhagen Metman L , Pal G , Slavin K ((2016) ) Surgical treatment of Parkinson’s disease. Curr Treat Options Neurol 18: , 49. |
[9] | Marks WJ Jr , Bartus RT , Siffert J , Davis CS , Lozano A , Boulis N , Vitek J , Stacy M , Turner D , Verhagen L , Bakay R , Watts R , Guthrie B , Jankovic J , Simpson R , Tagliati M , Alterman R , Stern M , Baltuch G , Starr PA , Larson PS , Ostrem JL , Nutt J , Kieburtz K , Kordower JH , Olanow CW ((2010) ) Gene delivery of AAV2-neurturin for Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol 9: , 1164–1172. |
[10] | Friedmann T , Roblin R ((1972) ) Gene therapy for human genetic disease? Science 175: , 949–955. |
[11] | Lang AE , Gill S , Patel NK , Lozano A , Nutt JG , Penn R , Brooks DJ , Hotton G , Moro E , Heywood P , Brodsky MA , Burchiel K , Kelly P , Dalvi A , Scott B , Stacy M , Turner D , Wooten VG , Elias WJ , Laws ER , Dhawan V , Stoessl AJ , Matcham J , Coffey RJ , Traub M ((2006) ) Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann Neurol 59: , 459–466. |
[12] | Christine CW , Starr PA , Larson PS , Eberling JL , Jagust WJ , Hawkins RA , VanBrocklin HF , Wright JF , Bankiewicz KS , Aminoff MJ ((2009) ) Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 73: , 1662–1669. |
[13] | LeWitt PA , Rezai AR , Leehey MA , Ojemann SG , Flaherty AW , Eskandar EN , Kostyk SK , Thomas K , Sarkar A , Siddiqui MS , Tatter SB , Schwalb JM , Poston KL , Henderson JM , Kurlan RM , Richard IH , Van Meter L , Sapan CV , During MJ , Kaplitt MG , Feigin A ((2011) ) AAV2-GAD gene therapy for advanced Parkinson’s disease: A double-blind, sham-surgery controlled, randomised trial. Lancet Neurol 10: , 309–319. |
[14] | Eberling JL , Jagust WJ , Christine CW , Starr P , Larson P , Bankiewicz KS , Aminoff MJ ((2008) ) Results from a phase I safety trial of hAADC gene therapy for Parkinson disease. Neurology 70: , 1980–1983. |
[15] | Lentz TB , Gray SJ , Samulski RJ ((2012) ) Viral vectors for gene delivery to the central nervous system. Neurobiol Dis 48: , 179–188. |
[16] | Abid Sheikh M , Saeed Malik Y , Xing Z , Guo Z , Tian H , Zhu X , Chen X ((2017) ) Polylysine-modified polyethylenimine (PEI-PLL) mediated VEGF gene delivery protects dopaminergic neurons in cell culture and in rat models of Parkinson’s Disease (PD). Acta Biomater 54: , 58–68. |
[17] | Martinez-Fong D , Bannon MJ , Trudeau LE , Gonzalez-Barrios JA , Arango-Rodriguez ML , Hernandez-Chan NG , Reyes-Corona D , Armendariz-Borunda J , Navarro-Quiroga I ((2012) ) NTS-Polyplex: A potential nanocarrier for neurotrophic therapy of Parkinson’s disease. Nanomedicine 8: , 1052–1069. |
[18] | Touchot N , Flume M ((2017) ) Early insights from commercialization of gene therapies in Europe. Genes (Basel) 8: (2). |
[19] | FDA, FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss, U.S. Food & Drug Administration, https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm589467.htm, Last updated December 21, 2018, Accessed 14 Feb, 2018. |
[20] | Lindahl M , Saarma M , Lindholm P ((2017) ) Unconventional neurotrophic factors CDNF and MANF: Structure, physiological functions and therapeutic potential. Neurobiol Dis 97: , 90–102. |
[21] | Kalinderi K , Bostantjopoulou S , Fidani L ((2016) ) The genetic background of Parkinson’s disease: Current progress and future prospects. Acta Neurol Scand 134: , 314–326. |
[22] | Nagatsu T , Nagatsu I ((2016) ) Tyrosine hydroxylase (TH), its cofactor tetrahydrobiopterin (BH4), other catecholamine-related enzymes, and their human genes in relation to the drug and gene therapies of Parkinson’s disease (PD): Historical overview and future prospects. J Neural Transm 123: , 1255–1278. |
[23] | Lees AJ ((1989) ) The on-off phenomenon. J Neurol Neurosurg Psychiatry Suppl: , 29–37. |
[24] | Mittermeyer G , Christine CW , Rosenbluth KH , Baker SL , Starr P , Larson P , Kaplan PL , Forsayeth J , Aminoff MJ , Bankiewicz KS ((2012) ) Long-term evaluation of a phase 1 study of AADC gene therapy for Parkinson’s disease. Hum Gene Ther 23: , 377–381. |
[25] | Muramatsu S , Fujimoto K , Kato S , Mizukami H , Asari S , Ikeguchi K , Kawakami T , Urabe M , Kume A , Sato T , Watanabe E , Ozawa K , Nakano I ((2010) ) A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson’s disease. Mol Ther 18: , 1731–1735. |
[26] | Eiden LE , Schafer MK , Weihe E , Schutz B ((2004) ) The vesicular amine transporter family (SLC18): Amine/proton antiporters required for vesicular accumulation and regulated exocytotic secretion of monoamines and acetylcholine. Pflugers Arch 447: , 636–640. |
[27] | Lee WY , Lee EA , Jeon MY , Kang HY , Park YG ((2006) ) Vesicular monoamine transporter-2 and aromatic L-amino acid decarboxylase gene therapy prevents development of motor complications in parkinsonian rats after chronic intermittent L-3,4-dihydroxyphenylalanine administration. Exp Neurol 197: , 215–224. |
[28] | Sanchez-Pernaute R , Harvey-White J , Cunningham J , Bankiewicz KS ((2001) ) Functional effect of adeno-associated virus mediated gene transfer of aromatic L-amino acid decarboxylase into the striatum of 6-OHDA-lesioned rats. Mol Ther 4: , 324–330. |
[29] | Sanftner LM , Rivera VM , Suzuki BM , Feng L , Berk L , Zhou S , Forsayeth JR , Clackson T , Cunningham J ((2006) ) Dimerizer regulation of AADC expression and behavioral response in AAV-transduced 6-OHDA lesioned rats. Mol Ther 13: , 167–174. |
[30] | Bankiewicz KS , Forsayeth J , Eberling JL , Sanchez-Pernaute R , Pivirotto P , Bringas J , Herscovitch P , Carson RE , Eckelman W , Reutter B , Cunningham J ((2006) ) Long-term clinical improvement in MPTP-lesioned primates after gene therapy with AAV-hAADC. Mol Ther 14: , 564–570. |
