The Synucleinopathies: Twenty Years On
Abstract
In 2017, it is two hundred years since James Parkinson provided the first complete clinical description of the disease named after him, fifty years since the introduction of high-dose D,L-DOPA treatment and twenty years since α-synuclein aggregation came to the fore. In 1998, multiple system atrophy joined Parkinson’s disease and dementia with Lewy bodies as the third major synucleinopathy. Here we review our work, which led to the identification of α-synuclein in Lewy bodies, Lewy neurites and Papp-Lantos bodies, as well as what has happened since. Some of the experiments described were carried out in collaboration with ML Schmidt, VMY Lee and JQ Trojanowski.
INTRODUCTION
Parkinson’s disease (PD) has a long and rich history. Although partial descriptions can be found in some ancient texts, the first complete clinical description of the disease was provided by James Parkinson in 1817 [1]. He captured the disease’s distinctive motor symptoms: “involuntary tremulous motion, with lessened muscular power, in parts not in action and even when supported; with a propensity to bend the trunk forward, and to pass from a walking to a running pace: the senses and intellect being uninjured.” Jean-Martin Charcot named the shaking palsy (or “paralysie agitante”) after Parkinson [2].
Close to one hundred years passed, before severe nerve cell loss in the pigmented pars compacta of the substantia nigra was identified as the most consistent pathological lesion of PD, together with the presence there and in other brainstem regions of what Konstantin Tretiakoff called “corps de Lewy” or Lewy bodies [3]. Friedrich Lewy first described these inclusions in the dorsal motor nucleus of the vagus nerve, the nucleus basalis of Meynert and some thalamic nuclei of PD patients [4]. Rolf Hassler showed that nerve cell loss in PD is most severe in the ventrolateral nerve cell group of the substantia nigra, with relative sparing of the more heavily pigmented dorsomedial group [5]. In the 1960 s, electron microscopy was used to show that the Lewy pathology is made of abnormal filaments [6].
The discovery of dopamine as a brain neurotransmitter [7, 8] and the finding that 80% of it is found in the striatum, unaccompanied by noradrenaline [9, 10], together with experiments showing that reserpine depletes brain dopamine and that levodopa increases dopamine levels [8], led Arvid Carlsson to propose that dopamine deficiency might underlie the motor symptoms of PD [11, 12]. Striatal dopamine deficiency was subsequently documented by Herbert Ehringer and Oleh Hornykiewicz in six cases of parkinsonism, four with post-encephalitic parkinsonism and two with PD [13]; the loss was greatest in post-encephalitic parkinsonism. Dopamine was also found to be reduced in the substantia nigra [14]. With the advent of the Falck-Hillarp fluorescence histochemical technique [15], dopamine-containing nerve cell bodies were found in the pars compacta of the substantia nigra, from where their nerve terminals ascend to the striatum [16]. Experiments in monkeys showed that lesioning the substantia nigra was followed by a loss of dopamine in the striatum [17].
This led to attempts at replacing the missing dopamine. Catecholamines are unable to cross the blood-brain barrier. The dopamine precursor 3,4-dihydroxyphenylalanine (DOPA), which crosses the blood-brain barrier and is decarboxylated to dopamine by aromatic amino acid decarboxylase, was therefore administered [18–20]. In the end, it took 150 years from Parkinson’s description of the disease to the development of what has been described as “the second coming” [21]: high-dose oral levodopa treatment [23–25]. In the 1967 paper [23], D-L-DOPA was used; however, since the D-isomer is not converted to dopamine in vivo, the 1969 studies [24] used the L-isomer of DOPA. In 1970, L-DOPA was approved by the US Food and Drug Administration as a treatment for PD.
Cotzias and colleagues believed that PD was caused by a reduction in neuromelanin; they administered high-dose levodopa in an attempt to re-pigment the substantia nigra, not to replace the depleted dopamine [25]. As a result, the symptoms of PD were much improved. Theirs seems to have been a case of obtaining the right result for the wrong reasons. There are currently no mechanism-based therapies for PD. The miraculous effects of levodopa are described in “Awakenings”, Oliver Sacks’ account of the treatment of patients with post-encephalitic parkinsonism [26]. His book inspired Harold Pinter’s play “A Kind of Alaska” [27] and the film “Awakenings” directed by Penny Marshall [28].
Despite these advances, it remained unclear what caused PD. Encouraged by William Langston’s report of sudden-onset parkinsonism in multiple patients following the injection of 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP) [29, 30], environmental factors were sought, and PD was mostly considered to be a sporadic disorder. David Marsden described PD as a Lewy body disease and wrote: “The crux of Parkinson’s disease is the Lewy body, but the origin of this inclusion is a mystery” [31]. Although a large number of proteins had been reported in Lewy bodies and Lewy neurites by immunohistochemistry, this work did not allow one to distinguish between integral components of the Lewy pathology and cellular constituents that get trapped in the abnormal filaments.
It was believed that it was possible to use biochemistry, as well as structural and molecular biology to unravel the composition of the inclusions that characterise the most common neurodegenerative diseases, including Alzheimer’s disease (AD) and PD, and that this would throw light on their aetiology and pathogenesis. As it happened, this was true of AD. In order to deconstruct the Lewy pathology of PD, genetics had to join biochemistry, structural biology and molecular biology.
HUMAN α-SYNUCLEIN AND β-SYNUCLEIN
Our findings on synucleinopathies grew out of work on tau, which we showed to be an integral component of the paired helical filaments of AD [32–34]. In the early 1990s, Ross Jakes, Maria Grazia Spillantini and Michel Goedert (Fig. 1), then at the MRC Laboratory of Molecular Biology in Cambridge, noticed that the partially characterized anti-tau antibody 11.57 [35] also labelled two proteins from a cytosolic extract of normal adult human brain, each with an apparent molecular mass of 19 kDa. Partial purification took advantage of the heat stability of both proteins and their separation by ion exchange chromatography. Complete purification required an additional HPLC step. The amino-termini were not amenable to Edman degradation (acetylated, formylated?). Analysis of peptides purified after cyanogen bromide cleavage allowed us to sequence up to 55 continuous residues by automated amino acid sequence analysis. From these successful results, we initially named the two proteins “Perfectin” (α-synuclein) and “Imperfectin” (β-synuclein). Polymerase chain reaction (PCR) products were then used to isolate full-length cDNA clones from a library of adult human hippocampus. The predicted proteins consisted of 140 (Perfectin) and 134 (Imperfectin) amino acids [36].
At the “Second International Symposium on Dementia” in Oiso (Japan) on November 16–19, 1993, we heard a talk from Tsunao Saitoh and realized that his precursor of the non-Aβ component of AD amyloid (NACP) was the same as our 140 amino acid protein. The paper describing Saitoh’s findings was published one month later [37]. The authors concluded that residues 61–95 of NACP (the peptide NAC) were an integral component of the Aβ plaques of AD. However, subsequent work using additional antibodies failed to confirm these findings [38, 39]. It is now clear that NAC/NACP is not an integral component of Aβ plaques.
In addition to its identity with NACP, we realized that the 140 amino acid protein was homologous to synucleins from the Pacific electric ray (Torpedo californica) and rat brain [40, 41], identifying it as a human synuclein. We also realized that the 134 amino acid protein from human brain was homologous to bovine phosphoneuroprotein-14. The latter had been identified as a new brain-specific protein that was present in synapses, but not glial cells [42, 43].
The previously unknown similarities between these proteins established the existence of a family of human brain synucleins. We named the 140 and 134 amino acid proteins α-synuclein and β-synuclein, respectively (the 140 amino acid protein was named α-synuclein, because it eluted before the 134 amino acid protein from a cation exchange HPLC column) [36]. By sequence alignment, β-synuclein was 61% identical to α-synuclein (Fig. 2). The 127 amino acid γ-synuclein was subsequently identified [44, 45]. Synucleins have a highly conserved amino-terminal repeat region, a hydrophobic middle region and a less well-conserved negatively charged carboxy-terminal region. An early study reported that NACP/α-synuclein is a natively unfolded protein [46].
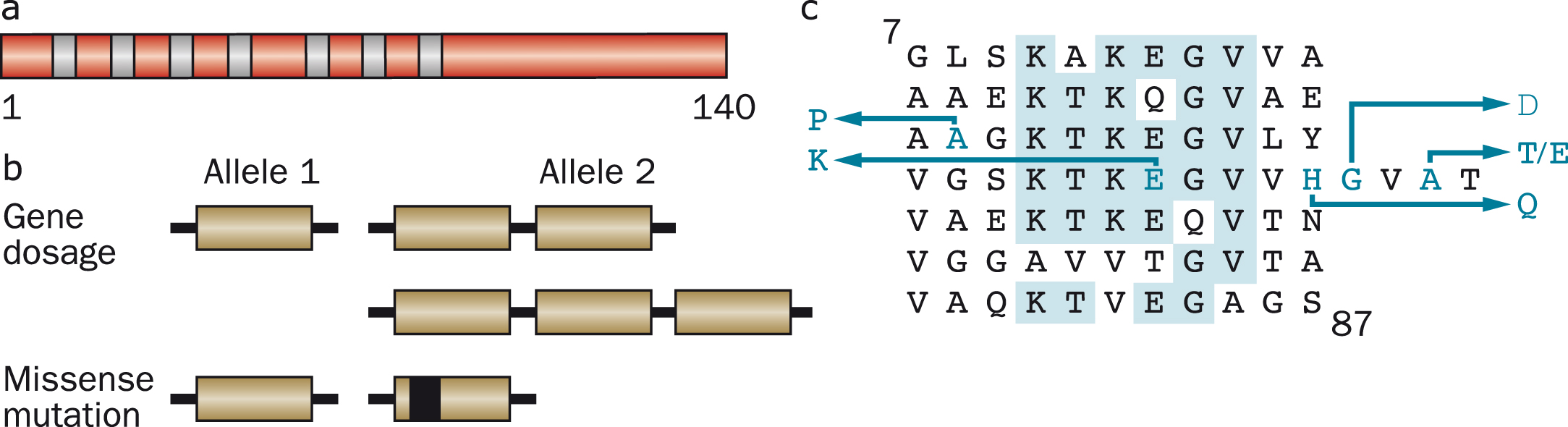
Over half the α-synuclein sequence is taken up by seven imperfect repeats of eleven amino acids, each having a conserved six amino acid core with the consensus sequence KTKEGV (residues 7–87). Core sequences of individual repeats are separated by five amino acids, with the exception of repeats four and five, which are separated by nine amino acids. We produced antibodies [PER (for Perfectin) 1–3] that were specific for either α- or β-synuclein and showed that both proteins are predominantly expressed in brain, where they are concentrated in nerve terminals. For historical reasons, we decided to keep the name “synuclein” [36].
We isolated genomic clones and analysed a panel of monochromosomal human-rodent somatic cell hybrids by PCR, followed by fluorescence in situ hybridization of metaphase spreads of human chromosomes. We found that SNCA (the α-synuclein gene) and SNCB (the β-synuclein gene) are encoded by genes that are located on the long arms of chromosomes 4 (region q21) and 5 (region q35), respectively [47]. The chromosomal location of SNCA to 4q21.3-q22 was also reported independently [48]. The γ-synuclein gene (SNCG) maps to the long arm of chromosome 10 (region q21) [49, 50].
α-SYNUCLEIN AND LEWY PATHOLOGY
Although much research on the pathogenesis of PD was focused on the search for environmental risk factors, inherited cases of PD were known. In 1990, an Italian-American family with early-onset PD and autopsy-confirmed Lewy pathology (the Contursi kindred) was described [51]. In 1996, Robert Nussbaum and colleagues identified genetic markers on the long arm of chromosome 4 (region q21-q23) that segregated with disease in this family [52]. This is the region that SNCA maps to.
In June 1997, Mihael Polymeropoulos and colleagues identified SNCA as the gene that is mutated in the Contursi kindred [53]. The mutation segregated with disease in the affected family and was identified as an alanine-to-threonine substitution at position 53 of the 140 amino acid protein. It was found not only in the Italian-American kindred, but also in three apparently unrelated Greek families with inherited PD. The pattern of inheritance was confirmed as autosomal-dominant. Surprisingly, A53 is only found in α-synuclein from humans and Old World primates, with T53 being present in rodents and several other mammalian species [54]. However, six additional amino acids differ between mouse and human α-synucleins.
