
Is the Enzyme ACMSD a Novel Therapeutic Target in Parkinson’s Disease?
Abstract
Several large genome wide association studies have identified a locus in close proximity to the gene encoding the enzyme aminocarboxymuconate-semialdehyde-decarboxylase (ACMSD) to be associated with the risk for Parkinson’s disease (PD), tentatively suggesting that this enzyme might influence PD pathogenesis. Further support for this comes from the recent identification of a disease-segregating stop codon mutation in ACMSD in a family with Parkinsonism, and a missense mutation in the ACMSD gene predicted to disrupt enzyme function in an individual with typical PD. ACMSD is part of the kynurenine pathway, responsible for the catalytic breakdown of tryptophan into NAD+, generating several neuroactive metabolites in the process. The enzyme is located at a key branch-point of the pathway, limiting the production of the neurotoxin quinolinic acid, which has excitotoxic and inflammatory properties. In this review, we discuss the genetic findings in light of the functions of ACMSD and its potential involvement in PD pathogenesis.
INTRODUCTION
Parkinson’s disease (PD) neuropathology is characterized by loss of dopaminergic neurons in the substantia nigra and the development of intracellular alpha-synuclein aggregates called Lewy bodies and Lewy neurites [1]. It is estimated that as many as 10 million individuals live with PD globally [2]. The classical motor symptoms include resting tremor, bradykinesia and rigidity, but patients frequently also exhibit a range of non-motor disturbances including constipation, hyposmia, depression and cognitive decline. The motor symptoms are believed to be largely related to the loss of nigral dopamine neurons. Although the pathogenic process leading to death of nigral dopamine neurons is not fully elucidated, alpha-synuclein aggregation [3] and neuroinflammation [4] both appear to play key roles, with mitochondrial dysfunction [5], oxidative stress [6] and excitotoxicity [7] also being implicated in the neurodegenerative process. By contrast, the neuroanatomical underpinnings of non-motor symptoms are less well defined but, once again, neuroinflammation and alpha-synuclein aggregates have been suggested to be important contributors [8]. Notably, measures of inflammation are reported to correlate to severity of non-motor symptoms in PD [9].
Over the last decade, several heritable (autosomal and recessive) disease-causing mutations have been identified for PD, providing more insight into the underlying pathogenesis in both familial and sporadic disease. However, these monogenic forms account for only a small fraction of familial PD cases, reflecting the multifactorial etiology of the disease [10]. Several large genome wide association studies (GWAS) have identified at least 41 loci harboring polymorphisms that are associated with altered PD risk. Although some of these loci are proximal to genes which have a clear role in neuronal function and PD (e.g., the genes encoding for alpha-synuclein and LRRK2), for the majority it remains unclear precisely which gene, or genes, are influenced by the polymorphisms. To add to the challenge, the normal functions of the proteins encoded by several of the genes in the vicinity of the PD associated polymorphisms are not known. Furthermore, because several of the polymorphisms appear to be in enhancer regions, they can influence multiple downstream genes that are either in cis (within 10 mega base-pairs of the polymorphism) or trans (more than one mega base-pair away, and potentially even on a different chromosome). Notwithstanding this complexity, in this context it is worth pointing out that in depth analyses have suggested that several of the polymorphisms that alter PD risk affect genes that are active in immune cells [11, 12]. Supporting this, a recent study showed a significant genetic overlap between PD and autoimmune disorders, suggesting shared mechanistic pathways in these disorders [13]. These recent observations have further increased the interest in research into protein aggregation and neuroinflammation.
Among the genetic loci that were confirmed in two large meta-analyses [14, 15] to significantly influence PD risk, one is located near the gene encoding the enzyme ACMSD, which is the focus of this review article. This enzyme has a well-described biological function in the kynurenine pathway, where it regulates and limits the formation of quinolinic acid. What makes the potential role of ACMSD in PD risk particularly intriguing is that quinolinic acid is an NMDA receptor agonist with excitotoxic properties that can also modulate inflammatory responses. Therefore, it is possible that ACMSD has an important functional role in PD pathogenesis, and that targeting the enzyme might provide a novel therapeutic strategy. The purpose of this review is threefold. First, we describe evidence from genetics (both from GWAS and mutations in the coding gene) and epigenetics (altered enhancer methylation in PD brain) which implicates ACMSD as a regulator of PD risk. Second, we review how ACMSD and quinolinic acid could influence PD pathogenesis by regulating excitotoxicity. Third, we discuss the potential interplay between a dysregulated kynurenine pathway and other pathogenic mechanisms that are strongly implicated in PD, such as neuroinflammation and protein aggregation.
THE GENETICS OF PARKINSON’S DISEASE AND ACMSD
In 2011, Singleton and coworkers utilized five GWAS data-sets to identify five novel loci that modify PD risk, at the same time confirming variations in previously reported genes such as SNCA, LRRK2, BST1, HLA-DRB5, GAK and MAPT [16]. One of the novel loci was ACMSD, where an SNP (rs6710823) proximal to the ACMSD gene was found to be significantly associated with PD risk. Several GWAS have been undertaken in different populations since then, in order to identify new, as well as confirm previously reported risk loci. At least four different SNPs in the same region in close proximity to ACMSD have been identified to be associated with PD risk, demonstrating that variation in this region genuinely affect PD risk [17–20]. In 2014, Nalls et al. verified and replicated 28 risk variants for PD across 24 loci, with an SNP (rs6430538) close to ACMSD being one among them (odds ratio (OR): 0.875), in a combined patient cohort size of 13,708 PD patients and 95,282 controls [15]. The most recent, very large study [14] compared 26,035 PD cases with 403,190 controls (including both novel cohorts and meta-analysis) and verified the same SNP, rs6430538, in proximity of ACMSD, to be significantly linked to PD risk (OR:0.88). The study refers to the locus as the TMEM163 locus because genetic variation at this site was associated with variation in TMEM163 gene expression in normal brain tissue (in the GTex eQTL database) [14]. Typically, promoters regulate the expression of multiple genes, both in cis and trans location, and therefore the same locus still might regulate ACMSD expression, as well as additional genes. Notably, as noted by the authors of this study [14], currently available eQTL data is insufficient to determine expression during diseases triggers and stages, as well as in multiple cell types of importance. In short, more in-depth transcriptomic and eQTL investigations in specific cell populations, under normal and disease states, are required to determine whether the implicated SNPs in this region increases PD risk via differential ACMSD expression.
Notably, the SNP identified in the two largest GWAS studies (rs6430538) [14, 15] is in a linkage disequilibrium (LD) block that is both proximal and upstream to the ACMSD gene (Fig. 1). In the adult substantia nigra, the LD block overlaps chromatin marks characteristic of active enhancers as well as DNAse hypersensitivity sites. This indicates that the chromatin is open, allowing active gene expression at this locus, in the adult nigra. Furthermore, chromatin conformation analysis by Hi-C (an unbiased, high throughput method to detect chromatin-looping interactions between all loci in the genome) supports that the enhancer elements at this PD-associated SNP interacts with the promoter area of both ACMSD and the other nearby gene TMEM163. Interestingly, in the substantia nigra there is a poised promoter at the ACMSD gene. A poised promoter state allows for rapid gene activation upon appropriate developmental and/or environmental cues. Hence ACMSD expression in the brain is likely to be transient and investigations (i.e., in cell models) examining allele-specific differences in ACMSD expression following PD-relevant triggers will be important for understanding the contributions of genetic variation at ACMSD to PD.
Fig.1
Gene regulatory effects of a PD-associated SNP near the ACMSD gene. The SNP rs6430538, located upstream of the ACMSD gene, was significantly associated with PD in two large GWAS meta-analyses [14, 15]. Eight SNPs (rs10928512, rs1942041, rs6741007, rs6751833, rs6753334, rs4953936, rs6759811, rs6758044) were found to be in linkage-disequilibrium (LD) with rs6430538, as determined by the rAggr program (http://raggr.usc.edu) with an R2≥0.8. The – Log10(P-value) represents the significance of each SNP in the LD block, as reported [15]. The LD block containing the PD-associated rs6430538 SNP exhibits 17 chromatin interactions, as identified by Hi-C analysis reported on the 4DGenome (https://4dgenome.research.chop.edu). Chromatin states in the adult brain substantia nigra (blue) and fetal brain (pink) tissue were determined by the NIH Epigenomics Roadmap.
