
Targeting AMPK Signaling as a Neuroprotective Strategy in Parkinson’s Disease
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disorder. It is characterized by the accumulation of intracellular α-synuclein aggregates and the degeneration of nigrostriatal dopaminergic neurons. While no treatment strategy has been proven to slow or halt the progression of the disease, there is mounting evidence from preclinical PD models that activation of 5′-AMP-activated protein kinase (AMPK) may have broad neuroprotective effects. Numerous dietary supplements and pharmaceuticals (e.g., metformin) that increase AMPK activity are available for use in humans, but clinical studies of their effects in PD patients are limited. AMPK is an evolutionarily conserved serine/threonine kinase that is activated by falling energy levels and functions to restore cellular energy balance. However, in response to certain cellular stressors, AMPK activation may exacerbate neuronal atrophy and cell death. This review describes the regulation and functions of AMPK, evaluates the controversies in the field, and assesses the potential of targeting AMPK signaling as a neuroprotective treatment for PD.
INTRODUCTION
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease, afflicting approximately 0.3% of the global population, with nearly 1 million Americans currently diagnosed. Its incidence is strongly correlated with age, tripling with each decade of life after age 60 [1]. Although the etiology of PD is still poorly defined, great progress has been made in characterizing the disease’s progression and manifestations. The neuropathological hallmarks of PD are the loss of dopaminergic (DA) neurons within the substantia nigra pars compacta (SNc) and the widespread occurrence of intracellular protein aggregates called Lewy bodies that consist primarily of accumulated α-synuclein (αSyn). These pathological features are associated with numerous motor and non-motor symptoms, including bradykinesia, rigidity, resting tremor, depression, cognitive impairment, and autonomic dysfunction. Although some of these symptoms can be therapeutically managed for many years after disease onset, PD pathology is progressive and there is still no proven treatment strategy to slow or halt the course of the disease. The development of effective treatments is thus one of the primary objectives of current PD research [1].
The vast majority of PD cases are sporadic, often with no readily identifiable cause. However, in some cases, PD can be directly linked to certain genetic mutations. PD-causing mutations, such as those in genes related to autophagy (e.g., LRRK2) and mitochondrial quality control (e.g., PINK1 and Parkin), have highlighted the relevance of these functions to the pathogenesis of PD. Various environmental factors have also been linked to PD. Exposure to neurotoxins such as the pesticides rotenone and paraquat or MPTP, which was inadvertently self-administered by a number of drug users in the 1980 s, can all produce PD-like pathology [2, 3]. Although exposure to such toxins may explain only a small subset of PD cases, their neurotoxic mechanisms have further informed the pathological underpinnings of PD, and they are now commonly used to reproduce the hallmark features of PD in preclinical models. Ultimately, most cases of PD likely arise from a combination of genetic susceptibility (including epigenetic changes), environmental factors, and aging processes; with the contribution of each of these factors varying among affected individuals [1, 4–6].
Accumulating research has pointed to several neuropathological factors that appear particularly relevant to the development and progression of PD. These factors include: mitochondrial dysfunction that may lead to bioenergetic failure and increased reactive oxygen species (ROS), proteostatic failure and the accumulation of αSyn aggregates, and neuroinflammation [1]. Various experimental treatment approaches for PD have sought to address these factors, often individually, but approaches that target them collectively may be more effective. Each of these factors is regulated to some extent by 5’-AMP-activated protein kinase (AMPK), a ubiquitous enzyme that is considered a master sensor of intracellular energy stress that plays a crucial role in adaptive responses to falling energy levels (e.g., from low nutrient availability or cellular stress) [7]. AMPK signaling declines with age, and increasing its activity is associated with extended lifespan in model organisms [8]. Activating AMPK has broad neuroprotective properties, increasing cell survival to diverse stressors, including starvation, hypoxia, ischemia, and excitotoxicity [9–12]. This review will briefly summarize the structure, regulation, and functions of AMPK and critically describe the results of several recent studies that have explored AMPK activation as a neuroprotective treatment strategy in models of PD.
STRUCTURE OF AMPK
AMPK is a heteromeric serine/threonine kinase comprised of three subunits: a catalytic α subunit and regulatory β and γ subunits. Multiple isoforms of each subunit exist that can assemble into a total of 12 potential AMPK complexes [13]. The differences between these individual complexes is still being investigated, but there is evidence that they can have differing inputs, outputs, functions, and subcellular localizations [14]. So, although AMPK is present in essentially all eukaryotic cells, differential expression of these isoforms may provide a degree of specificity and tissue-specific metabolic regulation [15]. Each subunit contains important catalytic or regulatory motifs essential for the overall function of AMPK. The α subunit contains the catalytic kinase domain within which resides AMPK’s primary activation site, known conventionally as Thr172 [16]. At least three upstream kinases are responsible for the phosphorylation of Thr172: liver kinase B1 (LKB1), calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2), and TGF-β activated kinase 1 (TAK1) [17–19]. The α subunit also contains a regulatory serine/threonine-rich loop (ST loop) near its C-terminus that negatively regulates AMPK activity [20]. The β subunit contains a carbohydrate-binding module (CBM), as well as a myristoylation site at its N-terminus that facilitates targeting of AMPK to cellular membranes, including lysosomes and mitochondria [21]. The γ subunit contains 4 nucleotide binding sites, of which three are functional in mammals (sites 1, 3, and 4); these sites bind AMP, ADP, and ATP interchangeably with varying selectivity [22]. In addition to these binding sites, AMPK also contains at least one additional allosteric site at the interface between the α and β subunits that has been termed the allosteric drug and metabolite (ADaM) site [23]. Although no endogenous ligand has yet been identified for this site, a number of exogenous activators have been identified or developed that bind there, providing an additional therapeutically relevant target for modulating AMPK [24].
REGULATION OF AMPK
The primary function of AMPK is to sense and respond to changes in cellular energy. Unsurprisingly then, the canonical regulator of AMPK is the ratio of AMP (and ADP) to ATP. The binding of adenine nucleotides at the γ subunit has three main functions in regulating the catalytic activity of AMPK [25]. The first is to modulate phosphorylation of Thr172. Until phosphorylated at this residue, AMPK is essentially inactive. Binding of AMP increases net phosphorylation by LKB1 [26], and AMP and ADP may promote phosphorylation by CaMKK2 as well [27], although this has been challenged [26]. Second, binding of AMP or ADP extends the functional “half-life” of AMPK by protecting pThr172 from dephosphorylation [13]. In the absence of such protection, pThr172 is rapidly dephosphorylated by phosphatases, including protein phosphatase 2A (PP2A) and 2C (PP2C) [28]. And third, in addition to promoting and protecting the phosphorylation of Thr172, AMP allosterically increases the catalytic activity of AMPK by as much as 10-fold [26]. These functions are achieved through a conformation change in the structure of AMPK. However, an opposing conformational change is induced by ATP binding, which decreases the catalytic activity of AMPK and increases the rate of dephosphorylation [29]. So, as the ratio of ATP to AMP increases within a cell, AMPK activity is inhibited.
In addition to its regulation by the adenine nucleotides, AMPK activity is also modified by certain hormones, cellular stressors, and other factors that do not necessarily alter the ratio of AMP to ATP. LKB1 was the first AMPK activating kinase to be identified [17]. It is constitutively active, but its phosphorylation of AMPK can be modulated by both AMP, as discussed above, and through a mechanism independent from AMP [30]. In response to glucose deprivation, a complex forms between AMPK and LKB1 at the lysosomal surface that facilitates Thr172 phosphorylation [31, 32]. CaMKK2, which may be the dominant AMPK activating kinase in neurons, is activated by increased intracellular calcium [18]. Intracellular calcium is increased as a second messenger by release from intracellular stores and through ion-channel mediated influx from the extracellular space in response to a multitude of upstream signals [33]. Through this signaling cascade, AMPK activity can be modulated by a wide range of factors including metabolic hormones like ghrelin [34] and neuronal activity [18]. The third upstream AMPK kinase, TAK1, is activated by various cytokine receptors, thus connecting AMPK activation to extracellular inflammatory, cell growth, and apoptotic signals [35, 36].
Regulatory regions within the α and β subunits of AMPK also modulate its activity. The ST loop can be phosphorylated at multiple residues by a number of different kinases, which results in a net decrease in Thr172 phosphorylation, possibly by interfering with upstream kinase binding [37]. It is through this mechanism that hormones such as insulin and leptin can decrease AMPK activity [20, 38]. It has also been proposed that the CBM within the β subunit of AMPK provides a mechanism for AMPK to directly sense the availability of stored cellular energy in the form of glycogen and that glycogen binding to the CBM decreases AMPK activity [39]. However, this idea remains controversial, as other studies have not observed an inhibitory effect of glycogen on AMPK function [29, 40]. Nitric-oxide (NO) can activate AMPK via inositol-requiring enzyme 1 (IRE1) [41], providing synchronization between NO production and AMPK activity [42, 43]. There is also some evidence that AMPK is regulated by ROS [44]. AMPK can be indirectly activated by the energy imbalance caused by ROS [45], but ROS may also directly affect AMPK activity via oxidative modifications at various AMPK cysteine residues [46, 47]. Ultimately, AMPK is sensitive to and regulated by a wide range of intra- and extracellular signals that allow it to respond to both local energy needs within cells and to synchronize whole-body energy homeostasis [48].
A number of exogenous small-molecule agents have been identified that can modulate the activity of AMPK, and many of these are already available for use in humans. Examples include anti-hyperglycemic medications such as metformin and pioglitazone, the dietary supplement β-guanidinopropionic acid (GPA), and a huge number of naturally derived phytochemicals, such as resveratrol, quercetin, and berberine [13, 49]. Each of these activates AMPK indirectly, either by increasing the ratio of AMP to ATP or by stimulating upstream regulators of AMPK [45]. Metformin, for example, activates AMPK via both methods [50]. It accumulates in mitochondria where it inhibits complex I of the electron transport chain, thereby increasing the ratio of AMP to ATP [51]. It also activates AMPK by promoting LKB1-mediated phosphorylation of AMPK at the lysosomal surface, facilitating formation of the same LKB1 complex that is normally triggered by glucose deprivation [52]. Resveratrol, a phytochemical found in the skin of grapes and various berries, also activates AMPK via two mechanisms. It inhibits ATP-synthase, increasing the ratio of AMP to ATP, and also stimulates SIRT1-mediated deacetylation of LKB1, thereby increasing AMPK activity [53]. AMPK and SIRT1 have an important reciprocal relationship that is critical for many of their downstream effects [54]. Unlike metformin and resveratrol, GPA activates AMPK without inhibiting the electron transport chain. Rather, it increases the ratio of AMP to ATP by decreasing the function of creatine kinase [55].
A number of pharmacological agents have also been developed that can activate AMPK directly [24]. The first of these was AICAR, which, when metabolized to the nucleotide ZMP, binds to and activates AMPK by mimicking the functions of AMP [56]. Other agents such as A769662 activate AMPK via the ADaM site [24, 57]. Intriguingly, it was found that A769662 could maintain allosteric activation of AMPK even in the absence of Thr172 phosphorylation [58], suggesting that AMPK could be pharmacologically regulated independent of upstream kinase activity. However, this finding may only apply to certain in vitro conditions [59]. Several newly developed synthetic activators of AMPK have differing affinities for the various AMPK isoforms, which may allow for more targeted and function specific activation of AMPK [60]. There are thus an increasing number of pharmacological methods to modulate the activity of AMPK that could be utilized therapeutically in patients with or at risk of developing PD.
FUNCTIONS OF AMPK AND THEIR RELEVANCE TO PD
Activating AMPK has a variety of effects that may be relevant to PD. This includes changes in cellular metabolism, promotion of autophagy, enhanced mitochondrial quality control, increased antioxidant capacity, and reduced inflammation (Fig. 1). Each of these functions and their relevance to PD are discussed below. Targeting the regulation of so many downstream pathways through a single kinase may be advantageous given the relevance that many of these functions have to PD. Yet this diversity also presents a challenge when attempting to isolate which downstream function(s) or target(s) of AMPK are responsible for the beneficial or detrimental effects that result from activating or inhibiting AMPK. Furthermore, while many of these pathways promote cell survival, AMPK may also have a role in mediating programmed cell death (PCD), particularly in cases of severe cellular stress or bioenergetic failure. Thus, the potentially positive effects of AMPK activation must be balanced with its potential for negative effects.