In August 1997, we showed that the Lewy bodies and Lewy neurites from the substantia nigra of six patients with idiopathic PD (Fig. 3) and four with dementia with Lewy bodies (DLB) were strongly immunoreactive for α-synuclein [55]. The same was true of the Lewy pathology from the cingulate cortex of DLB. Two different anti-α-synuclein antibodies (PER1 and PER2), which are specific for the amino- and carboxy-termini, respectively, of recombinantly expressed α-synuclein, stained the inclusions equally well, consistent with the presence of the whole molecule. An antibody specific for β-synuclein (PER3) failed to stain the inclusions of PD and DLB. These findings showed that brainstem-type and cortical Lewy pathologies were immunoreactive for α-synuclein. We concluded that both PD and DLB could be α-synuclein diseases. This work was a collaboration with Marie-Luise Schmidt, Virginia Lee and John Trojanowski. Abundant α-synuclein inclusions are also characteristic of the diseases caused by SNCA mutations [56, 57].
In May 1998, we reported that Lewy neurites were more abundant in PD and DLB than hitherto believed (Fig. 4) [58]. The staining of intraneuritic Lewy bodies helped to reinforce the view that Lewy pathology is unlikely to be benign. Prior to this, ubiquitin staining had been the most sensitive means of detecting Lewy pathology [59], but it lacked in specificity. We showed that staining for α-synuclein was more extensive than staining for ubiquitin, indicating that accumulation of α-synuclein precedes ubiquitination. Similar findings have subsequently been reported [60, 61]. Together, they suggest that ubiquitination of α-synuclein occurs after fibril formation and may be an attempt by cells to promote clearance of aggregated α-synuclein by targeting it for degradation by the proteasome.
We also confirmed that β-synuclein did not accumulate in the Lewy pathology and showed that the same was true of γ-synuclein. Of the three mammalian synucleins, only α-synuclein is found in the Lewy pathology. These findings suggested, but did not prove, that α-synuclein is a major component of the abnormal filaments that make up Lewy bodies and Lewy neurites.
In collaboration with Tony Crowther and Masato Hasegawa, we studied sarkosyl-insoluble filaments extracted from cingulate cortex of patients with DLB by immunoelectron microscopy [58]. Antibody PER4, which recognizes the carboxy-terminal region of α-synuclein, labelled filaments with a diameter of 5–10 nm and a length of 200–600 nm, indicating that they contained α-synuclein as a major component (Fig. 3). Antibody PER1, which was raised against residues 11–34 of α-synuclein, only labelled one end of each filament (Fig. 3). This suggested that the PER1 epitope is buried in the body of the filament and that the filaments are polar structures. We subsequently reported similar findings on filaments extracted from the substantia nigra of PD patients [62]. Some of the above findings were quickly confirmed [63–67]. Staining for α-synuclein is now used routinely for identifying Lewy bodies and Lewy neurites in tissue sections and PD and DLB.
α-SYNUCLEIN AND PAPP-LANTOS BODIES
Multiple system atrophy (MSA) is a neurodegenerative disease characterized by a combination of autonomic, cerebellar, parkinsonian and pyramidal symptoms [68, 69]. Depending on the motor phenotype, it is divided into parkinsonian (MSA-P) and cerebellar (MSA-C) variants. MSA-P is only poorly L-DOPA-responsive. MSA comprises what used to be called olivopontocerebellar atrophy, striatonigral degeneration and Shy-Drager syndrome. In most countries, MSA-P is more common than MSA-C. Like PD, the onset of MSA is usually in the sixth decade of life, but it progresses faster than PD. The mean survival time of MSA from the onset of symptoms is only 6–10 years.
Proteinaceous oligodendroglial cytoplasmic inclusions (Papp-Lantos bodies) are the major histological hallmark of MSA [70]. Less often, oligodendroglial nuclear inclusions are present, as are neuronal cytoplasmic and nuclear inclusions. Schwann cell cytoplasmic inclusions are also a common feature [71].
We teamed up with Nigel Cairns and Peter Lantos at the Institute of Psychiatry of King’s College London, and showed in July 1998 that glial and neuronal inclusions of MSA contained α-synuclein (Fig. 5) [72]. The inclusions were stained by antibodies recognizing the amino- and carboxy-termini of α-synuclein. Double-labelling showed that staining for α-synuclein was more extensive than staining for ubiquitin, indicating that the accumulation of α-synuclein preceded ubiquitination. Antibodies directed against β- or γ-synuclein failed to stain glial or neuronal inclusions. Similar findings were reported by others at about the same time [73, 74].
Isolated, sarkosyl-insoluble filaments extracted from MSA brains had a diameter of 5–18 nm and were strongly labelled by PER4. Antibody PER1 only labelled one filament end, as was the case in PD and DLB (Fig. 5). This work revealed an unexpected molecular link between MSA and Lewy pathology disorders. However, unlike PD and DLB, where α-synuclein filaments are present predominantly in the cytoplasm of nerve cells in the form of Lewy bodies and Lewy neurites, in MSA, α-synuclein-positive inclusions are found in the cytoplasm and nuclei of both nerve cells and glial cells.
With Crowther, we then showed that filaments assembled from bacterially expressed human α-synuclein were structurally and antigenically similar to those extracted from PD, DLB and MSA brains [75]. Since 1998 [76], PD, DLB and MSA have frequently been referred to as “synucleinopathies”.
TWENTY YEARS ON
The work described above identified synucleinopathies as a new group of human neurodegenerative diseases, with PD, DLB and MSA as the most common. Lewy pathology is also the defining feature of several rarer diseases, including pure autonomic failure, in which pathology is mostly restricted to the peripheral nervous system (PNS) [77]. In incidental Lewy body disease, a condition characteristic of 5–10% of individuals over the age of 60, which may be a preclinical form of PD [78–80], Lewy bodies and Lewy neurites are present in the absence of clinical symptoms. At the time of clinical diagnosis, around 30% of the dopaminergic neurons of the substantia nigra and 50–60% of their axon terminals have been lost [81], consistent with a centripetal mechanism of aggregate formation and neurodegeneration. In incidental Lewy body disease, no statistically significant nerve cell loss was observed in the substantia nigra [79]. However, a separate study reported that significant nerve cell loss occurred in the absence of Lewy pathology [82].
α-Synuclein inclusions are also found in a proportion of cases with other neurodegenerative diseases. Thus, in inherited and sporadic AD, approximately 60% have Lewy pathology, especially in the amygdala [83, 84]. However, not all cases of parkinsonism are characterized by the presence of α-synuclein inclusions. Thus, abundant tau aggregates of the Alzheimer type, in the absence of α-synuclein inclusions, are typical of post-encephalitic parkinsonism [85–88].
The clinical Parkinson’s syndrome, or parkinsonism, is defined as bradykinesia that worsens over time, in conjunction with at least one of three additional features: rigidity, resting tremor or gait disturbance [89, 90]. Full diagnostic certainty of PD is impossible during life, with 75–95% of patients having their diagnosis confirmed at autopsy. Supporting features are important for the clinical definition; they include response to L-DOPA, asymmetry and, more recently, the presence of non-motor symptoms. The latter has given rise to the concept of prodromal PD [91].
Physiological function of α-synuclein
The physiological function of α-synuclein is incompletely understood. It binds to acidic phospholipids through its amino-terminal repeats [92, 93], when it multimerizes and becomes α-helical [94–96]. α-Synuclein appears to be able to remodel membranes [97]. About 3,500 α-synuclein molecules exist for an average of 300 synaptic vesicles in synaptic boutons from rat brain [98]. Mitochondria have been found to fragment upon α-synuclein expression [99, 100], despite the fact that in nerve cells α-synuclein is concentrated in nerve terminals, whereas most mitochondria localize to nerve cell bodies and dendrites. It is possible that α-synuclein localizes to mitochondria when overexpressed [101].
Loss of SNCA does not lead to a neurodegenerative phenotype. [102]. Mice without α-synuclein showed increased release of striatal dopamine. Conversely, mice overexpressing α-synuclein showed reduced neurotransmitter release [103]. It appears that α-synuclein attenuates neurotransmitter release, possibly through multimerisation at synapses, resulting in the redistribution of SNARE proteins and the clustering of synaptic vesicles [104, 105]. A developmental function of α-synuclein for dopaminergic neurons has also been proposed [106], even though the SNCA locus was found to be deleted in a subset of inbred C57BL/6J mice [107].
The existence of three synucleins raised the possibility that redundancy might account for the relatively modest phenotype of α-synuclein knockout mice. Mice lacking α-,β- and γ-synucleins were subsequently produced [108, 109]. They showed an increase in striatal dopamine release beyond that of single knockouts, probably because of an increased tendency of synaptic vesicles to fuse with the presynaptic membrane. However, the overall phenotype was relatively mild. No synuclein homologues have been identified in C. elegans or Drosophila.
Perhaps the strongest effect of α-synuclein in vivo is its ability to rescue the loss of cysteine string protein (CSPα), a presynaptic chaperone. In mice, knockout of CSPα leads to progressive synaptic degeneration and death within 2 months [110]. Remarkably, overexpression of α-synuclein greatly delayed the synaptic degeneration resulting from loss of CSPα, whereas the loss of α-synuclein exacerbated the CSPα knockout phenotype [111]. These findings suggested that α-synuclein may be a chaperone, like CSPα. However, the mechanism by which α-synuclein rescues the loss of CSPα remains uncertain. It may be related to its ability to bind acidic phospholipids, because A30P α-synuclein, which has a greatly reduced lipid-binding ability [93], failed to rescue neurodegeneration resulting from the loss of CSPα.
α-Synuclein inclusions
Within pathological inclusions from human brain, the core of the filament extends over approximately 70 amino acids, which comprises the repeat region of α-synuclein (residues 30–110) [112, 113]. The crystal structure of residues 68–78 of human α-synuclein showed paired β-sheets with parallel β-strands in each sheet and anti-parallel β-strands between sheets. The zipper structure that marked the region between the paired sheets was longer than in other structures, and each pair of β-sheets contained two water molecules instead of being dry [114]. In a high-resolution structure obtained by solid-state nuclear magnetic resonance, scanning transmission electron microscopy and X-ray diffraction, core residues of the α-synuclein fibril were arranged in parallel, in-register β-sheets with the topology of a Greek key [115].
Over 90% of aggregated α-synuclein is phosphorylated at serine 129, while only about 4% of α-synuclein from normal brain is phosphorylated at this site [116, 117]. It remains to be seen if phosphorylated α-synuclein assembles into abnormal filaments. Antibodies that recognize α-synuclein phosphorylated at S129 are sensitive and specific tools for identifying the inclusions of PD, DLB and MSA. α-Synuclein can be phosphorylated at this site in vitro by multiple protein kinases, including G-protein coupled receptor kinases, casein kinases and polo-like kinases [118–120].
Recombinant mammalian α-synuclein readily assembles into filaments that share many of the morphological and ultrastructural characteristics of filaments from human brain (Fig. 6) [75, 121]. Upon assembly, α-synuclein adopts structures rich in β-sheet [122]. Assembly is a nucleation-dependent process that occurs through sequences in the amino-terminal 100 amino acids of α-synuclein. Deletion of residues 71–82 abolished the ability of α-synuclein to assemble into filaments [123]. Similarly, deletion of residues 66–74 also prevented assembly [124]. By contrast, the carboxy-terminal region of α-synuclein inhibited assembly [75].
In parallel experiments, human β- and γ-synucleins failed to assemble into filaments and remained in a natively unfolded conformation. β-Synuclein lacks a hydrophobic region that spans residues 73–83 of α-synuclein, suggesting that this difference may explain the proteins’ different propensities to assemble into filaments. However, deletion of residues 73–83 from α-synuclein did not abolish filament assembly. Moreover, a protein consisting of β-synuclein with residues 73–83 of α-synuclein inserted had a far lower fibrillogenic propensity than wild-type α-synuclein [125]. The fibrillogenic propensity of α-synuclein was determined by amino acid composition and β-strand contiguity. Mutants of β-synuclein could be produced that assembled into filaments [126].