![Gene regulatory effects of a PD-associated SNP near the ACMSD gene. The SNP rs6430538, located upstream of the ACMSD gene, was significantly associated with PD in two large GWAS meta-analyses [14, 15]. Eight SNPs (rs10928512, rs1942041, rs6741007, rs6751833, rs6753334, rs4953936, rs6759811, rs6758044) were found to be in linkage-disequilibrium (LD) with rs6430538, as determined by the rAggr program (http://raggr.usc.edu) with an R2≥0.8. The – Log10(P-value) represents the significance of each SNP in the LD block, as reported [15]. The LD block containing the PD-associated rs6430538 SNP exhibits 17 chromatin interactions, as identified by Hi-C analysis reported on the 4DGenome (https://4dgenome.research.chop.edu). Chromatin states in the adult brain substantia nigra (blue) and fetal brain (pink) tissue were determined by the NIH Epigenomics Roadmap.](https://content.iospress.com:443/media/jpd/2017/7-4/jpd-7-4-jpd171240/jpd-7-jpd171240-g001.jpg)
MUTATIONS OF ACMSD AND PARKINSONISM
Two recent studies have indicated that mutations in the ACMSD gene can be associated with neurological disease with features of parkinsonism. First, Marti-Masso et al. used whole exome sequencing to determine genetic variations in a Spanish family with a history of focal myoclonic tremor, parkinsonism and epilepsy. Upon screening individuals from three generations they identified a disease-segregating mutation in the ACMSD gene (p.Trp26Stop codon), which was predicted to be pathogenic using two well validated computational techniques [21]. Although ACMSD is normally expressed in the liver, kidney and brain, the disease primarily manifests as a neurological disorder in this family, suggesting an important role for ACMSD in the brain. Second, a novel p.Glu298Lys mutation in ACMSD was detected in a single individual with late onset (74 year-old) sporadic PD [22]. The individual was negative for other disease-causing mutations and had no first degree family history of PD. Using software that predicts the pathogenicity of amino acid substitutions (MutPred and SIFT, i.e., Sorting Tolerant From Intolerant) algorithm and mutation taster, the group predicted the mutation in the ACMSD gene to be pathogenic as it is located at a site that is expected to disrupt enzyme structure and function. Taken together, the two reports of mutations in the ACMSD gene that are associated with primary PD and PD-like symptoms suggest that reduced ACMSD function might confer PD risk. The fact that one disrupted allele of ACMSD might be sufficient to cause idiopathic PD in a patient is particularly strong evidence, implicating a role for this enzyme in PD pathogenesis [22]. However, there may be additional, unknown risk factors that might have contributed to PD pathology in this patient. Genetic studies need to be followed by functional assays to characterize the in-vivo role of this enzyme in the brain in order to draw conclusions about causative mechanisms.
FUNCTION OF ACMSD
ACMSD is part of the kynurenine pathway (Fig. 2), which is comprised of a series of enzymes responsible for the metabolism of tryptophan. These enzymes are expressed in immune cells and different tissues; including the liver, kidney, placenta and brain [23–25]. The first step of the kynurenine pathway results in the conversion of tryptophan into kynurenine via N-formyl kynurenine, catalyzed by the enzymes indoleamine 2,3-dioxygenase (IDO) 1 or 2, or tryptophan 2,3-dioxygenase (TDO). Kynurenine is next metabolized into 3–hydroxykynurenine by kynurenine monoxygenase (KMO), to anthranilic acid by kynureninase, or to kynurenic acid by kynurenine amino transferase (KAT) (Fig. 2). Interestingly, the enzymes of the kynurenine pathway are expressed in different cell types in the CNS. For example, microglia cells are known to produce quinolinic acid [26], whereas astrocytes contain the enzymes KAT, making them capable of diverting the metabolite production from the generation of quinolinic acid to kynurenic acid [27], via an early branch in the pathway (Fig. 2). However, the knowledge about the tissue- and disease specific expression and activity of the kynurenine pathway enzymes is still in its infancy.
Fig.2
Simplified diagram of the enzymes and metabolites of the kynurenine pathway. IDO, indoleamine-2,3-dioxygenase; TDO, tryptophan-2,3-dioxygenase; KATs, kynurenine aminotransferases; KMO, kynurenine-3-monooxygenase; 3-HAO, 3-hydro-xyanthranilate-3,4-dioxygenase; ACMSD, aminocarboxymuconate-semialdehyde decarboxylase; QPRT, quinolinate phosphoribosyltransferase; NAD, nicotinamide adenine dinucleotide.
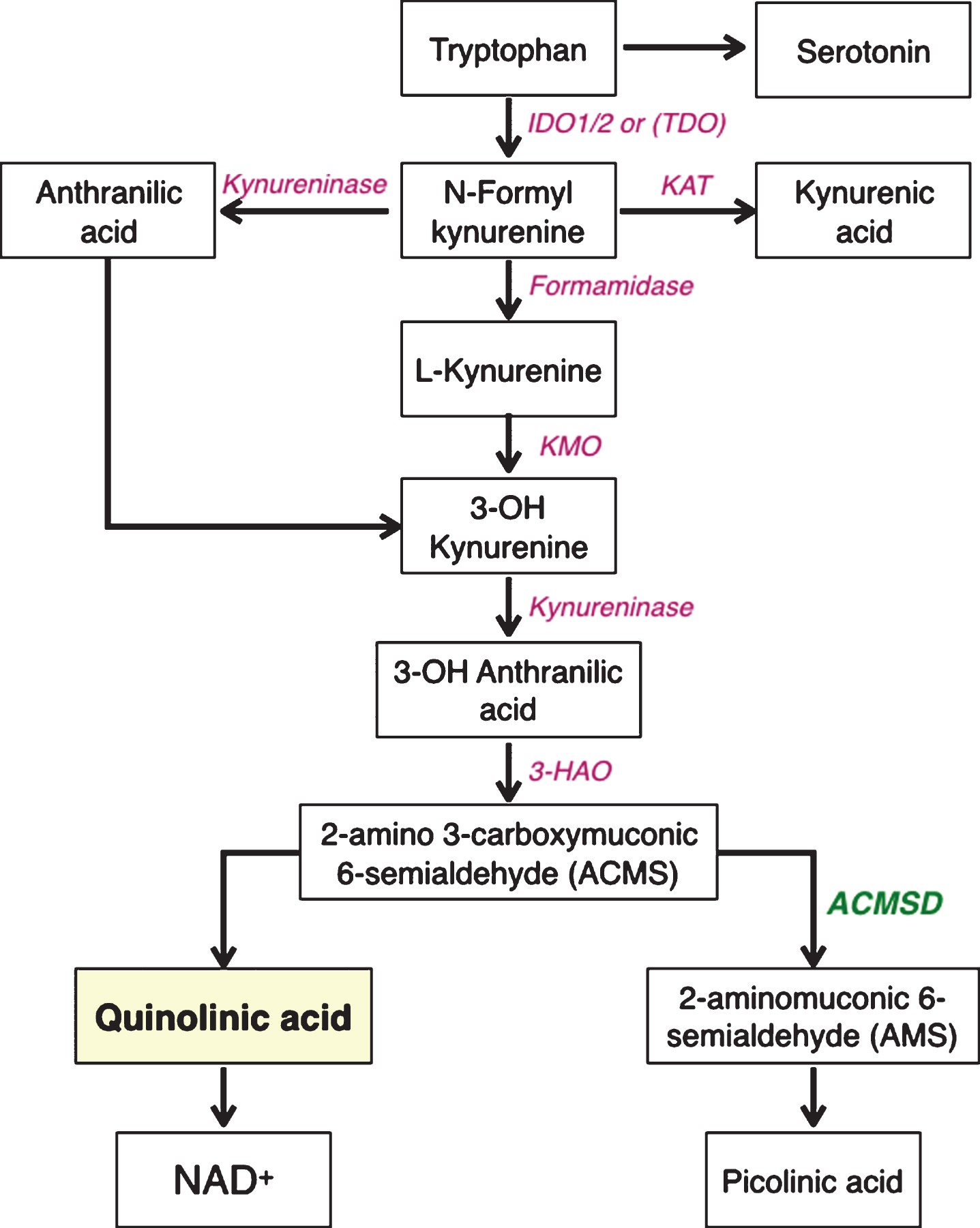