[31] | Forsayeth JR , Eberling JL , Sanftner LM , Zhen Z , Pivirotto P , Bringas J , Cunningham J , Bankiewicz KS ((2006) ) A dose-ranging study of AAV-hAADC therapy in Parkinsonian monkeys. Mol Ther 14: , 571–577. |
[32] | Daadi MM , Pivirotto P , Bringas J , Cunningham J , Forsayeth J , Eberling J , Bankiewicz KS ((2006) ) Distribution of AAV2-hAADC-transduced cells after 3 years in Parkinsonian monkeys. Neuroreport 17: , 201–204. |
[33] | Bankiewicz KS , Eberling JL , Kohutnicka M , Jagust W , Pivirotto P , Bringas J , Cunningham J , Budinger TF , Harvey-White J ((2000) ) Convection-enhanced delivery of AAV vector in parkinsonian monkeys; in vivo detection of gene expression and restoration of dopaminergic function using pro-drug approach. Exp Neurol 164: , 2–14. |
[34] | Parkinson’s News Today, Voyager Recruits Patients for Phase 1 Trial Testing VY-AADC01 Gene Therapy Delivery, Parkinson News Today, https://parkinsonsnewstoday.com/2017/11/21/advanced-parkinsons-disease-patients-recruited-voyager-therapeutics-vy-aadc-phase-1-trial-r-d-day/, Last updated November 21, 2017, Accessed on February 14, (2018) . |
[35] | Rafaloff G , Buff S ((2017) ) Review of Parkinson’s Disease Ongoing and Upcoming Clinical Trials for Disease-Modifying Treatments: Update 4-2017. |
[36] | Nagatsu T , Nagatsu I ((2016) ) Tyrosine hydroxylase (TH), its cofactor tetrahydrobiopterin (BH4), other catecholamine-related enzymes, and their human genes in relation to the drug and gene therapies of Parkinson’s disease (PD): Historical overview and future prospects. J Neural Transm 123: , 1255–1278. |
[37] | Kaplitt MG , Leone P , Samulski RJ , Xiao X , Pfaff DW , O’Malley KL , During MJ ((1994) ) Long-term gene expression and phenotypic correction using adeno-associated virus vectors in the mammalian brain. Nat Genet 8: , 148–154. |
[38] | During MJ , Samulski RJ , Elsworth JD , Kaplitt MG , Leone P , Xiao X , Li J , Freese A , Taylor JR Jr , Roth RH , Sladek JR , O’Malley KL , Redmond DE Jr ((1998) ) In vivo expression of therapeutic human genes for dopamine production in the caudates of MPTP-treated monkeys using an AAV vector. Gene Ther 5: , 820–827. |
[39] | Azzouz M , Martin-Rendon E , Barber RD , Mitrophanous KA , Carter EE , Rohll JB , Kingsman SM , Kingsman AJ , Mazarakis ND ((2002) ) Multicistronic lentiviral vector-mediated striatal gene transfer of aromatic L-amino acid decarboxylase, tyrosine hydroxylase, and GTP cyclohydrolase I induces sustained transgene expression, dopamine production, and functional improvement in a rat model of Parkinson’s disease. J Neurosci 22: , 10302–10312. |
[40] | Cederfjall E , Sahin G , Kirik D , Bjorklund T ((2012) ) Design of a single AAV vector for coexpression of TH and GCH1 to establish continuous DOPA synthesis in a rat model of Parkinson’s disease. Mol Ther 20: , 1315–1326. |
[41] | Fan DS , Ogawa M , Fujimoto KI , Ikeguchi K , Ogasawara Y , Urabe M , Nishizawa M , Nakano I , Yoshida M , Nagatsu I , Ichinose H , Nagatsu T , Kurtzman GJ , Ozawa K ((1998) ) Behavioral recovery in 6-hydroxydopamine-lesioned rats by cotransduction of striatum with tyrosine hydroxylase and aromatic L-amino acid decarboxylase genes using two separate adeno-associated virus vectors. Hum Gene Ther 9: , 2527–2535. |
[42] | Imaoka T , Date I , Ohmoto T , Nagatsu T ((1998) ) Significant behavioral recovery in Parkinson’s disease model by direct intracerebral gene transfer using continuous injection of a plasmid DNA-liposome complex. Hum Gene Ther 9: , 1093–1102. |
[43] | Li XG , Okada T , Kodera M , Nara Y , Takino N , Muramatsu C , Ikeguchi K , Urano F , Ichinose H , Metzger D , Chambon P , Nakano I , Ozawa K , Muramatsu S ((2006) ) Viral-mediated temporally controlled dopamine production in a rat model of Parkinson disease. Mol Ther 13: , 160–166. |
[44] | Shen Y , Muramatsu SI , Ikeguchi K , Fujimoto KI , Fan DS , Ogawa M , Mizukami H , Urabe M , Kume A , Nagatsu I , Urano F , Suzuki T , Ichinose H , Nagatsu T , Monahan J , Nakano I , Ozawa K ((2000) ) Triple transduction with adeno-associated virus vectors expressing tyrosine hydroxylase, aromatic-L-amino-acid decarboxylase, and GTP cyclohydrolase I for gene therapy of Parkinson’s disease. Hum Gene Ther 11: , 1509–1519. |
[45] | Sun M , Kong L , Wang X , Holmes C , Gao Q , Zhang GR , Pfeilschifter J , Goldstein DS , Geller AI ((2004) ) Coexpression of tyrosine hydroxylase, GTP cyclohydrolase I, aromatic amino acid decarboxylase, and vesicular monoamine transporter 2 from a helper virus-free herpes simplex virus type 1 vector supports high-level, long-term biochemical and behavioral correction of a rat model of Parkinson’s disease. Hum Gene Ther 15: , 1177–1196. |
[46] | Sun M , Zhang GR , Kong L , Holmes C , Wang X , Zhang W , Goldstein DS , Geller AI ((2003) ) Correction of a rat model of Parkinson’s disease by coexpression of tyrosine hydroxylase and aromatic amino acid decarboxylase from a helper virus-free herpes simplex virus type 1 vector. Hum Gene Ther 14: , 415–424. |
[47] | Fan D , Shen Y , Kang D , Nakano I , Ozawa K ((2001) ) Adeno-associated virus vector-mediated triple gene transfer of dopamine synthetic enzymes. Chin Med J 114: , 1276–1279. |
[48] | Muramatsu S , Fujimoto K , Ikeguchi K , Shizuma N , Kawasaki K , Ono F , Shen Y , Wang L , Mizukami H , Kume A , Matsumura M , Nagatsu I , Urano F , Ichinose H , Nagatsu T , Terao K , Nakano I , Ozawa K ((2002) ) Behavioral recovery in a primate model of Parkinson’s disease by triple transduction of striatal cells with adeno-associated viral vectors expressing dopamine-synthesizing enzymes. Hum Gene Ther 13: , 345–354. |
