
Is Exenatide a Treatment for Parkinson’s Disease?
Abstract
There is growing interest in the use of glucagon-like peptide-1 agonists as treatments for Parkinson’s disease following the recent publication of the results of the Exenatide-PD trial. In this randomized, double-blind, placebo controlled trial, patients with moderate stage Parkinson’s disease treated with once-weekly subcutaneous injections of exenatide 2 mg (Bydureon) for 48 weeks, had a 3.5-point advantage over the placebo group in the Movement Disorders Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) motor subscale (Part 3) in the practically defined OFF medication state, 12 weeks after cessation of the trial drug. In this article, we discuss some of the important issues of relevance to this trial, with regards to trial design, patient selection, choice of outcome measure and also place into context the implications these results have for patients with Parkinson’s disease and the wider research community.
Exenatide is a licensed drug for the treatment of type 2 diabetes mellitus. It is a Glucagon like peptide 1 (GLP-1) receptor agonist, which improves glucose control by stimulating insulin release from the pancreas and inhibiting glucagon release. In laboratory models of several neurological conditions, including some of Parkinson’s disease (PD), it has been shown to protect neurons and induce beneficial neuroplastic changes [1]. In an open label trial that was published in 2013, patients treated with exenatide for 1 year had a clinical advantage, compared to patients not receiving injections, in terms of motor severity and cognitive function that persisted even 1 year after cessation of the drug [2, 3]. It is therefore of major interest as a potential disease modifying agent in PD.
This interest has now extended further following the recent publication of the results of a phase 2 double blind placebo control trial (Exenatide–PD) [4] (In press). In this study 60 participants with “mid-stage” PD who were already on dopaminergic replacement therapy were randomized to self-administer exenatide 2 mg as “Bydureon” (a slow release, injectable formulation of exenatide) or matched placebo on a once weekly basis for a period of 48 weeks. The primary outcome was the severity of PD motor symptoms using the MDS-UPDRS part 3 in the “Practically defined OFF medication state” at the 60-week time-point, i.e., after a 12-week washout period. The primary outcome was met and, in line with the earlier open label trial, patients using exenatide exhibited better motor function compared with those on placebo. The difference adjusted for baseline scores was 4.3 points at the end of 48 weeks of exenatide exposure. Most importantly, this difference persisted (3.5 points) after the 12-week washout period, at which point exenatide was no longer detectable in the serum (Table 1). In addition, the drug was well tolerated in this patient group, who reported common adverse gastrointestinal effects and injection site reactions in similar frequencies to previously reported diabetes trials. Whether exenatide impacts the underlying pathophysiology of PD, influences compensatory responses/behaviours or simply induces long lasting symptomatic effects remains uncertain, however these results represent a major new avenue for investigation in the treatment of PD.
Table 1
Summary of outcomes between Baseline and Week 60 according to randomisation allocation. *Higher scores reflect improved status
| Domain | Baseline | 48 weeks | Change Baseline | Adjusted difference, | 60 weeks | Change Baseline | Adjusted difference, | |
| to 48 weeks | baseline to 48 weeks | to 60 weeks | baseline to 60 weeks | |||||
| Mean (SD) | Mean (SD) | Mean (95% CI) | Mean (95% CI) | Mean (SD) | Mean | Mean (95% CI) | ||
| p value | (95% CI) | p value | ||||||
| Motor | MDS-UPDRS Part 3 (OFF medication) | |||||||
| function | Exenatide | 32.8 (9.7) | 30.2 (11.1) | –2.3 (–4.1, –0.7) | –4.3 (–7.1, –1.6) | 31.9 (12.0) | –1.0 (–2.6, 0.7) | –3.5 (–6.7, –0.3) |
| Placebo | 27.1 (10.3) | 28.8 (10.8) | 1.7 (–0.6, 4.0) | 0.0026 | 29.2 (12.0) | 2.1 (–0.6, 4.8) | 0.0318 | |
| Cognition | Mattis Dementia Rating scale | |||||||
