
Best Practices for Generating and Using Alpha-Synuclein Pre-Formed Fibrils to Model Parkinson’s Disease in Rodents
Abstract
Parkinson’s disease (PD) is the second most common neurodegenerative disease, affecting approximately one-percent of the population over the age of sixty. Although many animal models have been developed to study this disease, each model presents its own advantages and caveats. A unique model has arisen to study the role of alpha-synuclein (aSyn) in the pathogenesis of PD. This model involves the conversion of recombinant monomeric aSyn protein to a fibrillar form—the aSyn pre-formed fibril (aSyn PFF)—which is then injected into the brain or introduced to the media in culture. Although many groups have successfully adopted and replicated the aSyn PFF model, issues with generating consistent pathology have been reported by investigators. To improve the replicability of this model and diminish these issues, The Michael J. Fox Foundation for Parkinson’s Research (MJFF) has enlisted the help of field leaders who performed key experiments to establish the aSyn PFF model to provide the research community with guidelines and practical tips for improving the robustness and success of this model. Specifically, we identify key pitfalls and suggestions for avoiding these mistakes as they relate to generating the aSyn PFFs from monomeric protein, validating the formation of pathogenic aSyn PFFs, and using the aSyn PFFs in vivo or in vitro to model PD. With this additional information, adoption and use of the aSyn PFF model should present fewer challenges, resulting in a robust and widely available model of PD.
INTRODUCTION
Parkinson’s disease (PD) affects approximately one in one hundred individuals over sixty years old today, with incidence projected to double in the coming decades [1]. Principally characterized as a motor disease, PD diagnosis is confirmed postmortem by two pathological hallmarks that have been linked to the motor dysfunction exhibited by patients—loss of dopaminergic projections in the nigrostriatal system associated with cell death in the substantia nigra pars compacta (SNpc) [2, 3], and presence of alpha-synuclein (aSyn) inclusions known as Lewy bodies and Lewy neurites [4–7]. The relationship between aSyn and PD was further strengthened by the identification of mutations and multiplications in the gene encoding aSyn, SNCA, leading to an autosomal dominant form of PD [5, 8, 9].
Although PD is the second most common neurodegenerative disease and the prevalence is expected to continue to rise, much progress still remains to be made in understanding the biology of PD and improving treatments for the disease. One hindrance to this progress is the fact that the majority of PD cases have no known cause and are not linked to a known genetic mutation. When combined with the fact that PD is a human-specific condition not present in other species, this makes research into the biology of the disease and testing of treatments difficult. As a result, various models are available for PD research with each model displaying key weaknesses in addition to its strengths.
COMMON METHODS FOR MODELING PARKINSON’S DISEASE IN RODENTS
Toxin models are the classical models for PD research, using injection of a toxin into the brain or periphery to eliminate the dopaminergic neurons of the SNpc. Toxin models—specifically 6-hydroxydopamine (6-OHDA) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)—were the first models of PD developed and are still widely in use today due to their ability to induce rapid degeneration of the SNpc, leading to robust, well-characterized motor deficits [10]. In addition, the spatiotemporal control and ability to induce a unilateral lesion in rodents and non-human primates with these toxins provides clear benefits for using these models to study nigrostriatal degeneration. However, although toxin models recapitulate one pathological hallmark of PD (nigrostriatal degeneration), they do not typically display the second pathological hallmark (aSyn and Lewy body pathology) [11]. In addition, the rapid degeneration of the nigrostriatal system does not seem particularly relevant to the human condition—which may begin decades before motor symptoms manifest—and does not allow for the testing of disease-modifying treatments during the pre-symptomatic phase [12, 13]. Regardless, toxin models are typically preferred for studies of sporadic PD where rapid degeneration of the nigrostriatal system is preferred or the consequences of nigrostriatal degeneration are studied.
Once familial forms of PD were identified, genetically-modified rodent models sought to provide a better model of PD that would more faithfully recapitulate the human condition. The most common genetic modifications modeled include the autosomal dominant mutations in the genes encoding aSyn [8, 9, 14] and leucine-rich repeat kinase 2 (LRRK2) [15, 16], and the autosomal recessive mutations in the genes encoding Parkin [17], PTEN-induced putative kinase 1 (PINK1) [18], and DJ-1 [19–21]. Genetically-modified models are advantageous in that they express genetic mutations linked to inherited forms of PD and express the implicated protein at moderate supra-physiological or near-physiological levels for long periods as occurs in the disease, allowing investigators to probe disease-relevant pathways and test therapeutic interventions for familial forms of PD [11–13, 20]. Despite these advantages, genetically-modified rodent models of PD are oftentimes not considered the best model of PD. Importantly, many of the genetically-modified rodent models of PD lack robust nigrostriatal degeneration and do not display consistent, reproducible motor deficits [11, 20, 21]. Furthermore, the nigrostriatal degeneration and motor phenotypes that have been observed are heavily dependent on the amount of protein expressed and the promoter used to drive expression of the transgene [20, 21]. Notwithstanding these drawbacks, genetically-modified rodent models of PD are useful models for PD research, particularly research into therapeutic strategies for familial forms of PD, non-motor symptoms of PD, and clinically-relevant pathways.
Viral vector-based models of PD have emerged more recently to model both pathological hallmarks of PD—degeneration of the nigrostriatal system and inclusions of abnormal, aggregated or phosphorylated aSyn. In these models, viral vectors are used as tools to transduce cell types of interest and overexpress or knockdown proteins related to PD, such as aSyn or LRRK2 [22]. Similar to toxin models, viral vectors overexpressing aSyn can induce a robust lesion of the nigrostriatal system leading to behavioral phenotypes in rodents and non-human primates using a system that allows spatiotemporal control and the ability to design studies with an internal control [23]. In addition, the level of protein overexpression can easily be altered to reduce the level of overexpression to sub-toxic levels or prolong the time before degeneration, if desired [14, 24, 25]. The main drawback of this model, however, is the supra-physiological level of overexpression required for pathology, leading to the question of generalizability between this model and the human condition. The viral vector-based model of PD requires fold-level increases of aSyn expression over endogenous to produce a robust PD phenotype, whereas patients with idiopathic PD do not exhibit higher levels of aSyn protein versus control patients [26] and even PD patients with the SNCA triplication mutation exhibit aSyn protein levels in the brain that are only double that of non-PD patients [27]. Furthermore, the aSyn aggregates produced in this model do not appear to be filamentous and therefore may not replicate many features of aSyn inclusions found in PD patients [28]. Aside from these drawbacks, the viral vector-based models of PD are still common models in instances where robust nigral degeneration, motor deficits, and aSyn pathology are desired.
A new model focusing on the misfolded, aggregated forms of aSyn found in Lewy bodies has recently arisen as an important tool in PD research. In this model, recombinant aSyn monomeric proteins are incubated under defined conditions to generate aggregated, amyloid pre-formed fibrils (aSyn PFFs) that are similar in structure to the building blocks of Lewy bodies and Lewy neurites. These aSyn fibrils are then sonicated to generate short fibrils which trigger endogenous aSyn to be hyperphosphorylated at the S129 site, ubiquitinated, insoluble in detergent, and 10–15 nm in length upon examination by electron microscopy when introduced in vitro and in vivo [29–31]. In addition to pS129 aSyn pathology, the aSyn PFFs trigger synaptic dysfunction, perturbations in cell excitability, and cell death in cell lines overexpressing disease-related proteins as well as in primary neuronal cultures from wild-type mice [32, 33]. In vivo, intracerebral injection of aSyn PFFs into the dorsal striatum results in dysregulation of striatal dopamine release, neurodegeneration in the SNpc, and behavioral deficits in rodents overexpressing disease-related proteins or non-transgenic rodents [29, 30, 34, 35]. The development and validation of this model has led to a new animal model for PD research that provides many advantages over other models.
Similar to the toxin- and viral vector-based models of PD, the aSyn PFF model of PD is an inducible model that allows for spatiotemporal control of the aSyn PFF introduction. This enables baseline measurements of behavior prior to induction of parkinsonian symptoms, an opportunity to study a prophylactic treatment, and a defined time course of degeneration for strategic intervention. Although the introduction of the aSyn PFFs generated from recombinant aSyn induces a model using supra-physiological levels of aSyn, the levels of aSyn are much closer to that of the human condition as compared to viral vector-based and some transgenic models. Importantly, in contrast to the viral vector-based model, the pathology exhibited in the aSyn PFF model is the result of PD-relevant pathological changes in endogenous aSyn initiated after aSyn PFF injection [30, 33, 34]. Furthermore, this model exhibits a more protracted time course of degeneration with early aSyn pathology in PD-relevant brain regions and development of dopamine dysfunction, nigral degeneration, and motor deficits months after induction [29, 30, 34]. This progression is more similar to the human condition whereby dopamine neuron dysfunction occurs in advance of overt motor symptoms that manifest only after a significant portion of the SNpc dopaminergic neurons have degenerated [36–40].
As with all preclinical models of PD, there are some considerations that must be acknowledged when using the aSyn PFF model. First, a unilateral injection of aSyn PFFs may produce bilateral pathology depending on the injection location and rodent model, although contralateral pathology develops at lower levels and at later time points than ipsilateral pathology [29, 30, 34, 41, 42]. Therefore, this model may not have an unaffected hemisphere to use as an internal control—although the uninjected hemisphere is often used as an internal comparator for pathology for the injected hemisphere. In addition, it still remains unclear if the recombinant aSyn PFFs are identical to the species of aSyn present in the pathology of the human condition. Independent of these two main caveats, this model is commonly used, well-validated, and important for the understanding of the pathophysiology and potential treatment of PD.
Recently, the popularity of this model has grown and many groups have attempted to establish this model in their labs to interrogate their specific research interests. Through the wide adoption of this model, it has become apparent that establishing this model in a lab presents unique challenges with regard to the aSyn PFFs. In particular, the ability to consistently generate pathogenic aSyn PFFs has arisen as an issue with this model, leading to concerns over reproducibility of findings and investment in a model with a time course of pathology that requires several months to develop. In consultation with the experts who initially developed, validated, and expanded this model, The Michael J. Fox Foundation for Parkinson’s Research (MJFF) has established a set of guidelines and recommendations to improve the usability of this model. These guidelines were developed through the accumulated experience and collaborative efforts of experts; the recommendations described in this manuscript have been tested and used across labs and batches of aSyn PFFs. These guidelines specifically aim to provide useful tips for generating, validating, and using aSyn PFFs to induce rodent models of PD. The guidelines described in this manuscript are intended be used as a starting point when adopting this model or when experiencing issues with aSyn PFFs. The protocols described within this manuscript may require tailoring and further validation based on the individual needs and experiences of the different labs.
GENERATION OF ALPHA-SYNUCLEIN PRE-FORMED FIBRILS FROM RECOMBINANT ALPHA-SYNUCLEIN MONOMERS
The first and most important step of the aSyn PFF model involves the generation of proper pathogenic PFFs from monomeric recombinant aSyn protein. This begins with choosing the proper starting material as not all monomeric aSyn protein preparations will aggregate into fibrils and not all aggregated aSyn protein will trigger pathology. For instance, some groups have reported issues generating aSyn PFFs using lyophilized protein or tagged aSyn protein [31]. However, these issues have been overcome by others [32, 43]. To increase the probability of success when first adopting this model, it is recommended that the investigator either generates and validates their own protein or purchases validated aSyn monomeric protein specifically-formulated to generate aSyn PFFs.
Recombinant aSyn monomers may be generated in house using protocols such as those described in Volpicelli-Daley, Luk, and Lee [31], Abdelmotilib et al. [35], or Fares et al. [44]. Monomeric aSyn protein specifically-formulated to generate aSyn PFFs may also be purchased from commercial sources (Supplementary Material). An added benefit of using a well-validated commercial source of aSyn is that it will provide the investigator with assurance that the protein should form the pathogenic PFF species if prepared correctly and will allow better comparisons between published studies. If the investigator is interested in purchasing pre-made aSyn PFFs rather than converting the aSyn monomers to PFFs in-house, it should be noted that not all aggregated aSyn protein is compatible with the aSyn PFF model and precaution should be taken to purchase the correct fibrillar material or generate aSyn PFFs in-house using validated protocols. It is not recommended to use aSyn PFFs from a previous batch to seed the conversion of aSyn monomers to PFFs in a new batch as this will lead to the generation of different types of aggregates and will lead to an increase in variation rather than enhancing reproducibility [45].