Idiopathic PD constitutes over 90% of PD cases. Studies of normal and diseased human brains have shown that α-synuclein inclusions emerge in a predictable order in nerve cells in different parts of the brain, making it possible to distinguish six stages of α-synuclein deposition [127, 128]. The first α-synuclein-positive structures in the brain occur in the olfactory bulb and/or the dorsal motor nucleus of the glossopharyngeal and vagal nerves (stage 1). Lewy pathology then develops in medulla oblongata and pontine tegmentum (stage 2), followed by amygdala and substantia nigra (stage 3). It is during stage 3 that the motor symptoms of PD begin to appear. The pathology worsens and reaches the temporal cortex (stage 4). During stages 5 and 6, Lewy bodies and neurites appear in the neocortex, accounting for many of the cognitive problems associated with advanced PD. Large numbers of α-synuclein-immunoreactive astrocytes appear in stages 4–6 [129, 130].
α-Synuclein inclusions may form early in the enteric nervous system, which is connected to the brain through the vagal nerve, whose cell bodies are located in the dorsal motor nucleus. They may also form early in the PNS. The mechanisms through which the disease process spreads remain unclear. It could start in the gut and move retrogradely to the brain through the vagal nerve; in support, vagotomy has been reported to be associated with a reduced risk of PD [131]. Alternatively, inclusion formation could begin in the vagal dorsal motor nucleus and move from there to the spinal cord and gut in an anterograde fashion [132], or it could start in the periphery at multiple autonomic sites and subsequently be transmitted to the spinal cord [133]. The distribution of Lewy pathology in the gut parallels the input from the vagal dorsal motor nucleus [134]. This staging scheme is consistent with the fact that most PD patients develop non-motor symptoms before motor dysfunction. Constipation, hyposmia, depression and rapid eye movement behaviour disorder can precede motor symptoms by many years [135], something Parkinson had already noticed [1]. Their appearance correlates with the systemic distribution of Lewy pathology [136]. Several studies support the view that α-synuclein aggregation begins at synapses [137].
Similar staging schemes are not available for MSA. However, a prodromal form has been described [138], akin to incidental Lewy body disease. An atypical form of MSA with abundant α-synuclein inclusions, which presented clinically and pathologically as frontotemporal lobar degeneration has been described in the presence of striatonigral degeneration and variable olivopontocerebellar degeneration, but in the absence of autonomic dysfunction [139, 140]. Besides MSA-C and MSA-P, MSA-FTLD may be a third form of disease.
Genetics of SNCA
Six dominantly inherited missense mutations in SNCA have been described as the cause of familial PD (Fig. 7). Besides A53T, they comprise A30P [141], E46K [142], H50Q [143, 144], G51D [145, 146] and A53E [147, 148]. The ages of disease onset can be variable, even within families, but mutations G51D, A53E and A53T give rise to the earliest ages of onset. Experimentally, mutations E46K, H50Q and A53T increase α-synuclein inclusion formation [122, 149, 150], whereas mutations A30P, G51D and A53E [151–154] reduce its rates of aggregation. Mutations A30P, G51D and A53E also lead to a reduced ability of mutant α-synuclein to interact with acidic phospholipids [94, 152, 153, 155]. These findings are consistent with other work, which has suggested the existence of an antagonistic relationship between lipid binding of α-synuclein and aggregation into cytotoxic species [156–158].
Dominantly inherited duplications and triplications of the chromosomal region comprising SNCA have also been found to cause PD [159–161]. In these cases, the sequence of α-synuclein is wild-type, showing that an increase in the protein levels rather than a change in its properties is sufficient to cause PD. Duplication of SNCA gives rise to a form of PD that is similar to the sporadic disorder in terms of age of onset and symptoms, but triplication causes a more severe phenotype, with an earlier age of onset and prominent cognitive impairment.
Individuals with the A53T mutation in SNCA developed a severe form of PD that was often accompanied by dementia. A clinical picture resembling DLB was characteristic of a family with the E46K mutation, whereas individuals from the family with the A30P mutation developed late-onset PD and had only mild dementia. Neuropathologically, some individuals, in particular those with mutations G51D and A53E, had features of both PD and MSA. This overlap of clinical and neuropathological characteristics supports the view that the aetiologies of PD, DLB and MSA are closely related.
Genome-wide association studies of risk in idiopathic PD showed that SNCA makes the largest contribution. The implicated polymorphisms lie outside the coding region and thus probably affect mRNA expression, resulting in increased expression of α-synuclein. [162, 163]. Variability in LRRK2 (leucine-rich repeat kinase 2), GAK (cyclin G-associated kinase) and MAPT (microtubule-associated protein tau) has been implicated. Variants in SNCA and MAPT are also risk factors for MSA [164–166].
Animal models of synucleinopathies
Transgenic mice overexpressing human wild-type or mutant α-synuclein in nerve cells can develop abundant α-synuclein filaments [167–169]. The formation of inclusions correlated with the development of a movement disorder, possibly because of abundant brainstem and spinal cord pathologies. In these and other models, a major difference with PD was the absence of significant pathology and neurodegeneration in dopaminergic nerve cells of the substantia nigra.
This problem has been partially solved by transgenic mouse lines that express carboxy-terminally truncated human α-synuclein under the control of the rat tyrosine hydroxylase promoter on a mouse α-synuclein null background [105, 170]. They developed α-synuclein aggregates, a striatal dopamine deficiency and reduced locomotion. The carboxy-terminal truncation of human α-synuclein promotes aggregation [75]. Aggregation is also promoted when full-length human α-synuclein is expressed on a mouse SNCA or SNCB knockout background [171]. When wild-type human α-synuclein was expressed in oligodendrocytes, the transgenic mice developed MSA-like degeneration in brain and spinal cord [172, 173]. A mouse line that inducibly expressed A53T α-synuclein in astrocytes developed widespread astrocytosis, microglial activation and nerve cell degeneration, as well as severe paralysis [174]. In inducible transgenic mouse models of neuronal synucleinopathy, which modelled DLB, suppression of α-synuclein expression in the limbic system led to the clearing of α-synuclein inclusions and the disappearance of synaptic defects [175].
Adeno-associated and lentiviral vectors have been used to express human wild-type and mutant α-synuclein in rodent and primate substantia nigra [176–178], leading to the formation of Lewy body-like inclusions and degeneration of nerve cells. Aggregation of α-synuclein promoted neurodegeneration [179, 180]. Membrane binding of α-synuclein inhibited aggregation, whereas blocking membrane binding enhanced aggregation [155]. Activation of autophagy mediated by Beclin 1 [181] or overexpression of transcription factor EB (TFEB) [182] protected against the aggregation of α-synuclein. Interestingly, dopaminergic neurons in the ventral tegmental area, which are quite resistant to the effects of virally expressed A53T α-synuclein [183], degenerated upon repression of TFEB [182]. The differential vulnerability of nigral and ventral tegmental dopaminergic neurons, which has been documented in PD [184], may be due to differences in lysosomal clearance [182] and/or a higher basal rate of oxidative phosphorylation and a more complex axonal arborisation [185]. This is reminiscent of the concept of pathoclisis, which was put forward by Cécile and Oskar Vogt in 1922 [186]. In contemporary parlance, pathoclisis amounts to selective vulnerability.
Expression of human α-synuclein in Drosophila led to the formation of filamentous Lewy body-like inclusions, age-dependent loss of some dopaminergic neurons, and locomotor deficits [187]. Aggregation of α-synuclein was necessary for neurodegeneration and these effects were modulated by chaperones [188, 189]. Similarly, overexpression of human α-synuclein in C. elegans also resulted in dopaminergic nerve cell loss and motor deficits [190].
Propagation of α-synuclein aggregates
Evidence for the existence of prion-like mechanisms in human brain has come from the development of scattered Lewy pathology in foetal human midbrain neurons that were therapeutically implanted into the striata of patients with advanced PD [191, 192]. Lewy pathology was detected in 2–5% of grafted cells in patients who had survived for 10 or more years, approximately the same percentage as that of neurons with Lewy pathology in the pars compacta of the substantia nigra in PD. After 24 years, 11–12% of grafted dopaminergic neurons exhibited α-synuclein- and ubiquitin-positive inclusions [193].
Over the past nine years, experimental studies have shown that the injection of α-synuclein inclusions into animals induces nerve cells to form intracellular inclusions at the injection sites, from where they can spread to distant brain regions [194]. By long-term in vivo imaging, aggregated recombinant α-synuclein could seed the ordered assembly of expressed α-synuclein [195]. Inclusion-bearing neurons degenerated, demonstrating that inclusion formation was linked to cellular toxicity. In the substantia nigra from PD patients, the proportion of Lewy body-bearing neurons is approximately 4% and it remains constant over time. The inclusions are believed to be eliminated when the neurons that bear them die. In a model, in which neurons are killed by the Lewy pathology, it has been estimated that the mean survival time of an eosinophilic Lewy body is of the order of six months [196].
Morphological differences between disease-associated α-synuclein filaments have been described [72]. Lewy pathology was positive by Campbell-Switzer silver staining and negative by Gallyas-Braak silver [197]. By contrast, the glial cytoplasmic inclusions of MSA were positive by both Campbell-Switzer and Gallyas-Braak. Brain extracts from MSA patients propagated in heterozygous mice transgenic for human A53T α-synuclein, in contrast to brain extracts from PD patients [198, 199]. However, unlike MSA, α-synuclein inclusions were exclusively neuronal.
Polymorphs of recombinant aggregated α-synuclein in the form of ribbons or fibrils have been described [200]. When injected into the rat substantia nigra, ribbons gave rise to Lewy pathology, whereas fibrils, which did not seed Lewy pathology, led to the loss of dopaminergic neurons [201]. It remains to be determined if ribbons and fibrils have their counterparts in human synucleinopathies. In separate work, some α-synuclein filaments seeded both tau and α-synuclein aggregation, whereas others only seeded α-synuclein aggregation [202]. These conformers of aggregated α-synuclein exhibited different properties after proteinase K digestion. They were similar to prion strains, in that they showed structural variations, differences in seeding properties and heritability of phenotypic traits.
Induced pluripotent stem cells
Induced pluripotent stem cell (iPSC)-derived neurons from SNCA mutation carriers are playing an increasingly important role as an experimental system [203, 204]. Their main advantage resides in the fact that they are human nerve cells. However, the application of iPSC technology to the modelling of adult-onset diseases that are believed to be caused by a gain of toxic function mechanism, as is the case of the synucleinopathies, remains challenging. In principle, the earliest pathological changes that lead to disease can be studied in these systems, but their correct interpretation may require end-stage pathology.
CONCLUSION
The abnormal aggregation of α-synuclein has proved to be the “rosetta stone” of PD, DLB and MSA. Understanding disease aetiology and pathogenesis is one thing, but developing safe and effective mechanism-based treatments is quite another. When mechanism-based therapies for PD, DLB and MSA will become available, they will probably interfere with the neurodegeneration caused by the aggregation of α-synuclein. Given the long preclinical phase, it may be possible to develop preventive strategies, provided at-risk individuals can be identified. In principle, the earliest pathological changes that lead to disease can be studied in model systems. However, their correct evaluation may require the development of end-stage pathology.
CONFLICT OF INTEREST
The authors have no conflict to report.
ACKNOWLEDGMENTS
MG is supported by the UK Medical Research Council (MC_U105184291) and the EU Joint Programme – Neurodegenerative Disease Research. MGS is supported by the UK Medical Research Council, Alzheimer’s Research UK, Gates International, Astra Zeneca, the Cambridge Biomedical Centre, Addenbrooke’s Hospital Trust and the National Centre for the Replacement, Refinement and Reduction of Animals in Research.