[49] | Sehara Y , Fujimoto KI , Ikeguchi K , Katakai Y , Ono F , Takino N , Ito M , Ozawa K , Muramatsu SI ((2017) ) Persistent expression of dopamine-synthesizing enzymes 15 years after gene transfer in a primate model of Parkinson’s disease. Hum Gene Ther Clin Dev 28: , 74–79. |
[50] | Jarraya B , Boulet S , Ralph GS , Jan C , Bonvento G , Azzouz M , Miskin JE , Shin M , Delzescaux T , Drouot X , Herard AS , Day DM , Brouillet E , Kingsman SM , Hantraye P , Mitrophanous KA , Mazarakis ND , Palfi S ((2009) ) Dopamine gene therapy for Parkinson’s disease in a nonhuman primate without associated dyskinesia. Sci Transl Med 1: , 2ra4. |
[51] | Palfi S , Gurruchaga JM , Ralph GS , Lepetit H , Lavisse S , Buttery PC , Watts C , Miskin J , Kelleher M , Deeley S , Iwamuro H , Lefaucheur JP , Thiriez C , Fenelon G , Lucas C , Brugieres P , Gabriel I , Abhay K , Drouot X , Tani N , Kas A , Ghaleh B , Le Corvoisier P , Dolphin P , Breen DP , Mason S , Guzman NV , Mazarakis ND , Radcliffe PA , Harrop R , Kingsman SM , Rascol O , Naylor S , Barker RA , Hantraye P , Remy P , Cesaro P , Mitrophanous KA ((2014) ) Long-term safety and tolerability of ProSavin, a lentiviral vector-based gene therapy for Parkinson’s disease: A dose escalation, open-label, phase 1/2 trial. Lancet 383: , 1138–1146. |
[52] | U.S. National Institutes of Health, Long Term Safety and Efficacy Study of ProSavin in Parkinson’s Disease, https://clinicaltrials.gov/ct2/show/NCT01856439?term=prosn&rank=1, Last updated June 10, 2014, Accessed on 28 Nov, 2017. |
[53] | Drouot X , Oshino S , Jarraya B , Besret L , Kishima H , Remy P , Dauguet J , Lefaucheur JP , Dolle F , Conde F , Bottlaender M , Peschanski M , Keravel Y , Hantraye P , Palfi S ((2004) ) Functional recovery in a primate model of Parkinson’s disease following motor cortex stimulation. Neuron 44: , 769–778. |
[54] | Singh A , Mewes K , Gross RE , DeLong MR , Obeso JA , Papa SM ((2016) ) Human striatal recordings reveal abnormal discharge of projection neurons in Parkinson’s disease. Proc Natl Acad Sci U S A 113: , 9629–9634. |
[55] | Baron MS , Wichmann T , Ma D , DeLong MR ((2002) ) Effects of transient focal inactivation of the basal ganglia in parkinsonian primates. J Neurosci 22: , 592–599. |
[56] | Luo J , Kaplitt MG , Fitzsimons HL , Zuzga DS , Liu Y , Oshinsky ML , During MJ ((2002) ) Subthalamic GAD gene therapy in a Parkinson’s disease rat model. Science 298: , 425–429. |
[57] | Lee B , Lee H , Nam YR , Oh JH , Cho YH , Chang JW ((2005) ) Enhanced expression of glutamate decarboxylase 65 improves symptoms of rat parkinsonian models. Gene Ther 12: , 1215–1222. |
[58] | Emborg ME , Carbon M , Holden JE , During MJ , Ma Y , Tang C , Moirano J , Fitzsimons H , Roitberg BZ , Tuccar E , Roberts A , Kaplitt MG , Eidelberg D ((2007) ) Subthalamic glutamic acid decarboxylase gene therapy: Changes in motor function and cortical metabolism. J Cereb Blood Flow Metab 27: , 501–509. |
[59] | Kaplitt MG , Feigin A , Tang C , Fitzsimons HL , Mattis P , Lawlor PA , Bland RJ , Young D , Strybing K , Eidelberg D , During MJ ((2007) ) Safety and tolerability of gene therapy with an adeno-associated virus (AAV) borne GAD gene for Parkinson’s disease: An open label, phase I trial. Lancet 369: , 2097–2105. |
[60] | Feigin A , Kaplitt MG , Tang C , Lin T , Mattis P , Dhawan V , During MJ , Eidelberg D ((2007) ) Modulation of metabolic brain networks after subthalamic gene therapy for Parkinson’s disease. Proc Natl Acad Sci U S A 104: , 19559–19564. |
[61] | Niethammer M , Tang CC , LeWitt PA , Rezai AR , Leehey MA , Ojemann SG , Flaherty AW , Eskandar EN , Kostyk SK , Sarkar A , Siddiqui MS , Tatter SB , Schwalb JM , Poston KL , Henderson JM , Kurlan RM , Richard IH , Sapan CV , Eidelberg D , During MJ , Kaplitt MG , Feigin A ((2017) ) Long-term follow-up of a randomized AAV2-GAD gene therapy trial for Parkinson’s disease. JCI Insight 2: , e90133. |
[62] | Collier TJ , Sortwell CE ((1999) ) Therapeutic potential of nerve growth factors in Parkinson’s disease. Drugs Aging 14: , 261–287. |
[63] | Airaksinen MS , Saarma M ((2002) ) The GDNF family: Signalling, biological functions and therapeutic value. Nat Rev Neurosci 3: , 383–394. |
[64] | Lei Z , Jiang Y , Li T , Zhu J , Zeng S ((2011) ) Signaling of glial cell line-derived neurotrophic factor and its receptor GFRalpha1 induce Nurr1 and Pitx3 to promote survival of grafted midbrain-derived neural stem cells in a rat model of Parkinson disease. J Neuropathol Exp Neurol 70: , 736–747. |
[65] | Zetterstrom RH , Solomin L , Jansson L , Hoffer BJ , Olson L , Perlmann T ((1997) ) Dopamine neuron agenesis in Nurr1-deficient mice. Science 276: , 248–250. |
[66] | Gill SS , Patel NK , Hotton GR , O’Sullivan K , McCarter R , Bunnage M , Brooks DJ , Svendsen CN , Heywood P ((2003) ) Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med 9: , 589–595. |
[67] | Slevin JT , Gerhardt GA , Smith CD , Gash DM , Kryscio R , Young B ((2005) ) Improvement of bilateral motor functions in patients with Parkinson disease through the unilateral intraputaminal infusion of glial cell line-derived neurotrophic factor. J Neurosurg 102: , 216–222. |
[68] | Ai Y , Markesbery W , Zhang Z , Grondin R , Elseberry D , Gerhardt GA , Gash DM ((2003) ) Intraputamenal infusion of GDNF in aged rhesus monkeys: Distribution and dopaminergic effects. J Comp Neurol 461: , 250–261. |