ACMSD is a decarboxylase that was first cloned in 2002 [28], and later confirmed to contain a zinc metal-dependent hydrolase and amidohydrolase domain [29]. Its expression in different tissue types and disease conditions has still not been fully characterized, although it is highly expressed in the liver, kidney and to a lower degree in brain under physiological conditions [30]. The enzyme is primarily cytosolic and is positioned at a critical branching point in the kynurenine pathway, wherein it influences the fate of the precursor 2-amino-3-carboxymuconic-6-semialdehyde (ACMS). Quinolinic acid results from the spontaneous conversion of ACMS. However, when the enzyme ACMSD is present, it instead catalyzes the conversion of ACMS to aminomuconic semialdehyde (AMS) [23]. Thus, ACMSD limits and regulates the formation of quinolinic acid (Fig. 2). The product of ACMSD, AMS, can enter another metabolic pathway, acting as a substrate for the formation of glutaryl-CoA and subsequently acetyl-CoA, which then enters the citric acid cycle [31]. Concordant with this, the expression of the enzyme ACMSD is known to be upregulated in response to metabolic conditions such as animal models of diabetes [32]. Picolinic acid is a stable end-product formed from AMS that is not further metabolized. This metabolite is a metal chelator that has been suggested to protect neurons against quinolinic acid-induced toxicity [33–35], but the underlying mechanism has not been determined. Thus, ACMSD is located at a key branching site of the kynurenine pathway, where it limits the metabolism of tryptophan into NAD+ by preventing the formation of quinolinic acid. Instead, ACMSD catalyzes the production of metabolites that subsequently enter the citric acid cycle, or can form picolinic acid.
Quinolinic acid is an NMDA receptor agonist with selectivity for receptors containing NR1, NR2A and NR1, NR2B subunits, with an affinity comparable to that of glutamate [36]. The distribution of these receptor subtypes is brain region specific [37, 38]. In the substantia nigra, NMDA receptors of the NR2B subunit type are present on dopaminergic neurons [39, 40]. Like NMDA, two molecules of quinolinic acid are required for receptor activation, and quinolinic acid binding to NMDA receptor evokes a calcium-dependent depolarizing current [41, 42]. Hyperactivation of NMDA receptors is an important mechanism for quinolinic acid-induced neurotoxicity. Sustained hyperactivation of NMDA receptors by quinolinic acid leads to increased calcium influx, with altered calcium flux within mitochondria and endoplasmic reticulum as immediate consequences, and elevated oxidative stress, apoptosis and neuronal death as downstream outcomes if the toxicity is sustained [43]. At physiologically high concentrations (0.3–10 mM) quinolinic acid also potentiates glutamate release from synapses, as well as inhibits glutamate uptake from astrocytes, events that could collectively contribute to overstimulation of NMDA receptors and excitotoxicity [44].
A POTENTIAL ROLE FOR ACMSD IN PD PATHOGENESIS
Genetic and environmental risk factors, as well as normal ageing, influence PD pathogenesis. All of these elements are believed to affect interdependent molecular mechanisms that cluster into four domains: protein aggregation, energy failure, oxidative stress and neuroinflammation. We propose that ACMSD may mitigate the PD pathogenesis by limiting the formation of quinolinic acid. The notion that the kynurenine pathway and quinolinic acid could be involved in PD is well established and the evidence for this has already been comprehensively reviewed in excellent articles [45, 46]. The novel feature that we are proposing is that genetic variations in ACMSD might play a pivotal role in PD pathology. In our conceptual model, ACMSD deficiency resulting from mutations in the gene or polymorphisms in putative regulatory elements will promote inflammation and excitotoxicity which in turn can drive mitochondrial failure and protein aggregation, ultimately leading to neuronal death (Fig. 3). These events can be initiated by exposure to environmental toxins, infections or a dysfunctional gut microbiome, and are exacerbated by age. Enhancing ACMSD function could serve to inhibit or delay these processes and as a result, reduce the vulnerability of nigral neurons (Fig. 4).
Fig.3
A. Schematic diagram depicting the interrelationships between putative triggers of Parkinson’s disease (PD) and three pathogenetic mechanism that are believed to contribute to the development of neuropathology. We suggest that these events are enhanced by aging. In this model, we propose that ACMSD activity can reduce the likelihood that potential disease triggers actually lead to PD. B. Physiological state.
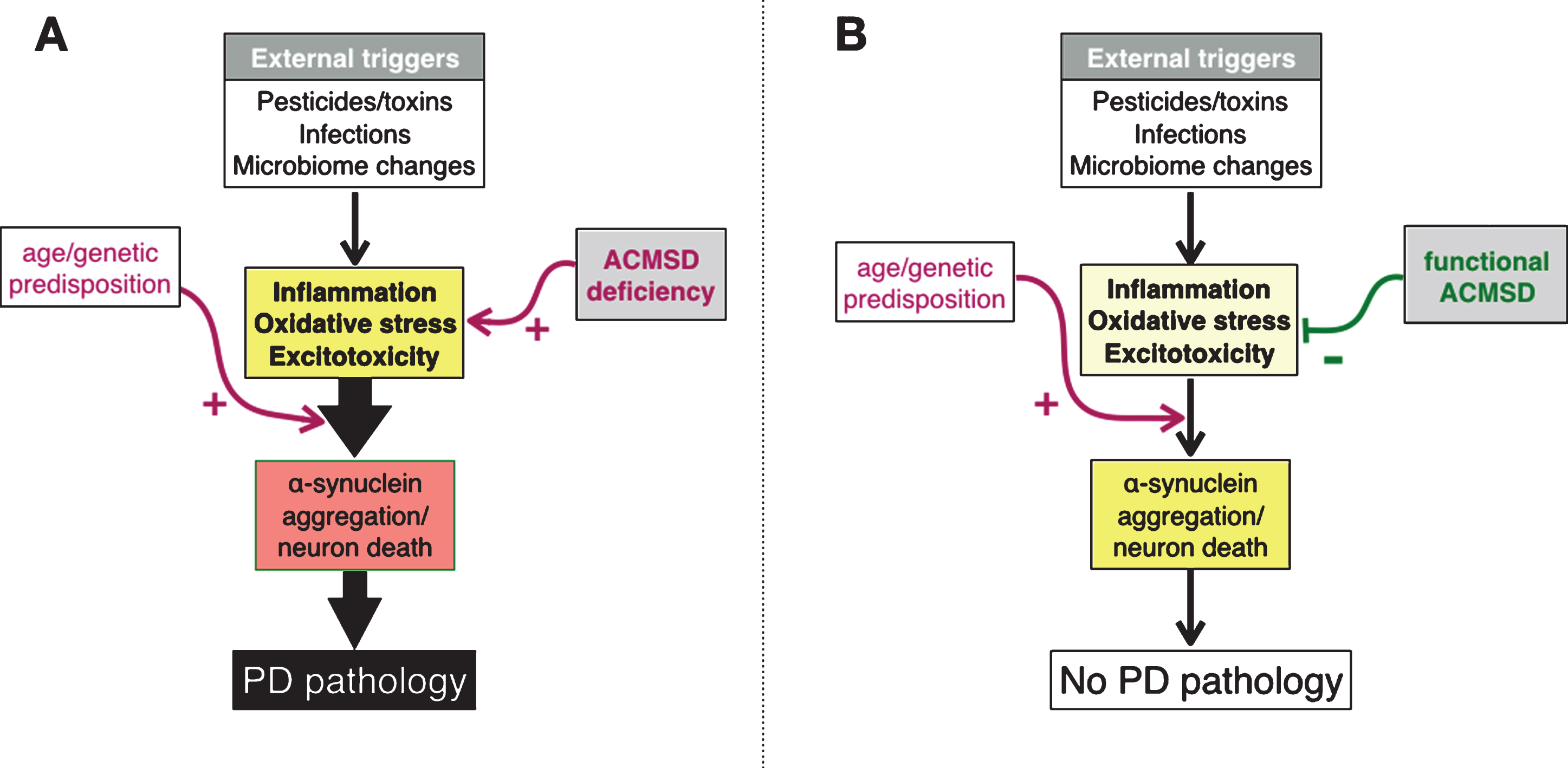
Fig.4
Schematic diagram describing a model for possible interactions between quinolinic acid and different cell types in the brain, when it comes to pathogenetic mechanisms that are highly relevant to Parkinson’s disease. In this model, we propose that a relative lack of ACMSD increases the levels of quinolinic acid and consequently elevates the risk for excitotoxicity and neuroinflammation. Maroon arrows depict an increase activity of a molecular pathway, and the green T-bar illustrates inhibition.