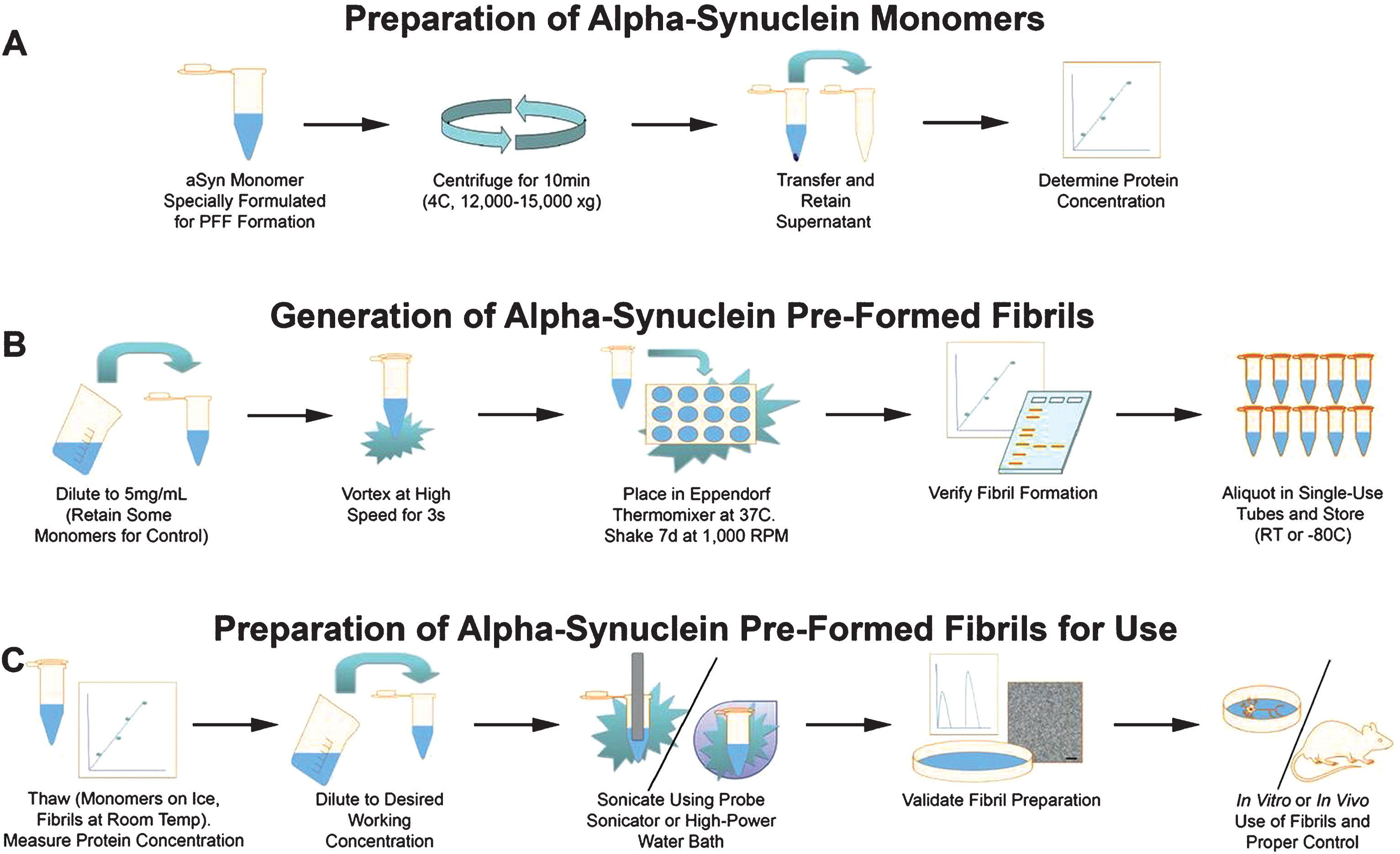
In consultation with experts in aSyn PFF generation and use, MJFF has developed and made available a protocol for generating aSyn PFFs from recombinant aSyn monomeric protein (Supplementary Material). The three stages of this protocol include preparing the monomers, generating PFFs from the monomers, and preparing the PFFs for use (see Fig. 1 for a schematic depiction of the protocol). Within each of these stages, considerations must be taken to ensure the monomers aggregate to form proper fibrils and the sonicated fibrils are the correct size for generating a pathological response in vitro or in vivo. Of particular importance are ensuring the buffers and the solutions are the correct pH and ionic strength, storing the fibrils properly, and using sonication parameters that generate sufficiently small fibrils.
Fig.1
Schematic depiction of the protocol for generating alpha-synuclein pre-formed fibrils (aSyn PFFs) from specially formulated monomers. The protocol for generating aSyn PFFs from monomers includes three main stages, (A) preparation of aSyn monomers, (B) generation of aSyn PFFs from monomers, and (C) preparation of aSyn PFFS for use. A) The protocol begins with the monomeric recombinant aSyn protein specially formulated for aSyn PFF development, which is then centrifuged for the supernatant to be transferred and protein concentration to be measured. B) To generate aSyn PFFs, the solution is diluted, vortexed, and incubated at 37°C shaking for 7 days before quality control and storage. Please note that a portion of the monomeric starting material should be set aside at the beginning of this step for use as a negative control in quality control experiments, with even larger quantities set aside if monomers are to be used as the control protein in model generation. C) On the day of use, an aliquot of aSyn PFFs should be thawed, diluted after the protein concentration has been re-measured, sonicated, and quality controlled prior to use. The protocol corresponding to this schematic can be found in the Supplementary Material. aSyn, alpha-synuclein; PFF, pre-formed fibril; s, seconds; d, days; RT, room temperature.
Optimizing buffer conditions for proper aSyn PFF formation
A common oversight in generating PFFs is using buffers that are not controlled for pH and/or ionic strength. Ionic strength and acidity are very important parameters to monitor when generating aSyn PFFs. These two parameters may affect PFF formation and should be analyzed prior to PFF induction. For optimal PFF formation, the pH should be between 7–8 (optimal around pH 7.4) and the salt concentration should be approximately 100 mM NaCl. These parameters directly affect aSyn aggregate formation and the development of different strains of aSyn aggregates that might not seed pathology efficiently has been reported under different buffer conditions [46].
In addition, an important consideration that should not be overlooked is the protocol used to convert the aSyn monomers to fibrils. A recent study by Tarutani and colleagues [47] showed that robust pathology in vitro and in vivo only develops when monomers are incubated at 37°C shaking for seven days. Specifically, samples of aSyn monomers incubated at 37°C, room temperature, or 4°C without shaking fail to form the elongated fibrillar aggregates of aSyn that adopt the beta-sheet conformation and amyloidogenic properties [47]. It is recommended to shake the aSyn monomers at 37°C in a sealed incubator or shaker with a temperature controlled lid to prevent evaporation, with verification of turbidity after incubation to confirm the presence of higher molecular weight species of aSyn such as the aSyn fibrils [31, 35, 47]. The MJFF protocol for fibril generation can be found in the Supplementary Material, with additional protocols in the cited literature [29–35, 41–48].
Storage considerations to promote aSyn monomer and PFF stability and activity
Proper storage of aSyn monomer and PFF samples is also an important factor to consider when using the aSyn PFF model. Sample concentration for storage, storage temperature, and thawing temperature can drastically affect the quality of the aSyn monomers and fibrils, preventing the formation of proper aggregates for use in the PFF model. It is recommended to store aSyn monomers at a concentration no higher than 7.5 mg/mL to enable proper PFF formation and sufficient quantity of sample for quality control and experimental use (higher concentrations may result in formation of aggregates). Monomeric aSyn protein should be stored at –80°C and thawed/kept on ice to prevent spontaneous aggregation that can occur at room temperature. Should monomers need to be shipped, aSyn monomers for PFF generation should be shipped frozen to avoid unnecessary freeze-thaw cycles and spontaneous aggregation. If monomers are to be used as the control, the monomeric solution may be aliquoted into single-use aliquots and stored at –80°C to prevent unnecessary freeze-thaw cycles that may degrade the sample.
Once the aSyn monomers have been converted to PFFs, the sample may be stored at room temperature or at –80°C; aSyn PFF samples should not be stored at 4°C or –20°C, as considerable dissociation may occur at these temperatures [46, 49] (Supplementary Figure 1A). Stocks of aSyn PFFs should be stored in single-use aliquots of 20–25 μL at a final concentration of 5 mg/mL, regardless if storing at –80°C or room temperature. It is generally recommended to make aSyn PFFs fresh before use and store aliquots at room temperature for a short duration before use in culture or in surgery. Stocks of aSyn PFFs have been shown to retain pathogenicity for up to 1–1.5 years at –80°C, although activity will decrease as compared to fresh aSyn PFFs (Fig. 2). Samples can be kept at room temperature for 1–2 weeks. Samples can be kept at room temperature over 2 weeks, however, sterile components must be used to prevent microbial contamination and it is generally not recommended to store aSyn PFFs at room temperature long-term as this risks sample contamination and degradation.
Fig.2
Impact of storage conditions on alpha-synuclein pre-formed fibril (aSyn PFF) structure and pathogenicity. A-C) Representative transmission electron microscopy (TEM) images of aSyn PFF samples stored at varying temperatures before sonication. A) Samples stored at room temperature for 3–4 weeks display long fibrillar structures characteristic of unsonicated aSyn PFFs. B) Samples stored at –80°C for 4–5 months display long fibril structures as well as fractured, smaller aggregates. C) Samples stored in liquid nitrogen for 4–5 months display a distinct morphology characterized by fractured fibrils and non-specific aggregates (arrowhead). D–F) Analysis of the impact of storage conditions on aSyn PFF pathogenicity in primary hippocampal neuron cultures. Primary hippocampal neurons were exposed to fibrils at 0.2 μg/mL and fixed 6 days later. D–E) Immunofluorescence was performed using an antibody to pS129 aSyn to visualize inclusions (green) or to neurofilament heavy chain (blue) to visualize axons. Scale bars = 50 μm. Fibrils were either (D) generated immediately before adding to primary neurons or (E) stored at –80°C for 6 months before use. F) Quantitation of the percent area occupied by pS129 aSyn immunoreactivity reveals a highly significant (p < 0.001) difference between fresh and frozen aSyn PFFs, with much greater levels of pS129 aSyn induced by fresh aSyn PFFs as compared to frozen aSyn PFFs. Graph depicts mean values with error bars denoting standard deviation. aSyn, alpha-synuclein; PFF, pre-formed fibril; RT, room temperature; liquid N2, liquid nitrogen; p-α-syn, pS129 aSyn.