| Exenatide | 138.0 (5.0) | 139.7 (4.1) | 1.7 (0.4, 2.9) | 0.4 (–1.0, 1.9) | 139.9 (3.6) | 1.9 (0.6, 3.1) | 0.8 (–0.9, 2.5) | |
| Placebo | 139.8 (3.7) | 140.2 (3.9) | 0.4 (–0.6, 1.5) | 0.57 | 140.2 (4.6) | 0.4 (–1.1, 1.8) | 0.32 | |
| Mood | MADRS | |||||||
| Exenatide | 4.1 (3.7) | 2.5 (2.7) | –1.6 (–3.4, 0.07) | –1.4 (–3.2, 0.5) | 2.1 (2.6) | –1.6 (–2.7, –0.4) | –0.9 (–2.2, 0.3) | |
| Placebo | 3.7 (3.0) | 3.8 (4.2) | 0.2 (–1.8, 2.2) | 0.15 | 2.8 (2.6) | –0.9 (–2.3, 0.5) | 0.15 | |
| Quality of | PDQ-39 Summary index | |||||||
| life | Exenatide | 19.9 (13.7) | 18.7 (12.7) | –1.2 (–4.7, 2.3) | –1.7 (–5.6, 2.1) | 18.4 (11.1) | –1.5 (–5.4, 2.4) | –3.3 (–8.0, 1.5) |
| Placebo | 21.1 (13.0) | 20.1 (12.8) | –1.1 (–4.2, 2.1) | 0.38 | 22.2 (14.8) | 0.3 (–3.4, 4.0) | 0.17 | |
The purpose of this article is to explain some of the decisions taken during the design of this trial and to provide a critique of the results and to discuss in further detail how they might be interpreted.
WHY WERE THE TRIALS DESIGNED IN MID STAGE PATIENTS?
Most previous trials that have sought to identify whether medications might alter the course of PD have recruited de novo/untreated PD patients and explored effects without contamination of the major clinical effects of pharmacological dopaminergic therapies [5]. Conceptually this is an appealing approach, however, it is limited because of the inevitable need for most patients to start on effective symptomatic medication within a couple of years of diagnosis. This leads to differential dropout (more severe patients dropout earlier and potentially more commonly among the group allocated placebo) and this issue limits the duration of follow up upon which to base conclusions. Alternatively, the difference in duration until previously untreated patients need to start on dopaminergic replacement can be used as an outcome, however this judgment is potentially very arbitrary and can lead to inconsistency between groups, as well as being a poor discriminator between symptomatic verses disease modifying effects of an experimental intervention. The likelihood of misdiagnosis of PD is also higher in early stages, which means that the results are more likely to be confounded by patients that have different underlying disease mechanisms and follow a progression that is atypical of PD. Furthermore, these trials have been very expensive and have (thus far) not identified any agent with a clear signal of effect [6]. As a result, commercial and academic funders of disease-modifying therapies for PD have become more cautious in their funding decisions requiring a high level of certainty of success before committing funds to expensive efficacy trials. Consequently, initially they are increasingly supporting “proof of concept” designs, as opposed to “proof of efficacy” designs.
In view of the relative rarity of incident (untreated) PD patients compared to the much larger pool of prevalent (treated) PD patients, recruitment of an untreated cohort takes longer and requires involvement of many more centres and therefore becomes more expensive to perform. Additionally, patients in the earlier phases of disease have more mild symptoms which (while being intuitively easier to treat) also limit the ability to assess a drug’s tolerability and any potential positive or negative effects on later emerging non-motor symptoms of PD.
For all of these reasons, the Exenatide-PD trial team chose a population of moderate-stage PD patients already on L-dopa treatment. Despite the ease of recruitment and the increased likelihood of detecting measurable changes on validated scales, some may argue these patients may have such advanced neurodegeneration that neuroprotective agents might be ‘too little, too late’ [7]. Nevertheless there will always be a need to identify neuroprotective treatments that are helpful in a population with clinically manifest disease.
To further reduce the noise introduced by assessments being conducted at multiple centres, all patients were recruited through a single large PD centre in London, UK. These decisions enabled rapid trial setup and recruitment and reduced the costs of the study.
WHY WAS THE “PRACTICALLY DEFINED OFF” USED AS THE PRIMARY OUTCOME?