[69] | Maswood N , Grondin R , Zhang Z , Stanford JA , Surgener SP , Gash DM , Gerhardt GA ((2002) ) Effects of chronic intraputamenal infusion of glial cell line-derived neurotrophic factor (GDNF) in aged Rhesus monkeys. Neurobiol Aging 23: , 881–889. |
[70] | Grondin R , Zhang Z , Yi A , Cass WA , Maswood N , Andersen AH , Elsberry DD , Klein MC , Gerhardt GA , Gash DM ((2002) ) Chronic, controlled GDNF infusion promotes structural and functional recovery in advanced parkinsonian monkeys. Brain 125: , 2191–2201. |
[71] | Patel NK , Bunnage M , Plaha P , Svendsen CN , Heywood P , Gill SS ((2005) ) Intraputamenal infusion of glial cell line-derived neurotrophic factor in PD: A two-year outcome study. Ann Neurol 57: , 298–302. |
[72] | Chen YH , Harvey BK , Hoffman AF , Wang Y , Chiang YH , Lupica CR ((2008) ) MPTP-induced deficits in striatal synaptic plasticity are prevented by glial cell line-derived neurotrophic factor expressed via an adeno-associated viral vector. FASEB J 22: , 261–275. |
[73] | Tian YY , Tang CJ , Wang JN , Feng Y , Chen XW , Wang L , Qiao X , Sun SG ((2007) ) Favorable effects of VEGF gene transfer on a rat model of Parkinson disease using adeno-associated viral vectors. Neurosci Lett 421: , 239–244. |
[74] | Lu-Nguyen NB , Broadstock M , Schliesser MG , Bartholomae CC , von Kalle C , Schmidt M , Yanez-Munoz RJ ((2014) ) Transgenic expression of human glial cell line-derived neurotrophic factor from integration-deficient lentiviral vectors is neuroprotective in a rodent model of Parkinson’s disease. Hum Gene Ther 25: , 631–641. |
[75] | Sterky FH , Pernold K , Harvey BK , Lindqvist E , Hoffer BJ , Olson L ((2013) ) Glial cell line-derived neurotrophic factor partially ameliorates motor symptoms without slowing neurodegeneration in mice with respiratory chain-deficient dopamine neurons. Cell Transplant 22: , 1529–1539. |
[76] | Espadas-Alvarez AJ , Bannon MJ , Orozco-Barrios CE , Escobedo-Sanchez L , Ayala-Davila J , Reyes-Corona D , Soto-Rodriguez G , Escamilla-Rivera V , De Vizcaya-Ruiz A , Eugenia Gutierrez-Castillo M , Padilla-Viveros A , Martinez-Fong D ((2017) ) Regulation of human GDNF gene expression in nigral dopaminergic neurons using a new doxycycline-regulated NTS-polyplex nanoparticle system. Nanomedicine 13: , 1363–1375. |
[77] | Yurek D , Hasselrot U , Sesenoglu-Laird O , Padegimas L , Cooper M ((2017) ) Intracerebral injections of DNA nanoparticles encoding for a therapeutic gene provide partial neuroprotection in an animal model of neurodegeneration. Nanomedicine 13: , 2209–2217. |
[78] | Eslamboli A , Cummings RM , Ridley RM , Baker HF , Muzyczka N , Burger C , Mandel RJ , Kirik D , Annett LE ((2003) ) Recombinant adeno-associated viral vector (rAAV) delivery of GDNF provides protection against 6-OHDA lesion in the common marmoset monkey (Callithrix jacchus). Exp Neurol 184: , 536–548. |
[79] | Johnston LC , Eberling J , Pivirotto P , Hadaczek P , Federoff HJ , Forsayeth J , Bankiewicz KS ((2009) ) Clinically relevant effects of convection-enhanced delivery of AAV2-GDNF on the dopaminergic nigrostriatal pathway in aged rhesus monkeys. Hum Gene Ther 20: , 497–510. |
[80] | Kells AP , Eberling J , Su X , Pivirotto P , Bringas J , Hadaczek P , Narrow WC , Bowers WJ , Federoff HJ , Forsayeth J , Bankiewicz KS ((2010) ) Regeneration of the MPTP-lesioned dopaminergic system after convection-enhanced delivery of AAV2-GDNF. J Neurosci 30: , 9567–9577. |
[81] | Eberling JL , Kells AP , Pivirotto P , Beyer J , Bringas J , Federoff HJ , Forsayeth J , Bankiewicz KS ((2009) ) Functional effects of AAV2-GDNF on the dopaminergic nigrostriatal pathway in parkinsonian rhesus monkeys. Hum Gene Ther 20: , 511–518. |
[82] | Su X , Kells AP , Huang EJ , Lee HS , Hadaczek P , Beyer J , Bringas J , Pivirotto P , Penticuff J , Eberling J , Federoff HJ , Forsayeth J , Bankiewicz KS ((2009) ) Safety evaluation of AAV2-GDNF gene transfer into the dopaminergic nigrostriatal pathway in aged and parkinsonian rhesus monkeys. Hum Gene Ther 20: , 1627–1640. |
[83] | Herzog CD , Dass B , Gasmi M , Bakay R , Stansell JE , Tuszynski M , Bankiewicz K , Chen EY , Chu Y , Bishop K , Kordower JH , Bartus RT ((2008) ) Transgene expression, bioactivity, and safety of CERE-120 (AAV2-neurturin) following delivery to the monkey striatum. Mol Ther 16: , 1737–1744. |
[84] | Warren Olanow C , Bartus RT , Baumann TL , Factor S , Boulis N , Stacy M , Turner DA , Marks W , Larson P , Starr PA , Jankovic J , Simpson R , Watts R , Guthrie B , Poston K , Henderson JM , Stern M , Baltuch G , Goetz CG , Herzog C , Kordower JH , Alterman R , Lozano AM , Lang AE ((2015) ) Gene delivery of neurturin to putamen and substantia nigra in Parkinson disease: A double-blind, randomized, controlled trial. Ann Neurol 78: , 248–257. |
[85] | Bartus RT , Baumann TL , Siffert J , Herzog CD , Alterman R , Boulis N , Turner DA , Stacy M , Lang AE , Lozano AM , Olanow CW ((2013) ) Safety/feasibility of targeting the substantia nigra with AAV2-neurturin in Parkinson patients. Neurology 80: , 1698–1701. |
[86] | U.S. National Institutes of Health, AAV2-GDNF for Advanced Parkinson s Disease, https://clinicaltrials.gov/ct2/show/NCT01621581?term=gdnf&rank=1, Last updated February 8, 2018, Accessed on February 21, 2017. |
[87] | Hagg T , Manthorpe M , Vahlsing HL , Varon S ((1988) ) Delayed treatment with nerve growth factor reverses the apparent loss of cholinergic neurons after acute brain damage. Exp Neurol 101: , 303–312. |
[88] | Gasmi M , Herzog CD , Brandon EP , Cunningham JJ , Ramirez GA , Ketchum ET , Bartus RT ((2007) ) Striatal delivery of neurturin by CERE-120, an AAV2 vector for the treatment of dopaminergic neuron degeneration in Parkinson’s disease. Mol Ther 15: , 62–68. |
