
Stomaching the Possibility of a Pathogenic Role for Helicobacter pylori in Parkinson’s Disease
Abstract
While a small subset of Parkinson’s disease cases have genetic causes, most cases are sporadic and may have an environmental contributor that has largely remained enigmatic. Remarkably, gastrointestinal symptoms in PD patients serve as a prodrome for the eventual motor dysfunctions. Herein, we review studies exploring a possible link between the gastric human pathogen Helicobacter pylori and PD. We provide plausible and testable hypotheses for how this organism might contribute to PD: 1) a toxin(s) produced by the bacteria; 2) disruption of the intestinal microbiome; 3) local inflammation that crosses the gut-brain axis, leading to neuroinflammation; and 4) manipulation of the pharmacokinetics of the PD drug levodopa by H. pylori, even in those not receiving exogenous levodopa. Key findings are: 1) people with PD are 1.5-3-fold more likely to be infected with H. pylori than people without PD; 2) H. pylori-infected PD patients display worse motor functions than H. pylori-negative PD patients; 3) eradication of H. pylori improves motor function in PD patients over PD patients whose H. pylori was not eradicated; and 4) eradication of H. pylori improves levodopa absorption in PD patients compared to that of PD patients whose H. pylori was not eradicated. Evidence is accumulating that H. pylori has a link with PD, but the mechanism is unclear. Future work should explore the effects of H. pylori on development of PD in defined PD animal models, focusing on the roles of H. pylori toxins, inflammation, levodopa absorption, and microbiome dysbiosis.
Parkinson’s disease (PD) is the second most common neurodegenerative disease in the world and causes motor dysfunction symptoms such as tremor, posture instability, rigidity, and bradykinesia. This neurodegenerative disease is characterized by loss of dopaminergic neurons in the brain and accumulation of neuronal cytoplasmic inclusions, Lewy bodies, composed of aggregated alpha-synuclein. A prodrome of PD is pathophysiology of the gut manifested by GI symptoms such as dyspepsia, hypersalivation from decreased swallowing, constipation, defecatory dysfunction, fecal incontinence, nausea, and abdominal pain [1–4]. Of these, constipation and defecatory dysfunction precede motor manifestations in PD patients [3], suggesting that the disease may initiate in the gut and subsequently spreads to the brain along the brain-gut axis. This progression was recently documented in rats, where injection of alpha-synuclein fibrils into the gut of healthy rats triggered pathology within the vagus nerve and brainstem [1, 5]. The loss in PD of myenteric dopaminergic neurons with presence of GI alpha-synuclein deposits and Lewy bodies, and the associated constipation and other gastrointestinal symptoms, suggest that the gastrointestinal tract is a key anatomical focus in the development of PD pathology [6, 7].
While genetic factors clearly contribute to the development of PD, much of the variance is likely due to environmental factors that have remained enigmatic. One intriguing environmental factor for PD that has gained attention in recent years is the gastric pathogen Helicobacter pylori [8–10]. H. pylori chronically infects half the world’s population, causing gastritis, ulcers, gastric adenocarcinoma and MALT lymphoma, accompanied by an array of gastrointestinal symptoms. The abundance of H. pylori in humans has prompted examination of the role of this pathogen in extra-gastric diseases [10].
Remarkably, the link between PD and ulcers predates the identification of H. pylori as the causative agent of gastritis and ulcer formation [11]. Schwab first described an increased occurrence of ulcers in patients with PD and suggested that these ulcer patients were more vulnerable to PD than ordinary people of the same age [12]. Since 1922 vagotomy surgeries were long of major importance in the treatment of peptic ulcer disease prior to the discovery of H. pylori and the advent of triple drug therapy for H. pylori infection [13]. Intriguingly, removal of part of the vagus nerve (truncal vagotomy) is also associated with a decreased risk for subsequent PD [13–15], suggesting a link beween PD and the gut along the vagus nerve. A follow-up study demonstrated that a group of 200 consecutive PD patients had 14% higher incidence of ulcers (determined by surgery or X-ray) than 200 age and sex-matched controls (4%), and ulceration preceded parkinsonian symptoms by about 10 years [16].
In the first epidemiologic study directly looking for H. pylori, PD patients (n = 33) had a three-fold elevated risk of testing seropositive for H. pylori versus controls (n = 78) [17]. Intriguingly, the non-PD siblings (n = 39) of PD patients had elevated risk for bradykinesia of gait and hands, abnormal posture and other PD-like symptoms and also had a 3-fold increased risk of H. pylori seropositivity [17]. The data suggest that H. pylori transmission within families can increase the risk of developing parkinsonian symptoms. Similarly, a larger study (n = 315) found a two-fold increased prevalence of H. pylori seropositivity in PD patients [18]. An extensive meta analysis involving 33,000 patients reported a 1.5-2-fold increased risk of developing PD if infected with H. pylori [19]. A more recent retrospective study involving 9,105 H. pylori infected and 9,105 matched uninfected controls found a similar 2-3-fold increased risk of PD in the H. pylori infected group [20]. Thus, chronic H. pylori infection may predispose people to idiopathic PD [18, 21–23]. The prevalence of H. pylori infection in PD patients is 32–70% in different studies involving ∼50–100 patients [24]. While there are clearly PD patients lacking H. pylori infection, it appears H. pylori may exacerbate PD symptoms and act as a potentiator of the disease.