During the first years of PD, most patients experience a honeymoon period during which many of their symptoms can be dramatically improved by the use of dopaminergic drugs. As the disease advances, motor fluctuations can occur, typically necessitating adjustment of the dose and frequency of L-dopa administration. With this approach, patients’ best functional performance can remain stable for several years despite underlying progression of disease. During the later stages of disease, L-dopa refractory symptoms emerge and these can be measurable despite optimal therapy.
The Exenatide-PD trial recruited patients that were on doses of pharmacological therapy that had been kept relatively stable, but who reported that they had experienced periods of suboptimal symptom control signaling the onset of motor fluctuations. This allowed a window to estimate the underlying severity of PD by performing assessments in the widely adopted “Practically defined OFF medication state”, i.e., after an overnight period free of all PD medication. The assessments were performed at the same time in the morning and each patient had stopped his or her medication the night before, or at least 24 hours beforehand if taking a long acting dopamine agonist. This consistent timing of assessment in a drug free period represents an informative way to measure PD severity in a manner that is less affected by changes in dopaminergic drugs.
This measure certainly does not equate to the consequences of exenatide on quality of life, nor on activities of daily living, nor the rate of evolution of L-dopa-refractory signs of PD. As such, it should simply be viewed as proof-of-concept that exenatide influences PD severity. Additionally, because the study duration was only 60 weeks, it also does not provide information on the effects of exenatide in the long term.
WHY WAS THE TRIAL SO SHORT AND WITH SO FEW PARTICIPANTS?
The costs for conducting this trial were generously supported by the Michael J. Fox Foundation, except for the provision of the trial drug and placebo which were provided by first Bristol Myers Squibb and later following an acquisition by Astra Zeneca. Given that the patent protection for exenatide was due to expire at the end of 2016, the commercial appeal of developing exenatide for neurodegenerative diseases was low already when the trial was set up in 2013. Therefore, commercial support was limited to the supply of drug and placebo for 48 weeks only, in a maximum of 60 participants. A formal power calculation was performed which identified that this number of patients had 90% power to detect a difference of 5.8 MDS UPDRS points between the 2 groups (assuming a SD of 13, and a correlation of 0.85 between baseline and end of trial scores and an overall type 1 error rate of 5%).
HOW ROBUST ARE THESE FINDINGS?
Patients treated with exenatide had positive effects on the practically defined OFF-medication motor scores in comparison to the placebo group, that were sustained beyond the period of exposure (a mean difference of 3.5 points) and these results reached statistical significance (the smaller effect size remained statistically significant because of a smaller variance in response than predicted). Furthermore this effect size is of potential major importance to patients with PD. However, it is important not to over-interpret the findings, which should still be regarded as “proof of concept” rather than “proof of efficacy”.
Importantly, there are a number of limitations of this trial, several of which are common to any small phase 2 trial. First, the relatively small number of participants meant that the trial only had the power to detect substantial effect sizes and only if there was little variation in the size of the effect between participants. While the trial indeed achieved its primary outcome, none of the secondary outcome measures reached the threshold for statistical significance (Table 1). Therefore, it is absolutely vital that these findings are reproduced in a much larger sample of patients across multiple sites to allow the scientific and health care communities, as well as regulatory agencies, to assess the consistency and reproducibility of these findings across the broader range of outcome measures, including those most relevant to patient function and quality of life.
A further issue which complicates trials with small numbers of participants is that even with randomization, differences between groups can occur at baseline and persist following statistical adjustments. It can be impossible to know if these baseline differences had an impact on any differences at trial end. This issue was present in Exenatide-PD. Specifically, at the start of the trial patients randomized to self-inject exenatide were on average almost 4 years older, had worse severity PD (5.7 points) and had lower levels of dopaminergic treatment (∼50 mg L-dopa equivalent dose; LED) than patients randomized to inject placebo. The statistical analysis for the end of trial results was pre-planned to adjust for baseline severity of PD, and was further adjusted for differences in baseline LED and/or the change in LED over the course of the trial. Importantly, these adjustments did not change the statistical effect favoring exenatide detected in the primary outcome. This adjustment does mitigate the risk that baseline differences may have contributed to differences detected in the final outcome, but does not absolutely exclude the possibility.