REFERENCES
[1] | Parkinson J ((1917) ) An Essay on the Shaking Palsy. Sherwood, Neely and Jones, London. |
[2] | Charcot JM ((1875) ) Leçons sur les maladies du système nerveux. Vol 1. Delhaye et Cie, Paris. |
[3] | Tretiakoff K ((1919) ) Contribution à l’étude de l’anatomie pathologique du locus niger de Soemmering avec quelques déductions relatives à la pathogénie des troubles du tonus musculaire et de la maladie de Parkinson. Thèse, Université de Paris. |
[4] | Lewy F ((1912) ) Paralysis agitans. I. Pathologische Anatomie. In Handbuch der Neurologie, Vol 3: , Lewandowsky M, Abelsdorff G, eds. Springer Verlag, Berlin, pp. 920–933. |
[5] | Hassler R ((1938) ) Zur Pathologie der Paralysis agitans und des postenzephalitischen Parkinsonismus. J Psychol Neurol, 48: , 387–455. |
[6] | Duffy PE , & Tennyson VM ((1965) ) Phase and electron microscopic observations of Lewy bodies and melanin granules in the substantia nigra and locus coeruleus in Parkinson’s disease. J Neuropathol Exp Neurol, 24: , 398–414. |
[7] | Montagu KA ((1957) ) Catechol compounds in rat tissues and in brains of different animals. Nature, 180: , 244–245. |
[8] | Carlsson A , Lindqvist M , Magnusson T , & Waldeck B ((1958) ) On the presence of 3-hydroxytyramine in brain. Science, 127: , 471. |
[9] | Bertler A , & Rosengren E ((1959) ) Occurrence and distribution of dopamine in brain and other tissues. Experientia, 15: , 10–11. |
[10] | Sano I , Gamo T , Kakimoto Y , Taniguchi K , Takesada M , & Nishinuma K ((1959) ) Distribution of catechol compounds in human brain. Biochim Biophys Acta, 32: , 586–587. |
[11] | Carlsson A ((1959) ) The occurrence, distribution and physiological role of catecholamines in the nervous system. Pharmacol Rev, 11: , 490–493. |
[12] | Carlsson A ((2001) ) A half-century of neurotransmitter research: Impact on neurology and psychiatry. In Les Prix Nobel 2000, Frangsmyr T, ed. Almqvist, Wiksell, pp. 303–322. |
[13] | Ehringer H , & Hornykiewicz O ((1960) ) Verteilung von Noradrenalin und Dopamin (3-Hydroxytyramin) im Gehirn des Menschen und ihr Verhalten bei Erkrankungen des extrapyramidalen Systems. Klin Wochenschr, 38: , 1236–1239. |
[14] | Hornykiewicz O ((1963) ) Die topische Lokalisation und das Verhalten von Noradrenalin und Dopamin (3-Hydroxytyramin) in der Substantia nigra des normalen und Parkinson-kranken Menschen. Wien Klin Wochenschr, 75: , 309–312. |
[15] | Falck B , Hillarp N-Å , Thieme G , & Torp A ((1962) ) Fluorescence of catecholamines and related compounds condensed with formaldehyde. J Histochem Cytochem, 10: , 348–354. |
[16] | Dahlström A , & Fuxe K ((1964) ) Evidence for the existence of monoamine-containing neurons in the central nervous system. I. Demonstration of monoamines in the cell bodies of brain stem neurons. Acta Physiol Scand, 62: (Suppl 232), 1–55. |
[17] | Poirier L , & Sourkes TL ((1965) ) Influence of the substantia nigra on the catecholamine content of the striatum. Brain, 88: , 181–192. |
[18] | Sano I ((1960) ) Biochemistry of the extrapyramidal system (translated from the Japanese). Shinkei Kenkyu No Shimpo, 5: , 42–48. |
[19] | Birkmayer W , & Hornykiewicz O ((1961) ) Der L-Dioxyphenylalanin (=DOPA) Effekt bei der Parkinson Akinese. Wien Klin Wochenschr, 73: , 787–788. |
[20] | Barbeau A , Sourkes TI , & Murphy GF ((1962) ) Les catécholamines dans la maladie de Parkinson. In Monoamines et Système Nerveux Central, De Ajuriaguerra J, ed. Georg et Cie, Genève, pp. 247–262. |
[21] | Foley PB ((2003) ) Beans, roots and leaves. A history of the chemical theory of parkinsonism. Tectum Verlag, Marburg. |
[22] | Cotzias GC , Van Woert MH , & Schiffer LM ((1967) ) Aromatic amino acids and modification of parkinsonism. N Engl J Med, 276: , 374–379. |
[23] | Cotzias GC , Van Woert MH , & Schiffer LM ((1969) ) Modification of parkinsonism – chronic treatment with L-dopa. N Engl J Med, 280: , 337–345. |
[24] | Yahr MD , Duvoisin RC , Schear MJ , Barrett RE , & Hoehn MM ((1969) ) Treatment of parkinsonism with levodopa. Arch Neurol, 21: , 343–354. |
[25] | Lees AJ , Tolosa E , & Olanow CW ((2015) ) Four pioneers of L-dopa treatment: Arvid Carlsson, Oleh Hornykiewicz, George Cotzias, and Melvin Yahr. Mov Disord, 30: , 19–36. |
[26] | Sacks O ((1990) ) Awakenings. Revised Edition. Harper Collins, New York. |
[27] | Pinter H ((1982) ) A Kind of Alaska (from Other Places). Askerdale Ltd, London. |
[28] | Marshall P , Director ((1990) ) Awakenings, a film with Robert De Niro and Robin Williams. Columbia Pictures. |
[29] | Davis GC , Williams AC , Markey SP , Ebert MH , Caine ED , Reichert CM , Reichert CM , & Kopin IJ ((1979) ) Chronic parkinsonism secondary to intravenous injection of meperidine analogues. Psychiatry Res, 1: , 249–254. |
[30] | Langston JW , Ballard P , Tetrud JW , & Irwin I ((1983) ) Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science, 219: , 979–980. |
[31] | Marsden CD ((1982) ) Basal ganglia disease. Lancet, 320: , 1141–1147. |
[32] | Goedert M , Wischik CM , Crowther RA , Walker JE , & Klug A ((1988) ) Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: Identification as the microtubule-associated protein tau. Proc Natl Acad Sci U S A, 85: , 4051–4055. |
[33] | Wischik CM , Novak M , Thøgersen HC , Edwards PC , Runswick MJ , Jakes R , Walker JE , Milstein C , Roth M , & Klug A ((1988) ) Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A, 85: , 4506–4510. |
[34] | Wischik CM , Novak M , Edwards PC , Klug A , Tichelaar W , & Crowther RA ((1988) ) Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc Natl Acad Sci U S A, 85: , 4884–4888. |
[35] | Bondareff W , Wischik CM , Novak M , & Roth M ((1991) ) Sequestration of tau by granulovacuolar degeneration in Alzheimer’s disease. Am J Pathol, 139: , 641–647. |
[36] | Jakes R , Spillantini MG , & Goedert M ((1994) ) Identification of two distinct synucleins from human brain. FEBS Lett, 345: , 27–32. |
[37] | Ueda K , Fukushima H , Masliah E , Xia Y , Iwai A , Yoshimoto M , Otero DA , Kondo J , Ihara Y , & Saitoh T ((1993) ) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A, 90: , 11282–11286. |
[38] | Culvenor JG , McLean CA , Cutt S , Campbell BCV , Maher F , Jäkälä P , Hartmann T , Beyreuther K , Masters CL , & Li QX ((1999) ) Non-Aβ component of Alzheimer’s disease amyloid (NAC) revisited. Am J Pathol, 155: , 1173–1181. |
[39] | Bayer TA , Jäkälä P , Hartmann T , Havas L , McLean C , Culvenor JG , Li QX , Masters CL , Falkai P , & Beyreuther K ((1999) ) α-Synuclein accumulates in Lewy bodies in Parkinson’s disease and dementia with Lewy bodies, but not in Alzheimer’s disease β-amyloid plaque cores. Neurosci Lett, 266: , 213–216. |
[40] | Maroteaux L , Campanelli JT , & Scheller RH ((1988) ) Synuclein: A neuron-secific protein localized to the nucleus and presynaptic terminal. J Neurosci, 8: , 2804–2815. |
[41] | Maroteaux L , & Scheller RH ((1991) ) The rat brain synucleins: Family of proteins transiently associated with neuronal membrane. Mol Brain Res, 11: , 335–343. |
[42] | Nakajo S , Omata K , Aiuchi T , Shibayama T , Okahashi I , Ochiai H , Nakai Y , Nakaya K , & Nakamura Y ((1990) ) Purification and characterization of a novel brain-specific 14-kDa protein. J Neurochem, 55: , 2031–2038. |
[43] | Nakajo S , Tsukada K , Omata K , Nakamura Y , & Nakaya K ((1993) ) A new brain-specific 14-kDa protein is a phosphoprotein. Eur J Biochem, 217: , 1057–1063. |
[44] | Ji H , Liu YE , Jia T , Wang M , Liu J , Xiao G , Joseph BK , Rosen C , & Shi YE ((1997) ) Identification of a breast cancer-specific gene, BCSG1, by direct differential cDNA sequencing. Cancer Res, 57: , 759–764. |
[45] | Buchman VL , Adu J , Pinon LGP , Ninkina NN , & Davies AM ((1998) ) Persyn, a member of the synuclein family, influences neurofilament network integrity. Nat Neurosci, 1: , 101–103. |
[46] | Weinreb PH , Zhen W , Poon AW , Conway KA , & Lansbury PT ((1996) ) NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry, 35: , 13709–13715. |
[47] | Spillantini MG , Divane A , & Goedert M ((1995) ) Assignment of human α-synuclein (SNCA) and β-synuclein (SNCB) genes to chromosomes 4q21 and 5q35. Genomics, 27: , 379–381. |
[48] | Chen X , Rohan de Silva HA , Pettenati MJ , Rao PN , St George-Hyslop P , Roses AD , Xia Y , Horsburgh K , Ueda K , & Saitoh T ((1995) ) The human NACP/α-synuclein gene: Chromosome assignment to 4q21.3-q22 and TaqI RFLP analysis. Genomics, 26: , 425–427. |
[49] | Ninkina NN , Alimova-Kost MV , Paterson JWE , Delaney L , Cohen BB , Imreh S , Gnuchev NV , Davies AM , & Buchman VL ((1998) ) Organization, expression and polymorphism of the human gene. Hum Mol Genet, 7: , 1417–1424. |
[50] | Lavedan C , Leroy E , Dehejia A , Buchholtz S , Dutra A , Nussbaum RL , & Polymeropoulos MH ((1998) ) Identification, localization and characterization of the human γ-synuclein gene. Hum Genet, 103: , 106–112. |
[51] | Golbe LI , Di Iorio G , Bonavita V , Miller DC , & Duvoisin RC ((1990) ) A large kindred with autosomal dominant Parkinson’s disease. Ann Neurol, 27: , 276–282. |
[52] | Polymeropoulos MH , Higgins JJ , Golbe LI , Johnson WG , Ide SE , Di Iorio G , Sanges G , Stenroos ES , Pho LT , Schaffer AA , Lazzarini AM , Nussbaum RL , & Duvoisin RC ((1996) ) Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science, 274: , 1197–1199. |
[53] | Polymeropoulos MH , Lavedan C , Leroy R , Ide SE , Dehejia S , Dutra A , Pike B , Root H , Rubenstein J , Boyer R , Stenroos ES , Chandrasekharappa S , Athanassiadou A , Papapetropoulos T , Johnson WG , Lazzarini AM , Duvoisin RC , Di Iorio G , Golbe LI , & Nussbaum RL ((1997) ) Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science, 276: , 2045–2047. |
[54] | Hamilton BA ((2004) ) α-Synuclein A53T substitution associated with Parkinson disease also marks the divergence of Old World and New World primates. Genomics, 83: , 739–742. |
[55] | Spillantini MG , Schmidt ML , Lee VMY , Trojanowski JQ , Jakes R , & Goedert M ((1997) ) α-Synuclein in Lewy bodies. Nature, 388: , 839–840. |
[56] | Spira PJ , Sharpe DM , Halliday G , Cavanagh J , & Nicholson GA ((2001) ) Clinical and pathological features of a parkinsonian syndrome in a family with an Ala53Thr α-synuclein mutation. Ann Neurology, 49: , 313–319. |
[57] | Pouloupoulos M , Levy OA , & Alcalay RN ((2012) ) The neuropathology of genetic Parkinson’s disease. Mov Disord, 27: , 831–842. |
[58] | Spillantini MG , Crowther RA , Jakes R , Hasegawa M , & Goedert M ((1998) ) α-Synuclein in filamentous inclusions of Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A, 95: , 6469–6473. |