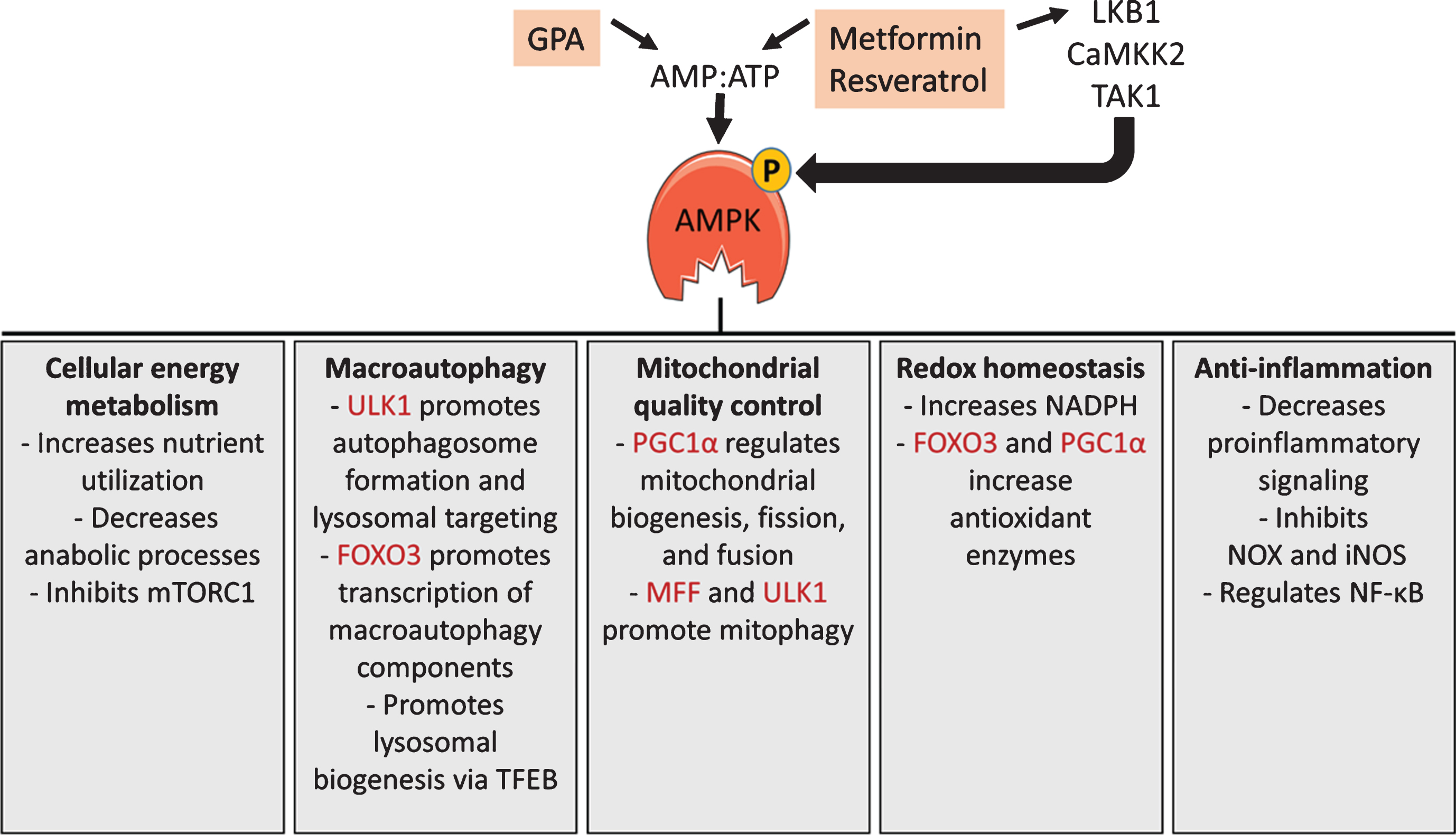
Fig.1
AMPK signaling. AMPK activity is regulated by the ratio of AMP to ATP and by at least 3 upstream kinases. Metformin, β-guanidinopropionic acid (GPA), and resveratrol activate AMPK by increasing the AMP:ATP ratio and/or by stimulating liver kinase B1 (LKB1) mediated phosphorylation of AMPK. AMPK has numerous functions, including changes to cellular energy metabolism, increased macroautophagy, enhanced mitochondrial quality control, redox homeostasis, and anti-inflammatory effects. A few specific functions within each area are listed, with direct phosphorylation targets of AMPK shaded. These functions are explained in more detail in the text.
Cellular energy metabolism
Impaired cellular energy metabolism is seen in patients with PD [61] and has been implicated in the progression of both sporadic and genetic forms of the disease [62–64]. In adult animals, the majority of ATP is derived from glucose through mitochondrial oxidative phosphorylation (OXPHOS) [65]. Aging, which is the most common risk factor for PD, is associated with decreased mitochondrial function, reduced energy metabolism, and lower AMPK function [66–68]. Decreased ATP generating capacity makes neurons more vulnerable to bioenergetic failure when faced with additional stressors that impair cellular energy metabolism, such as αSyn or environmental toxins [43, 69, 70]. The neuronal populations that degenerate in PD share some common characteristics that make them particularly vulnerable to bioenergetic failure [71, 72]. For instance SNc DA neurons, which are inextricably linked with PD, have high energy demands and low spare respiratory capacity [73]. Neurons in general consume a substantial amount of energy in order to maintain the ionic gradient across their plasma membranes, and because they do not store glycogen they are especially sensitive to fluctuations in energy demand and reliant on neighboring astrocytes to provide nutrients [74]. Unfortunately SNc DA neurons have relatively few surrounding astrocytes to provide this supplementary energy [75]. They also have long unmyelinated axons and extensive arborization making their energy demands higher than most neurons [76]. To maintain basal DA tone across the expansive region of their innervation, they fire tonically with pacemaker activity driven by L-type calcium channels [77]. These channels are associated with higher ROS production, greater mitochondrial dysfunction, increased energy demand, and heightened vulnerability to PD-related toxins [77–79]. These features make SNc DA neurons particularly vulnerable to compromised bioenergetic status and may explain their relatively selective degeneration in PD [80–83].
AMPK plays a critical role in sensing and responding to changing energy needs within cells. Upon activation, AMPK functions broadly to inhibit processes that consume energy and promote processes that generate energy [25]. Increasing the activity of AMPK may therefore be a viable strategy to increase cellular energy and stave off bioenergetic failure in vulnerable neurons. AMPK stimulates the uptake of glucose into cells through the translocation of glucose transporters to the plasma membrane and through the removal of glucose from stored glycogen [7, 84]. AMPK also increases utilization of other OXPHOS substrates such as glutamine and fatty acids (FAs) [85]. FAs are a particularly important source of ATP during periods of calorie restriction. AMPK promotes uptake and transport of FAs for catabolism by β-oxidation, while simultaneously inhibiting the energy consuming processes of synthesizing FAs, cholesterol, and triglycerides [86]. In addition to these acute effects, AMPK also mediates long-term metabolic reprogramming by activating transcriptional regulators such as forkhead box transcription factors (FOXO) and peroxisomal proliferator-activated receptor γ coactivator 1α (PGC-1α), which facilitate numerous functions, including lipid metabolism and mitochondrial function [15, 54].
Cell growth and the synthesis of new proteins require a considerable expenditure of energy and can be inhibited by AMPK to conserve cellular ATP. Mammalian target of rapamycin complex 1 (mTORC1) integrates growth factor and nutrient signals to promote protein synthesis and cell growth [25]. AMPK inhibits the activity of mTORC1 via activation of its negative regulator Tuberous Sclerosis Complex 2 (TSC2) [87] and by inhibiting Raptor, a subunit of mTORC1 [88]. AMPK also decreases protein synthesis by suppressing ribosomal RNA synthesis [89]. Collectively these processes increase ATP production through OXPHOS, while limiting anabolic processes that consume ATP.
Macroautophagy
Autophagy is a housekeeping and nutrient recycling process in which cellular components, debris, and other macromolecules are delivered to or engulfed by lysosomes for degeneration [25]. Impaired autophagy is central to the progression of PD [90]. Deletion of genes essential for autophagy, such as ATG7, produces PD-like neurodegeneration in mice [91]. Accumulation of αSyn is one of the pathological hallmarks of PD and may be both a cause and a consequence of impaired autophagy [90]. αSyn is normally cleared by a combination of the ubiquitin-proteasome system (UPS), chaperone-mediated autophagy (CMA), and macroautophagy [92]. However, oligomerization of αSyn prevents clearance by UPS and CMA, increasing the necessity of macroautophagy-mediated clearance, which involves the formation of an autophagosome to sequester cytoplasmic constituents and transport them to lysosomes. [93, 94]. Yet, there is evidence that macroautophagy is also impaired in PD [95]. Transcription factor EB (TFEB), which mediates lysosomal biogenesis and promotes macroautophagy, is sequestered by αSyn in both human PD brain tissue and in a rat PD model [96]. In the rat model, overexpressing αSyn is associated with dysfunctional macroautophagy, increased SNc DA neurodegeneration, and impaired motor function. However, these deficits can all be ameliorated by overexpressing TFEB or Beclin-1 (another autophagy regulator), suggesting that macroautophagy can be upregulated in PD to reduce αSyn aggregation and neurodegeneration [96, 97].
AMPK promotes autophagy by initiating the macroautophagic cascade through direct activation of unc-51 like autophagy activating kinase (ULK1) [98] and indirectly through its inhibition of mTORC1, which is an inhibitor of ULK1 [99]. ULK1 initiates autophagosome formation and maturation via phosphorylation of Beclin-1 [100]. AMPK promotes lysosomal biogenesis via increased TFEB activity [102] and activates FOXO3 mediated transcription of proteins necessary for macroautophagy [101]. AMPK is thus well positioned as a pharmacological target to stimulate macroautophagy as a treatment strategy in PD [103].
Mitochondrial quality control
Mitochondria are critical organelles that provide most of the energy needed for cellular functions through the production of ATP by OXPHOS. As a byproduct of OXPHOS, mitochondria also produce ROS that over time can damage mitochondria and impair their function [77]. This decreases cellular energy production and can result in chronic ROS overproduction, creating a cycle of increasing mitochondrial dysfunction [63]. It is therefore essential that cells maintain a healthy mitochondrial pool through the quality control mechanisms of mitochondrial biogenesis, fission-fusion dynamics, and mitophagy [104]. However, there is evidence that mitochondrial quality control declines with age and especially in patients with PD [105, 106]. This may explain the increased mitochondrial deficits observed in these groups, such as the accumulation of mitochondrial DNA deletions [107–109]. The aggregation of αSyn may also play a crucial role in disrupting mitochondrial dynamics, leading to increased mitochondrial dysfunction in PD [110, 111]. Mitochondrial dysfunction is associated with bioenergetic failure, increased ROS, and cell death [70]. By facilitating mitochondrial quality control, AMPK activation may be an effective strategy to ameliorate mitochondrial dysfunction and prevent neurodegeneration.
AMPK promotes mitochondrial biogenesis through transcriptional and post-translational activation of PGC-1α, a transcriptional coactivator that has emerged as a master regulator of mitochondrial biogenesis [112, 113]. PGC-1α function is down-regulated in Parkinson’s disease [114], and genes controlling cellular bioenergetics and mitochondrial biogenesis regulated by PGC-1α are underexpressed in PD [115]. Interestingly, the death of cultured DA neurons through overexpression of αSyn or treatment with rotenone can be prevented by overexpressing PGC-1α, suggesting that increased PGC-1α activity is sufficient to ameliorate certain PD-like pathologies [115]. Furthermore, PGC-1α may be necessary for the beneficial effects of AMPK in models of PD [116].
Mitochondrial dynamics, orchestrated by ongoing fission and fusion events, contribute to the dilution and sequestration of mitochondrial damage. Mitochondrial fusion and fission are regulated through the expression and phosphorylation of Mitofusin proteins and Drp1, respectively. AMPK promotes mitochondrial fusion, producing larger and more highly branched mitochondrial networks, through activation of PGC-1α which preferentially increases the transcription of Mitofusin proteins over Drp1 [117, 118]. AMPK may also inhibit mitochondrial fission through inhibition of mTORC1 [119]. However, AMPK has also been shown to facilitate fission through direct phosphorylation of mitochondrial fission factor, which recruits Drp1 to the outer mitochondrial membrane (OMM) [120]. Thus, AMPK may have a dynamic role mediating fission and fusion depending on cellular energy status. During mild energy shortage AMPK may promote fusion to maximize energy production, but in response to more severe cellular stress fission may be promoted to facilitate mitophagy [121].
Mitophagy is the autophagic process leading to lysosomal degradation of damaged mitochondria, which accumulate during aging and under oxidative stress conditions. The main form of mitophagy is mediated by PINK1 and parkin. Under normal conditions in healthy mitochondria, PINK1 is taken up by mitochondria through the OMM, into the inner membrane where it is cleaved and degraded. However, in damaged mitochondria, PINK1 becomes stably associated with the OMM and escapes cleavage. Parkin is then recruited to the OMM and activated by PINK1. Upon efficient stabilization and activation on the OMM, parkin ubiquitinates proteins on the OMM initiating recruitment to the autophagosome [122]. During energy stress, AMPK facilitates mitophagy through increased fission and by promoting autophagosome initiation and lysosomal targeting through phosphorylation of ULK1 [123]. Thus, AMPK has a key role in mitochondrial homeostasis, coupling fission to mitophagy and signaling the nucleus to initiate biogenesis of new mitochondria to replace damaged ones (Fig. 2).