The widely used PD drug levodopa (L-dopa) is converted to the neurotransmitter dopamine by dopa decarboxylase, thereby elevating dopamine levels in the brain. Dopamine has gastroprotective affects in humans and in animal models, protecting against ulcer formation [25–27]. Interestingly, H. pylori positive patients (n = 53) showed poorer motor responses to levodopa treatment than H. pylori negative patients (n = 48), as assessed by the Unified Parkinson’s Disease Rating Scale scores (UPDRS-III) [28, 29]. Moreover, H. pylori eradication elevates levodopa in plasma of PD patients [30]. It was observed that H. pylori adhesins directly bind to levodopa, sequestering it away from human brain absorption, thereby reducing effectiveness of levodopa treatment for PD [31–34]. Remarkably, H. pylori uses levodopa in vitro as a growth supplement in an iron-restricted medium [34], suggesting H. pylori converts this phenylalanine/tyrosine derivative into a functional amino acid for its metabolism. Thus, the pharmacokinetics of levodopa may be altered by H. pylori, though the mechanism is not well understood [35].
PD patients (n = 33) that are already infected with H. pylori have worse motor functions than H. pylori negative PD patients (n = 69) [36]. In several clinical studies, eradication of H. pylori with triple drug therapy improved motor responses in PD patients [28, 29, 33, 37–41]. For example, Dobbs, et al., found that stride increased in PD patients eradicated for H. pylori over that of PD patients given placebo, and in routine practice [38, 39]. In double-blind trials of biopsy-proven H. pylori eradiation [39], improved hypokinesia was independent of any (stable, long t
The mechanism of the association between H. pylori and PD is not well understood but is likely multifactorial. First, H. pylori may produce bacterial factors that are CNS toxic. For example, Altschuler speculated that H. pylori could be synthesizing a PD-inducing toxin, akin to methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [8], a compound long used to induce PD in animal models. H. pylori is known to produce several toxins, such as the
The second possibility is that H. pylori triggers a massive inflammatory response in the stomach and this response may become systemic and cross the BBB, leading to exacerbation of PD symptoms and pathology [53]. The immune response to H. pylori antigens that molecularly mimic those found on host cells could lead to autoantibody responses [53, 54]. For example, H. pylori lipopolysaccharide contains fucosylated Lewis antigens that molecularly mimic human cell surface glycoconjugates, and these bacterial antigen levels change in response to culturing the organism in cholesterol [47]. Indeed, a recent study demonstrated that H. pylori positive PD patients (n = 30) contained 13 autoantibodies (eight up-regulated, five down-regulated) lacking in age and gender-matched H. pylori negative PD controls (n = 30) [55]. H. pylori chronic infection thus may lead to bacterial antigens that trigger an autoimmune response that ultimately signals to the brain immune system; alternatively, antigens may gain direct access to the brain, causing direct neuronal damage [6].
First reported by James Parkinson in the early nineteen century, post-mortem and neuroimaging studies have collectively suggested that chronic neuroinflammation characterized by microglial activation and pro-inflammatory mediators (cytokines and chemokines) is associated with the pathophysiology of PD [56]. Furthermore, microglial activation is always tightly related to alpha-synuclein pathology [57, 58], even in the absence of neurodegeneration. In multiple genetic animal models of PD, early microglial activation has been consistently observed, accompanied by progression of neuronal dysfunction and non-motor symptoms, and always precedes motor deficits and nigra dopaminergic neuron degeneration [59–61]. It has been proposed that systemic inflammation can cause the activation of the local neuroimmune system in the diseased brain. However, its impact on disease progression remains controversial. Recently, there is accumulating evidence that BBB dysfunction is involved in the course of PD [62, 63]. During systemic inflammation, inflammatory cytokines and chemokines are quickly secreted from blood monocytes and macrophages. These proinflammatory cytokines and neurotoxic substances could enter the brain and stimulate microglia to initiate neuroinflammatory reactions. Therefore, it is possible that H. pylori chronic infection may lead to a “leaky” BBB that stimulates microglia activation, contributing to PD disease progression.
The third possibility is that H. pylori occasionally can reside intracellularly [64] and may hitch a ride inside monocytes or other cell types that cross the BBB, directly seeding the bacteria into the brain. However, thus far, no evidence exists that H. pylori is found in the brain, as the bacteria rarely are found in the bloodstream, and only scant evidence links the bacterium with meningitis [42].