[89] | Gasmi M , Brandon EP , Herzog CD , Wilson A , Bishop KM , Hofer EK , Cunningham JJ , Printz MA , Kordower JH , Bartus RT ((2007) ) AAV2-mediated delivery of human neurturin to the rat nigrostriatal system: Long-term efficacy and tolerability of CERE-120 for Parkinson’s disease. Neurobiol Dis 27: , 67–76. |
[90] | Oiwa Y , Yoshimura R , Nakai K , Itakura T ((2002) ) Dopaminergic neuroprotection and regeneration by neurturin assessed by using behavioral, biochemical and histochemical measurements in a model of progressive Parkinson’s disease. Brain Res 947: , 271–283. |
[91] | Kordower JH , Herzog CD , Dass B , Bakay RA , Stansell J, 3rd , Gasmi M , Bartus RT ((2006) ) Delivery of neurturin by AAV2 (CERE-120)-mediated gene transfer provides structural and functional neuroprotection and neurorestoration in MPTP-treated monkeys. Ann Neurol 60: , 706–715. |
[92] | Herzog CD , Dass B , Holden JE , Stansell J, 3rd , Gasmi M , Tuszynski MH , Bartus RT , Kordower JH ((2007) ) Striatal delivery of CERE-120, an AAV2 vector encoding human neurturin, enhances activity of the dopaminergic nigrostriatal system in aged monkeys. Mov Disord 22: , 1124–1132. |
[93] | Marks WJ Jr , Ostrem JL , Verhagen L , Starr PA , Larson PS , Bakay RA , Taylor R , Cahn-Weiner DA , Stoessl AJ , Olanow CW , Bartus RT ((2008) ) Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: An open-label, phase I trial. Lancet Neurol 7: , 400–408. |
[94] | Bartus RT , Herzog CD , Chu Y , Wilson A , Brown L , Siffert J , Johnson EM, Jr , Olanow CW , Mufson EJ , Kordower JH ((2011) ) Bioactivity of AAV2-neurturin gene therapy (CERE-120): Differences between Parkinson’s disease and nonhuman primate brains. Mov Disord 26: , 27–36. |
[95] | Bartus RT , Kordower JH , Johnson EM, Jr , Brown L , Kruegel BR , Chu Y , Baumann TL , Lang AE , Olanow CW , Herzog CD ((2015) ) Post-mortem assessment of the short and long-term effects of the trophic factor neurturin in patients with alpha-synucleinopathies. Neurobiol Dis 78: , 162–171. |
[96] | Bartus RT , Brown L , Wilson A , Kruegel B , Siffert J , Johnson EM, Jr , Kordower JH , Herzog CD ((2011) ) Properly scaled and targeted AAV2-NRTN (neurturin) to the substantia nigra is safe, effective and causes no weight loss: Support for nigral targeting in Parkinson’s disease. Neurobiol Dis 44: , 38–52. |
[97] | Runeberg-Roos P , Piccinini E , Penttinen AM , Matlik K , Heikkinen H , Kuure S , Bespalov MM , Peranen J , Garea-Rodriguez E , Fuchs E , Airavaara M , Kalkkinen N , Penn R , Saarma M ((2016) ) Developing therapeutically more efficient Neurturin variants for treatment of Parkinson’s disease. Neurobiol Dis 96: , 335–345. |
[98] | Benedetti F , Frisaldi E , Carlino E , Giudetti L , Pampallona A , Zibetti M , Lanotte M , Lopiano L ((2016) ) Teaching neurons to respond to placebos. J Physiol 594: , 5647–5660. |
[99] | Decressac M , Mattsson B , Bjorklund A ((2012) ) Comparison of the behavioural and histological characteristics of the 6-OHDA and alpha-synuclein rat models of Parkinson’s disease. Exp Neurol 235: , 306–315. |
[100] | Venda LL , Cragg SJ , Buchman VL , Wade-Martins R ((2010) ) alpha-Synuclein and dopamine at the crossroads of Parkinson’s disease. Trends Neurosci 33: , 559–568. |
[101] | Decressac M , Ulusoy A , Mattsson B , Georgievska B , Romero-Ramos M , Kirik D , Bjorklund A ((2011) ) GDNF fails to exert neuroprotection in a rat alpha-synuclein model of Parkinson’s disease. Brain 134: , 2302–2311. |
[102] | Kirik D , Rosenblad C , Burger C , Lundberg C , Johansen TE , Muzyczka N , Mandel RJ , Bjorklund A ((2002) ) Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci 22: , 2780–2791. |
[103] | Jankovic J , Chen S , Le WD ((2005) ) The role of Nurr1 in the development of dopaminergic neurons and Parkinson’s disease. Prog Neurobiol 77: , 128–138. |
[104] | Le W , Pan T , Huang M , Xu P , Xie W , Zhu W , Zhang X , Deng H , Jankovic J ((2008) ) Decreased NURR1 gene expression in patients with Parkinson’s disease. J Neurol Sci 273: , 29–33. |
[105] | Oh SM , Chang MY , Song JJ , Rhee YH , Joe EH , Lee HS , Yi SH , Lee SH ((2016) ) Combined Nurr1 and Foxa2 roles in the therapy of Parkinson’s disease. EMBO Mol Med 8: , 171. |
[106] | Dong J , Li S , Mo JL , Cai HB , Le WD ((2016) ) Nurr1-based therapies for Parkinson’s disease. CNS Neurosci Ther 22: , 351–359. |
[107] | Apostolou A , Shen Y , Liang Y , Luo J , Fang S ((2008) ) Armet, a UPR-upregulated protein, inhibits cell proliferation and ER stress-induced cell death. Exp Cell Res 314: , 2454–2467. |
[108] | Glembotski CC , Thuerauf DJ , Huang C , Vekich JA , Gottlieb RA , Doroudgar S ((2012) ) Mesencephalic astrocyte-derived neurotrophic factor protects the heart from ischemic damage and is selectively secreted upon sarco/endoplasmic reticulum calcium depletion. J Biol Chem 287: , 25893–25904. |
[109] | Matlik K , Yu LY , Eesmaa A , Hellman M , Lindholm P , Peranen J , Galli E , Anttila J , Saarma M , Permi P , Airavaara M , Arumae U ((2015) ) Role of two sequence motifs of mesencephalic astrocyte-derived neurotrophic factor in its survival-promoting activity. Cell Death Dis 6: , e2032. |
[110] | Wang M , Kaufman RJ ((2014) ) The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat Rev Cancer 14: , 581–597. |
[111] | Mercado G , Valdes P , Hetz C ((2013) ) An ERcentric view of Parkinson’s disease. Trends Mol Med 19: , 165–175. |