[59] | Kuzuhara S , Mori H , Izumiyama N , Yoshimura M , & Ihara Y ((1988) ) Lewy bodies are ubiquitinated. A light and electron microscopic immunocytochemical study. Acta Neuropathol, 75: , 345–353. |
[60] | Hasegawa M , Fujiwara H , Nonaka T , Wakabayashi K , Takahashi H , Lee VMY , Trojanowski JQ , Mann D , & Iwatsubo T ((2002) ) Phosphorylated α-synuclein is ubiquitinated in α-synucleinopathy lesions. J Biol Chem, 277: , 49071–49076. |
[61] | Sampathu DM , Giasson BI , Pawlyk AC , Trojanowski JQ , & Lee VMY ((2003) ) Ubiquitination of α-synuclein is not required for formation of pathological inclusions in α-synucleinopathies. Am J Pathol, 163: , 91–100. |
[62] | Crowther RA , Daniel SE , & Goedert M ((2000) ) Characterisation of isolated α-synuclein filaments from substantia nigra of Parkinson’s disease brain. Neurosci Lett, 292: , 128–130. |
[63] | Wakabayahi K , Matsumoto K , Takayama K , Yoshimoto H , & Takahashi H ((1997) ) NACP, a presynaptic protein, immunoreactivity in Lewy bodies in Parkinson’s disease. Neurosci Lett, 239: , 45–48. |
[64] | Takeda A , Mallory M , Sundsmo M , Honer W , Hansen L , & Masliah E ((1998) ) Abnormal accumulation of NACP/α-synuclein in neurodegenerative disorders. Am J Pathol, 152: , 367–372. |
[65] | Baba M , Nakajo S , Tu PH , Tomita T , Nakaya K , Lee VMY , Trojanowski JQ , & Iwatsubo T ((1998) ) Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am J Pathol, 152: , 879–884. |
[66] | Irizarry MC , Growdon W , Gomez-Isla T , Newell K , George JM , Clayton DF , & Hyman BT ((1998) ) Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson’s disease and cortical Lewy body disease contain α-synuclein immunoreactivity. J Neuropathol Exp Neurol, 57: , 334–337. |
[67] | Mezey E , Dehejia AM , Harta G , Tresser N , Suchy SF , Nussbaum RL , Brownstein MJ , & Polymeropoulos MH ((1998) ) α-Synuclein is present in Lewy bodies in sporadic Parkinson’s disease. Mol Psychiatry, 3: , 493–499. |
[68] | Fanciulli A , & Wenning GK ((2015) ) Multiple-system atrophy. N Engl J Med, 372: , 249–263. |
[69] | Quinn N ((2015) ) A short clinical history of multiple system atrophy. Clin Auton Res, 25: , 3–7. |
[70] | Papp MI , Kahn JE , & Lantos PL ((1989) ) Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome). J Neurol Sci, 94: , 79–100. |
[71] | Nakamura K , Mori F , Kon T , Tanji K , Miki Y , Tomiyama M , Kurotaki H , Toyoshima Y , Kakita A , Takahashi H , Yamada M , & Wakabayashi K ((2015) ) Filamentous aggregations of phosphorylated α-synuclein in Schwann cells (Schwann cell cytoplasmic inclusions) in multiple system atrophy. Acta Neuropathol Commun, 3: , 29. |
[72] | Spillantini MG , Crowther RA , Jakes R , Cairns NJ , Lantos PL , & Goedert M ((1998) ) Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett, 251: , 205–208. |
[73] | Wakabayashi K , Yoshimoto M , Tsuji S , & Takahashi H ((1998) ) α-Synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett, 249: , 180–182. |
[74] | Tu P , Galvin JE , Baba M , Giasson B , Tomita T , Leight S , Nakajo S , Iwatsubo T , Trojanowski JQ , & Lee VM ((1998) ) Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble α-synuclein. Ann Neurol, 44: , 415–422. |
[75] | Crowther RA , Jakes R , Spillantini MG , & Goedert M ((1998) ) Synthetic filaments assembled from C-terminally truncated α-synuclein. FEBS Lett, 436: , 309–312. |
[76] | Goedert M , & Spillantini MG ((1998) ) Lewy body diseases and multiple system atrophy as α-synucleinopathies. Mol Psychiatry, 3: , 462–465. |
[77] | Arai K , Kato K , Kashiwado K , & Hattori T ((2000) ) Pure autonomic failure in association with human α-synucleinopathy. Neurosci Lett, 296: , 171–173. |
[78] | Fearnley JM , & Lees AJ ((1991) ) Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain, 114: , 2283–2301. |
[79] | Dickson D , Fujishiro H , DelleDonne A , Menke J , Ahmed Z , Klos KJ , Josephs KA , Frigerio R , Burnett M , Parisi JE , & Ahlskog JE ((2008) ) Evidence that incidental Lewy body disease is pre-symptomatic Parkinson’s disease. Acta Neuropathol, 115: , 437–444. |
[80] | Beach TG , Adler CH , Sue LI , Peirce JB , Bachalakuri J , Dalsing-Hernandez J , Lue LF , Caviness JN , Connor DJ , Sabbagh MN , & Walker DG ((2008) ) Reduced striatal tyrosine hydroxylase in incidental Lewy body disease. Acta Neuropathol, 115: , 445–451. |
[81] | Cheng HC , Ulane CM , & Burke RE ((2010) ) Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol, 67: , 715–725. |
[82] | Milber JM , Noorigian JV , Morley JF , Petrovitch H , White L , Ross GW , & Duda JE ((2012) ) Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology, 79: , 2307–2314. |
[83] | Lippa CF , Fujiwara H , Mann DM , Giasson B , Baba M , Schmidt ML , Nee LE , O’Connell B , Pollen DA , St George-Hyslop P , Ghetti B , Nochlin D , Bird TD , Cairns NJ , Lee VM , Iwatsubo T , & Trojanowski JQ ((1998) ) Lewy bodies contain altered α-synuclein in brains of many familial Alzheimer’s disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol, 153: , 1365–1370. |
[84] | Hamilton RL ((2000) ) Lewy bodies in Alzheimer’s disease: A neuropathological review of 145 cases using α-synuclein immunohistochemistry. Brain Pathol, 10: , 378–384. |
[85] | Fényes I ((1932) ) Alzheimer’sche Fibrillenveränderung im Hirnstamm einer 28-jährigen Postencephalitikerin. Arch Psychiatr Nervenkr, 69: , 700–717. |
[86] | Geddes JF , Hughes AJ , Lees AJ , & Daniel SE ((1993) ) Pathological overlap in cases of parkinsonism associated with neurofibrillary tangles. Brain, 116: , 281–302. |
[87] | Buée-Scherrer V , Buée L , Leveugle B , Perl DP , Vermersch P , Hof PR , & Delacourte A ((1997) ) Pathological tau protein in post-encephalitic parkinsonism: Comparison with Alzheimer’s disease and other neurodegenerative disorders. Ann Neurol, 42: , 356–359. |
[88] | Josephs KA , Parisi JE , & Dickson DW ((2002) ) α-Synuclein studies are negative in postencephalitic parkinsonism of von Economo. Neurology, 59: , 645–646. |
[89] | Jenner P , Morris HR , Robbins TW , Goedert M , Hardy J , Ben-Shlomo Y , Bolam P , Burn D , Hindle JV , & Brooks D ((2013) ) Parkinson’s disease – the debate on the clinical phenomenology, aetiology, pathology and pathogenesis. J Parkinsons Dis, 3: , 1–11. |
[90] | Postuma RB , Berg D , Stern M , Poewe W , Olanow CW , Oertel W , Obeso J , Marek K , Litvan I , Lang AE , Halliday G , Goetz CG , Gasser T , Dubois B , Chan P , Bloem BR , Adler CH , & Deuschl G ((2015) ) MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord, 30: , 1591–1599. |
[91] | Berg D , Postuma RB , Adler CH , Bloem BR , Chan P , Dubois B , Gasser T , Goetz CG , Halliday G , Joseph L , Lang AE , Liepelt-Scarfone I , Litvan I , Marek K , Obeso J , Oertel W , Olanow CW , Poewe W , Stern M , & Deuschl G ((2015) ) MDS research criteria for prodromal Parkinson’s disease. Mov Disord, 30: , 1600–1609. |
[92] | Davidson WS , Jonas A , Clayton DF , & George JM ((1998) ) Stabilization of α-synuclein secondary structure upon binding to synthetic membranes. J Biol Chem, 273: , 9443–9449. |
[93] | Jensen PH , Nielsen MS , Jakes R , Dotti CG , & Goedert M ((1998) ) Binding of α−synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J Biol Chem, 273: , 26292–26294. |
[94] | Chandra S , Chen X , Rizo J , Jahn R , & Südhof TC ((2003) ) A broken alpha-helix in folded α-synuclein. J Biol Chem, 278: , 15313–15318. |
[95] | Jao CC , Hegde BG , Chen J , Haworth IS , & Langen R ((2008) ) Structure of membrane-bound α-synuclein from site-directed spin labelling and computational refinement. Proc Natl Acad Sci U S A, 105: , 19666–19671. |
[96] | Ulmer TS , Bax A , Cole NB , & Nussbaum RL ((2005) ) Structure and dynamics of micelle-bound human α-synuclein. J Biol Chem, 280: , 9595–9603. |
[97] | Varkey J , Isas JM , Mizuno N , Jensen MB , Bhatia VK , Jao CC , Petrlova J , Voss JC , Stamou DG , Steven AC , & Langen R ((2010) ) Membrane curvature induction and tubulation are common features of α-synuclein inclusion formation during aging. J Biol Chem, 285: , 32486–32493. |
[98] | Wilhelm BG , Mandad S , Truckenbrodt S , Kröhnert K , Schäfer C , Rammner B , Koo SJ , Claßen GA , Krauss M , Haucke V , Urlaub H , & Rizzoli SO ((2014) ) Composition of isolated synaptic boutons reveals the amounts of vesicle trafficking proteins. Science, 344: , 1023–1028. |
[99] | Kamp F , Exner N , Lutz AK , Wender N , Hegermann J , Brunner B , Nuscher B , Bartels T , Giese A , Beyer K , Eimer S , Winklhofer KF , & Haass C ((2010) ) Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J, 29: , 3571–3589. |
[100] | Nakamura K , Nemani VM , Azerbal F , Skibinski G , Levy JM , Egami K , Munishkina L , Zhang J , Gardner B , Wakabayashi J , Sesaki H , Cheng Y , Finkbeiner S , Nussbaum RL , Masliah E , & Edwards RH ((2011) ) Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein α-synuclein. J Biol Chem, 286: , 20710–20726. |
[101] | Bendor JT , Logan TP , & Edwards RH ((2013) ) The function of α-synuclein. Neuron, 79: , 1044–1066. |
[102] | Abeliovich A , Schmitz Y , Farinas I , Choi-Lundberg D , Ho WH , Castillo PE , hinsky N , Verdugo JM , Armanini M , Ryan A , Hynes M , Phillips H , Sulzer D , & Rosenthal A ((2000) ) Mice lacking α-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron, 25: , 239–252. |
[103] | Nemani VM , Lu W , Berge V , Nakamura K , Onoa B , Lee MK , Chaudhry FA , Nicoll RA , & Edwards RH ((2010) ) Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle re-clustering after endocytosis. Neuron, 65: , 66–79. |
[104] | Wang L , Das U , Scott DA , Tang Y , McLean PJ , & Roy S ((2014) ) α-Synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr Biol, 24: , 2319–2326. |
[105] | Garcia-Reitböck P , Anichtchik O , Bellucci A , Iovino M , Ballini C , Fineberg E , Ghetti B , Della Corte L , Spano P , Tofaris GK , Goedert M , & Spillantini MG ((2010) ) SNARE protein redistribution and synaptic failure in a transgenic mouse model of Parkinson’s disease. Brain, 133: , 2032–2044. |
[106] | Garcia-Reitböck P , Anichtchik O , Dalley JW , Ninkina N , Tofaris GK , Buchman VL , & Spillantini MG ((2013) ) Endogenous α-synuclein influences the number of dopaminergic neurons in mouse substantia nigra. Exp Neurol, 248: , 541–545. |
[107] | Specht CG , & Schoepfer R ((2001) ) Deletion of the α-synuclein locus in a subpopulation of C57BL/6J inbred mice. BMC Neurosci, 2: , 11. |
[108] | Greten-Harrison B , Polydoro M , Morimoto-Tomita M , Diao L , Williams AM , Nie EH , Makani S , Tian N , Castillo PE , Buchman VL , & Chandra SS ((2010) ) αβγ-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A, 107: , 19573–19578. |
[109] | Anwar S. Peters O. Millership S. Ninkina NN. Doig N. Connor-Robson N. Threlfell S. Kooner G. Deacon RM. Bannerman DM. Bolam JP. Chandra SS. Cragg SJ. Wade-Martins R, & Buchman VL (2011) Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J Neurosci, 31: , 7264–7274. |