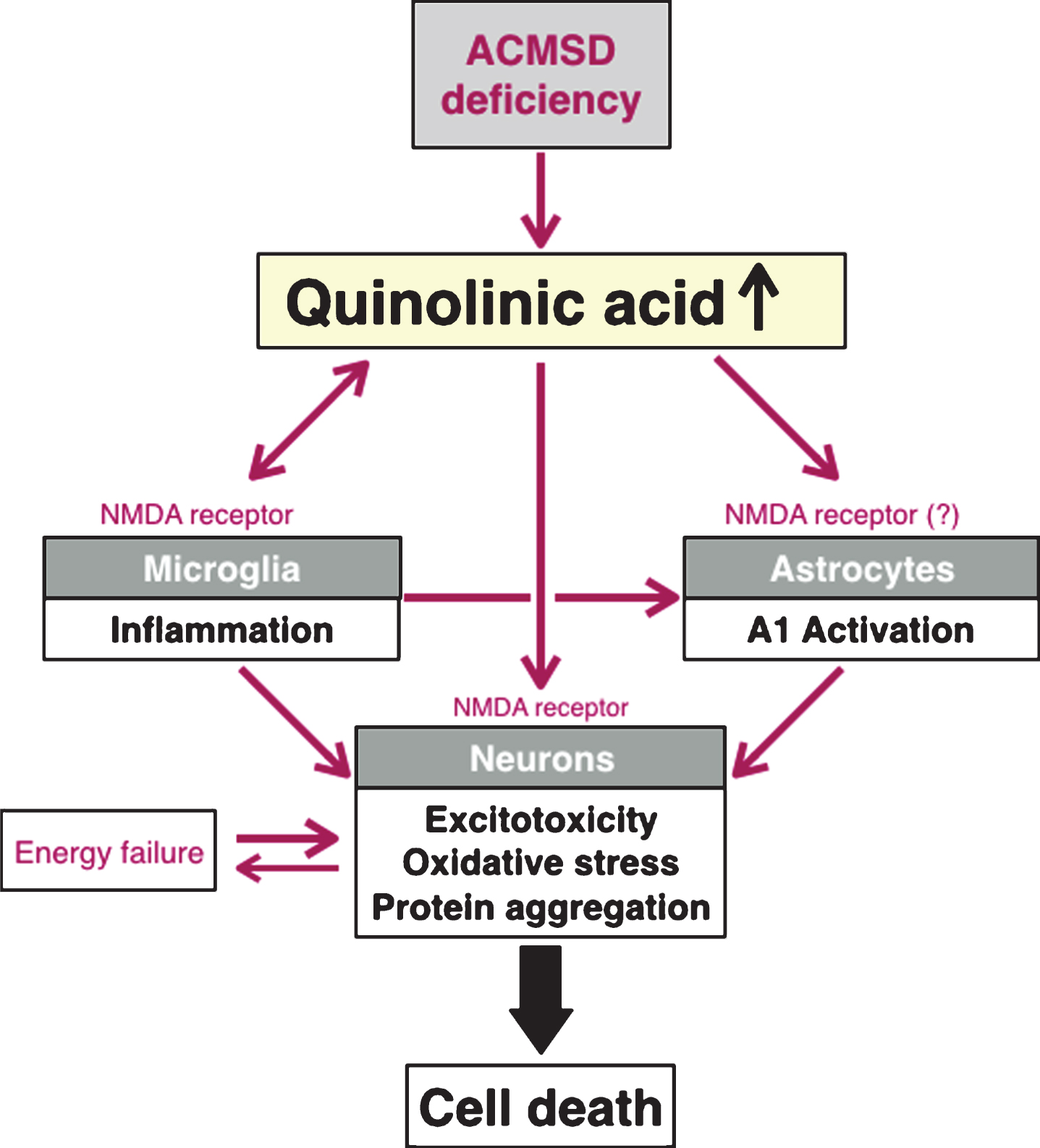
We propose that there are at least three distinct avenues through which altered ACMSD levels, and concomitant changes in the formation of quinolinic acid, can influence the development and progression of PD (Fig. 3). First, increased levels of quinolinic acid, due to reduced ACMSD activity, could potentially act as a primary trigger of excitotoxic neuronal death in PD [7, 47, 48], or augment an ongoing cell death process that is the consequence of other triggers (described in more detail below). Second, quinolinic acid can directly trigger neuroinflammation by activating microglial cells [49]. Neuroinflammation can also arise as a consequence of initial cell damage due to excitotoxicity, and contribute to establish a vicious cycle with further increased production of quinolinic acid, exacerbated by ACMSD deficiency. Third, recently the activation of microglia was shown to trigger (through the release of Il-1α, TNF-α and C1q) the conversion of quiescent astrocytes into an activated form (recently named A1 astrocytes) that is neurotoxic to neighboring neurons [50, 51]. Through these three pathways, it is possible that reduced ACMSD activity either initiates a primary neuroinflammatory response or exacerbates an ongoing detrimental inflammation, secondary to e.g., neuronal death, in the brain. In the following sections, we highlight prior work suggesting that excitotoxicity is important in PD as well as studies showing that neuroinflammation is a salient feature of the neurodegenerative process in PD, and we highlight the role of ACMSD and quinolinic acid in these processes.
A POTENTIAL ROLE FOR QUINOLINIC ACID AND NEUROTOXICITY IN PD
For decades, excitotoxicity has been discussed as a possible contributing factor to the pathogenesis of idiopathic PD. The evidence for this has been reviewed extensively elsewhere [7, 47, 48, 52]. Briefly, excitotoxicity involves impaired calcium homeostasis. Elevated intracellular calcium levels activates nitric oxide synthase and reactive oxygen species which impacts ATP synthesis via impairment of the electron transport chain, resulting in mitochondrial damage and cell death. Quinolinic acid is a direct agonist of the NMDA receptor, binding to the glutamate site. The NMDA receptors are ionotropic, ligand-dependent and responsible for fast excitatory neurotransmission, and NMDA receptors containing the NR2B subunit, which binds quinolinic acid and is implicated in excitotoxicity, are present on dopaminergic neurons in the substantia nigra [53]. Excess activation of the NMDA receptor leads to surplus calcium entry into neurons, triggering production of ROS, which in turn inactivates mitochondrial complex I activity. These events collectively impair mitochondrial function and trigger oxidative stress, which has been suggested to cause dopaminergic neuron death in PD [54]. Several lines of evidence from clinical neuropathology of PD [55–57], as well as toxin [58–62] and genetic [53] models of PD implicate excitotoxicity in the death of dopamine neurons. It is beyond the scope of this review to describe each of these findings in detail, so we refer the reader to the original articles cited above, as well as to other recent reviews that describe this topic in detail [7, 47, 52].