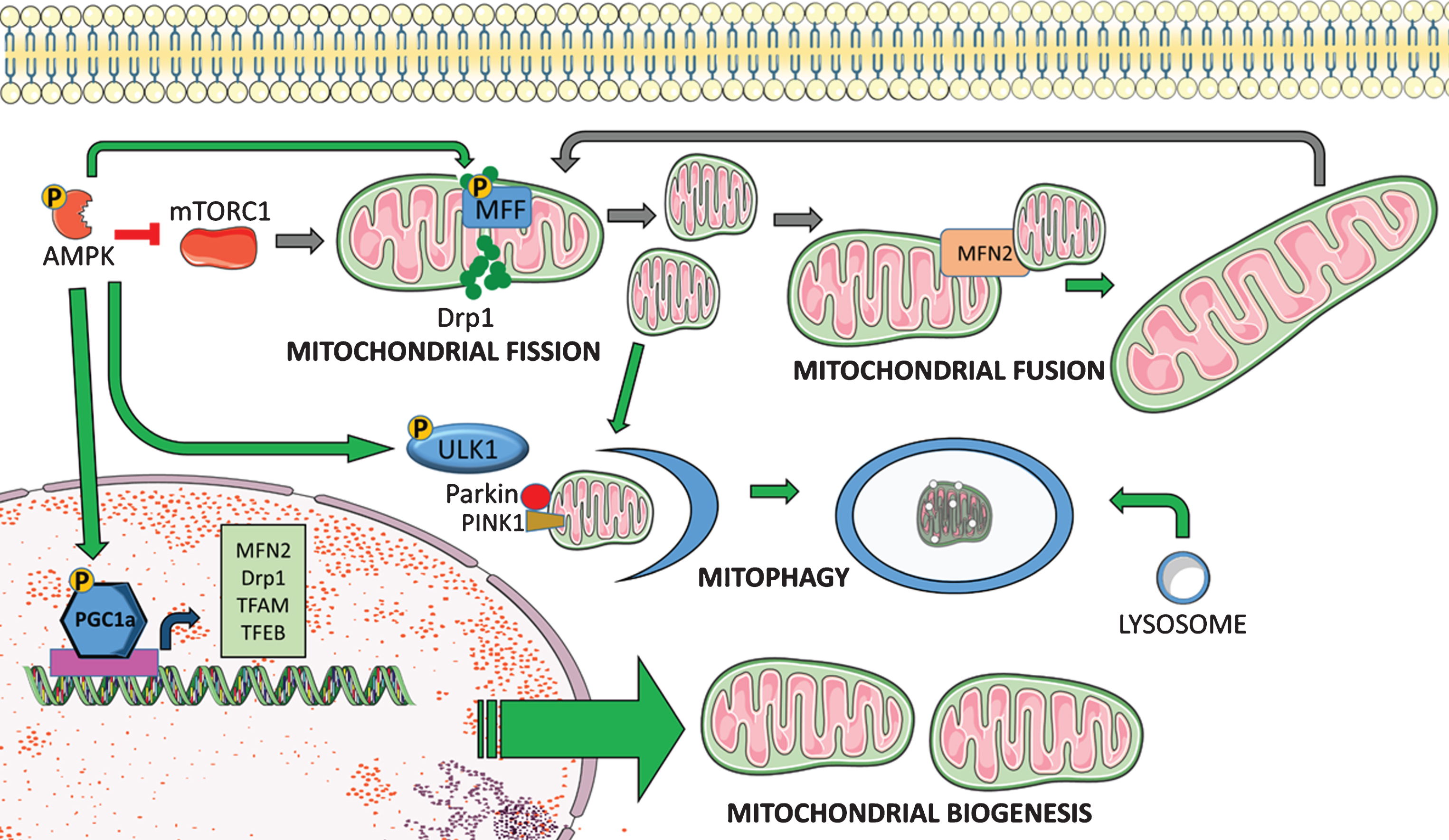
Fig.2
AMPK regulates mitochondrial quality control. AMPK facilitates mitochondrial quality control through direct phosphorylation of target proteins and transcriptional regulation of relevant genes. AMPK promotes mitochondrial biogenesis via increased transcription and post-translational phosphorylation of PGC-1α. PGC-1α is a master regulator of mitochondrial biogenesis that activates mitochondrial transcription factor A (TFAM), which drives transcription and replication of mitochondrial DNA. PGC-1α also facilitates mitochondrial fission and fusion through expression of Drp1 and Mitofusin2 (MFN2), and it promotes mitophagy and lysosomal biogenesis via activation of transcription factor EB (TFEB). AMPK can further promote mitophagy through phosphorylation of ULK1, which facilitates autophagosome formation and targeting of damaged mitochondria to lysosomes. Additionally, phosphorylation of mitochondrial fission factor (MFF) facilitates mitophagy through increased fission.
Redox homeostasis
Within cells ROS are generated both deliberately (e.g., NADPH oxidase) and as a byproduct of other cellular processes (e.g., OXPHOS), and they have many important biological roles, such as immune defense and cellular signaling. In healthy cells their production is tightly controlled and matched with antioxidant enzymes (glutathione peroxidase (GPX), catalase, and superoxide dismutase (SOD)) and transporter proteins (e.g., uncoupling protein 2 (UCP2)). However, when unchecked ROS can produce oxidative damage whereby they extract electrons from and destabilize other molecules, including proteins, lipids, and DNA [63]. As individuals age, antioxidant capabilities decline and mitochondrial dysfunctions accumulate leading to increased ROS [124]. As the primary source of ROS, mitochondria are especially vulnerable to oxidative stress. Damaged mitochondria produce more ROS creating a feed-forward cycle or oxidative stress and cellular damage that compounds across an organism’s lifespan [70]. ROS are strongly implicated in the pathogenesis of PD [125], and heightened oxidative stress is seen in the brains of PD patients relative to age-matched controls [126].
AMPK activation can decrease ROS though multiple mechanisms. As discussed above, AMPK can curtail the overproduction of ROS by increasing mitochondrial quality control. It can also decrease ROS by increasing cellular antioxidant defenses. AMPK increases production of NADPH (which is necessary for recycling the antioxidant cofactor GSH) by increasing glucose utilization through the pentose phosphate pathway, inhibiting FA synthesis, and stimulating FA β-oxidation [70, 127]. AMPK can also decrease ROS by activating PGC-1α and FOXO3 thereby increasing transcriptional regulation of antioxidant defenses including GPX, catalase, SOD, and UCP2 [128–132]. Activating AMPK may therefore be a viable strategy to decrease the production of ROS and increase cellular antioxidant capabilities, thereby reducing the potential for oxidative stress in vulnerable neurons such as the nigrostriatal DA system.
Anti-inflammation
Microglia are the resident macrophages of the central nervous system (CNS). In their “resting” state known as M2, they help to maintain CNS homeostasis and participate in cellular repair. However, upon activation, M2 microglia can rapidly switch to their “reactive” form known as M1 [133]. Microglia are activated by pathogens and a number of different extracellular signals including glutamate, proinflammatory cytokines, and ROS. Reactive microglia aggressively respond to these inflammatory signals by generating ROS through NADPH oxidase (NOX) and NO through inducible nitric-oxide synthase (iNOS) to help degrade pathogens or damaged cells [133, 134]. As opposed to acute reactivity, chronic inflammation has increasingly detrimental effects and has been strongly implicated in PD [135]. Chronic neuroinflammation increases with age and is especially elevated in the brains of PD patients [70, 136, 137]. Interestingly, increased chronic inflammation coincides with the gradual age-related decline of AMPK function, suggesting a potential correlation [8, 138].
Indeed, there is a strong negative association between inflammation and AMPK activity. AMPK exerts powerful anti-inflammatory effects, including inhibition of NOX-mediated ROS production [139, 140], iNOS-mediated NO production [141], and NF-κB-mediated production of proinflammatory cytokines such as IL-1 and TNFα [28, 142]. Furthermore, the transition from M2 to M1, involves a critical metabolic switch from OXPHOS to glycolysis. AMPK may exert an anti-inflammatory effect by counteracting this metabolic switch [133, 138]. The anti-inflammatory effects of AMPK activation are extremely robust and consistent across models, cell types, and different AMPK activators [143, 144]. Given the purported role of neuroinflammation in the pathogenesis of PD, AMPK activation may be a means to promote neuroprotection [145].
Programmed cell death
Activation of AMPK may promote cell survival by some combination of all the above mechanisms. However, AMPK may also play a role in mediating programmed cell death (PCD) under severe stress conditions. There are multiple overlapping pathways that mediate PCD, including intrinsic caspase-dependent apoptosis, autophagic cell death, and parthanatos. A detailed discussion of these pathways is beyond the scope of this review, but each has been implicated in PD neurodegeneration [146]. AMPK is sensitive to falling cellular energy levels and apoptotic signals, and may promote PCD by providing the necessary ATP [147]. This may be a protective strategy to prevent uncontrolled necrotic cell death that could damage neighboring cells. There is also some evidence that AMPK activation can promote PCD via FOXO3 mediated transcription of proapoptotic proteins [148]. FOXO3 mediates multiple conflicting transcriptional programs, many of which promote cell survival and are associated with increased cell viability and lifespan [149–152]. However, during cellular stress, c-Jun N-terminal kinase (JNK) can phosphorylate FOXO3 at serine 574 producing a proapoptotic transcriptional program [153]. Specifically, FOXO3 can increase transcription of Bim, facilitating formation of the BAX/BAK mediated mitochondrial outer membrane pore and release of apoptotic initiators cytochrome c and apoptosis-inducing factor (AIF), and this has been linked to prolonged AMPK activation [154, 155]. Neocortical neurons transfected with constitutively active AMPK or treated with a high concentration of AICAR, display increased apoptotic markers in a Bim dependent manner. Down regulation of AMPK with siRNA attenuated NMDA-induced cell death [154]. However, AMPK also promotes transcription of anti-apoptotic proteins that prevent PCD [36], and other studies have suggested that AMPK activation protects cells from PCD by restoring energy balance through increased mitochondrial metabolism [156, 157]. The effects of AMPK on PCD are thus complex and likely depend on multiple factors. Understanding the conditions in which AMPK activation promotes versus inhibits neuronal survival will be critical if it is to be a viable neuroprotective treatment for PD.
AMPK ACTIVATION AS A TREATMENT STRATEGY FOR PD
Given the many functions of AMPK and their relevance to the biochemical changes occurring in PD, it is not surprising that a substantial number of studies have investigated the neuroprotective effects of activating AMPK in models of PD. However, it remains unclear whether AMPK activation using available approaches will translate into a viable treatment strategy for PD [158]. The two cardinal pathologies of PD are the degeneration of SNc DA neurons and the accumulation of αSyn aggregates in Lewy bodies. In both cases, contradictory findings have been reported regarding the effects of AMPK activation, which are discussed in detail below.
Protection of nigrostriatal DA neurons
As discussed above, AMPK regulates numerous cellular functions that might facilitate neuroprotection in PD, including energy homeostasis, macroautophagy, inflammation, antioxidant defenses, and mitochondrial quality control. The selective degeneration of SNc DA neurons and the ensuing cascade of motor-behavior deficits that characterize PD can be replicated in model systems via the administration of dopaminergic toxins such as MPTP, or through expression of PD-associated genetic mutations. Numerous studies have evaluated AMPK in these models to determine if increasing AMPK activity can provide neuroprotection and ameliorate these deficits.
Across many different model systems, including rodents, flies, and cultured cells, AMPK activation can produce significant neuroprotection. Increasing AMPK activity through its transfectional overexpression in SH-SY5Y cells ameliorated cell loss induced by MPP+ (the active metabolite of MPTP), whereas decreasing AMPK activity with the AMPK inhibitor compound C or expressing a dominant-negative form of AMPK (AMPK-DN) exacerbated cell loss [159]. Mutations in LRRK2 and Parkin are the most common causes of heritable PD, and in Drosophila these mutations produce a clear age-related PD phenotype, with loss of DA neurons, motor deficits, and mitochondrial dysfunction. Each of these abnormalities is rescued by expression of a constitutively active form of AMPK (AMPK-CA) and exacerbated by expression of AMPK-DN or AMPK knockdown with siRNA [160]. The neuroprotective effects of genetic AMPK manipulations are informative, but for clinical viability these effects should ideally be replicable with pharmacological AMPK activators. And indeed, a number of AMPK activating compounds do have similar neuroprotective effects [160]. Of the available pharmacological activators, resveratrol, GPA, and metformin have particular clinical relevance because they have each been tested or are used clinically (although not currently for PD) and are well tolerated in humans [55, 161, 162]. However, these agents also have AMPK-independent effects, so it is not always clear whether their effects are related to AMPK activation [49].
Treatment with resveratrol for 10 weeks attenuated 6-OHDA-induced degeneration of SNc DA neurons and decreased pro-inflammatory cytokines in rats [163]. In mice, daily administration of resveratrol for 1-2 weeks prevented MPTP-induced depletion of striatal dopamine (DA) and tyrosine hydroxylase (TH) and loss of DA neurons in the SNc [164]. Many of resveratrol’s potentially therapeutic effects are mediated through AMPK activation, including increased macroautophagy, mitochondrial biogenesis, and expression of antioxidant genes [165, 166]. However, neither of the above PD-related studies verified whether AMPK was involved in the observed protection of SNc DA neurons. GPA appears to have similar neuroprotective effects. It extends lifespan in Drosophila via activation of AMPK [167], and it is neuroprotective in MPTP treated mice [168]. A diet of 1% GPA for 4 weeks robustly increased AMPK activity and mitochondrial respiratory capacity in the striatum, and fully prevented MPTP-induced loss of DA neurons in the SNc and ameliorated DA depletion in the striatum. These neuroprotective effects were hypothesized to be AMPK-dependent but this was not directly tested [168].
Compared to resveratrol and GPA, metformin has been more extensively tested in models of PD. Metformin is the most widely prescribed medication for type-2 diabetes (T2D) and has well-established effects in the liver but is also taken up in the brain [169]. Treatment of mice with metformin for 5 weeks significantly ameliorated MPTP-induced degeneration of SNc DA neurons, partially restored striatal DA content, and fully restored normal motor behavior on the rotarod test [170]. Metformin also increased macroautophagy, reduced αSyn, decreased reactive microglia, and attenuated expression of proinflammatory cytokines. These findings were further tested in SH-SY5Y cells, where metformin similarly increased cell survival and attenuated lactate dehydrogenase (LDH) release following treatment with MPP+. Metformin also increased markers of macroautophagy, decreased ROS, and lowered the number of dysfunctional mitochondria. Each of these effects was prevented by the AMPK inhibitor compound C and by the macroautophagy inhibitor 3-Methyladenine (3-MA). These findings suggest that AMPK activation via metformin is neuroprotective through multiple mechanisms, including decreased neuroinflammation, clearance of αSyn, and increased mitochondrial quality control [170]. Similar results were observed in mice treated with MPTP for 1 week followed by 1 week of metformin treatment [171]. Metformin ameliorated the loss of SNc DA neurons, restored striatal DA content, attenuated motor deficits, and decreased reactive astrogliosis in the striatum and SNc. Metformin also increased expression of the neurotrophic factor BDNF. In MPP+ treated SH-SY5Y cells, inhibiting the BDNF receptor (TrkB) blocked the neuroprotective effects of metformin, suggesting that BDNF signaling might be a critical pro-survival mechanism of metformin treatment [171].
Increased BDNF was also observed in mice treated with metformin for 21 days, concurrent with MPTP for the first 5 days [172]. Interestingly, after 4 days of treatment, motor deficits were equivalent between MPTP and MPTP+metformin treated mice, indicating that metformin did not attenuate the acute toxicity of MPTP. However, by day 20 motor function was significantly improved in the metformin treated mice, while remaining unchanged in the MPTP-only group. Metformin treated mice also had increased expression of the antioxidant enzymes SOD and catalase, decreased evidence of oxidative stress, and attenuated loss of SNc DA neurons [172]. These findings suggest that the chronic treatment regimen of metformin may have promoted neuronal repair, with BDNF possibly playing an important role.