[112] | Petrova P , Raibekas A , Pevsner J , Vigo N , Anafi M , Moore MK , Peaire AE , Shridhar V , Smith DI , Kelly J , Durocher Y , Commissiong JW ((2003) ) MANF: A new mesencephalic, astrocyte-derived neurotrophic factor with selectivity for dopaminergic neurons. J Mol Neurosci 20: , 173–188. |
[113] | Lindholm P , Voutilainen MH , Lauren J , Peranen J , Leppanen VM , Andressoo JO , Lindahl M , Janhunen S , Kalkkinen N , Timmusk T , Tuominen RK , Saarma M ((2007) ) Novel neurotrophic factor CDNF protects and rescues midbrain dopamine neurons in vivo. Nature 448: , 73–77. |
[114] | Back S , Peranen J , Galli E , Pulkkila P , Lonka-Nevalaita L , Tamminen T , Voutilainen MH , Raasmaja A , Saarma M , Mannisto PT , Tuominen RK ((2013) ) Gene therapy with AAV2-CDNF provides functional benefits in a rat model of Parkinson’s disease. Brain Behav 3: , 75–88. |
[115] | Ren X , Zhang T , Gong X , Hu G , Ding W , Wang X ((2013) ) AAV2-mediated striatum delivery of human CDNF prevents the deterioration of midbrain dopamine neurons in a 6-hydroxydopamine induced parkinsonian rat model. Exp Neurol 248: , 148–156. |
[116] | Cordero-Llana O , Houghton BC , Rinaldi F , Taylor H , Yanez-Munoz RJ , Uney JB , Wong LF , Caldwell MA ((2015) ) Enhanced efficacy of the CDNF/MANF family by combined intranigral overexpression in the 6-OHDA rat model of Parkinson’s disease. Mol Ther 23: , 244–254. |
[117] | Hao F , Yang C , Chen SS , Wang YY , Zhou W , Hao Q , Lu T , Hoffer B , Zhao LR , Duan WM , Xu QY ((2017) ) Long-term protective effects of AAV9-mesencephalic astrocyte-derived neurotrophic factor gene transfer in parkinsonian rats. Exp Neurol 291: , 120–133. |
[118] | Patapoutian A , Reichardt LF ((2001) ) Trk receptors: Mediators of neurotrophin action. Current Opinion in Neurobiology 11: , 272–280. |
[119] | Ding YX , Xia Y , Jiao XY , Duan L , Yu J , Wang X , Chen LW ((2011) ) The TrkB-positive dopaminergic neurons are less sensitive to MPTP insult in the substantia nigra of adult C57/BL mice. Neurochem Res 36: , 1759–1766. |
[120] | Hyman C , Hofer M , Barde YA , Juhasz M , Yancopoulos GD , Squinto SP , Lindsay RM ((1991) ) BDNF is a neurotrophic factor for dopaminergic neurons of the substantia nigra. Nature 350: , 230–232. |
[121] | Pencea V , Bingaman KD , Wiegand SJ , Luskin MB ((2001) ) Infusion of brain-derived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. J Neurosci 21: , 6706–6717. |
[122] | Zuccato C , Cattaneo E ((2009) ) Brain-derived neurotrophic factor in neurodegenerative diseases. Nat Rev Neurol 5: , 311–322. |
[123] | Garraway SM , Huie JR , ((2016) ) Spinal plasticity and behavior: BDNF-induced neuromodulation in uninjured and injured spinal cord. Neural Plast 2016: , 9857201. |
[124] | Parain K , Murer MG , Yan Q , Faucheux B , Agid Y , Hirsch E , Raisman-Vozari R ((1999) ) Reduced expression of brain-derived neurotrophic factor protein in Parkinson’s disease substantia nigra. Neuroreport 10: , 557–561. |
[125] | Porritt MJ , Batchelor PE , Howells DW ((2005) ) Inhibiting BDNF expression by antisense oligonucleotide infusion causes loss of nigral dopaminergic neurons. Exp Neurol 192: , 226–234. |
[126] | Tsukahara T , Takeda M , Shimohama S , Ohara O , Hashimoto N ((1995) ) Effects of brain-derived neurotrophic factor on 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in monkeys.733-739; discussion. Neurosurgery 37: , 739–741. |
[127] | Klein RL , Muzyczka LM , Meyer N , EM ((1999) ) Prevention of 6-hydroxydopamine-induced rotational behavior by BDNF somatic gene transfer. Brain Res 847: , 314–320. |
[128] | Sun M , Kong L , Wang X , Lu XG , Gao Q , Geller AI ((2005) ) Comparison of the capability of GDNF, BDNF, or both, to protect nigrostriatal neurons in a rat model of Parkinson’s disease. Brain Res 1052: , 119–129. |
[129] | Hernandez-Chan NG , Bannon MJ , Orozco-Barrios CE , Escobedo L , Zamudio S , De la Cruz F , Gongora-Alfaro JL , Armendariz-Borunda J , Reyes-Corona D , Espadas-Alvarez AJ , Flores-Martinez YM , Ayala-Davila J , Hernandez-Gutierrez ME , Pavon L , Garcia-Villegas R , Nadella R , Martinez-Fong D ((2015) ) Neurotensin-polyplex-mediated brain-derived neurotrophic factor gene delivery into nigral dopamine neurons prevents nigrostriatal degeneration in a rat model of early Parkinson’s disease. J Biomed Sci 22: , 59. |
[130] | Lapchak PA , Beck KD , Araujo DM , Irwin I , Langston JW , Hefti F ((1993) ) Chronic intranigral administration of brain-derived neurotrophic factor produces striatal dopaminergic hypofunction in unlesioned adult rats and fails to attenuate the decline of striatal dopaminergic function following medial forebrain bundle transection. Neuroscience 53: , 639–650. |
[131] | Ferrara N , Gerber HP , LeCouter J ((2003) ) The biology of VEGF and its receptors. Nat Med 9: , 669–676. |
[132] | Dvorak HF , Brown LF , Detmar M , Dvorak AM ((1995) ) Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 146: , 1029–1039. |
[133] | Neufeld G , Cohen T , Shraga N , Lange T , Kessler O , Herzog Y ((2002) ) The neuropilins: Multifunctional semaphorin and VEGF receptors that modulate axon guidance and angiogenesis. Trends Cardiovasc Med 12: , 13–19. |
[134] | Falk T , Gonzalez RT , Sherman SJ ((2010) ) The yin and yang of VEGF and PEDF: Multifaceted neurotrophic factors and their potential in the treatment of Parkinson’s Disease. Int J Mol Sci 11: , 2875–2900. |
[135] | Yasuhara T , Shingo T , Kobayashi K , Takeuchi A , Yano A , Muraoka K , Matsui T , Miyoshi Y , Hamada H , Date I ((2004) ) Neuroprotective effects of vascular endothelial growth factor (VEGF) upon dopaminergic neurons in a rat model of Parkinson’s disease. Eur J Neurosci 19: , 1494–1504. |