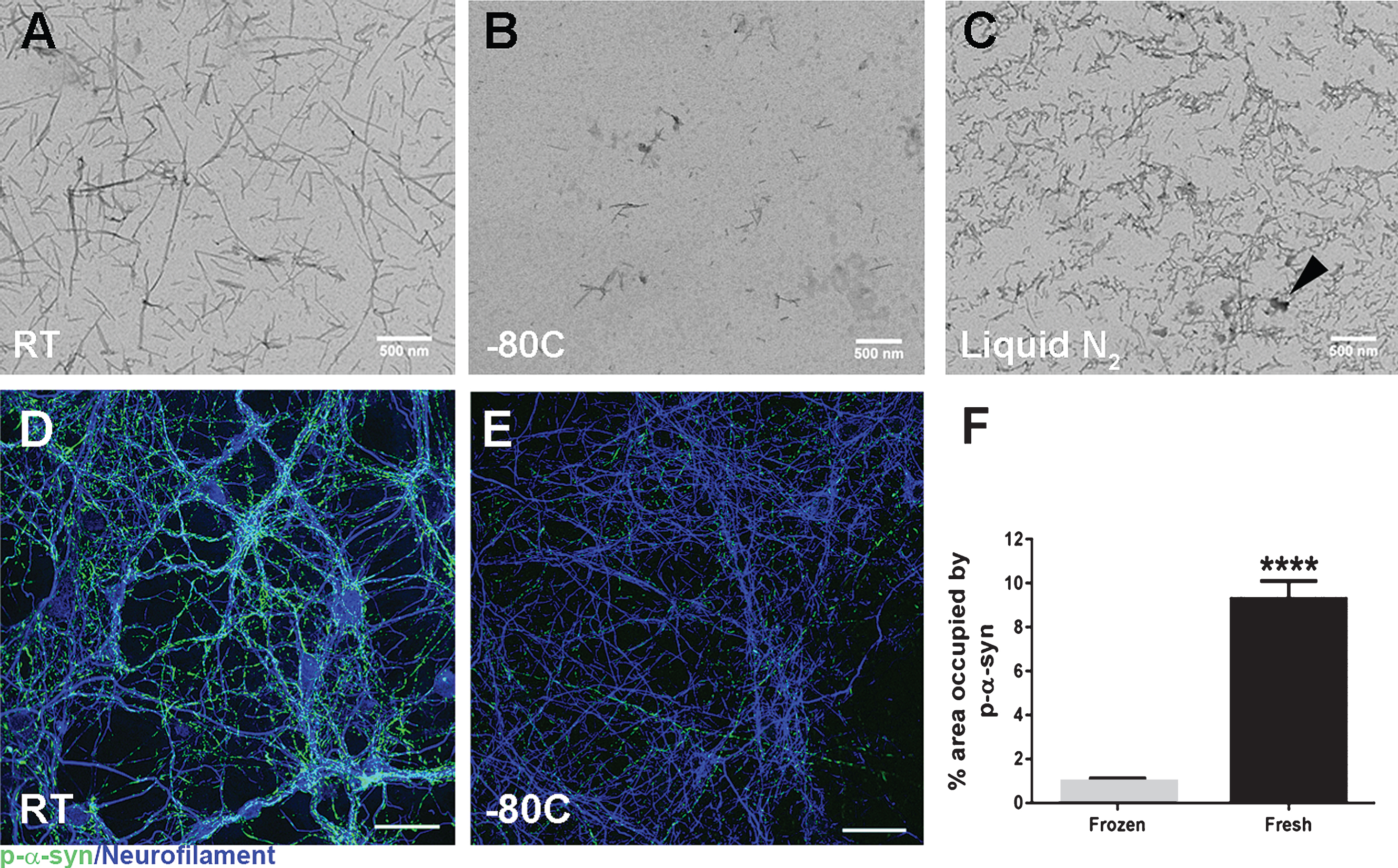
As aSyn PFFs are very prone to alterations with freeze-thaw cycles, it is important to properly freeze and thaw aliquots of aSyn PFFs. Before samples are placed at –80°C, aliquots of aSyn PFFs need to be gradually frozen on dry ice to prevent non-specific aggregations that might occur with snap-freezing. Although the exact pathogenic conformation within the aSyn PFFs samples is still unclear, the additional variations in aSyn fibril size, shape, and heterogeneity introduced by the freeze-thaw cycle can lead to added variation and therefore should be prevented to enhance reproducibility. An analysis of the impact of different storage parameters on fibril structure found large variations when comparing between aSyn PFFs stored at room temperature, frozen on dry ice and stored at –80°C, or snap frozen and stored in liquid nitrogen (Fig. 2). Importantly, freezing aSyn PFF samples with liquid nitrogen led to the development of non-specific, larger protein aggregates that could impact the concentration of protein measured and used (arrow in Fig. 2C). In addition to altering fibril structure (Fig. 2B), freezing and storing aSyn PFF samples at –80°C can also negatively impact the pathogenicity of the PFFs leading to a reduction in inclusion formation as evidenced by reduced pS129 aSyn staining following treatment with PFFs frozen at –80°C as compared to PFFs made fresh and stored at room temperature (Fig. 2D–F). Systematic studies investigating whether these alterations are due to the freeze-thaw cycle or duration of freezing are still lacking. Therefore, it is strongly recommended to validate frozen aSyn PFF samples—regardless of storage time—before use to confirm pathogenicity of the sample.
When freezing, single-use aliquots of aSyn PFFs must be stored in a freezer box at the back of the freezer to prevent degradation with freeze-thaw. When using frozen samples, aliquots should be thawed and kept at room temperature. The freeze-thaw cycle causes dissociation of fibrils and a product that differs from what was analyzed prior to freezing (Fig. 2). As a result, protein concentration should be re-measured after thawing and quality control experiments analyzing fibril size and seeding capacity should be performed after thawing a representative aliquot designated for quality control or the aliquot that will be used for experimental purposes (Fig. 1). Furthermore, it is important to highlight that the alterations during the freeze-thaw cycle necessitate sonication immediately prior to use—sonicated aliquots of aSyn PFFs should never be stored at any temperature below room temperature (Supplementary Figure 1).
Sonication of aSyn PFF samples to generate pathogenic species of PFFs
A common pitfall leading to lack of pathology and issues in reproducibility in the aSyn PFF model is the failure to sufficiently and consistently sonicate the aSyn fibrils generated from monomers. Immediately before use, the aSyn PFF aliquot should be diluted to the desired working concentration and sonicated to produce the short fibrillar version of aSyn that seeds pathology in vitro and in vivo (Figs. 1 and 3–5). Importantly, the sonicated aSyn PFF samples are heterogenous in nature and it remains unclear which specific form of aggregated aSyn—be it the short fibrils or oligomers generated during sonication—are seeding the majority of the pathology. However, previous studies have shown that sonication must result in majority of aSyn PFFs at 50 nm or smaller to consistently seed pS129 aSyn pathology in culture or after injection [35, 47, 48]. Failure to sonicate samples prior to use or failure to sufficiently sonicate the large fibrils to smaller components will reduce pathogenicity or even result in a lack of pathology [35, 47, 48]. To prevent this issue, it is suggested to test multiple sonication parameters when first establishing the model in one’s lab.
Fig.3
Analysis of the size and morphology of alpha-synuclein pre-formed fibrils (aSyn PFFs) after efficient sonication. A-B) Transmission electron microscopy (TEM) images of aSyn PFFs (A) pre-sonication and (B) post-sonication. A) aSyn PFFs pre-sonication are long fibrils with beta-sheet structure. Scale bars = 50 nm. B) aSyn PFFs that have been efficiently sonicated are short fibrils that keep the beta-sheet conformation. Scale bars = 50 nm. C) Atomic force microscopy (AFM) analysis of sonicated aSyn PFFs to determine size of the sonicated PFFs. D) Size distribution of the aSyn PFFs after sonication as determined by values obtained from statistical analysis of the aggregates identified in the atomic force microscopy images (C). aSyn, alpha-synuclein; PFF, pre-formed fibril.
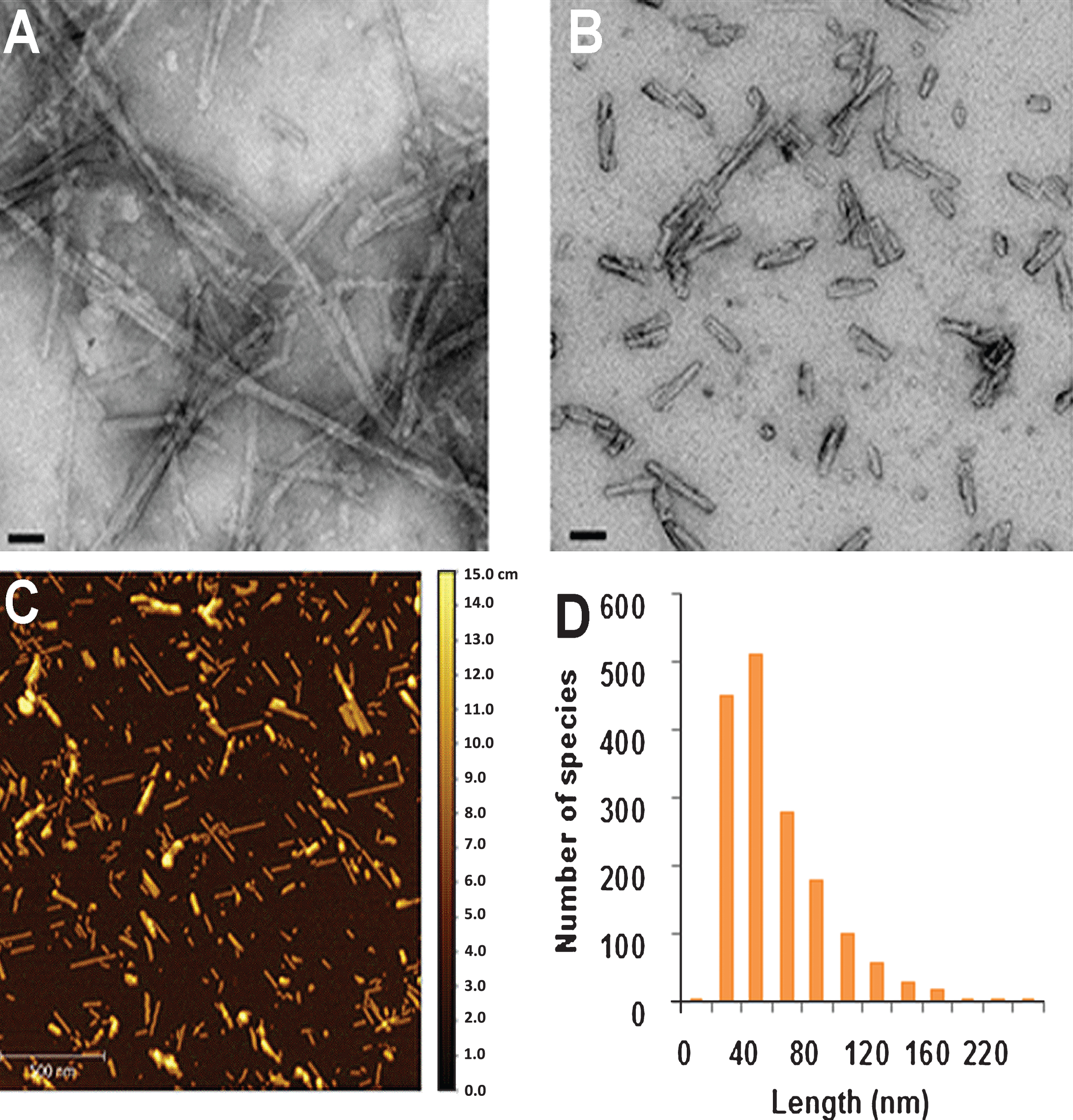
Fig.4
Schematic depiction of the workflow for generating, validating, and using alpha-synuclein pre-formed fibrils (aSyn PFFs) as the experimental protein and aSyn monomers as the control protein. When using aSyn PFFs as the experimental protein and aSyn monomers as the control, the first step for preparation and use of both samples is to defrost the specially-formulated aSyn monomers for PFF generation on ice and to preform endotoxin cleanup, if required (Green box). Once thawed, the sample should be divided in two, with half designated for use as the monomeric control protein (Blue boxes) and the other half destined for generation of PFFs (Orange boxes). Protocols for generating aSyn PFFs are located in the Supplementary Material, Fig. 1, and the cited literature. After the PFFs have been generated, aliquot into single use tubes of 5 mg/ml and store at –80°C for long-term storage or room temperature for short term storage. Use one aliquot to validate proper fibril formation (see Fig. 5A–C). Once fibril formation has been confirmed, sonication parameters must be validated. For this, use one aliquot (if stored at –80°C, defrost and keep at room temperature), dilute to the desired working concentration, and sonicate. After sonication, the fibril size and pathogenicity must be validated (see Fig. 5D–G). Similarly, aSyn monomers for the control samples should be aliquotted into single-use tubes at ≤7.5 mg/ml, validated alongside the aSyn PFFs as the control, and stored at –80°C. These validation steps should be performed in advance of use in order to confirm proper sample composition and to verify sonication parameters. On the day of use, defrost one aliquot of the aSyn PFFS (thaw at room temperature if frozen) and aSyn monomers (thaw on ice). Prepare using the validated dilutions and—in the case of the aSyn PFFs—sonication parameters tested previously. At this step it may be wise to again confirm sample composition via electron microscopy before use. Keep the aSyn PFFs at room temperature and the aSyn monomers on ice during use. Mix solutions between injections to ensure the larger aggregates do not pellet and lead to sample heterogeneity. A sample can be used for four hours before it should be replaced by a new sample. aSyn, alpha-synuclein; PFF, pre-formed fibrils; RT, room temperature; EM, electron microscopy; hrs, hours.