However, it is unclear whether all of these neuroprotective effects are due to increased AMPK activity. In mice treated with metformin for 2 weeks, increased PGC-1α expression was necessary for the attenuation of MPTP-induced SNc DA neuronal loss. Although AMPK activity was not assessed in vivo, metformin increased cell viability and PGC-1α expression in MPP+ treated SH-SY5Y cells, and there was no evidence that AMPK activity was increased [173]. Similarly, a phosphoproteomic analysis in mice found that 2 weeks of metformin treatment (at doses similar to those used in the neuroprotective studies above) up- or downregulated over 60 neuronal phosphoproteins but had no effect on AMPK [174]. Metformin has numerous functions independent from AMPK activation [175], so it is conceivable that some of the neuroprotective benefits of metformin observed in models of PD are not related to increased AMPK activity. Indeed, AMPK knockout in DA neurons did not diminish the neuroprotective effects of metformin in MPTP-treated mice [176]. Metformin treatment ameliorated SNc DA neuron loss and decreased reactive gliosis to a similar extent in both WT and KO mice. This seemingly indicates that metformin’s neuroprotective effects were AMPK-independent. However, AMPK was still functional in glial cells and other non-DA cells, so AMPK may still have mediated these effects (potentially by decreasing neuroinflammation), but additional studies will be necessary to clarify the precise role of AMPK in these models.
The lack of specificity among the pharmacological tools used to activate and inhibit AMPK presents a challenge for determining whether their effects are due to AMPK or some other off-target effect. Most known AMPK activating drugs have substantial AMPK-independent effects [177]. For instance, at high concentrations AICAR can induce apoptosis via an AMPK-independent mechanism [178]. The selectivity of AMPK inhibitors is also problematic. The most frequently used small molecule inhibitor of AMPK, compound C, potently inhibits other kinases and has numerous AMPK-independent effects, including antiproliferative effects and cytotoxicity [179]. Thus, conclusions about the effects of AMPK based on these modulators must be considered with caution. There is a significant need to develop more targeted approaches to manipulate AMPK activity, both for research purposes and as potential therapeutics.
Clearance of αSyn
The accumulation of αSyn into intraneuronal aggregates termed Lewy bodies is a pathological hallmark of PD that is associated with numerous detrimental effects, including impaired gene expression, increased ROS, mitochondrial dysfunction, and cell death [102]. Overexpression of αSyn is cytotoxic in vivo and in vitro [96, 180], and mutations of SNCA that lead to overexpression of αSyn in humans are sufficient to cause PD [1]. Post-mortem examinations of PD brain tissue have also noted evidence of impaired macroautophagy. Specifically, there is evidence of defective autophagic flux, whereby autophagosomes accumulate but are not cleared effectively by lysosomes [181]. Treatments that promote macroautophagy initiation and completion may therefore be neuroprotective in PD [182, 183]. As discussed above, AMPK promotes macroautophagy through its direct stimulation of ULK1, its indirect suppression of mTORC1, and through transcriptional regulation of autophagosome formation and lysosomal biogenesis. Decreasing the accumulation of αSyn may therefore contribute to the beneficial effects of AMPK activation in models of PD.
Several studies have successfully used AMPK activating agents to facilitate clearance of αSyn and promote neuronal survival [103]. Overexpression of the stress response protein sestrin2 stimulated macroautophagy, reduced αSyn accumulation, and attenuated cytotoxicity in rotenone treated Mes 23.5 cells [184]. These protective effects were found to be AMPK-dependent, and treatment with metformin was sufficient to restore neuroprotective effects in sestrin2-knockdown cells [184]. Similarly, treatment of SH-SY5Y cells with resveratrol activated AMPK and facilitated macroautophagy, reducing αSyn aggregation, cell death, and markers of PCD following treatment with rotenone. These pro-survival effects were prevented by inhibiting AMPK with compound C [185]. Further illustrating the importance of AMPK-mediated macroautophagy in PD, Clk1 deficient mice, which have impaired AMPK function and decreased autophagy, are more sensitive to MPTP-induced neurodegeneration. Treatment with metformin increased macroautophagy and ameliorated MPTP-induced motor deficits and loss of SNc DA neurons in these mice and increased cell viability in cultured MN9D cells [186].
Similar findings linking AMPK activity with αSyn expression have been noted in models with genetically-induced αSyn overexpression. In PC12 cells overexpressing αSyn, resveratrol reduced αSyn, and this effect was blocked by knockdown of Beclin-1 or lysosomal inhibition, suggesting that increased macroautophagy was essential for resveratrol-induced clearance of αSyn [185]. Similarly, PC12 cells incubated in cell media from αSyn overexpressing cells also had decreased viability and lower AMPK activity, but they could be rescued by activating AMPK with AICAR or metformin [187]. In rats, αSyn overexpression decreased the number of SNc DA neurons, while overexpression of the α subunit of AMPK or AMPK-CA ameliorated neuronal loss and deceased αSyn [188]. Likewise, αSyn overexpression in retinoic acid differentiated SH-SY5Y cells increased cell death and decreased AMPK activity [187]. Treatment with AICAR or metformin was neuroprotective in these cells and in cells incubated with cell media containing secreted αSyn or recombinant αSyn oligomers. Conversely, cells with AMPK knocked down via shRNA were more vulnerable to αSyn-induced cell death [187].
The above studies all suggest that AMPK activation can decrease αSyn and ameliorate the deficits caused by its aggregation. However, the relationship between AMPK and αSyn is not straightforward. Some studies have noted that αSyn decreases AMPK phosphorylation at Thr172 [187], while others have observed that αSyn increases it [189]. One possible explanation for these contradictory findings is that αSyn and phosphorylated αSyn differentially affect AMPK. Under normal physiological conditions, αSyn is mostly unphosphorylated. However, in PD αSyn becomes increasingly phosphorylated at serine 129 (pS129), and over 90% of αSyn within Lewy bodies is phosphorylated [190, 191]. Unphosphorylated αSyn activates the phosphatase PP2A, which promotes the dephosphorylation of both αSyn and AMPK, whereas pS129-αSyn inhibits PP2A [192, 193]. Furthermore, pS129-αSyn was found to sequester PIKE-L, an inhibitor of AMPK, leading to increased AMPK activity in mice and in SH-SY5Y cells overexpressing αSyn [189]. Thus, unphosphorylated αSyn may inhibit AMPK function through PP2A, but as αSyn becomes increasingly phosphorylated this inhibition declines and other AMPK regulating proteins become inhibited, leading to increased AMPK activity [193]. Yet increased AMPK in these conditions may not necessarily correspond with normal physiological AMPK activation. There is evidence that AMPK is highly dysregulated in cultured neurons overexpressing αSyn and in human cells with Lewy body pathology, with AMPK accumulating with αSyn outside the nucleus or around the rim of Lewy bodies [194]. Increasing AMPK activity further in this case may be detrimental to both cell survival and αSyn clearance. In mouse primary neuronal cultures, overexpression of αSyn or treatment with lactate increased pS129-αSyn and AMPK activity (as measured by pThr172). Overexpression of AMPK-DN or AMPK knockdown prevented lactate induced increases in pS129 and total αSyn and ameliorated the decreased neurite length in cells overexpressing αSyn or treated with lactate. Conversely, AMPK-CA expression increased αSyn, decreased neurite length, and impaired αSyn turnover in 3D5 cells [194].
The role of pS129 in αSyn aggregation, seeding, and toxicity is controversial [191]. There is some evidence that pS129 reduces αSyn toxicity and promotes its macroautophagic clearance [195]. However, other studies have observed that pS129 increases the toxicity of αSyn and promotes aggregation [171, 193]. The pathological role of pS129 may change across the course of PD. Early in disease progression pS129 may be protective, but as the severity of PD increases, pS129 may become increasingly detrimental [190]. Interestingly, AMPK may be one of the endogenous kinases that phosphorylates αSyn. Expression of AMPK-CA in vitro increased pS129-αSyn, while expression of AMPK-DN deceased it [189, 194]. However, treatment with metformin either in vitro or in vivo has repeatedly been shown to decrease phosphorylation of αSyn, and this effect is apparently through an AMPK-independent mechanism [171, 174, 196, 197]. Metformin increases the activity of PP2A, thereby decreasing pS129-αSyn, and this effect was necessary for the protective action of metformin in MPP+ treated SH-SY5Y cells [171]. Thus, the effects of metformin on αSyn phosphorylation and clearance might be distinct from the effects of AMPK itself, with differing neuroprotective effects depending on the state of disease progression (Fig. 3). A pure AMPK activator may be more beneficial in the early stages of PD or as a preventative, whereas metformin may be more beneficial later in the disease progression. Further research will be necessary to clarify whether AMPK activation promotes or inhibits the clearance of αSyn and whether the beneficial effects of AMPK activators such as metformin are due to AMPK.
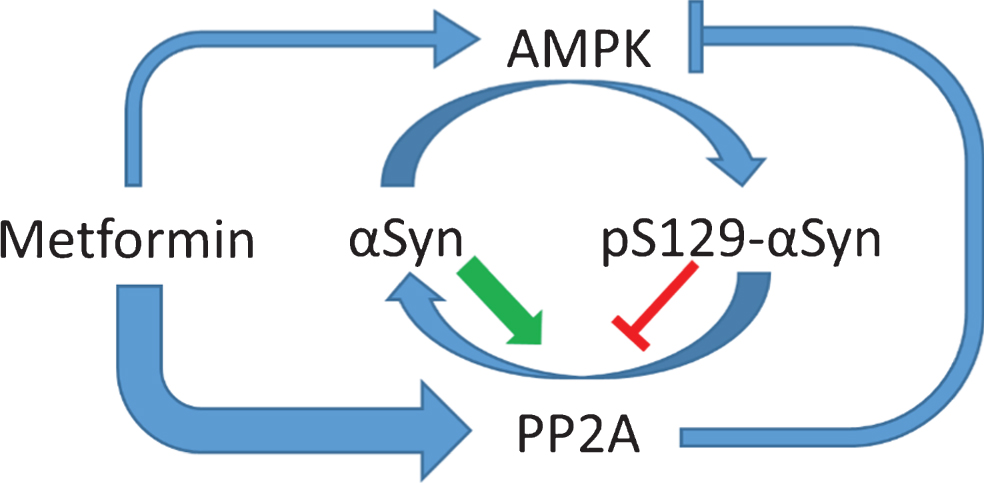
Fig.3
Metformin and AMPK have divergent effects on alpha-synuclein S129 phosphorylation. αSyn becomes increasingly phosphorylated at S129 in patients with PD. Multiple kinases are responsible for αSyn phosphorylation, including AMPK. pS129 may promote macroautophagic clearance of aggregated αSyn, but it may also increase the toxicity αSyn. Thus, the pathological role of pS129 in PD is complex and may change depending on disease severity. pS129 is dephosphorylated by PP2A, and unphosphorylated αSyn increases the activity of PP2A, while pS129-αSyn decreases PP2A activity. PP2A can also dephosphorylate and inhibit AMPK at pThr172. Thus, AMPK may be regulated in part by the phosphorylation state of αSyn. Metformin activates PP2A via an AMPK independent mechanism with higher potency than it activates AMPK, at least in vitro, and thereby promotes the dephosphorylation of pS129-αSyn.
Atrophy and cell death: A dark side of AMPK?
The majority of studies that have evaluated AMPK activity in models of PD have reported increased neuroprotection. However, several well-designed studies have conversely noted that AMPK activation facilitated neuronal atrophy and death in response to PD-related toxins. Perhaps the most obvious mechanism by which AMPK might facilitate neurodegeneration is through excessive macroautophagy and impaired protein synthesis that limits cellular regrowth, both of which could be mediated partly through AMPK’s inhibition of mTORC1 [95]. In a study of cultured murine midbrain DA neurons, cell loss induced by MPP+ was exacerbated by treatment with the canonical mTORC1 inhibitor rapamycin, while treatments that inhibited macroautophagy were neuroprotective [198]. Likewise, 6-OHDA increased autophagy, ROS, and apoptotic cell death in SH-SY5Y cells, and inhibiting macroautophagy with 3-MA or an AMPK inhibiting neurotrophic factor ameliorated these deficits and increased cell viability [199]. MPP+ similarly increased macroautophagy and markers of PCD in SH-SY5Y cells, and expression of AMPK-DN attenuated these effects, while 3-MA prevented cell death as measured by LDH release [189]. In both primary neurons and PC12 cells, administration of 6-OHDA, rotenone, or MPP+ each increased AMPK activity, decreased cell viability, and promoted apoptosis. Cell viability was improved by inhibiting AMPK with compound C, expressing AMPK-DN, or by inhibiting macroautophagy through overexpression of mTORC1 [200].
Detrimental effects of AMPK activation have also been observed in vivo. Lentiviral expression of AMPK-DN in the SNc of mice decreased MPTP-induced macroautophagy, ameliorated TH loss in the striatum and SNc, and attenuated motor deficits [189]. Treatment of mice with metformin for 1-week reduced MPTP-induced reactive microglia and pro-inflammatory cytokines in the SNc (indicating a strong anti-inflammatory effect), but metformin treatment did not attenuate the loss of SNc DA neurons, and it actually worsened MPTP-induced DA and DOPAC depletion. In an in vitro follow up, metformin decreased cell viability in response to MPP+, increased necrosis and ROS, and further exacerbated ATP loss [201]. Similarly, treatment of mice with metformin following 6-OHDA, increased nuclear translocation of AIF, decreased TH intensity in the SNc, increased cellular atrophy, and decreased the number of SNc DA neurons. Expression of AMPK-DN ameliorated all of these effects, substantially reducing neuronal atrophy and modestly decreasing PCD [202].