Adjustments to medication were allowed during the trial follow up period. Even though patients were consistently assessed in the OFF medication state, it could be argued that such changes in LED might have “long duration” effects that persisted even in the practically defined OFF state. To explore this further, the mean size of medication adjustment was calculated at each of the follow up visits, and it was explored whether the exact times of these drug adjustments were linked in time to when there were changes in the MDS UPDRS part 3 OFF scores. However, there was no clear relationship between the timing of LED drug changes and measured changes in MDS UPDRS part 3 OFF scores.
A further concern that has to be raised is the possibility that adverse effects of exenatide might have led to inadvertent unblinding of the randomization allocation and thus led to an unwitting placebo effect or observer bias. However, adverse events were recorded by individuals in the trial team separately from the rating of the clinical outcome measures, reducing the risk of observer bias. On the other hand, given some of the well documented gastrointestinal effects of exenatide unblinding of patients might theoretically have occurred in some participants, leading to an enhanced placebo effect. To investigate this further, a chi squared test was performed to see if the frequency of gastrointestinal adverse effects differed between exenatide and placebo groups. This revealed no significant differences between the groups regarding nausea, abdominal pain, loss of appetite and weight loss. Nevertheless the degree of weight loss experienced by the exenatide treated group was numerically greater and one can speculate that, as a result of weight loss, patients felt fitter and were able to move more easily. Therefore, a direct comparison was made between degree of weight loss and change in MDS UPDRS part 3 OFF scores and revealed no correlation between the two parameters. Furthermore when the primary analysis was again repeated adjusting for the degree of weight loss, there was no reduction in the statistical advantage favouring the exenatide treated group. Despite the small sample size, to the best of their ability, the trial authors could not find evidence that the difference in the primary outcome was explicable by unblinding due to adverse events.
Another issue that warrants discussion is that while the primary outcome was met, there were no statistically significant differences in any of the clinical secondary outcome measures, despite the direction of effect favoring exenatide for the majority of these. Perhaps most unexpectedly, the objective timed motor tests, performed at the same time as the MDS UPDRS part 3 (i.e., in the practically defined OFF state), did not show that exenatide treated patients performed better than controls. The timed tests provide an objective measure of bradykinesia [8], but do not capture the wider range of measures included in MDS UPDRS part 3 (tremor, rigidity, bradykinesia and axial features such as gait disturbance). It requires further study to clarify whether MDS UPDRS part 3 or the objective timed motor tests is the more accurate reflection of the overall impact of exenatide.
A further tool used to objectively measure change was DaTscan imaging. Each participant was imaged at baseline and again at the end of the trial after exenatide washout. Using statistical parametric modeling [9], patients in the exenatide group exhibited increased dopamine transporter binding in the right caudate and right and left putamen compared to the placebo group. Given the number of voxels compared between the groups, the statistical results need to be adjusted for multiple comparisons. The three areas (right caudate, and right and left putamen) with the biggest differences between exenatide and placebo treated patients all suggested (at p < 0.003 uncorrected) better preserved uptake in the exenatide treated group. However this did not reach the threshold for statistical significance after correction for multiple comparisons and therefore should not be regarded as definitive evidence for an effect of exenatide. Notably, there were no regions where the placebo treated group displayed better preserved dopamine transporter levels, even at uncorrected statistical thresholds.
IF EXENATIDE HAS A REAL EFFECT, WHAT MIGHT BE THE MECHANISM?
Taking into account all the preceding caveats, it is important to discuss how exenatide might be influencing the MDS UPDRS part 3 OFF scores. The impact of exenatide may be interpreted as a simple symptomatic effect. This does not necessarily mean that exenatide is directly stimulating dopamine receptors, but perhaps it might be impacting on dopaminergic signaling in other ways – one proposal is that it might have a previously unrecognized effect on L-dopa pharmacokinetics.
In support of this type of symptomatic mechanism of action, the exenatide trials have indicated that clinical effects are detectable within the first 12 weeks of treatment. It is hard to imagine that this early acute effect relates to slowing down of a neurodegenerative process when the typical rate of progression in PD is approximately only 3 UPDRS points per year. Furthermore, the magnitude of the difference between exenatide and placebo groups was greater during the period of continued exposure than 12 weeks after exenatide withdrawal. Therefore exenatide seems to have at least some symptomatic effect in PD, although this might require concomitant L-Dopa therapy.