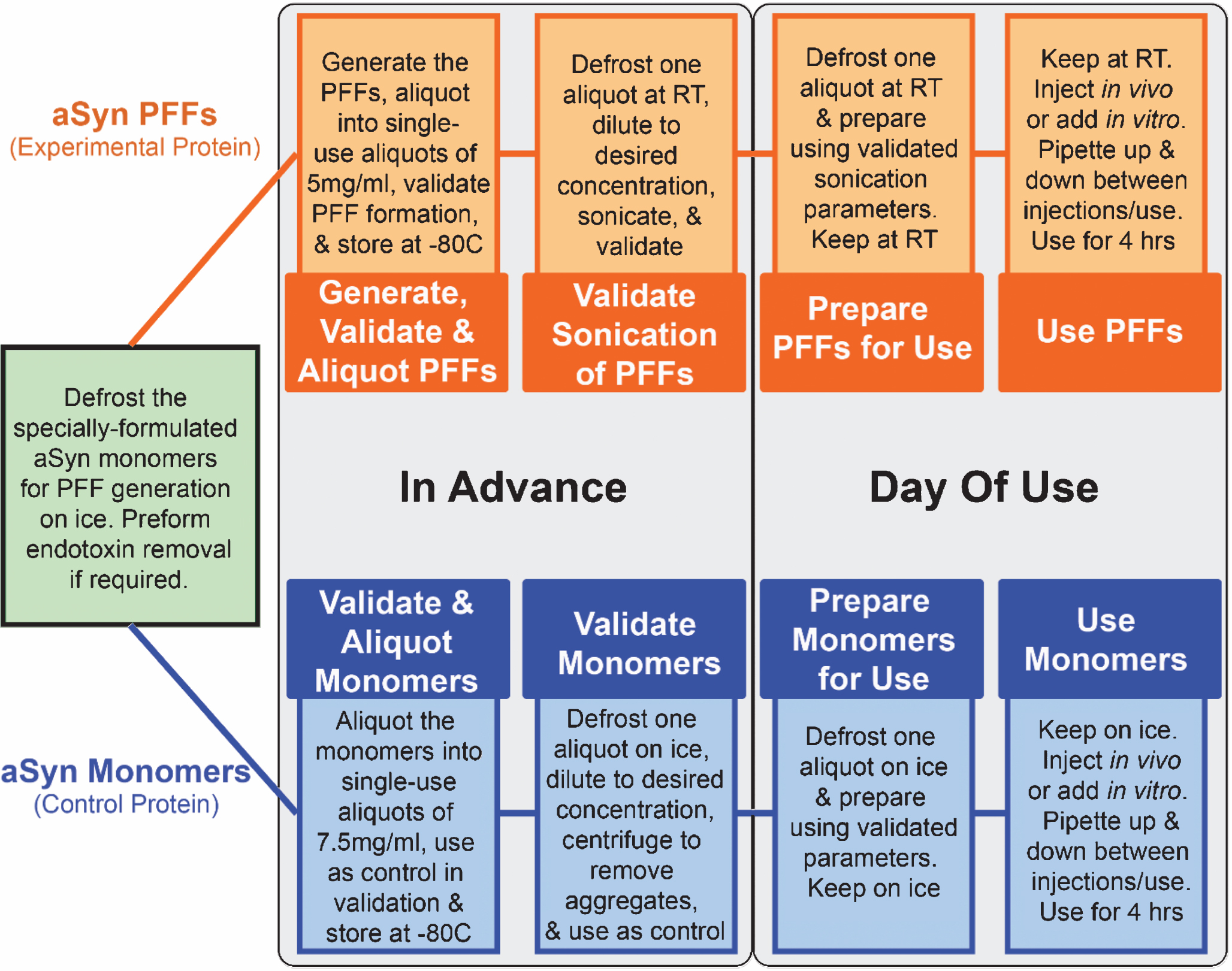
Fig.5
Recommended quality control experiments to verify proper alpha-synuclein (aSyn) fibril formation and preparation. Depiction of recommended quality control experiments (denoted with *) or supplemental quality control experiments with expected results. A–C) After the aSyn PFFs are generated from the monomeric starting material (Fig. 1B), samples should visually appear opaque and should be validated to confirm (A) amyloid conformation of the fibrils and (B–C) formation of long fibrillar protein aggregates. These validation efforts should be performed with every new batch of aSyn PFFs. A) The thioflavin T (ThT) assay is recommended to confirm presence of beta-sheet structures. Expected results include high levels of ThT fluorescent signal with PFFs as compared to monomers. It should be noted that human aSyn PFFs will elicit a stronger ThT signal than mouse aSyn PFFs [51]. B) The sedimentation assay is recommended to confirm aggregate formation in aSyn PFF samples. Expected results include substantially more protein in the pellet (pel) fraction as compared to the supernatant (sup) fraction for aSyn PFFs and the opposite result for monomeric aSyn protein. C) If sedimentation assays are not feasible or additional quality control is desired, electron microscopy is recommended to visualize the aSyn PFFs. Electron microscopy results should primarily show elongated fibrils. Scale bars = 50 nm. D–H) aSyn PFFs should again be validated post-sonication (Fig. 1C) to confirm (D–E) seeding capacity, (F–G) proper size of sonicated aSyn PFFs, and (H) in vivo pathogenicty. These validation efforts are at a minimum recommended when establishing or changing sonication parameters or before long-term in vivo studies. D–E) Seeding capacity should be confirmed using either (D) in vitro seeding experiments or (E) the ThT kinetic assay. D) In vitro seeding experiments are recommended for confirming the pathogenicity of aSyn fibrils as this will model the conversion of endogenous aSyn into pS129 aSyn in neurons after incubation with aSyn PFFs. Expected results include high levels of pS129 staining post-incubation with aSyn PFFs but no appreciable pS129 aSyn staining post-incubation with monomers or inadequately sonicated PFFs. Scale bar = 50 μm. E) If in vitro seeding assays are not feasible, the ThT kinetic assay may be used to confirm seeding capacity of aSyn PFF samples. Expected results include no increase in ThT fluorescence with addition of monomers and an increasing rate of ThT fluorescence with increased sonication time, indicating more pathogenic fibrils [35]. This assay is recommended when comparing different sonication parameters or when used with a positive control as the rate of increase or peak fluorescence levels may vary between runs and comparing only sonicated PFFs to monomers will not be informative. F–G) Average fibril size should also be confirmed post-sonication and before use by either (F) electron microscopy or (G) dynamic light scattering (DLS). F) Electron microscopy is recommended for visualizing sonicated aSyn PFFs to confirm size and uniformity of aggregates. The majority of fibrils analyzed should be 50 nm or smaller to elicit high levels of pathology (graph inset indicates average fibril size). Scale bars = 50 nm. G) If electron microscopy is not feasible or additional quality control is desired, DLS may be used to analyze fibril size [35]. Again, the majority of fibrils should be 50 nm or smaller to properly seed pathology. This graph depicts DLS data for fibrils separated by size. H) If using aSyn PFFs in a long-term in vivo study, it is highly recommended to perform a short term in vivo pilot study to verify in vivo pathogenicity and injection parameters. Thirty days post-injection may be sufficient to visualize early aSyn pathology by pS129 aSyn staining in the mouse [51] or rat [34] brain following striatal injection of mouse aSyn PFFs, although longer time points may be required. sup, supernatant; pel, pellet; aSyn, alpha-synuclein; PFF, pre-formed fibril; ThT, thioflavin T; AU, arbitrary units; sec, seconds; p-α-syn, alpha-synuclein phosphorylated at S129; M.W., molecular weight.
![Recommended quality control experiments to verify proper alpha-synuclein (aSyn) fibril formation and preparation. Depiction of recommended quality control experiments (denoted with *) or supplemental quality control experiments with expected results. A–C) After the aSyn PFFs are generated from the monomeric starting material (Fig. 1B), samples should visually appear opaque and should be validated to confirm (A) amyloid conformation of the fibrils and (B–C) formation of long fibrillar protein aggregates. These validation efforts should be performed with every new batch of aSyn PFFs. A) The thioflavin T (ThT) assay is recommended to confirm presence of beta-sheet structures. Expected results include high levels of ThT fluorescent signal with PFFs as compared to monomers. It should be noted that human aSyn PFFs will elicit a stronger ThT signal than mouse aSyn PFFs [51]. B) The sedimentation assay is recommended to confirm aggregate formation in aSyn PFF samples. Expected results include substantially more protein in the pellet (pel) fraction as compared to the supernatant (sup) fraction for aSyn PFFs and the opposite result for monomeric aSyn protein. C) If sedimentation assays are not feasible or additional quality control is desired, electron microscopy is recommended to visualize the aSyn PFFs. Electron microscopy results should primarily show elongated fibrils. Scale bars = 50 nm. D–H) aSyn PFFs should again be validated post-sonication (Fig. 1C) to confirm (D–E) seeding capacity, (F–G) proper size of sonicated aSyn PFFs, and (H) in vivo pathogenicty. These validation efforts are at a minimum recommended when establishing or changing sonication parameters or before long-term in vivo studies. D–E) Seeding capacity should be confirmed using either (D) in vitro seeding experiments or (E) the ThT kinetic assay. D) In vitro seeding experiments are recommended for confirming the pathogenicity of aSyn fibrils as this will model the conversion of endogenous aSyn into pS129 aSyn in neurons after incubation with aSyn PFFs. Expected results include high levels of pS129 staining post-incubation with aSyn PFFs but no appreciable pS129 aSyn staining post-incubation with monomers or inadequately sonicated PFFs. Scale bar = 50 μm. E) If in vitro seeding assays are not feasible, the ThT kinetic assay may be used to confirm seeding capacity of aSyn PFF samples. Expected results include no increase in ThT fluorescence with addition of monomers and an increasing rate of ThT fluorescence with increased sonication time, indicating more pathogenic fibrils [35]. This assay is recommended when comparing different sonication parameters or when used with a positive control as the rate of increase or peak fluorescence levels may vary between runs and comparing only sonicated PFFs to monomers will not be informative. F–G) Average fibril size should also be confirmed post-sonication and before use by either (F) electron microscopy or (G) dynamic light scattering (DLS). F) Electron microscopy is recommended for visualizing sonicated aSyn PFFs to confirm size and uniformity of aggregates. The majority of fibrils analyzed should be 50 nm or smaller to elicit high levels of pathology (graph inset indicates average fibril size). Scale bars = 50 nm. G) If electron microscopy is not feasible or additional quality control is desired, DLS may be used to analyze fibril size [35]. Again, the majority of fibrils should be 50 nm or smaller to properly seed pathology. This graph depicts DLS data for fibrils separated by size. H) If using aSyn PFFs in a long-term in vivo study, it is highly recommended to perform a short term in vivo pilot study to verify in vivo pathogenicity and injection parameters. Thirty days post-injection may be sufficient to visualize early aSyn pathology by pS129 aSyn staining in the mouse [51] or rat [34] brain following striatal injection of mouse aSyn PFFs, although longer time points may be required. sup, supernatant; pel, pellet; aSyn, alpha-synuclein; PFF, pre-formed fibril; ThT, thioflavin T; AU, arbitrary units; sec, seconds; p-α-syn, alpha-synuclein phosphorylated at S129; M.W., molecular weight.](https://content.iospress.com:443/media/jpd/2018/8-2/jpd-8-2-jpd171248/jpd-8-jpd171248-g005.jpg)
Tarutani and colleagues [47] and Abdelmotilib and colleagues [35] compared various probe sonication parameters and analyzed the effect of extended sonication times on fibril size, fibril structure, amyloid conformation of the fibrils, and fibril seeding capacity. Both groups reported no negative impact of longer sonication times on any readout. No difference was observed in amyloid confirmation or fibril structure (apart from size) between sonication for 0 s and up to 240 s, indicating that extended sonication times will not impair the amyloid properties of the fibrils or create new forms of aSyn aggregates. Furthermore, extended sonication times produced smaller aSyn fibrils on average, and these shorter fibrils were more efficient than longer fibrils at seeding the formation of new fibrils when incubated with monomeric aSyn or introduced in vitro or in vivo [35, 47]. It should be noted, however, that samples sonicated longer than 180 s have not been evaluated for in vitro or in vivo pathogenicity. In addition, heat generated during sonication may negatively impact fibril stability and should be avoided when probe sonicating, as was done in these two studies. To control for heat when probe sonicating for an extended period, it is recommended to probe sonicate in 15 s intervals with a 1 s sonication pulse and a 1 s wait, resting the solution at room temperature for 2 min to dissipate heat between each 15 s interval [35]. A detailed sonication protocol can also be found in Volpicelli-Daley, Luk, and Lee [31].