Although increased macroautophagy may facilitate αSyn clearance, nutrient recycling, and help promote mitophagy, the above data suggest that increasing macroautophagy as a neuroprotective strategy may have limits [203]. Excessive macroautophagy may cannibalize important cellular machinery, or cellular stress may irreparably impair the macroautophagic process such that autophagy cannot be completed successfully or safely. For example, high levels of ROS can impair the membrane integrity of lysosomes, leading to protease leakage that can damage other cellular components [181]. If lysosomal function is compromised, stimulating macroautophagy via AMPK activation may lead to a pathological accumulation of autophagosomes that cannot be effectively cleared [183]. The inhibition of cell growth pathways by AMPK may also promote neurodegeneration. Sustained AMPK activation may be detrimental by locking cells into a state of atrophy and preventing cellular regrowth following a severe but not necessarily lethal stressor [202].
More research is necessary to clarify the conditions in which AMPK activation is neuroprotective versus detrimental. Variables such as cell type, bioenergetic status, the specific cellular stressors (including baseline cellular stress), and the length and intensity of AMPK activation are all likely relevant factors [48, 53]. For example, the relationship between AMPK activity and cell death under some conditions may be biphasic, such that at certain levels AMPK activation promotes cell survival, but at higher intensities or over a longer duration it promotes PCD [155]. Determining the optimal treatment regimen and the appropriate level of AMPK activation to maximize its neuroprotective benefits, while limiting its potential adverse effects, will be essential. The neuroprotective effects of AMPK activators, such as metformin, appear to have a narrow effective dose ranges in both in vivo and in vitro models [171, 204]. There is evidence from studies in mice that a chronic low level of AMPK activity may be most beneficial [188]. Furthermore, the therapeutic efficacy of AMPK activation may depend on the stage and severity of PD. Several researchers have suggested that AMPK could be neuroprotective during the early stages of PD, but become less effective or potentially detrimental during more progressed stages of the disease [181, 188, 205].
The widespread clinical use of metformin for diabetes has provided a window into the effects of an AMPK activating agent on PD risk in humans. Several large cohort studies have tracked the development of PD in patients receiving metformin. In a Taiwanese T2D population, treatment with metformin and a sulfonylurea significantly reduced the risk of developing PD compared to patients not receiving oral anti-hyperglycemic treatment. Sulfonylureas alone increased PD risk while metformin had no effect alone [206]. However, a more recent cohort study from Taiwan found that T2D patients treated with metformin had a significantly higher cumulative incidence of PD and dementia compared to other T2D patients, and PD risk increased with higher doses of metformin and longer treatment durations [207]. These results suggest that metformin is unlikely to be a panacea for treating PD, and it may actually have an adverse effect. However, the effects of metformin on PD incidence in diabetic patients may be distinct from its effects in non-diabetics. And these results may not necessarily apply to other AMPK activating drugs. In a Norwegian diabetic population, the incidence of PD was compared between metformin users and glitazone users (pioglitazone and/or rosiglitazone) across a 10-year period. Although glitazones are potent peroxisome proliferator-activated receptor γ (PPARγ) agonists, they also activate AMPK [49]. Glitazone use was associated with a significantly lower incidence of PD compared to metformin use [208]. Although no other comparison groups were included in this study, the annual incidence rate of PD among the general Norwegian population is 12.6/100,000 people [209], which is noticeably higher than that observed in the glitazone treated diabetic patients (8.2/100,000 patient-years) and marginally lower than in metformin treated patients (14.2/100,000 patient-years) [208]. These clinical observations further suggest that the relationship between AMPK and PD may be complex. Further preclinical research should be undertaken to better understand the potential positive and negative effects of AMPK activation and to identify the best drug candidates for future clinical trials.
CONCLUSIONS
The development of therapeutic strategies to decrease the accumulation and spread of αSyn and slow or halt the degeneration of DA neurons in PD is a major unmet medical need. Many of the core mechanisms that underlie the pathology of PD can be regulated, as least in part, through the activation of AMPK. Although contradictory findings regarding the effects of AMPK activation in various models of PD provide reason for caution in deploying this strategy clinically, future preclinical studies will hopefully clarify the basis of these discrepancies. Finding the appropriate level of AMPK activation and developing more selective pharmacological activators may be key for effective therapeutic utilization of AMPK activation and may decrease the incidence of undesirable effects that have been observed in some prior studies. Alternatively, given the vast range of AMPK’s regulatory effects, identifying the specific downstream pathways that are most beneficial for treating PD may be necessary so that these pathways can be more selectively targeted without activating pathways responsible for the detrimental effects. Through additional careful research and more targeted pharmacotherapeutics, activating PD-relevant AMPK-dependent pathways may emerge as a powerful strategy to decrease neurodegeneration and ultimately curtail the progression of PD.
CONFLICTS OF INTEREST
The authors have no conflicts of interest to report.
ACKNOWLEDGMENTS
This work was supported by the National Institute on Aging (NIH R01AG048918 to John D. Elsworth).
REFERENCES
[1] | Poewe W , Seppi K , Tanner CM , Halliday GM , Brundin P , Volkmann J , Schrag AE , Lang AE ((2017) ) Parkinson disease. Nat Rev Dis Primers 3: , 1–21. |
[2] | Goldman SM ((2014) ) Environmental toxins and Parkinson’s disease. Annu Rev Pharmacol Toxicol 54: , 141–164. |
[3] | Brown TP , Rumsby PC , Capleton AC , Rushton L , Levy LS ((2006) ) Pesticides and Parkinson’s disease - Is there a link? Environ Health Perspect 114: , 156–164. |
[4] | Ali SF , Binienda ZK , Imam SZ ((2011) ) Molecular aspects of dopaminergic neurodegeneration: Gene-environment interaction in parkin dysfunction. Int J Environ Res Public Health 8: , 4702–4713. |
[5] | Gao J , Nalls MA , Shi M , Joubert BR , Hernandez DG , Huang X , Hollenbeck A , Singleton AB , Chen H ((2012) ) An exploratory analysis on gene-environment interactions for Parkinson disease. Neurobiol Aging 33: , 2528.e1–6. |
[6] | Habibi E , Masoudi-Nejad A , Abdolmaleky HM , Haggarty SJ ((2011) ) Emerging roles of epigenetic mechanisms in Parkinson’s disease. Funct Integr Genomics 11: , 523–537. |
[7] | Hardie DG , Ross FA , Hawley SA ((2012) ) AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13: , 251–262. |
[8] | Salminen A , Kaarniranta K ((2012) ) AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev 11: , 230–241. |
[9] | Culmsee C , Monnig J , Kemp BE , Mattson MP ((2001) ) AMP-activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. J Mol Neurosci 17: , 45–58. |
[10] | Li J , Zeng Z , Viollet B , Ronnett GV , McCullough LD ((2007) ) Neuroprotective effects of adenosine monophosphate-activated protein kinase inhibition and gene deletion in stroke. Stroke 38: , 2992–2999. |
[11] | Ashabi G , Khalaj L , Khodagholi F , Goudarzvand M , Sarkaki A ((2015) ) Pre-treatment with metformin activates Nrf2 antioxidant pathways and inhibits inflammatory responses through induction of AMPK after transient global cerebral ischemia. Metab Brain Dis 30: , 747–754. |
[12] | Kuramoto N , Wilkins ME , Fairfax BP , Revilla-Sanchez R , Terunuma M , Tamaki K , Iemata M , Warren N , Couve A , Calver A , Horvath Z , Freeman K , Carling D , Huang L , Gonzales C , Cooper E , Smart TG , Pangalos MN , Moss SJ ((2007) ) Phospho-dependent functional modulation of GABA(B) receptors by the metabolic sensor AMP-dependent protein kinase. Neuron 53: , 233–247. |
[13] | Kurumbail RG , Calabrese MF ((2016) ) Structure and Regulation of AMPK. EXS 107: , 3–22. |
[14] | Ross FA , MacKintosh C , Hardie DG ((2016) ) AMP-activated protein kinase: A cellular energy sensor that comes in 12 flavours. FEBS J 283: , 2987–3001. |
[15] | Cantó C , Auwerx J ((2010) ) AMP-activated protein kinase and its downstream transcriptional pathways. Cell Mol Life Sci 67: , 3407–3423. |
[16] | Hawley SA , Davison M , Woods A , Davies SP , Beri RK , Carling D , Hardie DG ((1996) ) Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem 271: , 27879–27887. |
[17] | Woods A , Johnstone SR , Dickerson K , Leiper FC , Fryer LGD , Neumann D , Schlattner U , Wallimann T , Carlson M , Carling D ((2003) ) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol 13: , 2004–2008. |
[18] | Hawley SA , Pan DA , Mustard KJ , Ross L , Bain J , Edelman AM , Frenguelli BG , Hardie DG ((2005) ) Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab 2: , 9–19. |
[19] | Momcilovic M , Hong SP , Carlson M ((2006) ) Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem 281: , 25336–25343. |
[20] | Hawley SA , Ross FA , Gowans GJ , Tibarewal P , Leslie NR , Hardie DG ((2014) ) Phosphorylation by Akt within the ST loop of AMPK-α1 down-regulates its activation in tumour cells. Biochem J 459: , 275–287. |
[21] | Sanz P , Rubio T , Garcia-Gimeno MA ((2013) ) AMPKβ subunits: More than just a scaffold in the formation of AMPK complex. FEBS J 280: , 3723–3733. |
[22] | Xiao B , Heath R , Saiu P , Leiper FC , Leone P , Jing C , Walker PA , Haire L , Eccleston JF , Davis CT , Martin SR , Carling D , Gamblin SJ ((2007) ) Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449: , 496–500. |
[23] | Langendorf CG , Kemp BE ((2015) ) Choreography of AMPK activation. Cell Res 25: , 5–6. |
[24] | Xiao B , Sanders MJ , Carmena D , Bright NJ , Haire LF , Underwood E , Patel BR , Heath RB , Walker PA , Hallen S , Giordanetto F , Martin SR , Carling D , Gamblin SJ ((2013) ) Structural basis of AMPK regulation by small molecule activators. Nat Commun 4: , 1–10. |
[25] | Garcia D , Shaw RJ ((2017) ) AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell 66: , 789–800. |
[26] | Gowans GJ , Hawley SA , Ross FA , Hardie DG ((2013) ) AMP is a true physiological regulator of amp-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab 18: , 556–566. |
[27] | Oakhill JS , Steel R , Chen Z-P , Scott JW , Ling N , Tam S , Kemp BE ((2011) ) AMPK is a direct adenylate charge-regulated protein kinase. Science 332: , 1433–1435. |
[28] | Peixoto CA , Oliveira WH de , Araújo SM da R , Nunes AKS ((2017) ) AMPK activation: Role in the signaling pathways of neuroinflammation and neurodegeneration. Exp Neurol 298: , 31–41. |
[29] | Li X , Wang L , Zhou XE , Ke J , De Waal PW , Gu X , Tan MHE , Wang D , Wu D , Xu HE , Melcher K ((2015) ) Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res 25: , 50–66. |
[30] | Hardie DG , Lin S-C ((2017) ) AMP-activated protein kinase - not just an energy sensor. F1000Research 6: , 1–11. |
[31] | Zhang YL , Guo H , Zhang CS , Lin SY , Yin Z , Peng Y , Luo H , Shi Y , Lian G , Zhang C , Li M , Ye Z , Ye J , Han J , Li P , Wu JW , Lin SC ((2013) ) AMP as a low-energy charge signal autonomously initiates assembly of axin-ampk-lkb1 complex for AMPK activation. Cell Metab 18: , 546–555. |
[32] | Zhang CS , Hawley SA , Zong Y , Li M , Wang Z , Gray A , Ma T , Cui J , Feng JW , Zhu M , Wu YQ , Li TY , Ye Z , Lin SY , Yin H , Piao HL , Hardie DG , Lin SC ((2017) ) Fructose-1,6-bisphosphate and aldolase mediate glucose sensing by AMPK. Nature 548: , 112–116. |
[33] | Clapham DE ((2007) ) Calcium signaling. Cell 131: , 1047–1058. |
[34] | Andrews ZB ((2011) ) Central mechanisms involved in the orexigenic actions of ghrelin. Peptides 32: , 2248–2255. |