The fourth possibility is that H. pylori infection causes dysregulation of the gut microbiota [65], thereby changing the microbiome signature, which in turn affects the gut metabolism, neuroendocrinology, and immune responses [1, 53]. This dysregulated gut microbiome may lead to altered inflammatory mediators that predispose to PD as part of the brain-gut axis interactions [1, 21, 22, 53, 66]. This dysregulation could occur through over-stimulation of innate immunity from gut microbe dysbiosis, changes in blood leukocyte subtype counts, or systemic inflammation mediated by small intestinal bacterial overgrowth, or activation of enteric neurons that contribute to alpha-synuclein aggregation and misfolding [1, 53, 67]. For example, brady/hypokinesia and flexor rigidity are worse the higher the natural killer (NK) cell count. Increased brady/hypokinesia was noted with Helicobacter-positivity, over and above that explained by NK count; tremor is worse with lower neutrophil counts [67].
The gut microbiota are known to affect BBB permeability [68]. A progressive mouse model of PD over-expressing full-length, human wild-type alpha-synuclein under the Thy-1 promoter [69] demonstrated that gut microbiota are required for alpha-synuclein pathology, GI pathology and motor dysfunction, while antibiotic treatment reversed these deficits [70]. Moreover, transplantation of gut flora from PD humans into mice enhances motor deficits not observed when gut flora from healthy controls is transplanted [70]. Others have also suggested gut microbiota dysfunctions in PD patients and a role of the gut microbiota in the brain-gut axis [4, 71–75], but the various studies do not reach a common consensus on what constitutes the PD gut microbiota signature. Many variables, such as diet, smoking, race, gender, and age influence the gut microbiome, complicating interpretation of the data. Additionally, specific gut species other than H. pylori have yet to be rigorously assessed for association with PD.
In a large metagenomic study involving 72 PD patients and 72 healthy controls, clear differences in gut microbiota were identified and associated with PD patients [76]. Indeed Dobbs, et al., found hydrogen-breath-test-positivity in PD to be inversely associated with Helicobacter positivity, and have reported incident overgrowth following H. pylori eradication [39, 67]. Additionally, the gut metabolome in PD patients, featuring changes in metabolites such as short-chain fatty acids, folate and vitamin B12, may alter the gut inflammatory milieu in favor of PD [53]. Taken together, strong data are emerging that the wrong gut microbiota can contribute to neurological disorders such as PD; H. pylori may be a major contributor of gut dysbiosis, but more evidence is needed.
CONCLUSION
Evidence for a strong association among H. pylori chronic infection, peptic ulceration and exacerabation of PD symptoms is accumulating. However, the hypotheses that H. pylori infection is a predisposing factor, disease progression modifier, or even a direct cause of PD remain largely unexplored. The key double-blind study of H. pylori eradication [39] does provide Grade 1b evidence of a causative link. Additional experimentation is clearly warranted to address these possibilities. H. pylori is a highly variable organism with numerous genotypic differences, and it is likely that only certain genotypes are associated with PD in humans, but this area remains almost entirely unexplored. Key experiments need to be conducted to examine: 1) the interaction of H. pylori with human neurons; 2) the role of H. pylori toxins such as VacA, CagA and cholesteryl glucosides in PD readouts in mice and human neurons; 3) the mechanisms underlying the interaction of H. pylori with L-dopa; 4) how the gut and systemic inflammatory responses to H. pylori infection contribute to PD in neurons and mouse models; 5) how H. pylori affects motor functions in established mouse models of PD; 6) the role of H. pylori in altering the gut microbiota towards a PD-like microbiota; and 7) the role of H. pylori in human or mouse parkinsonian motor functions, L-dopa absorption and reversal upon H. pylori eradication.