[110] | Fernández-Chacón R , Wölfel M , Nishimune H , Tabares L , Schmitz F , Castellano-Munoz M , Rosenmund C , Montesinos ML , Sanes JR , Schneggenburger R , & Südhof TC ((2004) ) The synaptic vesicle protein CSPα prevents presynaptic degeneration. Neuron, 42: , 237–251. |
[111] | Chandra S , Gallardo G , Fernández-Chacón R , Schlüter OM , & Südhof TC ((2005) ) α-Synuclein cooperates with CSPα in preventing neurodegeneration. Cell, 123: , 383–396. |
[112] | Miake H , Mizusawa H , Iwatsubo T , & Hasegawa M ((2002) ) Biochemical characterization of the core structure of α-synuclein filaments. J Biol Chem, 277: , 19213–19219. |
[113] | Der-Sarkissian A , Jao CC , Chen J , & Langen R ((2003) ) Structural organization of α-synuclein fibril structure studied by site-directed spin labelling. J Biol Chem, 278: , 24970–24979. |
[114] | Rodriguez JA , Ivanova MI , Sawaya MR , Cascio D , Reyes FE , Shi D , Sangwan S , Guenther EL , Johnson LM , Zhang M , Jiang L , Arbing MA , Nannenga BL , Hattne J , Whitelegge J , Brewster AS , Messerschmidt M , Boutet S , Sauter NK , Gonen T , & Eisenberg DS ((2015) ) Structure of the toxic core of α-synuclein from invisible crystals. Nature, 525: , 486–490. |
[115] | Tuttle MD , Comellas G , Nieuwkoop AJ , Covell DJ , Berthold DA , Kloepper KD , Courtney JM , Kim JK , Barclay AM , Kendall A , Wan W , Stubbs G , Schwieters CD , Lee VM , George JM , & Rienstra CM ((2016) ) Solid-state NMR structure of a pathogenic fibril of full-length human α-synuclein. Nat Struct Mol Biol, 23: , 409–415. |
[116] | Fujiwara H , Hasegawa M , Dohmae N , Kawashima A , Masliah E , Goldberg MS , Shen J , Takio K , & Iwatsubo T ((2002) ) α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol, 4: , 160–164. |
[117] | Anderson JP , Walker DE , Goldstein JM , de Laat R , Banducci K , Caccavello RJ , Barbour R , Huang J , Kling K , Lee M , Diep L , Keim PS , Shen X , Chataway T , Schlossmacher MG , Seubert P , Schenk D , Sinha S , Gai WP , & Chilcote TJ ((2006) ) Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J Biol Chem, 281: , 29739–29752. |
[118] | Pronin AN , Morris AJ , Surguchov A , & Benovic JL ((2000) ) Synucleins are a novel class of substrates for G protein-coupled receptor kinases. J Biol Chem, 275: , 26515–26522. |
[119] | Ishii A , Nonaka T , Taniguchi S , Saito T , Arai T , Mann D , Iwatsubo T , Hisanaga S , Goedert M , & Hasegawa M ((2007) ) Casein kinase 2 is the major enzyme in brain that phosphorylates Ser129 of human α-synuclein: Imlication for α-synucleinopathies. FEBS Lett, 581: , 4711–4717. |
[120] | Mbefo MK , Paleologou KE , Bucharaba A , Oueslati A , Schell H , Fournier M , Olschewski D , Yin G , Zweckstetter M , Masliah E , Kahle PJ , Hirling H , & Lashuel HA ((2010) ) Phosphorylation of synucleins by members of the Polo-like kinase family. J Biol Chem, 285: , 2807–2822. |
[121] | Conway KA , Harper JD , & Lansbury PT ((1998) ) Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat Med, 4: , 1318–1320. |
[122] | Serpell LC , Berriman J , Jakes R , Goedert M , & Crowther RA ((2000) ) Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-β conformation. Proc Natl Acad Sci U S A, 97: , 4897–4902. |
[123] | Giasson BI , Murray IV , Trojanowski JQ , & Lee VMY ((2001) ) A hydrophobic stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J Biol Chem, 276: , 2380–2386. |
[124] | Du HN , Tang L , Luo XY , Li HT , Hu J , Zhou JW , & Hu HY ((2003) ) A peptide motif consisting of glycine, alanine, and valine is required for the fibrillization and cytotoxicity of human α-synuclein. Biochemistry, 42: , 8870–8878. |
[125] | Zibaee S , Jakes R , Fraser G , Serpell LC , Crowther RA , & Goedert M ((2007) ) Sequence determinants for amyloid fibrillogenesis of human α-synuclein. J Mol Biol, 374: , 454–464. |
[126] | Zibaee S , Fraser G , Jakes R , Owen D , Serpell LC , Crowther RA , & Goedert M ((2010) ) Human β-synuclein rendered fibrillogenic by designed mutations. J Biol Chem, 285: , 38555–38567. |
[127] | Braak H , Del Tredici K , Rüb U , de Vos RA , Jansen Steur EN , & Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging, 24: , 197–211. |
[128] | Goedert M , Spillantini MG , Del Tredici K , & Braak H ((2013) ) 100 years of Lewy pathology. Nat Rev Neurol, 9: , 13–24. |
[129] | Arai T , Ueda K , Ikeda K , Akiyama H , Haga C , Kondo H , Kuroki N , Niizato K , Iritani S , & Tsuchiya K ((1999) ) Argyrophilic glial inclusions in the midbrain of patients with Parkinson’s disease and diffuse Lewy body disease are immunopositive for NACP/α-synuclein. Neurosci Lett, 259: , 83–86. |
[130] | Braak H , Sastre M , & Del Tredici K ((2007) ) Development of α-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol, 114: , 231–241. |
[131] | Svensson E , Horváth-Puhó E , Thomsen RW , Djurhuus JC , Pedersen L , Borghammer P , & Sørensen HT ((2015) ) Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol, 78: , 522–529. |
[132] | Del Tredici K , & Braak H ((2012) ) Spinal cord lesions in sporadic Parkinson’s disease. Acta Neuropathol, 124: , 643–654. |
[133] | Braak H , Sastre M , Bohl JR , de Vos RA , & Del Tredici K ((2007) ) Parkinson’s disease: Lesions in dorsal horn layer I, involvement of parasympathetic and sympathetic pre- and postganglionic neurons. Acta Neuropathol, 113: , 421–429. |
[134] | Annerino DM , Arshad S , Taylor GM , Adler CH , Beach TG , & Greene JG ((2012) ) Parkinson’s disease is not associated with gastrointestinal myenteric ganglion neuron loss. Acta Neuropathol, 124: , 665–680. |
[135] | Schapira AH , & Tolosa E ((2010) ) Molecular and clinical prodrome of Parkinson disease: Implications for treatment. Nat Rev Neurol, 6: , 309–317. |
[136] | Adler CH , & Beach TG ((2016) ) Neuropathological basis of nonmotor manifestations of Parkinson’s disease. Mov Disord, 31: , 1114–1119. |
[137] | Hunn BH , Cragg SJ , Bolam JP , Spillantini MG , & Wade-Martins R ((2015) ) Impaired intracellular trafficking defines early Parkinson’s disease. Trends Neurosci, 38: , 178–188. |
[138] | Fujishiro H , Ahn TB , Frigerio R , DelleDonne A , Josephs KA , Parisi JE , Eric Ahlskog J , & Dickson DW ((2008) ) Glial cytoplasmic inclusions in neurologically normal elderly: Prodromal multiple system atrophy? Acta Neuropathol, 116: , 269–275. |
[139] | Aoki N , Boyer PJ , Lund C , Lin WL , Koga S , Ross OA , Weiner M , Lipton A , Powers JM , White CL 3rd , & Dickson DW ((2015) ) Atypical multiple system atrophy is a new subtype of frontotemporal lobar degeneration: Frontotemporal lobar degeneration associated with α-synuclein. Acta Neuropathol, 130: , 93–105. |
[140] | Rohan Z , Rahimni J , Weis S , Kapas I , Auff E , Mitrovic N , Liberski PP , Sikorska B , Matej R , & Kovacs GG ((2015) ) Screening for α-synuclein immunoreactive neuronal inclusions in the hippocampus allows identification of atypical MSA (FTLD-synuclein). Acta Neuropathol, 130: , 299–301. |
[141] | Krüger R , Kuhn W , Müller T , Woitalla D , Graeber M , Kösel S , Przuntek H , Epplen JT , Schöls L , & Riess O ((1998) ) Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat Genet, 18: , 106–108. |
[142] | Zarranz JJ , Alegre J , Gómez-Esteban JC , Lezcano E , Ros R , Ampuero I , Vidal L , Hoenicka J , Rodriguez O , Atarés B , Llorens V , Gomez Tortosa E , del Ser T , Muñoz DG , & de Yebenes JG ((2003) ) The new mutation, E46K, of α-synuclein causes Parkinson and Lewy body dementia. Ann Neurol, 55: , 164–173. |
[143] | Appel-Cresswell S , Vilarino-Guell C , Encamacion M , Sherman H , Yu I , Shah B , Weir D , Thompson C , Szu-Tu C , Trinh J , Aasly JO , Rajput A , Rajput AH , Jon Stoessl A , & Farrer MJ ((2013) ) Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord, 28: , 811–813. |
[144] | Proukakis C , Dudzik CG , Brier T , MacKay DS , Cooper JM , Millhauser GL , Houlden H , & Schapira AH ((2013) ) A novel α-synuclein missense mutation in Parkinson disease. Neurology, 80: , 1062–1064. |
[145] | Kiely AP , Asi YT , Kara E , Limousin P , Ling H , Lewis P , Proukakis C , Quinn N , Lees AJ , Hardy J , Revesz T , Houlden H , & Holton JL ((2013) ) α-Synucleinopathy associated with G51Dmutation: A link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol, 125: , 753–769. |
[146] | Lesage S , Anheim M , Letournel F , Bousset L , Honoré A , Rozas N , Pieri L , Madiona K , Dürr A , Melki R , Verny C , & Brice A ; French Parkinson’s Disease Genetics Study Group ((2013) ) G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol, 73: , 459–471. |
[147] | Pasanen P , Myllykangas L , Siitonen M , Raunio A , Kaakola S , Lyytinen J , Tienari PJ , Pöyhönen M , & Paetau A ((2014) ) A novel α-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol Aging, 25: , 2180.e1–2180.e5. |
[148] | Martikainen M , Päivärinta M , Hietala M , & Kaasinen V ((2015) ) Clinical and imaging findings in Parkinson disease associated with the A53E mutation. Neurol Genet, 1: , e27. |
[149] | Choi W , Zibaee S , Jakes R , Serpell LC , Davletov B , Crowther RA , & Goedert M ((2004) ) Mutation E46K increases phospholipid binding and assembly into filaments of human α-synuclein. FEBS Lett, 576: , 363–368. |
[150] | Ghosh D , Mondal M , Mohite GM , Singh PK , Rankan P , Anoop A , Ghosh S , Jha NN , Kumar A , & Maji SK ((2013) ) The Parkinson’s disease-associated H50Q mutation accelerates α-synuclein aggregation in vitro. Biochemistry, 52: , 6925–6927. |
[151] | Narhi L , Wood SJ , Steavenson S , Jiang Y , Wu GM , Anafi D , Kaufman SA , Martin F , Sitney K , Denis P , Louis JC , Wypych J , Biere AL , & Citron M ((1999) ) Both familial Parkinson’s disease mutations accelerate α-synuclein aggregation. J Biol Chem, 274: , 9843–9846. |
[152] | Bilal-Fares M , Alt-Bouziad N , Dikiy I , Mbefo MK , Jovicic A , Kiely A , Holton JL , Lee SJ , Gitler AD , Eliezer D , & Lashuel HA ((2014) ) The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of α-synuclein, and enhances its secretion and nuclear localization in cells. Hum Mol Genet, 23: , 4491–4509. |
[153] | Ghosh D , Sahay S , Ranjan P , Salot S , Mohite GM , Singh PK , Dwivedi S , Carvalho E , Banerjee R , Kumar A , & Maji SK ((2014) ) The newly discovered Parkinson’s disease associated Finnish mutation (A53E) attenuates α-synuclein aggregation and membrane binding. Biochemistry, 53: , 6419–6421. |
[154] | Rutherford NJ , Moore BD , Golde TE , & Giasson BI ((2014) ) Divergent effects of the H50Q and G51Dmutations on the aggregation of α-synuclein. J Neurochem, 131: , 859–867. |