[35] | Inokuchi-Shimizu S , Park EJ , Roh YS , Yang L , Zhang B , Song J , Liang S , Pimienta M , Taniguchi K , Wu X , Asahina K , Lagakos W , Mackey MR , Akira S , Ellisman MH , Sears DD , Olefsky JM , Karin M , Brenner DA , Seki E ((2014) ) TAK1-mediated autophagy and fatty acid oxidation prevent hepatosteatosis and tumorigenesis. J Clin Invest 124: , 3566–3578. |
[36] | Kim SY , Jeong S , Jung E , Baik K-H , Chang MH , Kim SA , Shim J-H , Chun E , Lee K-Y ((2012) ) AMP-activated protein kinase-α1 as an activating kinase of TGF-β-activated kinase 1 has a key role in inflammatory signals. Cell Death Dis 3: , 1–13. |
[37] | Craig PM , Moyes CD , LeMoine CMR ((2017) ) Sensing and responding to energetic stress: Evolution of the AMPK network. Comp Biochem Physiol B Biochem Mol Biol. doi: 10.1016/j.cbpb.2017.11.001 |
[38] | Dagon Y , Hur E , Zheng B , Wellenstein K , Cantley LC , Kahn BB ((2012) ) P70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake. Cell Metab 16: , 104–112. |
[39] | McBride A , Ghilagaber S , Nikolaev A , Hardie DG ((2009) ) The glycogen-binding domain on the AMPK β subunit allows the kinase to act as a glycogen sensor. Cell Metab 9: , 23–34. |
[40] | Polekhina G , Gupta A , Michell BJ , van Denderen B , Murthy S , Feil SC , Jennings IG , Campbell DJ , Witters LA , Parker MW , Kemp BE , Stapleton D ((2003) ) AMPK beta subunit targets metabolic stress sensing to glycogen. Curr Biol 13: , 867–871. |
[41] | Meares GP , Hughes KJ , Naatz A , Papa FR , Urano F , Hansen PA , Benveniste EN , Corbett JA ((2011) ) IRE1-dependent activation of AMPK in response to nitric oxide. Mol Cell Biol 31: , 4286–4297. |
[42] | Tong X , Kono T , Evans-Molina C ((2015) ) Nitric oxide stress and activation of AMP-activated protein kinase impair β-cell sarcoendoplasmic reticulum calcium ATPase 2b activity and protein stability. Cell Death Dis 6: , 1–12. |
[43] | Anandhan A , Lei S , Levytskyy R , Pappa A , Panayiotidis MI , Cerny RL , Khalimonchuk O , Powers R , Franco R ((2017) ) Glucose metabolism and AMPK signaling regulate dopaminergic cell death induced by gene (α-Synuclein)-environment (Paraquat) interactions. Mol Neurobiol 54: , 3825–3842. |
[44] | Cardaci S , Filomeni G , Ciriolo MR ((2012) ) Redox implications of AMPK-mediated signal transduction beyond energetic clues. J Cell Sci 125: , 2115–2125. |
[45] | Hawley SA , Ross FA , Chevtzoff C , Green KA , Evans A , Fogarty S , Towler MC , Brown LJ , Ogunbayo OA , Evans AM , Hardie DG ((2010) ) Use of cells expressing gamma subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab 11: , 554–565. |
[46] | Shao D , Oka SI , Liu T , Zhai P , Ago T , Sciarretta S , Li H , Sadoshima J ((2014) ) A redox-dependent mechanism for regulation of AMPK activation by thioredoxin1 during energy starvation. Cell Metab 19: , 232–245. |
[47] | Zmijewski JW , Banerjee S , Bae H , Friggeri A , Lazarowski ER , Abraham E ((2010) ) Exposure to hydrogen peroxide induces oxidation and activation of AMP-activated protein kinase. J Biol Chem 285: , 33154–33164. |
[48] | Amato S , Man H ((2011) ) Bioenergy sensing in the brain: The role of AM-activated protein kinase in neuronal metabolism, development and neurological diseases. Cell Cycle 10: , 3452–3460. |
[49] | Kim J , Yang G , Kim Y , Kim J , Ha J ((2016) ) AMPK activators: Mechanisms of action and physiological activities. Exp Mol Med 48: , 1–12. |
[50] | Rena G , Hardie DG , Pearson ER ((2017) ) The mechanisms of action of metformin. Diabetologia 60: , 1577–1585. |
[51] | Owen MR , Doran E , Halestrap AP ((2000) ) Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 348: , 607–614. |
[52] | Zhang CS , Li M , Ma T , Zong Y , Cui J , Feng JW , Wu YQ , Lin SY , Lin SC ((2016) ) Metformin activates AMPK through the lysosomal pathway. Cell Metab 24: , 521–522. |
[53] | Lan F , Weikel KA , Cacicedo JM , Ido Y ((2017) ) Resveratrol-induced AMP-activated protein kinase activation is cell-type dependent: Lessons from basic research for clinical application. Nutrients 9: , 1–14. |
[54] | Cantó C , Gerhart-Hines Z , Feige JN , Lagouge M , Noriega L , Milne JC , Elliott PJ , Puigserver P , Auwerx J ((2009) ) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458: , 1056–1060. |
[55] | Karamat FA , Horjus DL , Haan YC , van der Woude L , Schaap MC , Oudman I , van Montfrans GA , Nieuwland R , Salomons GS , Clark JF , Brewster LM ((2017) ) The acute effect of beta-guanidinopropionic acid versus creatine or placebo in healthy men (ABC-Trial): A randomized controlled first-in-human trial. Br J Clin Pharmacol 83: , 2626–2635. |
[56] | Corton JM , Gillespie JG , Hawley SA , Hardie DG ((1995) ) 5-aminoimidazole-4-carboxamide ribonucleoside. A specific method for activating AMP-activated protein kinase in intact cells? Eur J Biochem 229: , 558–565. |
[57] | Calabrese MF , Rajamohan F , Harris MS , Caspers NL , Magyar R , Withka JM , Wang H , Borzilleri KA , Sahasrabudhe PV , Hoth LR , Geoghegan KF , Han S , Brown J , Subashi TA , Reyes AR , Frisbie RK , Ward J , Miller RA , Landro JA , Londregan AT , Carpino PA , Cabral S , Smith AC , Conn EL , Cameron KO , Qiu X , Kurumbail RG ((2014) ) Structural basis for AMPK activation: Natural and synthetic ligands regulate kinase activity from opposite poles by different molecular mechanisms. Structure 22: , 1161–1172. |
[58] | Scott JW , Ling N , Issa SMA , Dite TA , O’Brien MT , Chen ZP , Galic S , Langendorf CG , Steinberg GR , Kemp BE , Oakhill JS ((2014) ) Small molecule drug A-769662 and AMP synergistically activate naive AMPK independent of upstream kinase signaling. Chem Biol 21: , 619–627. |
[59] | Willows R , Sanders MJ , Xiao B , Patel BR , Martin SR , Read J , Wilson JR , Hubbard J , Gamblin SJ , Carling D ((2017) ) Phosphorylation of AMPK by upstream kinases is required for activity in mammalian cells. Biochem J 474: , 3059–3073. |
[60] | Cameron KO , Kurumbail RG ((2016) ) Recent progress in the identification of adenosine monophosphate-activated protein kinase (AMPK) activators. Bioorganic Med Chem Lett 26: , 5139–5148. |
[61] | Henchcliffe C , Beal FM ((2008) ) Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol 4: , 600–609. |
[62] | Poliquin PO , Chen J , Cloutier M , Trudeau LÉ , Jolicoeur M ((2013) ) Metabolomics and in-silico analysis reveal critical energy deregulations in animal models of Parkinson’s disease. PLoS One 8: , 1–9. |
[63] | Franco-Iborra S , Vila M , Perier C ((2016) ) The Parkinson disease mitochondrial hypothesis: Where are we at? Neuroscientist 22: , 266–277. |
[64] | Bose A , Beal MF ((2016) ) Mitochondrial dysfunction in Parkinson’s disease. J Neurochem 139: , 216–231. |
[65] | Berndt N , Bulik S , Holzhütter H-G ((2012) ) Kinetic modeling of the mitochondrial energy metabolism of neuronal cells: The impact of reduced α-ketoglutarate dehydrogenase activities on ATP production and generation of reactive oxygen species. Int J Cell Biol 2012: , 1–11. |
[66] | Yin F , Boveris A , Cadenas E ((2014) ) Mitochondrial energy metabolism and redox signaling in brain aging and neurodegeneration. Antioxid Redox Signal 20: , 353–371. |
[67] | Reznick RM , Zong H , Li J , Morino K , Moore IK , Yu HJ , Liu ZX , Dong J , Mustard KJ , Hawley SA , Befroy D , Pypaert M , Hardie DG , Young LH , Shulman GI ((2007) ) Aging-associated reductions in AMP-activated protein kinase activity and mitochondrial biogenesis. Cell Metab 5: , 151–156. |
[68] | Reeve A , Simcox E , Turnbull D ((2014) ) Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res Rev 14: , 19–30. |
[69] | Devi L , Raghavendran V , Prabhu BM , Avadhani NG , Anandatheerthavarada HK ((2008) ) Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem 283: , 9089–9100. |
[70] | Anandhan A , Jacome MS , Lei S , Hernandez-Franco P , Pappa A , Panayiotidis MI , Powers R , Franco R ((2017) ) Metabolic dysfunction in Parkinson’s disease: Bioenergetics, redox homeostasis and central carbon metabolism. Brain Res Bull 133: , 12–30. |
[71] | Goldberg JA , Guzman JN , Estep CM , Ilijic E , Kondapalli J , Sanchez-Padilla J , Surmeier DJ ((2012) ) Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson’s disease. Nat Neurosci 15: , 1414–1421. |
[72] | Chan CS , Gertler TS , Surmeier DJ ((2009) ) Calcium homeostasis, selective vulnerability and Parkinson’s disease. Trends Neurosci 32: , 249–256. |
[73] | Surmeier DJ , Schumacker PT ((2013) ) Calcium, bioenergetics, and neuronal vulnerability in Parkinson’s disease. J Biol Chem 288: , 10736–10741. |
[74] | Poels J , Spasić MR , Callaerts P , Norga KK ((2009) ) Expanding roles for AMP-activated protein kinase in neuronal survival and autophagy. BioEssays 31: , 944–952. |
[75] | Mena MA , García de Yébenes J ((2008) ) Glial cells as players in parkinsonism: The “good,” the “bad,” and the “mysterious” glia. Neuroscience 14: , 544–560. |
[76] | Pacelli C , Giguère N , Bourque MJ , Lévesque M , Slack RS , Trudeau LÉ ((2015) ) Elevated mitochondrial bioenergetics and axonal arborization size are key contributors to the vulnerability of dopamine neurons. Curr Biol 25: , 2349–2360. |
[77] | Surmeier DJ , Schumacker PT , Guzman JD , Ilijic E , Yang B , Zampese E ((2017) ) Calcium and Parkinson’s disease. Biochem Biophys Res Commun 483: , 1013–1019. |
[78] | Chan CS , Guzman JN , Ilijic E , Mercer JN , Rick C , Tkatch T , Meredith GE , Surmeier DJ ((2007) ) “Rejuvenation” protects neurons in mouse models of Parkinson’s disease. Nature 447: , 1081–1086. |
[79] | Guzman JN , Sanchez-Padilla J , Wokosin D , Kondapalli J , Ilijic E , Schumacker PT , Surmeier DJ ((2010) ) Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468: , 696–700. |
[80] | Pissadaki EK , Bolam JP ((2013) ) The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front Comput Neurosci 7: , 1–17. |
[81] | Hang L , Thundyil J , Lim KL ((2015) ) Mitochondrial dysfunction and Parkinson disease: A Parkin–AMPK alliance in neuroprotection. Ann N Y Acad Sci 1350: , 37–47. |
[82] | Bolam JP , Pissadaki EK ((2012) ) Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov Disord 27: , 1478–1483. |
[83] | Duda J , Pötschke C , Liss B ((2016) ) Converging roles of ion channels, calcium, metabolic stress, and activity pattern of Substantia nigra dopaminergic neurons in health and Parkinson’s disease. J Neurochem 139: , 156–178. |
[84] | Wu N , Zheng B , Shaywitz A , Dagon Y , Tower C , Bellinger G , Shen C-H , Wen J , Asara J , McGraw TE , Kahn BB , Cantley LC ((2013) ) AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol Cell 49: , 1167–1175. |
[85] | Chaube B , Bhat MK ((2016) ) AMPK, a key regulator of metabolic/energy homeostasis and mitochondrial biogenesis in cancer cells. Cell Death Dis 7: , 1–2. |
[86] | Hardie DG , Pan DA ((2002) ) Regulation of fatty acid synthesis and oxidation by the AMP-activated protein kinase. Biochem Soc Trans 30: , 1064–1070. |
[87] | Inoki K , Zhu T , Guan K-L ((2003) ) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115: , 577–590. |
[88] | Gwinn DM , Shackelford DB , Egan DF , Mihaylova MM , Mery A , Vasquez DS , Turk BE , Shaw RJ ((2008) ) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30: , 214–226. |
[89] | Hoppe S , Bierhoff H , Cado I , Weber A , Tiebe M , Grummt I , Voit R ((2009) ) AMP-activated protein kinase adapts rRNA synthesis to cellular energy supply. Proc Natl Acad Sci U S A 106: , 17781–17786. |
[90] | Lynch-Day MA , Mao K , Wang K , Zhao M , Klionsky DJ ((2012) ) The role of autophagy in Parkinson’s disease. Cold Spring Harb Perspect Med 2: , 1–13. |
[91] | Komatsu M , Waguri S , Chiba T , Murata S , Iwata JI , Tanida I , Ueno T , Koike M , Uchiyama Y , Kominami E , Tanaka K ((2006) ) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441: , 880–884. |
[92] | Vogiatzi T , Xilouri M , Vekrellis K , Stefanis L ((2008) ) Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem 283: , 23542–23556. |
[93] | Cuervo AM , Stefanis L , Fredenburg R , Lansbury PT , Sulzer D ((2004) ) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305: , 1292–1295. |
[94] | Ciechanover A , Kwon YT ((2015) ) Degradation of misfolded proteins in neurodegenerative diseases: Therapeutic targets and strategies. Exp Mol Med 47: , 1–16. |
[95] | Xilouri M , Stefanis L ((2011) ) Autophagic pathways in Parkinson disease and related disorders. Expert Rev Mol Med 13: , 1–21. |
[96] | Decressac M , Mattsson B , Weikop P , Lundblad M , Jakobsson J , Björklund A ((2013) ) TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc Natl Acad Sci U S A 110: , E1817–E1826. |