Important safety considerations must be taken when sonicating aSyn PFF samples as it remains unclear if aSyn PFFs pose a biosafety hazard. If probe sonicating aSyn PFF samples, sonication should be performed in a BSL-2 level fume hood to prevent inhalation of aerosolized aSyn PFFs [50]. Bath sonication can also be performed in lieu of probe sonication as this provides the advantage of sonicating in closed tubes with better temperature regulation, thereby eliminating inhalation risks and preventing heat-related damage to the sample. Where bath sonication is the preferred method, a high-powered water bath sonicator (such as the Bioruptor) should be used to ensure sufficient sonication. If the temperature of the water bath is not regulated or the sonicator is not sufficiently powered, aggregates may fail to fragment appropriately and the aSyn PFFs may show reduced ability to seed pathology. When performed correctly, bath sonication leads to highly reproducible results and eliminates the inhalation hazard. Regardless of sonication time or method, the protocol and fibrils should always be validated before use.
VALIDATION OF PROPER ALPHA-SYNUCLEIN PRE-FORMED FIBRIL FORMATION PRIOR TO USE
An important and often overlooked step in generating aSyn PFFs is validating proper fibril formation and activity before use. The pathogenicity of the aSyn PFFs in vitro and in vivo is dependent on the structure/conformation and size of the aggregates [35, 45–48]. In addition, the structure and size of aSyn PFFs are highly dependent on buffer conditions, sonication efficiency, and storage—parameters that are prone to change between labs or batches, leading to reproducibility issues. Furthermore, failing to validate proper fibril formation and size before intracerebral injection could lead to a lack of pathology in vivo and failure of a long-term, costly experiment. As a result, it is extremely important to validate successful conversion of aSyn monomers to fibrils and successful preparation of aSyn fibrils for experimental use. It is highly recommended to perform extensive validation when first establishing the PFF model in one’s lab, and again before initiating a long-term or costly study.
Herein we describe various experimental techniques that can be used to validate proper biophysical and biochemical properties of aSyn PFFs pre- and post-sonication. It is recommended to incorporate these quality control steps into experiments using aSyn PFFs to confirm certain properties of the aSyn PFFs. Passing these quality control steps does not guarantee the aSyn PFFs will generate pathology in vitro or in vivo. Rather, these quality control experiments should be used as general pass/fail criteria for the PFFs whereby failure to pass the quality control step indicates the need to re-make the aSyn PFFs but passing the quality control step does not necessarily ensure in vivo pathogenicity. In vivo pilot studies are the only method for validating the in vivo pathogenicity of aSyn PFF batches prior to long term in vivo studies.
Validating successful conversion of aSyn monomers to fibrils
To ensure aSyn monomers have successfully aggregated into fibrils, it is recommended to perform basic quality control experiments to verify resulting fibrils exhibit the proper biophysical and biochemical properties, as described below. Recommended quality control experiments at this stage include, but are not limited to, measuring protein concentration to ensure accurate dilutions for formation and use, the sedimentation assay to verify high molecular weight species of aSyn, and the thioflavin T (ThT) assay to confirm the beta sheet conformation that characterize the amyloid-like PFFs. These assays are simple, low-cost methods to quickly confirm aSyn concentration and the successful formation of fibrils, and should always be performed when generating a new batch of aSyn PFFs (Figs. 1B, 4, and 5A, B).
The protein concentration used in the PFF model can greatly impact the extent of pathology induced in the model [35]. Therefore, the exact protein concentration must be known in order to generate a robust, reproducible model. Protein concentration should be measured at multiple stages during the aSyn PFF generation and use protocol (Fig. 1). The first measurement of protein concentration should occur prior to dilution and incubation of the monomer. This measurement is important to verify the protein concentration reported in the datasheet and to accurately dilute the solution to the 5 mg/mL final concentration. Protein concentration should be re-measured after sample storage and prior to dilution, sonication, and use as protein levels have been shown to diminish with long-term storage or freeze-thaw. For protein determination, it is recommended to use a spectrophotometer for direct A280 measurement on a nanodrop device with Beer’s law to calculate the concentration of aSyn within the sample. To measure the concentration of synuclein, ɛ for synuclein is 5960 M–1 cm–1 for human synuclein and 7450 M–1 cm–1 for mouse synuclein. The BCA protein assay can also be used to measure protein concentration. If this assay is used, it is recommended to use three dilutions of protein (in triplicate for each dilution) to obtain accurate measurements. Please note, the aggregated sample would need to be disaggregated in order to accurately measure the protein concentration; for this, denaturation agents such guanidine chloride are recommended (Supplementary Figure 2).
After the seven day incubation to convert the aSyn monomers to fibrils, successful conversion should be analyzed using the sedimentation and ThT assays (Figs. 1B and 5A, B). A protocol for the sedimentation assay can be found in the Supplementary Material and in Volpicelli-Daley, Luk, and Lee [31]. When aSyn monomers have successfully assembled into fibrils, the sedimentation assay should result in the majority of protein in the pellet fraction compared to the supernatant (Fig. 5B). If the sedimentation assay results in higher protein levels in the soluble fraction than the pellet, this may indicate a failure to generate aSyn aggregates. In this case, it is recommended to repeat the sedimentation assay to verify results and discard the samples if greater protein levels are again found in the solute vs the pellet.
Whereas the sedimentation assay indicates the general presence of higher molecular weight species of aSyn, the ThT assay provides slightly more information on the higher molecular weight species of aSyn. Specifically, the ThT assay analyzes the structure of the higher molecular weight species to determine whether they formed the typical cross-beta structure. When aSyn fibrils have formed, the ThT assay should result in readings 20–100 folder higher for human aSyn PFFs as compared to human aSyn monomers (Fig. 5A). Human aSyn PFFs will also result in higher levels of ThT than murine aSyn PFFs, leading to greater subtlety in differences between monomers and PFFs in murine aSyn as compared to human aSyn [51]. A protocol for the ThT assay can also be found in the Supplementary Material.
Additional quality control experiments to validate successful conversion of aSyn monomers to fibrils include an imaging technique to visualize the size and morphology of aggregates such as transmission electron microscopy (TEM) or atomic force microscopy (AFM) (Figs. 3 and 5C), circular dichroism spectroscopy, or fourier-transform infrared spectroscopy to verify structural changes in the aSyn protein upon fibril formation [35]. When performing any experiment to validate successful conversion of aSyn monomers to fibrils, the fibrils should always be compared to the monomeric starting material (Fig. 4). Generally, outcome measures analyzing successful conversion of aSyn monomers to fibrils will result in binary results as the PFF protein is only compared to the negative control monomeric protein. Adding an aliquot of a previously-successful batch of aSyn PFFs may increase the reproducibility across batches and experiments by providing a positive control.
Validating successful preparation of aSyn PFFs for experimental use
As mentioned previously, quality control experiments to validate successful conversion of aSyn monomers to PFFs should be performed regularly with each new batch of PFFs. In contrast, the necessity of performing experiments to validate successful sonication and preparation of aSyn PFFs may vary based on the end use of the aSyn PFFs or the investigator’s experience with the PFF generation protocol. In general, experiments to validate successful preparation of aSyn PFFs should be performed exhaustively when first establishing a PFF generation protocol or when altering any step of the protocol. It is also advisable to test multiple sonication parameters to determine which parameters perform best for that lab, as sonication effectiveness may differ based on the sonicator, sample volume, buffers, protocol, or other factors. Although an investment, these early optimization and validation experiments could prevent future issues that may be costly and time-consuming. Once a protocol is established and successful preparation and sonication parameters have been repeatedly validated the need for continued, exhaustive quality control experiments is reduced and may be skipped for short-term, low-risk experiments.
Validating successful preparation of aSyn PFFs for experimental use occurs after dilution and sonication of an aSyn PFF aliquot (Figs. 1C and 4) and should seek to confirm aSyn PFF size and seeding capacity (Fig. 5D–G). Similar to the earlier quality control experiments, the monomeric aSyn starting material should be used as the negative control and an aliquot of a previous lot of aSyn PFFs that has demonstrated success may be used as the positive control. Examples of recommended techniques to analyze the average size of the aSyn PFFs include TEM (Fig. 5F), AFM (Fig. 3), and dynamic light scattering (Fig. 5G), which can be performed when optimizing sonication parameters or validating an aSyn PFF sample before use in a long-term study. Examples of recommended experiments to analyze seeding capacity include the in vitro seeding assay (Fig. 5D), which can be performed when optimizing sonication parameters or validating a sample before use, or the ThT kinetic seeding assay (Fig. 5E), which is recommended when testing multiple sonication parameters.
The length of the fibrils is extremely important for the pathogenicity of the aSyn PFFs. Studies have demonstrated that aSyn PFFs 50 nm or smaller seed the most pathology in vitro and in vivo [35, 47]. If the average size of the aSyn PFFs in a sample is greater than 50 nm, pathology will be reduced or absent. Therefore, adequate sonication and confirmation of small fibrils are extremely important before embarking on a long-term in vivo study or a study investigating a costly therapeutic intervention or outcome measure. The gold standard method for confirming sonication and fibril size is the statistical analysis of TEM/AFM images (Fig. 5F). This outcome measure provides a direct measure of average fibril size and provides the additional advantage of allowing analysis of fibril shape, morphology, and sample purity.
If access to TEM/AFM facilities are limited and analysis pre-injection is not feasible, it is recommended to retain a portion of the sample used for injection and perform the sample imaging analysis shortly after the surgical session. In that case, the PFF sample should be kept at room temperature and analyzed within one week of use. However, samples should be analyzed shortly before/after use as the sonicated fibrils can change over time (Supplementary Figure 1). An alternative outcome measure for analyzing fibril size when TEM/AFM is not available is dynamic light scattering (Fig. 5G). Similarly, it is recommended to perform dynamic light scattering on a portion of the sonicated PFF samples prior to use and to compare the results of the PFF sample to the monomeric starting material and the unsonicated fibrillar sample. Although this method does not allow direct visualizing of fibril size, morphology, or purity, it will give an indication of sonication efficiency [35]. Regardless of the method used to analyze fibril size post-sonication, confirmation that sonication resulted in the majority of fibrils 50 nm or smaller is an important quality control measure and should not be overlooked.
Similarly, analyzing seeding capacity of the prepared aSyn PFF sample is an important step in validating a sample or protocol before embarking on a long-term or expensive study. This quality control step is highly recommended when first establishing or when altering the PFF generation protocol to verify the formation of aSyn PFFs that have the ability to seed pathology. There are multiple methods for analyzing seeding capacity of PFF samples. These include in vitro seeding assays in primary neuron cultures, in vitro seeding assays in aSyn-expressing or -overexpressing cell lines, and ThT kinetic seeding assays (Fig. 5D, E). With all three methods, aSyn PFFs should be compared to the monomeric aSyn starting material for a negative control and can be compared to a previously successful batch of aSyn PFFs for a positive control.