ACMSD AND NEUROINFLAMMATION IN PD
Inflammation is another prominent biological feature of PD with activated microglia present in several brain areas, e.g., such as the olfactory bulb, basal ganglia and cortex [63–67]. Furthermore, interleukin-2 (IL-2) and infiltrating immune cells are increased in PD brain [67] and IL-6 is elevated in plasma [68]. High levels of pro-inflammatory cytokines in CSF are associated with more severe fatigue, cognitive difficulties and depression in PD [9]. Thus, ample evidence links intracerebral and systemic inflammation to PD. In addition, numerous animal models of PD also exhibit inflammation [69]. Importantly, inflammation and the kynurenine pathway are directly linked. For example, interferon-γ (IFN-γ) and IL-6 increase the activity of IDO (indoleamine-2, 3 dioxygenase), the rate-limiting enzyme of the pathway. Elevated IDO activity promotes conversion of tryptophan to kynurenine, which is further metabolized to quinolinic acid (see Fig. 1). Because ACMSD limits the formation of quinolinic acid by catalyzing the production of another metabolite, ACMSD can potentially reduce neurodegeneration that occurs during neuroinflammation. High levels of quinolinic acid have also been observed during neurotropic infections [70–73]. Interestingly, epidemiological studies have linked neurotrophic infections and PD [74, 75]. Notably, alpha-synuclein is suggested to serve to protect neurons against pathogen infections [76, 77]. Viral infection of neurons can result in increased alpha-synuclein expression and genetic deletion of alpha-synuclein can lead to increased viral titer in the brain and exacerbated neuronal injury in experimental models of viral encephalitis [78]. Induction of alpha-synuclein by viral pathogens is not just restricted to the central nervous system. Stolzenberg et al. demonstrated that alpha-synuclein expression within the enteric nervous system was induced by norovirus and associated with mucosal inflammation in the small intestine [79]. Additionally, alpha-synuclein exhibits both antibacterial and anti-fungal activity, and it can protect neurons against Escherichia coli, Staphylococcus aureus and several strains of fungi [80]. Of direct relevance to these findings, the gut microbiome is increasingly implicated in PD pathogenesis [81]. Alterations in the gut microbiota has been suggested to initiate a chronic immune response and increase trafficking of alpha-synuclein aggregates via the vagus nerve to the brain stem, thereby triggering PD pathology in the brain via a prion-like mechanism [82, 83]. Recently, transfer of gut microbiome from PD patients was found to accelerate the development of microglial activation, alpha-synuclein pathology and motor deficits in mice overexpressing alpha-synuclein [84]. Altogether, these studies connect inflammation and alpha-synuclein aggregation pathology, and suggest that microbial pathogens might play a role as triggers of PD.
SUMMARY
This review highlights novel genetic findings linking ACMSD to the risk for PD, and describes how it may exert a critical regulatory effect on excitotoxic and inflammatory pathways in the CNS. Furthermore, neuroinflammation and excitotoxicity are involved in other neurological and neuropsychiatric conditions such as Huntington’s disease, multiple sclerosis, depression and suicide, and as such specific roles for the kynurenine pathway enzymes have been proposed in the pathogenesis of these conditions [85–87]. Therefore, future studies should evaluate the potential role of ACMSD in these disorders.
We here propose that by limiting the formation of quinolinic acid, a well-known neurotoxin, ACMSD plays a key role specifically under circumstances of challenge, such as inflammation, infection, or exposure to environmental toxins. Under such conditions, ACMSD may serve as a “brake”, limiting the damaging effects of inflammation, excitotoxicity and ROS formation. Importantly, the role of quinolinic acid and ACMSD in PD is yet to be fully characterized. Studies are warranted in order to determine quinolinic acid levels in the plasma, CSF and brain from PD patients as well as to characterize the ACMSD expression in PD brains. Furthermore, disease relevant animal models are required to help understand the molecular mechanisms associated with ACMSD gene regulation in neurodegeneration, and will help validate the role of ACMSD as a relevant therapeutic target in PD.
CONFLICT OF INTEREST
PB has received commercial support as a consultant from Renovo Neural, Inc., Roche, Teva, Lundbeck, AbbVie, NeuroDerm, Cellular Dynamics International Inc and Axial Biotherapeutics. Additionally, he has received commercial support for grants/research from Renovo, Teva and Lundbeck. PB has ownership interests in Acousort AB and Parkcell AB.
ACKNOWLEDGMENTS
Lena Brundin has relevant grant support from the National Institutes of Health (R01MH104622) and The Michael J. Fox Foundation for Parkinson’s Research. Viviane Labrie is supported by grants from the Alzheimer’s Society of Canada (16 15), the Scottish Rite Charitable Foundation of Canada (15110) and the Brain and Behavior Research Foundation (23482). Patrik Brundin reports relevant grants from National Institutes of Health (R01DC016519-01 and R21NS093993-02), Department of Defense (W81XWH-17-1-0534), The Michael J. Fox Foundation for Parkinson’s Research and Cure Parkinson’s Trust.
REFERENCES
[1] | Dickson DW ((2017) ) Neuropathology of Parkinson disease. Parkinsonism Relat Disord, 17: , 30280–30288. |
[2] | Parkinson Disease Foundation webpage, Statistics on Parkinson’s Disease, Accessed 20 September, 2017. |
[3] | Goedert M , Jakes R , & Spillantini MG ((2017) ) The synucleinopathies: Twenty years on. J Parkinsons Dis, 7: , S53–S71. |
[4] | Tansey MG , & Goldberg MS ((2010) ) Neuroinflammation in Parkinson’s disease: Its role in neuronal death and implications for therapeutic intervention. Neurobiol Dis, 37: , 510–518. |
[5] | Bose A , & Beal MF ((2016) ) Mitochondrial dysfunction in Parkinson’s disease. J Neurochem, 139: , 216–231. |
[6] | Dias V , Junn E , & Mouradian MM ((2013) ) The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis, 3: , 461–491. |
[7] | Ambrosi G , Cerri S , & Blandini F ((2014) ) A further update on the role of excitotoxicity in the pathogenesis of Parkinson’s disease. J Neural Transm (Vienna), 121: , 849–859. |
[8] | Jellinger KA ((2015) ) Neuropathobiology of non-motor symptoms in Parkinson disease. J Neural Transm (Vienna), 122: , 1429–1440. |
[9] | Lindqvist D , Hall S , Surova Y , Nielsen HM , Janelidze S , Brundin L , & Hansson O ((2013) ) Cerebrospinal fluid inflammatory markers in Parkinson’s disease – Associations with depression, fatigue, and cognitive impairment. Brain Behav Immun, 33: , 183–189. |