In considering the potential mechanistic options in more detail; if exenatide had an impact on L-dopa pharmacokinetics, e.g., akin to a COMT inhibitor, we might expect that peak doses of L-dopa would rise and dyskinesia scores would worsen. Possible changes in L-dopa pharmacokinetics have, so far, not been studied in people receiving exenatide injections. Against this proposal, however, is that neither dyskinesia rating scale scores nor MDS UPDRS part 3 ON medication scores differed between exenatide and placebo groups in either trial, which casts doubt on changes in L-dopa pharmacokinetics as a proposed mechanism of action explaining the changes seen in the Exenatide-PD trial.
The persisting effect of exenatide even after drug washout cannot exclude the possibility of a disease-modifying effect of the drug. Samples of serum, urine and CSF were taken from trial participants at multiple time-points during trial follow up and these will be used to assess whether differences in insulin signaling and the subsequent levels of intracellular downstream molecules are detectable in a direction consistent with this putative neuroprotective mechanism of action.
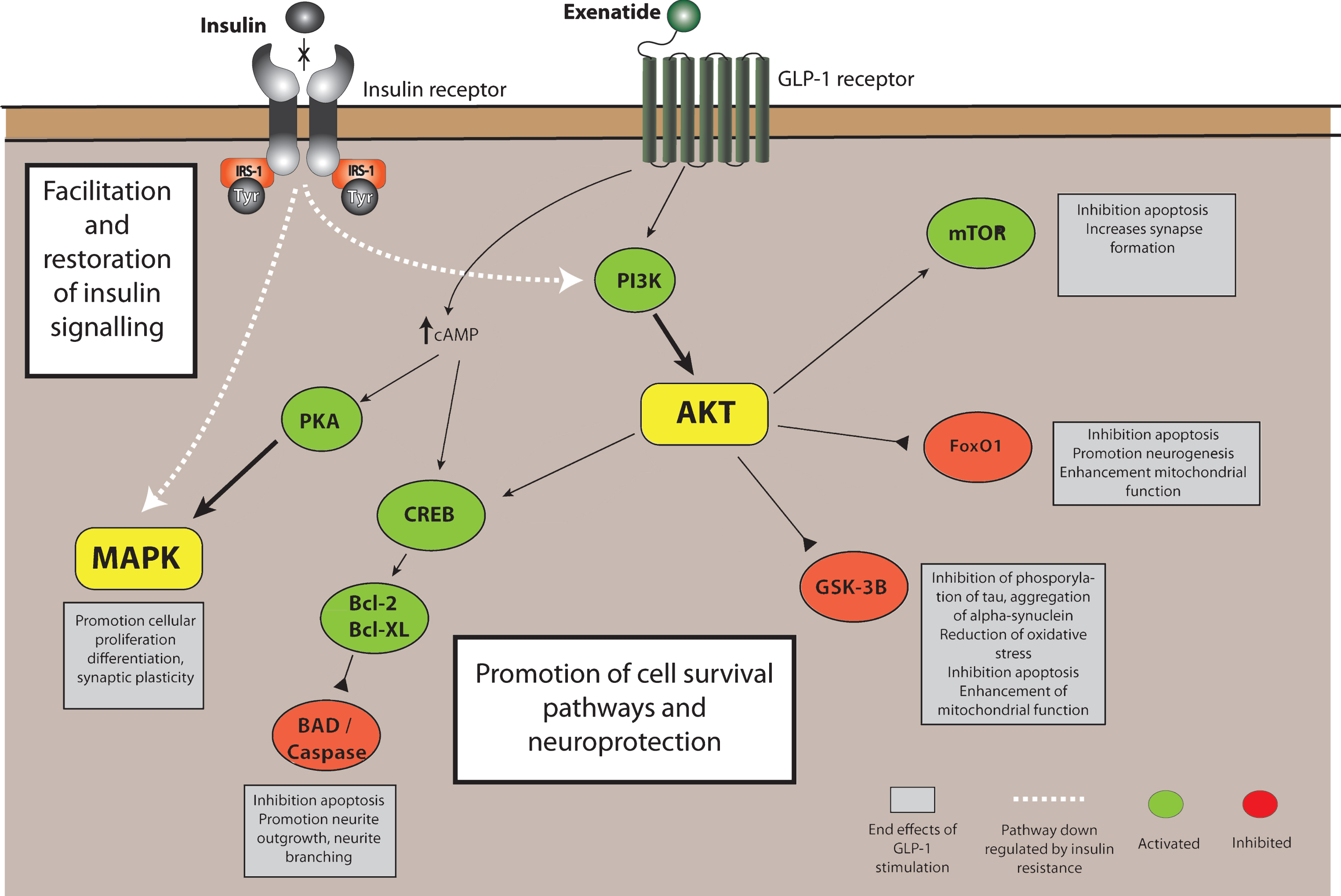
Data from in vitro and in vivo models suggest that exenatide may, through its action on the GLP-1 receptor, have a role on influencing resistance to neuronal insulin signaling and consequences on AKT (protein kinase B) and downstream substrates including mTOR (mechanistic target of rapamycin) and GSK3β (glycogen synthase kinase 3β) which together influence neuronal survival pathways, mitochondrial function and neuroinflammation [10] (Fig. 1). Specifically there are also data to suggest that exenatide has an effect on MAPK (Mitogen associated protein kinase) activity as a downstream consequence of GLP-1 receptor activation [11]. Activation of MAPK has effects on long term potentiation and synaptic plasticity as well as stabilization of dendritic spines [12, 13]. One explanation of the early effects of exenatide on OFF medication scores may therefore be via improved functioning of surviving dopaminergic neurons as a result of stabilization of dendritic spines. The primary outcome was the difference in MDS UPDRS part 3 OFF scores after a 12-week washout period. At this point, exenatide levels were no longer detectable in the serum and yet there were still significant differences observed. This replicates