[136] | Yasuhara T , Shingo T , Date I ((2004) ) The potential role of vascular endothelial growth factor in the central nervous system. Rev Neurosci 15: , 293–307. |
[137] | Xu YQ , Long L , Yan JQ , Wei L , Pan MQ , Gao HM , Zhou P , Liu M , Zhu CS , Tang BS , Wang Q ((2013) ) Simvastatin induces neuroprotection in 6-OHDA-lesioned PC12 via the PI3K/AKT/caspase 3 pathway and anti-inflammatory responses. CNS Neurosci Ther 19: , 170–177. |
[138] | Hedman M , Muona K , Hedman A , Kivelä A , Syvänne M , Eränen J , Rantala A , Stjernvall J , Nieminen M , Hartikainen J ((2009) ) Eight-year safety follow-up of coronary artery disease patients after local intracoronary VEGF gene transfer. Gene Ther 16: , 629–634. |
[139] | Yasuhara T , Shingo T , Muraoka K , wen Ji Y , Kameda M , Takeuchi A , Yano A , Nishio S , Matsui T , Miyoshi Y , Hamada H , Date I ((2005) ) The differences between high and low-dose administration of VEGF to dopaminergic neurons of in vitro and in vivo Parkinson’s disease model. Brain Res 1038: , 1–10. |
[140] | Yu M , Suo H , Liu M , Cai L , Liu J , Huang Y , Xu J , Wang Y , Zhu C , Fei J , Huang F ((2013) ) NRSF/REST neuronal deficient mice are more vulnerable to the neurotoxin MPTP. Neurobiol Aging 34: , 916–927. |
[141] | Wu P , Zuo X , Deng H , Liu X , Liu L , Ji A ((2013) ) Roles of long noncoding RNAs in brain development, functional diversification and neurodegenerative diseases. Brain Res Bull 97: , 69–80. |
[142] | Decressac M , Pain S , Chabeauti PY , Frangeul L , Thiriet N , Herzog H , Vergote J , Chalon S , Jaber M , Gaillard A ((2012) ) Neuroprotection by neuropeptide Y in cell and animal models of Parkinson’s disease. Neurobiol Aging 33: , 2125–2137. |
[143] | Malva JO , Xapelli S , Baptista S , Valero J , Agasse F , Ferreira R , Silva AP ((2012) ) Multifaces of neuropeptide Y in the brain–neuroprotection, neurogenesis and neuroinflammation. Neuropeptides 46: , 299–308. |
[144] | Cannizzaro C , Tel BC , Rose S , Zeng BY , Jenner P ((2003) ) Increased neuropeptide Y mRNA expression in striatum in Parkinson’s disease. Brain Res Mol Brain Res 110: , 169–176. |
[145] | Duarte-Neves J , Pereira de Almeida L , Cavadas C ((2016) ) Neuropeptide Y (NPY) as a therapeutic target for neurodegenerative diseases. Neurobiol Dis 95: , 210–224. |
[146] | Settembre C , Di Malta C , Polito VA , Garcia Arencibia M , Vetrini F , Erdin S , Erdin SU , Huynh T , Medina D , Colella P , Sardiello M , Rubinsztein DC , Ballabio A ((2011) ) TFEB links autophagy to lysosomal biogenesis. Science 332: , 1429–1433. |
[147] | Dehay B , Bove J , Rodriguez-Muela N , Perier C , Recasens A , Boya P , Vila M ((2010) ) Pathogenic lysosomal depletion in Parkinson’s disease. J Neurosci 30: , 12535–12544. |
[148] | Decressac M , Mattsson B , Weikop P , Lundblad M , Jakobsson J , Bjorklund A ((2013) ) TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A 110: , E1817-E1826. |
[149] | Diester I , Kaufman MT , Mogri M , Pashaie R , Goo W , Yizhar O , Ramakrishnan C , Deisseroth K , Shenoy KV ((2011) ) An optogenetic toolbox designed for primates. Nat Neurosci 14: , 387–397. |
[150] | Wang F , Belanger E , Paquet ME , Cote DC , De Koninck Y ((2016) ) Probing pain pathways with light. Neuroscience 338: , 248–271. |
[151] | Tung JK , Berglund K , Gross RE ((2016) ) Optogenetic approaches for controlling seizure activity. Brain Stimul 9: , 801–810. |
[152] | Heshmati M , Russo SJ ((2015) ) Anhedonia and the brain reward circuitry in depression. Curr Behav Neurosci Rep 2: , 146–153. |
[153] | Entcheva E ((2013) ) Cardiac optogenetics. Am J Physiol Heart Circ Physiol 304: , H1179–H1191. |
[154] | Duebel J , Marazova K , Sahel JA ((2015) ) Optogenetics. Curr Opin Ophthalmol 26: , 226–232. |
[155] | Sanders TH , Jaeger D ((2016) ) Optogenetic stimulation of cortico-subthalamic projections is sufficient to ameliorate bradykinesia in 6-ohda lesioned mice. Neurobiol Dis 95: , 225–237. |
[156] | Gradinaru V , Mogri M , Thompson KR , Henderson JM , Deisseroth K ((2009) ) Optical deconstruction of parkinsonian neural circuitry. Science 324: , 354–359. |
[157] | Yoon HH , Park JH , Kim YH , Min J , Hwang E , Lee CJ , Suh JK , Hwang O , Jeon SR , ((2014) ) Optogenetic inactivation of the subthalamic nucleus improves forelimb akinesia in a rat model of Parkinson disease. Neurosurgery 74: , 540–531; 533-540; discussion. |
[158] | Yoon HH , Min J , Hwang E , Lee CJ , Suh JK , Hwang O , Jeon SR , ((2016) ) Optogenetic inhibition of the subthalamic nucleus reduces levodopa-induced dyskinesias in a rat model of Parkinson’s disease. Stereotact Funct Neurosurg 94: , 41–53. |
[159] | Kravitz AV , Freeze BS , Parker PR , Kay K , Thwin MT , Deisseroth K , Kreitzer AC ((2010) ) Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature 466: , 622–626. |
[160] | Freeze BS , Kravitz AV , Hammack N , Berke JD , Kreitzer AC ((2013) ) Control of basal ganglia output by direct and indirect pathway projection neurons. J Neurosci 33: , 18531–18539. |
[161] | Moon HC , Won SY , Kim EG , Kim HK , Cho CB , Park YS ((2017) ) Effect of optogenetic modulation on entopeduncular input affects thalamic discharge and behavior in an AAV2-alpha-synuclein-induced hemiparkinson rat model. Neurosci Lett 662: , 129–135. |
[162] | Chuong AS , Miri ML , Busskamp V , Matthews GA , Acker LC , Sorensen AT , Young A , Klapoetke NC , Henninger MA , Kodandaramaiah SB , Ogawa M , Ramanlal SB , Bandler RC , Allen BD , Forest CR , Chow BY , Han X , Lin Y , Tye KM , Roska B , Cardin JA , Boyden ES ((2014) ) Noninvasive optical inhibition with a red-shifted microbial rhodopsin. Nat Neurosci 17: , 1123–1129. |