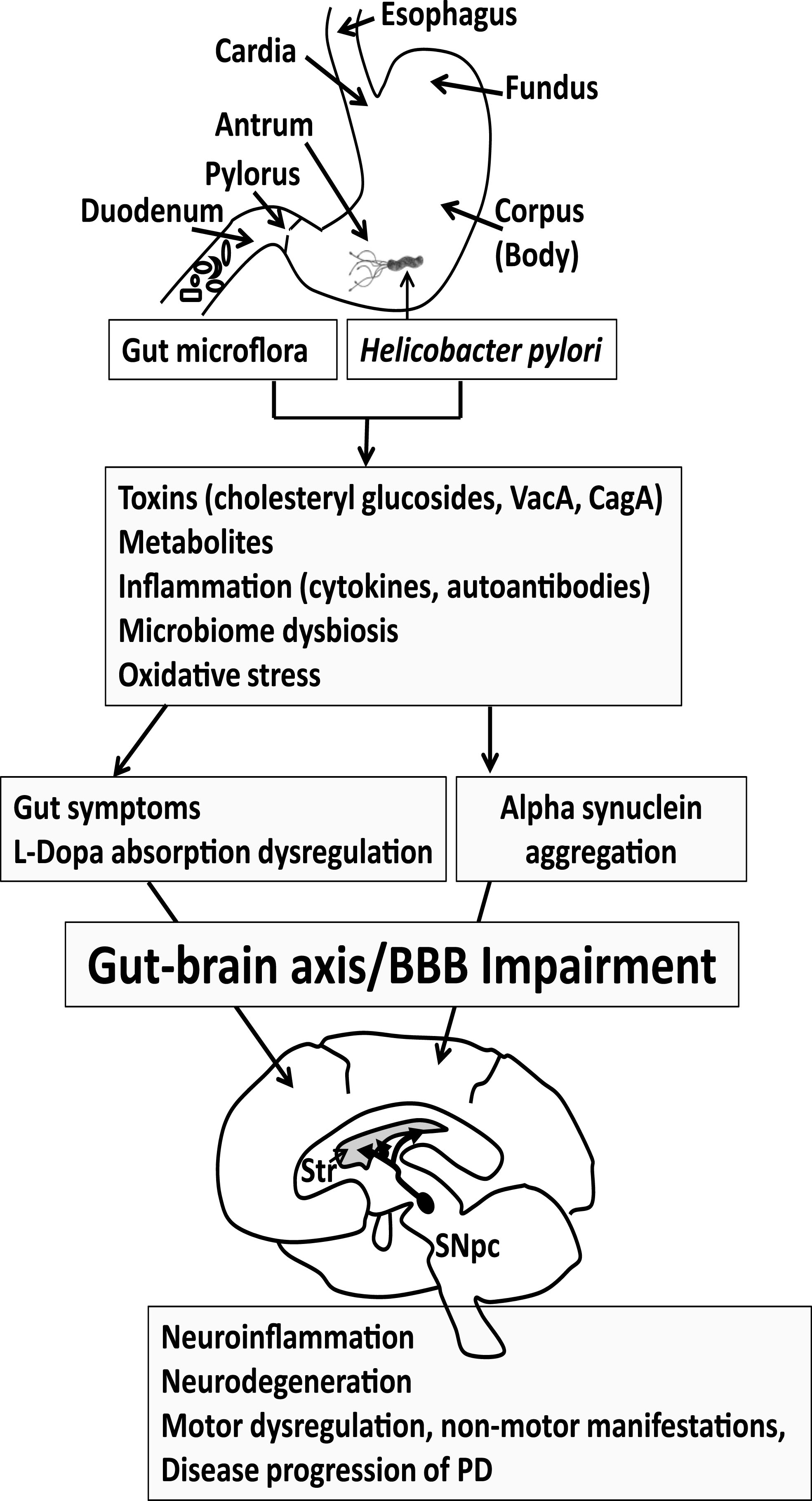
The gut is being increasingly considered as a critical focal point in the pathology of PD. This gut pathology may be multifactorial, involving H. pylori, intestinal microflora, inflammation, misfolding of alpha-synuclein in the gut and brain, cholesterol and other metabolites, and potential neurotoxins from bacteria or dietary sources (Fig. 1). Eradication of H. pylori or return of the gut microflora to the proper balance in PD patients may ameliorate gut symptoms, L-dopa absorption and motor functions.
Fig.1
Progression of Parkinson’s disease (PD). Gut microflora and Helicobacter pylori may produce toxins and metabolites and trigger an immune response that features cytokines and autoantibodies. The gut microbiome may become dysregulated. Reactive oxygen species may also contribute to the pathogenesis of PD. Collectively this may lead to GI symptoms and altered L-dopa absorption in PD patients taking L-dopa medication, reducing effectiveness of the medication. The GI symptoms and inflammatory process may become systemic to compromise the BBB, eliciting neuroinflammation in the brain. Alpha synuclein aggregation, which is observed both in the gut and brain, leads to the spreading of neuropathology. The loss of dopaminergic neurons in the substantial nigra pars compacta (SNpc) manifests as motor symptoms observed in PD patients. Thus, eradication of H. pylori or return of the gut microflora to the proper balance may ameliorate gut symptoms, L-dopa absorption and motor functions in PD patients, which could delay PD disease progression. Str, Striatum.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Mulak A , Bonaz B ((2015) ) Brain-gut-microbiota axis in Parkinson’s disease. World J Gastroenterol 21: , 10609–10620. |
[2] | Poirier AA , Aube B , Cote M , Morin N , Di Paolo T , Soulet D ((2016) ) Gastrointestinal dysfunctions in Parkinson’s disease: Symptoms and treatments. Parkinsons Dis, 6762–528. |
[3] | Cersosimo MG , Raina GB , Pecci C , Pellene A , Calandra CR , Gutierrez C , Micheli FE , Benarroch EE (2013) Gastrointestinal manifestations in Parkinson’s disease: Prevalence and occurrence before motor symptoms. J Neurol 260: , 1332–1338. |
[4] | Nair AT , Ramachandran V , Joghee NM , Antony S , Rama-lingam G ((2018) ) Gut microbiota dysfunction as reliable non-invasive early diagnostic biomarkers in the patho-physiology of Parkinson’s disease: A critical review. J Neurogastroenterol Motil 24: , 30–42. |
[5] | Holmqvist S , Chutna O , Bousset L , Aldrin- Kirk P , Li W , Bjorklund T , Wang ZY , Roybon L , Melki R , Li JY ((2014) ) Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol 1: 805–820. |
[6] | Dobbs SM , Dobbs RJ , Weller C , Charlett A Link between Helicobacter pylori infection and idi parkinsonism. Med Hypotheses 55: , 93–98. |
[7] | Punsoni M , Friedman JH , Resnick M , Donahu DF , Stopa EG ((2017) ) Enteric pathologic manii alpha-synucleinopathies. Appl Immunohistoc phol. doi: 10.1097/PAI.0000000000000613 |
[8] | Altschuler E ((1996) ) Gastric Helicobacter as a cause of idiopathic Parkinson dise anterior optic ischemic neuropathy 413–414. |
[9] | Dobbs RJ , Dobbs SM , Weller C , Charlett A , Bjarnason IT , Curry A , Ellis DS , Ibrahim MA , McCrossan MV , O’Donohue J , Owen RJ , Oxlade NL , Price AB , Sanderson JD , Sudhanva M , Williams J ((2008) ) Helicobacter hypothesis for idiopathic parkinsonism: Before and beyond. Helicobacter 13: , 309–322. |
[10] | Testerman TL , Morris J ((2014) ) Beyond the stomach: An updated view of Helicobacter pylori pathogenesis, diagnosis, and treatment. World J Gastroenterol 20: , 12781–12808. |
[11] | Warren JR , Marshall B ((1983) ) Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1: , 1273–1275. |