[10] | Klein C , & Westenberger A ((2012) ) Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med, 2: , a008888. |
[11] | Coetzee SG , Pierce S , Brundin P , Brundin L , Hazelett DJ , & Coetzee GA ((2016) ) Enrichment of risk SNPs in regulatory regions implicate diverse tissues in Parkinson’s disease etiology. Sci Rep, 6: , 30509. |
[12] | Gagliano SA , Pouget JG , Hardy J , Knight J , Barnes MR , Ryten M , & Weale ME ((2016) ) Genomics implicates adaptive and innate immunity in Alzheimer’s and Parkinson’s diseases. Ann Clin Transl Neurol, 3: , 924–933. |
[13] | Witoelar A , Jansen IE , Wang YP , Desikan RS , Gibbs JR , Blauwendraat C , Thompson WK , Hernandez DG , Djurovic S , Schork AJ , Bettella F , Ellinghaus D , Franke A , Lie BA , McEvoy LK , Karlsen T , Lesage S , Morris HR , Brice A , Wood NW , Heutink P , Hardy J , Singleton AB , Dale AM , Gasser T , Andreassen OA , Sharma M , Conso IPsDG, N ABEC, & Co UKBE ((2017) ) Genome-wide pleiotropy between Parkinson disease and autoimmune diseases. JAMA Neurol, 74: , 780–792. |
[14] | Chang D , Nalls MA , Hallgrimsdottir IB , Hunkapiller J , van der Brug M , Cai F , International Parkinson’s Disease Genomics C, and Me Research T, Kerchner GA , Ayalon G , Bingol B , Sheng M , Hinds D , Behrens TW , Singleton AB , Bhangale TR , & Graham RR ((2017) ) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet, 49: , 1511–1516. |
[15] | Nalls MA , Pankratz N , Lill CM , Do CB , Hernandez DG , Saad M , DeStefano AL , Kara E , Bras J , Sharma M , Schulte C , Keller MF , Arepalli S , Letson C , Edsall C , Stefansson H , Liu X , Pliner H , Lee JH , Cheng R , International Parkinson’s Disease Genomics Consortium (IPDGC), Parkinson’s Study Group (PSG) Parkinson’s Research: The Organized GENetics Initiative (PROGENI) 23andMe, GenePD, NeuroGenetics Research Consortium (NGRC), Hussman Institute of Human Genomics (HIHG), Ashkenazi Jewish Dataset Investigator, Cohorts for Health and Aging Research in Genetic Epidemiology (CHARGE), North American Brain Expression Consortium (NABEC), United Kingdom Brain Expression Consortium (UKBEC), Greek Parkinson’s Disease Consortium, Alzheimer Genetic Analysis Group, Ikram MA , Ioannidis JP , Hadjigeorgiou GM , Bis JC , Martinez M , Perlmutter JS , Goate A , Marder K , Fiske B , Sutherland M , Xiromerisiou G , Myers RH , Clark LN , Stefansson K , Hardy JA , Heutink P , Chen H , Wood NW , Houlden H , Payami H , Brice A , Scott WK , Gasser T , Bertram L , Eriksson N , Foroud T , & Singleton AB ((2014) ) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet, 46: , 989–993. |
[16] | International Parkinson Disease Genomics Consortium, Nalls MA , Plagnol V , Hernandez DG , Sharma M , Sheerin UM , Saad M , Simón-Sánchez J , Schulte C , Lesage S , Sveinbjörnsdóttir S , Stefánsson K , Martinez M , Hardy J , Heutink P , Brice A , Gasser T , Singleton AB , & Wood NW ((2011) ) Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet, 377: , 641–649. |
[17] | Lill CM , Roehr JT , McQueen MB , Kavvoura FK , Bagade S , Schjeide BM , Schjeide LM , Meissner E , Zauft U , Allen NC , Liu T , Schilling M , Anderson KJ , Beecham G , Berg D , Biernacka JM , Brice A , DeStefano AL , Do CB , Eriksson N , Factor SA , Farrer MJ , Foroud T , Gasser T , Hamza T , Hardy JA , Heutink P , Hill-Burns EM , Klein C , Latourelle JC , Maraganore DM , Martin ER , Martinez M , Myers RH , Nalls MA , Pankratz N , Payami H , Satake W , Scott WK , Sharma M , Singleton AB , Stefansson K , Toda T , Tung JY , Vance J , Wood NW , Zabetian CP , 23andMe Genetic Epidemiology of Parkinson’s Disease Consortium, International Parkinson’s Disease Genomics Consortium, Parkinson’s Disease GWAS Consortium, Wellcome Trust Case Control Consortium 2), Young P , Tanzi RE , Khoury MJ , Zipp F , Lehrach H , Ioannidis JP , & Bertram L ((2012) ) Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: The PDGene database. PLoS Genet, 8: , e1002548. |
[18] | Sharma M , Ioannidis JP , Aasly JO , Annesi G , Brice A , Van Broeckhoven C , Bertram L , Bozi M , Crosiers D , Clarke C , Facheris M , Farrer M , Garraux G , Gispert S , Auburger G , Vilariño-Güell C , Hadjigeorgiou GM , Hicks AA , Hattori N , Jeon B , Lesage S , Lill CM , Lin JJ , Lynch T , Lichtner P , Lang AE , Mok V , Jasinska-Myga B , Mellick GD , Morrison KE , Opala G , Pramstaller PP , Pichler I , Park SS , Quattrone A , Rogaeva E , Ross OA , Stefanis L , Stockton JD , Satake W , Silburn PA , Theuns J , Tan EK , Toda T , Tomiyama H , Uitti RJ , Wirdefeldt K , Wszolek Z , Xiromerisiou G , Yueh KC , Zhao Y , Gasser T , Maraganore D , Krüger R , & GEO-PD Consortium ((2012) ) Large-scale replication and heterogeneity in Parkinson disease genetic loci. Neurology, 79: , 659–667. |
[19] | Pihlstrom L , Axelsson G , Bjornara KA , Dizdar N , Fardell C , Forsgren L , Holmberg B , Larsen JP , Linder J , Nissbrandt H , Tysnes OB , Ohman E , Dietrichs E , & Toft M ((2013) ) Supportive evidence for 11 loci from genome-wide association studies in Parkinson’s disease. Neurobiol Aging, 34: , 1708 e1707–1713. |
[20] | Bandres-Ciga S , Price TR , Barrero FJ , Escamilla-Sevilla F , Pelegrina J , Arepalli S , Hernandez D , Gutierrez B , Cervilla J , Rivera M , Rivera A , Ding JH , Vives F , Nalls M , Singleton A , & Duran R ((2016) ) Genome-wide assessment of Parkinson’s disease in a Southern Spanish population. Neurobiol Aging, 45: , 213 e213–219. |
[21] | Marti-Masso JF , Bergareche A , Makarov V , Ruiz-Martinez J , Gorostidi A , Lopez de Munain A , Poza JJ , Striano P , Buxbaum JD , & Paisan-Ruiz C ((2013) ) The ACMSD gene, involved in tryptophan metabolism, is mutated in a family with cortical myoclonus, epilepsy, and parkinsonism. J Mol Med (Berl), 91: , 1399–1406. |
[22] | Vilas D , Fernandez-Santiago R , Sanchez-Rodriguez E , Azcona LJ , Santos-Montes M , Casquero P , Argandona L , Tolosa E , & Paisan-Ruiz C ((2017) ) A Novel p.Glu298Lys Mutation in the ACMSD Gene in Sporadic Parkinson’s Disease. J Parkinsons Dis, 7: , 459–463. |
[23] | Schwarcz R , Bruno JP , Muchowski PJ , & Wu HQ ((2012) ) Kynurenines in the mammalian brain: Whenhysiology meets pathology. Nat Rev Neurosci, 13: , 465–477. |
[24] | Ligam P , Manuelpillai U , Wallace EM , & Walker D ((2005) ) Localisation of indoleamine 2,3-dioxygenase and kynurenine hydroxylase in the human placenta and decidua: Implications for role of the kynurenine pathway in pregnancy. Placenta, 26: , 498–504. |
[25] | Guillemin GJ , Smith DG , Smythe GA , Armati PJ , & Brew BJ ((2003) ) Expression of the kynurenine pathway enzymes in human microglia and macrophages. Adv Exp Med Biol, 527: , 105–112. |
[26] | Espey MG , Chernyshev ON , Reinhard JF , Namboodiri MAA , & Colton CA ((1997) ) Activated human microglia produce the excitotoxin quinolinic acid. NeuroReport, 8: , 431–434. |