[155] | Ysselstein D , Joshi M , Mishra V , Griggs AM , Asiago JM , McCabe GP , Stanciu LA , Post CB , & Rochet JC ((2015) ) Effects of impaired membrane interactions on α-synuclein aggregation and neurotoxicity. Neurobiol Dis, 79: , 150–163. |
[156] | Burré J , Sharma M , & Südhof TC ((2015) ) Definition of a molecular pathway mediating α-synuclein neurotoxicity. J Neurosci, 35: , 5221–5232. |
[157] | Iljina M , Tosatto L , Choi ML , Sang JC , Ye Y , Hughes CD , Bryant CE , Gandhi S , & Klenerman D ((2016) ) Arachidonic acid mediates the formation of abundant helical multimers of α-synuclein. Sci Rep, 6: , 33928. |
[158] | Cremades N , Cohen SIA , Deas E , Abramov AY , Chen AY , Orte A , Sandal M , Clarke RW , Dunne P , Aprile FA , Bertoncini CW , Wood NW , Knowles TP , Dobson CM , & Klenerman D ((2012) ) Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell, 149: , 1048–1059. |
[159] | Singleton AB , Farrer M , Johnson J , Singleton A , Hague S , Kachergus J , Hulihan M , Peuralinna T , Dutra A , Nussbaum R , Lincoln S , Crawley A , Hanson M , Maraganore D , Adler C , Cookson MR , Muenter M , Baptista M , Miller D , Blancato J , Hardy J , & Gwinn-Hardy K ((2003) ) α-Synuclein locus triplication causes Parkinson’s disease. Science, 302: , 841. |
[160] | Chartier-Harlin MC , Kachergus J , Roumier C , Mouroux V , Douay X , Lincoln S , Levecque C , Larvor L , Andrieux J , Hulihan M , Waucquier N , Defebvre L , Amouyel P , Farrer M , & Destée A ((2004) ) α-Synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet Neurol, 364: , 1167–1169. |
[161] | Ibánez P , Bonnet AM , Débarges B , Lohmann E , Tison F , Pollak P , Agid Y , Dürr A , & Brice A ((2003) ) Causal relation between α-synuclein gene duplication and familial Parkinson’s disease. Lancet Neurol, 364: , 1169–1171. |
[162] | Satake W , Nakabayahi Y , Mizuta I , Hirota Y , Ito C , Kubo M , Kawaguchi T , Tsunoda T , Watanabe M , Takeda A , Tomiyama H , Nakashima K , Hasegawa K , Obata F , Yoshikawa T , Kawakami H , Sakoda S , Yamamoto M , Hattori N , Murata M , Nakamura Y , & Toda T ((2009) ) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet, 41: , 1303–1307. |
[163] | Simón-Sánchez J , Schulte C , Bras JM , Sharma M , Gibbs JR , Berg D , Paisan-Ruiz C , Lichtner P , Scholz SW , Hernandez DG , Krüger R , Federoff M , Klein C , Goate A , Perlmutter J , Bonin M , Nalls MA , Illig T , Gieger C , Houlden H , Steffens M , Okun MS , Racette BA , Cookson MR , Foote KD , Fernandez HH , Traynor BJ , Schreiber S , Arepalli S , Zonozi R , Gwinn K , van der Brug M , Lopez G , Chanock SJ , Schatzkin A , Park Y , Hollenbeck A , Gao J , Huang X , Wood NW , Lorenz D , Deuschl G , Chen H , Riess O , Hardy JA , Singleton AB , & Gasser T ((2009) ) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet, 41: , 1308–1312. |
[164] | Scholz SW , Houlden H , Schulte C , Sharma M , Li A , Berg D , Melchers A , Paudel R , Gibbs JR , Simon-Sanchez J , Paisan-Ruiz C , Bras J , Ding J , Chen H , Traynor BJ , Arepalli S , Zonozi RR , Revesz T , Holton J , Wood N , Lees A , Oertel W , Wüllner U , Goldwurm S , Pellecchia MT , Illig T , Riess O , Fernandez HH , Rodriguez RL , Okun MS , Poewe W , Wenning GK , Hardy JA , Singleton AB , Del Sorbo F , Schneider S , Bhatia KP , & Gasser T ((2009) ) variants are associated with increased risk for multiple system atrophy. Ann Neurol, 65: , 610–614. |
[165] | Al-Chalabi A , Dürr A , Wood NW , Parkinson MH , Camuzat A , Hulot JS , Morrison KE , Renton A , Sussmuth SD , Landwehrmeyer BG , Ludolph A , Agid Y , Brice A , Leigh PN , & Bensimon G ; NNIPPS Genetic Study Group ((2009) ) Genetic variants of the α-synuclein gene are associated with multiple system atrophy. PLoS One, 4: , e7114. |
[166] | Vilarino-Güell C , Soto-Ortolaza AI , Rajput A , Mash DC , Papapetropoulos S , Pahwa R , Lyons KE , Uitti RJ , Wszolek ZK , Dickson DW , Farrer MJ , & Ross OA ((2011) ) H1 haplotype is a risk factor for essential tremor and multiple system atrophy. Neurology, 76: , 670–672. |
[167] | Masliah E , Rockenstein E , Veinbergs I , Mallory M , Hashimoto M , Takeda A , Sagara Y , Sisk A , & Mucke L ((2000) ) Dopaminergic loss and inclusion body formation in α-synuclein mice: Implications for neurodegenerative disorders. Science, 287: , 1265–1269. |
[168] | Giasson BI , Duda JE , Quinn SM , Zhang B , Trojanowski JQ , & Lee VMY ((2002) ) Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron, 34: , 521–533. |
[169] | Emmer KL , Waxman EA , Covy JP , & Giasson BI ((2011) ) E46K human α-synuclein transgenic mice develop Lewy-like and tau pathology associated with age-dependent, detrimental motor impairment. J Biol Chem, 286: , 35104–35118. |
[170] | Tofaris GK , Reitböck PG , Humby T , Lambourne SL , O’Connell M , Ghetti B , Gossage H , Emson PC , Wilkinson LS , Goedert M , & Spillantini MG ((2006) ) Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human α-synuclein (1-120): Implications for Lewy body disorders. J Neurosci, 25: , 3942–3950. |
[171] | Fares MB , Maco B , Oueslati A , Rockenstein E , Ninkina N , Buchman VL , Masliah E , & Lashuel HA ((2016) ) Induction of de novo α-synuclein fibrillization in a neuronal model for Parkinson’s disease. Proc Natl Acad Sci U S A, 113: , E912–E921. |
[172] | Yazawa I , Giasson BI , Sasaki R , Zhang B , Joyce S , Uryu K , Trojanowski JQ , & Lee VM ((2005) ) Mouse model of multiple system atrophy α-synuclein expression in oligodendrocytes causes glial and neuronal degeneration. Neuron, 45: , 847–859. |
[173] | Shults CW , Rockenstein E , Crews L , Adame A , Mante M , Larrea G , Hashimoto M , Song D , Iwatsubo T , Tsuboi K , & Masliah E ((2005) ) Neurological and neurodegenerative alterations in a transgenic mouse model expressing human α-synuclein under oligodendrocyte promoter: Implications for multiple system atrophy. J Neurosci, 25: , 10689–10699. |
[174] | Gu XL , Long CX , Sun L , Xie C , Lin X , & Cai H ((2010) ) Astrocytic expression of Parkinson’s disease-related A53T α-synuclein causes neurodegeneration in mice. Mol Brain, 3: , 12. |
[175] | Lim Y , Kehm VM , Lee EB , Soper JH , Li C , Trojanowski JQ , & Lee VM ((2011) ) α-Syn suppression reverses synaptic and memory defects in a mouse model of dementia with Lewy bodies. J Neurosci, 31: , 10076–10087. |
[176] | Kirik D , Rosenblad C , Burger C , Lundberg C , Johansen TE , Muzyczka N , Mandel RJ , & Björklund A ((2002) ) Parkinson-like neurodegeneration induced by targeted overexpression of α-synuclein in the nigrostriatal system. J Neurosci, 22: , 2780–2791. |
[177] | Lo Bianco C , Ridet JL , Schneider BL , Déglon N , & Aebischer P ((2002) ) α-Synucleinopathy and selective dopaminergic neuron loss in a rat lentiviral-based model of Parkinson’s disease. Proc Natl Acad Sci U S A, 99: , 10813–10818. |
[178] | Klein RL , King MA , Hamby ME , & Meyer EM ((2002) ) Dopaminergic cell loss induced by human A30P α-synuclein gene transfer to the rat substantia nigra. Ther, 13: , 605–612. |
[179] | Taschenberger G , Garrido M , Tereshchenko Y , Bähr M , Zweckstetter M , & Kügler S ((2012) ) Aggregation of α-synuclein promotes progressive in vivo neurotoxicity in adult rat dopaminergic neurons. Acta Neuropathol, 123: , 671–683. |
[180] | Burré J , Sharma M , & Südhof TC ((2012) ) Systematic mutagenesis of α-synuclein reveals distinct sequence requirements for physiological and pathological activities. J Neurosci, 32: , 15227–15242. |
[181] | Spencer B , Potkar R , Trejo M , Rockenstein E , Patrick C , Gindi R , Adame A , Wyss-Coray T , & Masliah E ((2009) ) Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci, 29: , 13578–13588. |
[182] | Decressac M , Mattson B , Weikop P , Lundblad M , Jakobsson , & Björklund A ((2013) ) TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc Natl Acad Sci U S A, 110: , E1817–E1826. |
[183] | Maingay M , Romero-Ramos M , Carta M , & Kirik D ((2006) ) Ventral tegmental area dopamine neurons are resistant to human mutant α-synuclein overexpression. Neurobiol Dis, 23: , 522–532. |
[184] | Hirsch E , Graybiel AM , & Agid YA ((1988) ) Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature, 334: , 345–348. |
[185] | Pacelli C , Giguère N , Bourque MJ , Lévesque M , Slack RS , & Trudeau LE ((2015) ) Elevated mitochondrial bioenergetics and axonal arborisation size are key contributors to the vulnerability of dopamine neurons. Curr Biol, 25: , 2349–2360. |
[186] | Vogt C , & Vogt O ((1922) ) Erkrankungen der Grosshirnrinde im Lichte der Topistik – Pathoklise und Pathoarchitektonik. Monatsschr Psychiatr Neurol, 29: , 9–171. |
[187] | Feany MB , & Bender WW ((2000) ) Amodel of Parkinson’s disease. Nature, 404: , 394–398. |
[188] | Auluck PK , Chan HY , Trojanowski JQ , Lee VMY , & Bonini NM ((2002) ) Chaperone suppression of α-synuclein toxicity in amodel for Parkinson’s disease. Science, 295: , 865–868. |
[189] | Périquet M , Fulga T , Myllykangas L , Schlossmacher MG , & Feany MB ((2007) ) Aggregated α-synuclein mediates dopaminergic neurotoxicity in vivo. J Neurosci, 27: , 3338–3334. |
[190] | Lakso M , Vartiainen S , Moilanen AM , Sirviö J , Thomas JH , Nass R , Blakely RD , & Wong G ((2003) ) Dopaminergic neuronal loss and motor deficits inoverexpressing human α-synuclein. J Neurochem, 86: , 165–172. |
[191] | Li J , Englund E , Holton JL , Soulet D , Hagell P , Lees AJ , Lashley T , Quinn NP , Rehncrona S , Björklund A , Widner H , Revesz T , Lindvall O , & Brundin P ((2008) ) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med, 14: , 501–503. |
[192] | Kordower JH , Chu Y , Hauser RA , Freeman TB , & Olanow CW ((2008) ) Lewy body-like pathology in long-term embryonic neural transplants in Parkinson’s disease. Nat Med, 14: , 504–506. |
[193] | Li W , Englund E , Widner H , Mattson B , van Westen D , Lätt J , Rehncrona S , Brundin P , Björklund A , Lindvall O , & Li JY ((2016) ) Extensive graft-derived dopaminergic innervation is maintained 24 years after transplantation in the degenerating parkinsonian brain. Proc Natl Acad Sci U S A, 113: , 6544–6549. |
[194] | Goedert M ((2015) ) Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science, 349: , 1255555. |
[195] | Osterberg VR , Spinelli KJ , Weston LJ , Luk KC , Woltjer RL , & Unni VK ((2015) ) Progressive aggregation of α-synuclein and selective degeneration of Lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep, 10: , 1252–1260. |
[196] | Greffard S , Verny M , Bonnet AM , Seilhean D , Hauw JJ , & Duyckaerts C ((2010) ) A stable proportion of Lewy body bearing neurons in the substantia nigra suggests a model in which the Lewy body causes neuronal death. Neurobiol Aging, 31: , 99–103. |