Evaluating the ability of aSyn PFFs to generate pS129 aSyn pathology in vitro in mouse primary neuronal cultures is the most relevant method for analyzing seeding capacity of an aSyn PFF sample prior to in vivo use (Fig. 5D). This assay is preferred over the other two methods as primary mouse neuronal cultures express endogenous forms of aSyn at normal levels, cells are not immortalized, and the cell species is similar or identical to the species of animal receiving the in vivo injection. A detailed protocol for analyzing the seeding capacity of aSyn PFFs in primary neuronal cultures is provided in Volpicelli-Daley, Luk, and Lee [31]. Additional considerations for using aSyn PFFs in primary neuronal cultures can be found in the subsequent section of this manuscript. If primary neuronal cultures are not feasible or desired, seeding capacity of aSyn PFFs can also be analyzed using in vitro cultures of cell lines overexpressing aSyn, such as SH-SY5Y cells [32, 46, 47] or HEK293 cells [32]. However, these cell lines provide a more artificial system in that they express aSyn at supraphysiological levels and they are not from the species that will receive the in vivo injection. Therefore, caution must be used when generalizing in vitro results from aSyn-overexpressing cell lines to predictions of in vivo seeding capacity of aSyn PFFs.
Another method for analyzing seeding capacity is the ThT kinetic seeding assay (Fig. 5E). In this assay, a small amount of aSyn PFFs are introduced to recombinant aSyn monomeric protein and the rate of ThT fluorescence intensity is recorded at various time points post-induction [35, 46, 47, 49]. This method is best used when comparing different sonication parameters or aSyn species rather than analyzing one PFF sample vs the monomeric control as there is no threshold the assay must reach in terms of rate of ThT intensity or level of ThT intensity to deem the reaction successful.
Additionally, one may consider a short, in vivo pilot study before embarking on a long-term in vivo study (Fig. 5H). This quality control step is especially important when altering or adopting new aSyn PFF generation protocols as the aSyn PFF model oftentimes requires months for a phenotype to develop, thereby resulting in a substantial loss of time and resources if the fibrils are not pathogenic. An in vivo pilot study is the only quality control method that will evaluate in vivo pathogenicity. Although the other quality control methods proposed will provide evidence for the presence of desirable traits in the fibrils (Fig. 5A–G), none of these completely correlate with in vivo success. Therefore, investigators should remember to use the proposed quality control methods in Fig. 5A–G as general pass/fail criteria whereby failure suggests the need to remake the PFFs and success suggests likely (but not guaranteed) pathology when introduced in vivo. For pilot studies, analysis at 30 days post-injection (dpi) is generally enough time to induce robust pS129 pathology in the rat [34] and mouse [51] brain after striatal injection, although rates of pathology may differ based on the species of aSyn PFFs, the rodent strain, the target structure, and the aSyn PFF dose, among other factors.
The aSyn PFF model is a robust, reproducible model when the PFF generation protocol has been validated and adapted for use in a lab. The need to extensively characterize the aSyn PFF samples before use decreases after a protocol is successfully used multiple times. In such cases, the ThT and sedimentation assays could suffice to validate successful PFF generation before use in vitro or in a short in vivo study. Before long-term/costly in vivo studies or after long-term storage of aSyn PFFs, however, it is still recommended to analyze aSyn PFF size, seeding capacity, and possibly in vivo performance before use even if the protocol has been validated previously. In these cases, an upfront investment in quality control and validation of aSyn PFFs may prevent the failure of a large, long-term in vivo study.
CONSIDERATIONS FOR THE USE OF ALPHA-SYNUCLEIN PRE-FORMED FIBRILS IN CULTURE OR INTRACEREBRAL INJECTION
After aSyn PFF samples have been generated, sonicated, and validated, they are ready for use in in vitro and in vivo experiments (Figs. 1 and 4). At this point, appropriate study designs and optimized protocols must be used to ensure a preclinical model is successfully generated for accurate modeling of aSyn biology and testing of therapeutic interventions. Important considerations for this step include choosing the appropriate control and experimental proteins, choosing an amenable cell line for in vitro studies, and using optimized surgical procedures for in vivo studies.
Choosing the appropriate species of alpha-synuclein pre-formed fibrils
Choosing the appropriate species of recombinant aSyn to generate PFFs is a critical step of designing and using the aSyn PFF model as the species of aSyn used can impact the level of pathology induced and the rate of progression of the pathological phenotypes. Generally, greater homology between the host and species of recombinant aSyn protein will result in greater pathology in vitro and in vivo. This has been demonstrated at the level of seeding recombinant protein as human aSyn PFFs are much more effective at templating amyloid-formation of human aSyn monomers than mouse aSyn monomers when measured by the ThT kinetic seeding assay [44, 51]. Similarly, mouse aSyn PFFs seed amyloid-structure formation in mouse aSyn monomers more effectively than human aSyn monomers [51]. The importance of species homology between the aSyn PFF and host was confirmed in vitro with higher levels of pS129 aSyn staining, soluble aSyn, and insoluble aSyn in mouse primary hippocampal neuron cultures following incubation with mouse aSyn PFFs versus human aSyn PFFs [51], although other studies have reported relatively similar levels of seeding between mouse and human aSyn PFFs in vitro [35].
In vivo, pathology is more severe and rapid with greater species homology. Intrastriatal injection of mouse aSyn PFFs in mice resulted in pS129 aSyn pathology at 30 dpi, whereas pathology was absent at this time point following injection of human aSyn PFFs [51]. At 180 dpi, mouse aSyn PFFs resulted in pS129 aSyn in 38% of dopaminergic neurons, whereas human aSyn PFFs resulted in pS129 aSyn in only 20% of dopaminergic neurons—affirming the ability of human aSyn PFFs to seed pathology in mice but to a lesser extent than mouse aSyn PFFs. Furthermore, significant dopaminergic neuron degeneration was observed in mice at 180 dpi of mouse aSyn PFFs (∼40% degeneration) but no significant decrease in dopaminergic neuron numbers was observed at 180 dpi of human aSyn PFFs [51]. This phenomenon may also occur in rats, as previous reports have indicated that mouse aSyn PFFs can trigger pathology in wildtype rats [34] whereas human aSyn PFFs require the overexpression of aSyn for adequate pathology to develop [52]. Taken together, these results highlight the importance of factoring in species differences between host species and recombinant aSyn protein when designing studies to ensure adequate levels of pS129 aSyn pathology and neuronal degeneration are obtained at the chosen time points.
Choosing the proper control for the alpha-synuclein pre-formed fibril model
Currently, the most highly recommended control for the aSyn PFF model is the aSyn monomeric protein starting material. If monomers are to be used as the control, it is recommended to remove endotoxins and add an additional centrifugation step before use as endotoxins and low levels of spontaneous aggregates in the monomeric protein sample may seed a small number of pS129 aSyn-positive aggregates in the SNpc at 6 months post-injection [34]. When prepared correctly by controlling for endotoxin units and spontaneous aggregates, aSyn monomers will fail to induce pathology in vitro and in vivo [34, 35, 47] and will provide the added benefit of serving as a control for injection of a foreign protein that is identical to the protein used to generate the aSyn PFFs. If using the aSyn monomeric starting material as the control, a large quantity of aSyn monomers should be ordered or generated, with a portion of that protein used to generate aSyn PFFs and a portion kept for use as the control protein (Fig. 4). This will ensure that the aSyn monomers and PFFs are from the same lot of protein to avoid any confounds that may arise with separate protein batches or shipping/handling.
Importantly, precautions should be taken to measure and minimize endotoxin in monomeric protein samples. In particular, aSyn monomers purified from bacterial cell expression systems such as E. coli may have high levels of endotoxin units (EUs) that can persist after PFF generation or if monomers are used as a control protein. Even at moderate levels, EUs can trigger an inflammatory response or induce toxicity in cells and therefore should be avoided in the control samples. It is recommended that EU levels should be <0.5 EU/mL or <0.05 EU/mg at 10 mg/mL protein. The Pierce High Capacity Endotoxin Removal Kit is a reliable method for removing endotoxin from monomers but may result in considerable loss of sample during the process. If endotoxin removal is required, it is recommended to generate or purchase extra monomeric protein to account for this potential loss and to perform endotoxin removal prior to fibril generation (Fig. 4). If using the aSyn monomeric protein as the control, it should also be noted that an additional centrifugation step before use—like that at the beginning of the protocol—should be considered as this will remove spontaneous aggregates and prevent low levels of pathology that may ensue (Fig. 1) [31].
Aside from aSyn monomers, other controls have been used in aSyn PFF experiments. The second most common control is PBS, as this control is guaranteed to not produce toxicity [29–31, 51]. However, the injection of saline does not control for the introduction of a foreign protein to the brain. Aggregation-incompetent forms of aSyn have also been suggested as a control for the aSyn PFF model. These forms of aSyn offer the advantage of allowing the sample to undergo the same pretreatment as the aSyn PFFs (heat, shaking, sonication) to control for the effects of these treatments on the protein. Although aggregation-incompetent forms of aSyn have shown promising results as a control in in vitro experiments [44], these proteins have not been tested in vivo. Serum albumin has also been suggested as a control for aSyn PFF experiments as this protein would control for the introduction of a larger protein in the brain and would therefore control for protein size and uptake. However, serum albumin may activate microglia in the local environment and should not be used as a control when microglial activation is an important variable at the injection site. With these benefits and caveats in mind, the aSyn monomeric starting material is still the most recommended control for the aSyn PFF model.
Use of aSyn PFFs for in vitro studies
As mentioned previously, there are multiple methods for studying aSyn PFF seeding and toxicity in vitro, including the use of primary neuronal cultures or immortalized human cell lines expressing aSyn. It is recommended to use primary neuronal cultures first and foremost, as these cultures express physiologically-relevant levels of aSyn and are not immortalized. The most effective primary neuronal cultures for aSyn PFF seeding are mouse hippocampal cultures. Rationale and a protocol for culturing mouse primary hippocampal neurons can be found in Volpicelli-Daley, Luk, and Lee [31]. Briefly, hippocampal neurons are more efficient in seeding aSyn pathology than mesencephalic neuronal cultures, are well-characterized for morphological and physiological characteristics in vitro, and hippocampal cultures produce enough neurons for multiple outcome measures [31]. Furthermore, mouse primary cultures are recommended over rat primary cultures as rat primary neuron cultures do not consistently seed mouse aSyn PFF or human aSyn PFF pathology well. It is unclear as to whether this is due to species homology issues, lower levels of endogenous aSyn expression, or other factors.
Use of aSyn PFFs for in vivo studies
When using aSyn PFFs for in vivo studies, it is recommended to use freshly-generated aSyn PFFs rather than frozen aliquots of aSyn PFFs. In this case, aSyn PFFs should be generated 1-2 weeks before the surgical session and stored in single-use aliquots at room temperature. Quality control experiments such as the ThT assay and sedimentation assay can be performed during this 1–2 week window using an aliquot of the prepared sample. Preparation of the aSyn PFFs and/or aSyn monomers for use—including dilution and sonication—should be performed using validated protocols on the day of surgery (Figs. 1C and 4). If an 8-hour surgical session is planned, it is recommended to prepare and use one aliquot of aSyn PFFs in the morning and to prepare and use a separate aliquot of aSyn PFFs from the same batch in the afternoon. Many groups advise against using a sonicated aSyn PFF aliquot for longer than four hours as the sonicated aSyn PFF samples may not remain stable over time (Supplementary Figure 1B). Nonetheless, there remains some debate as to fibril stability so this recommendation is made primarily out of caution as this storage duration has been validated (Supplementary Figure 1B). If using aSyn PFF samples longer than 4 hours, it is recommended to perform a time course experiment to determine acceptable post-sonication sample viability.