[12] | Schwab RS ((1961) ) Symptomatology and medical treatment of Parkinson’s disease. Int J Neurol 2: , 61–75. |
[13] | Lagoo J , Pappas TN , Perez A ((2014) ) A relic or still relevant: The narrowing role for vagotomy in the treatment of peptic ulcer disease. Am J Surg 207: , 120–126. |
[14] | Liu B , Fang F , Pedersen NL , Tillander A , Ludvigsson JF , Ekbom A , Svenningsson P , Chen H , Wirdefeldt K ((2017) ) Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study. Neurology 88: , 1996–2002. |
[15] | Svensson E , Horvath-Puho E , Thomsen RW , Djurhuus JC , Pedersen L , Borghammer P , Sorensen HT ((2015) ) Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol 78: , 522–529. |
[16] | Strang RR ((1965) ) The association of gastro-duodenal ulceration and Parkinson’s disease. MedJAust 1: , 842–843. |
[17] | Charlett A , Dobbs RJ , Dobbs SM , Weller C , Brady P , Peterson DW ((1999) ) Parkinsonism: Siblings share Helicobacter pylori seropositivity and facets of syndrome. Acta Neurol Scand 99: , 26–35. |
[18] | Dobbs RJ , Charlett A , Dobbs SM , Weller C , Peterson DW ((2000) ) Parkinsonism: Differential age-trend in Helicobacter pylori antibody. Aliment Pharmacol Ther 14: , 1199–1205. |
[19] | Shen X , Yang H , Wu Y , Zhang D , Jiang H ((2017) ) Meta-analysis: Association of Helicobacter pylori infection with Parkinson’s diseases. Helicobacter 22: , hel.12398. |
[20] | Huang HK , Wang JH , Lei WY , Chen CL , Chang CY , Liou LS ((2018) ) Helicobacter pylori infection is associated with an increased risk of Parkinson’s disease: A population-based retrospective cohort study. Parkinsonism Relat Disord 47: , 26–31. |
[21] | Weller C , Charlett A , Oxlade NL , Dobbs SM , Dobbs RJ , Peterson DW , Bjarnason IT ((2005) ) Role of chronic infection and inflammation in the gastrointestinal tract in the etiology and pathogenesis of idiopathic parkinsonism. Part 3: Predicted probability and gradients of severity of idio-pathic parkinsonism based on H. pylori antibody profile. Helicobacter 10: , 288–297. |
[22] | Weller C , Oxlade N , Dobbs SM , Dobbs RJ , Charlett A , Bjar-nason IT ((2005) ) Role of inflammation in gastrointestinal tract in aetiology and pathogenesis of idiopathic parkinson-ism. FEMS Immunol Med Microbiol 44: , 129–135. |
[23] | Nielsen HH , Qiu J , Friis S , Wermuth L , Ritz B ((2012) ) Treatment for Helicobacter pylori infection and risk of Parkinson’s disease in Denmark. Eur J Neurol 19: , 864–869. |
[24] | Camci G , Oguz S ((2016) ) Association between Parkinson’s Disease and Helicobacter pylori. J Clin Neurol 12: ,147–150. |
[25] | Glavin GB ((1991) ) Dopamine and gastroprotection. The brain-gut axis. Dig Dis Sci 36: , 1670–1672. |
[26] | Cross JM , Mercer DW , Gunter J , Miller TA ((1997) ) Effects of dopamine and alpha-2 adrenoreceptor blockade on L-dopa and cholecystokinin-induced gastroprotection. J Gastroin-test Surg 1: , 257–265; discussion 265. |
[27] | Szabo S ((1986) ) Experimental basis for a role for sulfhydryls and dopamine in ulcerogenesis: A primer for cytoprotection-organoprotection. Klin Wochenschr 64: (Suppl 7), 116–122. |
[28] | Lee WY , Yoon WT , Shin HY , Jeon SH , Rhee PL ((2008) ) Heli-cobacter pylori infection and motor fluctuations in patients with Parkinson’s disease. Mov Disord 23: , 1696–1700. |
[29] | Mridula KR , Borgohain R , Reddy VC , Bandaru VCS , Suryaprabha T ((2017) ) Association of Helicobacter pylori with Parkinson’s Disease. J Clin Neurol 13: , 181–186. |
[30] | Pierantozzi M , Pietroiusti A , Galante A , Sancesario G , Lunardi G , Fedele E , Giacomini P , Stanzione P ((2001) ) Helicobacter pylori-induced reduction of acute levodopa absorption in Parkinson’s disease patients. Ann Neurol 50: , 686–687. |