[27] | Guidetti P , Hoffman GE , Melendez-Ferro M , Albuquerque EX , & Schwarcz R ((2007) ) Astrocytic localization of kynurenine aminotransferase II in the rat brain visualized by immunocytochemistry. Glia, 55: , 78–92. |
[28] | Fukuoka S , Ishiguro K , Yanagihara K , Tanabe A , Egashira Y , Sanada H , & Shibata K ((2002) ) Identification and expression of a cDNA encoding human alpha-amino-beta-carboxymuconate-epsilon-semialdehyde decarboxylase (ACMSD). A key enzyme for the tryptophan-niacine pathway and “quinolinate hypothesis”. J Biol Chem, 277: , 35162–35167. |
[29] | Huo L , Liu F , Iwaki H , Li T , Hasegawa Y , & Liu A ((2015) ) Human alpha-amino-beta-carboxymuconate-epsilon-semialdehyde decarboxylase (ACMSD): A structural and mechanistic unveiling. Proteins, 83: , 178–187. |
[30] | Pucci L , Perozzi S , Cimadamore F , Orsomando G , & Raffaelli N ((2007) ) Tissue expression and biochemical characterization of human 2-amino 3-carboxymuconate 6-semialdehyde decarboxylase, a key enzyme in tryptophan catabolism. FEBS J, 274: , 827–840. |
[31] | Houtkooper RH , Canto C , Wanders RJ , & Auwerx J ((2010) ) The secret life of NAD(+): An old metabolite controlling new metabolic signaling pathways. Endocr Rev, 31: , 194–223. |
[32] | Tanabe A , Egashira Y , Fukuoka SI , Shibata K , & Sanada H ((2002) ) Expression of rat hepatic 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase is affected by a high protein diet and by streptozotocin-induced diabetes. J Nutr, 132: , 1153–1159. |
[33] | Beninger RJ , Colton AM , Ingles JL , Jhamandas K , & Boegman RJ ((1994) ) Picolinic acid blocks the neurotoxic but not the neuroexcitant properties of quinolinic acid in the rat brain: Evidence from turning behaviour and tyrosine hydroxylase immunohistochemistry. Neuroscience, 61: , 603–612. |
[34] | Kalisch BE , Jhamandas K , Boegman RJ , & Beninger RJ ((1994) ) Picolinic acid protects against quinolinic acid-induced depletion of NADPH diaphorase containing neurons in the rat striatum. Brain Res, 668: , 1–8. |
[35] | Chen Y , Brew BJ , & Guillemin GJ ((2011) ) Characterization of the kynurenine pathway in NSC-34 cell line: Implications for amyotrophic lateral sclerosis. J Neurochem, 118: , 816–825. |
[36] | DeCarvalho LP , Bochet P , & Rossier J ((1996) ) The endogenous agonist quinolinic acid and the non endogenous homoquinolinic acid discriminate between NMDAR2 receptor subunits. Neurochem Int, 28: , 445–452. |
[37] | Monyer H , Burnashev N , Laurie DJ , Sakmann B , & Seeburg PH ((1994) ) Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron, 12: , 529–540. |
[38] | Wenzel A , Scheurer L , Kunzi R , Fritschy JM , Mohler H , & Benke D ((1995) ) Distribution of NMDA receptor subunit proteins NR2A, 2B, 2C and 2D in rat brain. Neuroreport, 7: , 45–48. |
[39] | Christoffersen CL , & Meltzer LT ((1995) ) Evidence for N-methyl-D-aspartate and AMPA subtypes of the glutamate receptor on substantia nigra dopamine neurons: Possible preferential role for N-methyl-D-aspartate receptors. Neuroscience, 67: , 373–381. |
[40] | Suarez F , Zhao Q , Monaghan DT , Jane DE , Jones S , & Gibb AJ ((2010) ) Functional heterogeneity of NMDA receptors in rat substantia nigra pars compacta and reticulata neurones. Eur J Neurosci, 32: , 359–367. |
[41] | Williams TL , Smith DA , Burton NR , & Stone TW ((1988) ) Amino acid pharmacology in neocortical slices: Evidence for bimolecular actions from an extension of the Hill and Gaddum-Schild equations. Br J Pharmacol, 95: , 805–810. |
[42] | Stone TW , & Burton NR ((1988) ) NMDA receptors and ligands in the vertebrate CNS. Prog Neurobiol, 30: , 333–368. |
[43] | Fan MM , & Raymond LA ((2007) ) N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington’s disease. Prog Neurobiol, 81: , 272–293. |
[44] | Tavares RG , Tasca CI , Santos CES , Alves LB , Porciuncula LO , Emanuelli T , & Souza DO ((2002) ) Quinolinic acid stimulates synaptosomal glutamate release and inhibits glutamate uptake into astrocytes. Neurochem Int, 40: , 621–627. |
[45] | Zinger A , Barcia C , Herrero MT , & Guillemin GJ ((2011) ) The involvement of neuroinflammation and kynurenine pathway in Parkinson’s disease. Parkinsons Dis, 2011: , 716–859. |
[46] | Lim CK , Fernandez-Gomez FJ , Braidy N , Estrada C , Costa C , Costa S , Bessede A , Fernandez-Villalba E , Zinger A , Herrero MT , & Guillemin GJ ((2017) ) Involvement of the kynurenine pathway in the pathogenesis of Parkinson’s disease. Prog Neurobiol, 155: , 76–95. |
[47] | Blandini F ((2010) ) An update on the potential role of excitotoxicity in the pathogenesis of Parkinson’s disease. Func Neurol, 25: , 65–71. |
[48] | Blandini F ((2013) ) Neural and immune mechanisms in the pathogenesis of Parkinson’s disease. J Neuroimm Pharmacol, 8: , 189–201. |
[49] | Feng W , Wang Y , Liu ZQ , Zhang X , Han R , Miao YZ , & Qin ZH ((2017) ) Microglia activation contributes to quinolinic acid-induced neuronal excitotoxicity through TNF-alpha. Apoptosis, 22: , 696–709. |
[50] | Liddelow SA , & Barres BA ((2017) ) Reactive astrocytes: Production, function, and therapeutic potential. Immunity, 46: , 957–967. |
[51] | Liddelow SA , Guttenplan KA , Clarke LE , Bennett FC , Bohlen CJ , Schirmer L , Bennett ML , Munch AE , Chung WS , Peterson TC , Wilton DK , Frouin A , Napier BA , Panicker N , Kumar M , Buckwalter MS , Rowitch DH , Dawson VL , Dawson TM , Stevens B , & Barres BA ((2017) ) Neurotoxic reactive astrocytes are induced by activated microglia. Nature, 541: , 481–487. |
[52] | Caudle WM , & Zhang J ((2009) ) Glutamate, excitotoxicity, and programmed cell death in Parkinson disease. Exp Neurol, 220: , 230–233. |
[53] | Yang J , Hertz E , Zhang X , Leinartaite L , Lundius EG , Li J , & Svenningsson P ((2016) ) Overexpression of alpha-synuclein simultaneously increases glutamate NMDA receptor phosphorylation and reduces glucocerebrosidase activity. Neurosci Lett, 611: , 51–58. |
[54] | Nguyen D , Alavi MV , Kim KY , Kang T , Scott RT , Noh YH , Lindsey JD , Wissinger B , Ellisman MH , Weinreb RN , Perkins GA , & Ju WK ((2011) ) A new vicious cycle involving glutamate excitotoxicity, oxidative stress and mitochondrial dynamics. Cell Death Dis, 2: , e240. |
[55] | Liang CL , Sinton CM , & German DC ((1996) ) Midbrain dopaminergic neurons in the mouse: Co-localization with Calbindin-D28K and calretinin. Neuroscience, 75: , 523–533. |
[56] | Damier P , Hirsch EC , Agid Y , & Graybiel AM ((1999) ) The substantia nigra of the human brain. I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D(28K) immunohistochemistry. Brain, 122: , 1421–1436. |
[57] | Damier P , Hirsch EC , Agid Y , & Graybiel AM ((1999) ) The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain, 122: , 1437–1448. |