the previous open label data but it is easily argued that the 12-week washout period was insufficient to allow symptomatic effects to wane completely. Changes in dendritic spines, with a protracted beneficial effect on neuronal function, could conceivably explain why the improvement in MDS UPDRS part 3 OFF scores are maintained after a 12-week washout. Previous studies have suggested that exenatide can affect striatal dopamine neurotransmission [14, 15], possibly via promotion of intracellular cyclic AMP [16], and this small but useful effect on nigrostriatal dopamine function, irrespective on any effect on neurodegeneration, is also of potential importance. This said, the focus on exenatide as a treatment for PD is primarily because of the preclinical evidence of its neuro-protective effects in animal models involving acute neurotoxic and pro-inflammatory lesions, and it is therefore worth considering how strong the trial results are in support of such an effect.
Fig.1
GLP-1 receptor activation in neurons. Stimulation of the GLP-1R leads to an increase in intracellular cAMP, activating protein kinase A (PKA), and phosphoinositide 3-kinase (PI3K), which phosphorylate and activates a variety of downstream signalling pathways that can be simplified into 2 branches: the Raf-1/MEK-MAP-K (mitogen associated protein kinase)/ERK (extracellular signal regulated kinase) and (PI3K)/protein kinase B(AKT) pathways which can modulate intracellular events such as activation of calcium channels, enhancing protein synthesis, cellular proliferation and mitochondrial biogenesis while inducing inhibition of apoptosis, inflammation and protein aggregation – promoting cell survival and neuroprotection. Either directly or indirectly, exenatide also induces facilitation and restoration of insulin signalling. AKT, Protein kinase B; Bcl-2, B-cell lymphoma 2; BAD, (Bcl-2) antagonist of death; Bcl-XL, B-cell lymphoma 2 extra-large; cAMP, Cyclic adenosine monophosphate; CREB, cAMP response element-binding protein; FoxO1/O3, Forkhead box O1/O3; GLP-1, Glucagon like peptide-1; GSK3B, Glycogen synthase 3 beta; IRS-1, Insulin receptor signalling substrate 1; MAPK, Mitogen associated protein kinase; mTOR, mechanistic target of rapamycin; NF-kB, nuclear factor kappa-light-chain-enhancer of activated B cells; PI3-K, Phosphoinositide 3-kinase; PKA, Protein kinase A; Tyr, Tyrosine residue.
WHAT FURTHER TRIAL EVIDENCE IS NEEDED?