[31] | Lahner E , Annibale B , Delle Fave G ((2009) ) Systematic review: Helicobacter pylori infection and impaired drug absorption. Aliment Pharmacol Ther 29: , 379–386. |
[32] | Niehues M , Hensel A ((2009) ) In-vitro interaction of L-dopa with bacterial adhesins of Helicobacter pylori: An explanation for clinical differences in bioavailability? J Pharm Pharmacol 61: , 1303–1307. |
[33] | Pierantozzi M , Pietroiusti A , Brusa L , Galati S , Stefani A , Lunardi G , Fedele E , Sancesario G , Bernardi G , Bergamaschi A , Magrini A , Stanzione P , Galante A ((2006) ) Helicobacter pylori eradication and l-dopa absorption in patients with PD and motor fluctuations. Neurology 66: , 1824–1829. |
[34] | Lyte M ((2010) ) Microbial endocrinology as a basis for improved L-DOPA bioavailability in Parkinson’s patients treated for Helicobacter pylori. Med Hypotheses 74: , 895–897. |
[35] | Narozanska E , Bialecka M , Adamiak-Giera U , Gawronska-Szklarz B , Soltan W , Schinwelski M , Robowski P , Madalinski MH , Slawek J ((2014) ) Pharmacokinetics of levodopa in patients with Parkinson disease and motor fluctuations depending on the presence of Helicobacter pylori infection. Clin Neuropharmacol 37: , 96–99. |
[36] | Tan AH , Mahadeva S , Marras C , Thalha AM , Kiew CK , Yeat CM , Ng SW , Ang SP , Chow SK , Loke MF , Vadivelu JS , Ibrahim N , Yong HS , Tan CT , Fox SH , Lang AE , Lim SY ((2015) ) Helicobacter pylori infection is associated with worse severity of Parkinson’s disease. Parkinsonism Relat Disord 21: , 221–225. |
[37] | Hashim H , Azmin S , Razlan H , Yahya NW , Tan HJ , Manaf MR , Ibrahim NM ((2014) ) Eradication of Helicobacterpylori infection improves levodopa action, clinical symptoms and quality of life in patients with Parkinson’s disease. PLoS One 9: , e112330. |
[38] | Dobbs SM , Charlett A , Dobbs RJ , Weller C , Iguodala O , Smee C , Lawson AJ , Taylor D , Bjarnason I ((2013) ) Antimicrobial surveillance in idiopathic parkinsonism: Indication-specific improvement in hypokinesia following Helicobacter pylori eradication and non-specific effec antimicrobials for other indications in worsening ri Helicobacter 18: , 187–196. |
[39] | Dobbs SM , Dobbs RJ , Weller C , Charlett A , Bjar Lawson AJ , Letley D , Harbin L , Price AB , Ib Oxlade NL , Bowthorpe J , Leckstroem D , Smee C , Plant JM , Peterson DW ((2010) ) Differential effect of Helicobacter pylori eradication on time-trends in brady/hypokinesia and rigidity in idiopathic parkinsonism. Helicobacter 15: , 279–294. |
[40] | Rahne KE , Tagesson C , Nyholm D ((2013) ) Motor fluctuations and Helicobacter pylori in Parkinson’s disease. J Neurol 260: , 2974–2980. |
[41] | Rees K , Stowe R , Patel S , Ives N , Breen K , Clarke CE , Ben-Shlomo Y ((2011) ) Helicobacter pylori eradication for Parkinson’s disease. Cochrane Database of Syst Rev, CD008453. |
[42] | Attallah AM , Toson EA , Ibrahim GG , Bakr NE ((2007) ) Detection of a Helicobacter pylori antigen in cere-brospinal fluid of patients with meningitis. J Immunoassay Immunochem 28: , 25–33. |
[43] | Wunder C , Churin Y , Winau F , Warnecke D , Vieth M , Lindner B , Zahringer U , Mollenkopf HJ , Heinz E , Meyer TF ((2006) ) Cholesterol glucosylation promotes immune evasion by Helicobacterpylori. Nat Med 12: , 1030–1038. |
[44] | Schulz JD , Hawkes EL , Shaw CA ((2006) ) Cycad toxins, Helicobacter pylori and parkinsonism: Cholesterol gluco-sides as the common denomenator. Med Hypotheses 66: , 1222–1226. |
[45] | Shen WB , McDowell KA , Siebert AA , Clark SM , Dugger NV , Valentino KM , Jinnah HA , Sztalryd C , Fishman PS , Shaw CA , Jafri MS , Yarowsky PJ ((2010) ) Environmental neurotoxin-induced progressive model of parkinsonism in rats. Ann Neurol 68: , 70–80. |