During the surgical session, aSyn PFFs and monomers must be kept at the correct temperature and precautions must be taken to ensure the sample remains homogeneous. Sonicated aSyn PFF samples must be kept at room temperature to prevent degradation [46, 49] (Supplementary Figure 1A). Prior to each injection, the aliquot of aSyn PFFs should be mixed by pipetting up and down to prevent aggregates from pelleting over time. It is not advisable to sonicate the aSyn PFF sample between injections as this will generate different sizes of aSyn PFFs and result in variations within the aSyn PFF-injected cohort. If aSyn monomers are used as the control for an in vivo study, aliquots of aSyn monomers should be kept on ice to prevent spontaneous aggregation.
For intracerebral injections, it is recommended to use a pulled pipette glass needle attached to a Hamilton syringe to minimize tissue injury and reduce non-target placement and infusion [34], although other groups have reported success using a Hamilton syringe directly [29, 30, 35, 42, 51]. When performing the injection, it is strongly recommended to slowly infuse the injectate at a rate of at least 0.5 μl/minute and to leave the needle in place 2–5 minutes after each injection to allow the solution to permeate the parenchyma and prevent the solution from being suctioned up the needle tract. By far, the striatum is the most common target for aSyn PFF injections [29, 30, 34, 35, 47, 51], with injection into the dorsal striatum leading to midbrain pathology specifically in the SNpc (Fig. 6). Other targets include the SNpc [35, 42], the cortex [41] and the cortex in conjunction with the striatum [29], among others. Injection coordinates are available in the published literature for these sites.
Fig.6
Optimized striatal injection coordinates for use in the rat alpha-synuclein pre-formed fibril (aSyn PFF) model. A–B) Comparison of pS129 aSyn expression in the ipsilateral midbrain after two site striatal injections using (A) original, traditionally-used coordinates [34] or (B) optimized coordinates guided by recent findings revealing distinct SNpc-striatal innervation patterns [54]. Neuroanatomical atlas images from Paxinos and Watson [58] depict the general coordinates used for injection. Representative low and high magnification images are shown for ipsilateral midbrain stained with cresyl violet (purple) and antibodies directed against pS129 aSyn (brown). A) Traditional injection coordinates involve injection into the dorsomedial striatum (DMS) and ventrolateral striatum (VLS). Injection into these coordinates induces robust pS129 aSyn expression in the SNpc as well as the ventral tegmental area (VTA). B) Optimized injection coordinates involve injection into the DMS as well as the dorsolateral striatum (DLS). Injection into these coordinates induces robust pS129 aSyn expression that is localized primarily to the SNpc and absent from the VTA. C–D) Quantitation of the percentage of pS129 aggregates in the (C) SNpc or (D) VTA following injection using the original (DMS + VLS) or optimized (DMS + DLS) injection coordinates. Injection using the original coordinates results in ∼80% of aggregates in the SNpc and ∼20% of aggregates in the VTA whereas injection using the optimized coordinates results in ∼90% of aggregates in the SNpc and ∼10% of aggregates in the VTA. Mean values are shown with error bars denoting standard error of the mean. aSyn, alpha-synuclein; PFF, pre-formed fibril; DMS, dorsomedial striatum; VLS, ventrolateral striatum; DLS, dorsolateral striatum; SNpc, substantia nigra pars compacta; VTA, ventral tegemental area; pS129 aSyn, alpha-synuclein phosphorylated at S129.
![Optimized striatal injection coordinates for use in the rat alpha-synuclein pre-formed fibril (aSyn PFF) model. A–B) Comparison of pS129 aSyn expression in the ipsilateral midbrain after two site striatal injections using (A) original, traditionally-used coordinates [34] or (B) optimized coordinates guided by recent findings revealing distinct SNpc-striatal innervation patterns [54]. Neuroanatomical atlas images from Paxinos and Watson [58] depict the general coordinates used for injection. Representative low and high magnification images are shown for ipsilateral midbrain stained with cresyl violet (purple) and antibodies directed against pS129 aSyn (brown). A) Traditional injection coordinates involve injection into the dorsomedial striatum (DMS) and ventrolateral striatum (VLS). Injection into these coordinates induces robust pS129 aSyn expression in the SNpc as well as the ventral tegmental area (VTA). B) Optimized injection coordinates involve injection into the DMS as well as the dorsolateral striatum (DLS). Injection into these coordinates induces robust pS129 aSyn expression that is localized primarily to the SNpc and absent from the VTA. C–D) Quantitation of the percentage of pS129 aggregates in the (C) SNpc or (D) VTA following injection using the original (DMS + VLS) or optimized (DMS + DLS) injection coordinates. Injection using the original coordinates results in ∼80% of aggregates in the SNpc and ∼20% of aggregates in the VTA whereas injection using the optimized coordinates results in ∼90% of aggregates in the SNpc and ∼10% of aggregates in the VTA. Mean values are shown with error bars denoting standard error of the mean. aSyn, alpha-synuclein; PFF, pre-formed fibril; DMS, dorsomedial striatum; VLS, ventrolateral striatum; DLS, dorsolateral striatum; SNpc, substantia nigra pars compacta; VTA, ventral tegemental area; pS129 aSyn, alpha-synuclein phosphorylated at S129.](https://content.iospress.com:443/media/jpd/2018/8-2/jpd-8-2-jpd171248/jpd-8-jpd171248-g006.jpg)
Optimization of injection parameters is recommended before embarking on a long-term study. At times, injection coordinates may need to be refined based on the dose/volume of aSyn PFFs used or the target structure. For instance, recent optimization efforts for striatal injections in rats have indicated that targeting the dorsal striatum in contrast to the ventral striatum results in better pS129 pathology in the SNpc while avoiding innervation to the nearby ventral tegmental area (VTA) (Fig. 6) [54]. By performing a short-term pilot study, investigators can analyze injection volume, PFF concentration, needle placement, infusion success, and verify pathogenicity of the aSyn PFFs in vivo.
Similar to species homology affecting the rate of aSyn pathology in vivo, the rodent strain may also affect the rate of pathology and phenotype. Mice from the following genetic backgrounds have been used successfully with the PFF-seeding paradigm: C57Bl/6J [55, 42], C57Bl/6/ C3H [30, 56], CD-1 [57], and C3H/HeJ [35]. Sprague-Dawley rats have also been used successfully for the aSyn PFF model [34, 52]. The rodent strain should be chosen based on the desired rate of pathology, extent of pathology, pathological markers, and outcome measures or techniques.
Safety considerations when using alpha-synulcein pre-formed fibrils
Importantly, the ability of the aSyn PFFs to seed pathology in vivo leads to concerns over safety precautions that must be taken when handling these proteins. Precautions must be taken when sonicating aSyn PFFs during preparation to reduce the risk of inhaling the aerosolized proteins. For this step, it is recommended to sonicate the aSyn PFF samples in a BSL-2 level safety hood that is externally ducted and does not re-circulate exhaust into the laboratory space. In addition, precautions must be taken when handling the aSyn PFF solution to prevent contamination of oneself and reusable laboratory materials. Proper safety attire should be worn when working with aSyn PFF samples to avoid coming in direct contact with the aSyn PFFs. Proper methods should be used to clean reusable materials that come in contact with the aSyn PFF solution. An effective detergent-based method for removing aSyn assemblies from plastic, glass, aluminum, and stainless steel surfaces is described in Bousset et al. [50].
CONCLUSIONS
Although a useful model with unique benefits, the aSyn PFF model presents key challenges that have made it occasionally difficult to adopt. By raising awareness to these challenges and presenting solutions to avoid these pitfalls, The Michael J. Fox Foundation strives to enhance the robustness and reproducibility of this model. With the help of experts and input from the broader scientific community, MJFF has identified common challenges/missteps in the generation, validation, and use of the recombinant aSyn PFF protein to generate the aSyn PFF model. A summary of the common mistakes and solutions presented in this manuscript can be found in Fig. 7. With this information, investigators should be able to avoid many of the difficulties that have been previously reported by others who have attempted to use the aSyn PFF model. Increasing the robustness of the aSyn PFF model should hopefully providing a new avenue to improve understanding in the biology of PD and develop new disease-modifying treatments for this neurodegenerative disease.
Fig.7
Common pitfalls in the alpha-synuclein pre-formed fibril (aSyn PFF) model and solutions for avoiding these mistakes. Six common issues that lead to lack of pathology or other issues in the aSyn PFF model include (A) using an incompatible protein as the monomeric starting material or injected material, (B) forming PFFs in an incompatible buffer, (C) storing or keeping solutions at the incorrect temperature, (D) inadequately sonicating the aSyn PFF sample, (E) failing to validate the aSyn PFFs have the proper biophysical and biochemical properties, and (F) choosing an unideal control or not accounting for the control when reserving aSyn monomeric protein. These are six very important factors to which attention should be paid and caution should be taken to avoid. Guidelines for how to avoid these pitfalls are included as well. aSyn, alpha-synuclein; PFF, pre-formed fibril; RT, room temperature.

CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
The authors would like to acknowledge individuals that were responsible for contributing thought leadership and experimental expertise to this manuscript. In particular, we would like to thank Dr. Andrew West (University of Alabama at Birmingham) for contribution of the dynamic light scattering data, Ms. Marta Castellana, Dr. Serene Chen, and Dr. Janet Kumita (University of Cambridge) for contribution of the TEM and AFM imaging analysis of fibril storage and PFFs size analysis, Ms. Samantha Decker (University of Pennsylvania) for ThT and sedimentation data, Ms. Amani Allen (Michigan State University) for assistance with intrastriatal surgical parameter optimization studies and Dr. Joseph Patterson (Michigan State University) for assistance with TEM analysis. Finally, we would like to thank all the Parkinson’s disease patients and our donors that continue to inspire us with their endless optimism.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: http://dx.doi.org/10.3233/JPD-171248.