[97] | Spencer B , Potkar R , Trejo M , Rockenstein E , Patrick C , Gindi R , Adame A , Wyss-Coray T , Masliah E ((2009) ) Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci 29: , 13578–13588. |
[98] | Egan DF , Chun MGH , Vamos M , Zou H , Rong J , Miller CJ , Lou HJ , Raveendra-Panickar D , Yang C-C , Sheffler DJ , Teriete P , Asara JM , Turk BE , Cosford NDP , Shaw RJ ((2015) ) Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Mol Cell 59: , 285–297. |
[99] | Kim J , Kundu M , Viollet B , Guan KL ((2011) ) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: , 132–141. |
[100] | Russell RC , Tian Y , Yuan H , Park HW , Chang YY , Kim J , Kim H , Neufeld TP , Dillin A , Guan KL ((2013) ) ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 15: , 741–750. |
[101] | Rosso P , Fioramonti M , Fracassi A , Marangoni M , Taglietti V , Siteni S , Segatto M ((2016) ) AMPK in the central nervous system: Physiological roles and pathological implications. Res Rep Biol 7: , 1–13. |
[102] | Phillipson OT ((2017) ) Alpha-synuclein, epigenetics, mitochondria, metabolism, calcium traffic, circadian dysfunction in Parkinson’s disease. An integrated strategy for management. Ageing Res Rev 40: , 149–167. |
[103] | Moors TE , Hoozemans JJM , Ingrassia A , Beccari T , Parnetti L , Chartier-Harlin M-C , van de Berg WDJ ((2017) ) Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol Neurodegener 12: , 1–18. |
[104] | Fischer F , Hamann A , Osiewacz HD ((2012) ) Mitochondrial quality control: An integrated network of pathways. Trends Biochem Sci 37: , 284–292. |
[105] | Shi R , Guberman M , Kirshenbaum LA ((2017) ) Mitochondrial quality control: The role of mitophagy in aging. Trends Cardiovasc Med. doi: 10.1016/j.tcm.2017.11.008 |
[106] | De Castro IP , Martins LM , Loh SHY ((2011) ) Mitochondrial quality control and parkinson’s disease: A pathway unfolds. Mol Neurobiol 43: , 80–86. |
[107] | Kraytsberg Y , Kudryavtseva E , McKee AC , Geula C , Kowall NW , Khrapko K ((2006) ) Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet 38: , 518–520. |
[108] | Bender A , Krishnan KJ , Morris CM , Taylor GA , Reeve AK , Perry RH , Jaros E , Hersheson JS , Betts J , Klopstock T , Taylor RW , Turnbull DM ((2006) ) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38: , 515–517. |
[109] | Carelli V , Maresca A , Caporali L , Trifunov S , Zanna C , Rugolo M ((2015) ) Mitochondria: Biogenesis and mitophagy balance in segregation and clonal expansion of mitochondrial DNA mutations. Int J Biochem Cell Biol 63: , 21–24. |
[110] | Kamp F , Exner N , Lutz AK , Wender N , Hegermann J , Brunner B , Nuscher B , Bartels T , Giese A , Beyer K , Eimer S , Winklhofer KF , Haass C ((2010) ) Inhibition of mitochondrial fusion by α-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J 29: , 3571–3589. |
[111] | Ordonez DG , Lee MK , Feany MB ((2018) ) α-synuclein induces mitochondrial dysfunction through spectrin and the actin cytoskeleton. Neuron 97: , 108–124. |
[112] | Scarpulla RC ((2011) ) Metabolic control of mitochondrial biogenesis through the PGC-1 family regulatory network. Biochim Biophys Acta 1813: , 1269–1278. |
[113] | Fernandez-Marcos PJ , Auwerx J ((2011) ) Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 93: , 884S–890S. |
[114] | Corona JC , Duchen MR ((2014) ) PPARγ and PGC-1α as therapeutic targets in Parkinson’s. Neurochem Res 40: , 308–316. |
[115] | Zheng B , Liao Z , Locascio JJ , Lesniak KA , Roderick SS , Watt ML , Eklund AC , Zhang-James Y , Kim PD , Hauser MA , Grünblatt E , Moran LB , Mandel SA , Riederer P , Miller RM , Federoff HJ , Wüllner U , Papapetropoulos S , Youdim MB , Cantuti-Castelvetri I , Young AB , Vance JM , Davis RL , Hedreen JC , Adler CH , Beach TG , Graeber MB , Middleton FA , Rochet J-C , Scherzer CR , Global PD Gene Expression (GPEX) Consortium ((2010) ) PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci Transl Med 2: , 1–14. |
[116] | Ng C-H , Basil AH , Hang L , Tan R , Goh K-L , O’Neill S , Zhang X , Yu F , Lim K-L ((2017) ) Genetic or pharmacological activation of the Drosophila PGC-1α ortholog spargel rescues the disease phenotypes of genetic models of Parkinson’s disease. Neurobiol Aging 55: , 33–37. |
[117] | Peng K , Yang L , Wang J , Ye F , Dan G , Zhao Y , Cai Y , Cui Z , Ao L , Liu J , Zou Z , Sai Y , Cao J ((2017) ) The interaction of mitochondrial biogenesis and fission/fusion mediated by PGC-1α regulates rotenone-induced dopaminergic neurotoxicity. Mol Neurobiol 54: , 3783–3797. |
[118] | Robb EL , Moradi F , Maddalena LA , Valente AJF , Fonseca J , Stuart JA ((2017) ) Resveratrol stimulates mitochondrial fusion by a mechanism requiring mitofusin-2. Biochem Biophys Res Commun 485: , 249–254. |
[119] | Morita M , Prudent J , Basu K , Goyon V , Katsumura S , Hulea L , Pearl D , Siddiqui N , Strack S , McGuirk S , St-Pierre J , Larsson O , Topisirovic I , Vali H , McBride HM , Bergeron JJ , Sonenberg N ((2017) ) mTOR controls mitochondrial dynamics and cell survival via MTFP1. Mol Cell 67: , 922–935. |
[120] | Toyama EQ , Herzig S , Courchet J , Lewis TL , Losón OC , Hellberg K , Young NP , Chen H , Polleux F , Chan DC , Shaw RJ ((2016) ) Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351: , 275–281. |
[121] | Zhang CS , Lin SC ((2016) ) AMPK promotes autophagy by facilitating mitochondrial fission. Cell Metab 23: , 399–401. |
[122] | Jin SM , Youle RJ ((2012) ) PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci 125: , 795–799. |
[123] | Laker RC , Drake JC , Wilson RJ , Lira VA , Lewellen BM , Ryall KA , Fisher CC , Zhang M , Saucerman JJ , Goodyear LJ , Kundu M , Yan Z ((2017) ) Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun 8: , 1–13. |
[124] | Hauser DN , Hastings TG ((2013) ) Mitochondrial dysfunction and oxidative stress in Parkinson’s disease and monogenic parkinsonism. Neurobiol Dis 51: , 35–42. |
[125] | Manoharan S , Guillemin GJ , Abiramasundari RS , Essa MM , Akbar M , Akbar MD ((2016) ) The role of reactive oxygen species in the pathogenesis of Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease: A mini review. Oxid Med Cell Longev 2016: , 1–15. |
[126] | Ayton S , Lei P ((2014) ) Nigral iron elevation is an invariable feature of Parkinson’s disease and is a sufficient cause of neurodegeneration. Biomed Res Int 2014: , 1–9. |
[127] | Jeon SM , Chandel NS , Hay N ((2012) ) AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485: , 661–665. |
[128] | Jeon S-M ((2016) ) Regulation and function of AMPK in physiology and diseases. Exp Mol Med 48: , 1–13. |
[129] | Zhao Y , Hu X , Liu Y , Dong S , Wen Z , He W , Zhang S , Huang Q , Shi M ((2017) ) ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol Cancer 16: , 1–12. |
[130] | Joo MS , Kim WD , Lee KY , Kim JH , Koo JH , Kim SG ((2016) ) AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol Cell Biol 36: , 1931–1942. |
[131] | Zimmermann K , Baldinger J , Mayerhofer B , Atanasov AG , Dirsch VM , Heiss EH ((2015) ) Activated AMPK boosts the Nrf2/HO-1 signaling axis - A role for the unfolded protein response. Free Radic Biol Med 88: , 417–426. |
[132] | Klotz LO , Sánchez-Ramos C , Prieto-Arroyo I , Urbánek P , Steinbrenner H , Monsalve M ((2015) ) Redox regulation of FoxO transcription factors. Redox Biol 6: , 51–72. |
[133] | O’Neill LAJ , Grahame Hardie D ((2013) ) Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493: , 346–355. |
[134] | Green SJ , Scheller LF , Marletta MA , Seguin MC , Klotz FW , Slayter M , Nelson BJ , Nacy CA ((1994) ) Nitric oxide: Cytokine-regulation of nitric oxide in host resistance to intracellular pathogens. Immunol Lett 43: , 87–94. |
[135] | Tansey MG , Goldberg MS ((2010) ) Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis 37: , 510–518. |
[136] | McGeer PL , Itagaki S , Boyes BE , McGeer EG ((1988) ) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38: , 1285–1291. |
[137] | Salminen A , Huuskonen J , Ojala J , Kauppinen A , Kaarniranta K , Suuronen T ((2008) ) Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res Rev 7: , 83–105. |
[138] | Salminen A , Hyttinen JMT , Kaarniranta K ((2011) ) AMP-activated protein kinase inhibits NF-κB signaling and inflammation: Impact on healthspan and lifespan. J Mol Med 89: , 667–676. |
[139] | Skonieczna M , Hejmo T , Poterala-Hejmo A , Cieslar-Pobuda A , Buldak RJ ((2017) ) NADPH oxidases: Insights into selected functions and mechanisms of action in cancer and stem cells. Oxid Med Cell Longev 2017: , 1–15. |
[140] | Balteau M , Van Steenbergen A , Timmermans AD , Dessy C , Behets-Wydemans G , Tajeddine N , Castanares-Zapatero D , Gilon P , Vanoverschelde J-L , Horman S , Hue L , Bertrand L , Beauloye C ((2014) ) AMPK activation by glucagon-like peptide-1 prevents NADPH oxidase activation induced by hyperglycemia in adult cardiomyocytes. Am J Physiol Heart Circ Physiol 307: , H1120–H1133. |
[141] | Pilon G , Dallaire P , Marette A ((2004) ) Inhibition of inducible nitric-oxide synthase by activators of AMP-activated protein kinase: A new mechanism of action of insulin-sensitizing drugs. J Biol Chem 279: , 20767–20774. |
[142] | Guma M , Wang Y , Viollet B , Liu-Bryan R ((2015) ) AMPK activation by A-769662 controls IL-6 expression in inflammatory arthritis. PLoS One 10: , 1–13. |
[143] | Zhao W , Ge H , Liu K , Chen X , Zhang J , Liu B ((2017) ) Nandinine, a derivative of berberine, inhibits inflammation and reduces insulin resistance in adipocytes via regulation of AMP-kinase activity. Planta Med 83: , 203–209. |
[144] | Wu J , Xu X , Li Y , Kou J , Huang F , Liu B , Liu K ((2015) ) Quercetin, luteolin and epigallocatechin gallate alleviate TXNIP and NLRP3-mediated inflammation and apoptosis with regulation of AMPK in endothelial cells. Eur J Pharmacol 745: , 59–68. |
[145] | Gao HM , Liu B , Zhang W , Hong JS ((2003) ) Novel anti-inflammatory therapy for Parkinson’s disease. Trends Pharmacol Sci 24: , 395–401. |
[146] | Venderova K , Park DS ((2012) ) Programmed cell death in Parkinson’s disease. Cold Spring Harb Perspect Med 2: , 1–23. |
[147] | Patel VA , Massenburg D , Vujicic S , Feng L , Tang M , Litbarg N , Antoni A , Rauch J , Lieberthal W , Levine JS ((2015) ) Apoptotic cells activate AMP-activated protein kinase (AMPK) and inhibit epithelial cell growth without change in intracellular energy stores. J Biol Chem 290: , 22352–22369. |
[148] | Greer EL , Oskoui PR , Banko MR , Maniar JM , Gygi MP , Gygi SP , Brunet A ((2007) ) The energy sensor AMP-activated protein kinase directly regulates the mammalian FOXO3 transcription factor. J Biol Chem 282: , 30107–30119. |
[149] | Greer EL , Banko MR , Brunet A ((2009) ) AMP-activated protein kinase and FoxO transcription factors in dietary restriction-induced longevity. Ann N Y Acad Sci 1170: , 688–692. |
[150] | Willcox BJ , Donlon TA , He Q , Chen R , Grove JS , Yano K , Masaki KH , Willcox DC , Rodriguez B , Curb JD ((2008) ) FOXO3A genotype is strongly associated with human longevity. Proc Natl Acad Sci U S A 105: , 13987–13992. |
[151] | Giannakou ME , Goss M , Jacobson J , Vinti G , Leevers SJ , Partridge L ((2007) ) Dynamics of the action of dFOXO on adult mortality in Drosophila. Aging Cell 6: , 429–438. |
[152] | Akasaki Y , Alvarez-Garcia O , Saito M , Caramés B , Iwamoto Y , Lotz MK ((2014) ) FoxO transcription factors support oxidative stress resistance in human chondrocytes. Arthritis Rheumatol 66: , 3349–3358. |
[153] | Li Z , Zhao J , Tikhanovich I , Kuravi S , Helzberg J , Dorko K , Roberts B , Kumer S , Weinman SA ((2016) ) Serine 574 phosphorylation alters transcriptional programming of FOXO3 by selectively enhancing apoptotic gene expression. Cell Death Differ 23: , 583–595. |
[154] | Concannon CG , Tuffy LP , Weisová P , Bonner HP , Dávila D , Bonner C , Devocelle MC , Strasser A , Ward MW , Prehn JHM ((2010) ) AMP kinase-mediated activation of the BH3-only protein Bim couples energy depletion to stressinduced apoptosis. J Cell Biol 189: , 83–94. |
[155] | Weisová P , Dávila D , Tuffy LP , Ward MW , Concannon CG , Prehn JHM ((2011) ) Role of 5’-adenosine monophosphate-activated protein kinase in cell survival and death responses in neurons. Antioxid Redox Signal 14: , 1863–1876. |