[46] | Hirai Y , Haque M , Yoshida T , Yokota K , Yasuda T , Oguma K ((1995) ) Unique cholesteryl glucosides in Helicobacter pylori: Composition and structural analysis. JBacteriol 177: , 5327–5333. |
[47] | Hildebrandt E , McGee DJ ((2009) ) Helicobacter pylori lipopolysaccharide modification, Lewis antigen expression, and gastric colonization are cholesterol-dependent. BMC Microbiol 9: , 258. |
[48] | Bjorkhem I , Lovgren-Sandblom A , Leoni V , Meaney S , Brodin L , Salveson L , Winge K , Palhagen S , Svennings-son P ((2013) ) Oxysterols and Parkinson’s disease: Evidence that levels of 24S-hydroxycholesterol in cerebrospinal fluid correlates with the duration of the disease. Neurosci Let 555: , 102–105. |
[49] | Ly PT , Pelech S , Shaw CA ((2008) ) Cholesteryl gluco-side stimulates activation of protein kinase B/Akt in the motor neuron-derived NSC34 cell line. Neurobiol Lipids 7: , 620081. |
[50] | Panov A , Kubalik N , Brooks BR , Shaw CA (2010) In vitro effects of cholesterol ß-D-glucoside, cholesterol and cycad phytosterol glucosides on respiration and reactive oxygen species generation in brain mitochondria. J MembrBiol 237: , 71–77. |
[51] | habazian I , Bains JS , Williams DE , Cheung J , Wilson JM , Pasqualotto BA , Pelech SL , Andersen RJ , Wang YT , Liu L , Nagai A , Kim SU , Craig UK , Shaw CA ((2002) ) Isolation of various forms of sterol beta-D-glucoside from the seed of Cycas circinalis: Neurotoxicity and implications for ALS-parkinsonism dementia complex. J Neurochem 82: , 516–528. |
[52] | Hawkes CH , Del Tredici K , Braak H ((2007) ) Parkinson’s disease: A dual-hit hypothesis. Neuropathol Appl Neurobiol 33: , 599–614. |
[53] | Dobbs SM , Dobbs RJ , Weller C , Charlett A , Augustin A , Taylor D , Ibrahim MA , Bjarnason I ((2016) ) Peripheral aetiopathogenic drivers and mediators of Parkinson’s disease and co-morbidities: Role of gastrointestinal micro-biota. J Neurovirol 22: , 22–32. |
[54] | Appelmelk BJ , Negrini R , Moran AP , Kuipers EJ ((1997) ) Molecular mimicry between Helicobacter pylori and the host. Trends Microbiol 5: , 70–73. |
[55] | Suwarnalata G , Tan AH , Isa H , Gudimella R , Anwar A , Loke MF , Mahadeva S , Lim SY , Vadivelu J ((2016) ) Augmentation of autoantibodies by Helicobacter pylori in Parkinson’s disease patients may be linked to greater severity. PLoS One 11: , e0153725. |
[56] | Sanchez-Guajardo V , Annibali A , Jensen PH , Romero-Ramos M ((2013) ) alpha-Synuclein vaccination prevents the accumulation of parkinson disease-like pathologic inclusions in striatum in association with regulatory T cell recruitment in a rat model. J Neuropathol Exp Neurol 72: , 624–645. |
[57] | Gomez-Isla T , Irizarry MC , Mariash A , Cheung B , Soto O , Schrump S , Sondel J , Kotilinek L , Day J , Schwarzschild MA , Cha JH , Newell K , Miller DW , Ueda K , Young AB , Hyman BT , Ashe KH ((2003) ) Motor dysfunction and gliosis with preserved dopaminergic markers in human alpha-synuclein A30Ptransgenic mice. Neurobiol Aging 24: , 245–258. |
[58] | Tofaris GK , Garcia Reitbock P , Humby T , Lambourne SL , O’Connell M , Ghetti B , Gossage H , Emson PC , Wilkinson LS , Goedert M , Spillantini MG ((2006) ) Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): Implications for Lewy body disorders. J Neurosci 26: , 3942–3950. |
[59] | Ginns EI , Mak SK , Ko N , Karlgren J , Akbarian S , Chou VP , Guo Y , Lim A , Samuelsson S , LaMarca ML , Vazquez-DeRose J , Manning-Bog AB ((2014) ) Neuroinflammation and alpha-synuclein accumulation in response to glu-cocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol Genet Metab 111: , 152–162. |