[163] | Conklin BR , Hsiao EC , Claeysen S , Dumuis A , Srinivasan S , Forsayeth JR , Guettier JM , Chang WC , Pei Y , McCarthy KD , Nissenson RA , Wess J , Bockaert J , Roth BL ((2008) ) Engineering GPCR signaling pathways with RASSLs. Nat Methods 5: , 673–678. |
[164] | Wess J , Nakajima K , Jain S ((2013) ) Novel designer receptors to probe GPCR signaling and physiology. Trends Pharmacol Sci 34: , 385–392. |
[165] | Jorgensen SH , Fitzpatrick CM , Gether U , Woldbye DPD , Sorensen AT ((2017) ) Chemogenetic modulation of G protein-coupled receptor signalling in visual attention research. Basic Clin Pharmacol Toxicol 121: , 373–381. |
[166] | Roth BL ((2016) ) DREADDs for neuroscientists. Neuron 89: , 683–694. |
[167] | Pienaar IS , Gartside SE , Sharma P , De Paola V , Gretenkord S , Withers D , Elson JL , Dexter DT ((2015) ) Pharmacogenetic stimulation of cholinergic pedunculopontine neurons reverses motor deficits in a rat model of Parkinson’s disease. Mol Neurodegener 10: , 47. |
[168] | Pienaar IS , Harrison IF , Elson JL , Bury A , Woll P , Simon AK , Dexter DT ((2015) ) An animal model mimicking pedunculopontine nucleus cholinergic degeneration in Parkinson’s disease. Brain Struct Funct 220: , 479–500. |
[169] | Polymeropoulos MH , Lavedan C , Leroy E , Ide SE , Dehejia A , Dutra A , Pike B , Root H , Rubenstein J , Boyer R , Stenroos ES , Chandrasekharappa S , Athanassiadou A , Papapetropoulos T , Johnson WG , Lazzarini AM , Duvoisin RC , Di Iorio G , Golbe LI , Nussbaum RL ((1997) ) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276: , 2045–2047. |
[170] | Healy DG , Falchi M , O’Sullivan SS , Bonifati V , Durr A , Bressman S , Brice A , Aasly J , Zabetian CP , Goldwurm S , Ferreira JJ , Tolosa E , Kay DM , Klein C , Williams DR , Marras C , Lang AE , Wszolek ZK , Berciano J , Schapira AH , Lynch T , Bhatia KP , Gasser T , Lees AJ , Wood NW ((2008) ) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: A case-control study. Lancet Neurol 7: , 583–590. |
[171] | Kang UB , Marto JA ((2017) ) Leucine-rich repeat kinase 2 and Parkinson’s disease. Proteomics 17: , 1–2. |
[172] | Follett J , Bugarcic A , Yang Z , Ariotti N , Norwood SJ , Collins BM , Parton RG , Teasdale RD ((2016) ) Parkinson disease-linked Vps35 R524W mutation impairs the endosomal association of retromer and induces alpha-synuclein aggregation. J Biol Chem 291: , 18283–18298. |
[173] | Girard M , Poupon V , Blondeau F , McPherson PS ((2005) ) The DnaJ-domain protein RME-8 functions in endosomal trafficking. J Biol Chem 280: , 40135–40143. |
[174] | Gustavsson EK , Trinh J , Guella I , Vilarino-Guell C , Appel-Cresswell S , Stoessl AJ , Tsui JK , McKeown M , Rajput A , Rajput AH , Aasly JO , Farrer MJ ((2015) ) DNAJC13 genetic variants in parkinsonism. Mov Disord 30: , 273–278. |
[175] | Schrag A , Schott JM ((2006) ) Epidemiological, clinical, and genetic characteristics of early-onset parkinsonism. Lancet Neurol 5: , 355–363. |
[176] | Lucking CB , Durr A , Bonifati V , Vaughan J , De Michele G , Gasser T , Harhangi BS , Meco G , Denefle P , Wood NW , Agid Y , Brice A ((2000) ) Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med 342: , 1560–1567. |
[177] | Kang UB , Marto JA ((2017) ) Leucine-rich repeat kinase 2 and Parkinson’s disease. Proteomics 17: . |
[178] | Ledford H ((2015) ) CRISPR, the disruptor. Nature 522: , 20–24. |
[179] | Singh V , Braddick D , Dhar PK ((2017) ) Exploring the potential of genome editing CRISPR-Cas9 technology. Gene 599: , 1–18. |
[180] | Cyranoski D ((2016) ) Chinese scientists to pioneer first human CRISPR trial. Nature 535: , 476–477. |
[181] | Yang W , Tu Z , Sun Q , Li XJ ((2016) ) CRISPR/Cas9: Implications for modeling and therapy of neurodegenerative diseases. Front Mol Neurosci 9: , 30. |
[182] | Freund HJ ((2005) ) Long-term effects of deep brain stimulation in Parkinson’s disease. Brain 128: , 2222–2223. |
[183] | Hadaczek P , Eberling JL , Pivirotto P , Bringas J , Forsayeth J , Bankiewicz KS ((2010) ) Eight years of clinical improvement in MPTP-lesioned primates after gene therapy with AAV2-hAADC. Mol Ther 18: , 1458–1461. |
[184] | Kordower JH , Emborg ME , Bloch J , Ma SY , Chu Y , Leventhal L , McBride J , Chen EY , Palfi S , Roitberg BZ , Brown WD , Holden JE , Pyzalski R , Taylor MD , Carvey P , Ling Z , Trono D , Hantraye P , Deglon N , Aebischer P ((2000) ) Neurodegeneration prevented by lentiviral vector delivery of GDNF in primate models of Parkinson’s disease. Science 290: , 767–773. |
[185] | Kozlowski DA , Bremer E , Redmond DE, Jr , George D , Larson B , Bohn MC ((2001) ) Quantitative analysis of transgene protein, mRNA, and vector DNA following injection of an adenoviral vector harboring glial cell line-derived neurotrophic factor into the primate caudate nucleus. Mol Ther 3: , 256–261. |
[186] | Palfi S , Leventhal L , Chu Y , Ma SY , Emborg M , Bakay R , Deglon N , Hantraye P , Aebischer P , Kordower JH ((2002) ) Lentivirally delivered glial cell line-derived neurotrophic factor increases the number of striatal dopaminergic neurons in primate models of nigrostriatal degeneration. J Neurosci 22: , 4942–4954. |
[187] | Emborg ME , Hurley SA , Joers V , Tromp do PM , Swanson CR , Ohshima-Hosoyama S , Bondarenko V , Cummisford K , Sonnemans M , Hermening S , Blits B , Alexander AL ((2014) ) Titer and product affect the distribution of gene expression after intraputaminal convection-enhanced delivery. Stereotact Funct Neurosurg 92: , 182–194. |