[197] | Uchihara T , Nakamura A , Mochizuki Y , Hayashi M , Orimo S , Isozaki E , & Mizutani T ((2005) ) Silver stainings distinguish Lewy bodies and glial cytoplasmic inclusions: Comparison between Gallyas-Braak and Campbell-Switzer methods. Acta Neuropathol, 110: , 255–260. |
[198] | Woerman AL , Stöhr J , Aoyagi A , Rampersaud R , Krejciova Z , Watts JC , Ohyama T , Patel S , Widjaja K , Oehler A , Sanders DW , Diamond MI , Seeley WW , Middleton LT , Gentleman SM , Mordes DA , Südhof TC , Giles K , & Prusiner SB ((2015) ) Propagation of prions causing synucleinopathies in cultured cells. Proc Natl Acad Sci U S A, 112: , E4949–E4958. |
[199] | Prusiner SB , Woerman AL , Mordes DA , Watts JC , Rampersaud R , Berry DB , Patel S , Oehler A , Lowe JK , Kravitz SN , Geschwind DH , Glidden DV , Halliday GM , Middleton LT , Gentleman SM , Grinberg LT , & Giles K ((2015) ) Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A, 112: , E5308–E5317. |
[200] | Bousset L , Peri L , Ruiz-Arlandis G , Gath J , Jensen PH , Habenstein B , Madiona K , Olieric V , Böckmann A , Meier BH , & Melki R ((2013) ) Structural and functional characterization of two α-synuclein strains. Nat Commun, 4: , 2575. |
[201] | Peelaerts W , Bousset L , Van der Perren A , Moskalyuk A , Pulizzi R , Giugliano M , Van den Haute C , Melki R , & Baekelandt V ((2015) ) α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature, 522: , 340–344. |
[202] | Guo JL , Covell DJ , Daniels JP , Iba M , Stieber A , Zhang B , Riddle DM , Kwong LK , Xu Y , Trojanowski JQ , & Lee VM ((2013) ) Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell, 154: , 103–117. |
[203] | Soldner F , Laganière J , Cheng AW , Hockemeyer D , Gao Q , Alagappan R , Khurana V , Golbe LI , Myers RH , Lindquist S , Zhang L , Guschin D , Fong LK , Vu BJ , Meng X , Urnov FD , Rebar EJ , Gregory PD , Zhang HS , & Jaenisch R ((2011) ) Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell, 146: , 318–331. |
[204] | Devine MJ , Ryten M , Vodicka P , Thomson AJ , Burdon T , Houlden H , Cavaleri F , Nagano M , Drummond NJ , Taanman JW , Schapira AH , Gwinn K , Hardy J , Lewis PA , & Kunath T ((2011) ) Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat Commun, 2: , 440. |
[205] | Goedert M ((2001) ) Alpha-synuclein and neurodegenerative diseases. Nat Rev Neurosci, 2: , 492–501. |
[206] | Goedert M , Spillantini MG ((2012) ) Synucleinopathies and tauopathies. In Basic Neurochemistry, 8th edition, Brady ST, Siegel GJ, Albers RW, Price DL, eds. Elsevier Academic Press pp. 829–843. |
Figures and Tables
Fig.1
Twenty years ago: Ross Jakes, Maria Grazia Spillantini and Michel Goedert (from left to right) in 1997.

Fig.2
Sequence comparison of human α-synuclein and β-synuclein. Amino acids were aligned and two gaps introduced to maximize homology. Amino acid identities between α-synuclein (α-Syn) and β-synuclein (β-Syn) are indicated by black bars. From Jakes et al. [36].
![Sequence comparison of human α-synuclein and β-synuclein. Amino acids were aligned and two gaps introduced to maximize homology. Amino acid identities between α-synuclein (α-Syn) and β-synuclein (β-Syn) are indicated by black bars. From Jakes et al. [36].](https://content.iospress.com:443/media/jpd/2017/7-s1/jpd-7-s1-jpd179005/jpd-7-jpd179005-g002.jpg)
Fig.3
The α-synuclein pathology of Parkinson’s disease. Lewy bodies and Lewy neurites in the substantia nigra and several other brain regions define Parkinson’s disease at a neuropathological level. They are shown here at light (a-c) and electron microscopic (d-g) levels, labelled by α-synuclein antibodies. (a), Two pigmented nerve cells, each containing an α-synuclein-positive Lewy body (red arrows). Lewy neurites (black arrows) are also immunopositive. Scale bar, 20μm. (b), Pigmented nerve cell with two α-synuclein-positive Lewy bodies. Scale bar, 8μm. (c) α-Synuclein-positive extracellular Lewy body. Scale bar, 4μm. (d-g), Isolated filaments from the substantia nigra of patients with Parkinson’s disease are decorated with an antibody directed against the carboxy-terminal (d and e) or the amino-terminal (f,g) region of α-synuclein. The gold particles conjugated to the second antibody appear as black dots. Note the uniform decoration (d,e), and the labelling of only one filament end (f,g). Scale bar, 100 nm. From Goedert [205].
![The α-synuclein pathology of Parkinson’s disease. Lewy bodies and Lewy neurites in the substantia nigra and several other brain regions define Parkinson’s disease at a neuropathological level. They are shown here at light (a-c) and electron microscopic (d-g) levels, labelled by α-synuclein antibodies. (a), Two pigmented nerve cells, each containing an α-synuclein-positive Lewy body (red arrows). Lewy neurites (black arrows) are also immunopositive. Scale bar, 20μm. (b), Pigmented nerve cell with two α-synuclein-positive Lewy bodies. Scale bar, 8μm. (c) α-Synuclein-positive extracellular Lewy body. Scale bar, 4μm. (d-g), Isolated filaments from the substantia nigra of patients with Parkinson’s disease are decorated with an antibody directed against the carboxy-terminal (d and e) or the amino-terminal (f,g) region of α-synuclein. The gold particles conjugated to the second antibody appear as black dots. Note the uniform decoration (d,e), and the labelling of only one filament end (f,g). Scale bar, 100 nm. From Goedert [205].](https://content.iospress.com:443/media/jpd/2017/7-s1/jpd-7-s1-jpd179005/jpd-7-jpd179005-g003.jpg)
Fig.4
The α-synuclein pathology of dementia with Lewy bodies. (a,b) α-Synuclein-positive Lewy bodies and Lewy neurites in substantia nigra. Scale bar, 100μm. (c,d) α-Synuclein-positive Lewy bodies and Lewy neurites in hippocampus. Scale bar, 80μm. (e) α-Synuclein-positive Lewy body inside a Lewy neurite in substantia nigra. Scale bar, 40μm. From Spillantini et al. [58].
![The α-synuclein pathology of
dementia with Lewy bodies. (a,b) α-Synuclein-positive Lewy bodies and Lewy neurites in substantia nigra.
Scale bar, 100μm. (c,d) α-Synuclein-positive Lewy bodies and Lewy neurites in hippocampus. Scale
bar, 80μm. (e) α-Synuclein-positive Lewy body inside a
Lewy neurite in substantia nigra. Scale bar, 40μm. From Spillantini et al. [58].](https://content.iospress.com:443/media/jpd/2017/7-s1/jpd-7-s1-jpd179005/jpd-7-jpd179005-g004.jpg)
Fig.5
The α-synuclein pathology of multiple system atrophy. Glial cytoplasmic inclusions in several brain regions define multiple system atrophy. Similar inclusions are also present in the nuclei of some glial cells, as well as in the cytoplasm and nuclei of some nerve cells, and in nerve cell processes. Inclusions are shown here at light (a-f) and electron microscopic (g-j) levels, labelled by α-synuclein antibodies. (a-d) α-Synuclein-immunoreactive oligodendrocytes and nerve cells in white matter of pons (a,b,d) and cerebellum (c,e,f). α-Synuclein-immunoreactive oligodendrocytes and nerve cells in grey matter of pons (e) and frontal cortex (f). Arrows identify the characteristic lesions: cytoplasmic, oligodendroglial inclusions (a,f), cytoplasmic nerve cell inclusions (b), nuclear oligodendroglial inclusions (c), neuropil threads (d) and nuclear nerve cell inclusions (e). Scale bar, 33μm in (e) and 50μm in (f). (g-j), Isolated filaments from the frontal cortex and cerebellum of patients with multiple system atrophy are decorated by antibodies specific for the carboxy-terminal (g,h) and amino-terminal (i,j) regions of α-synuclein. The gold particles conjugated to the secondary antibody appear as black dots. Note the uniform decoration in (g,h) and the labelling of only one filament end in (i,j). A twisted filament is shown in (g), whereas (h) shows a straight filament. Scale bar, 100 nm. From Goedert [205].
![The α-synuclein pathology of multiple system atrophy. Glial cytoplasmic inclusions in several brain regions define multiple system atrophy. Similar inclusions are also present in the nuclei of some glial cells, as well as in the cytoplasm and nuclei of some nerve cells, and in nerve cell processes. Inclusions are shown here at light (a-f) and electron microscopic (g-j) levels, labelled by α-synuclein antibodies. (a-d) α-Synuclein-immunoreactive oligodendrocytes and nerve cells in white matter of pons (a,b,d) and cerebellum (c,e,f). α-Synuclein-immunoreactive oligodendrocytes and nerve cells in grey matter of pons (e) and frontal cortex (f). Arrows identify the characteristic lesions: cytoplasmic, oligodendroglial inclusions (a,f), cytoplasmic nerve cell inclusions (b), nuclear oligodendroglial inclusions (c), neuropil threads (d) and nuclear nerve cell inclusions (e). Scale bar, 33μm in (e) and 50μm in (f). (g-j), Isolated filaments from the frontal cortex and cerebellum of patients with multiple system atrophy are decorated by antibodies specific for the carboxy-terminal (g,h) and amino-terminal (i,j) regions of α-synuclein. The gold particles conjugated to the secondary antibody appear as black dots. Note the uniform decoration in (g,h) and the labelling of only one filament end in (i,j). A twisted filament is shown in (g), whereas (h) shows a straight filament. Scale bar, 100 nm. From Goedert [205].](https://content.iospress.com:443/media/jpd/2017/7-s1/jpd-7-s1-jpd179005/jpd-7-jpd179005-g005.jpg)
Fig.6
Filaments extracted from the brains of patients with dementia with Lewy bodies (a) and multiple system atrophy (b) or assembled from bacterially expressed human α-synuclein (c) were decorated by an anti-α-synuclein antibody. The gold particles conjugated to the secondary antibody appear as black dots. From Goedert and Spillantini [206].
![Filaments extracted from the brains of patients with dementia with Lewy bodies (a) and multiple system atrophy (b) or assembled from bacterially expressed human α-synuclein (c) were decorated by an anti-α-synuclein antibody. The gold particles conjugated to the secondary antibody appear as black dots. From Goedert and Spillantini [206].](https://content.iospress.com:443/media/jpd/2017/7-s1/jpd-7-s1-jpd179005/jpd-7-jpd179005-g006.jpg)
Fig.7
Human α-synuclein and its disease-causing mutations. (a), Diagram of the 140 amino acid human α-synuclein protein. The core regions of the seven amino-terminal repeats are shown as blue bars. (b), An increase in gene dosage (duplication or triplication) of the chromosomal region containing SNCA or missense mutations in SNCA cause dominantly inherited forms of Parkinson’s disease and dementia with Lewy bodies. (c), The repeats (residues 7–87) of human α-synuclein are shown, with disease-causing missense mutations (A30P, E46K, H50Q, G51D, A53E and A53T) given as blue letters. Amino acids that are identical in at least five of the seven repeats are shaded in blue.