To provide irrefutable evidence that exenatide confers a therapeutic advantage to patients with PD requires a larger multi-site replication study. Given that most patients do not spend the majority of their time in the OFF-medication state, identifying a meaningful advantage will require a comparison of the severity of PD symptoms during optimal dopaminergic replacement therapy. This will require long term follow up to evaluate the emergence of dopa refractory motor and non-motor symptoms. This type of study is called a Long term simple design and is expensive to perform because of its long duration and the need to include large numbers of patients to compensate for potential dropout of some trial participants. Inclusion of an additional “OFF medication” assessment also adds to the complexity and the expense. However, given that all patients will receive optimal dopaminergic replacement therapy, dropout will hopefully be minimized. This type of study will answer the fundamental question whether PD patients are indeed advantaged in terms of their quality of life and functional ability when given exenatide, but would not necessarily reveal an underlying mechanism of action.
From the scientific perspective, the other priority is to perform a trial specifically designed to distinguish whether any effects of exenatide on PD are mediated through a symptomatic or disease-modifying action. One suggestion has been to use a Randomized Withdrawal design. In this type of study, patients are randomized to either placebo or active disease groups at baseline, then after a 6 month period, half of the active treatment group switch to placebo. After a further 6 months period, any difference between the 2 groups on placebo predominantly represents the disease modifying effect resulting from 6 months treatment exposure, while the difference between those still on active treatment and those that switched to placebo represents predominantly the symptomatic effect. This type of (relatively short term) study should build on what we already know by recruiting de novo patients (i.e., naive to dopaminergic drugs) and therefore be able to judge more sensitively any symptomatic or disease-modifying effects without the noise introduced by dopaminergic treatment. Given the relative rarity of de novo patients, this type of trial will still require multiple recruiting centres to ensure recruitment in a timely fashion.
WHAT IMPLICATIONS DO THESE RESULTS HAVE FOR PwP NOW?
The effects observed in the Exenatide-PD trial, although unlikely, might conceivably still be explicable by a 1 in 20 chance finding. Therefore, it is urgent to perform a replication trial that either supports or refutes the current findings. The data from the Exenatide-PD trial are arguably consistent with either a symptomatic effect and/or a disease modifying effect. If the effect turns out to be entirely symptomatic, then it is unique in that it persists 12 weeks after termination of the drug therapy. While relatively well tolerated, exenatide causes weight loss and gastrointestinal upset. It also has the potential to interfere with L-dopa absorption from the GI tract. As such it is easily argued that it is premature to recommend that patients with PD seek prescription of this drug. This aside, it may be that in a few years time, definitive proof emerges that exenatide has a beneficial disease modifying action. Patients might therefore feel that the risk of not immediately taking this drug (and potentially suffering from deterioration in PD) outweighs the risk of taking it. We would argue that currently the relative pros and cons of starting exenatide requires individual patient discussions that must include an evaluation of their existing symptom control and its rate of progression, their weight, their comorbidity (particularly gastrointestinal symptoms) and the availability of appropriately skilled and motivated team to support this course of action, as well as the availability of properly regulated randomized trials of this agent.
WHAT IMPLICATIONS DOES THIS HAVE FOR PD SCIENTIFIC COMMUNITY?
The findings of this study underscore the importance that the relationship between GLP-1 receptor stimulation and neurodegeneration is studied in more detail. Simple clinical, metabolic and genetic observations of patients with PD compared with age matched controls patients with other neurodegenerative disorders can help shed light on the relationship between insulin resistance and neurodegeneration. Data from novel laboratory models such as iPS cell lines or inducible neurons can also confirm mechanisms of action of this class of drug. This will be further illuminated by the imminent analyses of serum, urine and CSF from patients treated with GLP-1 agonists in previous and future interventional trials.
DISCLOSURES
DA is a co-investigator on the Exenatide-PD trial and has no conflicts of interest. RW has no conflicts of interest. PB has received commercial support as a consultant from Renovo Neural, Inc., Roche, Teva/Lundbeck, AbbVie and Axial Biotherapeutics, Inc. Additionally, he has received commercial support for grants/research from Renovo and Teva/Lundbeck, and has ownership interests in Acousort AB and Parkcell AB. TF has received honoraria from Profile Pharma, BIAL, Abbvie, Genus, Medtronic, and St Jude Medical and is lead investigator on the Exenatide-PD trial. PB consults for NeuroDerm and Cellular Dynamics International.