[58] | Brouillet E , & Beal MF ((1993) ) NMDA antagonists partially protect against MPTP induced neurotoxicity in mice. Neuroreport, 4: , 387–390. |
[59] | Sonsalla PK , Albers DS , & Zeevalk GD ((1998) ) Role of glutamate in neurodegeneration of dopamine neurons in several animal models of parkinsonism. Amino Acids, 14: , 69–74. |
[60] | Hong J , Sha S , Zhou L , Wang C , Yin J , & Chen L ((2015) ) Sigma-1 receptor deficiency reduces MPTP-induced parkinsonism and death of dopaminergic neurons. Cell Death Dis, 6: , e1832. |
[61] | Massari CM , Castro AA , Dal-Cim T , Lanznaster D , & Tasca CI ((2016) ) In vitro 6-hydroxydopamine-induced toxicity in striatal, cerebrocortical and hippocampal slices is attenuated by atorvastatin and MK-801. Toxicol In Vitro, 37: , 162–168. |
[62] | Silva-Adaya D , Pérez-de la Cruz V , Villeda-Hernández J , Carrillo-Mora P , González-Herrera IG , García E , Colín-Barenque L , Pedraza-Chaverrí J , & Santamaría A ((2011) ) Protective effect of L-kynurenine and probenecid on 6-hydroxydopamine-induced striatal toxicity in rats: Implications of modulating kynurenate as a protective strategy. Neurotoxicol Teratol, 33: , 303–312. |
[63] | Imamura K , Hishikawa N , Sawada M , Nagatsu T , Yoshida M , & Hashizume Y ((2003) ) Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol, 106: , 518–526. |
[64] | Doorn KJ , Goudriaan A , Blits-Huizinga C , Bol JG , Rozemuller AJ , Hoogland PV , Lucassen PJ , Drukarch B , van de Berg WD , & van Dam AM ((2014) ) Increased amoeboid microglial density in the olfactory bulb of Parkinson’s and Alzheimer’s patients. Brain Pathol, 24: , 152–165. |
[65] | Mogi M , Harada M , Riederer P , Narabayashi H , Fujita K , & Nagatsu T ((1994) ) Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett, 165: , 208–210. |
[66] | McGeer PL , Itagaki S , Boyes BE , & McGeer EG ((1988) ) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology, 38: , 1285–1291. |
[67] | Rydbirk R , Elfving B , Andersen MD , Langbol MA , Folke J , Winge K , Pakkenberg B , Brudek T , & Aznar S ((2017) ) Cytokine profiling in the prefrontal cortex of Parkinson’s disease and multiple system atrophy patients. Neurobiol Dis, 106: , 269–278. |
[68] | Lindqvist D , Kaufman E , Brundin L , Hall S , Surova Y , & Hansson O ((2012) ) Non-motor symptoms in patients with Parkinson’s disease – correlations with inflammatory cytokines in serum. PLoS One, 7: , e47387. |
[69] | Joers V , Tansey MG , Mulas G , & Carta AR ((2017) ) Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog Neurobiol, 155: , 57–75. |
[70] | Eastman CL , Urbanska E , Love A , Kristensson K , & Schwarcz R ((1994) ) Increased brain quinolinic acid production in mice infected with a hamster neurotropic measles virus. Exp Neurol, 125: , 119–124. |
[71] | Holtze M , Asp L , Schwieler L , Engberg G , & Karlsson H ((2008) ) Induction of the kynurenine pathway by neurotropic influenza A virus infection. J Neurosci Res, 86: , 3674–3683. |
[72] | Heyes MP , Ellis RJ , Ryan L , Childers ME , Grant I , Wolfson T , Archibald S , Jernigan TL , & Center HGHNR ((2001) ) Elevated cerebrospinal fluid quinolinic acid levels are associated with region-specific cerebral volume loss in HIV infection. Brain, 124: , 1033–1042. |
[73] | Formisano S , Hornig M , Yaddanapudi K , Vasishtha M , Parsons LH , Briese T , Lipkin WI , & Williams BL ((2017) ) Central nervous system infection with borna disease virus causes kynurenine pathway dysregulation and neurotoxic quinolinic acid production. J Virol, 91: , pii: e00673–17. |
[74] | Boyd JT , Wangensteen KJ , Krawitt EL , & Hamill RW ((2016) ) Hepatitis C virus infection as a risk factor for Parkinson disease: A nationwide cohort study. Neurology, 87: , 342–342. |
[75] | Bu XL , Wang X , Xiang Y , Shen LL , Wang QH , Liu YH , Jiao SS , Wang YR , Cao HY , Yi X , Liu CH , Deng B , Yao XQ , Xu ZQ , Zhou HD , & Wang YJ ((2015) ) The association between infectious burden and Parkinson’s disease: A case-control study. Parkinsonism Relat Disord, 21: , 877–881. |
[76] | Massey AR , & Beckham JD ((2016) ) Alpha-synuclein, a novel viral restriction factor hiding in plain sight. DNA Cell Biol, 35: , 643–645. |
[77] | Labrie V , & Brundin P ((2017) ) Alpha-synuclein to the rescue: Immune cell recruitment by alpha-synuclein during gastrointestinal infection. J Innate Immun, 9: , 437–440. |
[78] | Beatman EL , Massey A , Shives KD , Burrack KS , Chamanian M , Morrison TE , & Beckham JD ((2015) ) Alpha-synuclein expression restricts RNA viral infections in the brain. J Virol, 90: , 2767–2782. |
[79] | Stolzenberg E , Berry D , Yang, Lee EY , Kroemer A , Kaufman S , Wong GCL , Oppenheim JJ , Sen S , Fishbein T , Bax A , Harris B , Barbut D , & Zasloff MA ((2017) ) A role for neuronal alpha-synuclein in gastrointestinal immunity. J Innate Immun, 9: , 456–463. |
[80] | Park SC , Moon JC , Shin SY , Son H , Jung YJ , Kim NH , Kim YM , Jang MK , & Lee JR ((2016) ) Functional characterization of alpha-synuclein protein with antimicrobial activity. Biochem Biophys Res Comm, 478: , 924–928. |
[81] | Houser MC , & Tansey MG ((2017) ) The gut-brain axis: Is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? NPJ Parkinsons Dis, 3: , 3. |
[82] | Holmqvist S , Chutna O , Bousset L , Aldrin-Kirk P , Li W , Bjorklund T , Wang ZY , Roybon L , Melki R , & Li JY ((2014) ) Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol, 128: , 805–820. |
[83] | Lema Tome CM , Tyson T , Rey NL , Grathwohl S , Britschgi M , & Brundin P ((2013) ) Inflammation and alpha-synuclein’s prion-like behavior in Parkinson’s disease–is there a link? Mol Neurobiol, 47: , 561–574. |
[84] | Sampson TR , Debelius JW , Thron T , Janssen S , Shastri GG , Ilhan ZE , Challis C , Schretter CE , Rocha S , Gradinaru V , Chesselet MF , Keshavarzian A , Shannon KM , Krajmalnik-Brown R , Wittung-Stafshede P , Knight R , & Mazmanian SK ((2016) ) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell, 167: , 1469–1480. |
[85] | Campesan S , Green EW , Breda C , Sathyasaikumar KV , Muchowski PJ , Schwarcz R , Kyriacou CP , & Giorgini F ((2011) ) The kynurenine pathway modulates neurodegeneration in a Drosophila model of Huntington’s disease. Curr Biol, 21: , 961–966. |
[86] | Lim CK , Bilgin A , Lovejoy DB , Tan V , Bustamante S , Taylor BV , Bessede A , Brew BJ , & Guillemin GJ ((2017) ) Kynurenine pathway metabolomics predicts and provides mechanistic insight into multiple sclerosis progression. Sci Rep, 7: , 41473. |
[87] | Brundin L , Sellgren CM , Lim CK , Grit J , Pålsson E , Landén M , Samuelsson M , Lundgren K , Brundin P , Fuchs D , Postolache TT , Traskman-Bendz L , Guillemin GJ , & Erhardt S ((2016) ) An enzyme in the kynurenine pathway that governs vulnerability to suicidal behavior by regulating excitotoxicity and neuroinflammation. Transl Psych, 6: , e865. |