REFERENCES
[1] | Dorsey ER , Constantinescu R , Thompson JP , Biglan KM , Holloway RG , Kieburtz K , Marshall FJ , Ravina BM , Schifitto G , Siderowf A , Tanner CM ((2007) ) Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 68: , 384–386. |
[2] | Graybiel AM , Hirsch EC , Agid Y ((1990) ) The nigrostriatal system in Parkinson’s disease. Adv Neurol 53: , 17–29. |
[3] | Burke RE , O’Malley K ((2013) ) Axon degeneration in Parkinson’s disease. Exp Neurol 246: , 72–83. |
[4] | Pollanen MS , Dickson DW , Bergeron C ((1993) ) Pathology and biology of the Lewy body. J Neuropathol Exp Neurol 52: , 183–191. |
[5] | Spillantini MG , Schmidt ML , Lee VM , Trojanowski JQ , Jakes R , Goedert M ((1997) ) Alpha-synuclein in Lewy bodies. Nature 388: , 839–840. |
[6] | Braak H , Del Tredici K , Bratzke H , Hamm-Clement J , Sandmann-Keil D , Rub U ((2002) ) Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages). J Neurol 249: (Supp 3), III/1–III/5. |
[7] | Olanow CW , Stern MB , Sethi K ((2009) ) The scientific and clinical basis for the treatment of Parkinson disease. Neurology 72: , S1–S136. |
[8] | Polymeropoulos MH , Lavedan C , Leroy E , Ide SE , Dehejia A , Dutra A , Pike B , Root H , Rubenstein J , Boyer R , Stenroos ES , Chandrasekharappa S , Athanassiadou A , Papapetropoulos T , Johnson WG , Lazzarini AM , Duvoisin RC , Di Iorio G , Golbe LI , Nussbaum RL ((1997) ) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276: , 2045–2047. |
[9] | Singleton AB1 , Farrer M , Johnson J , Singleton A , Hague S , Kachergus J , Hulihan M , Peuralinna T , Dutra A , Nussbaum R , Lincoln S , Crawley A , Hanson M , Maraganore D , Adler C , Cookson MR , Muenter M , Baptista M , Miller D , Blancato J , Hardy J , Gwinn-Hardy K ((2003) ) Alpha-synuclein locus triplication causes Parkinson’s disease. Science 302: , 841. |
[10] | Schober A ((2004) ) Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res 318: , 215–224. |
[11] | Jackson-Lewis V , Blesa J , Przedborski S ((2012) ) Animal models of Parkinson’s disease. Parkinsonism Relat Disord 18: (Supp 1), S183–S185. |
[12] | Gonera EG1 , van’t Hof M , Berger HJ , van Weel C , Horstink MW ((1997) ) Symptoms and duration of the prodromal phase in Parkinson’s disease. Mov Disord 12: , 871–876. |
[13] | Salat D , Noyce AJ , Schrag A , Tolosa E ((2016) ) Challenges of modifying disease progression in prediagnostic Parkinson’s disease. Lancet 15: , 637–648. |
[14] | Visanji NP , Brotchie JM , Kalia LV , Koprich JB , Tandon A , Watts JC , Lang AE ((2016) ) Alpha-synuclein-based animal models of Parkinson’s disease: Challenges and opportunities in a new era. Trends Neurosci 39: , 750–762. |
[15] | Zimprich A1 , Biskup S , Leitner P , Lichtner P , Farrer M , Lincoln S , Kachergus J , Hulihan M , Uitti RJ , Calne DB , Stoessl AJ , Pfeiffer RF , Patenge N , Carbajal IC , Vieregge P , Asmus F , Müller-Myhsok B , Dickson DW , Meitinger T , Strom TM , Wszolek ZK , Gasser T ((2004) ) Mutation s in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44: , 601–607. |
[16] | Volta M , Melrose H ((2017) ) LRRK2 mouse models: Dissecting the behavior, striatal neurochemistry and neurophysiology of PD pathogenesis. Biochem Soc Trans 45: , 113–122. |
[17] | Yokochi M , Mizuno Y , Shimizu N ((1998) ) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: , 605–608. |
[18] | Valente EM1 , Abou-Sleiman PM , Caputo V , Muqit MM , Harvey K , Gispert S , Ali Z , Del Turco D , Bentivoglio AR , Healy DG , Albanese A , Nussbaum R , González-Maldonado R , Deller T , Salvi S , Cortelli P , Gilks WP , Latchman DS , Harvey RJ , Dallapiccola B , Auburger G , Wood NW ((2004) ) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304: , 1158–1160. |
[19] | Bonifati V , Rizzu P , van Baren MJ , Schaap O , Breedveld GJ , Krieger E , Dekker MC , Squitieri F , Ibanez P , Joosse M , van Dongen JW , Vanacore N , van Swieten JC , Brice A , Meco G , van Duijn CM , Oostra BA , Heutink P ((2003) ) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299: , 256–259. |
[20] | Lee Y , Dawson VL , Dawson TM ((2012) ) Animal models of Parkinson’s disease: Vertebrate genetics. Cold Spring Harb Perspect Med 2: , a009324. |
[21] | Blesa J , Przedborski S ((2014) ) Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front Neuroanat 8: , 155. |
[22] | Van der Perren A , Van den Haute C , Baekelandt V ((2015) ) Viral vector-based models of Parkinson’s disease. Curr Top Behav Neurosci 22: , 271–301. |
[23] | Low K , Aebischer P ((2012) ) Use of viral vectors to create animal models for Parkinson’s disease. Neurobiol Dis 48: , 189–201. |
[24] | Koprich JB1 , Johnston TH , Huot P , Reyes MG , Espinosa M , Brotchie JM ((2011) ) Progressive neurodegeneration or endogenous compensation in an animal model of Parkinson’s disease produced by decreasing doses of alpha-synuclein. PLoS One 6: , e17698. |
[25] | Gombash SE1 , Manfredsson FP , Kemp CJ , Kuhn NC , Fleming SM , Egan AE , Grant LM , Ciucci MR , MacKeigan JP , Sortwell CE ((2013) ) Morphological and behavioral impact of AAV2/5-mediated overexpression of human wildtype alpha-synuclein in the rat nigrostriatal system. PLoS One 8: , e81426. |
[26] | Zhou J1 , Broe M , Huang Y , Anderson JP , Gai WP , Milward EA , Porritt M , Howells D , Hughes AJ , Wang X , Halliday GM ((2011) ) Changes in the solubility and phosphorylation of alpha-synuclein over the course of Parkinson’s disease. Acta Neuropathol 121: , 695–704. |
[27] | Miller DW , Hague SM , Clarimon J , Baptista M , Gwinn-Hardy K , Cookson MR , Singleton AB ((2004) ) Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62: , 1835–1838. |
[28] | Volpicelli-Daley L , Kirik D , Stoyka LE , Standaert DG , Harms AS ((2016) ) How can rAAV-alpha-synuclein and the fibril alpha-synuclein models advance our understanding of Parkinson’s disease? J Neurochem 139: (Suppl 1), 131–155. |
[29] | Luk KC , Kehm VM , Zhang B , O’Brien P , Trojanowski JQ , Lee VM-Y ((2012) ) Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med 209: , 975–986. |
[30] | Luk KC1 , Kehm V , Carroll J , Zhang B , O’Brien P , Trojanowski JQ , Lee VM-Y ((2012) ) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338: , 949–953. |
[31] | Volpicelli-Daley LA , Luk KC , Lee VM-Y ((2014) ) Addition of exogenous alpha-synuclein pre-formed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc 9: , 2135–2146. |
[32] | Luk KC1 , Song C , O’Brien P , Stieber A , Branch JR , Brunden KR , Trojanowski JQ , Lee VM-Y ((2009) ) Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc Natl Acad Sci U S A 106: , 20051–20056. |
[33] | Volpicelli-Daley LA1 , Luk KC , Patel TP , Tanik SA , Riddle DM , Stieber A , Meaney DF , Trojanowski JQ , Lee VM ((2011) ) Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72: , 57–71. |
[34] | Paumier KL , Luk KC , Manfredsson FP , Kanaan NM , Lipton JW , Collier TJ , Steece-Collier K , Kemp CJ , Celano S , Schulz E , Sandoval IM , Fleming S , Dirr E , Polinski NK , Trojanowski JQ , Lee VM , Sortwell CE ((2015) ) Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis 82: , 185–199. |
[35] | Abdelmotilib H , Maltbie T , Delic V , Liu Z , Hu X , Fraser KB , Moehle MS , Stoyka L , Anabtawi N , Krendelchtchikova V , Volpicelli-Daley LA , West A ((2017) ) Alpha-synuclein fibril-induced inclusion spread in rats and mice correlates with dopaminergic neurodegeneration. Neurobiol Dis 105: , 84–98. |
[36] | Fearnley JM , Lees AJ ((1991) ) Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain 114: (Pt 5), 2283–2301. |
[37] | Ma SY , Roytta M , Rinne JO , Collan Y , Rinne UK ((1997) ) Correlation between neuromorphometry in the substantia nigra and clinical features in Parkinson’s disease using disector counts. J Neurol Sci 151: , 83–87. |
[38] | Bezard E1 , Dovero S , Prunier C , Ravenscroft P , Chalon S , Guilloteau D , Crossman AR , Bioulac B , Brotchie JM , Gross CE ((2001) ) Relationship between the appearance of symptoms and the level of nigrostriatal degeneration in a progressive 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of Parkinson’s disease. J Neurosci 21: , 6853–6861. |
[39] | Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease ((2013) ) . Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136: , 2419–2431. |
[40] | Tagliaferro P , Burke RE ((2016) ) Retrograde axonal degeneration in Parkinson’s disease. J Parkinsons Dis 6: , 1–15. |
[41] | Osterberg VR , Spinelli KJ , Weston LJ , Luk KC , Woltjer RL , Unni VK ((2015) ) Progressive aggregation of alpha-synuclein and selective degeneration of Lewy inclusion-bearing neurons in a mouse model of Parkinsonism. Cell Reports 10: , 1252–1260. |
[42] | Masuda-Suzukake M , Nonaka T , Hosokawa M , Oikawa T , Arai T , Akiyama H , Mann DMA , Hasegawa M ((2013) ) Prion-like spreading of pathological alpha-synuclein in brain. Brain 136: , 1128–1138. |
[43] | Karpowicz RJ , Haney CM , Mihaila TS , Sandler RM , Petersson EJ , Lee VM-Y ((2017) ) Selective imaging of internalized proteopathic alpha-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J Biol Chem 292: , 13482–13497. |
[44] | Fares MB , Maco B , Oueslati A , Rockenstein E , Ninkina N , Buchman VL , Masliah E , Lashuel HA ((2016) ) Induction of de novo alpha-synuclein fibrillization in a neuronal model for Parkinson’s disease. Proc Natl Acad Sci U S A 113: , E912–921. |
[45] | Guo JL , Covell DJ , Daniels JP , Iba M , Stieber A , Zhang B , Riddle DM , Kwong LK , Xu Y , Trojanowski JQ , Lee VM ((2013) ) Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 154: , 103–117. |
[46] | Bousset L , Pieri L , Ruiz-Arlandis G , Gath J , Jensen PH , Habenstein B , Madiona K , Olieric V , Böckmann A , Meier BH , Melki R ((2013) ) Structural and functional characterization of two alpha-synuclein strains. Nat Commu 4: , 2575. |
[47] | Tarutani A , Suzuki G , Shimozawa A , Nonaka T , Akiyama H , Hisanaga S , Hasegawa M ((2016) ) The effect of fragmented pathogenic alpha-synuclein seeds on prion-like propagation. J Biol Chem 291: , 18675–18688. |
[48] | Pieri L , Madiona K , Melki R ((2016) ) Structural and functional properties of prefibrillar alpha-synuclein oligomers. Sci Rep 6: , 24526. |
[49] | Ikenoue T , Lee YH , Kardos J , Saiki M , Yagi H , Kawata Y , Goto Y ((2014) ) Cold denaturation of alpha-synuclein amyloid fibrils. Angew Chem Int Ed Engl 53: , 7799–7804. |
[50] | Bousset L , Brundin P , Bockmann A , Meier B , Melki R ((2016) ) An efficient procedure for removal and inactivation of alpha-synuclein assemblies from laboratory materials. J Parkinsons Dis 6: , 143–151. |
[51] | Luk KC , Covell DJ , Kehm VM , Zhang B , Song IY , Byrne MD , Pitkin RM , Decker SC , Trojanowski JQ , Lee VM ((2016) ) Molecular and biological compatibility with host alpha-synuclein influences fibril pathogenicity. Cell Reports 16: , 3373–3387. |
[52] | Thakur P , Breger LS , Lundblad M , Wan OW , Mattsson B , Luk KC , Lee VMY , Trojanowski JQ , Björklund A ((2017) ) Modeling Parkinson’s disease pathology by combination of fibril seeds and alpha-synuclein overexpression in the rat brain. Proc Natl Acad Sci U S A 114: , E8284–E8293. |
[53] | Chen SW , Cremades N ((2018) ) Preparation of alpha-synuclein amyloid assemblies for toxicity experiments. Amyloid Proteins: Methods and Protocols, Third Edition, in press. |
[54] | Lerner TN , Shilyansky C , Davidson TJ , Evans KE , Beier KT , Zalocusky KA , Crow AK , Malenka RC , Luo L , Tomer R , Deisseroth K ((2015) ) Intact-brain analyses reveal distinct information carried by SNc dopamine subcircuits. Cell 162: , 635–647. |
[55] | Rey NL , Steiner JA , Maroof N , Luk KC , Madaj Z , Trojanowski JQ , Lee VM-Y , Brundin P ((2016) ) Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med 213: , 1759–1778. |
[56] | Karampetsou M , Ardah MT , Semitekolou M , Polissidis A , Samiotaki M , Kalomoiri M , Majbour N , Xanthou G , El-Agnaf OMA , Vekrellis K ((2017) ) Phosphorylated exogenous alpha-synuclein fibrils exacerbate pathology and induce neuronal dysfunction in mice. Sci Rep 7: , 16533. |
[57] | Nouraei N , Mason DM , Miner KM , Carcella MA , Bhatia TN , Dumm BK , Soni D , Johnson DA , Luk KC , Leak RK ((2018) ) Critical appraisal of pathology transmission in the alpha-synuclein fibril model of Lewy body disorders. Exp Neurol 299: (Pt A), 172–196. |
[58] | Paxinos G , Watson C ((2013) ) The rat brain in stereotaxic coordinates: Seventh edition, Academic Press, Sydney. |