REFERENCES
[1] | Hölscher C ((2014) ) Central effects of GLP-1: New opportunitiesfor treatments of neurodegenerative diseases. JEndocrinol, 221: , T31–41. |
[2] | Aviles-Olmos I , Dickson J , Kefalopoulou Z , Djamshidian A , Ell P , Soderlund T , Whitton P , Wyse R , Isaacs T , Lees A , Limousin P , & Foltynie T ((2013) ) Exenatide and the treatment of patients with Parkinson’s disease. J Clin Invest, 123: , 2730–2736. |
[3] | Aviles-Olmos I , Dickson J , Kefalopoulou Z , Djamshidian A , Kahan J , Fmedsci PE , Whitton P , Wyse R , Isaacs T , Lees A , Limousin P , & Foltynie T ((2014) ) Motor and cognitive advantages persist 12 months after Exenatide exposure in Parkinson’s disease. J Parkinsons Dis, 4: , 337–344. |
[4] | Athauda D , Maclagan K , Skene SS , Bajwa-joseph M , Letchford D , Chowdhury K , Hibbert S , Budnik N , Zampedri L , Dickson J , Li Y , Aviles-olmos I , Warner TT , Limousin P , Lees AJ , Greig NH , & Tebbs S ((2017) ) A randomised, double-blind, placebo controlled trial of Exenatide once-weekly in Parkinson’s disease. Lancet, in press. |
[5] | Kalia LV , Kalia SK , & Lang AE ((2015) ) Disease-modifying strategiesfor Parkinson’s disease. Mov Disord, 30: , 1442–1450. |
[6] | Athauda D , & Foltynie T ((2014) ) The ongoing pursuit of neuroprotective therapies in Parkinson disease. Nat Rev Neurol, 11: , 25–40. |
[7] | Kordower JH , Olanow CW , Dodiya HB , Chu Y , Beach TG , Adler CH , Halliday GM , & Bartus RT ((2013) ) Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain, 136: , 2419–2431. |
[8] | Noyce AJ , Nagy A , Acharya S , Hadavi S , Bestwick JP , Fearnley J , Lees AJ , & Giovannoni G ((2014) ) Bradykinesia-akinesia incoordination test: Validating an online keyboard test of upper limb function. PLoS One, 9: , e96260. |
[9] | Kas A , Payoux P , Habert M-O , Malek Z , Cointepas Y , Fakhri G El , Chaumet-Riffaud P , Itti E , & Remy P ((2007) ) Validation of a standardized normalization template for statistical parametric mapping analysis of 123 I-FP-CIT images. J Nucl Med, 48: , 1459–1467. |
[10] | Athauda D , & Foltynie T ((2016) ) Insulin resistance and Parkinson’s disease: A new target for disease modification? Prog Neurobiol, 145: , 98–120. |
[11] | Baggio LL , & Drucker DJ ((2007) ) Biology of incretins: GLP-1 and GIP. Gastroenterology, 132: , 2131–2157. |
[12] | Goldin M , & Segal M ((2003) ) Protein kinase C and ERK involvement in dendritic spine plasticity in cultured rodent hippocampal neurons. Eur J Neurosci, 17: , 2529–2539. |
[13] | Thomas GM , & Huganir RL ((2004) ) MAPK cascade signalling andsynaptic plasticity. Nat Rev Neurosci, 5: , 173–183. |
[14] | Harkavyi A , Abuirmeileh A , Lever R , Kingsbury AE , Biggs CS , & Whitton PS ((2008) ) Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson’s disease. J Neuroinflammation, 5: , 19. |
[15] | Ventorp F , Bay-Richter C , Nagendra AS , Janelidze S , Matsson VS , Lipton J , Nordström U , Westrin Åsa , Brundin P , &Brundin L ((2017) ) Exendin-4 treatment improves LPS-induceddepressive-like behavior without affecting pro-inflammatorycytokines. J Parkinsons Dis, 7: , 263–273. |
[16] | Kim KS , Park DH , Wessel TC , Song B , Wagner JA , & Joh TH ((1993) ) Adual role for the cAMP-dependent protein kinase in tyrosinehydroxylase gene expression. Proc Natl Acad Sci U S A, 90: , 3471–3475. |