[156] | Anilkumar U , Weisová P , Düssmann H , Concannon CG , König HG , Prehn JHM ((2013) ) AMP-activated protein kinase (AMPK)-induced preconditioning in primary cortical neurons involves activation of MCL-1. J Neurochem 124: , 721–734. |
[157] | Huang CT , Huang DY , Hu CJ , Wu D , Lin WW ((2014) ) Energy adaptive response during parthanatos is enhanced by PD98059 and involves mitochondrial function but not autophagy induction. Biochim Biophys Acta 1843: , 531–543. |
[158] | Arbeláez-Quintero I , Palacios M ((2017) ) To use or not to use metformin in cerebral ischemia: A review of the application of metformin in stroke rodents. Stroke Res Treat 2017: , 1–13. |
[159] | Choi JS , Park C , Jeong JW ((2010) ) AMP-activated protein kinase is activated in Parkinson’s disease models mediated by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Biochem. Biophys Res Commun 391: , 147–151. |
[160] | Ng C-H , Guan MSH , Koh C , Ouyang X , Yu F , Tan E-K , O’Neill SP , Zhang X , Chung J , Lim K-L ((2012) ) AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in Drosophila models of Parkinson’s disease. J Neurosci 32: , 14311–14317. |
[161] | Barzilai N , Crandall JP , Kritchevsky SB , Espeland MA ((2016) ) Metformin as a tool to target aging. Cell Metab 23: , 1060–1065. |
[162] | Berman AY , Motechin RA , Wiesenfeld MY , Holz MK ((2017) ) The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis Oncol 1: , 1–9. |
[163] | Jin F , Wu Q , Lu Y-F , Gong Q-H , Shi J-S ((2008) ) Neuroprotective effect of resveratrol on 6-OHDA-induced Parkinson’s disease in rats. Eur J Pharmacol 600: , 78–82. |
[164] | Blanchet J , Longpré F , Bureau G , Morissette M , DiPaolo T , Bronchti G , Martinoli MG ((2008) ) Resveratrol, a red wine polyphenol, protects dopaminergic neurons in MPTP-treated mice. Prog Neuropsychopharmacol Biol Psychiatry 32: , 1243–1250. |
[165] | Ferretta A , Gaballo A , Tanzarella P , Piccoli C , Capitanio N , Nico B , Annese T , Di Paola M , Dell’Aquila C , De Mari M , Ferranini E , Bonifati V , Pacelli C , Cocco T ((2014) ) Effect of resveratrol on mitochondrial function: Implications in parkin-associated familiar Parkinson’s disease. Biochim Biophys Acta 1842: , 902–915. |
[166] | de Oliveira MR , Nabavi SF , Manayi A , Daglia M , Hajheydari Z , Nabavi SM ((2016) ) Resveratrol and the mitochondria: From triggering the intrinsic apoptotic pathway to inducing mitochondrial biogenesis, a mechanistic view. Biochim Biophys Acta 1860: , 727–745. |
[167] | Yang S , Long LH , Li D , Zhang JK , Jin S , Wang F , Chen JG ((2015) ) β-Guanidinopropionic acid extends the lifespan of Drosophila melanogaster via an AMP-activated protein kinase-dependent increase in autophagy. Aging Cell 14: , 1024–1033. |
[168] | Horvath TL , Erion DM , Elsworth JD , Roth RH , Shulman GI , Andrews ZB ((2011) ) GPA protects the nigrostriatal dopamine system by enhancing mitochondrial function. Neurobiol Dis 43: , 152–162. |
[169] | Łabuzek K , Suchy D , Gabryel B , Bielecka A , Liber S , Okopień B ((2010) ) Quantification of metformin by the HPLC method in brain regions, cerebrospinal fluid and plasma of rats treated with lipopolysaccharide. Pharmacol Rep 62: , 956–965. |
[170] | Lu M , Su C , Qiao C , Bian Y , Ding J , Hu G ((2016) ) Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson’s disease via autophagy and mitochondrial ROS clearance. Int J Neuropsychopharmacol 19: , 1–11. |
[171] | Katila N , Bhurtel S , Shadfar S , Srivastav S , Neupane S , Ojha U , Jeong GS , Choi DY ((2017) ) Metformin lowers α-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson’s disease. Neuropharmacology 125: , 396–407. |
[172] | Patil SP , Jain PD , Ghumatkar PJ , Tambe R , Sathaye S ((2014) ) Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience 277: , 747–754. |
[173] | Kang H , Khang R , Ham S , Jeong GR , Kim H , Jo M , Lee BD , Lee Y Il , Jo A , Park C , Kim H , Seo J , Paek SH , Lee Y-S , Choi J-Y , Lee Y , Shin J-H ((2017) ) Activation of the ATF2/CREB-PGC-1α pathway by metformin leads to dopaminergic neuroprotection. Oncotarget 8: , 48603–48618. |
[174] | Khang R , Park C , Shin JH ((2014) ) The biguanide metformin alters phosphoproteomic profiling in mouse brain. Neurosci Lett 579: , 145–150. |
[175] | Deschemin J-C , Foretz M , Viollet B , Vaulont S ((2017) ) AMPK is not required for the effect of metformin on the inhibition of BMP6-induced hepcidin gene expression in hepatocytes. Sci Rep 7: , 1–6. |
[176] | Bayliss JA , Lemus MB , Santos VV , Deo M , Davies JS , Kemp BE , Elsworth JD , Andrews ZB ((2016) ) Metformin prevents nigrostriatal dopamine degeneration independent of AMPK activation in dopamine neurons. PLoS One 11: , 1–15. |
[177] | Vincent EE , Coelho PP , Blagih J , Griss T , Viollet B , Jones RG ((2015) ) Differential effects of AMPK agonists on cell growth and metabolism. Oncogene 34: , 3627–3639. |
[178] | López JM , Santidrián AF , Campàs C , Gil J ((2003) ) 5-Aminoimidazole-4-carboxamide riboside induces apoptosis in Jurkat cells, but the AMP-activated protein kinase is not involved. Biochem J 370: , 1027–1032. |
[179] | Liu X , Chhipa RR , Nakano I , Dasgupta B ((2014) ) The AMPK inhibitor compound C is a potent AMPK-independent antiglioma agent. Mol Cancer Ther 13: , 596–605. |
[180] | Vekrellis K , Xilouri M , Emmanouilidou E , Stefanis L ((2009) ) Inducible over-expression of wild type α-synuclein in human neuronal cells leads to caspase-dependent non-apoptotic death. J Neurochem 109: , 1348–1362. |
[181] | Button RW , Luo S , Rubinsztein DC ((2015) ) Autophagic activity in neuronal cell death. Neurosci Bull 31: , 382–394. |
[182] | Dadakhujaev S , Noh HS , Jung EJ , Cha JY , Baek SM , Ha JH , Kim DR ((2010) ) Autophagy protects the rotenone-induced cell death in alpha-synuclein overexpressing SH-SY5Y cells. Neurosci Lett 472: , 47–52. |
[183] | Gómez-Suaga P , Luzón-Toro B , Churamani D , Zhang L , Bloor-Young D , Patel S , Woodman PG , Churchill GC , Hilfiker S ((2012) ) Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum Mol Genet 21: , 511–525. |
[184] | Hou Y-S , Guan J-J , Xu H-D , Wu F , Sheng R , Qin Z-H ((2015) ) Sestrin2 protects dopaminergic cells against rotenone toxicity through AMPK-dependent autophagy activation. Mol Cell Biol 35: , 2740–2751. |
[185] | Wu Y , Li X , Zhu JX , Xie W , Le W , Fan Z , Jankovic J , Pan T ((2011) ) Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. NeuroSignals 19: , 163–174. |
[186] | Yan Q , Han C , Wang G , Waddington JL , Zheng L , Zhen X ((2017) ) Activation of AMPK/mTORC1-mediated autophagy by metformin reverses Clk1 deficiency-sensitized dopaminergic neuronal death. Mol Pharmacol 92: , 640–652. |
[187] | Dulovic M , Jovanovic M , Xilouri M , Stefanis L , Harhaji-Trajkovic L , Kravic-Stevovic T , Paunovic V , Ardah MT , El-Agnaf OMA , Kostic V , Markovic I , Trajkovic V ((2014) ) The protective role of AMP-activated protein kinase in alpha-synuclein neurotoxicity in vitro. Neurobiol Dis 63: , 1–11. |
[188] | Bobela W , Nazeeruddin S , Knott G , Aebischer P , Schneider BL ((2017) ) Modulating the catalytic activity of AMPK has neuroprotective effects against α-synuclein toxicity. Mol Neurodegener 12: , 1–19. |
[189] | Kang SS , Zhang Z , Liu X , Manfredsson FP , He L , Iuvone PM , Cao X , Sun YE , Jin L , Ye K ((2017) ) α-Synuclein binds and sequesters PIKE-L into Lewy bodies, triggering dopaminergic cell death via AMPK hyperactivation. Proc Natl Acad Sci U S A 114: , 1183–1188. |
[190] | Stewart T , Sossi V , Aasly JO , Wszolek ZK , Uitti RJ , Hasegawa K , Yokoyama T , Zabetian CP , Leverenz JB , Stoessl AJ , Wang Y , Ginghina C , Liu C , Cain KC , Auinger P , Kang UJ , Jensen PH , Shi M , Zhang J ((2015) ) Phosphorylated α-synuclein in Parkinson’s disease: Correlation depends on disease severity. Acta Neuropathol Commun 3: , 1–11. |
[191] | Oueslati A ((2016) ) Implication of alpha-synuclein phosphorylation at S129 in synucleinopathies: What have we learned in the last decade? J Parkinsons Dis 6: , 39–51. |
[192] | Lou H , Montoya SE , Alerte TNM , Wang J , Wu J , Peng X , Hong CS , Friedrich EE , Mader SA , Pedersen CJ , Marcus BS , McCormack AL , Di Monte DA , Daubner SC , Perez RG ((2010) ) Serine 129 phosphorylation reduces the ability of α-synuclein to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. J Biol Chem 285: , 17648–17661. |
[193] | Chen M , Yang W , Li X , Li X , Wang P , Yue F , Yang H , Chan P , Yu S ((2016) ) Age- and brain region-dependent α-synuclein oligomerization is attributed to alterations in intrinsic enzymes regulating α-synuclein phosphorylation in aging monkey brains. Oncotarget 7: , 8466–8480. |
[194] | Jiang P , Gan M , Ebrahim AS , Castanedes-Casey M , Dickson DW , Yen SHC ((2013) ) Adenosine monophosphate-activated protein kinase overactivation leads to accumulation of α-synuclein oligomers and decrease of neurites. Neurobiol Aging 34: , 1504–1515. |
[195] | Tenreiro S , Reimão-Pinto MM , Antas P , Rino J , Wawrzycka D , Macedo D , Rosado-Ramos R , Amen T , Waiss M , Magalhães F , Gomes A , Santos CN , Kaganovich D , Outeiro TF ((2014) ) Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of Parkinson’s disease. PLoS Genet 10: , 1–23. |
[196] | Pérez-Revuelta BI , Hettich MM , Ciociaro A , Rotermund C , Kahle PJ , Krauss S , Di Monte DA ((2014) ) Metformin lowers Ser-129 phosphorylated α-synuclein levels via mTOR-dependent protein phosphatase 2A activation. Cell Death Dis 5: , 1–10. |
[197] | Kickstein E , Krauss S , Thornhill P , Rutschow D , Zeller R , Sharkey J , Williamson R , Fuchs M , Kohler A , Glossmann H , Schneider R , Sutherland C , Schweiger S ((2010) ) Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci U S A 107: , 21830–21835. |
[198] | Chu CT , Zhu J , Dagda R ((2007) ) Beclin 1-independent pathway of damage-induced mitophagy and autophagic stress: Implications for neurodegeneration and cell death. Autophagy 3: , 663–666. |
[199] | Zhang J , Cai Q , Jiang M , Liu Y , Gu H , Guo J , Sun H , Fang J , Jin L ((2017) ) Mesencephalic astrocyte-derived neurotrophic factor alleviated 6-OHDA-induced cell damage via ROS-AMPK/mTOR mediated autophagic inhibition. Exp Gerontol 89: , 45–56. |
[200] | Xu Y , Liu C , Chen S , Ye Y , Guo M , Ren Q , Liu L , Zhang H , Xu C , Zhou Q , Huang S , Chen L ((2014) ) Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson’s disease. Cell Signal 26: , 1680–1689. |
[201] | Ismaiel AAK , Espinosa-Oliva AM , Santiago M , García-Quintanilla A , Oliva-Martín MJ , Herrera AJ , Venero JL , de Pablos RM ((2016) ) Metformin, besides exhibiting strong in vivo anti-inflammatory properties, increases mptp-induced damage to the nigrostriatal dopaminergic system. Toxicol Appl Pharmacol 298: , 19–30. |
[202] | Kim TW , Cho HM , Choi SY , Suguira Y , Hayasaka T , Setou M , Koh HC , Mi Hwang E , Park JY , Kang SJ , Kim HS , Kim H , Sun W ((2013) ) (ADP-ribose) polymerase 1 and AMP-activated protein kinase mediate progressive dopaminergic neuronal degeneration in a mouse model of Parkinson’s disease. Cell Death Dis 4: , 1–10. |
[203] | Yonekawa T , Thorburn A ((2013) ) Autophagy and cell death. Essays Biochem 55: , 105–117. |
[204] | Ashabi G , Khodagholi F , Khalaj L , Goudarzvand M , Nasiri M ((2014) ) Activation of AMP-activated protein kinase by metformin protects against global cerebral ischemia in male rats: Interference of AMPK/PGC-1α pathway. Metab Brain Dis 29: , 47–58. |
[205] | Domise M , Vingtdeux V ((2016) ) AMPK in neurodegenerative diseases. EXS 107: , 153–177. |
[206] | Wahlqvist ML , Lee M-S , Hsu C-C , Chuang S-Y , Lee J-T , Tsai H-N ((2012) ) Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson’s disease occurring with Type 2 diabetes in a Taiwanese population cohort. Parkinsonism Relat Disord 18: , 753–758. |
[207] | Kuan YC , Huang KW , Lin CL , Hu CJ , Kao CH ((2017) ) Effects of metformin exposure on neurodegenerative diseases in elderly patients with type 2 diabetes mellitus. Prog Neuropsychopharmacol Biol Psychiatry 79: , 77–83. |
[208] | Brakedal B , Flønes I , Reiter SF , Torkildsen Ø , Dölle C , Assmus J , Haugarvoll K , Tzoulis C ((2017) ) Glitazone use associated with reduced risk of Parkinson’s disease. Mov Disord 32: , 1594–1599. |
[209] | Alves G , Müller B , Herlofson K , HogenEsch I , Telstad W , Aarsland D , Tysnes O-B , Larsen JP , Norwegian ParkWest study group ((2009) ) Incidence of Parkinson’s disease in Norway: The Norwegian ParkWest study. J Neurol Neurosurg Psychiatry 80: , 851–857. |