[60] | L’Episcopo F , Tirolo C , Caniglia S , Testa N , Serra PA , Impagnatiello F , Morale MC , Marchetti B ((2010) ) Combining nitric oxide release with anti-inflammatory activity preserves nigrostriatal dopaminergic innervation and prevents motor impairment in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson’s disease. J Neuroinflammation 7: , 83. |
[61] | Watson MB , Richter F , Lee SK , Gabby L , Wu J , Masliah E , Effros RB , Chesselet MF ((2012) ) Regionally-specific microglial activation in young mice over-expressing human wildtype alpha-synuclein. Exp Neurol 237: , 318–334. |
[62] | Gray MT , Woulfe JM ((2015) ) Striatal blood-brain barrier permeability in Parkinson’s disease. J Cereb Blood Flow Metab 35: , 747–750. |
[63] | Kortekaas R , Leenders KL , van Oostrom JC , Vaalburg W , Bart J , Willemsen AT , Hendrikse NH ((2005) ) Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol 57: , 176–179. |
[64] | Necchi V , Candusso ME , Tava F , Luinetti O , Ventura U , Fiocca R , Ricci V , Solcia E ((2007) ) Intracellular, intercellular, and stromal invasion of gastric mucosa, preneoplastic lesions, and cancer by Helicobacter pylori. Gastroenterol 132: , 1009–1023. |
[65] | Noto JM , Peek RM ((2017) ) The gastric microbiome, its int action with Helicobacter pylori, and its potential role in progression to stomach cancer. PLoS Pathog 13: , e1006573 |
[66] | Fasano A , Bove F , Gabrielli M , Petracca M , Zocco Ragazzoni E , Barbaro F , Piano C , Fortuna S , Tortora A Di Giacopo R , Campanale M , Gigante G , Lauritano EC , Navarra P , Marconi S , Gasbarrini A , Bentivoglio AR ((2013) ) The role of small intestinal bacterial overgrowth in Parkinson’s disease. Mov Disord 28: , 1241–1249. |
[67] | Dobbs RJ , Charlett A , Dobbs SM , Weller C , Ibrahim MAA , Iguodala O , Smee C , Plant JM , Lawson AJ , Taylor D , Bjarnason I ((2012) ) Leukocyte-subset counts in idiopathic parkinsonism provide clues to a pathogenic pathway involving small intestinal bacterial overgrowth. A surveillance study. Gut Pathog 4: , 12. |
[68] | Braniste V , Al-Asmakh M , Kowal C , Anuar F , Abbaspour A , Toth M , Korecka A , Bakocevic N , Ng LG , Kundu P , Gulyas B , Halldin C , Hultenby K , Nilsson H , Hebert H , Volpe BT , Diamond B , Pettersson S ((2014) ) The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med 6: , 263ra158. |
[69] | Chesselet MF , Richter F , Zhu C , Magen I , Watson MB , Subramaniam SR ((2012) ) A progressive mouse model of Parkinson’s disease: The Thy1-aSyn ("Line 61") mice. Neu-rotherapeutics 9: , 297–314. |
[70] | Sampson TR , Debelius JW , Thron T , Janssen S , Shastri GG , Ilhan ZE , Challis C , Schretter CE , Rocha S , Gradinaru V , Chesselet MF , Keshavarzian A , Shannon KM , Krajmalnik-Brown R , Wittung-Stafshede P , Knight R , Mazmanian SK ((2016) ) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167: , 1469–1480. |
[71] | Collins SM , Surette M , Bercik P ((2012) ) The interplay between the intestinal microbiota and the brain. Nat Rev Microbiol 10: , 735–742. |
[72] | Cryan JF , Dinan TG ((2012) ) Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci 13: , 701–712. |
[73] | Mayer EA , Tillisch K , Gupta A ((2015) ) Gut/brain axis and the microbiota. J Clin Invest 125: , 926–938. |
[74] | Sampson TR , Mazmanian SK ((2015) ) Control of brain development, function, and behavior by the microbiome. Cell Host Microbe 17: , 565–576. |
[75] | Unger MM , Spiegel J , Dillmann KU , Grundmann D , Philippeit H , Burmann J , Fasbender K , Schwiertz A , Schafer KH ((2016) ) Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord 32: , 66–72. |
[76] | Scheperjans F , Aho V , Pereira PA , Koskinen K , Paulin L , Pekkonen E , Haapaniemi E , Kaakkola S , Eerola-Rautio J , Pohja M , Kinnunen E , Murros K , Auvinen P ((2015) ) Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord 30: , 350–358. |


